
Abstract
Background:
Incurable hereditary diseases such as Duchenne muscular dystrophy (DMD), Huntington’s disease (HD), and myotonic dystrophy type 1 (DM1) fall into the nucleotide repeat expansion disorder (NRED) category. The discovery of CRISPR-Cas genome editing has paved the way toward hopeful strategies for accurate DNA-level repair. This systematic review presents preclinical data on the efficacy, molecular effects, and limitations of CRISPR-based treatments for NREDs.
Methods:
As per Preferred Reporting Items for Systematic Reviews and Meta-Analyses 2020 guidelines, systematic PubMed, Scopus, and Embase searches up to June 2025 identified studies that evaluated CRISPR-Cas systems in human-derived in vitro models of NREDs. Methodological Index for Non-Randomized Studies tool was used to score eligible studies by methodological quality. CRISPR platforms, delivery systems, gene targets, molecular endpoints, and functional rescue data were extracted and synthesized descriptively.
Results:
Twenty-four out of 6510 records screened were included. They employed most of them to target specific DMD (n = 9), HD (n = 6), and DM1 (n = 3) with patient-derived induced pluripotent stem cells or differentiated myogenic/neuronal cells. Streptococcus pyogenes CRISPR-associated protein 9 as a nuclease was the most frequently used, although engineered Cas9 enzymes and dCas9 fusion proteins were also utilized to control transcription. Delivery was achieved through viral vectors (adeno-associated virus, lentivirus) and nonviral routes (plasmid, lipofection, electroporation). Uniform genomic editing, transcript rescue, and protein restoration were seen in CRISPR-mediated editing studies, and functional restoration was demonstrated for splicing correction and dystrophin restoration. Methodological flaws such as the absence of blinding, failure to follow up, and lack of full reporting of off-target effects limited robustness.
Conclusion:
CRISPR-Cas systems exhibit reproducible molecular and functional correction in NRED models with their translational potential. Methodological strength, whole safety profiling, and in vivo verification remain a necessity, however, before clinical translation.
Keywords
Introduction
Nucleotide repeat expansion disorders (NREDs) are a phenotypically and genetically heterogeneous collection of hereditary diseases caused by pathological expansion of simple deoxyribonucleic acid (DNA) repeats, leading to progressive neurodegeneration and progressive disability (Bhuiyan, 2024). The most important NREDs are Huntington’s disease (HD), myotonic dystrophy type 1 (DM1), spinal and bulbar muscular atrophy, and fragile X syndrome (FXS), among several others. Pathogenic processes of NREDs involve toxic ribonucleic acid (RNA) and protein aggregate production, which cause disarray in cellular function and lead to disease progression (Atienzar-Aroca et al., 2024; Malik et al., 2021).
The traditional therapeutic methods have been symptomatic, aiming to treat disease manifestation instead of finding and fixing the causative underlying genetic issue. The development of CRISPR-Cas9 gene-editing technology has introduced new possibilities for curative treatment owing to the capability of making targeted alterations at the DNA level. CRISPR-Cas9 allows efficacious editing of specific genomic sequences and the elimination or fixing of the expanded repeats responsible for NREDs (Babačić et al., 2019).
Recent preclinical studies have demonstrated that repeat expansion can be intervened upon employing CRISPR-based techniques. In HD models, for instance, CRISPR-Cas9-directed silencing of the mutant huntingtin allele normalized striatal neurotoxicity. Base editing strategies have also been employed to reduce trinucleotide repeat expansions in patient cells and animal models of HD and Friedreich’s ataxia and, in turn, lower somatic repeat instability (Matuszek et al., 2025). Although such promising preclinical results exist, clinical use of CRISPR-based therapies is afflicted by several challenges. Some of the key challenges are to confirm the specificity and efficiency of gene editing, minimize off-target effects, and develop efficient delivery carriers to penetrate the central nervous system. Overcoming these challenges is also required to efficiently use CRISPR-based therapies to treat NREDs (Babačić et al., 2019; Matuszek et al., 2025).
This systematic review aims to critically review the current state of the art concerning CRISPR-transferred gene-editing platforms for NREDs, synthesizing evidence from preclinical and clinical studies to establish their potential as a treatment and what remains to be investigated.
Methods
Protocol approval and registration
This systematic review was designed in accordance with the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) 2020 guidelines (Supplementary Table S1) (PRISMA, 2020). The protocol for this study was registered in the International Prospective Register of Systematic Reviews (PROSPERO) under the registration number CRD420251119476. As this investigation involved secondary analysis of previously published data, ethical approval and informed consent were not necessary.
