Renewable resources based hydrovanilloin [1,2-bis(4-hydroxy-3-methoxyphenyl)-1,2-ethanediol] was synthesized in 86% yield by electrochemical dimerization of vanillin in aqueous NaOH. This symmetrical bis-phenol monomer was then used for the preparation of urethane polymers by two different methods. In the first method a 1:2 mole ratio mixture of hydrovanilloin and diisocyanate was polymerized in DMF using 1,4-diazabicyclo[2,2,2]octane as the catalyst at 60°C, for 1 h to give poly(hydrovanilloin–urethane)s. In the second method diisocyanates were first reacted with polyethylene glycol-400 to give pre-polymers. Then prepolymers were reacted with equivalent amount of hydrovanilloin at 60°C for 4 days to produce poly(hydrovanilloin-ethylene glycol-urethane)s. The first method resulted hard poly(hydrovanilloin–urethane)s showing Tg values in the range of 121–172°C. The second method yielded softer poly(hydrovanilloin-ethylene glycol-urethane)s and these polymers failed to show distinct glass transition temperatures in the DSC analysis. However, poly(hydrovanilloin-ethylene glycol-urethane)s showed better thermal stabilities than polymers without polyethylene glycol units.
The current interest in the use of renewable feedstocks in the polymer industry has resulted a rapid growth in research activities on lignocellulosic biomass as well as fermentation derived monomers.1-3 The lignocellulosic biomass derived monomers can be divided into two groups of polysaccharide derived and lignin derived monomers. Among the two groups the polysaccharide derived monomers such furan and levulinic acid derivatives have attracted more attention as renewable feedstocks than lignin derived compounds. The lignin fraction of the lignocellulosic biomass is a rich source for aromatic feedstocks, however the structural complexity of lignin is a major obstacle in the processing of lignin to aromatic feedstocks.4,5 Currently vanillin (4-hydroxy-3-methoxybenzaldehyde, 1, Figure 1)6-8 as well as the related compound ferulic acid9,10 are the two main aromatic feedstocks derived from lignin. Among a number of potential biomass derived polymer feedstocks, vanillin has attracted attention of polymer chemists due to multiple functional groups –OH and –CHO as well as the activated aromatic ring. The recent advancements in the production of vanillin by metal catalyzed air oxidation of abundant lignin have further endorsed the standing of this phenolic-aldehyde to a favorable renewable feedstock for the next generation renewable polymer industry.11,12 A verity of high lignin containing woods have been used in the production of vanillin and the related compound syringaldehyde (4-hydroxy-3,5-dimethoxybenzaldehyde). The air-oxidation of powdered wood in an alkaline medium at 170–200°C has been used in this process and typically a 17–31% combined yield of vanillin and syringaldehyde can be achieved in this process.13 Additionally biotechnology based routes using genetically engineered microorganisms, fungi, bacteria, plant cells are also currently under study for the industrial production of renewable vanillin.14,15 Since this lignin based phenol-aldehyde is becoming readily accessible, a number of research groups have studied the potential of this versatile renewable building block as a monomer for the next generation polymer industry.7,16,17 The multifunctional vanillin can be modified to make monomers in a number of ways. One of the early approaches is the use of Knoevenagel condensation to prepare ferulic acid from vanillin and then catalytic hydrogenation to dihydroferulic acid. This monomer was then polymerized as the acetylated derivative: acetyldihydroferulic acid