
Abstract
Angiomatoid fibrous histiocytoma is a rare intermediate soft tissue tumor with uncertain differentiation and prone to misdiagnosis because of atypical morphology and aberrant biomarker expression. Herein, we report a unique case of occipital scalp angiomatoid fibrous histiocytoma in a 33-year-old female, which presented with rapid tumor growth, marked nuclear pleomorphism, elevated mitotic activity, and atypical positive expression of CDK4 and MDM2 immunohistochemically, leading to an initial misdiagnosis of dedifferentiated liposarcoma. Preoperative routine blood tests, biochemical examinations, and inflammatory biomarker detection, including C-reactive protein, erythrocyte sedimentation rate, and interleukin-6, were performed, and all hematological indicators were within normal reference ranges without systemic inflammatory or hematological abnormalities. The patient underwent complete marginal resection of the tumor, and no adjuvant radiotherapy or chemotherapy was administered postoperatively because of the absence of high-risk malignant features, residual lesions, and distant metastasis. Molecular testing confirmed EWSR1 gene rearrangement without CDK4 or MDM2 gene amplification, which confirmed the final diagnosis of angiomatoid fibrous histiocytoma. The patient achieved a favorable prognosis with no recurrence or metastasis during 20 months of postoperative follow-up. Distinct from previously reported conventional angiomatoid fibrous histiocytoma cases with low proliferative activity and negative liposarcoma-related markers, the present case is characterized by atypical dual positive expression of CDK4/MDM2 and a markedly elevated Ki-67 proliferation index, which fills the clinical data gap of atypical angiomatoid fibrous histiocytoma with liposarcoma-mimicking immunophenotype. This study further clarifies the differential diagnostic criteria between angiomatoid fibrous histiocytoma and dedifferentiated liposarcoma, verifies that isolated CDK4/MDM2 immunohistochemical positivity cannot independently support the diagnosis of liposarcoma, and highlights the irreplaceable value of molecular testing in the definitive diagnosis of atypical angiomatoid fibrous histiocytoma, providing a reliable reference for pathologists to avoid diagnostic errors in similar rare soft tissue tumors.
Background
Angiomatoid fibrous histiocytoma (AFH), first reported in 1979, is a rare type of soft tissue neoplasm accounting for ~0.3% of all soft tissue tumors. 1 According to the 2020 World Health Organization classification of soft tissue tumors, AFH is categorized as an intermediate (rarely metastasizing) tumor of uncertain differentiation. This tumor exhibits a broad age distribution, predominantly affecting children and adolescents (with a mean age at presentation of ~20 years) and becoming less common after the fourth decade of life. It shows a slight female predominance. The most frequent site of involvement is the superficial soft tissue of the limbs, although it can also arise in various other locations, including the head and neck, trunk, intracranial compartment, and retroperitoneum.2,3 Owing to its wide range of lesion sites, variable histological morphology, and lack of specific immunohistochemical markers, AFH is prone to diagnostic misinterpretation by clinicians and pathologists. This article reports a rare case of AFH expressing CDK4 and MDM2, which was initially misdiagnosed as dedifferentiated liposarcoma. By combining its clinicopathological features, immunophenotype, molecular findings, and relevant literature review, this study aims to improve diagnostic recognition of AFH and provide a reference for its accurate clinical diagnosis.