Search strategy
A comprehensive literature search was performed across three major electronic databases: PubMed, Scopus, and Embase. The search included all publications available up to June 22, 2025 (Supplementary Table S2). The strategy combined MeSH terms and free-text keywords relating to CRISPR-Cas gene editing and the target neuromuscular or NREDs. Keywords included: “CRISPR,” “CRISPR-Cas,” “gene editing,” “genome editing,” “Duchenne Muscular Dystrophy,” “Becker Muscular Dystrophy,” “Huntington’s Disease,” “Myotonic Dystrophy,” “Fragile X Syndrome,” “Spinocerebellar Ataxia,” “Friedreich’s Ataxia,” “Facioscapulohumeral Dystrophy,” “Limb-Girdle Muscular Dystrophy,” and “Congenital Muscular Dystrophy.” Boolean operators were applied to ensure a sensitive and comprehensive search. In addition, the reference lists of all eligible studies and related systematic reviews were manually screened to identify additional relevant publications.
Study selection criteria
Studies were considered eligible if they were peer-reviewed original research articles, including randomized controlled trials, cohort studies, case–control studies, or cross-sectional studies, that explicitly described their experimental design. Only in vitro or ex vivo studies utilizing human-derived cells or tissues were included, and the use of CRISPR-Cas as the primary gene-editing method was required. Eligible studies had to focus on at least one of the neuromuscular or NREDs as mentioned above. Studies were excluded if they involved animal models or animal-derived cells; were reviews, editorials, commentaries, conference abstracts, or notes; did not use CRISPR-Cas as the main editing method; did not focus on the listed target diseases; or lacked a clearly defined experimental design, sufficient extractable data, or were not in English. The detailed Patient/Population, Intervention, Comparison, Outcome (PICO) framework used for study selection is provided in Supplementary Table S3.
Screening
All records retrieved from the database search were imported into Rayyan, a web-based software for systematic review management (Rayyan, n.d). Two reviewers (S.H. and A.H.S.) independently screened titles and abstracts to identify potentially eligible studies. Full-text screening of selected articles was then performed independently by the same reviewers. Any disagreements regarding inclusion were resolved through discussion, and a third reviewer (H.I.T.) was consulted when necessary to reach consensus.
Data extraction and risk of bias assessment
Data extraction was performed independently by two researchers (K.M.A. and Z.H.A.) using a predesigned standardized form. Extracted information included: study title, authors’ names, year of publication, country, journal, study design, disease model investigated, source of human-derived cells or tissues, specific CRISPR-Cas system applied, target gene(s) or mutation (s), editing strategy, delivery method, experimental outcomes, and key findings. The risk of bias was assessed independently using appropriate tools according to the study design. The Cochrane Risk of Bias 2.0 tool was applied for randomized controlled trials, while the Methodological Index for Non-Randomized Studies (MINORS) was used for observational studies (Cochrane Methods, 2017; Dent, 2003). Any disagreements during data extraction or bias assessment were resolved through discussion or consultation with a third reviewer (H.I.T.). Studies deemed to be at high or critical risk of bias were excluded from the final synthesis to maintain methodological rigor.
Outcome measures
The primary outcomes of interest were the efficacy and precision of CRISPR-Cas-mediated gene editing, assessed through measures such as successful gene correction, deletion, or disruption of pathogenic mutations, restoration of normal gene or protein expression, and phenotypic rescue at the cellular level. Secondary outcomes included the efficiency of delivery methods, off-target effects, editing fidelity, and safety-related findings, as well as functional improvements in human-derived cells or tissues relevant to disease pathology. Where applicable, subgroup analyses were planned based on the type of CRISPR system employed (e.g., Cas9, Cas12, base editors, prime editors), disease subtype [e.g., Duchenne muscular dystrophy (DMD) vs. HD vs. FXS], and the nature of the human-derived model used (primary patient-derived cells vs. induced pluripotent stem cell–derived models).
Results
A total of 6510 records were identified through database searches including PubMed, Scopus, and EMBASE. After removing 2615 duplicates, 3895 records were screened by title and abstract. Of these, 3844 records were excluded due to irrelevance to the research question. The remaining 51 full-text articles were assessed for eligibility. Following full-text review, 27 studies were excluded for the following reasons: conference abstracts missing full text and irrelevant outcomes. Finally, 24 studies met the inclusion criteria and were included in the systematic review (Fig. 1).
