
Abstract
Introduction:
Optimal cryopreservation of testicular tissue is essential for species research and conservation, enabling long-term storage of genetic resources through vitrification or slow freezing. Comparing responses from different taxonomic groups to these techniques is crucial for refining protocols and improving cryopreservation outcomes.
Objectives:
This study evaluated the effects of cryopreservation on cell viability, morphology, mitochondrial activity, and proliferative potential to optimize testicular tissue preservation strategies for wildlife conservation.
Methods:
We assessed testicular tissue from Felidae and Cervidae, including domestic cats (Felis catus) and Neotropical deer species, white-tailed deer (Odocoileus virginianus), Brazilian red brocket deer (Mazama nana), and gray brocket deer (Subulo gouazoubira). Experimental groups included control (no cryopreservation), slow freezing using Mr. Frosty or progressive temperature decrease, and conventional vitrification.
Results:
All methods preserved live cells and normal tissue morphology; however, compared with fresh tissue, cryopreservation significantly reduced tissue viability, mitochondrial membrane potential, and the proportion of intact seminiferous tubules. Species-specific differences emerged, with vitrification being most effective for domestic cats, while slow freezing yielded better results for Neotropical deer. Despite lower viability scores, vitrification could still be an acceptable option for cervids due to its rapid processing and minimal equipment requirements. In addition, post-cryopreservation tissue culture increased cell abnormalities, highlighting the need to optimize culture conditions for different species.
Conclusion:
This comparative study advances reproductive tissue preservation techniques and emphasizes the importance of tailored cryopreservation as well as in vitro culture protocols for diverse taxonomic groups.
Introduction
Cryobanking of testicular tissue plays a crucial role in biodiversity preservation and genetic resource accessibility.1–2 Standardized cryopreservation protocols, often developed using model species, retain genetic and cytogenetic information and store live germ cells at various developmental stages within the preserved tissue.3–6 These tissues enable cytogenetic studies and sperm production through transplantation or in vitro gametogenesis.7–8 This is particularly valuable for species with taxonomic uncertainties, such as Neotropical deer, in which cryopreserved tissues aid in karyotypic characterization and species delimitation.9–10
Tissue cryopreservation can be performed using two main methods, slow freezing and vitrification. In slow freezing, tissue fragments are equilibrated in a low concentration of cryoprotectants. 11 The freezing process typically follows one of two protocols: (1) gradual freezing at a rate of 1°C/min to reach −80°C before plunging into liquid nitrogen, or (2) cooling tissues to 4°C over a few hours, followed by exposure to liquid nitrogen vapor (under 30 minutes) before final immersion.1,10–12 Slow freezing is widely used in biobanking due to its requirement for lower concentrations of cryoprotectants, thereby reducing cytotoxicity. 13 However, slow freezing often leads to ice crystal formation, causing cellular damage. 14 This limitation can be mitigated by vitrification, in which tissues are exposed to higher concentrations of multiple cryoprotectants and then rapidly plunged into liquid nitrogen. 11 This method mitigates ice crystal formation through ultrarapid cooling.2,11–13 Vitrification also offers several advantages over slow freezing, including greater time efficiency, simplicity, and minimal equipment requirements, making it a more viable option in field settings where quick sampling and processing are necessary. 14 However, the high concentrations of cryoprotectants required for vitrification may exert cytotoxic effects, which is the main disadvantage of this method. 15
Previous studies on gonadal tissues highlight both functional and practical considerations of cryopreservation methods. Thuwanut and Chatdarong 16 found two-step slow freezing to be a simpler and less cytotoxic alternative to vitrification in cats, while Dumont et al. 17 demonstrated that a vitrification protocol with dimethyl sulfoxide (DMSO), ethylene glycol (EG), and sucrose effectively preserved testicular structure and enhanced sperm yield in mice. Similar cryoprotectant combinations improved tissue preservation in cats, 18 and simplified vitrification tools, such as the needle-immersion method for ovarian tissue, 19 supporting the emphasis on accessible cryopreservation techniques in both laboratory and field settings. Despite recent advances in vitrification and slow freezing techniques in domestic species, there is a critical need to characterize tissue cryopreservation in nontraditional species, particularly to understand species-specific responses to different cryopreservation methods.11,20 Most studies have focused on a single species, limiting our understanding of how biological differences influence cryopreservation outcomes. Comparative studies across multiple species and developmental stages are essential to refine protocols and improve the preservation of genetic resources for conservation efforts. To address this gap, our study evaluated tissue viability, cellular response, and replication potential in testicular tissues following slow freezing and conventional vitrification. We compared two families, Felidae and Cervidae, using domestic cat and white-tailed deer as model species due to the availability of tissue collection, non-endangered status, and prior use in reproductive cryopreservation studies.18,21 To further assess interspecific variability in response to cryopreservation within the same family, we examined Brazilian dwarf brocket deer (Mazama nana) and gray brocket deer (Subulo gouazoubira). These comparisons generated crucial data to enhance cryopreservation strategies across diverse species and developmental stages, ultimately aiding wildlife conservation efforts. The limited number of samples analyzed from each species, particularly from deer, highlights the need for broader comparative assessments in future studies to strengthen species-specific applications of cryopreservation protocols.
Materials and Methods
All media and solutions were purchased from Thermo Fisher Scientific, Hudson, NH, USA, and all other chemicals were purchased from Sigma-Aldrich®, St. Louis, MO, USA, unless otherwise indicated.
