
Abstract
POEMS syndrome is a rare plasma cell disorder that is characterized by polyneuropathy, organomegaly, endocrinopathy, monoclonal plasma cell disorder, and skin changes, most commonly affecting middle-aged men. Due to its rarity and clinical overlap with other conditions, diagnosis relies on established mandatory, major, and minor criteria. We report a case of POEMS syndrome in a 17-year-old female. This case contributes to the limited literature on pediatric and adolescent presentations of POEMS syndrome and underscores clinical features that may aid in earlier recognition and diagnosis.
Introduction
A 17-year-old previously healthy female presented to the emergency department (ED) with a one-week history of generalized abdominal pain, shortness of breath, and body aches. Her physical exam was unremarkable. Complete blood count (CBC) and comprehensive metabolic panel (CMP) revealed mild hypoalbuminemia and microcytic anemia. Chest radiography (CXR) and computed tomographic (CT) angiography of the chest were negative for acute pathology, including pulmonary embolism. Her symptoms improved after receiving intravenous fluids. She was discharged with iron supplementation for presumed iron deficiency anemia and with plans for outpatient follow-up.
Two and a half weeks later, the patient presented again to the ED with recurrence of symptoms as well as new-onset palpitations. Physical examination was now notable for nonpitting edema extending to the mid-calf, tachycardia, mild abdominal distension, and swelling of the hands. Testing, including CBC, CMP, CXR, troponin, thyroid function tests, urinalysis, pregnancy test, and urine drug screen, was obtained. The results revealed persistent hypoalbuminemia, worsening microcytic anemia, proteinuria, and an elevated C-reactive protein and erythrocyte sedimentation rate. The patient was subsequently admitted to the general pediatrics floor for further management and workup.
Upon admission, the patient received a packed red blood cell transfusion and furosemide with significant improvement in symptoms and exam findings. Further workup identified nutritional deficiencies in iron, folate, vitamin D, and vitamin B12. Given concerns for a malabsorptive process, gastroenterology was consulted. Stool guaiac, celiac panel, and fecal alpha-1 antitrypsin were found to be unremarkable. Magnetic resonance enterography demonstrated omental and peritoneal thickening, raising concern for malignancy (Fig. 1); however, subsequent abdominal CT findings revealed colonic wall thickening more suggestive of an inflammatory rather than neoplastic process.
