
Abstract
Significance:
Refractory wounds are complicated multistep biological processes that can lead to severe complications in patients. Selective autophagy plays a crucial role in precisely controlling the quality of intracellular components and regulating biological behavior. This review explores the features and underlying mechanisms of various types of selective autophagy and highlights their implications in burn injury and wound healing.
Recent Advances:
In-depth studies have underscored the critical role of selective autophagy, including mitophagy, endoplasmic reticulum (ER)-phagy, pexophagy, xenophagy, lysophagy, ferritinophagy, and lipophagy, in effectively controlling the quality of intracellular components and regulating biological behavior, which may enhance wound-healing process.
Critical Issues:
Autophagy is a housekeeping and self-renewal process that utilizes lysosomal machinery to degrade and recycle cellular components, thereby enhancing cellular adaptability to stressful conditions. In addition to nonselective bulk degradation, autophagy selectively recycles specific cell constituents, including mitochondria, ER, peroxisomes, pathogens, lysosomes, lipid droplets, and ferritin. The effective management of the quality of cellular components during wound healing remains a challenge in clinical practice.
Future Directions:
Understanding the basic mechanisms and intricate crosstalk underlying selective autophagy may facilitate the development of comprehensive strategies and therapeutic targets for wound healing.
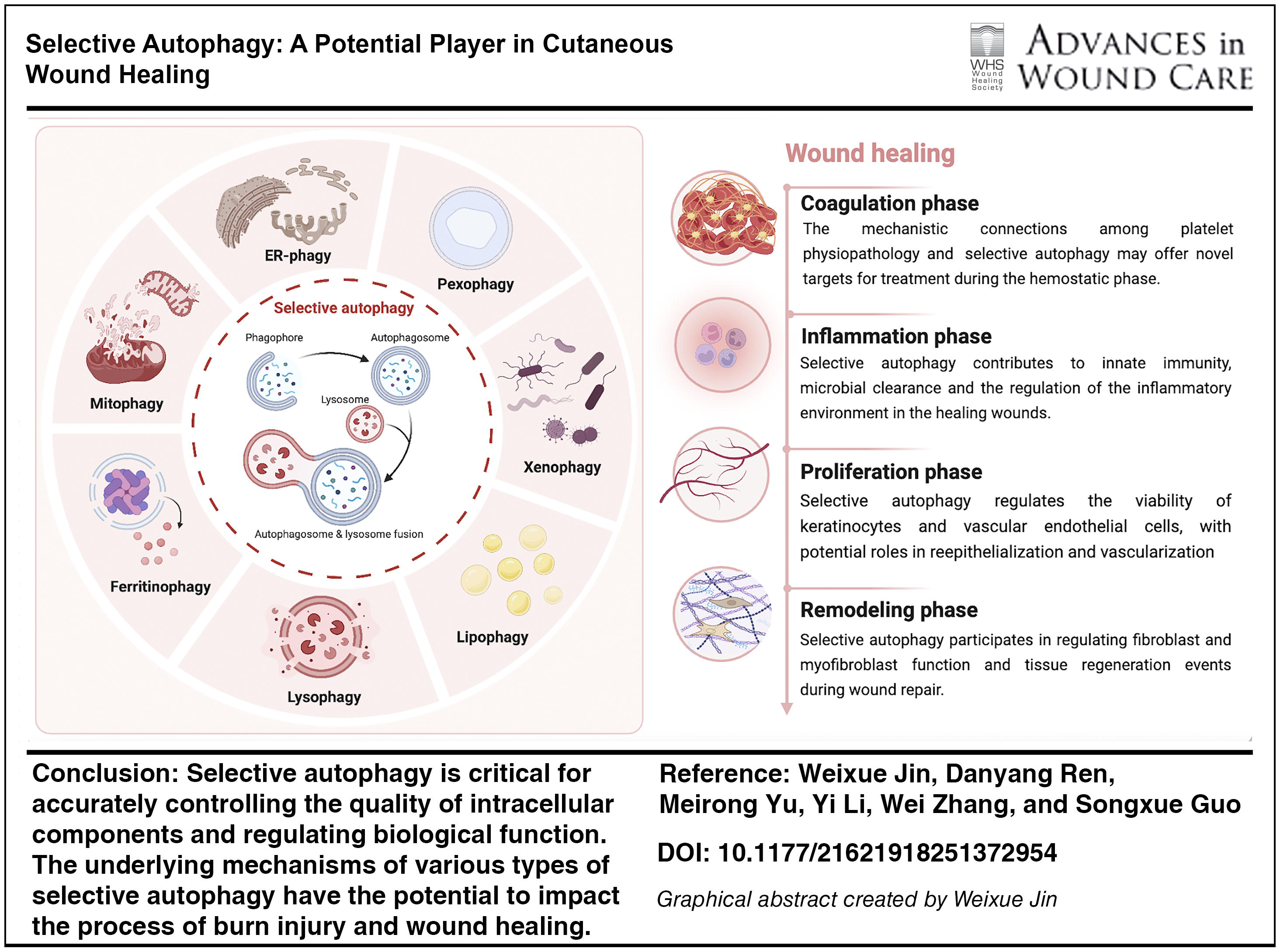
SCOPE AND SIGNIFICANCE
Selective autophagy offers precise control over the quality of intracellular components and regulation of biological behavior, ultimately benefiting the wound-healing process. This review recapitulates the features and underlying mechanisms of various types of selective autophagy and their implications in burn injuries and wound healing. Additionally, we discuss the clinical potential of selective autophagy in wound healing, which may provide insights for future research and improve the quality of life of patients with refractory wounds.
Songxue Guo, MD

TRANSLATIONAL RELEVANCE
Managing wound healing is a critical concern that affects the quality of life of patients, society, and health care systems. Understanding the fundamental mechanisms and crosstalk underlying selective autophagy may facilitate the development of comprehensive agents and therapeutic targets to enhance wound healing.
CLINICAL RELEVANCE
A retrospective analysis in 2019 revealed that chronic wounds affect approximately 10.5 million Medicare beneficiaries in the United States, substantially affecting the quality of life in approximately 2.5% of Americans. 1 Refractory wounds result from disruptions in normal wound-associated cellular processes. Selective autophagy offers precise regulation of intracellular component quality and biological behavior and provides a promising avenue for improved wound healing. Research on the role of selective autophagy in the wound-healing process and its potential targets holds the potential to accelerate healing and reduce health care costs.
BACKGROUND
Wound management accounts for over half of the community health nurse resources in European settings and imposes a substantial health economic burden on the UK’s National Health Service (£5.0 billion).2,3 In China, 1.7% of patients require hospitalization for chronic wounds, 42.3% of patients pay for their own treatment, 25.0% do so through social medical insurance, and 27.9% receive free medical care. 4 Comprehending and managing wound-healing problems remain a major challenge that considerably impacts society, clinical practice, and health care systems. Wound healing is a complex process that requires the coordinated involvement of various cell lineages, extracellular matrix (ECM) molecules, and cytokines. Wounds can range from minor breaches in the skin epithelium to severe lesions that extend into the dermis or subcutaneous tissue or deeper structures, including muscles, arteries, and organs (such as the cornea, lungs, and intestines). 5 Effective wound healing relies on a well-orchestrated process involving the overlapping molecular and biological activities of cell migration, reepithelialization, angiogenesis, ECM deposition, and remodeling. 6 Following injury, coagulation cascades involving platelets and fibrin are activated to form clots, which release bioactive substances that attract inflammatory cells. 7 Antimicrobial agents produced by leukocytes (neutrophils, monocytes, T cells, and macrophages) trigger an immunological response. 8 Keratinocyte proliferation and epithelial migration continue until the wound is completely covered. Fibroblasts and myofibroblasts contribute to the formation of granulation tissue and ECM.9,10 Disruptions at any stage can result in either underhealing or overhealing, leading to chronic nonhealing wounds or excessive scarring and fibrosis.11–13 An internet survey of 11,100 individuals conducted between April and May 2020 revealed that approximately 28.9% of patients with recent scars experienced discomfort, followed by burning (23.7%), itching (35.0%), and redness (44.1%). 14 Chronic wounds can result from disruption of wound-associated cellular behaviors, influenced by conditions, such as diabetes, infection, aging, cardiovascular problems, and sensory neuropathies.15–17 Diabetic foot ulcers affect approximately 18.6 million individuals worldwide annually, with mortality reported in approximately 30% of patients within 5 years. 18 Moreover, burns may cause skin contractures, fibroproliferative scarring, or chronic wounds that often require prolonged healing time and lack standardized diagnostic techniques.19,20 These challenges underscore the importance of advancing research into the wound-healing process and identifying novel therapeutic targets. Therefore, the development of safer and more efficient treatment strategies is imperative.