Case presentation
A 33-year-old female patient initially noted a nodule on her occipital scalp 3 years prior, measuring ~0.5 cm in maximum diameter. The lesion remained asymptomatic, and no specific intervention was undertaken. Over the past 3 months, the nodule enlarged rapidly, reaching a maximum diameter of about 2.2 cm. It was moderately firm in consistency and not associated with pain, pruritus, fever, fatigue, or other systemic symptoms. Preoperative laboratory blood examinations were completed, including routine blood tests, liver and renal function, and inflammatory markers (C-reactive protein, erythrocyte sedimentation rate, and interleukin-6 (IL-6)). All detected indicators were within normal ranges, with no anemia, elevated inflammatory status, or visceral dysfunction, and no typical paraneoplastic hematological abnormalities associated with AFH were observed. The patient subsequently presented to our hospital, where the mass was completely excised. Postoperative pathological examination revealed the following features: The tumor was predominantly situated within the dermis (Figure 1(a)), adjacent to the subcutaneous adipose tissue, with focally ill-defined borders. Central cystic degeneration was observed, with the cystic wall containing abundant red blood cells and no clearly identifiable endothelial lining. In the peripheral areas, tumor cells were seen surrounding sweat gland ducts (Figure 1(b)). Lymphocyte infiltration was observed both surrounding and within the tumor, with focal formation of lymphoid follicles. The tumor cells were arranged in a disorganized pattern and consisted mainly of oval to short-spindle forms. Some cells were distributed in sheets, while others showed focal vortex patterns (Figure 1(c)). Some cells exhibited signet-ring morphology or contained multiple vacuoles, resembling adipocytes (Figure 1(d)). In some areas, the nuclei were significantly enlarged, hyperchromatic, and exhibited marked pleomorphism (Figure 1(e)). Mitotic figures were readily identified (Figure 1(f)), with a hotspot density of ~20/2 mm2.