Tissue collection
Domestic cat (Felis catus) testes from adult (n = 4) and juvenile (n = 4) males were collected from routine orchiectomy at local veterinary hospitals. Age classification was based on clinical records and physical examination, with juveniles defined as younger than 6 months and adults over 1 year old. White-tailed deer (Odocoileus virginianus) testes (n = 2) were obtained from local butchers in Maryland, USA. Because cat and white-tailed deer testes were collected as byproducts from routine procedures, no approval was required from the Animal Care and Use Committee of the Smithsonian’s National Zoo and Conservation Biology Institute. Testes from adult Brazilian dwarf brocket deer (Mazama nana) (n = 3, age 2 to 5 years) and adult gray brocket deer (Subulo gouazoubira) (n = 2, age 3 and 4 years) were obtained from routine orchiectomy at the Deer Research and Conservation Center (NUPECCE) in Brazil. For the brocket deer samples, the study was approved by the Ethics and Animal Welfare Committee of the School of Agricultural and Veterinary Studies, Jaboticabal, SP, Brazil (protocol n°005433/19). After collection, the testes were washed in Dulbecco’s phosphate-buffered saline (DPBS) and transported to the laboratory at 4°C within 24 hours. Testes were washed in dissection solution (Ham’s F10, supplemented with 25 mM HEPES, 1 mM pyruvate, 2 mM
Experimental design
As shown in Figure 1, to evaluate the different cryopreservation techniques on testicular tissue from domestic cats and Neotropical deer species, four treatment groups were established: conventional vitrification, slow freezing using Mr. Frosty, slow progressive freezing, and fresh tissue without cryopreservation (control group). Each testicular tissue sample was dissected into multiple fragments, with eight fragments allocated per treatment group. Within each treatment group, four fragments were analyzed immediately after thawing/warming, while the remaining four were subjected to in vitro culture for 24 hours before analysis. Since all deer tissue samples were from adult individuals, only adult domestic cat samples were included in the main comparative analyses. Accordingly, all data and analyses derived from juvenile cat samples are presented as Supplementary Data.

Schematic of the experimental design. Testicular tissues from domestic cats, white-tailed deer, Brazilian dwarf brocket deer, and gray brocket deer were transported at 4°C, dissected, cryopreserved (vitrification or slow freezing), and allocated to cultured (24 hours at 38.5°C) or non-cultured groups. (*) Mr. Frosty freezing was not used in brocket deer. (•) All cryopreservation protocols were applied to all species.
Conventional vitrification
Testicular tissue fragments were held on a 30-G needle (BD Precision Glide needle, Fischer Scientific, Waltham, MA, USA) 19 and immersed in an equilibrium solution containing 1.4 M DMSO, 1.4 M glycerol, 0.25 M sucrose, and Ham’s F10 for 10 minutes at room temperature (20°C–22°C). Then, tissues were exposed to a vitrification solution containing 2.8 M of each cryoprotectant, 0.50 M sucrose, Ham’s F10, and 10% FBS for 5 minutes at room temperature (20°C–22°C). 22 After placing the tissues on an aseptic absorbent filter to remove excess vitrification solution, they were plunged directly into liquid nitrogen and stored in cryotubes for at least 1 week. 22
Slow freezing using Mr. Frosty
Fragmented testicular tissues from domestic cats and white-tailed deer were placed in 2.0 mL cryovials containing 2.0 mL of slow freezing solution (1.4 M DMSO, 1.4 M glycerol, 0.50 M sucrose, Ham’s F10, and 10% FBS) at room temperature (20°C –22°C). Each cryovial was transferred to a Nalgene freezing container (Mr. Frosty, Thermo Scientific Nalgene, Rochester, NY, USA) filled with isopropyl alcohol at 25°C and kept in a freezer at −80°C for 12 hours. This system usually ensures a cooling rate of about 1°C/min. After this period, samples were plunged into a liquid nitrogen container for a 1-week storage period. 11 This method was not tested on brocket deer tissues due to lack of equipment at NUPECCE.
Slow freezing progressive
Fragmented testicular tissues were placed in 2.0 mL cryovials containing 2.0 mL of slow freezing solution (1.4 M DMSO, 1.4 M glycerol, 0.50 M sucrose, Ham’s F10, and 10% FBS) at room temperature (20°C–22°C). Each cryovial was kept at 4°C for 3 hours, then transferred to a liquid nitrogen vapor system where the samples were kept 2 cm above liquid nitrogen for 30 minutes before being plunged into a liquid nitrogen container for at least 1 week.
Thawing/warming of cryopreserved tissue fragments
Slow-freezing samples were transferred to a container with liquid nitrogen, the cryovials were removed, and they were immersed in a water bath at 37°C for 2 minutes. Vitrified samples were warmed by direct immersion in DPBS at 50°C for 5 seconds, following a protocol adapted from Lima et al. 18 The samples were then immersed in Ham’s F10 and 10% FBS with decreasing concentrations of sucrose (0.5, 0.25, 0.15 M) at 37°C for 5 minutes each, following a protocol adapted from Silva et al. 23
Testicular tissue culture
Fresh or thawed/warmed tissue fragments were placed on 1 cm3 blocks of 1.5% agarose gel, which had been pre-equilibrated overnight in culture medium (Ham’s F10, supplemented with 10% FBS). Two fragments were positioned per gel block, ensuring no contact between them. Each block was placed in a well of a 4-well culture dish containing 0.4 mL culture medium, creating a gas–liquid interface system. The culture dishes were incubated at 38.5°C in a humidified atmosphere with 5% CO2 for 24 hours before analysis.
Cell death and viability assay
Cells were dissociated and isolated from tissue fragments by enzymatic digestion. 24 Briefly, fragments were incubated in Liberase (Sigma-Aldrich®, St. Louis, MO, USA) solution (1:30—Liberase/1 × 3 DPBS) at 38°C for 30 minutes (mixing every 10 minutes). The samples were then added to a cold stop solution (1:9—1 × 3 DPBS/FBS) and centrifuged at 300 × g, for 5 minutes. The supernatant was removed, and the pellets were incubated in a live/dead reagent mixture of 1 µL propidium iodide (Sigma-Aldrich®, St. Louis, MO, USA) (PI, 0.5 mg/mL in DPBS) and 4 µL of Calcein-AM (Invitrogen™, Thermo Fisher Scientific, Waltham, MA, USA) in 500 µL DPBS at 38°C for 30 minutes in a dark room. To visualize DNA,10 µL Hoechst 33342 (1:100, Sigma Aldrich®, St. Louis, MO, USA) was added and incubated for 10 minutes. Samples were transferred to slides and evaluated using an Olympus BX41 epifluorescence microscope to detect viable testicular cells (green, stained with Calcein), dead testicular cells (red, stained with PI), and to count the total number of cells (blue, nuclear stained with Hoechst 33342). For each sample, fluorescent images were captured using Zen 3.6 software and analyzed using ImageJ software (v 1.48, NIH, Bethesda, MD, USA). A total of 1000 Hoechst-stained nuclei were counted per sample, and the proportions of viable (Calcein+) and dead (PI+) cells were calculated and expressed as percentages relative to the total cell count. 25
Mitochondrial activity
The method from Faure et al. 26 was adapted to assess mitochondrial activity. A volume of 5 µL (500 µmol/L) of MitoTracker™ Green (Invitrogen™, Thermo Fisher Scientific, Waltham, MA, USA) was added to each cell suspension, briefly dissociated as described for the viability assay, and incubated for 15 minutes at 37°C. The cells were then examined using standard epifluorescent microscopy. The fluorescent intensity of the mitochondrial staining was quantified through pixel count, using ImageJ. The intensity of fresh samples was used as a reference. Values obtained in each treatment group were divided by the intensity of fresh samples to obtain relative expression levels (arbitrary fluorescence units).