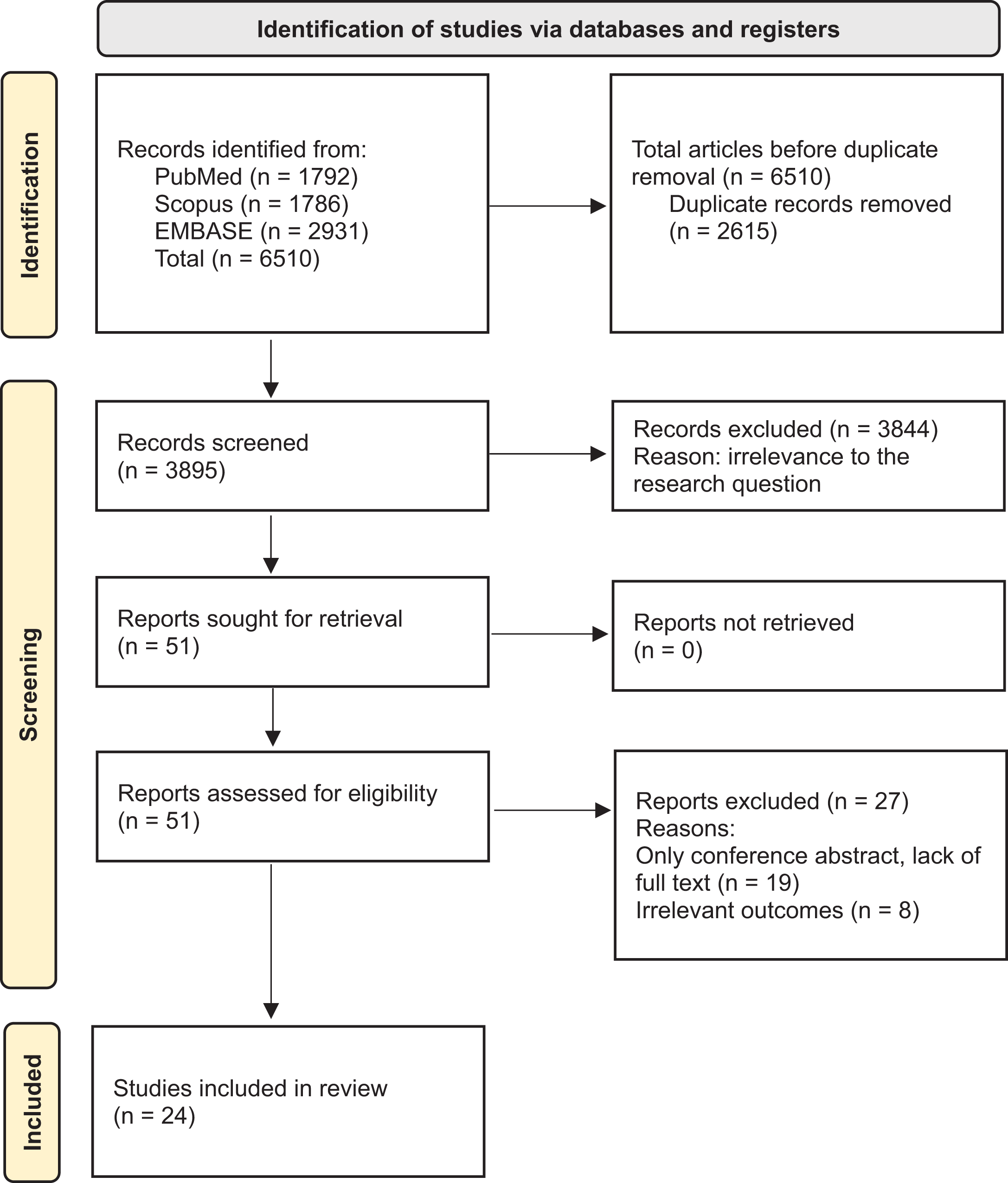
Preferred Reporting Items for Systematic Reviews and Meta-Analyses flow diagram of study identification and screening.
General characteristics of included studies
A total of 24 in vitro experimental studies investigating CRISPR/Cas-based genome editing for neuromuscular and neurodegenerative disorders were included in this systematic review (Table 1). These studies span a broad geographical distribution, with contributions from the United States (n = 10), Poland (n = 3), Japan (n = 2), China (n = 2), and one each from France, Italy, Spain, and Turkey. The most commonly targeted diseases included DMD (n = 9), HD (n = 6), and DM1 (n = 3), reflecting the prioritization of monogenic disorders with clearly defined mutational hotspots for CRISPR correction. Other diseases addressed were limb-girdle muscular dystrophy, facioscapulohumeral muscular dystrophy, dysferlinopathy, FXS, and Becker muscular dystrophy.
