
Abstract
There is a current lack of harmonized regulatory guidance in evaluating the genotoxic potential of oligonucleotide-based therapeutics (ONTs). In particular, guidance has not established the circumstances under which it is acceptable to deviate from the standard test battery. In this study, we analyzed genotoxicity testing strategies and supporting rationales for 91 noncoding ONTs receiving European Scientific Advice between 2004 and 2024. While the standard test battery was performed for the majority of ONTs, reduced test approaches were proposed for 10 products. Furthermore, we examined both the positions of applicants and corresponding European Union (EU) regulatory opinions to identify critical considerations in evaluating genotoxicity. Our findings show that EU regulators see opportunities to deviate from the standard test battery for ONTs if sufficient evidence for class experience can be demonstrated. This was confirmed for several ONTs with well-characterized chemical modifications (ie, phosphorothioate, 2′-methoxyethyl, and 2′-Omethyl), making the standard battery redundant in these cases. Although all reported genotoxicity tests have been uniformly negative, uncertainty remains for future modifications. Ideally, what constitutes sufficient evidence for class experience should be defined in the upcoming International Council for Harmonisation guideline addressing the nonclinical safety evaluation of ONTs (ICH S13), which would allow regulators to accept reduced testing. Together with the industry sharing more knowledge and underlying data that support growing class experience, this development can promote a harmonized approach for future genotoxicity testing of noncoding ONTs.
Introduction
Oligonucleotide-based therapeutics (ONTs) are a unique class of pharmaceuticals composed of chemically synthesized, short single- or double-stranded nucleic acid-based sequences. The ability of ONTs to interfere at the pre-translation level allows them to target and intervene in previously “undruggable” diseases. Combined with the versatility of sequence design, this feature has made them valuable modalities for treating rare diseases. Expertise in ONT development and clinical experience has grown over the last two decades, and the landscape of ONTs is currently further expanding to a wider range of diseases. 1 To date, over 19 antisense oligonucleotides (ASOs) and small interfering RNAs (siRNAs) have received market approval in Europe and/or the United States.2,3
Nowadays, most ONTs are chemically modified on their phosphate backbone [eg, phosphorothioate (PS)], pentose sugar moieties [eg, 2′-Omethyl (2′OMe) and 2′-methoxyethyl (2′MOE)], or bases (eg, 5-methylcytosine), with different modifications associated with different ONT classes. For instance, 2′MOE is a commonly applied modification to improve nuclease resistance and affinity for ASOs, whereas 2′OMe and 2′-fluoro modifications (2′F) are more commonly found in siRNA designs.4,5 In general, these chemical modifications improve ONT stability and efficacy as pharmaceuticals, resulting in a prolonged pharmacological effect while having a short plasma half-life, and differentiate ONTs from endogenous nucleic acids.4,6,7 However, they may also introduce toxicological effects that must be adequately evaluated. Additionally, conjugate and linker moieties attached to ONTs [eg, N-acetyl galactosamine (GalNAc)] also require safety assessment based on their characteristics. While pharmaceutical companies and regulators have traditionally approached the nonclinical safety evaluation of ONTs by treating them as small molecules due to their chemical synthesis, ONTs also share characteristics with biologics, resulting in a unique safety profile.8,9 As such, they do not fit neatly within the internationally accepted guidelines designed for small molecules or biologics.
A crucial stage during nonclinical development is conducting a genotoxicity risk assessment, in which a substance’s potential to cause genetic damage is evaluated. As described in the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) Guidelines M3(R2) 10 and S2(R1), 11 a standard test battery must be conducted to detect chromosomal damage and gene mutations that may result from interactions of small molecules with DNA. This standard battery usually consists of a bacterial assay (Ames test) to evaluate mutagenicity, an in vitro mammalian cell assay to evaluate chromosomal damage, and a rodent in vivo micronucleus study [ICH S2(R1)]. 11 In contrast to traditional small molecules, biologics (eg, peptides and proteins) generally do not warrant genotoxicity testing since direct interaction with DNA is not expected [ICH S6(R1)]. 12 In 2016, the Oligonucleotide Safety Working Group (OSWG) 13 genotoxicity subcommittee, consisting of industry, academic, and regulatory stakeholders, elaborated on the hypothetical mechanisms by which ONTs may induce genotoxicity and provided recommendations to address these ONT-specific concerns. 14 The primary genotoxic concern originates from chemically modified monomers that arise from ONT degradation, which upon incorporation into newly synthesized DNA could cause strand breakage or gene mutations. Other theoretical concerns are considered unlikely, such as nucleotide pool imbalance and sequence-dependent triple helix formation between the ONT and double-stranded genomic DNA. 14
Regional regulatory guidance, such as that of the Food and Drug Administration (FDA) and Pharmaceuticals and Medical Devices Agency (PMDA), and the OSWG recommendation paper agree that the standard battery as laid out in ICH S2(R1) is generally adequate to address ONT-specific genotoxicity concerns. However, these authorities and the OSWG genotoxicity subcommittee also concur that genotoxicity testing is not necessary for ONTs composed solely of natural nucleic acids.14–16 Even so, regional regulatory differences remain regarding whether deviations from the standard battery are considered acceptable under certain circumstances. In 2005, the reflection paper of the European Committee for Medicinal Products for Human Use [CHMP/Safety Working Party (SWP)] suggested that the genotoxic potential of new PS ONTs could be extrapolated from previously submitted negative standard battery test results of one assessed PS ONT with evidence for uptake and exposure deeming standard battery testing no longer necessary for this chemical class. 17 Furthermore, the concept of using class experience—in other words, informing on the negligible genotoxic potential for a new ONT based on previous negative test outcomes within the same chemical class—also appears in the recent FDA draft guidance. This guidance mentions the possibility of using class experience from approved products to inform about the genotoxic potential of “well-characterized” ONTs with certain class characteristics (ie, consistent size, structure, modifications, and impurity profile) differing in sequence only. 15 In contrast, the Japanese PMDA recommends following ICH S2(R1) for all “chemically modified nucleic acid drugs,” without mentioning any exceptions. 16 To harmonize regulatory expectations globally, the ICH has committed to drafting a new guideline that addresses the nonclinical safety evaluation of ONTs (ICH S13), 18 which is expected to be finalized in 2027.