to give poly(dihydroferulic acid) by Miller and co-workers.18 Furthermore, they have found that the new poly(dihydroferulic acid) displays thermal and mechanical properties similar to the widely used polyethylene terephthalate (PET).18 Another approach of upgrading vanillin to a monomer is the dimerization to hydrovanilloin (2, Figure 1). Vanillin can be reductively dimerized to hydrovanilloin by electrochemical coupling as described in the Pearl’s 1952 publication or by McMurry coupling reaction.19,20 In a recent application, Harvey et al. have prepared cyanate ester resins and polycarbonate thermoplastics from vanillin by using this dimer, hydrovanilloin as the key renewable feedstock.20 In another example, Nikafshar et al. reported the preparation of epoxy resins from vanillin; in this case vanillin was first converted to methoxyhydroquinone by using an equivalent amount of sodium percarbonate as the oxidant to prepare the monomer.21
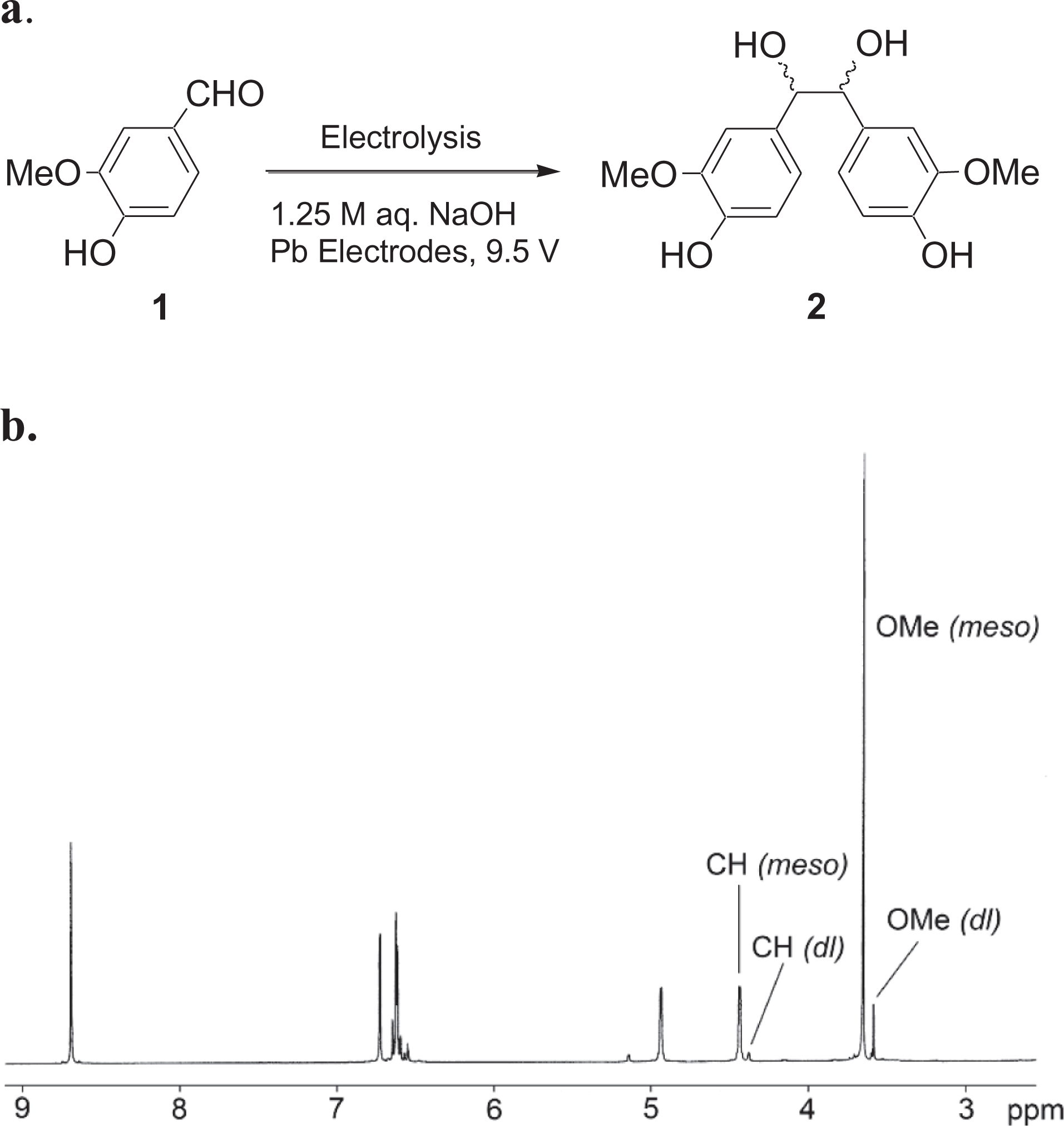
a. Synthesis of meso/dl-hydrovanilloin (2). b.1H NMR spectrum (DMSO-d6) of meso/dl-hydrovanilloin (2, 94% meso) produced by electrolysis of vanillin. 0.164 M vanillin, 1.25 M aqueous NaOH, Pb electrodes, 9.5 V, 3 hr., 25°C.
In our efforts in the development of new generation renewable materials and feedstock chemicals we have studied the potential of vanillin in the synthesis of new polymeric materials.22-27 For instance, we first reported the synthesis of poly(vanillin) by electrochemical polymerization of divanillin; which was prepared via enzymatic oxidative coupling of vanillin using horse radish peroxidase.28 In another example we have shown that Schiff base polymers with a degree of polymerization (DP) ∼ 25–32 can be prepared by condensation of the dialdehyde, divanillin with alkyl diamines.29 Furthermore, these divanillin Schiff base polymers have demonstrated transition metal chelation properties, with potential applications in water purifications and metal ion separations.29 In a more recent effort we have shown that hydrovanilloin synthesized by electrochemical dimerization of vanillin can be used as a renewable substitute for bisphenol A for the preparation of epoxy resins.30 The reaction of equivalent amounts of the disodium salt of hydrovanilloin with epichlorohydrin gave a hydrovanilloin–diglycidyl ether phenoxy resin with Tg 146°C. On the other hand, two equivalents of epichlorohydrin with hydrovanilloin gave a curable oligomer of hydrovanilloin–diglycidyl ether with 2.1 repeating units. Furthermore, this oligomer could be cured with various aliphatic diamines to give hard epoxy resins with Tg values in the range of 116–149°C.30
The use of vanillin based monomers for the preparation of polyurethanes is a relatively unexplored area with only two reports. In the first case, N,N′-bis(4-hydroxy-3-methoxy benzylidene)-o-tolidine synthesized by condensation of vanillin with o-tolidine has been used as the monomer for the synthesis of polyurethanes with various diisocyanates.31 In the second and more recent example, Gang et al. used divanillin to prepare a diol chain extender by reacting with two equivalents of ethanolamine.32 This diol together with polytetrahdrofuran has been used later in the preparation of high performance polyurethane elastomers.32 As far as we are aware the vanillin dimer hydrovanilloin has not been used as a monomer in polyurethane preparations. This easily accessible symmetrical vanillin dimer has certain advantages over earlier reported vanillin based monomers as hydrovanilloin is functionalized with two alcoholic and two phenolic groups, all reacting with isocyanate group, making it an excellent cross-linking unit.