Histomorphology of testicular tissue fragments
Histomorphology of tissue fragments from control and cryopreserved groups were evaluated by hematoxylin and eosin (H&E) staining. The fragments were fixed in Bouin solution overnight at 4°C, then dehydrated in 50% and 70% ethanol before embedding in paraffin blocks. The blocks were sectioned at a thickness of 5 µm, mounted, deparaffinized, rehydrated, and stained with a solution of hematoxylin and eosin. Histological analysis was performed using a light microscopy (Olympus CX 31 RBSFA, Tokyo, Japan) at 200× magnification. For each sample, three randomly selected areas of testicular tissue were analyzed, totaling 30 seminiferous tubules per sample. Evaluation focused on epithelial preservation and included the following criteria: attachment of germ cells to the basal membrane, absence of swelling or shrinkage of the lamina propria, no stromal disruption, and preserved cellular organization. Tubules that exhibited none of these alterations were considered histologically normal. The percentage of normal tubules was calculated relative to the total number evaluated per sample. This methodology was adapted from previously published studies Milazzo et al. 27
Cell proliferative potential in testicular tissue fragments
Using a silver staining technique previously reported, 23 the proliferative capacity potential was evaluated by detecting nucleolar organizer regions (NORs) in spermatogonia and Sertoli cells. Cells were identified through their morphological features inside the tubules. Spermatogonia were characterized as the round cells localized next to the basement membrane with dark and oval nuclei, whereas Sertoli cells were located near to the basolateral portion of the tubule with large, tightly linked morphology and indented nucleus. Tissue sections mounted on slides were exposed to a silver solution composed of 1 part of 2% gelatin (in 1% aqueous formic acid) and 2 parts of 50% aqueous silver nitrate solution for 30 minutes in a dark room. After extensive washing in 5% thiosulfate solution for 10 minutes, 28 the number of NOR dots were counted within the nucleoli of spermatogonia and Sertoli cells. Observations were made in 10 randomly selected nuclei in 10 different fields at 1000× magnification. 29
Statistical analysis
Values were first tested for normality (Shapiro–Wilk test) and homoscedasticity (Levene test). The effects of the cryopreservation technique on testicular parameters of different species were analyzed by two-way ANOVA, and pairwise comparisons of significant differences (p-values <0.05) among treatment groups were assessed using Tukey’s test. Control groups were compared with the cryopreservation protocols using Dunnett’s Test. All analyses were performed using R Studio.
Results
Cell viability