The histopathological features of this case. (a) The tumor mass is primarily located in the dermis, adjacent to the subcutaneous adipose tissue, with central cystic change. (b) Tumor cells are seen surrounding sweat gland ducts. (c) The tumor is composed of oval and short spindle-shaped cells, partially arranged in a vortex pattern. (d) Some cells exhibit a signet-ring cell-like appearance, while others contain multiple vacuoles, resembling an adipocyte. (e) Focal areas show markedly enlarged, hyperchromatic, and pleomorphic nuclei. (f) Mitotic figures are readily identifiable.
Immunohistochemical analysis demonstrated moderate-to-strong nuclear positivity for CDK4 in ~80% of tumor cells (Figure 2(a)). MDM2 expression was observed in about 50% of cells, with staining intensity varying from weak to strong (Figure 2(b)). The Ki-67 proliferation index reached ~20% in hotspot regions (Figure 2(c)), and some tumor cells were positive for CD68 (Figure 2(d)). Staining for CD21, S-100, CK-P, SMA, EMA, and Desmin was negative. Combined with the significant pleomorphism of tumor cells, active mitotic activity, high Ki-67 expression, and positive expression of CDK4 and MDM2, dedifferentiated liposarcoma (with the dedifferentiated component being polymorphic undifferentiated sarcoma) was initially considered. It was noted, however, that the lesion occurred in an atypical anatomical location and was associated with a prolonged clinical history. Consequently, fluorescence in situ hybridization (FISH) for CDK4 and MDM2 was recommended to further clarify the diagnosis.
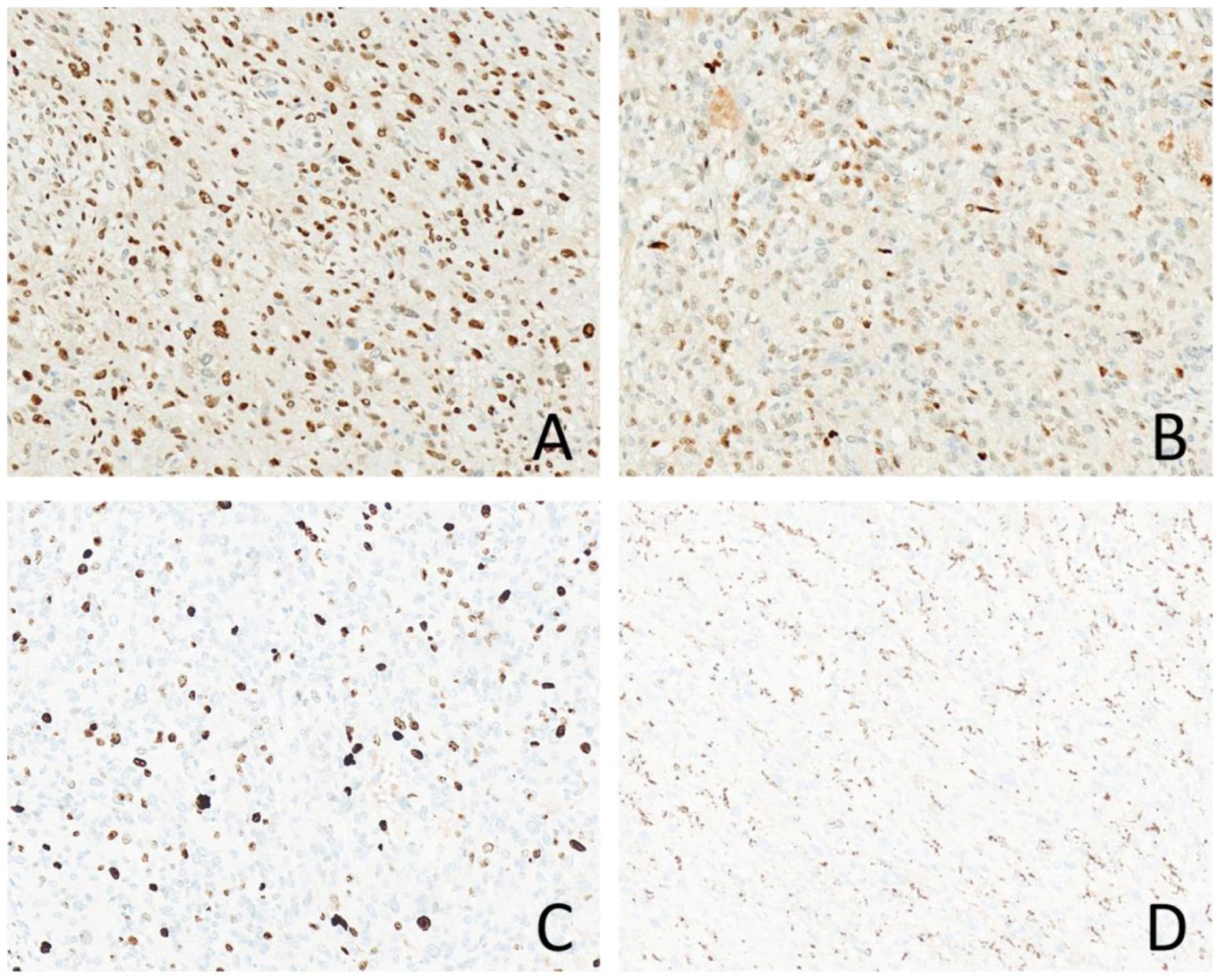
Immunohistochemical findings of this case. (a) Approximately 80% of tumor cells show moderate-to-strong positivity for CDK4. (b) About 50% of tumor cells exhibit variably positive staining for MDM2. (c) The Ki-67 proliferation index is ~20% in hotspot areas. (d) Some tumor cells are positive for CD68.
The patient was subsequently consulted at an external hospital, where additional FISH analysis for CDK4, MDM2, DDIT3, and EWSR1 was performed. The results revealed rearrangement of the EWSR1 gene only, with no aberrations detected in the other genes. A final diagnosis of AFH (ICD-O code: 8836/1) was established. The patient recovered well postoperatively and has remained recurrence- and metastasis-free during 20 months of regular follow-up.