Characteristics of Included Clustered Regularly Interspaced Short Palindromic Repeats-Based Gene Editing Studies (n = 24)
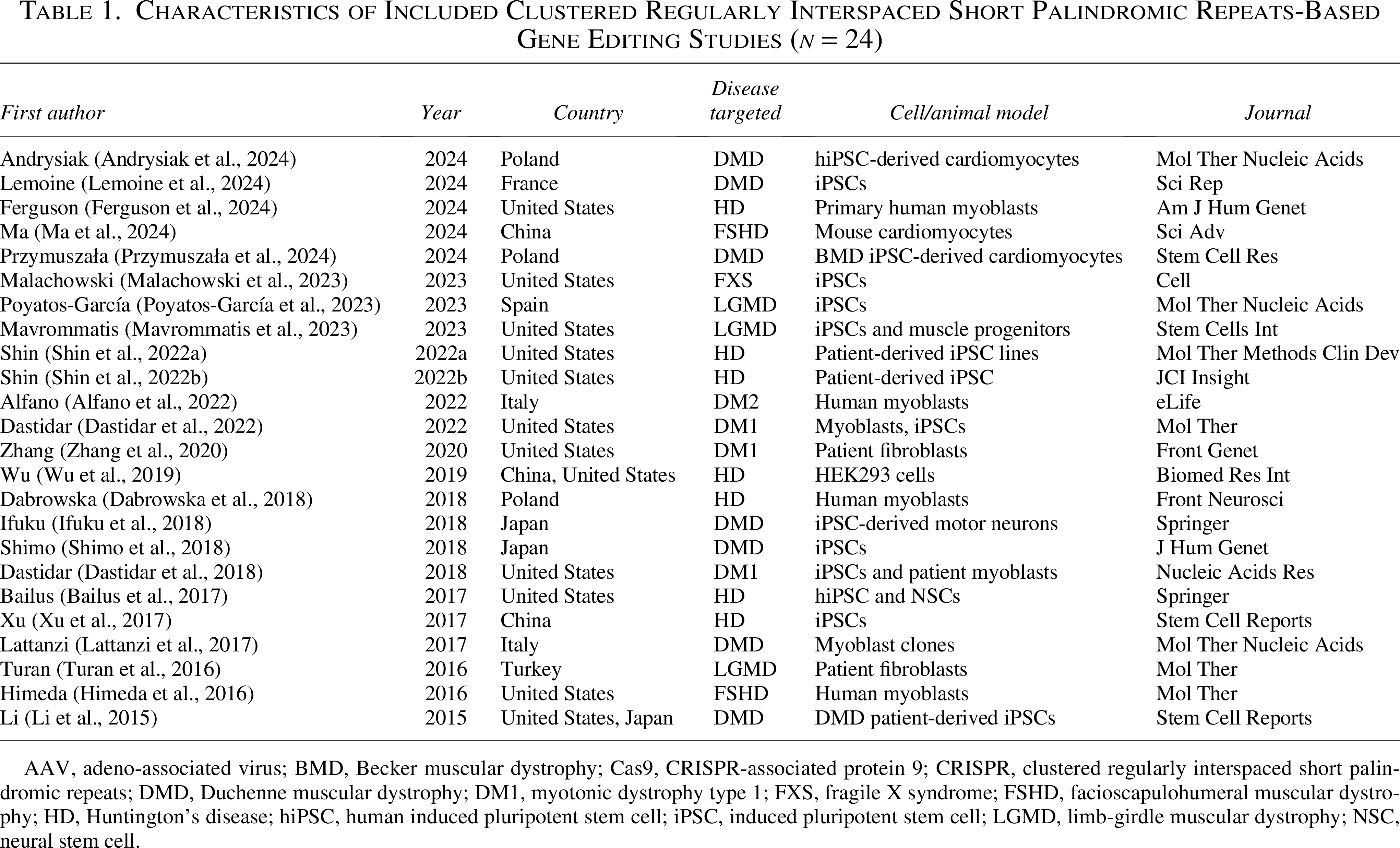
AAV, adeno-associated virus; BMD, Becker muscular dystrophy; Cas9, CRISPR-associated protein 9; CRISPR, clustered regularly interspaced short palindromic repeats; DMD, Duchenne muscular dystrophy; DM1, myotonic dystrophy type 1; FXS, fragile X syndrome; FSHD, facioscapulohumeral muscular dystrophy; HD, Huntington’s disease; hiPSC, human induced pluripotent stem cell; iPSC, induced pluripotent stem cell; LGMD, limb-girdle muscular dystrophy; NSC, neural stem cell.
All studies were performed using in vitro models, employing either induced pluripotent stem cells (iPSCs) derived from patients or primary cell lines, including myoblasts, fibroblasts, motor neurons, and cardiomyocytes. Notably, human induced pluripotent stem cell-derived cardiomyocytes and muscle-specific progenitors were frequently utilized in DMD models, providing disease-relevant readouts for dystrophin restoration. Two studies incorporated dual models, combining iPSCs with primary differentiated derivatives to evaluate editing outcomes at both transcriptional and protein levels.
All included studies adopted a preclinical in vitro experimental design, focusing on proof-of-concept genome editing, transcriptomic rescue, or epigenetic modulation. Delivery systems primarily consisted of plasmid transfection, electroporation, lipid-based delivery, or adeno-associated virus (AAV)-mediated transduction, aligning with clinically translatable approaches. The selected studies were published in reputable peer-reviewed journals including Molecular Therapy, Cell Stem Cell, Stem Cell Reports, Nature Communications, and Human Gene Therapy, ensuring methodological rigor and scientific impact.
This curated dataset provides a comprehensive cross-sectional view of global CRISPR research efforts focused on monogenic neuromuscular and neurodevelopmental disorders, revealing consistent use of iPSC-based platforms and diverse targeting strategies across disease models.