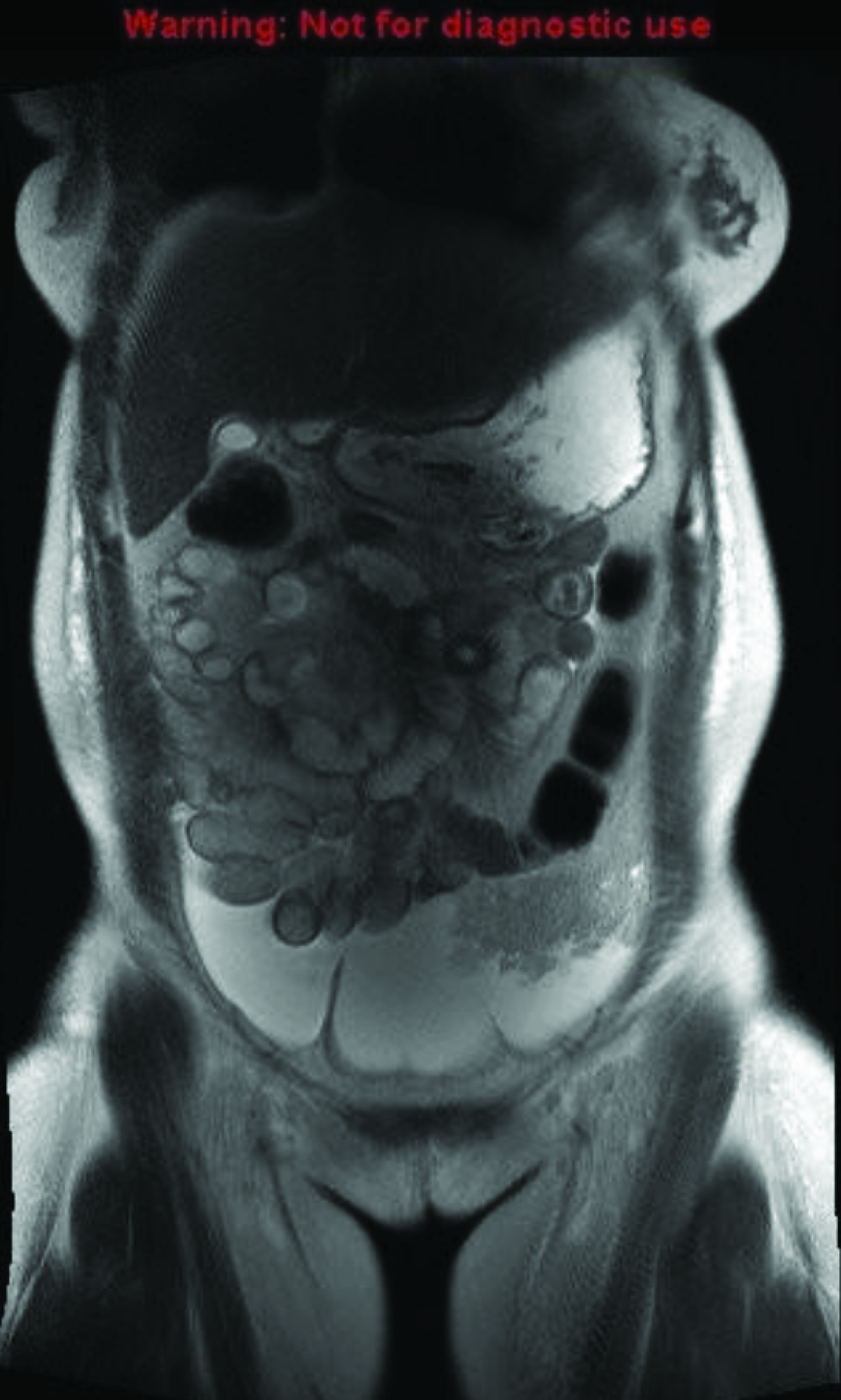
Magnetic resonance enterography showing ascites, bilateral small pleural effusions, and omental thickening of the left hemiabdomen with peritoneal thickening along the paracolic gutters.
On further history, the patient reported having a limited diet primarily consisting of fish, potato chips, lentil or vegetable soup, and watermelon. Her clinical presentation and lab abnormalities were initially attributed to a combination of poor nutritional intake and suspected underlying inflammatory bowel disease with malabsorption. However, outpatient esophagogastroduodenoscopy and colonoscopy with biopsies showed mild pill-induced gastritis without other significant abnormalities. With no inflammatory findings identified, the patient was referred to surgery for the omental biopsy to rule out malignancy.
Several weeks later, prior to the biopsy, the patient returned to the ED with reports of difficulty concentrating and sleeping due to paresthesias in her feet bilaterally. She also endorsed early satiety secondary to abdominal fullness. Her physical exam revealed a 7-kilogram weight increase from discharge, facial hyperpigmentation, abdominal distension, and nonpitting edema up to her ankles. The patient was re-admitted for further workup and management. Labs were again significant for persistent microcytic anemia, hypoalbuminemia, and proteinuria. A CT scan of the chest, abdomen, and pelvis showed gallbladder sludge, mild splenomegaly, increased ascites, and mesenteric edema (Fig. 2). A diagnostic paracentesis yielded three liters of fluid with a low serum-ascites albumin gradient, raising concern for an infectious or malignant etiology or, less commonly, a nephrotic process, rather than portal hypertension. Peritoneal cultures, including those for fungal and acid-fast bacilli, were negative, and cytology was negative for malignant cells. An inpatient omental biopsy was performed and showed chronic inflammation and reactive mesothelial hyperplasia with no evidence of malignancy.