Discussion
The clinical characteristics of AFH
AFH exhibits a broad age distribution, predominantly occurring in children and adolescents, with a mean age at onset of ~20 years. The condition is uncommon after the age of 40, and the oldest reported age of onset in the literature is 70 years. AFH shows a slight female predilection. The tumor most frequently arises in the superficial soft tissues of the extremities, typically within the deep dermis or subcutaneous layer. However, it has also been documented in a wide range of other anatomical sites, including the head and neck, trunk, intracranial cavity, 4 retroperitoneum, and other sites. With the accumulation of case reports, AFH has been increasingly recognized in various locations throughout the body, such as the lung, 5 mediastinum, 6 bone,7,8 and ovary.9,10 When AFH occurs in these unusual or rare sites, it is clinically prone to misdiagnosis as other diseases.11,12 Definitive diagnosis, therefore, depends on histopathological examination complemented by molecular testing. Clinically, AFH typically manifests as a slow-growing, painless mass. Its indolent progression frequently leads to misinterpretation as benign lesions, such as hematomas or hemangiomas. In addition, some patients may present with systemic symptoms, including fever, anemia, fatigue, and weight loss, accompanied by elevated inflammatory markers such as C-reactive protein, IL-6, and erythrocyte sedimentation rate. These systemic symptoms may be associated with the tumor’s characteristic EWSR1::CREB1 fusion gene.13,14 AFH exhibits non-specific radiological findings. On PET–CT(positron emission tomography–computed tomography), the tumor shows intense 18F-FDG uptake, closely resembling malignancies, such as lung cancer, limiting the diagnostic value of SUVmax (maximum standardized uptake value). Nevertheless, PET–CT plays a significant role in clinical staging and monitoring response to therapy.15,16
The histopathological features of AFH
Histologically,17,18, classic AFH exhibits the following three characteristic morphological features: (1) Pseudoangiomatous spaces: Approximately 70% of AFH cases show slit-like spaces surrounded by tumor cells. These structures are not true blood vessels and often contain erythrocytes and hemosiderin deposits, the latter resulting from hemoglobin degradation following intratumoral hemorrhage; (2) Lymphocytic cuff: Approximately 80% of AFH cases are surrounded by a lymphocytic cuff composed of infiltrating lymphocytes and plasma cells. Lymphoid follicles may occasionally form and merge with the outer fibrous pseudocapsule, giving the appearance of a lymph node-like structure at low magnification; (3) Histiocytoid cells: The tumor cells display morphological diversity, with a predominance of spindle-shaped forms, though polygonal, epithelioid, or histiocytoid morphologies may also be observed. Cellular atypia is absent or only mild. Some cases may contain a variable number of multinucleated giant cells. In addition to the classic morphology, AFH can also present as several special subtypes: (1) Solid type: This subtype exhibits less cystic change or hemorrhagic spaces, with predominantly solid tumor tissue. Pseudoangiomatous spaces are not prominent, and the inflammatory infiltrate is reduced compared to the classic type; (2) Sclerotic type: Characterized by prominent fibrous stroma with sparse cellularity and abundant collagen deposition, resembling a fibroma or sclerosing lesion; (3) Myxoid type: The stroma contains abundant myxoid matrix, with tumor cells arranged in cords or nests, morphologically resembling myxoid chondrosarcoma or acral fibromyxoma. However, it is often accompanied by lymphocytic infiltration and pseudoangiomatous spaces; (4) Pleomorphic cell type: Tumor cells exhibit significant pleomorphism, including variation in size, prominent nucleoli, and an increased mitotic rate, though mitoses generally do not exceed 5/2 mm2. Classic features such as pseudoangiomatous spaces and lymphocytic cuffing are still present, necessitating differentiation from undifferentiated pleomorphic sarcoma. In this case, focal areas of the AFH showed marked cellular pleomorphism with a mitotic count reaching 20/2 mm2, which significantly deviates from the typical morphological profile of classic AFH. This feature was a principal factor contributing to the initial misdiagnosis as a malignant neoplasm. Nevertheless, when considered alongside its characteristic findings – including lymphocytic cuffing, pseudoangiomatous spaces, histiocytoid cellular morphology, and histiocytoid morphology – the possibility of AFH should still be considered.