Given these evolving and sometimes divergent regulatory perspectives, there is a clear need to understand how genotoxicity testing strategies for ONTs have been applied in practice. To address this issue, the present study analyzed genotoxicity test strategies from European Union (EU)-authorized ONTs as well as ONTs under development over the past 20 years, providing insights into testing approaches and their underlying rationales. We combined this approach with a review of EU Scientific Advice (SA) reports to identify critical aspects and perspectives on ONT genotoxicity evaluation from both the European regulatory authority [ie, CHMP, part of the European Medicines Agency (EMA)] and pharmaceutical developers (ie, applicants) to provide valuable insights that support the development of ICH S13.
Methods
Please refer to Supplementary Data for a more detailed description of the methods.
Selection of ONTs and corresponding nonclinical and regulatory documentation
In this study, we identified ONTs through the EMA internal tool Scientific Explorer, searching for all SA procedures with an ONT-specific “Substance/Product class” assigned to them from 1995 to 2024. Noncoding ONTs, such as ASOs and siRNAs, were considered within the scope of this study. We excluded aptamers and generic products even though they belong to the noncoding ONT family. ONTs targeting genomic DNA, as well as coding ONTs (including RNA- or DNA-based vaccines and gene therapies), were also excluded. The search strategy applied in the Scientific Explorer tool is presented in Supplementary Data.
Subsequently, we retrieved all identified SA reports and the corresponding nonclinical documentation (ie, briefing documents and/or investigator’s brochures) from EMA’s internal databases (IRIS and MMD). For ONTs with initial marketing authorization applications (MAAs) in Europe, we retrieved nonclinical MAA documentation (ie, Module 2.6) from the Medicines Evaluation Board’s (MEB) internal database.
Selection of nonclinical SA procedures addressing genotoxicity assessment
SA reports and their question-and-answer (Q&A) sections (containing the applicant’s questions and positions and the CHMP responses) were screened, and those with nonclinical or multidisciplinary advice, including nonclinical topics, were included (Supplementary Fig. S1; Dataset 2A).
All relevant Q&A sections were compiled into a single document, which was subsequently searched using genotoxicity-specific keywords to identify SA reports that discussed genotoxicity testing for ONTs (Dataset 2B). We reviewed the text surrounding keyword hits to confirm their relevance. The selection process was performed by C.S., with uncertainties discussed and resolved with B.D. and C.L.E.S.
Data extraction and analysis
From SA reports addressing genotoxicity testing, the topics of the questions along with the full-text CHMP and applicant statements were extracted for analysis (Dataset 2B). Unless mentioned otherwise, related SA procedures were treated independently (ie, in case there were multiple SA procedures for one product). However, for the analysis of test strategies and CHMP suitability decisions, related SA procedures were considered parts of a final test strategy and/or suitability decision, with the most recent SA procedure reflecting the conclusive outcome. To avoid duplicating advice in SA procedures that contained multiple Q&A sections on genotoxicity for the same ONT, we included only the most detailed and representative Q&A section for further analysis.
From the nonclinical documentation (Dataset 1), we extracted the number and types of tests performed per ONT to determine the genotoxicity test strategies that the applicants followed or proposed. Additionally, we determined the chemical composition of ONTs by extracting information on chemical modifications, conjugates, and linker structures.
Content analysis of genotoxicity-related aspects and test strategies discussed in SA reports
We performed multiple systematic text analyses on the SA reports, combining qualitative coding and quantitative frequency assessment, in order to evaluate diverse ONT genotoxicity aspects (Dataset 2B; Supplementary Fig. S1; Dataset 2C). To define the initial categories, one or two researchers (C.S. and B.D.) used a combined deductive–inductive coding approach. We started with preconceived themes related to the anticipated content, then performed an iterative review of the textual data from CHMP and the applicant. Next, uncertainties were discussed within the team (C.S., B.D., and C.L.E.S.) and discrepancies resolved by consensus. Finally, categories were compared, related categories merged, and the final classification discussed within the team until consensus was reached.
Unless indicated otherwise, one researcher (C.S.) reviewed and assigned all relevant Q&A sections with full-text CHMP and applicant statements to the final set of categories. All categorized text was descriptively analyzed using methods appropriate to the structure and content of the data. Deviations from this general procedure and additional methodological details are provided for each analysis in Supplementary Data.
Results
ONTs are predominantly assessed in the standard test battery
Our initial search of the EMA databases for the years 1995–2024 identified 238 SA procedures (containing advice on quality and clinical and/or nonclinical aspects) corresponding to 107 unique ONTs. For a total of 91 noncoding ONTs, including 62 ASOs, 27 siRNAs, and 2 other ONTs, we were able to extract the nonclinical testing strategies (Dataset 1). Out of the initial 238 SA procedures, 113 SA reports contained advice on nonclinical aspects (280 unique Q&A sections) for 82 ONTs (Dataset 2A). Out of 113 SA reports, 78 mentioned genotoxicity testing and were considered relevant for our study (Dataset 2B; Fig. 1A).