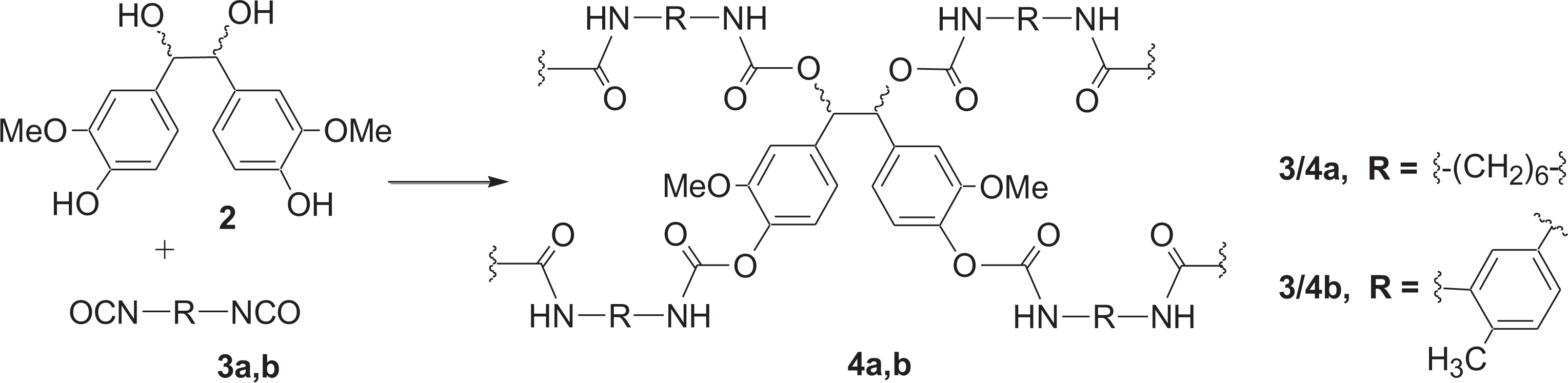
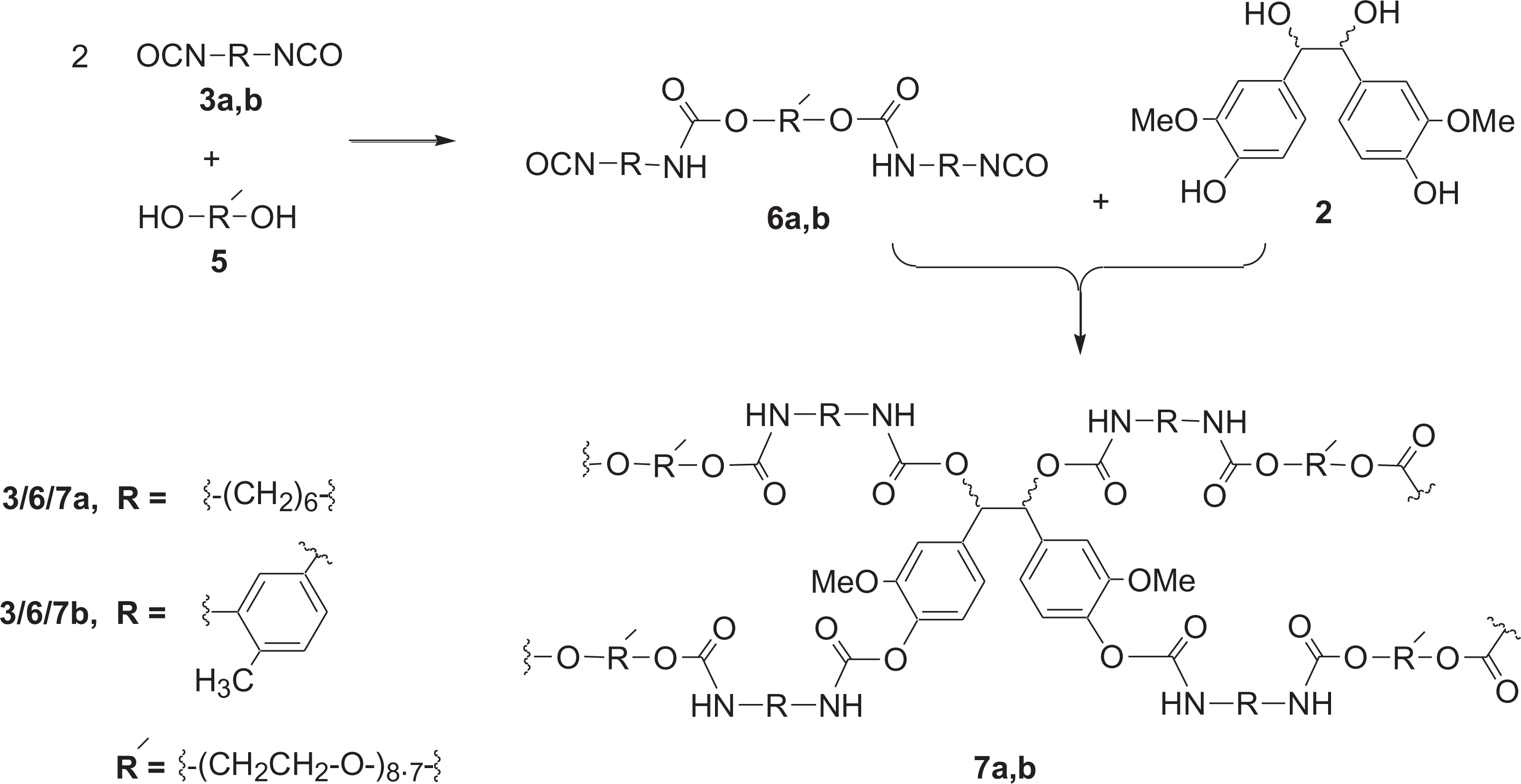
In continuation of our efforts in this field, we have investigated the direct use of meso/dl-hydrovanilloin (2) obtained from electrochemical dimerization of vanillin as shown in Figure 1a. for the preparation of polyurethanes. 1H NMR spectrum of meso/dl-hydrovanilloin (2, 94% meso) produced by electrolysis of vanillin is shown in Figure 1b. The In this publication, we report two simple, scalable methods for the preparation of polyurethanes from hydrovanilloin. In the first method hydrovanilloin was reacted with equivalent amounts of diisocyanates (3a, b) in the presence of a tertiary amine catalyst to give cross-linked polyurethanes (4a, b) as shown in Figure 2. In the second method, diisocyanates were first reacted with polyethylene-glycol 400 (5) to prepare pre-polymers (6a, b) and then polymerized with hydrovanilloin (2) to give poly(hydrovanilloin-ethylene glycol-urethane) 7a, b as shown in Figure 3.
Synthesis of poly(hydrovanilloin–urethane) 4a, b.
Synthesis of poly(hydrovanilloin-ethylene glycol-urethane) 7a, b.
Experimental
Materials and instrumentation
Vanillin (>99%), hexamethylene diisocyanate (99%), toluene-2,4-diisocyanate (98%), polyethylene glycol 400 (>99%), 1,4-diazabicyclo[2,2,2]octane and NaOH were purchased from Aldrich Chemical Co. The hydrovanilloin (2, Figure 1) was prepared by electrochemical coupling of vanillin according to the previously published procedure in 86% yield and was analyzed by 1H NMR to determine the meso and dl hydrovanilloin composition as 94% meso.19,281H NMR Spectra were recorded in CDCl3 or in DMSO-d6 on a Varian Mercury plus spectrometer operating at 400 MHz, and chemical shifts are given in ppm downfield from TMS (δ = 0.00). Attenuated total reflection infrared (ATR-IR) spectra of polymers were recorded in the 650–4000 cm−1 range on a Smiths IdentifyIR spectrometer with diamond ATR (Danbury, CT, USA). Thermogravimetric (TG) analysis was carried out in air using TA instruments TGA 2050 system. The mass of the polymer used for a scan was approximately 5 mg, and Pt crucibles were used. The TG curves were recorded in 23–600°C range using a scanning rate of 10°C/ min, in air. Temperatures reported from TG data are onset temperatures (Tonset), as determined by the step tangent method.33 The derivative TG analysis (DTG) data are the peaks in the derivative of the weight % curve correspond to weight loss steps in the TG curve. Differential Scanning Calorimetry (DSC) analysis was performed on a Mettler Toledo DSC 3 STAR system. STARE-E software was incorporated to evaluate the glass transition and onset temperatures. The mass of the samples was approximately 5 mg and placed in aluminum pan. The difference between the power applied to the investigated sample is measured as a function of temperature. DSC experiments were carried out at a heating and cooling rate of 10°C/min. Hardness of the polymers were measured using a D-type Shore Durometer (ASTM 2240). The number average molecular weight and polydispersity index (PDI) of the polymer samples were measured on a Waters gel permeation chromatography (GPC) system with Waters Styragel column, a UV detector, and using dimethylformamide (DMF) as the solvent.
General procedure for the synthesis of poly(hydrovanilloin–urethane) 4a, b
A mixture of hydrovanilloin (0.31 g, 1.0 mmol) and 1,4-diazabicyclo[2,2,2]octane (0.02 g, 0.18 mmol) was prepared in 2 mL of N,N′-dimethylformamide (DMF). Then diisocyanate (3a, b; 2.0 mmol) was added and heated in an oil bath at 60°C for 1 h. The solid formed was washed with water, allowed to dry at room temperature for 2 days. 4a, pale yellow solid: 0.53 g; 4b, pale yellow solid: 0.35 g. The polymers 4a and 4b were analyzed by FT-IR, DSC and TGA/DTGA; FT-IR data are shown in Table 1. The thermal characteristics (DSC and TGA/TGA) data of poly(hydrovanilloin–urethane) 4a, b is shown in Table 2.