The immunohistochemical and molecular genetic characteristics of AFH
The lineage differentiation of AFH remains unclear, and its immunohistochemical profile is complex without specific markers. It is hypothesized that AFH may show histiocytic or myofibroblastic differentiation. Vimentin is the only marker consistently expressed in AFH. Desmin shows positivity in 50%–67% of cases, often presenting a dendritic staining pattern that may aid in diagnosis. There is no consensus on the positive rates of CD99, EMA, and CD68. According to existing studies, 19 CD99 is positive in ~50% of cases, EMA in about 40%, while CD68 positivity varies more widely, ranging from 15% to 50%. Focal expression of myogenic markers such as SMA and MyoD1 may be observed in some cases. Typically, AFH is negative for markers including CD34, S-100, and CK-P. In classical AFH, the Ki-67 proliferation index typically ranges from 2% to 4%. Literature reports indicate that nuclear positivity for SOX9 can assist in the diagnosis of AFH. 20 AFH frequently harbors EWSR1::CREB1/ATF1 or FUS::ATF1 fusion genes. As a transcription factor, SOX9 is specifically expressed in EWSR1-rearranged tumors, such as AFH and clear cell sarcoma. 21 Its high expression aids in distinguishing AFH from morphologically similar tumors such as Ewing sarcoma. In addition, literature reports indicate that AFH can diffusely express ALK.22,23 This immunoreactivity may be related to EWSR1 gene rearrangement, which could lead to ALK protein overexpression via cis-regulation or intergenic interactions. Consequently, AFH must be distinguished from ALK-positive inflammatory myofibroblastic tumor (IMT) and anaplastic large cell lymphoma. Co-expression of pan-TRK may also be observed in AFH. 24 Both the promoter regions of the ALK and NTRK genes contain binding sites for the CREB family of transcription factors, which can be directly activated by EWSR1/FUS::CREB fusion proteins. Approximately 85.7% of AFH cases are ALK-positive, and 64.2% are pan-TRK-positive; however, none of these cases show corresponding gene rearrangements or amplifications. Therefore, molecular testing is essential to differentiate AFH from other spindle cell tumors harboring NTRK rearrangements, among others. Study found MUC4 expression in 22.2% of AFH cases (4/18) with variable cytoplasmic staining, but did not address its mechanism of regulation. 25 Notably, MUC4 is a specific diagnostic marker for low-grade fibromyxoid sarcoma (LGFMS) and sclerosing epithelioid fibrosarcoma (SEF),26,27 tumors driven by FUS::CREB3L2 or FUS::CREB3L1 fusions. 28 Therefore, in MUC4-positive AFH cases, careful diagnostic evaluation is required to avoid misdiagnosis as LGFMS or SEF. Pan et al. reported 29 that TLE1 showed moderate-to-strong positivity in all 36 AFH cases (100%, 36/36), while BCOR showed similar positivity in 24 of the 36 AFH cases (67%, 24/36). Consequently, AFH also needs to be differentiated from tumors characterized by TLE1 and/or BCOR expression, such as synovial sarcoma and BCOR-rearranged sarcomas.
In this case of AFH, the Ki-67 proliferation index reached 20%, markedly higher than that typically reported in classical AFH. Furthermore, abnormal expression of CDK4 and MDM2 was observed, which has not been documented in existing literature. Clinically, CDK4/MDM2 immunohistochemical positivity is conventionally regarded as a typical biomarker signature of well-differentiated and dedifferentiated liposarcoma, usually accompanied by corresponding gene amplification. However, accumulating evidence demonstrates that such aberrant protein expression is not exclusive to liposarcoma and can be detected in multiple intermediate and malignant soft tissue tumors without underlying CDK4/MDM2 gene amplification. Accordingly, isolated CDK4/MDM2 immunohistochemical positivity cannot serve as an independent diagnostic criterion for liposarcoma and is insufficient to mandate a definitive differential diagnosis of dedifferentiated liposarcoma. Such marker positivity only necessitates tentative differential consideration, and the final diagnosis must be comprehensively determined by morphological features and molecular genetic results to avoid overdiagnosis and diagnostic bias. Overexpression of MDM2 and CDK4 is associated with tumors such as dedifferentiated liposarcoma, well-differentiated liposarcoma, low-grade central osteosarcoma, and parosteal osteosarcoma, frequently accompanied by corresponding gene amplification. As a proto-oncogene, MDM2 inhibits the tumor-suppressor function of p53 and promotes cell proliferation. The mechanism underlying MDM2 expression in this AFH case may be related to transcriptional dysregulation resulting from EWSR1 gene rearrangement, which could indirectly affect MDM2 transcription or protein stability, leading to its overexpression. In this case, ~50% of tumor cells showed variably positive staining for MDM2. Whether this pattern reflects wild-type protein expression (analogous to p53) remains to be determined. CDK4, a key kinase regulating the G1/S phase transition of the cell cycle, can drive uncontrolled proliferation when amplified or overexpressed. In AFH, EWSR1 rearrangement may disrupt cell cycle checkpoints, potentially synergizing with the proliferative effect mediated by CDK4. Moreover, fusion genes such as EWSR1::CREB1/ATF1 may indirectly activate CDK4 transcription or stabilize its expression through abnormal transcriptional regulation, which may account for the markedly elevated Ki-67 proliferation index observed here. Therefore, CDK4 expression here may not stem from genetic alterations in the CDK4 gene itself.