Risk of bias assessment
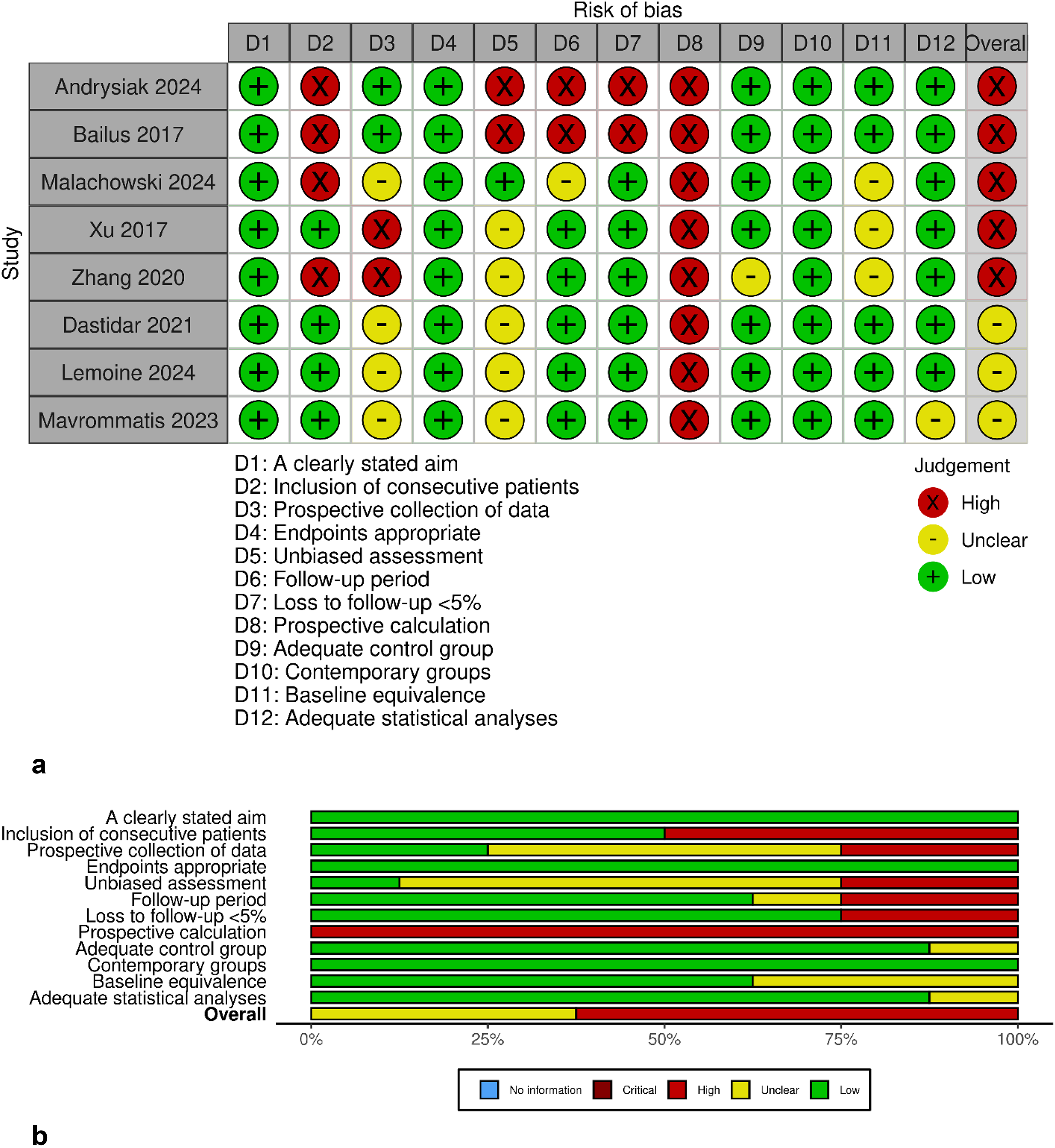
The MINORS tool was applied to assess the methodological quality of the included non-randomized comparative studies (n = 8) (Fig. 2a and b). Most studies had clearly defined aims and selected appropriate endpoints, with adequate control groups reported. However, multiple studies demonstrated a high risk of bias, particularly in domains related to blinding of outcome assessment, short or undefined follow-up periods, and absence of prospective sample size calculation. Loss to follow-up was often unreported, and baseline equivalence between groups was not always confirmed. Based on these findings, six out of eight studies were rated as having a high overall risk of bias, highlighting common methodological gaps in current CRISPR-Cas gene editing research.