Autophagy is a self-renewal process that involves lysosomal degradation of damaged or harmful cellular components. After degradation, the products are returned to the cytoplasm and recycled for various cellular functions. 21 Autophagy promotes wound healing by inhibiting inflammation, enhancing angiogenesis and reepithelization, and facilitating tissue repair.22–28 In addition to nonselective forms, autophagy can target specific organelles and cellular components selectively. 29 Selective autophagy promotes housekeeping activity by eliminating pathogens that infiltrate cells, misfolded or aggregated proteins, and damaged organelles such as mitochondria, endoplasmic reticulum (ER), and peroxisomes. 30 It relies on soluble or membrane-bound cargo receptors that identify cargo and interact with lipidated autophagy-related gene 8 (ATG8) family proteins anchored in the autophagosome’s membrane to form the autophagosome at the cargo. 31 Conventional wound management strategies have focused on developing proregenerative treatments targeting specific skin cell activities such as fibroblast and keratinocyte functions. 32 The likelihood of pigmentary abnormalities and atrophic scarring is increased by traditional procedures such as biological stenting, skin grafting, skin flap transplantation, and laser therapy. 33 Although stem cells can support wound healing by promoting angiogenesis, the proliferation and differentiation of fibroblasts and keratinocytes, and the production of antifibrotic cytokines, they are associated with storage and transportation issues, as well as with the hazards of cancer and deformity. New insights have been provided by three-dimensional (3D) printing, decellularization of allogeneic or xenogeneic skin, and genetic modification of wound beds. Several tissue engineering techniques are being investigated with an emphasis on growth factors, such as acellular scaffolds, cell transplants, matrix materials, skin substitutes, and temporary wound coverings to enhance the wound milieu. Yet, cell signaling, cellular interactions, and the pathophysiological requirements of distinct wound conditions have not been fully investigated.34,35 None of the aforementioned scientific efforts have yet found their way into standardized clinical application and have not produced adequate outcomes. Selective autophagy plays a crucial role in maintaining organelle quality and regulating cell fate. 36 It regulates cellular signaling and pathophysiological processes in the wound microenvironment. Therefore, summarizing recent research on the potential role of selective autophagy in wound healing provides valuable insights into its potential applications. Although the precise mechanisms underlying selective autophagy remain unclear, its therapeutic potential is promising.
DISCUSSION
Autophagy and selective autophagy
Autophagy
Autophagy is a key biochemical process that maintains cellular homeostasis and involves the transport of cellular materials to lysosomes for breakdown, facilitating the basal turnover of cellular constituents, and providing energy and macromolecular precursors. Morphologically, autophagy is classified into three categories: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA). 37 Macroautophagy involves the formation of double-membrane vesicles called autophagosomes, which engulf damaged organelles and cellular proteins and deliver them to lysosomes. 38 ATGs play key roles in autophagosome assembly and turnover. The process involves the following stages: initialization, autophagosome nucleation, elongation and extension, autophagosome closure, lysosome fusion, and degradation of intravesicular products. 39 The UNC51-like kinase 1 (ULK1; also known as ATG1) complex, which is composed of ATG101, FIP200 (also called RB1CC1), ULK1, ULK2, and ATG13, is activated first. This triggers vesicle nucleation, which is facilitated by a class III phosphoinositide 3-kinase (PI3K) complex that includes beclin 1, ultraviolet radiation resistance-associated gene protein (UVRAG; also called p63), VPS15, VPS34 (also called PIK3C3), ATG14, and activating molecule in BECN1-regulated autophagy protein 1 (AMBRA1). Two ubiquitin-like conjugation mechanisms mediate vesicle elongation, ultimately forming autophagosomes. In the first system, phosphatidylethanolamine is conjugated with cytoplasmic light chain 3 (LC3)-I to generate the lipidated form LC3-II (also called MAP1LC3B). This process is facilitated by the E1-like enzyme ATG7 and the protease ATG4B, with LC3II subsequently integrated into the developing membrane. ATG7 and the E2-like enzyme ATG10 mediate the second conjugation mechanism, yielding an ATG5-ATG12 conjugate. The soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) protein syntaxin 17 facilitates autophagosome fusion with lysosomes, resulting in autophagolysosome formation.38,40 Nonselective microautophagy is a catabolic process characterized by the direct engulfment of dysfunctional or superfluous cytoplasmic constituents into lysosomes via tubular membrane invagination. 21 Mechanistically, microautophagy is mediated by the endosomal sorting complex required for transport (ESCRT). SNARE proteins and the basic autophagy apparatus are necessary for certain forms of microautophagy.41,42 Microautophagy selectively degrades organelles via the interaction of organelle proteins with surface proteins in vacuoles, lysosomes, and late endosomes. Heat shock cognate 71 kDa protein (Hsc70; also called HSPA8), a cytosolic chaperone, regulates selective protein degradation via microautophagy.43,44 CMA targets KFERQ-like motif-bearing proteins, which are selectively degraded by the chaperone Hsc70 and cochaperones, including HSP40 (also called DNABJ1), carboxyl terminus of Hsc70-interacting protein, and HSP70-HSP90 organizing protein. These proteins are internalized in lysosomes through lysosome-associated membrane protein (LAMP) type 2A (LAMP2A) 44 (Fig. 1).
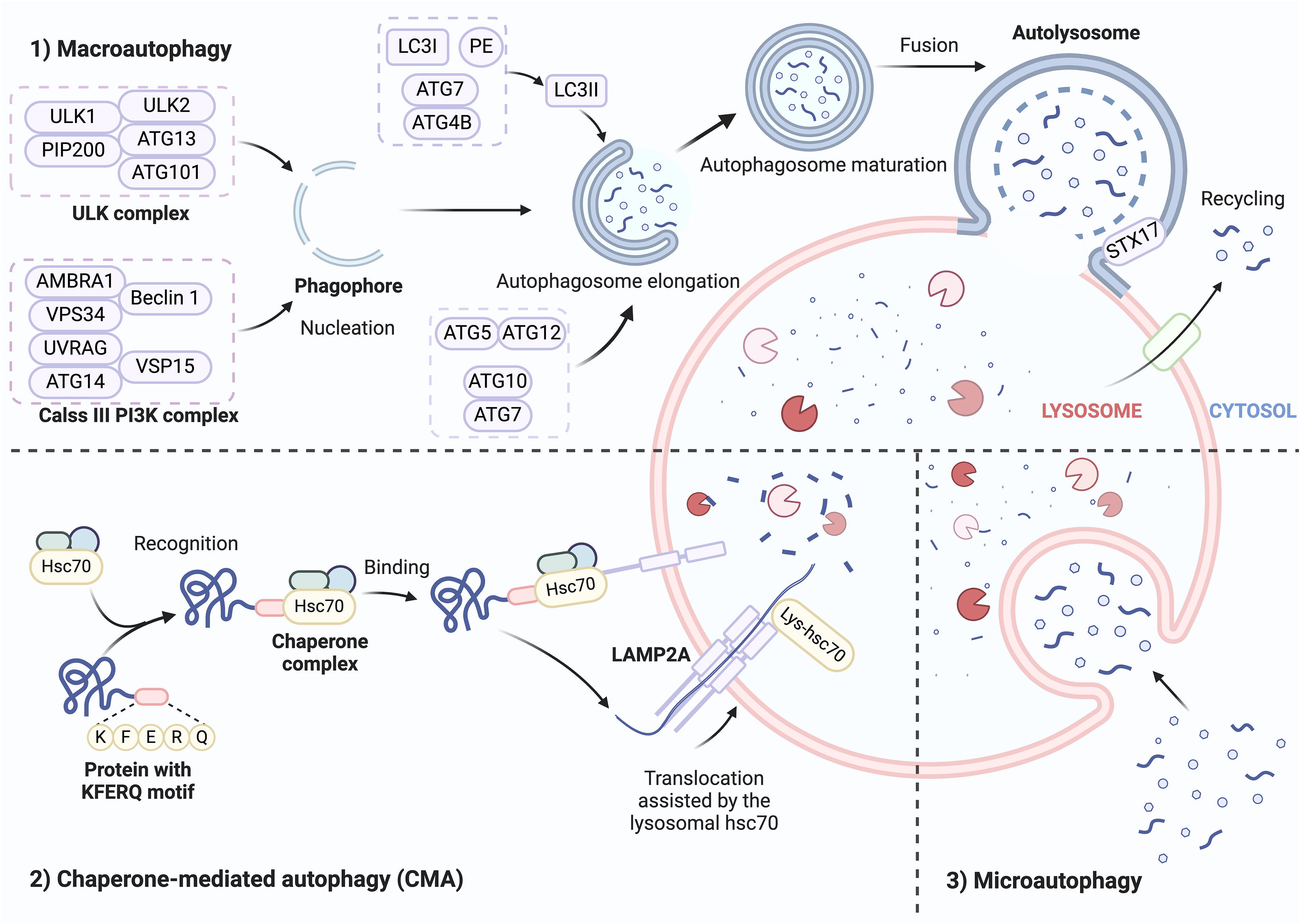
Three main types of autophagy. Autophagy can be morphologically categorized into three types: macroautophagy, microautophagy, and CMA. Macroautophagy involves the synthesis of autophagosomes, which are double-membrane vesicles that absorb organelles and cellular proteins and transfer them to lysosomes. The procedures are as follows: initiation, autophagosome nucleation, elongation and extension, autophagosome closure, lysosome fusion, and ultimately, the degradation of intravesicular products. The Hsc70 chaperone identifies proteins with a pentapeptide KFERQ-like sequence during CMA. It subsequently binds to the integral lysosomal membrane protein LAMP2A, causing it to oligomerize. This event initiates the translocation of the bound protein into the lysosomal interior. Microautophagy is characterized by the direct engulfment of malfunctioning constituents in the cytoplasm via invagination of the tubular membrane into lysosomes. ATG, autophagy-related; Hsc70, heat shock cognate 71-kDa protein; LAMP2A, lysosome-associated membrane protein type 2A; LC3, light chain 3; PE, phosphatidylethanolamine; STX17, syntaxin 17; ULK, UNC51-like kinase.