Molecular studies17,30–32 have established EWSR1 gene rearrangement as a characteristic genetic alteration in AFH. More than 90% of cases harbor EWSR1-related translocations, most commonly EWSR1::CREB1 fusion (t(2;22)(q33;q12)), followed by EWSR1::ATF1 fusion (t(12;22)(q13;q12)). Detection of EWSR1 rearrangement by FISH or RT-PCR is considered the diagnostic gold standard for AFH. However, a negative result for EWSR1 does not exclude AFH, as a minority of cases (<10%) harbor a FUS::ATF1 fusion (t(12;16)(q13;p11)). In summary, the diagnosis of AFH is challenging due to its uncertain differentiation and the absence of definitive immunohistochemical markers. Consequently, it must be distinguished from a range of other soft tissue tumors, and definitive diagnosis ultimately relies on molecular testing.
Most previously reported AFH cases present typical indolent clinical behaviors, low mitotic activity, negative CDK4/MDM2 expression, and a low Ki-67 proliferation index ranging from 2% to 4%. The present case firstly reports an atypical AFH characterized by concurrent high Ki-67 proliferative activity (20%) and dual aberrant CDK4/MDM2 positivity, establishing a novel atypical immunophenotype of AFH. This finding breaks the traditional understanding of stable biomarker expression in AFH, confirms the non-specificity of CDK4/MDM2 markers in the differential diagnosis of soft tissue sarcomas, and resolves the clinical diagnostic dilemma between high-grade atypical AFH and dedifferentiated liposarcoma. Moreover, this case verifies that AFH with significantly elevated mitotic activity and focal invasive growth still achieves an excellent prognosis after complete surgical resection, supplementing the prognostic data of aggressive atypical AFH. This study provides a critical clinical and pathological reference for pathologists to improve diagnostic accuracy and reduce misdiagnosis of rare mimicking soft tissue tumors.
Differential diagnosis of AFH
AFH must be differentiated from various soft tissue tumors that share overlapping morphological features, immunophenotypes, or molecular alterations. The principal distinguishing characteristics are outlined as follows:
Aneurysmal fibrous histiocytoma: This tumor typically arises in the skin and exhibits the classic histological appearance of dermatofibroma. It lacks the thick fibrous capsule and lymphoplasmacytic infiltration zone, and is negative for both Desmin and EMA expression.
Malignant fibrous histiocytoma (undifferentiated pleomorphic sarcoma): Tumor cells show significant pleomorphism and cytological atypia, with easily identifiable mitotic figures. Histological findings often include Touton-type giant cells and a storiform growth pattern, accompanied by frequent hemorrhage and necrosis. In contrast to AFH, it does not display pseudovascular spaces and lacks EWSR1 gene rearrangements.