In adult domestic cat tissues, viability of testicular tissue after cryopreservation remained >90% for all tested methods (Fig. 2A). Vitrification yielded the highest percentage of viability (100%) and Mr. Frosty slow freezing the lowest (94.7%); while the control group showed 95%; statistical analysis did not detect significant differences among these values (p = 0.073). Culture influenced cell viability after cryopreservation of adult samples (p = 0.004), with the proportion of viable cells decreasing to less than 70% in all cultured tissues. Samples subjected to conventional vitrification were the most affected after tissue culture, with an increase in cell death (29.3%; Fig. 2A) (p = 0.003). In juvenile tissues, viability remained statistically similar between control and cryopreserved groups (p = 0.066). After culture, the proportion of viable cells decreased to 78% in the progressive slow freezing group (p = 0.041), whereas cultured tissues from other cryopreservation methods retained cell viability above 80% (p ≥ 0.074; Supplementary Fig. S1A).
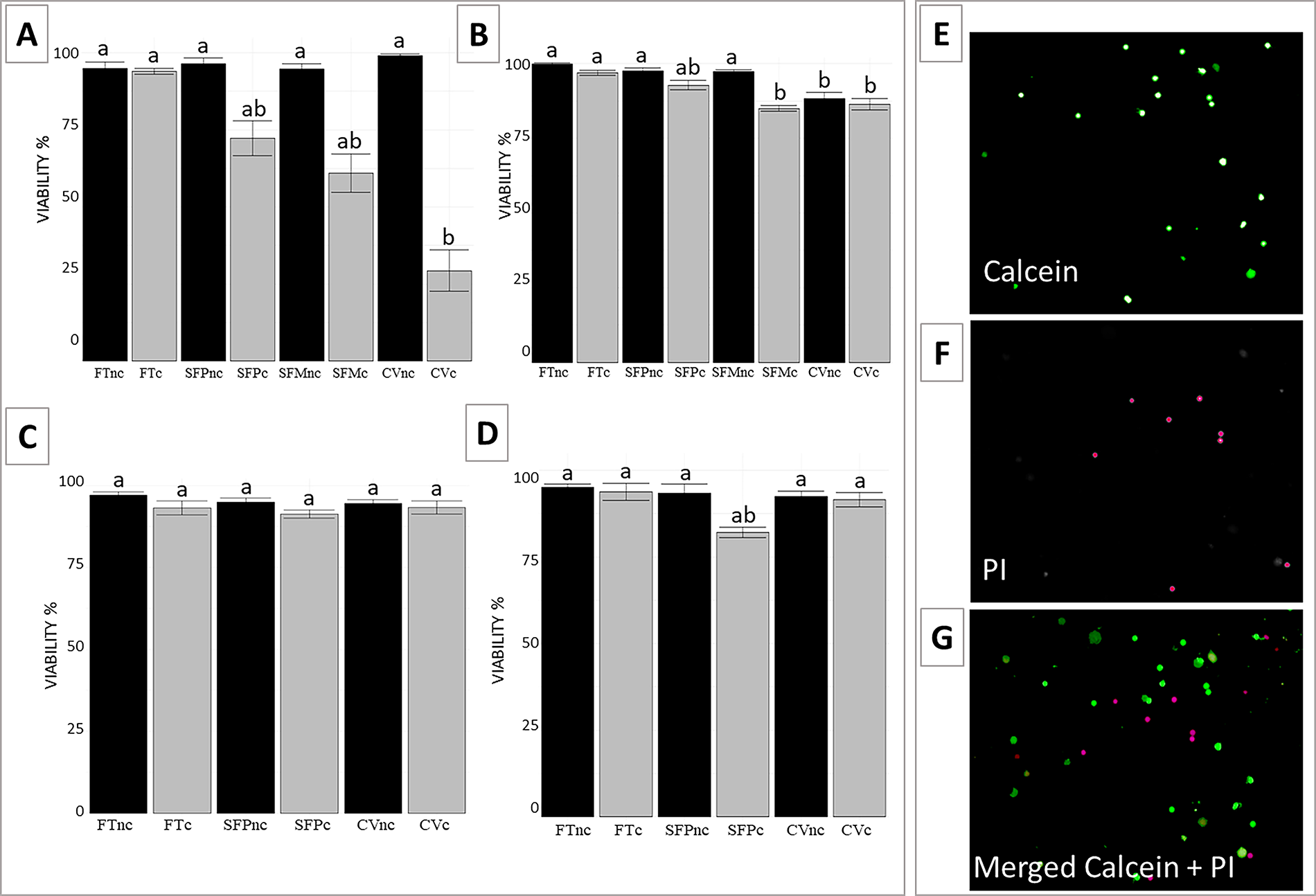
Viability (%) of testicular cells from domestic cats
In deer tissues, over 88% of viable cells were recovered after all cryopreservation methods tested (Fig. 2B–D). In white-tailed deer, a significant decline in viability was observed after vitrification (88.3%, p = 0.016), whereas progressive and Mr. Frosty slow freezing maintained values near 97% (p = 0.112). In brocket deer species, no statistically significant difference in cell viability was found between protocols. After culture, only white-tailed deer tissues subjected to Mr. Frosty freezing showed a significant viability reduction (p = 0.01), while all other groups maintained values above 80%.
Mitochondrial activity
Fluorescent intensity of MitoTracker Green, reflecting mitochondrial membrane potential, was set at 100% in fresh tissues (control group). In non-cultured domestic cat tissues, relative intensity decreased after progressive slow freezing (86.1%), vitrification (80.5%), and Mr. Frosty slow freezing (75.2%, p = 0.011) (Fig. 3A). In non-cultured juvenile tissues, relative intensity was lower following progressive slow freezing (71.2%), vitrification (73.4%), and Mr. Frosty slow freezing (74.2%) compared with fresh tissues (p = 0.021) (Supplementary Fig. S1B). In both adult and juvenile tissues, viability remained high after 24 hours of culture in the fresh control group (100%). However, cryopreserved samples showed a further decline in viability following culture, with values ranging from 65% to 70% (p = 0.006 for juvenile tissues and p = 0.049 for adult tissues) in intensity values.
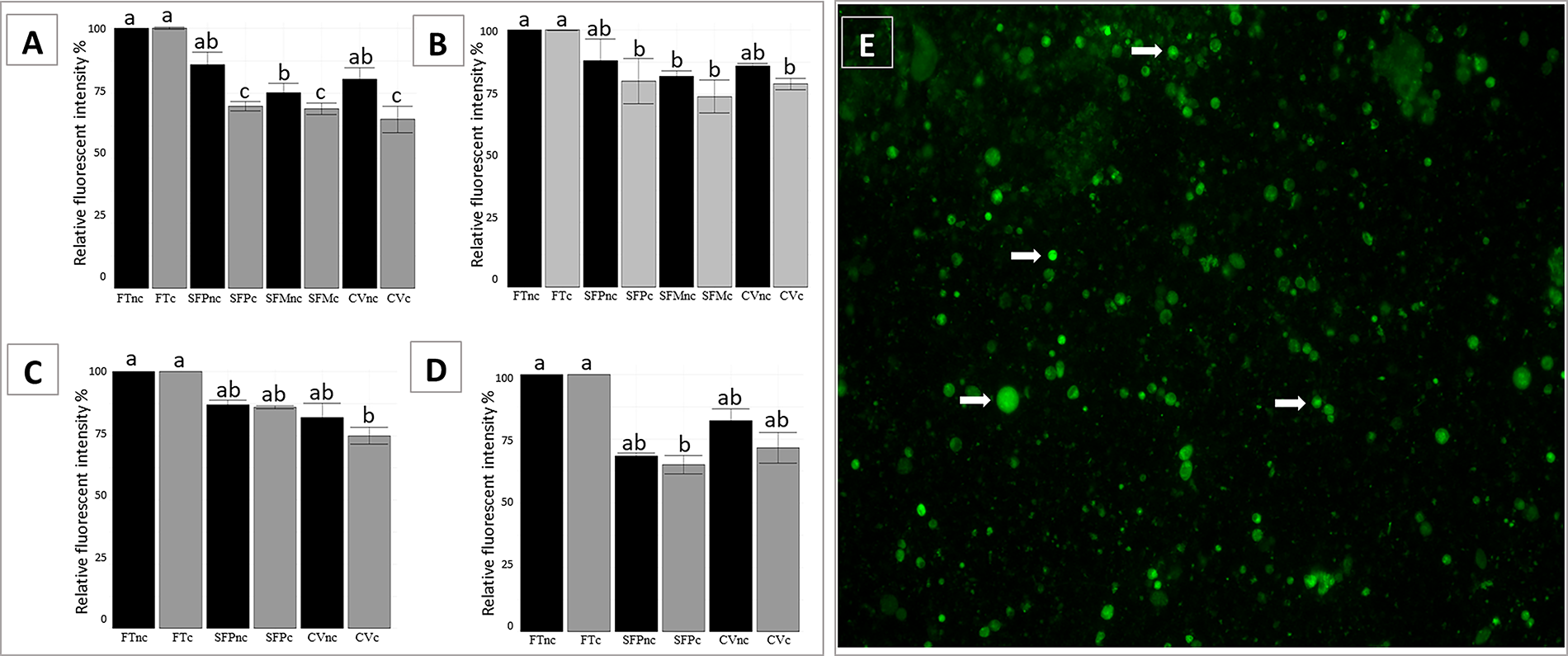
Mitochondrial membrane potential in testicular tissues from domestic cats
Although decline of mitochondrial membrane potential was also observed in cryopreserved deer tissues, relative intensity differed among deer species. In white-tailed and Brazilian dwarf brocket, mitochondrial membrane potential values did not differ significantly between progressive slow freezing (88% and 87%, respectively) and vitrification (86% and 82%, respectively) (p = 0.396 and p = 0.476, respectively) (Fig. 3B, C). Conversely, in gray brocket there was no significant difference in values between vitrification (75%) and progressive slow freezing (68%) (p > 0.230) (Fig. 3D). In white-tailed deer, in which Mr. Frosty slow freezing was also performed, this protocol resulted in significantly lower values (82%) compared with the fresh control (p = 0.043). No statistically significant change in intensity values was observed in cultured testicular tissues from deer species after culture (p ≥ 0.12805; Fig. 3B–D).