Selective autophagy
In addition to their nonspecific degradation in the cytosol, lysosomes can selectively target specific components for degradation through the action of chaperones, degradation tags, and complex mechanisms that enable their translocation across the lysosomal membrane. This selective degradation of intracellular components was first demonstrated by CMA. 44 Several specific types of macroautophagy, such as mitophagy (involving mitochondria), reticulophagy (involving the ER), lysophagy (involving lysosomes), aggrephagy (involving protein aggregates), pexophagy (involving peroxisomes), and xenophagy (involving microbes), have been investigated.45,46 Upon binding to degradation targets, receptors or adaptors interact with the scaffold protein ATG11 in yeast or FIP200 in mammals to recruit core ATG proteins and trigger the formation of autophagosomes (Table 1).47–57,60,61,63–65,71 The ATG8 family interacting motif or LC3-interacting region (LIR) is a common motif used by macroautophagy receptors to bind to ATG8 family proteins attached to the isolating membrane for efficient autophagosome formation. 45 Additionally, various forms of selective microautophagy that target endosomes, nuclei, peroxisomes, ER, and mitochondria have recently received considerable attention.41,74 Herein, we briefly summarize the characteristics and underlying mechanisms of various types of selective autophagy.
An overview of selective autophagy adaptors or receptors
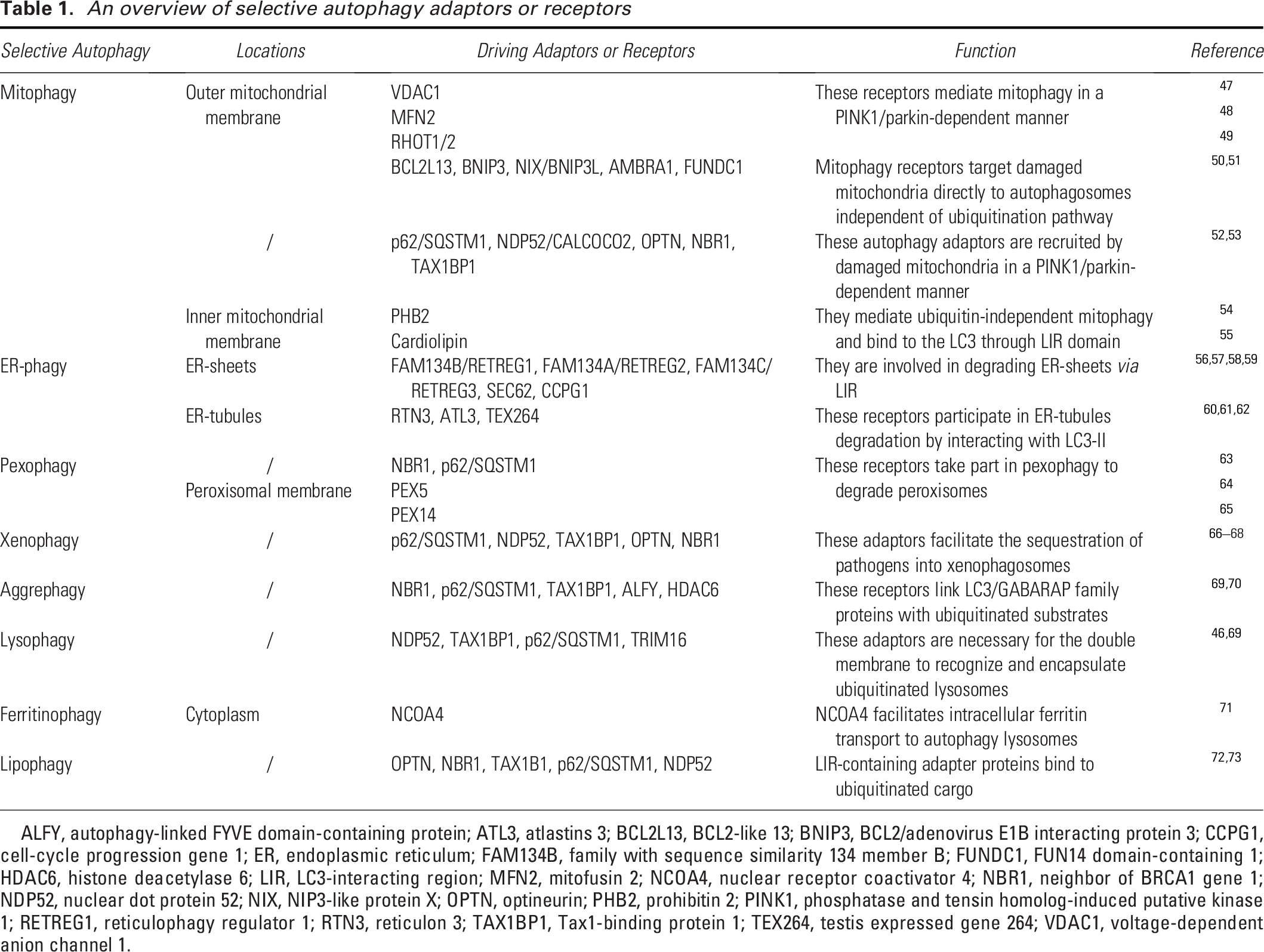
ALFY, autophagy-linked FYVE domain-containing protein; ATL3, atlastins 3; BCL2L13, BCL2-like 13; BNIP3, BCL2/adenovirus E1B interacting protein 3; CCPG1, cell-cycle progression gene 1; ER, endoplasmic reticulum; FAM134B, family with sequence similarity 134 member B; FUNDC1, FUN14 domain-containing 1; HDAC6, histone deacetylase 6; LIR, LC3-interacting region; MFN2, mitofusin 2; NCOA4, nuclear receptor coactivator 4; NBR1, neighbor of BRCA1 gene 1; NDP52, nuclear dot protein 52; NIX, NIP3-like protein X; OPTN, optineurin; PHB2, prohibitin 2; PINK1, phosphatase and tensin homolog-induced putative kinase 1; RETREG1, reticulophagy regulator 1; RTN3, reticulon 3; TAX1BP1, Tax1-binding protein 1; TEX264, testis expressed gene 264; VDAC1, voltage-dependent anion channel 1.