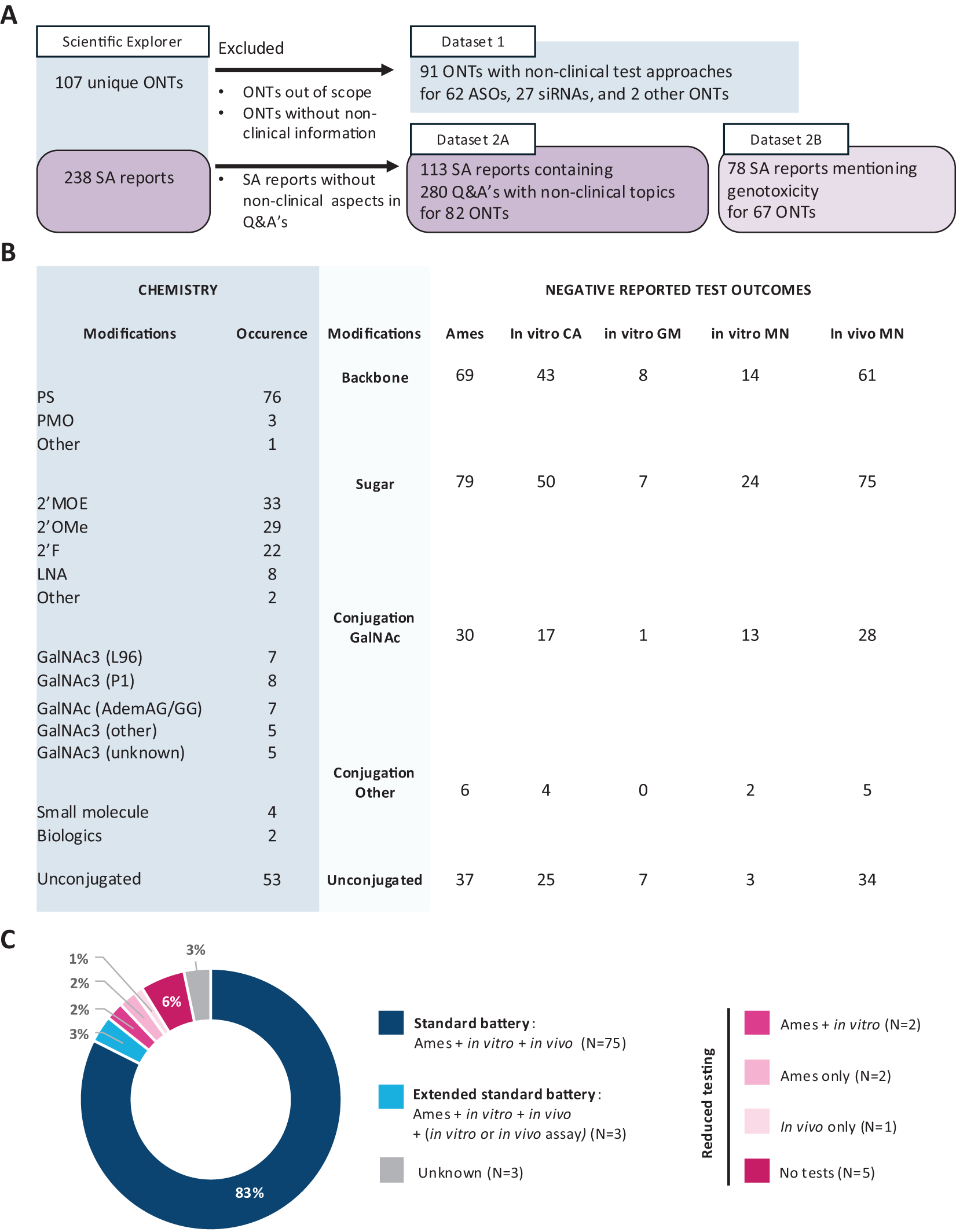
Identification and characterization of noncoding ONTs and their corresponding genotoxicity testing strategy.
The vast majority of ONTs in our dataset contained one or more types of chemical modifications; the most frequently occurring were PS linkages (n = 76), followed by 2′OMe (n = 33), 2′MOE (n = 29), and 2′F (n = 22) sugar modifications (Fig. 1B). While most ONTs were unconjugated (n = 53), GalNAc conjugates with different linker molecules were present in 22 out of 27 siRNAs. Only two ONTs consisted solely of naturally occurring nucleotides, while the chemistry of four ONTs was not disclosed by the applicant.
Most ONTs were tested, or the applicants planned to test them, using a standard test battery (n = 75, 83%) (Fig. 1C). The choice for the standard test battery appeared to be independent of the chemical composition or conjugation status of the ONT product (Fig. 1B, C). For 3% of ONTs, the standard test battery was further supplemented with an additional test, either an in vivo Comet assay (n = 1) or an in vitro mammalian cell assay (n = 2). In contrast, for 11% of ONTs, a reduced test package, or even no tests, was performed or planned (Fig. 1C). Finally, no dedicated genotoxicity studies for only the linker or conjugate moieties were reported. Importantly, for 72 ONTs with a known outcome of the genotoxicity tests, the results were reported uniformly negative by the applicant.
Triple helix formation and incorporation of chemically modified monomers are the main ONT-specific genotoxic concerns addressed in SA reports
To examine which ONT-specific genotoxic concerns were frequently addressed by applicants and CHMP in SA reports, we classified ONT-specific genotoxic concerns mentioned in relevant literature (Supplementary Data), which ultimately resulted in six final categories: “triple helix formation,” “incorporation of chemically modified monomers,” “imbalance of nucleotide pool,” “related to primary and/or secondary pharmacology,” “related to conjugation and linker,” and “related to impurities” (Supplementary Table S1).
In 44 out of 78 SA reports that mentioned genotoxicity testing, we identified a discussion by the applicant and/or the CHMP on genotoxicity test approaches for ONTs (Supplementary Fig. S1; Dataset 2C). In the remaining 34 reports, genotoxicity tests and/or their outcomes were mentioned solely as part of the overview of the nonclinical program or the applicant’s carcinogenicity assessment strategy (Supplementary Fig. S1).
The most frequently addressed genotoxic concern was triple helix formation, mentioned in 37% of SA reports (n = 29), followed by incorporation of chemically modified monomers, which was addressed in 12% (n = 9) (Fig. 2A). Notably, the concern for triple helix formation occurred less often in applicant statements (n = 11, 14%) than in CHMP statements (n = 18, 23%). The final recurrent genotoxic concern mentioned in SA reports was the concern for genotoxicity arising from process-related impurities, which was primarily addressed by applicants (n = 4, 5%) (Fig. 2A). The remaining three predefined genotoxic concern categories were not mentioned in any SA report.