FT-IR absorption peaks of poly(hydrovanilloin–urethane) 4a, b and poly(hydrovanilloin-ethylene glycol-urethane) 7a, b.
The thermal characteristics (DSC, TGA), Shore hardness, molecular weight and polydispersity index (PDI) data of poly(hydrovanilloin–urethane) 4a, b and poly(hydrovanilloin-ethylene glycol-urethane) 7a, b. Polymers 7a, b did not show any clear peaks in the DSC curves. Shore hardness was measured by taking six readings at room temperature (23°C) using a D-type Shore Durometer (ASTM 2240).
Poly-urethane
DSC
TGA
Hardness (Shore, type D)
(g/mole)
PDI
Tg (°C)
Tonset (°C)
▵H (J g−1)
Tonset (°C)
DTG (°C)
4a
172
162
10
210
365, 455, 552
50 ± 1
26,300
1.54
4b
121
118
11
200
290, 390, 564
36 ± 2
24,500
1.63
7a
—
—
—
255
325, 460, 520
22 ± 2
37,100
1.49
7b
—
—
—
265
328, 390, 520
22 ± 2
34,900
1.61
General procedure for the synthesis of poly(hydrovanilloin-ethylene glycol-urethane) 7a, b
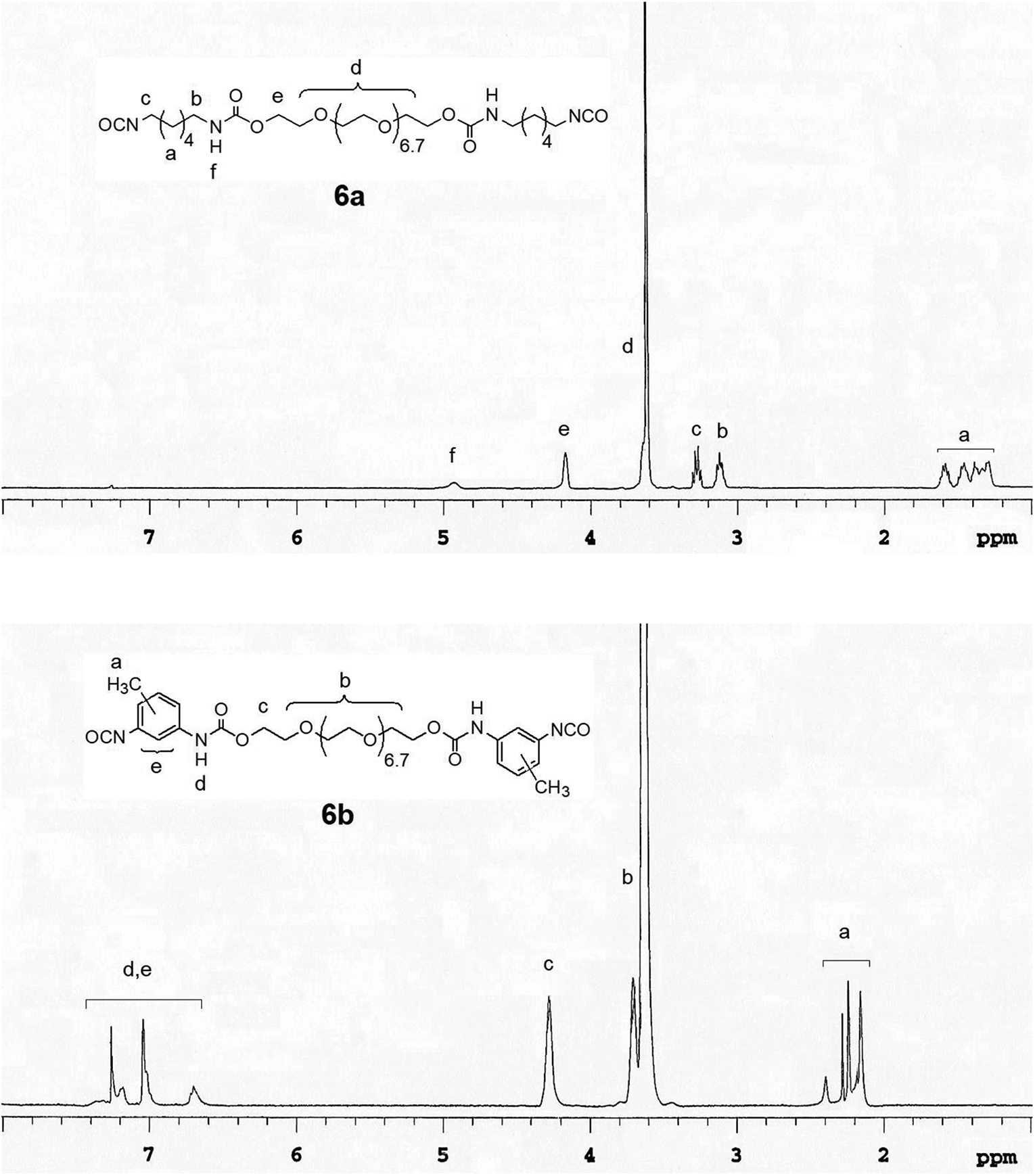
A mixture of polyethyleneglycol-400 (5, 0.40 g, 1.0 mmol) and 1,4-diazabicyclo (2,2,2) octane (0.02 g, 0.18 mmol) was prepared and then diisocyanate (3a, b; 2.0 mmol) was added and heated in an oil bath at 60°C for 1 h. to prepare the prepolymer (6a, b). The structure of the prepolymers was confirmed by withdrawing a sample (5 mg) and recording the 1H NMR in CDCl3. The NMR spectra of prepolymers 6a and 6b are shown in Figure 4. Then hydrovanilloin (2, 0.15 g, 0.5 mmol) dissolved in 2 ml of warm DMF (80°C) was added to the prepolymer. The resulting mixture was heated in an oil bath at 60°C for 4 days. The solid formed was washed with water, allowed to dry at room temperature for 2 days. 7a, pale yellow solid: 0.62 g; 7b, pale yellow solid: 0.93 g. The polymers 7a and 7b were analyzed by FT-IR, DSC and TGA/DTGA; FT-IR data are shown in Table 1. The thermal characteristics (DSC and TGA/TGA) data of poly(hydrovanilloin-ethylene glycol-urethane) 7a, b is shown in Table 2.