Mitophagy
Mitochondria are double-membraned subcellular organelles responsible for essential functions such as phospholipid synthesis and transport, iron homeostasis, calcium signaling, and adenosine triphosphate (ATP) generation. 75 Because mitochondria generate reactive oxygen species (ROS), they are constantly at risk of oxidative stress, which can lead to dysfunction and disrupt cellular homeostasis. 76 To maintain mitochondrial quality and function, cells employ various mechanisms, including mitophagy, mitochondrial fusion and fission, and protein refolding/degradation. 77 Mitophagy, a key process conserved from yeast to humans, selectively degrades mitochondria to regulate both their quantity and quality. By eliminating dysfunctional mitochondria, mitophagy contributes to various biological processes, including inflammation, apoptosis, and cell differentiation. 53
ATG32, a single-pass transmembrane protein in the outer mitochondrial membrane (OMM), primarily mediates mitophagy in the budding yeast Saccharomyces cerevisiae. During the stationary phase growth or nitrogen deprivation, ATG32 accumulates on the OMM to form a complex with ATG11 and ATG8, which is anchored to the isolation membrane on the surface of mitochondria. 53 In mammals, the primary role of phosphatase and tensin homolog-induced putative kinase 1 (PINK1) is to detect damaged mitochondria, activate the E3 ubiquitin ligase parkin, and facilitate its translocation from the cytoplasm to damaged mitochondria. PINK1 initiates mitophagy by phosphorylating and activating parkin, which activates ubiquitin chains and triggers a signaling cascade, leading to the engulfment of mitochondria by autophagosomes. 78 During autophagosome formation, microtubule-associated protein 1 (MAP1) LC3, a homolog of yeast ATG8, is covalently linked to phosphatidylethanolamine, enabling it to integrate into and shape the expanding isolation membrane and participate in cargo recruitment. 79 Autophagy adaptors, through their ubiquitin-binding domains and LC3/GABAA receptor-associated protein (GABARAP), which are connected to autophagosomal membranes via an LIR, concurrently bind to ubiquitin chains on cargo. 52 This places the autophagy apparatus close to the ubiquitinated payloads. During PINK1/parkin mitophagy, five distinct autophagy receptors are translocated to the mitochondria: p62/SQSTM1, nuclear dot protein 52 (NDP52)/CALCOCO2, optineurin (OPTN), neighbor of BRCA1 gene 1 (NBR1), and Tax1-binding protein 1 (TAX1BP1). Damaged mitochondria recruit these autophagy adaptors in a PINK1/parkin-dependent manner.52,53 Among them, only NDP52 and OPTN are essential for inducing mitophagy, which recruits key components such as ULK1, double FYVE domain-containing protein, and WD repeat domain, phosphoinositide interacting 1, which are involved in the formation of autophagosomal membranes. 80 The effective recruitment of OPTN and NDP52 to ubiquitinated mitochondria depends on TBK1 activity, which phosphorylates all known autophagy receptors during PINK1/Parkin-mediated mitophagy. 53 In addition, there exist ubiquitin-dependent mechanisms that do not require parkin. PINK1 can ubiquitylate the autophagy receptors OPTN and NDP52, directly attracting them to mitochondria and promoting autophagy biogenesis. 80 Furthermore, other E3 ubiquitin ligases, such as Gp78, mitochondrial E3 ubiquitin protein ligase 1, and small mother against decapentaplegic (SMAD) ubiquitination regulatory factor-1, can ubiquitinate mitochondrial proteins and induce mitophagy. 50
In addition to the ubiquitination pathway, mitophagy receptors directly target damaged mitochondria to the autophagosomes via the LIR motif. Mitophagy receptors include BCL2-like 13 (BCL2L13), BCL2/adenovirus E1B interacting protein 3 (BNIP3), NIP3-like protein X (NIX)/BNIP3L, AMBRA1, FUN14 domain-containing 1 (FUNDC1), and cardiolipin.50,51 A study in yeast revealed that BCL2L13 may act as a mammalian ATG32 functional counterpart. 81 NIX, BNIP3, and FUNDC1 are OMM proteins that function as mitophagy receptors, facilitating mitochondrial elimination in response to various stressors. Under hypoxic conditions, both BNIP3 and FUNDC1 are critical for efficient mitochondrial turnover. 50 FUNDC1 orchestrates mitophagy and mitochondrial morphology under stress conditions by mediating DRP1 recruitment and promoting mitochondrial fragmentation, as it translocates to ER-mitochondrion contact sites in hypoxic environments. Additionally, FUNDC1 may function as an adaptor for ULK1, because its interaction promotes ULK1 translocation to the mitochondria and de novo phagophore formation. 82 BNIP3 and NIX are homologous proteins that integrate into the OMM and form stable homodimers via their C-terminal transmembrane domains in response to stress signals. 83 NIX causes selective mitochondrial degradation during erythroid development. 84 Intriguingly, BNIP3 and NIX interact with the PINK1/parkin signaling pathway, contributing to parkin accumulation in the OMM85,86 (Fig. 2).
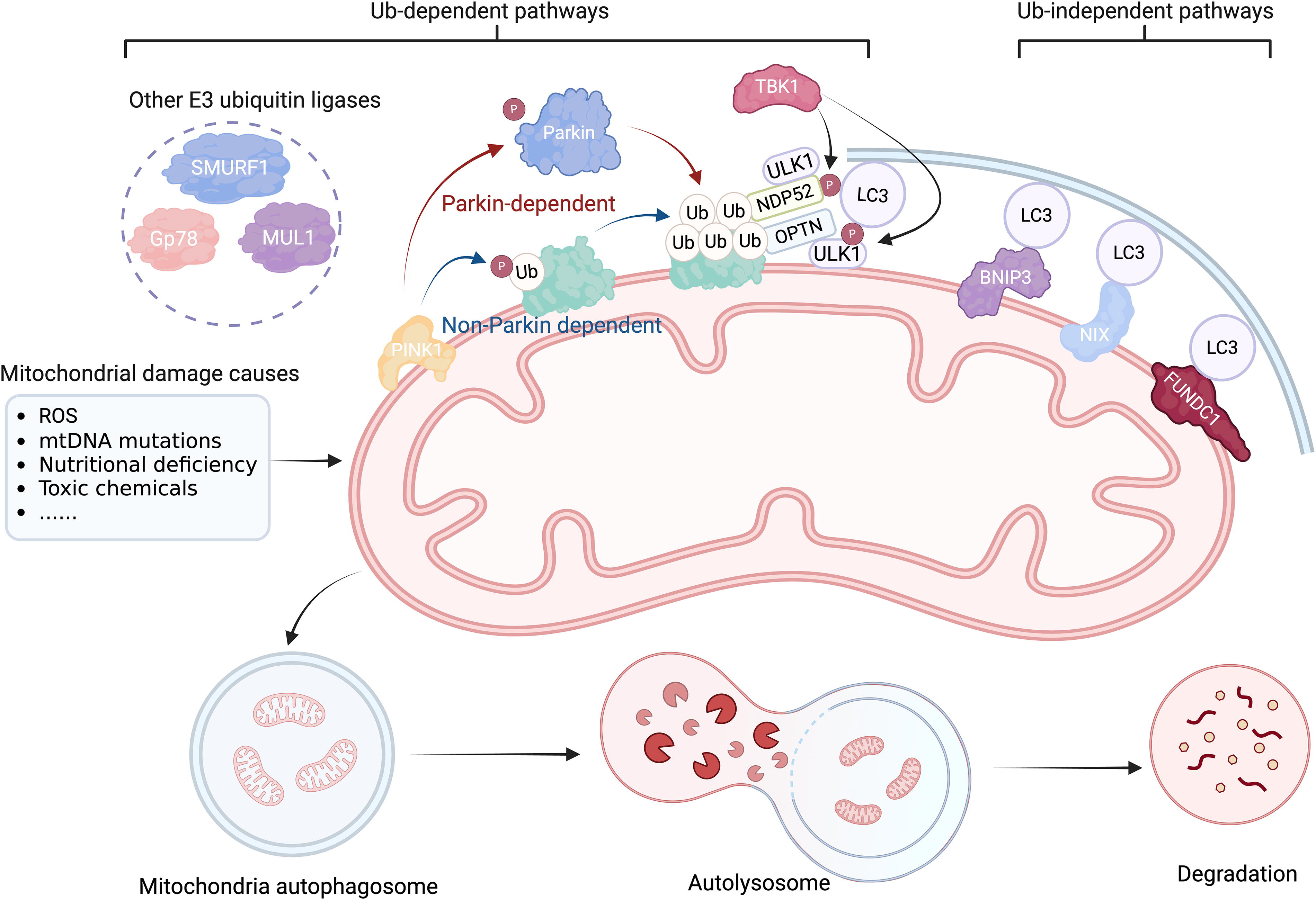
The process of mitophagy. When external stimuli such as ROS, mtDNA mutations, nutritional deficiency, toxic chemicals, infection, and cell aging cause damage to the mitochondria, the damaged mitochondria depolarize and lose their outer membrane potential. PINK1 detects damaged mitochondria and facilitates Parkin’s transfer from the cytoplasm to injured mitochondria. PINK1 triggers mitophagy by phosphorylating and activating Parkin, an E3 ubiquitin ligase that activates ubiquitin chains and amplifies a signaling cascade that results in the engulfment of mitochondria by autophagosomes. NDP52 and OPTN are essential for mitophagy induction. The effective recruitment of OPTN and NDP52 to ubiquitinated mitochondria depends on TBK1 activity, which phosphorylates all known autophagy receptors during PINK1/Parkin-mediated mitophagy. Independent of Parkin, PINK1 may ubiquitin phosphorylate the autophagy receptors OPTN and NDP52, attracting them directly to the mitochondria and boosting autophagy. In addition to the ubiquitination pathway, mitophagy receptors, such as BNIP3, NIX, and FUNDC1, can initiate mitophagy by employing the LIR motif to directly deliver damaged mitochondria to autophagosomes. BNIP3, BCL2/adenovirus E1B interacting protein 3; FUNDC1, FUN14 domain-containing 1; mtDNA, mitochondrial DNA; MUL1, mitochondrial E3 ubiquitin protein ligase 1; NDP52, nuclear dot protein 52; NIX, NIP3-like protein X; OPTN, optineurin; ROS, reactive oxygen species; SMURF1, SMAD ubiquitination regulatory factor-1; Ub, ubiquitin; ULK1, UNC51-like kinase 1.