Histomorphology of testicular tissue
The proportion of intact seminiferous tubules in adult domestic cat tissues in fresh samples was 86.3%. There was no statistically significant difference in the proportion observed after progressive slow freezing (85%), vitrification (82.5%), and slow freezing with Mr. Frosty (77.5%) (p = 0.104) (Fig. 4A). After culture, a significant reduction in tissue integrity was observed across all groups (p = 0.003), with fresh tissues retaining 60% integrity and cryopreserved samples dropping to 13.8% after Mr. Frosty slow freezing, 10.0% after progressive slow freezing, and 13.8% after vitrification (Fig. 4A), with notable damage such as shrinkage, stromal disruption, and reduced cellularity (Fig. 5A–F). In juvenile cats, the proportion of intact seminiferous tubules was 93.7% in fresh tissues, and it was largely maintained after progressive slow freezing (85.5%; p = 0.732), slow freezing with Mr. Frosty (87.5%; p = 0.588), and conventional vitrification (90.0%; p = 0.943) (Supplementary Fig. S2). In juvenile cultured samples, the proportion of intact seminiferous tubules was 92% in fresh controls. However, a decrease was observed in samples that underwent either of the three cryopreservation procedures (p = 0.014) (Supplementary Fig. S2).
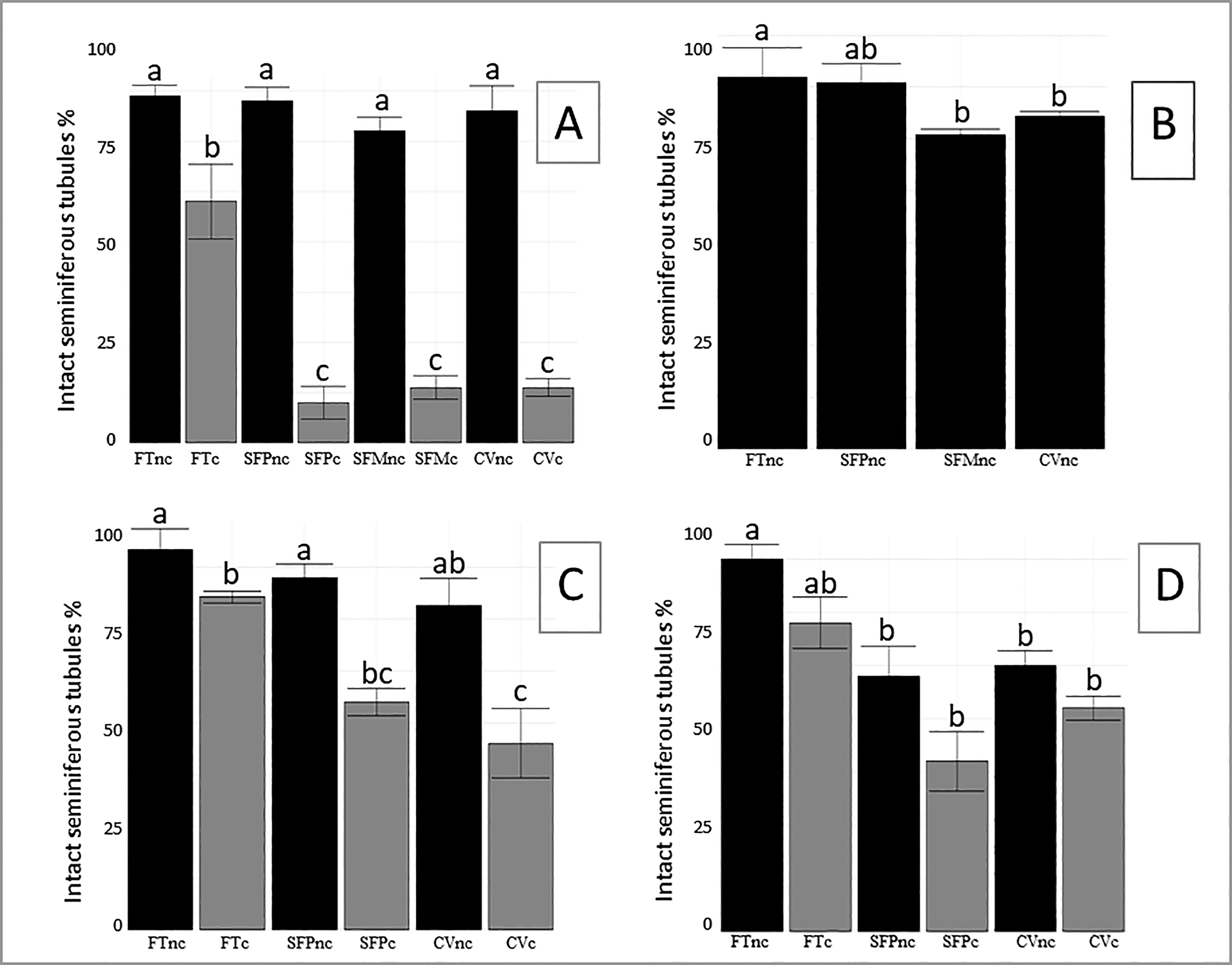
Percentage of intact seminiferous tubules in testicular tissue from domestic cat
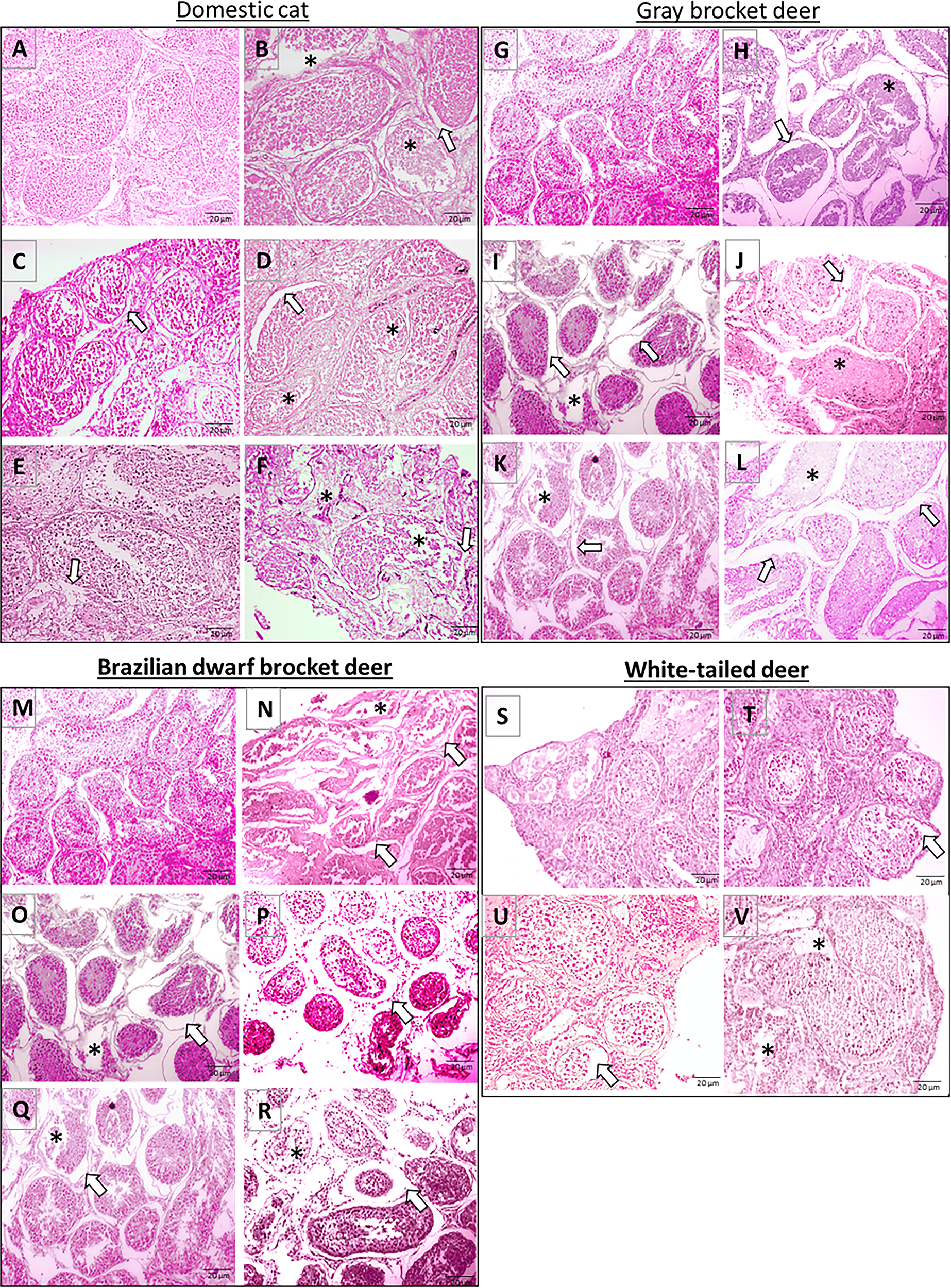
Histomorphology of testicular tissue from domestic cat
In deer, the percentage of normal seminiferous tubules decreased after all cryopreservation methods compared with fresh tissues (Fig. 4B–D). Specifically, in white-tailed deer, vitrification and Mr. Frosty methods caused significant reduction (p = 0.016), while progressive freezing maintained tissue integrity (p = 0.657) (Fig. 4B). Brazilian dwarf brocket showed moderate reductions (p = 0.0561) after