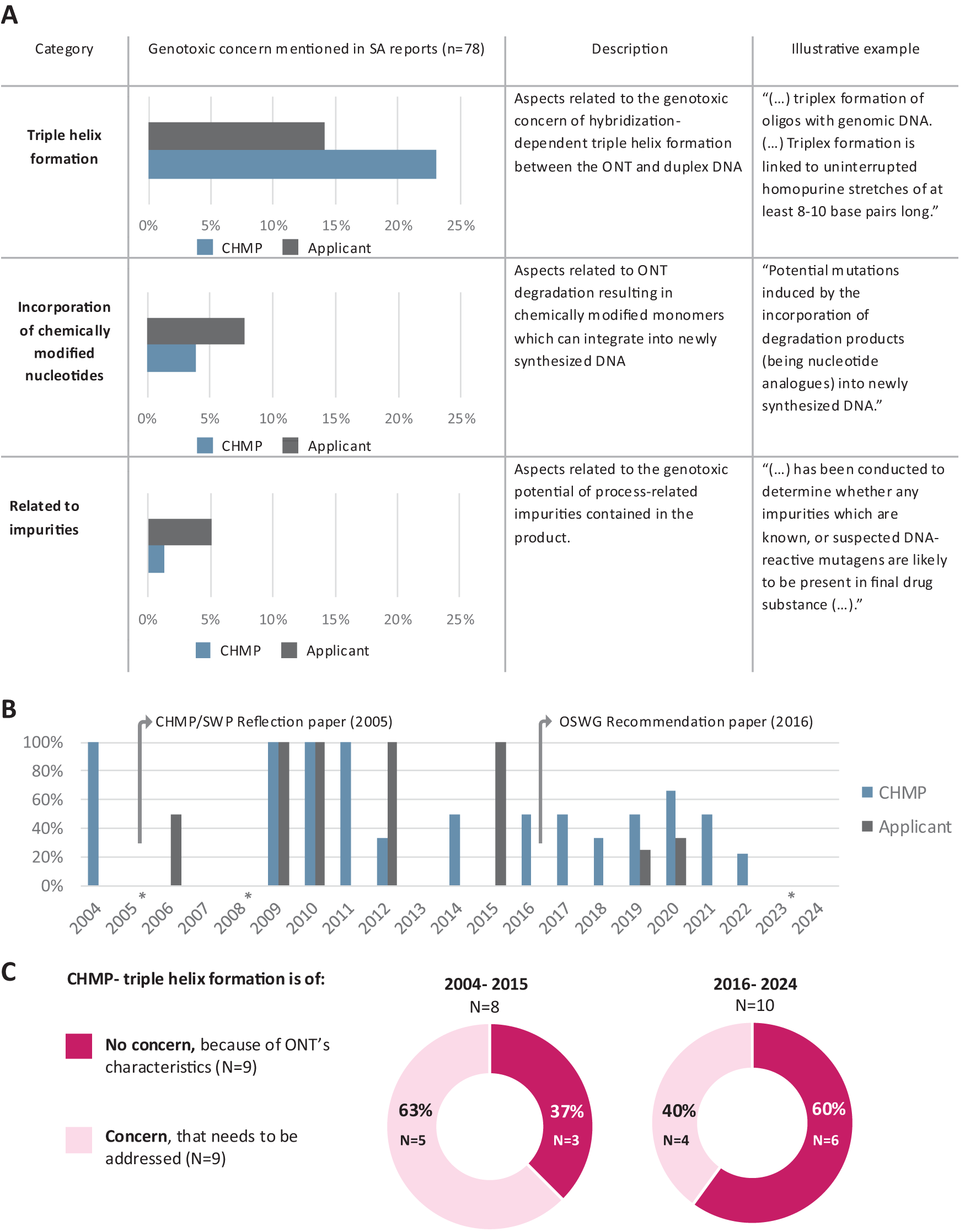
Concerns related to the incorporation of chemically modified monomers and triple helix formation were most frequently mentioned in SA reports.
To further evaluate whether the perspectives of applicants and CHMP regarding triple helix formation changed over time, we analyzed the frequency with which triple helix formation was mentioned in the European SA reports between 2004 and 2024. Generally, we found that triple helix formation was less frequently mentioned in both applicant and CHMP statements written after 2016 compared with reports written before 2016, especially by the applicants (Fig. 2B). To further assess the context in which the CHMP mentioned triple helix formation in these SA reports, CHMP statements were assigned to two iteratively derived categories: “concern, which needs to be addressed” and “no concern, because of the ONT’s characteristics.” In earlier SA reports from 2004 to 2015, the CHMP frequently regarded triple helix formation as a concern that should be addressed (n = 5, 63%), while in more recent SA reports (2016–2022), the CHMP’s perspective changed. In the majority of SA reports written after 2015, the CHMP no longer considered triple helix formation a concern, based on the absence of noninterrupted purine and pyrimidine nucleotide stretches in the presented ONT sequence (Fig. 2C).
The standard test battery is considered adequate, but it can be reduced when sufficient class experience exists
To provide insight into the EU regulatory perspective regarding the suitability of different test approaches for the genotoxicity evaluation of ONTs, the applicant’s argumentation for the test approach was classified into “no additional arguments provided” or “argues for class experience.” The corresponding classification of CHMP decisions on the suitability of the proposed test approaches resulted in five categories: “suitable—no comment,” “suitable—comment,” “unsuitable—no comment,” “unsuitable—comment,” and “unknown.”
In the vast majority of SA reports that mentioned genotoxicity testing, the CHMP agreed with the applicant and considered the standard battery a suitable test approach (45 out of 54 ONTs) (Table 1). Importantly, the CHMP mostly silently agreed with the performed standard battery tests, often as part of an agreement with the general nonclinical program or the presented carcinogenicity risk assessment, without discussing the reasons for their positive suitability decision for the presented genotoxicity test strategy. The CHMP opinion regarding the suitability of the standard battery testing remained unclear for five ONTs in the SA reports (Table 1). However, for four ONTs, the CHMP deemed the standard battery not suitable (Table 1). More precisely, for one ONT without modifications and three ONTs containing 2′MOE and PS modifications, the CHMP commented that performing the standard test battery was redundant. The CHMP justified this advice by suggesting that the genotoxic potential of the presented ONTs is negligible, based on sufficient evidence for class experience (Fig. 3A). Consequently, they recommended that the applicant reconsider the test strategy and refrain from performing any additional genotoxicity tests (Fig. 3A). Opposed to the standard test battery, reduced (n = 8) or no (n = 4) genotoxicity testing was the strategy chosen by applicants for 12 ONTs in corresponding SA reports (Table 1). In most cases, the applicant justified the lack of testing or a reduced test approach by referring to class experience (n = 7), which the CHMP deemed suitable for six ONTs (Fig. 3B). Additionally, the CHMP considered the proposed in vitro testing suitable for ONTs in two other cases, even though the applicant provided no direct evidence for class experience in their position, and instead, the CHMP addressed existing class experience in their answer (Fig. 3B). In contrast, for one ONT, the CHMP regarded the proposed reduced test strategies as inadequate because the applicant provided insufficient justification or evidence for the negligible genotoxic potential (Fig. 3B). The corresponding SA report indicated that the applicant provided class experience for only one modification (ie, PS), although the ONT appeared to contain additional modifications. As a result, the CHMP recommended that the applicant either perform a standard battery or provide a more detailed discussion of the genotoxic risks. In another case, the applicant did not provide any justification for performing only an Ames test; thus, the CHMP recommended that they conduct additional in vitro tests. However, in a follow-up procedure, the applicant referred to class experience for PS ONTs as a justification for their test approach, which the CHMP acknowledged, thereby deeming the reduced testing suitable.