ER-phagy/reticulophagy
The ER functions as a quality-controlled organelle that maintains protein and lipid homeostasis, which is essential for cell survival. ER-phagy occurs physiologically within cells to maintain ER homeostasis and membrane abundance. Various external and internal stressors, including hypoxia, nutrient deprivation, oxidative stress, and calcium flux imbalance, can lead to the accumulation of unfolded or misfolded proteins, a condition termed “ER stress.”56,87 Persistent ER stress due to the accumulation of misfolded proteins can trigger the unfolded protein response (UPR), an adaptive cellular response. 88 Stress-induced misfolded proteins are subsequently released from the ER into the cytoplasm and degraded by the ubiquitin-proteasome system in a cascade of processes known as ER-associated degradation. ER-phagy can be enhanced in stressful conditions, such as ER stress, the recovery phase from ER stress, as well as in the context of development, immunity, and disease pathogenesis, to satisfy elevated demands to preserve ER proteostasis. 56
ER stress is specifically associated with reticulophagy. The UPR interacts with autophagy through AKT1-rapamycin (mTOR), MAPK8 transduction, and adenosine monophosphate-activated protein kinase (AMPK). 89 To date, 11 ER-phagy receptors have been identified in mammals: p62, CDK5RAP3, atlastins 3 (ATL3), cell-cycle progression gene 1 (CCPG1), SEC62, testis expressed gene 264 (TEX264), family with sequence similarity 134 member B (FAM134B)/reticulophagy Regulator 1, FAM134A, FAM134C, reticulon 3 (RTN3), and CALCOCO1.56,90,91 FAM134B and reticulon 3 long (RTN3L) contain reticulon homology domains (RHDs) that connect to regions involved in substrate recognition and LC3 binding. FAM134B and RTN3L are implicated in autophagy-mediated membrane turnover of ER sheets and tubules, respectively, during nutrient deprivation.60,92 Transmembrane reticulophagy receptors devoid of RHDs are as follows: SEC62 and CCPG1 are predominantly found in ER sheets, whereas TEX264 and ATL3 are predominantly found in ER tubules. 58 Similar to RTN3L, ATL3 facilitates autophagic degradation of the tubular ER during nutrient deprivation. 61 SEC62 is identified as a recovER-phagy receptor, which is essential for the reestablishment of prestress levels of ER proteins. 93 In addition to supporting recovER-phagy, CCPG1 is engaged in the degradation of peripheral ER. 57
FAM134B, the first known macro-ER-phagy receptor in mammals, recognizes misfolded substrate proteins attached to the ER transmembrane chaperone calnexin.58,92 The RHD of FAM134B preferentially binds to positively curved membranes, promoting the accumulation of FAM134B along the edges of ER sheets. This localization induces deformation and fission of ER regions. 94 Posttranslational modifications, including phosphorylation, ubiquitination, and acetylation, converge to fine tune the activation and cluster assembly of FAM134B. 95 The mTOR pathway regulates phosphorylation, which involves downstream kinases including ataxia-telangiectasia mutant (ATM), ATR, Akt, Chk1, and CK2. CK2 has been demonstrated to phosphorylate FAM134B at critical serine residues within its RHD. 96 In addition, the calcium/calmodulin-dependent protein kinase CAMK2B phosphorylates the FAM134B RHD, triggering its oligomerization, which is crucial for ER fission. 97 Phosphorylation within the RHDs directs the ubiquitin machinery to target them for modification. 95 Ubiquitination causes structural rearrangements in the RHD domains, allowing interactions between nearby RHD-containing proteins, which are required for membrane curvature and fission. 98 ARL6IP1 and the E3 ligase autocrine motility factor receptor (AMFR) have been demonstrated to catalyze the ubiquitination of FAM134B.98,99 CBP acetyltransferase-mediated acetylation of FAM134B promotes CAMK2-mediated phosphorylation, strengthening its role in driving ER degradation via the lysosomal route. 100 In parallel, transcription factor MiT/TFE regulates the SESTRIN2-XBP1 axis and following TFEB/TFE3 phosphorylation to dynamically adjust ER-phagy to meet the cell’s needs.101,102 Transcriptional circuits ensure a rapid response to environmental cues, balancing ER turnover with cellular homeostasis amid nutritional variations and stress. RTN3 primarily triggers ER-phagy in response to oxygen or energy deprivation. RTN3L forms clusters at ER-isolation membrane contact sites by interacting with ATG8 family proteins. These clusters mediate ER fission and sequestration in the autophagosomes. Unlike FAM134B, RTN3 localizes primarily to the tubular ER and initiates the autophagic degradation of this ER subdomain. Both RTN3 and FAM134B function exclusively as ER-phagy receptors at locations where they are present. 60 Macro-ER-phagy mediated by ATL3, a dynamin-like GTPase, depends on its specific interaction with GABARAP through two GABARAP-interacting motifs. 61 A recovery phase is triggered by stress resolution, during which excess ER membranes are removed and ER proteins are restored to their prestress levels. 93 SEC62 is a recovER-phagy receptor necessary for the restoration of prestress levels of ER proteins, such as protein disulfide isomerases, HSPA5/BiP, and HSP90B1/Grp94. After binding to lipidated LC3B, SEC62 triggers microautophagy, which depends on the ATPase CHMP2A/VPS2A, which provides the energy required for direct endolysosomal engulfment of SEC62-containing ER-derived vesicles and ESCRT-III-mediated membrane remodeling.58,93 CCPG1, a transmembrane protein within the ER, is activated in response to the substantial accumulation of aggregated proteins in the ER. By interacting with FIP200, CCPG1 recruits the ULK1 complex, promoting autophagosome formation in the ER and initiating macroautophagic degradation of the ER. It can also bind GABARAP, LC3B, and LC3C through LIR on the cytosolic amino terminus. 57 In contrast, the carboxy-terminal lumenal domain of CCPG1 identifies ER-lumenal cargo through numerous cargo-interacting regions. 103 TEX264 has a long intrinsically disordered region (IDR) between the LIR motif and a gyrase inhibitor-like domain located after the transmembrane domain. This IDR is crucial for the LIR motif to navigate the dense array of ribosomes associated with the ER, enabling it to interact with ATG8 family proteins on the isolation membrane. 62 Although ER-phagy serves as a protective mechanism against ER stress, persistent ER stress can be harmful. Persistent ER stress activates the three branches of the UPR, resulting in cell death via a complex comprising procaspase-8 and an fas-associated protein with a new death domain 104 (Fig. 3).

Multiple forms of selective autophagy. RETREG1, SEC62, CCPG1, ALT3, and RTN3 facilitate the sequestration of isolated ER cargo into autophagosomes, which is necessary for the initiation of reticulophagy upon interaction with LC3-II. NCOA4-mediated ferritinophagy transfers intracellular ferritin to autophagolysosomes for the degradation and release of free iron. Pexophagy has great potential for peroxisome quality control and may be induced by the following mechanisms: PEX3 overexpression, activation of the ATG9A-TNKS/2-PEX14 complex, the ABCD3-dependent NBR1-LC3-II pathway, and phosphorylation and mono-ubiquitination of PEX5 via ATM and the PEX2–PEX10–PEX12 complex, which is recognized by SQSTM1. Different xenophagy adaptors, including p62, NDP52, TAX1BP1, TOLLIP, and OPTN, recognize ubiquitination, which leads to the trapping of xenophagosome infections. TRIM16 is an E3 ubiquitin ligase that binds Gal3 in injured lysosomes. TRIM16 initiates the autophagic machinery by ubiquitinating autophagy-associated molecules, including ULK1 and ATG16L1. Another E3 ubiquitin ligase, FBXO27, collaborates with Gal3 to perform lysophagy and regulate the recruitment of the autophagic machinery in SQSTM1-LC3-II pathway. ALT3, atlastins 3; ATG, autophagy-related; ATM, ataxia-telangiectasia mutant; CCPG1, cell-cycle progression gene 1; ER, endoplasmic reticulum; NBR1, neighbor of BRCA1 gene 1; NCOA4, nuclear receptor coactivator 4; RTN3, reticulon 3; RETREG1, reticulophagy regulator 1; TAX1BP1, Tax1-binding protein 1; Ub, ubiquitin; ULK1, UNC51-like kinase 1.