vitrification and progressive slow freezing (Fig. 4C). In gray brocket, the proportion of intact seminiferous tubules showed a reduction after both methods, vitrification and progressive slow freezing (p = 0.041) (Fig. 4D). Following culture, testicular tissue integrity further decreased significantly compared with non-cultured tissues (p < 0.05) (Figs. 4B–D and 5G–V). The proportion of intact tubules dropped to 55%−45% in Brazilian dwarf brocket and 40%−52.5% in gray brocket; no significant differences were observed between species or cryopreservation protocols (p ≥ 0.05) (Figs. 4B–D and 5G–V).
Cell proliferative potential
NOR staining was performed in non-cultured testicular tissues, in both fresh and after freezing protocols (Fig. 6). In fresh domestic cat tissues, the mean number of NORs (Table 1) was 2.9 per cell nucleus. There were no significant differences in the number of NOR dots per cell among fresh control, vitrification, slow freezing with Mr. Frosty, and progressive slow freezing (p = 0.675). In juvenile domestic cats, the mean number of NORs dots per cell was 3.2 in the fresh control. Tissues subjected to vitrification exhibited comparable values of 2.9, whereas Mr. Frosty and progressive slow freezing showed 2.7 and 2.6, respectively (p > 0.684; Supplementary Table S1).

NOR staining in testicular cells from domestic cat
Number of Nucleolar Organizer Regions (NOR) Dots in Testicular Cells of Domestic Cats, White-Tailed Deer, Brazilian Dwarf Brocket, and Gray Brocket Deer

Data are expressed as mean ± SD. Different superscript letters (a, b) in the columns indicate a significant statistical difference between treatments.
p-value indicate the significance of the differences between groups as determined by ANOVA followed by Tukey’s post hoc test.
FT, fresh tissue; SFM, slow freezing Mr. Frosty; SFP, slow freezing progressive; CV, conventional vitrification.