1H NMR spectra of pre-polymers 6a and 6b in CDCl3.
Shore hardness measurement of poly(hydrovanilloin–urethane) 4a, b and poly(hydrovanilloin-ethylene glycol-urethane) 7a, b
The polymer samples with ∼ 4 mm thickness and 15 mm diameter were used in hardness measurements. The Shore hardness was measured by taking six readings at room temperature (23°C) using a D-type Shore Durometer (ASTM 2240). The average hardness data of poly(hydrovanilloin–urethane) 4a, b and poly(hydrovanilloin-ethylene glycol-urethane) 7a, b are shown in Table 2.
Results and discussion
Synthesis of poly(hydrovanilloin–urethane) 4a, b
In the direct synthesis of poly(hydrovanilloin–urethane) 4a, b, a 1:2 mole ratio of hydrovanilloin:diisocyanate was used to ensure the reactions at both phenol and alcohol functional groups of hydrovanilloin. The tertiary amine 1,4-diazabicyclo[2,2,2]octane was used as the catalyst for the reaction. The poly(hydrovanilloin–urethane) 4a and b are insoluble in water and in all common organic solvents. As these polymers are highly insoluble, all attempts to collect molecular weight information were not successful. The FT-IR data of these polymers are shown in Table 1. The hydrogen bonded carbonyl groups of the two polymers appear at 1656 cm−1, whereas the hydrogen bonded NH absorptions were found as broad absorptions in the region of 3300–3315 cm−1.34,35
Synthesis of poly(hydrovanilloin-ethylene glycol-urethane) 7a, b
In the synthesis of poly(hydrovanilloin-ethylene glycol-urethane) 7a, b first the prepolymers 6a and 6b were prepared by reacting polyethylene glycol-400 with the diisocyanates (2a, b) in 1:2 mole ratio as shown in Figure 3. The tertiary amine 1,4-diazabicyclo[2,2,2]octane was used as the catalyst for these reactions as well. The relatively smaller molecular weight linear pre-polymers 6a and 6b are soluble in organic solvents. 1H NMR spectra of 6a and 6b were recorded in CDCl3 to confirm the structures and the NMR spectra are shown in Figure 4. The broad multiplets in the 1.22–1.64 ppm region of the NMR spectrum of 6a was assigned to four inner methylene groups of the diisocyanate derived unit. The multiplets at 3.12 and 3.27 ppm were assigned to -CH2-NH- and -CH2-NCO methylene groups. The broad absorption at 4.17 ppm is due to the -CH2-O-CO- methylene units. The largest peak in the spectrum around 3.62 ppm was assigned to the inner polyether methylene units. The 1H NMR spectrum of the prepolymer 6b is also shown in Figure 4. The broad peaks in the 2.16–2.28 ppm high field region was assigned to methyl groups. The peaks at 3.60–3.71 ppm region were assigned to polyethylene inner methylene groups. The broad absorption at 4.18 ppm is due to the -CH2-O-CO- methylene units. The peaks in the low field region 6.60–7.26 ppm was assigned to the aromatic and NH protons.
Next the prepolymers (6a, b) were reacted with diisocyanates (2a, b) to give poly(hydrovanilloin-ethylene glycol-urethane) 7a, b as shown in Figure 3. The polymers 7a, b is both insoluble in water and in all common organic solvents, and all attempts to collect molecular weight information were not successful. The carbonyl groups of the two polymers 7a and 7b appear at 1701 and 1712 cm−1 respectively. The NH absorptions of 7a and 7b were observed as broad absorptions at 3341 and 3289 cm−1 respectively.