Pexophagy
Peroxisomes are dynamic and heterogeneous organelles that vary in quantity, size, morphology, protein composition, and function depending on the cell type and metabolic state. These adaptable organelles are primarily involved in ROS metabolism and fatty acid breakdown, particularly of polyunsaturated and very long-chain fatty acids.105,106 Peroxisomes maintain homeostasis by regulating the rates of organelle production and degradation, ensuring optimal abundance and quality. 107 Pexophagy preferentially destroys excess or damaged peroxisomes, thereby preserving redox equilibrium and adapting to environmental changes, such as hypoxia or amino acid deficiency. 108
In mammalian cells, p62 and NBR1 play key roles in pexophagy. 63 These autophagy receptors facilitate the sequestration of ubiquitinated peroxisomes into autophagosomes by interacting with their LIR motifs and binding ubiquitinated residues on peroxisomes via their ubiquitin-associated domains. 109 The ATM regulates pexophagy by inhibiting mTOR and activating ULK1, enhancing autophagic flux. During oxidative stress, the peroxisome import receptor PEX5 recruits ATM to the peroxisomes. ATM phosphorylates PEX5, promotes its ubiquitination, and subsequently targets p62 to trigger autophagosome formation. 64 PEX14 binds directly to LC3, indicating that it acts as a ubiquitin-independent pexophagy receptor. NBR1 and p62 promoted the interaction between PEX14 and LC3-II by inducing conformational changes in PEX14 65 (Fig. 3).
Xenophagy
Xenophagy, a specialized form of autophagy, involves various defense mechanisms to target and eliminate pathogens such as bacteria, viruses, and protozoa. Additionally, the degradation of pathogens by lysosomes improves cellular antigen presentation and initiates a supplementary immune response. 110 However, certain pathogens can prevent the formation of autophagosomes or even exploit the xenophagy system for replication and survival. Despite these challenges, xenophagy remains a vital survival mechanism that eliminates several antibiotic-resistant pathogens, including Salmonella spp. and Group A Streptococcus (GAS). 46
Certain bacteria evade entrapment in endosomes by lysing them. Rupture of host membranes often acts as a signal for xenophagy activation. Bacteria within cells are ubiquitinated when they come in contact with the cytoplasm of their host. Ubiquitination serves as a recognition signal for various xenophagy adaptors, including p62, NDP52, TAX1BP1, and OPTN. These adaptors facilitate the sequestration of pathogens into xenophagosomes.66–68 For example, galectins bind to polysaccharides on bacterial surfaces and recruit NDP52 to facilitate autophagosome formation. Galectin 8 specifically attracts NDP52 to invasive bacteria, promoting the binding of NDP52 to bacterial ubiquitin. 111 Moreover, ubiquitylation is not always a prerequisite for xenophagy. 46 Although the bulk of ubiquitin signaling on bacterial membranes surrounding invaders occurs during xenophagy, ubiquitylation occurs on both host components and bacterial carbohydrates 112 (Fig. 3).
Aggrephagy
Protein aggregation is the accumulation of unfolded or misfolded proteins that clump together to form insoluble aggregates that can damage cells. To maintain protein quality, cells employ three primary mechanisms: chaperone-assisted folding, proteasome-dependent degradation, and aggrephagy, a type of selective autophagy that degrades ubiquitinated aggregates. 113 The pathway activated for aggregate breakdown depends on the size and solubility of the aggregated proteins. Aggrephagy is essential to eliminate potentially hazardous inclusions. 114
Ubiquitination of misfolded proteins is a key mediator in the identification and elimination of protein aggregates through aggrephagy. Aggrephagy is dependent on adaptors such as NBR1, SQSTM/p62, TAX1BP1, autophagy-linked FYVE domain-containing protein (ALFY), and histone deacetylase 6 (HDAC6).69,70 These receptors link LC3/GABARAP family proteins with ubiquitinated substrates. 115 WDR81 and ALFY, members of the BEACH domain-containing protein family, facilitate p62 interaction with ubiquitin and recruit LC3/GABARAP to ubiquitylated proteins. 116 HDAC6 sorts polyubiquitinated misfolded proteins for axonal retrograde transport via the dynein-snapin pathway. 117 By directly interacting with FIP200 via an intact LIR, p62 attracts the ULK1 complex to ubiquitylated proteins, mediating autophagosome formation on protein aggregates. 118 TBK1 also contributes to aggrephagy by phosphorylating p62, which enhances its binding with ubiquitin and facilitates receptor homo-oligomerization into filamentous polymers. 119 Moreover, TBK1 engages various ubiquitin-binding receptors, including NDP52, TAX1BP1, and OPTN, possibly enabling localized activation of aggregated condensates. However, whether TBK1 regulates the interaction between FIP200 and p62 remains unclear. 46
Others
Lysophagy
Lysosomes play a central role in maintaining cellular homeostasis by receiving cargo from the phagocytic, autophagic, and endocytic pathways. 120 Therefore, optimal lysosomal function is crucial for maintaining cellular homeostasis. However, extracellular substances that enter cells, such as cholesterol, uric acid crystals, asbestos, and silica, can damage lysosomal membranes. 121 To prevent cell death caused by hydrolytic enzymes, cells initiate repair mechanisms to isolate or repair the damage. Lysosomes can be repaired via ESCRT machinery if the damage is minimal. More extensive damage often leads to lysosomal tagging with ubiquitin to activate lysophagy. 121
Ubiquitylation occurs within lysosomal lumen. The ability of the double membrane to recognize and encapsulate ubiquitinated lysosomes depends on the actions of adaptors, such as NDP52, TAX1BP1, and SQSTM1/p62.46,69 UBE2QL1 is crucial for the effective ubiquitination of lysosomes following chemically induced damage. Loss of UBE2QL1-mediated ubiquitination decreases the interaction between the autophagy receptor SQSTM1/p62 and the LC3-decorated phagophore and impairs the recruitment of ubiquitin-targeted AAA-ATPase VCP/p97, which promotes lysophagy. 122 TRIM16, an E3 ubiquitin ligase, binds to damaged lysosomes through galectin 3 (Gal3). In the early stages of lysophagy, it serves as a bridge between damaged lysosomes and ATG proteins, recruiting ATG proteins to damaged lysosomes and facilitating autophagosome membrane formation. 123 Another E3 ubiquitin ligase, FBXO27, interacts with Gal3 to induce lysophagy. In cells overexpressing FBXO27, lysosomal damage results in the ubiquitylation of the key lysosomal membrane proteins, LAMP1 and LAMP2 124 (Fig. 3).
Ferritinophagy
Although iron is necessary for several biological functions, excess free iron can produce ROS, leading to ferroptosis. Ferritinophagy is the selective autophagic degradation of ferritin, which results in the accumulation of cytosolic iron (Fe2+). 125 Nuclear receptor coactivator 4 (NCOA4) functions as a receptor by binding to ferritin heavy chain-1, facilitating intracellular ferritin transport to autophagosomes and subsequent release of free iron. 71 HERC2, an E3 ubiquitin ligase, mediates the ubiquitin-dependent degradation of NCOA4 to reduce ferritinophagic flux, thereby regulating iron levels. 126 Intriguingly, NCOA4 and TAX1BP1 interact to increase ferritin transport to lysosomes in the absence of FIP200, indicating that the ULK1 complex is not required for NCOA4-TAX1BP1-mediated ferritinophagy. 127 This raises the question of how membrane separation can be achieved (Fig. 3).
Lipophagy
Lipid droplets (LDs) are composed of a single phospholipid layer surrounding a neutral lipid core, mainly comprising triacylglycerols and sterol esters. Lipophagy refers to the autophagic breakdown of LDs and is crucial for cellular energy metabolism. Given that there is a size limit for LDs that can be engulfed by autophagosomes, only smaller droplets are subjected to lipophagy, whereas lipases break down larger droplets via lipolysis.46,73 Because LDs are substantially larger (approximately 200 μm) than lysosomes are (0.1–1 μm), the autophagosome membrane usually develops on the surface of LDs, pinching off sections of the LD membrane to generate autolysosomes. 128 PKA phosphorylates perilipins, which are then degraded by the proteasome during lipophagy. After identification by the heat shock cognate protein HSPA8/Hsc70, perilipins bind to the lysosomal membrane receptor LAMP2A, facilitating their relocation to the lysosome. 73 A functional small interfering RNA screen for lipophagy identified several ATGs, including ATG5 and UVRAG, and the selective autophagy receptors SQSTM1/p62, NBR1, and OPTN. 72 The potential role of ubiquitylation in recruiting autophagy receptors to LDs remains unclear. However, inhibiting autophagy results in p62 and ubiquitin recruitment to LDs, indicating that ubiquitylation is necessary for lipophagy. 72
Crosstalk between multiple types of selective autophagy
The ER and mitochondria are closely associated organelles that are essential for maintaining equilibrium between Ca2+ and lipids. Mitochondria-associated ER membranes (MAMs) are regions where the ER membrane contacts the mitochondrial membrane and play pivotal roles in autophagy, inflammation, ER stress, oxidative stress, and mitochondrial fusion and fission. MAMs function as crucial scaffolds for crosstalk between the mitochondria and ER, facilitating several signaling pathways that enable the quick exchange of biological molecules vital for cellular survival.129–131 Active Ca2+ transfer between the mitochondria and ER caused by IP3R opening during ER stress activates the NLRP3 inflammasome.131,132 The close proximity between the ER and mitochondria in hypoxic environments attracts several autophagy-associated proteins such as ATG5 and ATG14.133,134 Moreover, hypoxia concurrently causes ER stress and functional alterations in the mitochondria, along with the activation of multiple organelle-specific autophagies. 135 Both PINK1 and BECN1 relocate to MAMs in response to mitophagic stimuli, enhancing mitophagy by promoting the synthesis of MAMs and autophagosomes. 136 BNIP3L, located in both the ER and mitochondria, mediates the crosstalk between the two organelles to regulate apoptosis. 137 Similarly, FUNDC1 accumulates in MAMs and initiates mitochondrial fission prior to mitophagy under hypoxic conditions. Through its indirect interactions with the ER-resident protein calnexin, FUNDC1 stimulates mitophagic activity. 82 Additionally, the PINK2/parkin pathway, which is associated with mitophagy, can be inhibited by eIF2α/ATF4 pathway knockout (KO). 138 During ER stress, the eIF2α/ATF4 pathway is crucial for the transcription of autophagy genes, such as p62, NBR1, ATG5, ATG7, ATG10, and GABARAP. 139 Therefore, the structural and functional integrity of MAMs is essential for ER–mitochondria interactions and mitophagy activation in response to various stimuli.