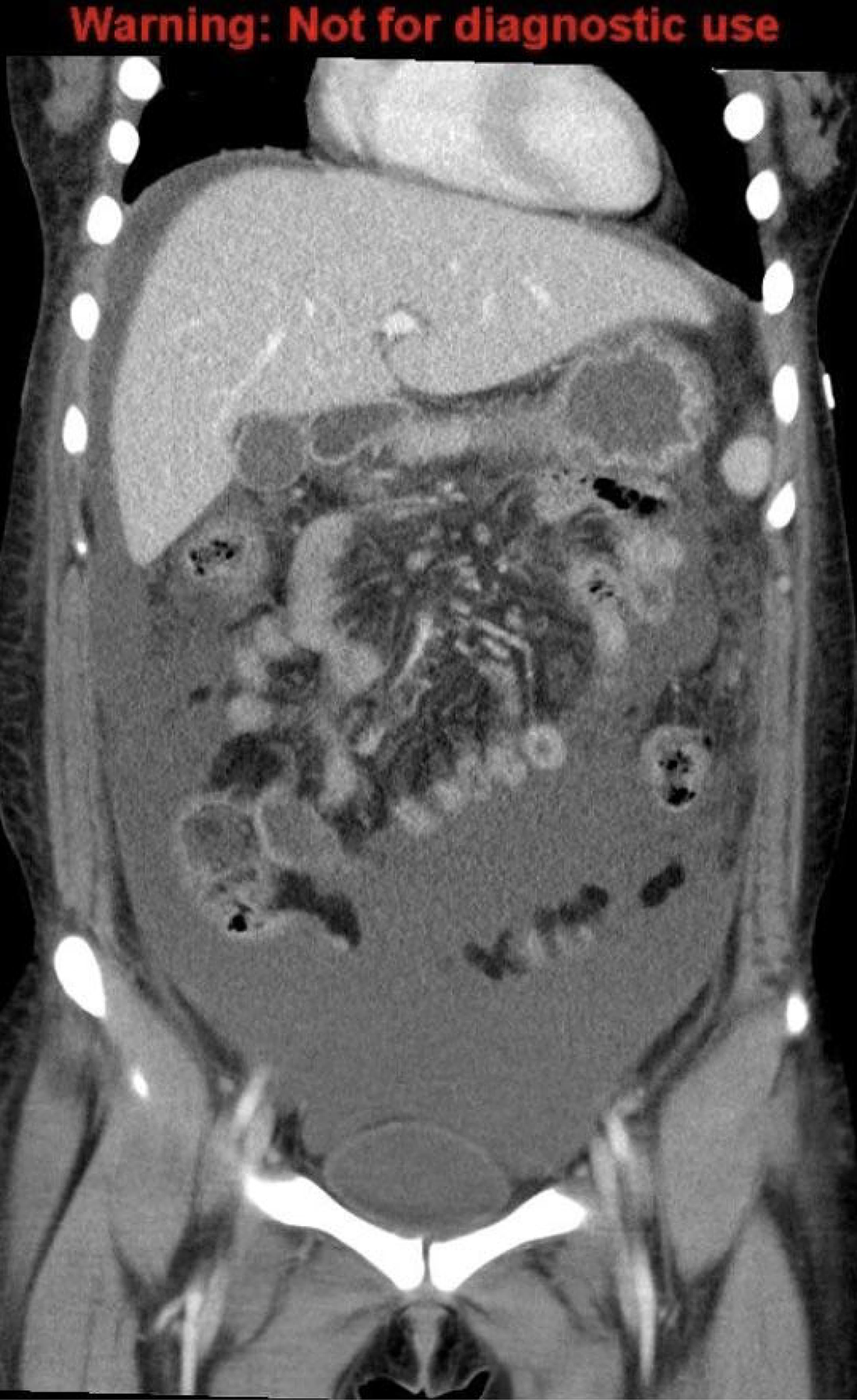
Computed tomography of the chest, abdomen, and pelvis one month after initial imaging showing increased ascites, increased pleural effusions, and diffuse mesenteric edema.
The patient was initiated on scheduled furosemide and antihypertensive therapy in consultation with nephrology. Rheumatology was also involved due to persistently elevated inflammatory markers. Although a comprehensive autoimmune panel was negative, serum protein electrophoresis (SPEP) with reflex to immunofixation electrophoresis (IFE) was recommended given her ongoing, unexplained proteinuria. This revealed elevated kappa and lambda free light chains, as well as a small, nonquantifiable paraprotein spike in the gamma region identified as monoclonal immunoglobulin G (IgG) lambda. A bone marrow biopsy was performed and revealed hypocellular marrow suggestive of iron deficiency anemia but was otherwise unremarkable. Flow cytometry showed no evidence of a monoclonal B-cell population or of T-cell abnormality.