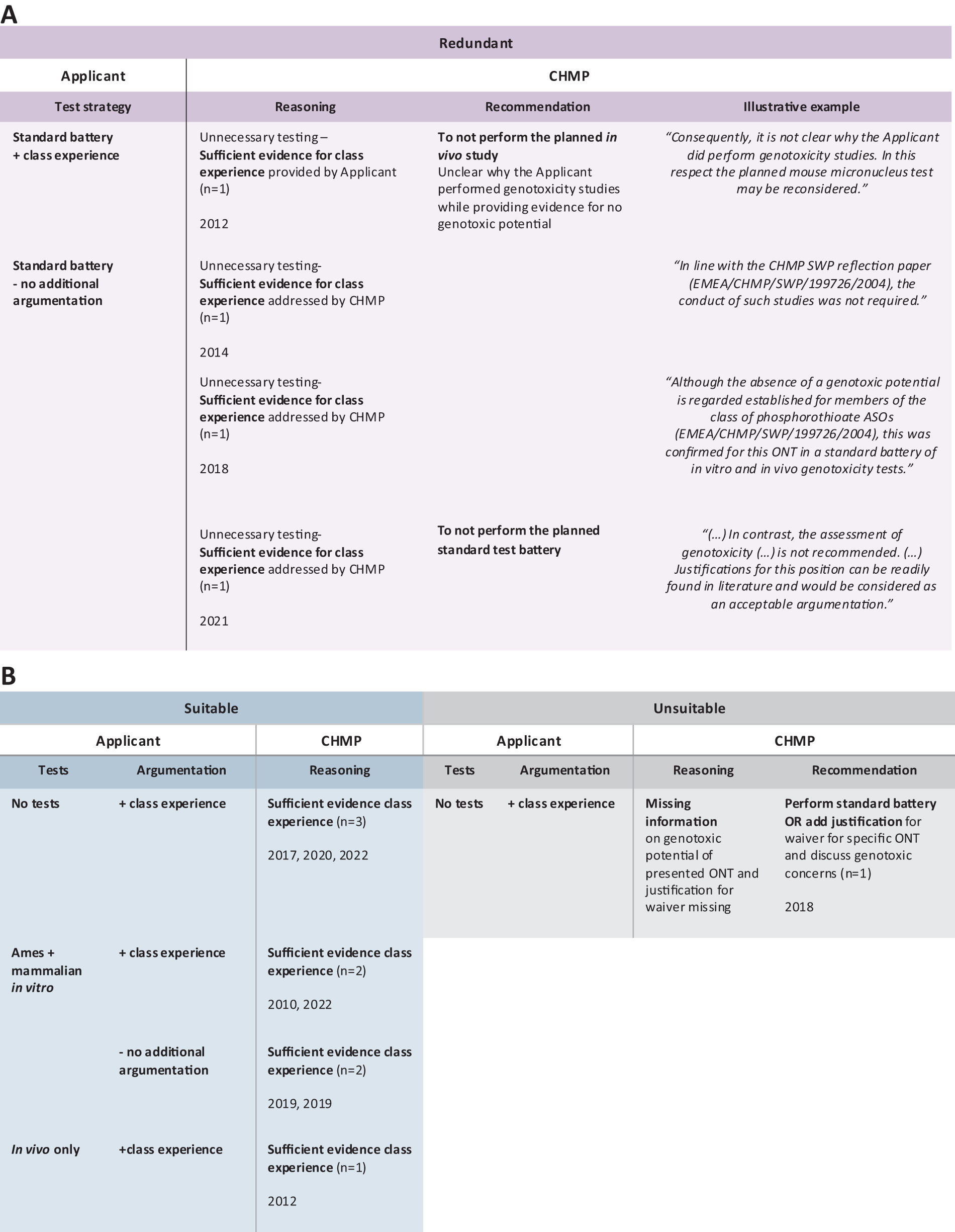
CHMP suitability decisions and reasoning regarding different genotoxicity test strategies.
Overview of European Committee for Medicinal Products for Human Use Suitability Decisions for Different Genotoxicity Test Strategies as Proposed in Scientific Advice Procedures
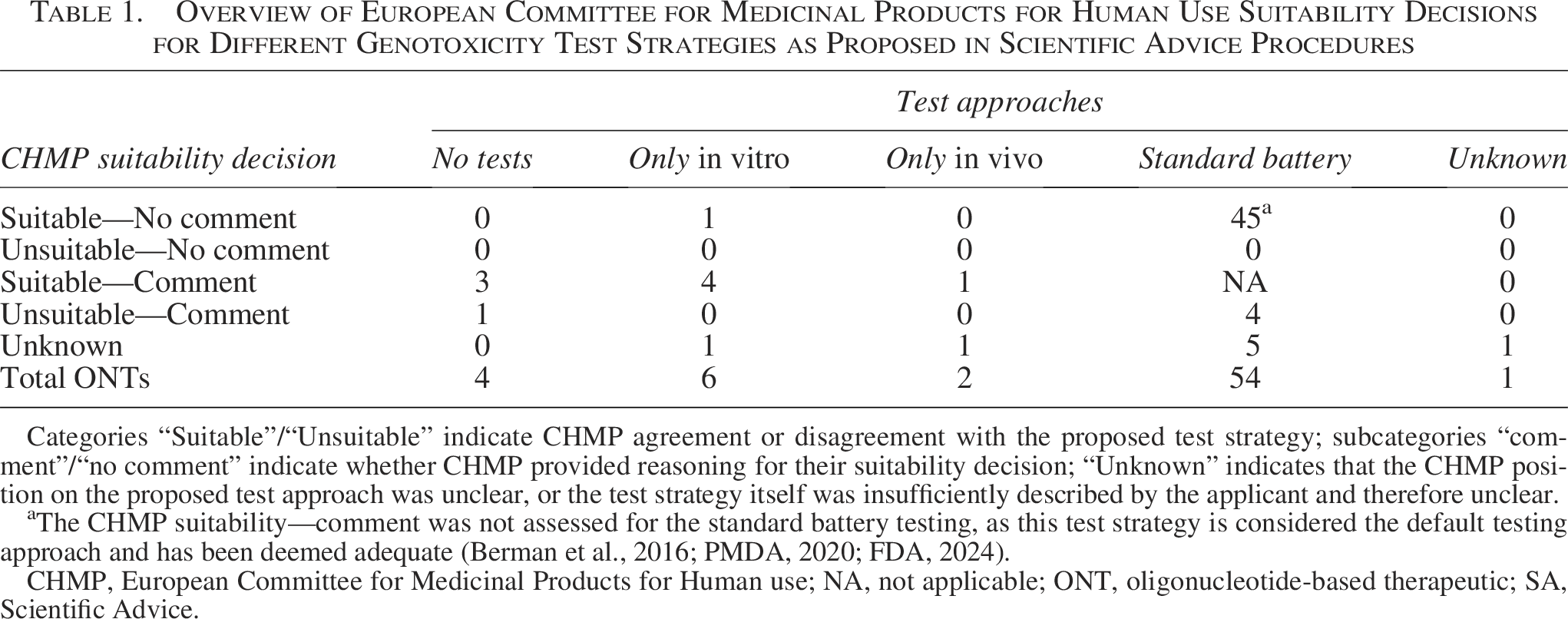
Categories “Suitable”/“Unsuitable” indicate CHMP agreement or disagreement with the proposed test strategy; subcategories “comment”/“no comment” indicate whether CHMP provided reasoning for their suitability decision; “Unknown” indicates that the CHMP position on the proposed test approach was unclear, or the test strategy itself was insufficiently described by the applicant and therefore unclear.
aThe CHMP suitability—comment was not assessed for the standard battery testing, as this test strategy is considered the default testing approach and has been deemed adequate (Berman et al., 2016; PMDA, 2020; FDA, 2024).