In deer, a statistical difference in NOR results was observed related to the species (p = 0.002) but not to the cryopreservation protocol (p = 0.581), with white-tailed deer cells containing the highest number of NOR dots per cell compared with the brocket deer species. The mean number of NOR dots observed in white-tailed deer cells was 4.3 in the fresh tissues, and comparable values were observed following slow freezing protocols (4.3) and vitrification (4.2) (p > 0.053; Table 1, Fig. 6) For the brocket species, the number of NOR dots per cell observed had an average of 3.0 and 4.0 for fresh control, 2.9 and 3.4 for the progressive slow freezing protocol, and 2.8 and 3.7 for the vitrification protocol in Brazilian dwarf brocket and gray brocket, respectively (Table 1, Fig. 6).
Discussion
Effects of cryopreservation on testicular cell viability, morphology, mitochondrial activity, and proliferative potential
Testicular tissue from all species analyzed here demonstrated varying levels of tissue viability and structural preservation depending on the method used. In general, slow freezing and vitrification protocols showed a decrease in tissue viability and proportion of intact seminiferous tubule compared with fresh tissues, as reported in previous studies.15,16,25 However, in non-cultured tissues, viable cells and well-preserved testicular cell morphology were recovered across all cryopreservation techniques, confirming their potential for functional tissue preservation. Cryopreserved tissues showed reduced mitochondrial activity, as indicated by lower fluorescence intensity of MitoTracker compared with fresh tissues, indicating the negative impact of cryopreservation on cell metabolism. This pattern, previously reported by Lima et al. 18 in vitrified domestic cat testicular tissues, was also observed with slow freezing, suggesting comparable effects on mitochondria, which are known to be sensitive to nonphysiological conditions. 30
Although NOR staining has not been widely applied in testicular cryopreservation studies, the variability observed in our study, with no significant differences across cryopreservation methods, suggests functional cell populations were retained regardless of protocol. In cats, NOR dot numbers per cell have already been studied, and an optimal proliferative potential of cells has been recovered after testicular tissue vitrification using DMSO/glycerol. 15 In that study, the average NOR dot number per cell in fresh tissues was 3.85 ± 1.81, and the DMSO/glycerol group showed no significant difference from the fresh control, while the DMSO/EG and EG/glycerol groups exhibited significantly lower values (p < 0.05). In our study, the NOR dot numbers per cell observed were within the range reported by Lima et al., 22 with our findings further supporting the efficacy of the DMSO/Glycerol vitrification protocol in preserving cellular proliferative potential. In deer, significant interspecies variation was observed, highlighting the need for future studies with larger sample sizes to establish species-specific NOR baselines for evaluating cryopreservation impact.
Comparison of cryopreservation techniques across species
Cryopreservation has been widely applied in preserving testicular tissues across species including rodents, domestic cats, ungulates, and wild carnivores.11,24,31,32 Both slow freezing and vitrification are viable options depending on species and cell type.12,20 For example, Peris-Frau et al. 20 evaluated testicular tissue cryopreservation methods in some species of artiodactyls, primates, carnivores, and rodents. Their data showed higher viability of spermatids/spermatozoa using vitrification, while viability of rounded germ cells was better preserved by slow freezing. In the same study, an increase in DNA damage after slow freezing was observed, suggesting that the reduction of mechanical cell injury with tissue vitrification could be an advantage of using this method to better preserve DNA integrity of wild artiodactyl cells. 20 The improvement in structural preservation, mitochondrial activity, and viability was particularly observed in juvenile domestic cat testicular tissues, while yielding similar results to slow freezing in adult samples, suggesting an age-related difference in tissue response to cryopreservation. Vitrification appears to maintain NOR integrity, supporting its role in preserving cellular activity, specifically in domestic cat. 22 Despite the observed trend of higher NOR dot numbers in vitrified samples, no significant age-related differences in proliferative potential were detected. This suggests that functional nucleolar activity can be retained across ages and cryopreservation methods. These results align with gene expression analyses suggesting age-dependent resilience in testicular tissue 33 and reinforce the utility of NOR as a complementary viability indicator. 29 Overall, our study suggests that domestic cat testicular tissues could be well-preserved after both vitrification and slow freezing, maintaining more than 70% seminiferous tubule integrity, a high percentage of viable cells, and no significant differences in the NORs dot number compared with fresh tissue. However, taking time efficiency, simplicity, and tissue preservation outcome into consideration, vitrification may have a slight advantage over other techniques.
On the other hand, slow freezing has been considered optimal for preserving the quality of Artiodactyla testicular tissue and has been indicated for cellular function preservation.11,12,20 Despite this, species-specific differences must be considered. In our study, slow freezing protocols were significantly more efficient in recovering viable cells than vitrification in white-tailed deer, but the difference was not observed in brocket deer samples. In general, the findings suggest that deer testicular cells retain more functional viability after slow freezing; however, vitrification is still a viable alternative. 20 For example, in our study, despite the significant decrease in white-tailed deer tissue viability after vitrification compared with fresh and slow-frozen tissues, this rapid-freezing protocol recovered over 80% of viable cells. This suggests that vitrification could also be considered for testicular preservation in this species, presenting it as a suitable methodology in fieldwork or other situations where the equipment needed for slow freezing is unavailable. Beyond cell viability, additional parameters such as mitochondrial membrane potential, seminiferous tubule integrity, and NOR dot number per cell provided further insight into the comparative efficiency of cryopreservation techniques across species. These analyses revealed subtle but important species-specific responses. Overall, progressive slow freezing tended to better preserve mitochondrial function and tissue morphology, particularly in white-tailed deer, where results were comparable with fresh tissues. In contrast, vitrification yielded higher mitochondrial activity in gray brocket deer, suggesting a potential species-dependent preference for this method. However, a study with a larger sample size is necessary to corroborate the effects of this type of cryopreservation on the testes of this species. The analysis of NOR dots per cell, an indicator of proliferative potential, also reflected interspecies variability, with white-tailed deer exhibiting higher values overall and better preservation following slow freezing. Taken together, these results provide preliminary evidence that slow freezing may be broadly effective for preserving cellular function and tissue architecture in white-tailed deer, while vitrification could potentially yield better outcomes for some brocket deer species. Vitrification has already been presented not only as an alternative but as the better protocol to recover sperm cells from testicular tissue from Rusa deer, Fea’s muntjac, and Sumatran serows. 34 This demonstrates that the effectiveness of a cryopreservation protocol is also influenced by species response, which could be associated with genetic particularities of each taxonomic group. 20 Furthermore, different species may require different freezing media, as observed by Peris-Frau et al., 20 in which the need for FBS in cryopreservation media varied among cervids. This reinforces that within the same order, different methodologies for slow freezing and vitrification may be required to achieve better tissue preservation.