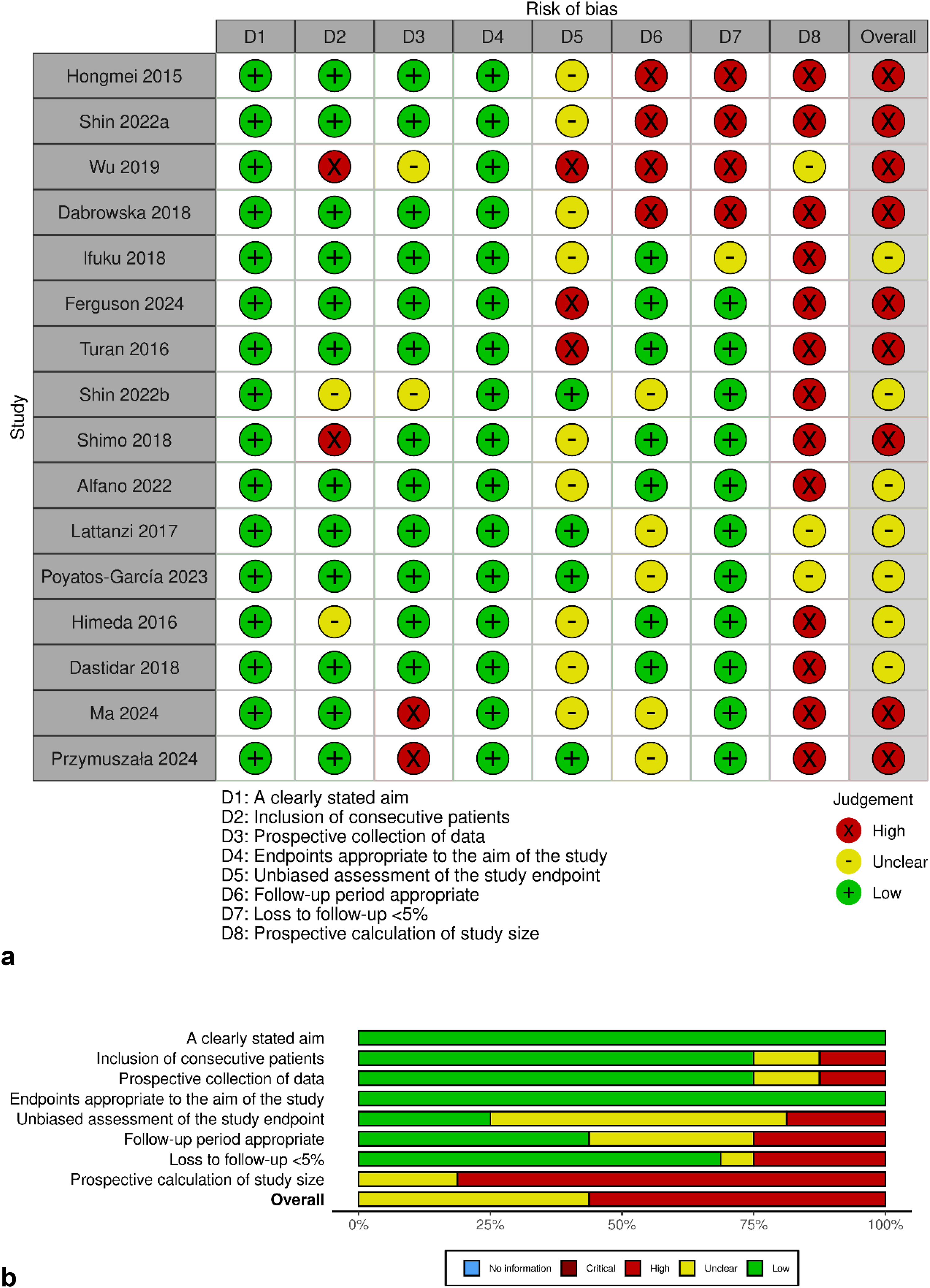
Risk of bias was assessed using the MINORS tool, specifically for non-comparative, non-randomized studies (n = 16) (Fig. 3a and b). Most studies clearly stated their objectives and selected endpoints appropriate to their aims. However, important methodological shortcomings were consistently observed. These included lack of blinding, unclear or absent follow-up periods, no information on loss to follow-up, and absence of prospective sample size calculation. Due to these limitations, 13 out of 16 studies were judged to have a high overall risk of bias.
CRISPR platforms, targeted genes, and delivery methods
The 24 studies included in this review demonstrated considerable heterogeneity in their choice of CRISPR systems, gene targets, and delivery strategies, reflecting the evolving technical landscape of genome editing in neuromuscular and neurodevelopmental research. Across these studies, DMD was the most frequently targeted gene (n = 9), followed by HTT (Huntingtin gene; n = 5). Other targets included DYSF, UTRN, SGCA, FMR1, DMPK, CAPN3, CHCHD2, DUX4, TNPO3, and CNBP, each representing critical loci implicated in monogenic muscle or neurodegenerative disorders (Table 2).