CHMP, European Committee for Medicinal Products for Human use; NA, not applicable; ONT, oligonucleotide-based therapeutic; SA, Scientific Advice.
Both the CHMP and the applicants mainly refer to class experience for the PS backbone modification in SA reports
The CHMP regarded sufficient class experience as a valid argument for conducting reduced testing. Therefore, we next investigated the extent to which the CHMP and the applicants acknowledged class experience for the same ONT modalities. Class experience for ONT modalities mentioned in CHMP and applicant statements could be categorized into five final categories: “general—chemistry not specified,” “known chemistry,” “conjugates,” “phosphate modifications,” and “sugar modifications.” When class experience for a certain modality was further specified, a subcategory was used to refer to specific ONT classes (ie, ASO), conjugations (ie, GalNAc3), or modifications (ie, PS) (Supplementary Fig. S2 and Supplementary Table S2).
Class experience was addressed in 33 out of 78 SA reports, regardless of the testing strategy (ie, in both standard and reduced testing strategies). Class experience was more frequently mentioned in applicants’ positions (39 mentions in 25 positions) than in CHMP opinions (27 mentions in 21 opinions), and it was predominantly addressed for specific chemical modifications [PS, 2′MOE, and 2′OMe (n = 43)] (Fig. 4A). CHMP and applicant statements in SA reports referred to general class experience for ONTs (n = 6) and ASOs (n = 9); however, no statement referred to general class experience for siRNAs (Fig. 4A and Supplementary Table S3). Ultimately, class experience was most frequently mentioned with regard to the PS modification (n = 27) and was equally addressed by both the CHMP (n = 12) and the applicants (n = 15) (Fig. 4A). In comparison, there were some mentions of class experience for ONT sugar modifications (n = 16), but they occurred less often than those associated with the PS modification. While the CHMP (n = 4) and applicants (n = 4) referred with equal frequency to class experience for the 2′OMe modification (n = 8), class experience for 2′MOE-modified ONTs was found to be referred to more often by applicants (n = 6) than by the CHMP (n = 2) (Fig. 4A).
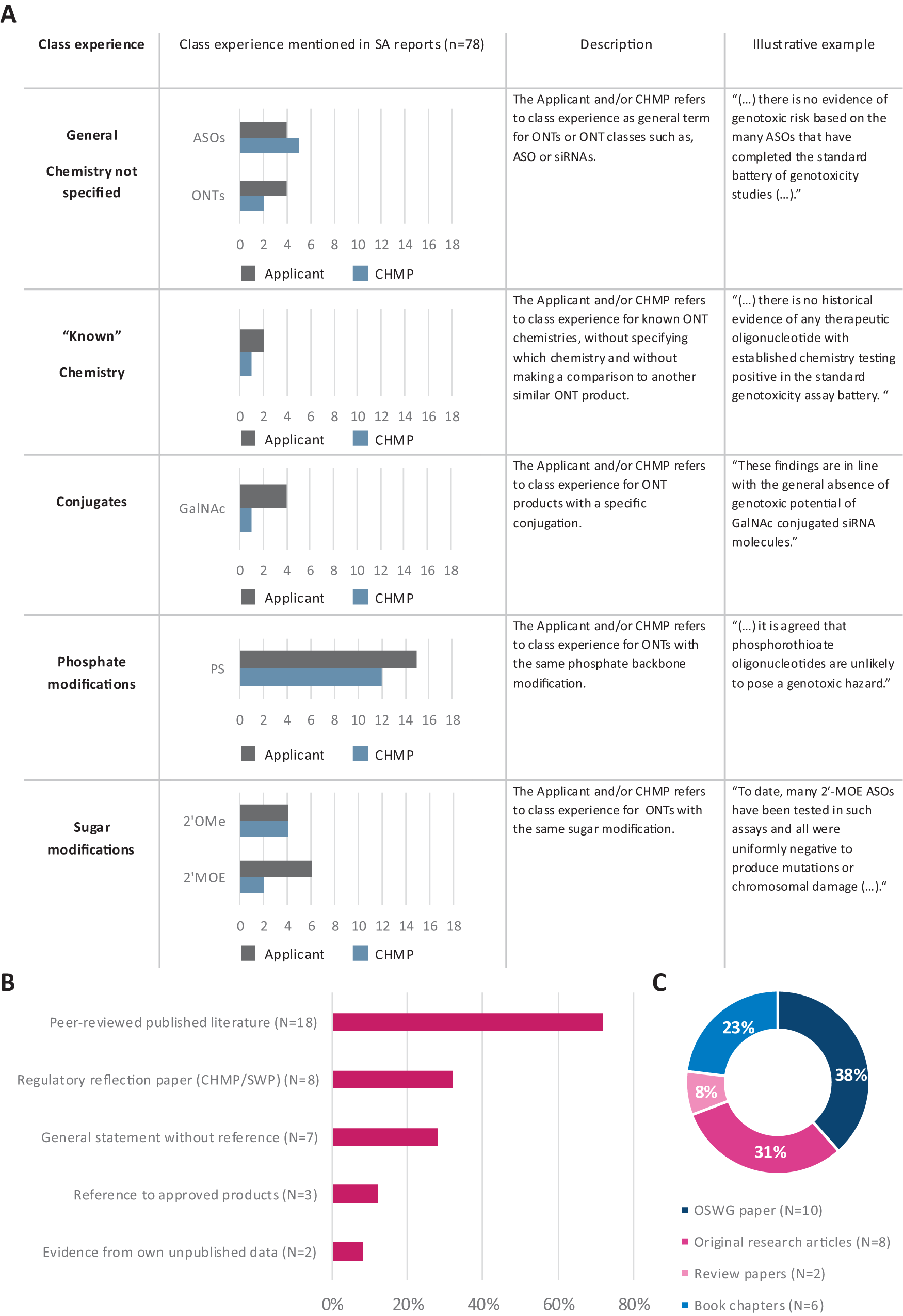
Class experience referring to PS backbone and 2′MOE and 2′OMe sugar modifications was mentioned most often in SA reports.