With the identification of an abnormal SPEP, the differential diagnosis broadened to include conditions more typical of adult populations, such as multiple myeloma, monoclonal gammopathy of undetermined significance, Waldenström macroglobulinemia, and polyneuropathy, organomegaly, endocrinopathy, monoclonal plasma cell disorder, and skin changes (POEMS) syndrome.
As paresthesias became the patient’s most prominent and debilitating symptom, electromyography (EMG) of the upper and lower extremities was performed and revealed severe, diffuse demyelinating motor and sensory polyneuropathy with secondary axonal injury. These findings prompted additional testing, including an elevated vascular endothelial growth factor (VEGF) level and ophthalmological exam, which revealed grade 3 optic disc edema, confirming the diagnosis of POEMS syndrome.
Discussion
This 17-year-old patient’s constellation of symptoms, including demyelinating polyneuropathy, biclonal gammopathy, elevated VEGF, organomegaly (splenomegaly), extravascular volume overload, skin changes (hyperpigmentation of the face), and papilledema, fulfilled the diagnostic criteria for POEMS syndrome. POEMS syndrome is a rare paraneoplastic disorder associated with an underlying plasma cell neoplasm. 1 It has an estimated incidence of 0.3 cases per 100,000 individuals and typically presents in males in the fifth or sixth decade of life. 1 Excluding the present case, only five pediatric cases of POEMS syndrome have been reported, occurring in patients age 6–17 years with a median age of 15, with only one case described in a female patient within this age group.2–6 As illustrated by our case, the diagnosis and management of POEMS syndrome require a multidisciplinary approach and a high index of suspicion.
The diagnosis of POEMS syndrome requires the presence of two mandatory criteria, one of three major criteria, and one of three minor criteria (Table 1). The first mandatory criterion for the diagnosis of POEMS syndrome is polyneuropathy. While neuropathy is often the initial manifestation in adult patients, clinical presentations in pediatric cases have demonstrated marked variability. 8 Among the five previously reported pediatric cases of POEMS syndrome, polyneuropathy was the presenting feature in only three and was notably absent in the remaining two.2–4 Of the two pediatric cases without polyneuropathy, one was considered an incomplete diagnosis, while the other reported leg pain despite a normal EMG study, with the authors suggesting that the absence or delayed presentation of these findings may have been related to the patient’s young age.2,4 The polyneuropathy seen in POEMS syndrome not only has variable clinical presentations but is also frequently misdiagnosed. One study found that up to 60% of patients with POEMS syndrome were initially misdiagnosed with chronic inflammatory demyelinating polyneuropathy. 8 In our patient, although polyneuropathy was a late manifestation, its severity and progression became her most prominent and debilitating symptom, which ultimately prompted additional workup. The neuropathy associated with POEMS syndrome is characteristically demyelinating, 7 as was demonstrated in our patient’s EMG showing severe, diffuse demyelinating motor and sensory polyneuropathy.
Criteria for the Diagnosis of POEMS Syndrome

Reference: Dispenzieri, 2021. 7
Note: Adapted with permission from the Journal of Hematology.