Thermal properties of poly(hydrovanilloin–urethane) 4a, b and poly(hydrovanilloin-ethylene glycol-urethane) 7a, b
The DSC data (Tg, Tonset, Tendset and ▵H) of the poly(hydrovanilloin–urethane) 4a, b are shown in Table 2. The poly(hydrovanilloin–urethane) 4a and 4b showed Tg values of 172 and 121°C respectively. The urethane polymer prepared using the symmetrical diisocyanate hexamethylene diisocyanate showed a higher Tg value in comparison to the polymer 4b prepared using unsymmetrical toluene-2,4-diisocyanate. This is probably due to more ordered packing in the polymer prepared using the symmetrical monomer units. The poly(hydrovanilloin-ethylene glycol-urethane) 7a, b with long flexible polyethylene oligomers as part of the polymer failed to show any distinct peaks in the DSC curves and well defined glass transition temperatures.
The thermogravimetric analysis decomposition onset temperatures (Tonset) and derivative thermogravimetric analysis (DTGA) data of poly(hydrovanilloin–urethane) 4a, b and poly(hydrovanilloin-ethylene glycol-urethane) 7a, b are also shown in Table 2. The poly(hydrovanilloin–urethane) 4a, b prepared without the polyethylene glycol-400 showed decomposition Tonset at 210 and 200°C. Whereas the 7a, b with polyethylene glycol-400 showed Tonset values 255 and 265°C. Therefore, this result shows that incorporation of polyethylene glycol-400 units to hydrovanilloin–urethane polymers improves the thermal stability of these polymers. All polyurethanes showed three peaks in the derivative thermogravimetric analysis (DTGA) curve indicating that these polymer materials decompose in similar steps at high temperatures in air.
Hardness measurements of polyurethanes
The Shore hardness data of polyurethanes 4a, b and 7a, b are shown in Table 2. The poly(hydrovanilloin–urethane) 4a, b prepared without the polyethylene glycol-400 showed Shore hardness values of 50 ± 1 and 36 ± 2. The poly(hydrovanilloin-ethylene glycol-urethane) 7a, b with polyethylene glycol-400 oligomer showed the same hardness value of 22 ± 2. This result clearly indicates that incorporation of polyethylene glycol-400 units to hydrovanilloin–urethane polymers gives softer polymers. In comparison of poly(hydrovanilloin–urethane) 4a and 4b the polymer prepared with symmetrical diisocyanate hexamethylene diisocyanate showed a higher hardness value of 50 ± 1, while the polymer 4b prepared using the unsymmetrical toluene-2,4-diisocyanate showed the lower hardness of 36 ± 2. This is also probably due to the more ordered packing in polymer with the symmetrical monomer units as similar trend is noticed with the glass transition temperatures of these polymers.
Conclusion
In conclusion, we have shown that easily accessible dimerized vanillin, hydrovanilloin can be used as a versatile renewable monomer unit for the preparation of polyurethanes. The hard thermoplastics of poly(hydrovanilloin–urethane) with Tg values in 121–172°C range are easily accessible by reacting hydrovanilloin with diisocyanates in DMF using 1,4-diazabicyclo[2,2,2]octane as the catalyst. However, the preparation of softer polymers with better thermal stabilities require the incorporation of more flexible polyethylene glycol units into the polymer structure. This type of co-polymers could be prepared by first reacting the diisocyanates with polyethylene-glycol 400 in 2:1 mole ratio to prepare prepolymers and then polymerization with hydrovanilloin to give poly(hydrovanilloin-ethylene glycol-urethane). The poly(hydrovanilloin–urethane)s and poly(hydrovanilloin-ethylene glycol-urethane)s prepared are insoluble in water and in all common organic solvents. The new poly(hydrovanilloin–urethane) materials developed may be useful in adhesives such as wood working glues due to structural compatibility with lignin.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the United States National Science Foundation (NSF) grants CBET-1336469, CBET-1704144 and HRD-1036593.
ORCID iD
Ananda S Amarasekara
References
1.
ZhuYRomainCWilliamsCK. Sustainable polymers from renewable resources. Nature (London, U. K.)2016; 540: 354–362.
2.
GandiniALacerdaTM. From monomers to polymers from renewable resources: recent advances. Prog Polym Sci2015; 48: 1–39.
3.
GandiniA. Polymers from renewable resources: a challenge for the future of macromolecular materials. Macromolecules2008; 41(24): 9491–9504.
4.
LaurichesseSAvérousL. Chemical modification of lignins: towards biobased polymers. Prog Polym Sci2014; 39: 1266–1290.
5.
AzadiPInderwildiORFarnoodR, et al.Liquid fuels, hydrogen and chemicals from lignin: a critical review. Renew Sustain Energ Rev2013; 21: 506–523.
6.
FacheMBoutevinBCaillolS. Vanillin, a key-intermediate of biobased polymers. Europ Polym J2015; 68: 488–502.
7.
FacheMDarromanEBesseV, et al.Vanillin, a promising biobased building-block for monomer synthesis. Green Chem2014; 16(4): 1987–1998.
8.
KayserLVHartiganEMArndtsenBA. Multicomponent coupling approach to cross-conjugated polymers from vanillin-based monomers. ACS Sustain Chem Eng2016; 4: 6263–6267.