Targeted Genes, Clustered Regularly Interspaced Short Palindromic Repeats Platforms, and Delivery Methods

AAV6, adeno-associated virus serotype 6; AAV9, adeno-associated virus serotype 9; CAPN3, calpain-3; Cas9n, Cas9 nickase; CHCHD2, coiled-coil-helix-coiled-coil-helix domain containing 2; CNBP, cellular nucleic acid-binding protein; DMPK, dystrophia myotonica protein kinase; DUX4, double homeobox 4; DYSF, dysferlin; electroporation, high-voltage mediated cell transfection method; FMR1, fragile X mental retardation 1; GFP, green fluorescent protein; HTT, Huntingtin; Lentivirus, integrating retroviral vector used for stable transduction; Lipofection, lipid-based transfection technique; MBNL1, muscleblind-like splicing regulator 1; PEI, polyethylenimine; PX458, SpCas9-GFP expression plasmid; SSO, splice-switching oligonucleotide; SpCas9, Streptococcus pyogenes Cas9; TALEN, transcription activator-like effector nuclease; TNPO3, transportin 3; UTRN, utrophin; VP64, Four tandem copies of VP16 activation domain.
The choice of the CRISPR platform was closely aligned with the editing strategy and the nature of the intended correction. The wild-type Cas9 endonuclease from Streptococcus pyogenes (SpCas9) was employed in 20 studies, underscoring its continued dominance as a precise and efficient DNA-cutting tool. Several studies also utilized Cas9 nickase (Cas9n) or nuclease-dead Cas9 (dCas9) fused to effector domains such as VP64 (for transcriptional activation) or MBNL1 (for splicing rescue). Notably, Bailus et al. (2017) and Zhang et al. (2020) leveraged dCas9 fusions for gene regulation without introducing double-strand breaks, highlighting the growing use of CRISPR interference and epigenetic modulation in preclinical models.
Delivery methods varied across studies and were tailored to the CRISPR construct and cell type. Lentiviral vectors were used in six studies to ensure stable expression in myoblasts or iPSCs, particularly when long-term assays or clonal analysis were required. AAV vectors, particularly AAV6 and AAV9, were frequently used for transient expression in post-mitotic cells and for disease-relevant delivery into muscle or cardiac models. Nonviral methods such as plasmid transfection, lipofection, nucleofection, and electroporation were widely used for transient expression, offering simplicity and low immunogenicity. Several studies combined these approaches (e.g., electroporation + AAV or CRISPR + SSO) to optimize delivery efficiency and targeting precision.
Overall, the distribution of target genes, CRISPR platforms, and delivery modalities across these studies reflects a detailed optimization of genome editing tools tailored to the biological and clinical nuances of each disease model.
Comprehensive molecular outcomes and functional rescues by CRISPR gene editing
Each study employed a multimodal approach to confirm successful genome editing, integrating molecular analyses across DNA, RNA, and protein levels. Validation techniques included genomic polymerase chain reaction (PCR), Sanger sequencing, reverse-transcription quantitative PCR, Western blotting, and immunocytochemistry, enabling comprehensive confirmation of editing efficiency and biological impact (Table 3).
Comprehensive Molecular Outcomes and Functional Rescues Across All 24 Clustered Regularly Interspaced Short Palindromic Repeats Gene Editing Studies

ATP, adenosine triphosphate; CRISPRa, CRISPR activation; dCas9, nuclease-dead Cas9; dCas9-VP64, dCas9 fused to VP64 activator domain; gRNA, guide RNA; HDR, homology-directed repair; qRT-PCR, quantitative reverse transcriptase PCR; RNA, ribonucleic acid; sgRNA, single guide RNA; WT, wild-type.
Most studies (n = 20) confirmed editing at all three levels: genomic DNA alteration, transcriptomic changes, and protein modulation, while four studies assessed RNA and protein levels without direct DNA confirmation. For example, Li et al. (2015), Ferguson et al. (2024), Wu et al. (2019), and Przymuszała et al. (2024) demonstrated clear restoration of dystrophin and HTT protein, respectively, following precise genomic editing confirmed by sequencing. Dabrowska et al. (2018) (Turan et al., 2016) observed protein-level reduction of mutant HTT despite unchanged messenger RNA (mRNA), supporting post-transcriptional mechanisms of regulation. Several studies such as Shimo et al. (2018), Dastidar et al. (2018), and Turan et al. (2016) demonstrated rescue of functional phenotypes, including splicing correction, dystrophin restoration, and foci resolution, indicating successful translation of gene editing to physiological recovery.
Expression outcomes were generally favorable. Many studies reported upregulation of therapeutic transcripts (e.g., Andrysiak et al., 2024: 4× increase in utrophin mRNA) or downregulation of toxic alleles (e.g., Shin et al., 2022: significant HTT mRNA knockdown using a 10-gRNA array). Protein-level rescue was explicitly reported in 17 studies, with several showing full-length or near-wild-type expression (e.g., Mavrommatis et al., 2023: restoration of full-length CAPN3 protein; Ifuku et al., 2018: dystrophin expression in motor neurons).