Applicants provide diverse sources of evidence when supporting their claim for class experience
Subsequently, we evaluated which sources of evidence applicants used in their statements to support their claim for class experience (n = 25). In total, five categories describing different sources of related evidence for class experience were derived: “peer-reviewed published literature,” “regulatory reflection paper (CHMP/SWP),” “general statement without reference,” “reference to approved products,” and “evidence from own unpublished data.” We found that in the majority (18 out of 25) of the SA reports, publicly available peer-reviewed literature was referenced (Fig. 4B). Furthermore, 32% of applicant statements (8 out of 25) referenced the CHMP/SWP reflection paper released in 2005, which discussed class experience for the PS backbone modification (Fig. 4B). The applicant referenced their own unpublished data only in two SA reports, whereas in seven SA reports, the applicant made a general assertion of class experience without providing a reference to substantiate this claim (Fig. 4B). Similarly, in three reports, the applicant referred to approved products without providing a specific reference to their documentation (Fig. 4B).
To provide more detailed insights into the peer-reviewed literature that was referenced, we further differentiated between referenced sources. The references to peer-reviewed published literature were diverse. Notably, the OSWG recommendation paper was referenced 10 times (38%) as a source to support class experience (Fig. 4C). This paper addresses class experience for ONTs with selected chemistries (eg, PS linkages, 2′MOE, and 2′OMe modifications). Original research articles were the second most cited source of peer-reviewed literature (31%); these articles presented negative test outcomes for ONTs with selected chemistries [eg, locked nucleic acid (LNA) and PS] and with confirmed cellular uptake and exposure into the test systems19,20 (Fig. 4C). Book chapters were referred to in 23% of cases, and 8% referred to review papers (Fig. 4C).
Discussion
Over the past two decades, EU regulators have generally considered the standard test battery suitable for assessing the genotoxicity of chemically modified noncoding ONTs, in agreement with industry and other regional regulatory viewpoints.14–16 However, when sufficient evidence for class experience was available for ONTs, the CHMP asserted that reduced test approaches or even no genotoxicity testing was adequate. This EU regulatory perspective aligns with the 2005 CHMP/SWP reflection paper and the recent FDA draft guidance describing the opportunity to deviate from the standard test battery under certain circumstances.15,17 However, this perspective diverges from the position presented in the 2020 Japanese PMDA guidance, which does not mention any possibility of deviating from the standard test battery for genotoxicity assessment of chemically modified ONTs. 16 The FDA definition for class-based experience and well-characterized ONT classes is currently restricted to evidence based on approved products, while our analysis of EU SA reports indicates that the EU regulator’s definition for class experience is not restricted to evidence for negligible genotoxic potential originating solely from approved products. Furthermore, the FDA’s definition of well-characterized ONT classes for genotoxicity assessment limits the use of prior information to nearly identical ONT compounds (ie, consistent size, structure, modifications, and impurity profile) that differ only in sequence. In contrast, the EU SA reports here analyzed indicate that evidence for well-characterized chemical modifications independent of other ONT characteristics was sufficient for EU regulators to acknowledge class experience for genotoxicity assessment. These discrepancies highlight that current regulatory expectations for genotoxicity assessment for ONTs differ between regional authorities. In addition to this difference in overall expectations, there is currently no clear consensus between different regulatory and industry stakeholders for which particular ONT chemistries sufficient class experience can be acknowledged.
Our study suggests that EU regulators acknowledge class experience for three chemical modifications, namely PS, 2′MOE, and 2′OMe. In the SA reports, the CHMP and applicants both equally acknowledged class experience for the PS modification. In contrast, class experience for those sugar modifications was less frequently mentioned in SA reports; this could be because fewer SA reports discussed genotoxicity testing for ONTs with 2′MOE (n = 17) or 2′OMe modifications (n = 13) compared with reports for ONTs with a PS modification (n = 39) (Supplementary Table S3). However, when these particular sugar modifications were discussed in the context of reduced testing, the testing strategies were considered suitable based on class experience, which shows that regulatory acknowledgment of class experience might actually exist for those modifications as well. Overall, EU regulators have acknowledged sufficient class experience for the PS, 2′MOE, and 2′OMe modifications in concordance with the OSWG recommendation published in 2016, which concluded that further genotoxicity testing for ONTs containing those chemistries is unnecessary. 14 Even though the FDA draft guidance on ONT safety assessment does not specify which ONT modifications are considered well-characterized, 15 an earlier FDA draft guidance for individualized ONTs for severely debilitating or life-threatening diseases explicitly mentions single-stranded PS or mixed PS/PO, 2′-MOE, and PMO oligonucleotides as well-characterized ONT classes due to “substantial nonclinical information and clinical experience” that they consider to be available from approved products. 21 Furthermore, Parry et al. argue that in addition to PS, 2′MOE, and 2′OMe modifications, sufficient class experience is also available for additional backbone [glycol nucleic acid (GNA), vinyl phosphonate (VP), and morpholino] and sugar modifications (2′F, LNA, and cEt) and emphasize that further genotoxicity testing for ONTs with these chemistries is unlikely to be informative. 22
Since the release of the CHMP Reflection Paper in 2005, which highlighted triple helix formation as a theoretical genotoxic concern warranting discussion or additional testing, 17 experience with a wide variety of ONTs has noticeably increased. More recent literature, such as the OSWG recommendation paper released in 2016, suggests that triple helix formation is no longer considered a concern for ONTs since they do not appear to contain the specific sequence characteristics known to facilitate this process. 14 We demonstrated that this change in perspective over time is reflected in CHMP and applicants’ positions presented in EU SA reports. Consistent with this, outcomes from the 2022 EFPIA survey demonstrated that companies either did not consider the risk of triple helix formation in their genotoxicity assessment or only did so through sequence analysis and discussion of evidence in the literature. 22 This latter assessment strategy, using sequence analysis and evidence from literature, aligns with the recommendations provided in the OSWG paper 14 and was considered a sufficient assessment strategy by the CHMP in the majority of SA reports released after 2015.