The amplification loop established by TBK1-mediated phosphorylation activates the molecular pathway responsible for mitophagy. Additionally, TBK1 amplifies signaling during xenophagy. However, the exact relationship between these two processes and whether they compete for shared molecular components remains to be fully elucidated. 140 Currently, it is uncertain whether defective mitophagy impairs xenophagy by disrupting TBK1 recruitment or whether an overactive xenophagy response inhibits mitophagy. BNIP3 is involved in lipophagy by promoting the lysosomal transport of LDs associated with mitochondria 141 (Fig. 4). Because LDs generate ubiquitous interorganelle contact sites, lipophagy potentially affects diverse selective autophagy mechanisms. 142 The associations between multiple autophagic processes and dysfunction, simultaneously or sequentially, in response to different stimuli warrant further exploration. This provides a new perspective on the therapeutic potential of focusing on interactions between specific types of autophagy.
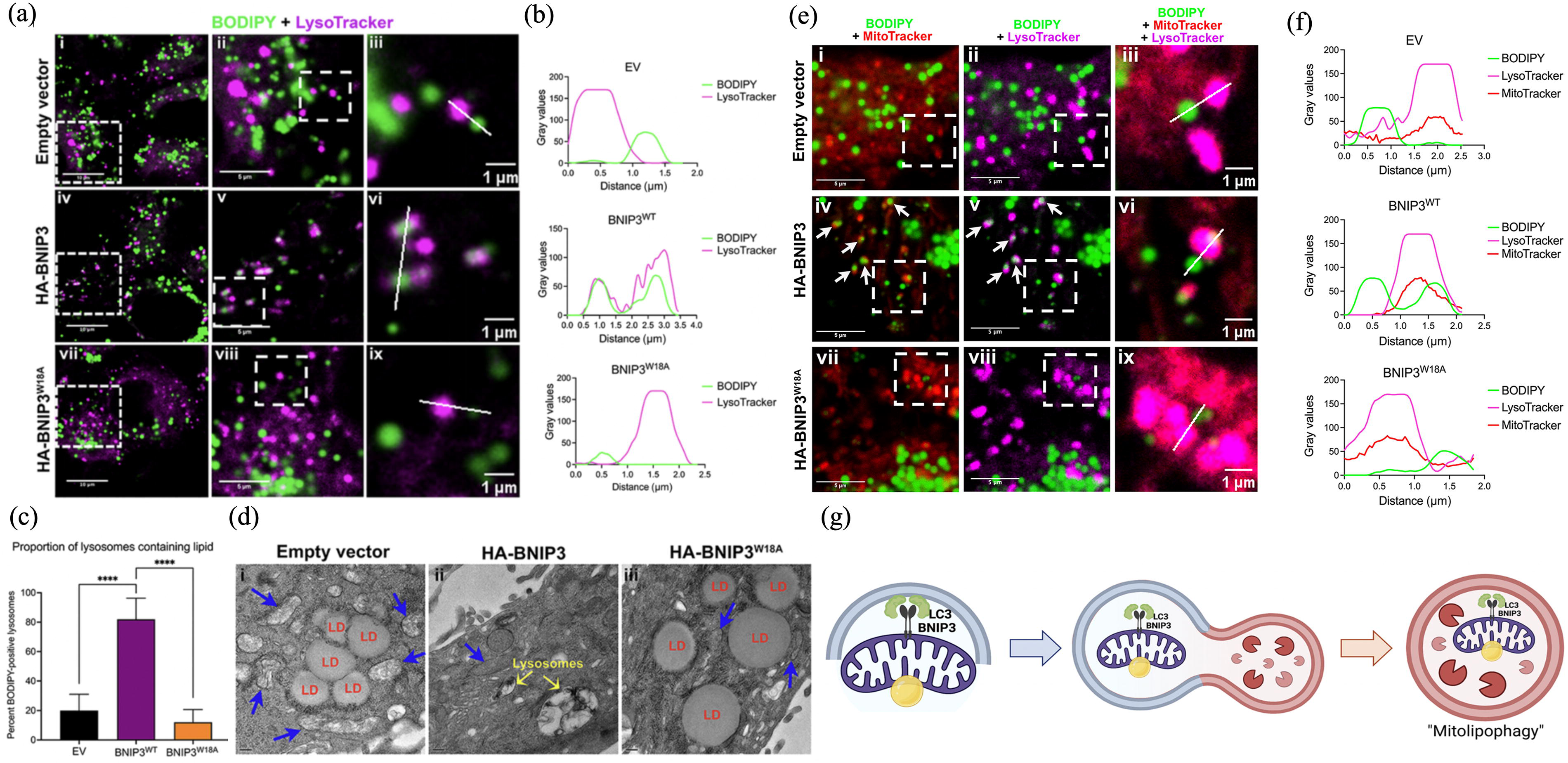
Coordinated autophagic degradation of LDs with mitochondria.
Role of selective autophagy in wound healing
Selective autophagy plays a key role in wound healing with various functions depending on the stage of wound healing. Given that both excessive and inadequate selective autophagy can be deleterious, future studies should focus on elucidating the mechanisms through which selective autophagy promotes tissue repair, thereby maximizing its protective benefits while minimizing potential damage.
Common wound-healing process
Wound healing is a complex process requiring the coordinated involvement of ECM components, cytokines, and several cell lineages. Following a burn injury, the body initiates a wound-healing response to restore tissue integrity. This process involves multiple coordinated actions by different cells. 143 The overlapping biological and molecular processes of coagulation, inflammation, reepithelialization, angiogenesis, and ECM remodeling must be orchestrated for optimal wound healing. 6 Maintaining selective autophagy homeostasis is crucial for the response to damage throughout the wound-healing process. Given its role in the components implicated in the wound-healing process, understanding the role of selective autophagy in promoting wound healing is essential (Table 2).
Selective autophagy as a therapeutic target for wound healing

CCCP, carbonyl cyanide m-chlorophenyl hydrazone; DAMP, damage-associated molecular pattern; EC, endothelial cell; EPC, endothelial progenitor cell; GSP, grape seed proanthocyanidin; HaCaT, human immortal keratinocyte cell line; HG, high glucose; HUVEC, human umbilical vein endothelial cell; IL, interleukin; ManN, N-acetyl-
Mitophagy regulates wound healing
Burn injuries can result in mitochondrial dysfunction. For example, fibrinogen, a damage-associated molecular pattern (DAMP), is elevated in the plasma of patients with burn injuries, indicating a poor prognosis. Elevated fibrinogen levels lower mitochondrial membrane potential and contribute to inflammation. 197 Mitophagy selectively eliminates damaged mitochondria and regulates mitochondrial quality. Elevated ROS levels trigger oxidative stress, which initiates tissue injury in a burn injury model. 144 ROS activates PINK1/parkin-dependent mitophagy. 145 In rats with burn injury, PINK1 and parkin interact to mediate the mitochondrial response. 146 In addition, hypoxia was observed 48 h after the burn and peaked on day 3 at the healing margins of the burn sites. Positive hypoxia-inducible factor (HIF)-1α staining was observed along the leading edge of the healing region in consecutive slices of the same tissue block. 198 Because increased mitophagy significantly reduces ROS generation, mTOR inhibition increases mitochondrial efficiency during acute hypoxia to ensure ATP synthesis. 199 DRP1 inhibits mitophagy by upregulating mito-Clec16a, which prevents parkin recruitment and hinders autophagosome formation in vascular tissues following ischemic damage. Furthermore, ischemia-induced DRP1 activation promotes apoptosis by triggering the translocation of BAX within the mitochondria, increasing cytochrome C release, and activating caspase-3/-9 signaling. 147 Our previous research revealed a strong correlation between the degree of mitophagy and HIF-1α expression in early burn wound development, with downstream involvement of parkin, BNIP3, and FUNDC1 148 (Fig. 5). However, the precise mechanism underlying hypoxia-mediated mitophagy in burn injuries remains unclear. A deeper understanding of the exact mechanisms driving mitophagy during burn wound progression is essential for understanding the function of mitophagy in burn damage.