9.
BarbaraIFlouratALAllaisF. Renewable polymers derived from ferulic acid and biobased diols via ADMET. Europ Polym J2015; 62: 236–243.
10.
TakeshimaHSatohKKamigaitoM. Bio-based functional styrene monomers derived from naturally occurring ferulic acid for poly(vinylcatechol) and poly(vinylguaiacol) via controlled radical polymerization. Macromolecules2017; 50: 4206–4216.
11.
AraújoJDGrandeCARodriguesAE. Vanillin production from lignin oxidation in a batch reactor. Chem Eng Res Des2010; 88: 1024–1032.
12.
VoitlTRohrPR. Demonstration of a process for the conversion of kraft lignin into vanillin and methyl vanillate by acidic oxidation in aqueous methanol. Ind Eng Chem Res2010; 49: 520–525.
13.
TarabankoVETarabankoN. Catalytic oxidation of lignins into the aromatic aldehydes: general process trends and development prospects. Int J Mol Sci2017; 18: 2421.
14.
KaurBChakrabortyD. Biotechnological and molecular approaches for vanillin production: a review. Appl Biochem Biotechnol2013; 169: 1353–1372.
15.
Havkin-FrenkelDDudaiN. Biotechnology in flavor production. Hoboken, NJ: John Wiley & Sons, 2009, pp. 83–103.
16.
HernandezEDBassettAWSadlerJM, et al.Synthesis and characterization of bio-based epoxy resins derived from vanillyl alcohol. ACS Sustain Chem Eng2016; 4: 4328–4339.
17.
FacheMViolaAAuvergneR, et al.Biobased epoxy thermosets from vanillin-derived oligomers. Europ Polym J2015; 68: 526–535.
18.
MialonLPembaAGMillerSA. Biorenewable polyethylene terephthalate mimics derived from lignin and acetic acid. Green Chem2010; 12: 1704–1706.
19.
PearlIA.Reactions of vanillin and its derived compounds. XVI.1 the synthesis of Vanillil2. J Am Chem Soc1952; 74: 4260–4262.
20.
HarveyBGGuenthnerAJMeylemansHA, et al.Renewable thermosetting resins and thermoplastics from vanillin. Green Chem2015; 17: 1249–1258.
21.
NikafsharSZabihiOHamidiS, et al.A renewable bio-based epoxy resin with improved mechanical performance that can compete with DGEBA. RSC Adv2017; 7: 8694–8701.
22.
AmarasekaraASHasanMA.Vanillin based polymers: III. Electrochemical dimerization of vanillin revisited and synthesis of hydrovanilloin–formaldehyde polymer. Polym Sci Ser B2016; 58: 307–312.
23.
AmarasekaraASHaUOkorieNC. Renewable polymers: synthesis and characterization of poly(levulinic acid–pentaerythritol). J Polym Sci Part A: Polym Chem2018; 56: 955–958.
24.
AmarasekaraASSinghTBLarkinE, et al.NaOH catalyzed condensation reactions between levulinic acid and biomass derived furan-aldehydes in water. Ind Crops Prod2015; 65: 546–549.
25.
AmarasekaraASHasanMA. Pd/C catalyzed conversion of levulinic acid to γ-valerolactone using alcohol as a hydrogen donor under microwave conditions. Catal Commun2015; 60; 5–7.
26.
AmarasekaraASGreenDWilliamsLD. Renewable resources based polymers: Synthesis and characterization of 2, 5-diformylfuran-urea resin. Europ Polym J2009; 45: 595–598.
27.
AmarasekaraASOwerehOS. Homogeneous phase synthesis of cellulose carbamate silica hybrid materials using 1-n-butyl-3-methylimidazolium chloride ionic liquid medium. Carbohyd Polym2009; 78: 635–638.
28.
AmarasekaraASWireduBRazzaqA.Vanillin based polymers: I. An electrochemical route to polyvanillin. Green Chem2012; 14: 2395–2397.
29.
AmarasekaraASRazzaqA. Vanillin-based polymers—part II: synthesis of Schiff base polymers of divanillin and their chelation with metal ions. ISRN Polym Sci2012; 2012: 532171.
IssamAMIsmailJ. New aromatic poly(azomethine urethane)s containing o-tolidine moiety in the polymer backbone. Des Monom Polym2006; 9, 237–246.
32.
GangHLeeDChoiKY, et al.Development of high performance polyurethane elastomers using vanillin-based green polyol chain extender originating from lignocellulosic biomass. ACS Sustain Chem Eng2017; 5: 4582–4588.
33.
NgoHLLeCompteKHargensL, et al.Thermal properties of imidazolium ionic liquids. Thermochim Acta2000; 357: 97–102.
34.
ReddyKRRaghuAVJeongHM. Synthesis and characterization of pyridine-based polyurethanes. Designed Monom Polym2009; 12: 109–118.
35.
YoonSCSungYKRatnerBD. Surface and bulk structure of segmented poly(ether urethanes) with perfluoro chain extenders. 4. Role of hydrogen bonding on thermal transitions. Macromolecules1990; 23: 4351–4356.