Although multiple industry-led publications have argued that there is sufficient class experience for a broader range of ONT chemistries,14,20,22–24 we found that reduced genotoxicity test approaches were only proposed for 10 ONTs with 4 different chemical modifications (ie, 2′OMe, 2′MOE, PS, and LNA). Overall, the majority of ONTs were still evaluated using the standard test battery, indicating that the knowledge of class experience had a minimal impact on the selection of the test strategies. This could be the result of risk-avoidant testing strategies by companies or regional differences in acceptance of reduced test approaches between regulatory authorities and can be seen as a missed opportunity.
Notably, the low occurrence of direct questions from applicants on genotoxicity testing strategies in EU SA reports may explain the CHMP’s predominant silent agreement with standard battery testing as part of the general nonclinical program, despite the acknowledgment of class experience for commonly used chemical modifications. In addition, most studies were already completed at the time the applicants reached out for regulatory advice. Consequently, if more explicit questions regarding the genotoxicity testing strategies had been raised, for example, earlier during development, this might have resulted in more cases where the EU regulators would have recommended a reduced testing strategy or commented on the redundancy of the standard test battery.
ONTs comprised of less-established chemistries (eg, GNA, VP, cET, and LNA) were considerably less represented in our dataset than in the dataset published by Parry et al., although both studies were conducted within the same timeframe. In Europe, applying for Scientific Advice is optional during drug development, and therefore, regulators have sparse access to data for drug candidates in early development, which limits the evidence on which they can base their opinions, as previously noted by Parry et al. 22 This could explain the lack of ONTs with more novel chemistries in our dataset.
In addition to the discrepancy between industry and regulatory stakeholders regarding which chemical modifications have sufficient class experience, there is currently no guidance on the type of evidence applicants would need to provide as proof of class experience. We have already demonstrated that applicants referenced diverse sources to support their claim for class experience. The majority of the references were made to sources that either discussed or presented negative standard test battery outcomes and highlighted or provided evidence for ONT uptake and exposure in the test system.14,17,19,20 These sources appear to be acceptable evidence for class experience, since reduced testing strategies supported by these sources were considered adequate by the CHMP.
Finally, the uniformly negative test outcomes reported for the ONTs within our dataset appear to support the idea that well-characterized ONT chemistries (ie, chemical modifications commonly used in ONTs for which there is substantial nonclinical information available) (eg, PS, 2′MOE, 2′OMe, and 2′F) have negligible genotoxic potential. This finding is consistent with previous publications.14,19,22,23 Notably, a recent industry survey showed that companies generally assume sufficient exposure occurs at ICH S2(R1)-defined limit doses and rarely confirm it experimentally. The authors of the survey further argued that regulators appear to accept this approach; as to date, many ONTs have proceeded into clinical development. 22 Similarly, we observed that the CHMP regarded the standard test battery as suitable in many SA reports, while uptake and exposure of the ONT in the test systems were rarely mentioned. The rare mention of such evidence in EU SA reports contrasts with the emphasis that previous publications placed on ONT uptake and exposure data for an adequate genotoxicity evaluation of ONTs.14,15 Although the OSWG genotoxicity subcommittee highlights the importance of uptake and exposure into the test systems, it also recommends to follow ICH S2(R1) regarding the selection of the top concentration for the in vitro tests. 14
Based on uniformly negative reported test outcomes for ONTs in this dataset and previously published evidence for exposure and uptake of chemically modified ONTs into standard battery test systems,19,20,23,25 the authors consider that an ONT chemistry can be considered well-characterized once negative outcomes from Good Laboratory Practice (GLP)-compliant standard battery tests conducted in accordance with the current genotoxicity guidelines [ICH S2(R1)] are available from at least two or three different ONT products containing the same chemical modifications and for which sufficient uptake and exposure has been demonstrated.
It must be noted that most ONTs within our dataset had already reached later phases of clinical development, which implies a favorable nonclinical safety profile. Less successful candidates (for instance, those with possibly positive genotoxicity signals) are therefore highly unlikely to be present in our dataset from the EMA. Nonetheless, a recent EFPIA industry survey revealed that companies reported no positive genotoxicity outcomes in either GLP or non-GLP studies for any ONT products, indicating that ONT candidates at earlier stages of development also lack positive genotoxicity signals. 22 To ensure that regulatory considerations regarding the acknowledgment of class experience and well-characterized ONT chemistries can be aligned with industry knowledge and experience, it may be necessary, or at least beneficial, for the industry to share new insights and the underlying data with regulators and through peer-reviewed publications.
In conclusion, EU regulatory perspectives indicate that the standard battery might become redundant for well-characterized ONTs for which sufficient class experience can be demonstrated. Although the genotoxic potential of ONTs currently appears low regardless of the chemical modifications used, some uncertainty persists regarding whether this will hold true for less-established or future modifications. The development of new ONT modalities with advanced chemistry and conjugations is expected to further warrant the use of the standard test battery in the near future. Hence, clear guidance is required to promote a harmonized and tailored approach for genotoxicity evaluation of future ONTs, particularly in the case of exceptions to the standard test battery for well-characterized ONTs. Importantly, the definition for well-characterized in the context of genotoxicity assessment is not necessarily applicable to other safety assessment endpoints, which might require separate considerations and definitions. Ideally, the upcoming ICH S13 guideline should define what constitutes sufficient evidence for class experience, warranting regulators’ acceptance for reduced testing. Companies can also promote reduced testing and accelerate the acknowledgment of class experience for novel ONT modalities by publishing peer-reviewed studies. Ultimately, these efforts can contribute to a harmonized approach for future genotoxicity testing of noncoding ONTs. Based on current evidence from EU SA procedures, it appears that standard genotoxicity testing has become redundant, at least for well-characterized ONT chemistries.
Footnotes
Acknowledgments
The authors want to thank the EMA for their assistance in finding and accessing briefing packages for relevant ONT products and corresponding SA procedures issued before 2016, which were not available through other platforms. During article preparation, ChatGPT (OpenAI) was used solely for writing assistance to refine the phrasing of some sentences. No AI was involved in data analysis or interpretation. The authors are fully responsible for the content. Finally, the authors would like to thank Peter van Meer for his critical review of their article.
Disclaimer
The views and opinions expressed in this article are those of the authors and do not necessarily represent the views, opinions, and/or policies of the MEB.
Author Disclosure Statement
All authors declare no conflicts of interest for this work. Christine L.E Siezen is the European Commission topic lead in the Expert Working Group for the ICH S13 guideline.
Funding Information
No external funding was received for this work.
Supplemental Material
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.