Dedifferentiated liposarcoma 33 : The tumor in this case was adjacent to the subcutaneous adipose tissue with focal, ill-defined borders. Microscopically, some cells exhibited signet ring-like morphology, while others contained multiple cytoplasmic vacuoles, morphologically resembling lipoblasts. Marked nuclear atypia was present, and mitotic figures were easily identifiable. Based on the above morphological features, the initial differential diagnosis included adipocytic malignant neoplasms, namely dedifferentiated liposarcoma without a well-differentiated liposarcoma component, pleomorphic liposarcoma, and myxoid liposarcoma. It has been reported in the literature34,35 that ~10% of dedifferentiated liposarcomas show no microscopically identifiable well-differentiated liposarcoma component (pure dedifferentiated type). However, this tumor typically arises in the retroperitoneum and deep soft tissues of the extremities, and is extremely rare in superficial locations. Immunohistochemically, it usually demonstrates diffuse and strong nuclear positivity for MDM2 and CDK4, and its molecular genetic hallmark is amplification of the MDM2 and CDK4 genes in the 12q13–15 region.36,37 In this case, although immunohistochemistry showed positivity for MDM2 and CDK4, FISH analysis revealed no amplification of the MDM2, CDK4, or DDIT3 genes, which did not support the diagnosis of dedifferentiated liposarcoma or myxoid liposarcoma. 38 Notably, AFH can occasionally show immunohistochemical positivity for MDM2 and CDK4, but without corresponding gene amplification, and characteristically harbors EWSR1 gene rearrangement, which serves as a key differentiating feature. FISH testing in this case confirmed the presence of the EWSR1 gene rearrangement. Based on the morphological features and literature review, the final diagnosis was AFH.
IMT: IMT commonly arises in deep soft tissues. Both IMT and AFH may show diffuse ALK positivity, but IMT is frequently associated with ALK/ROS1 protein abnormalities and corresponding gene rearrangements.22,39,40 In contrast, AFH lacks ALK rearrangement but is characterized by EWSR1 rearrangement.
LGFMS: This entity can be confused with AFH when AFH exhibits atypical morphology and expresses MUC4. LGFMS is characterized by the presence of FUS::CREB3L1/CREB3L2 fusion, whereas AFH typically features EWSR1::CREB1 fusion (less commonly EWSR1::ATF1 fusion).
Soft tissue clear cell sarcoma: Usually occurring in the extremities of children, this tumor also carries EWSR1::ATF1 fusions. However, its tumor cells show significant cytologic atypia, and melanin granules are present in approximately two-thirds of cases. Melanocytic markers such as HMB45 and S-100 are positive, in contrast to AFH, which is consistently negative for these markers. It is important to note that the EWSR1::CREB1 or EWSR1::ATF1 fusion genes are not exclusive to AFH; they are also found in malignant gastrointestinal neuroectodermal tumors, soft tissue clear cell sarcoma, and primary pulmonary myxoid sarcoma. Therefore, accurate diagnosis requires comprehensive correlation of histomorphologic, immunohistochemical, and clinical features.
Treatment and prognosis of AFH
Surgical resection is the cornerstone of treatment for AFH. The goal of surgery is to achieve complete, wide excision of the tumor and avoid residual disease, thereby reducing the risk of local recurrence. Overall, patients with AFH have a favorable prognosis, but the local recurrence rate can be as high as 15%, and the distant metastasis rate is <5%. 41 Therefore, long-term follow-up is particularly important. Even after metastasis, patients may still survive for a prolonged period. Key prognostic factors include tumor size, tumor site, and completeness of surgical resection; incomplete resection significantly increases the probability of local recurrence.
Surgical treatment considerations
The extent of surgical resection should be individualized. For small (⩽3 cm), superficial AFH with well-defined borders, no deep invasion, and mild histologic atypia, complete marginal excision with negative margins is sufficient. 42 For tumors with large volume (>5 cm), ill-defined borders, deep invasion, marked cellular pleomorphism, extremely high mitotic activity, or recurrent lesions, wide resection with adequate negative margins is strongly recommended to eliminate potential microscopic residual tumor cells.3,43 For tumors located in deep anatomical sites, adjuvant postoperative radiotherapy may help reduce the risk of recurrence. 44 In the present case, the tumor was located in the superficial scalp dermis, measured 2.2 cm in maximum diameter, with no deep invasion or high-risk features; thus, complete marginal excision with negative margins was sufficient, and extensive wide resection was not required to avoid unnecessary tissue damage and cosmetic impairment.