The second mandatory criterion is an elevation in monoclonal antibodies, most commonly of the lambda light chain type. 7 In our patient, SPEP with reflex to IFE revealed elevated levels of both lambda and kappa light chains. Expression of kappa light chains is exceedingly rare in patients of all ages with POEMS syndrome, with only one other pediatric case documented in the literature. 2 Although clonal plasma cells can be identified by bone marrow biopsy using flow cytometry, immunohistochemistry, or immunofluorescence, free light chains are more reliably assessed through serum or urine assays, as illustrated in this case where the bone marrow biopsy was unremarkable despite abnormal SPEP with reflex to IFE and clinically significant disease. 9
The three major criteria for POEMS syndrome include Castleman disease, osteosclerotic bone lesions, and elevated VEGF levels. 7 At least one of these findings must be present to meet the diagnostic criteria for POEMS syndrome. Castleman disease, a rare condition characterized by benign lymph node overgrowth, is often associated with POEMS syndrome. 5 Osteosclerotic bone lesions are seen in about 95% of POEMS cases, typically in bone marrow-producing areas such as the pelvis, spine, ribs, and proximal extremities, and can be detected on CT or positron emission tomography (PET). 5 Although a PET scan performed after our patient’s diagnosis as well as multiple CT scans obtained during her hospitalizations did not reveal osteosclerotic lesions or features suggestive of Castleman disease, prior pediatric cases of POEMS syndrome have documented both findings.5,6 In our patient, the only observed finding that met a major criterion was an elevated VEGF level. Chronic overproduction of VEGF is believed to play a key role in the disease process of POEMS syndrome, contributing to increased vascular permeability, peripheral edema, ascites, pleural effusions, neovascularization, and polyneuropathy. 5
At least one minor criterion is required for the diagnosis of POEMS syndrome and may include organomegaly, extravascular volume overload, endocrinopathies, skin changes, papilledema, and thrombocytosis or polycythemia. 7 Organomegaly typically affects the spleen, liver, and lymph nodes. In our patient, imaging revealed mild splenomegaly, and clinically she exhibited significant extravascular volume overload characterized by peripheral edema and recurrent ascites that required diuretic therapy. Adrenal, thyroid, pituitary, gonadal, parathyroid, and pancreatic endocrinopathies have been previously reported in patients with POEMS syndrome. 7 Due to a lack of clinical signs of endocrinopathy, endocrine evaluation was performed only after our patient's diagnosis of POEMS syndrome was established and was negative. Common skin changes include hyperpigmentation—which was present on our patient’s second presentation—as well as hypertrichosis, glomeruloid hemangiomata, plethora, acrocyanosis, flushing, and white nails. 7 Furthermore, our patient’s ophthalmological exam revealed grade 3 papilledema, although neither thrombocytosis nor polycythemia was observed. Other signs and symptoms include clubbing, weight loss, hyperhidrosis, pulmonary hypertension/restrictive lung disease, thrombotic diatheses, diarrhea, and low vitamin B12 values. 7
There is no standard treatment for POEMS syndrome. Interestingly, although elevated VEGF levels are common and one of the major criteria in POEMS syndrome, the disease does not typically respond to anti-VEGF therapy. 7 The most successful outcomes are associated with therapies that target the underlying clonal plasma cell disorder and include radiation, chemotherapy, and hematopoietic stem cell transplant. 7 Although data remain limited, autologous stem cell transplantation has shown the highest response rates, with significant clinical improvement observed in 100% of surviving patients compared with approximately 50% of patients treated with corticosteroids alone or a combination of cyclophosphamide and dexamethasone. 7 Our patient experienced near-complete resolution of symptoms with chemotherapy consisting of cyclophosphamide and mesna, followed by autologous stem cell transplant. Additionally, her SPEP with reflex to IFE demonstrated a reduction in both kappa and lambda free light chains from the time of diagnosis to 2 weeks after initiation of cyclophosphamide and mesna, with further normalization observed 2 months following autologous stem cell transplantation (Table 2). The small paraprotein spike in the gamma region of the SPEP, identified as monoclonal IgG lambda, also became undetectable following treatment.