In terms of editing strategy, the number and design of guide RNAs (gRNAs) varied depending on the target region and complexity of the mutation. The majority of studies used 1–2 gRNAs, sufficient for inducing deletions or frameshift corrections. However, multiplexing strategies using up to 10 gRNAs (as in Shin et al., 2022 and Wu et al., 2019) were applied for gene silencing, targeting expanded repeats, or disrupting long coding sequences. Precision was further enhanced in some studies by combining CRISPR editing with splicing-modulatory elements or homology-directed repair templates (e.g., Przymuszała et al., 2024).
In summary, the included studies consistently demonstrated not only molecular confirmation of CRISPR activity but also translational restoration of transcript and protein function, reinforcing the therapeutic viability of genome editing approaches in neuromuscular and neurodegenerative disease models.
Discussion
This systematic review of 24 preclinical studies underlines important developments in the use of CRISPR/Cas-mediated genome editing for neuromuscular and neurodegenerative diseases, with specific advancement observed in DMD, HD, and DM1. Across the studies reviewed, there was consistent success in targeting specific genes, correcting molecular defects at both RNA and protein levels, and developing clinically relevant cell models. While wild-type SpCas9 remains the most widely used editing tool, newer applications using modified Cas9 proteins for gene regulation highlight the growing potential of CRISPR-based therapies (Gillmore et al., 2021; Tabebordbar et al., 2016).
These results are consistent with key studies like Nelson et al. (2016), who used CRISPR to remove mutation-bearing exons and achieved durable dystrophin expression in X-linked muscular dystrophy (mdx) mice and patient-derived myotubes. The sustained gene expression and functional improvement observed highlight the therapeutic potential of CRISPR. Similar outcomes across most DMD studies in this review further support its promise in treating monogenic diseases (Nelson et al., 2016; Tabebordbar et al., 2016). Moreover, the practical translation of these findings is supported by advanced delivery approaches, including AAV serotypes (notably AAV6 and AAV9) and lentiviral vectors employed in several included studies, paralleling strategies associated with clinical-stage gene editing research (Gillmore et al., 2021).
For neurodegenerative diseases such as HD, Xu et al. in 2021 demonstrated that CRISPR targeting of the mutant HTT gene significantly reduced abnormal transcripts and protein levels in patient-derived iPSCs, along with reversal of disease-related cellular phenotypes. Similar findings across several HD and DM1 studies in our review highlight the promise and specificity of allele-selective editing in polyglutamine expansion disorders (Shin et al., 2016; Xu et al., 2021). Notably, approaches using multiple gRNAs and finely tuned nuclease strategies show that careful design of the CRISPR system is crucial, not only for limiting off-target effects but also for achieving meaningful therapeutic benefit (Shin et al., 2016).
Despite the progress, consistent issues, such as lack of blinding, absence of sample size calculations, and limited follow-up, continue to introduce bias, as reflected in elevated MINORS scores. These challenges mirror a broader pattern in genome editing research, where many early-phase studies fall short in design quality, reproducibility, and long-term evaluation (Schwank et al., 2013). Recent studies are making progress by validating findings across DNA, RNA, and protein levels and by focusing on outcomes that are more relevant to clinical practice. This reflects a growing shift in the field toward more translational, patient-focused research (Gillmore et al., 2021).
In conclusion, this systematic review highlights that CRISPR/Cas-based genome editing consistently achieves both molecular correction and functional improvement across in vitro models of neuromuscular and neurodegenerative diseases. However, ongoing shortcomings in methodological rigor and inconsistent reporting limit the strength of clinical translation. Overall, the precision, adaptability, and rapid advancement of this technology underscore its significant therapeutic potential. Moving forward, progress will depend on stronger study designs, reduction of off-target effects, and vigorous in vivo validation.
Conclusion
This systematic review highlights the significant progress achieved by CRISPR-based genome editing in correcting pathogenic mutations underlying NREDs. Preclinical studies consistently demonstrated restoration of normal gene expression and partial functional recovery, particularly in DMD, HD, and DM1 models. Despite these advances, the evidence base is constrained by methodological biases, short follow-up durations, and limited exploration of off-target effects. As the field advances, rigorous study designs, scalable delivery strategies, and translational in vivo validation will be crucial. CRISPR holds substantial therapeutic potential, but its clinical application for NREDs requires careful optimization to ensure both efficacy and safety.
Authors’ Contributions
All authors made significant contributions to this research in the form of study design, acquisition of information, drafting, revising, and critically reviewing the article.
Footnotes
Acknowledgment
The authors would like to thank Dr. F.E.H. for her valuable time and contribution.
Consent for Publication
All authors approve the publication of the final version of the article.
Data Availability
Data sharing is not applicable to this article, as no new data were created or analyzed in this study.
Author Disclosure Statement
No conflicts of interest, financial or otherwise, is declared by the authors.
Funding Information
No funding was received for this article.
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.