Adjuvant therapy considerations
Adjuvant radiotherapy or chemotherapy is not routinely recommended for primary AFH after complete resection. 45 Radiotherapy may be considered in specific scenarios, including positive margins, incomplete resection, recurrent disease, deep anatomical location, tumor size >5 cm, or high-grade histologic features (e.g. necrosis, high mitotic activity, and invasive growth), as it can effectively eliminate microscopic residual disease.44,46 Chemotherapy is reserved for advanced disease with distant metastasis, unresectable multiple recurrent lesions, or progressively aggressive AFH.45,47,48 Conventional adjuvant chemotherapy provides no survival benefit. In the present case, the surgical margins were negative, with no residual disease, no metastasis, and no high-risk factors; therefore, no adjuvant radiotherapy or chemotherapy was administered postoperatively.
Treatment of recurrent/metastatic disease and targeted therapy
For patients with recurrent or metastatic AFH, treatment options include repeat surgical resection, chemotherapy, and radiotherapy. In the realm of precision medicine, the ALK-targeted agent crizotinib is effective in some ALK-positive patients. 49 The IL‑6 inhibitor tocilizumab may be used in patients harboring the EWSR1::CREB1 fusion, with favorable responses reported in some cases. 13 In addition, MUC4 represents a potential therapeutic target for AFH, warranting further investigation (Supplemental Material).
Conclusion
AFH is a rare intermediate mesenchymal tumor with uncertain differentiation. Classic cases can be readily diagnosed based on characteristic histopathological features and molecular alterations. However, when the morphology is atypical (such as significant pleomorphism and increased mitotic activity), and immunohistochemistry demonstrates aberrant expression of other tumor-related markers such as ALK, MUC4, MDM2, and CDK4, misdiagnosis becomes highly likely. Immunohistochemical detection of SOX9 shows some preliminary screening value in the diagnosis of AFH. However, definitive diagnosis relies on molecular confirmation of the EWSR1–CREB1/ATF1 fusion gene. It should be noted that this molecular alteration is not specific to AFH, and therefore, a comprehensive assessment incorporating multiple diagnostic indicators is required. Complete surgical resection constitutes the primary treatment modality, and the prognosis of patients is generally favorable; however, long-term follow-up is necessary to monitor for potential recurrence. Accurate diagnosis is the key to avoiding misdiagnosis (such as differentiation from LGFMS, dedifferentiated liposarcoma, etc.) and formulating individualized treatment plans. This case report offers a valuable reference for the diagnosis of AFH with uncommon phenotypic features.
Supplemental Material
sj-pdf-1-sco-10.1177_2050313X261460565 – Supplemental material for Angiomatoid fibrous histiocytoma with CDK4 and MDM2 expression misdiagnosed as dedifferentiated liposarcoma: The first case report and literature review
Supplemental material, sj-pdf-1-sco-10.1177_2050313X261460565 for Angiomatoid fibrous histiocytoma with CDK4 and MDM2 expression misdiagnosed as dedifferentiated liposarcoma: The first case report and literature review by Jiaodi Cai, Feng Jiang, Li Xiao and Wenqin Zhang in SAGE Open Medical Case Reports
Footnotes
Ethical considerations
Ethical review and approval were waived for this study because it was a case report of a single case.
Consent for publication
Informed consent was obtained from the patient to publish this paper.
Author contributions
Conceptualization: Wenqin Zhang and Jiaodi Cai. Histological examination: Li Xiao. Original draft preparation: Feng Jiang and Li Xiao. Writing – review and editing: Wenqin Zhang and Jiaodi Cai. All authors read and approved the final manuscript.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
The data presented in this study are available on request from the corresponding author due to privacy.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.