Serum Protein Electrophoresis with Reflex to Immunofixation Electrophoresis
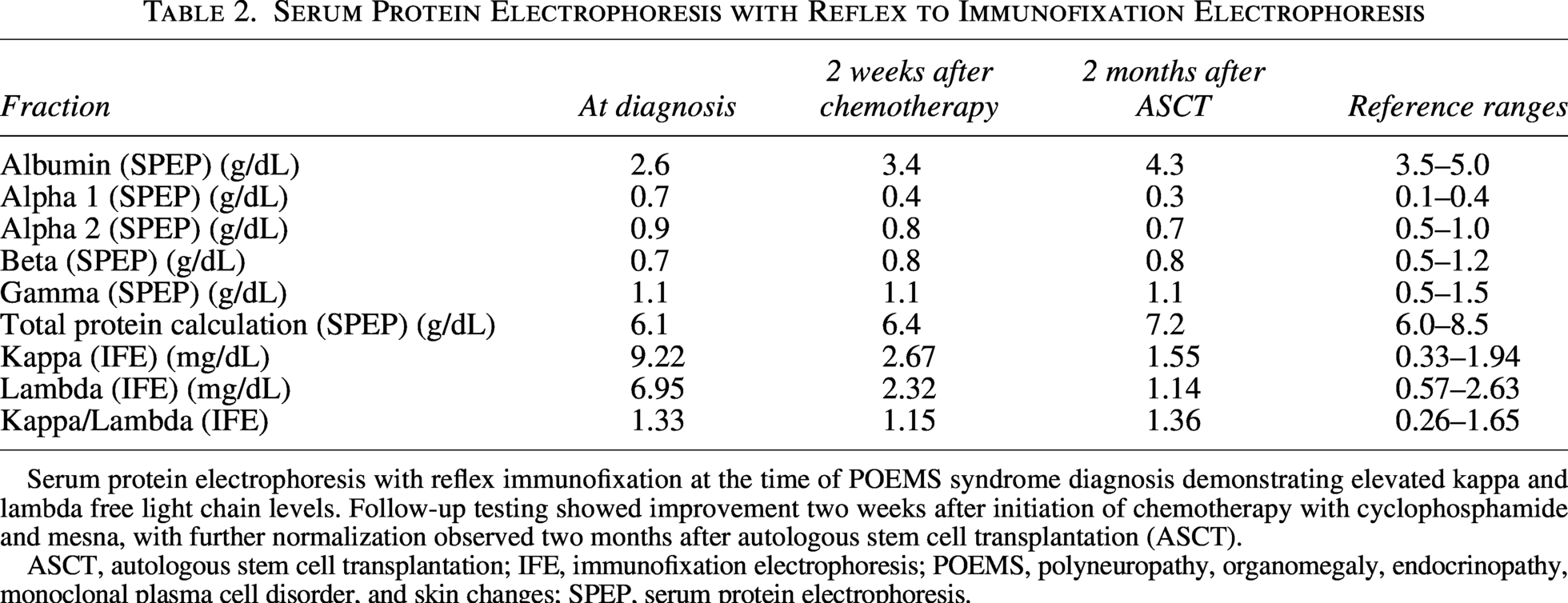
Serum protein electrophoresis with reflex immunofixation at the time of POEMS syndrome diagnosis demonstrating elevated kappa and lambda free light chain levels. Follow-up testing showed improvement two weeks after initiation of chemotherapy with cyclophosphamide and mesna, with further normalization observed two months after autologous stem cell transplantation (ASCT).
ASCT, autologous stem cell transplantation; IFE, immunofixation electrophoresis; POEMS, polyneuropathy, organomegaly, endocrinopathy, monoclonal plasma cell disorder, and skin changes; SPEP, serum protein electrophoresis.
Conclusion
To the best of our knowledge, only five cases of POEMS syndrome have been previously documented in the pediatric population.2–6 Given its rarity and significant clinical overlap with more common neurological, hematological/oncologic, and gastrointestinal conditions, POEMS syndrome poses a considerable diagnostic challenge in this age group. Polyneuropathy, a defining feature of the syndrome, may present later in the disease course or demonstrate variable manifestations in children and adolescents. Therefore, POEMS syndrome should be considered in pediatric patients with multisystem involvement and evidence of monoclonal gammopathy, even in the absence of polyneuropathy. Additionally, negative bone marrow findings do not exclude the diagnosis, as monoclonal free light chains may be more reliably detected through serum or urine assays. Early consideration of the diagnosis in pediatric patients with multisystem involvement—with or without polyneuropathy—may facilitate earlier recognition, appropriate diagnostic evaluation, and timely initiation of therapy.
Authors’ Contributions
C.S.P. and C.M.: Writing—original draft and writing—review and editing. C.C.: Conceptualization, supervision, writing—review and editing. All authors approved the final article as submitted and agree to be accountable for all aspects of the work.
Footnotes
Statement of Patient Consent
Not applicable.
Disclosure Statement
All authors have disclosed no financial relationships relevant to this article. This commentary does not contain a discussion of an unapproved/investigative use of a commercial product/device.
Funding Information
This project was done with no specific support.