Effects of short-term culture after cryopreservation
Culture condition in the present study was adequate to maintain viability and mitochondrial potential in the fresh testicular tissue. However, when followed by culture, the impacts of cryopreservation on testicular tissues were more pronounced, particularly in adult cats, suggesting that culture conditions to maintain live cells and structural integrity and to facilitate cellular recovery from stressed conditions are not yet ideal. 18 Therefore, the pronounced decline in the evaluated parameters may be attributed to cryopreservation-induced alterations in molecular components critical for maintaining cellular function and viability.
Although exposure to higher temperatures during warming has been suggested as an optimal strategy to improve post-culture integrity, 18 the results obtained in this study reveals that its effectiveness may vary across species and developmental stages. Evidence of cell injuries during culture 35 was specifically observed in adult domestic cat testicular tissues, where cell viability decreased significantly from >90% to 29.3%–72.3%, contrasting with juvenile samples in which tissues retained above 70% viable cells after cryopreservation and culture. This may reflect differences in membrane composition or stress response pathways. 33 The reduced connections between cells and the extracellular matrix in adult testicular tissue might be linked to the age-related difference in tissue response, as discussed by Amelkina et al., 33 who reported a detrimental effect of vitrification and 24-hour culture on the transcriptomic dynamics of adult domestic cat testicular tissues compared with juveniles. In addition, the proportion of intact seminiferous tubules in adult testicular tissue was highly reduced after cryopreservation, warming, and short-term culture, reinforcing the increased fragility of adult tissues under these conditions. 33 Regarding mitochondrial activity, the relative intensity in cryopreserved tissues declined further after culture in both adult and juvenile cat samples, indicating that culture conditions can exacerbate cellular stress, regardless of age. In deer tissues, a similar trend was observed with cell viability and mitochondrial activity decreasing after culture. However, the reduction was mostly not statistically significant, with viability and relative fluorescent intensity remaining above 80% and 65%, respectively. In Brazilian dwarf brocket deer tissues, the proportion of intact seminiferous tubules declined significantly, suggesting that while overall cell viability was preserved, structural damages to the tissue were more pronounced after culture. Thus, the finding from both domestic cats and brocket deer suggests that cryopreservation protocols alone did not fully preserve tissue integrity when combined with culture, underscoring the need for species-specific optimization of post-thaw culture protocols. Furthermore, the comparative analysis of domestic cat and brocket deer testicular tissues emphasizes the importance of adapting cryopreservation and culture methods based on taxonomic group, age, and tissue type.20,33
Study limitations and next steps
In this study, we evaluated the effects of slow freezing and vitrification on testicular tissues from felids and cervids, focusing on structural integrity, viability, mitochondrial activity, and proliferative potential. While results provided valuable insights, several limitations should be considered. First, the number of individuals per species was limited due to the difficulties of obtaining wildlife samples. This reduced statistical power and may explain why some comparisons were not significant. Consequently, species-specific patterns should be regarded as preliminary and interpreted with caution, as individual variability could have influenced the results. Second, our analyses concentrated on short-term endpoints. Although we assessed tissue morphology and cellular quality after cryopreservation, we did not evaluate long-term functionality, such as extended culture time or fertility potential of the gametes. Therefore, the further outcomes for future reproductive use remain to be examined. Third, while we reported differences in cryopreservation responses between felids and cervids, the underlying mechanisms (e.g., tissue composition, extracellular matrix architecture, cryoprotectant permeability, or warming kinetics) were not investigated. Understanding these mechanistic aspects will be crucial to explain taxon-specific differences and to design more effective protocols. Despite these limitations, our comparative approach represents a contribution to the development of cryopreservation strategies in wildlife species. Future studies will expand the sample size through multi-institutional collaborations, investigate mechanistic factors underlying taxonomic differences, and test the functional potential of preserved tissues. Such work will be essential to refine cryopreservation protocols and to maximize their contribution to biodiversity conservation.
Conclusion
This study provides a comprehensive evaluation of cryopreservation techniques for preserving testicular tissues from domestic cats and Neotropical deer species, focusing on the effects of slow freezing and vitrification on cell viability, morphology, mitochondrial activity, and proliferative potential. The findings demonstrated that both vitrification and slow freezing are effective protocols for domestic cat, with slow freezing as the most suitable option for Neotropical deer testicular tissue. In cervids, despite the lower values obtained with vitrification, this method was also effective and could be advantageous in situations requiring time efficiency and minimal equipment for tissue cryopreservation. These results highlight a species-dependent response to cryopreservation, evident in the differential effectiveness of vitrification versus slow freezing across taxa, the age-related resilience observed in domestic cats, the greater post-culture impact on Felidae tissues compared with Cervidae, and the species-specific variations in proliferative potential. Nonetheless, given the small sample sizes, these findings are encouraging but should be interpreted cautiously and confirmed in further studies. Our work emphasizes the value of comparative studies across taxa to better understand the biological factor influencing tissue preservation. It also lays the groundwork for adapting these protocols to other Neotropical cervids and rare or endangered mammals for conservation purposes.
Authors’ Contributions
E.D.P.S., P.L., and P.C.: Conceptualization and investigation. E.D.P.S. and P.L.: Data curation and methodology. E.D.P.S., P.L., P.C., and J.M.B.D.: Writing—review and editing. P.C. and J.M.B.D.: Resources and supervision.
Footnotes
Acknowledgments
The authors thank the Deer Research and Conservation Center (NUPECCE/UNESP) and the Smithsonian Institution for providing all the structure for the development of this work.
Author Disclosure Statement
The authors declare that there are no competing interests.
Funding Information
This study was supported by the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) process n°2017/07014-8 and 2024/04621-4.
Supplemental Material
Supplemental Material
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.