Mitophagy and HIF-1α protect against burn wound progression.
Atypical autophagy reduces the platelet count and inhibits platelet aggregation. 200 Mitophagy inhibits platelet apoptosis and plays dual roles in platelet activation: under hypoxia or oxidative stress, it prevents platelet aggregation; however, during reperfusion, it may stimulate it.171,201 Hypoxic preconditioning triggers FUNDC1-dependent mitophagy in platelets, reducing ischemia/reperfusion (I/R)-induced heart damage; however, PINK1 is not necessary for platelet function and does not modulate basal platelet mitophagy.149,202 However, studies on Kawasaki disease revealed that thymic stromal lymphopoietin (TSLP) increases platelet mitophagy by activating a new TSLP receptor/parkin/voltage-dependent anion channel 1 pathway, which enhances platelet activation.203,204 Furthermore, NIX plays a platelet-autonomous role in platelet activation—that is, it acts only on platelets and not on other cells. NIX genetic ablation reduces platelet activation and mitochondrial quality without changing the expression of platelet glycoproteins (GPs), such as GPIb, GPVI, and αIIbβ3. Intriguingly, NIX deficiency prolongs platelet survival in vivo, possibly by blocking the autophagic degradation of the mitochondrial protein Bcl-xL. 151 NIX regulates platelet inflammasome activity. 205 Additionally, peroxisome proliferator-activated receptor-γ (PPARγ) loss is associated with FUNDC1 dephosphorylation and mitophagy activation, resulting in enhanced respiratory function, ATP generation, and mitochondrial electron transport chain complex activity. 150 Increased mitochondrial activity is a major factor in platelet aggregation and microthromboses. 149 Melatonin inhibits platelet activation and protects against I/R damage via the PPARγ/FUNDC1/mitophagy axis. Melatonin effectively prevents FUNDC1-dependent mitophagy and platelet hyperactivity by restoring the PPARγ content in platelets. 150 Although mitophagy is required for platelet activation, excessive mitochondrial clearance may be harmful because platelet activation is significantly decreased following hypoxia and carbonyl cyanide m-chlorophenyl hydrazone (CCCP) therapy, both of which induce strong mitophagy. 151 However, the role of other mitophagic mechanisms in platelet activation remains unclear. The mechanistic connections among platelet physiopathology, mitophagy, and platelet lifespan may offer novel targets for treatment during the hemostatic phase.
During tissue damage or infection, pattern recognition receptors (PRRs) such as toll-like receptors (TLRs) and nod-like receptors (NLRs) detect pathogen-associated molecular patterns (PAMPs) or DAMPs, which trigger inflammation. 206 This activates chemical signaling cascades that induce leukocyte chemotaxis from the general circulation to the wound site. The recruited leukocytes subsequently produce cytokines that amplify the inflammatory response.206,207 PAMPs inhibit mitophagy, which enhances mitochondrial ROS (mtROS) production and macrophage activation in the early stages of wound healing, exacerbating the severity of bacterial infections and sepsis in vivo by degrading PINK1/parkin via caspase 1.152,153 Mitophagy can prevent DAMPs generation and protect organelle integrity, suggesting that mitophagy may mitigate inflammation. 154 Parkin- and PINK1-mediated mitophagy mitigates stimulator of interferon gene (STING)-induced inflammation and inhibits innate immunity in mutant mice. 155 Macrophage activation may be associated with mitophagy. The M1 phenotype triggers the production of inflammatory mediators in response to various stimuli. However, prolonged M1 activity delays healing. M2 macrophages aid in tissue repair and remodeling and inhibit inflammation by releasing substantial amounts of transforming growth factor (TGF)-β and interleukin (IL)-10.208,209 IL-10 blocks the mTOR signaling pathway, facilitating mitophagy, and its absence leads to dysregulated NLRP3 inflammasome activation and IL-1β production. 210 Aging reduces mitophagic flow, allowing macrophage mitochondrial DNA (mtDNA) to enter the cytosol and activate STING. Mechanistically, aged macrophages exhibit a decrease in mitochondrial polyubiquitination mediated by PINK1/parkin. 156 M1 macrophages exhibit elevated NIX expression, and NIX deletion reduces M1 macrophage polarization. 157 These findings imply that NIX-mediated mitophagy is essential for macrophage activation. Further research is necessary to determine whether other mitophagy-related receptors are involved in macrophage activation because the precise underlying mechanisms remain unclear. Additionally, mitophagy may play a crucial role in the development of sepsis. For example, hemin reduces mitochondrial lipid peroxidation and mitochondrial dysfunction while enhancing mitophagy, as shown by an increase in parkin levels in a mouse model of sepsis. 158 In summary, mitophagy has both pro- and anti-inflammatory functions, contributing to microbial clearance and tissue healing in the early and later stages of wound healing, respectively, by regulating inflammation during wound healing.
Endothelial cells (ECs) initiate angiogenesis, whereas epithelial cells proliferate and migrate to restore the epidermal layers over the wound, a clinical indicator of healing. 9 The epidermal barrier is continuously replenished by differentiating keratinocytes through cornification, during which autophagy helps remove organelles and the nucleus.159,211 Gsdma1/a3 activation disrupts mitophagy in vivo, promoting mitochondrial turnover and epidermal cornification. A lack of Gsdma1/a3 impairs stratum corneum maturation and tight junction formation. 159 Mitophagy protects the human immortal keratinocyte cell line (HaCaT) against benzo[a]pyrene-induced cell death by activating beclin-1-dependent autophagy via the AMPK/mTOR pathway. 160 Dasatinib inhibits HMGB1-mediated mitophagy, leading to keratinocyte death. The generation of mtROS results in DNA damage and cell death. 161 Exposure to ultraviolet B (UVB) radiation inhibits manganese-containing superoxide dismutase, resulting in mitochondrial damage and mitophagy via the ROS-mediated mTORC2 pathway in keratinocytes. 212 However, UVB damages HaCaT cells by suppressing PINK1/parkin-mediated mitophagy, which leads to cytosolic leakage of mtDNA and subsequent activation of cyclic GMP-AMP synthase (cGAS)-STING, which can be rescued by 1,25(OH)2D3 and collagen I.213–215 These findings highlight the importance of mitophagy in regulating keratinocyte fate. Although MAP4 promotes hypoxia-induced wound healing through depolymerizing microtubules in a phosphorylation-dependent manner, 216 mitophagy-associated self-degradation of p-MAP4 is crucial for keratinocytes’ migratory and proliferative responses to hypoxia. 162 Therefore, investigating the potential involvement of BNIP3L and FUNDC1, key HIF-1α transcriptional targets, in keratinocyte vitality is promising. Ectopic NIX expression accelerates keratinocyte differentiation and triggers mitochondrial fragmentation via DRP1, whereas NIX depletion impairs mitochondrial elimination and hinders epidermal maturation. 163 MiR-30a specifically targets BNIP3L and is overexpressed in the aged epidermis. Interestingly, a 3D organotypic model of the reconstructed human epidermis overexpressing miR-30a showed a substantial decrease in BNIP3L expression in the granular layer. Both human primary keratinocytes in vitro and epidermal sections of skin biopsies showed a similar trend, whereas BNIP3L expression decreased with age, along with mitochondrial preservation. 164 Additionally, stimulating FUNDC1 is crucial for enhancing the migration, proliferation, and odontoblastic differentiation of human dental pulp cells under hypoxic conditions. 217 This suggests that FUNDC1 represents an alternative mitophagic regulator involved in epithelial and granulation tissue formation. In addition to epidermal barrier formation, mitophagy regulates vascular and EC functions. Disruptions in PINK1/parkin-mediated mitophagy in the arteries of ethanol-treated mice led to vascular dysfunction, which was reversed by CCCP. 165 HOXD9 controls downstream PINK1-mediated mitophagy and enhances angiogenesis and the migratory capabilities of endothelial progenitor cells (EPCs). 166 DRP1 overexpression in arterial ECs causes dysregulated mitophagy and mitochondrial fission. Mitophagy plays a key role in DRP1/Bak/BNIP3-dependent ECs apoptosis. 218 Therefore, the intricate and crucial regulation of mitophagy under physiological and pathological conditions may contribute to vascularization, which requires further investigation. Grape seed proanthocyanidins (GSPs) facilitate wound healing in mice by enhancing angiogenesis, collagen deposition, and granulation tissue formation. Mechanistically, GSPs reduced ROS levels and promoted the synthesis of antioxidant proteins by upregulating p-JNK/FOXO3a protein expression, which subsequently regulated mitophagy-associated proteins in ECs 167 (Fig. 6). These findings offer novel insights into the potential role of mitophagy in wound healing; however, further research is necessary to elucidate the underlying mechanisms.
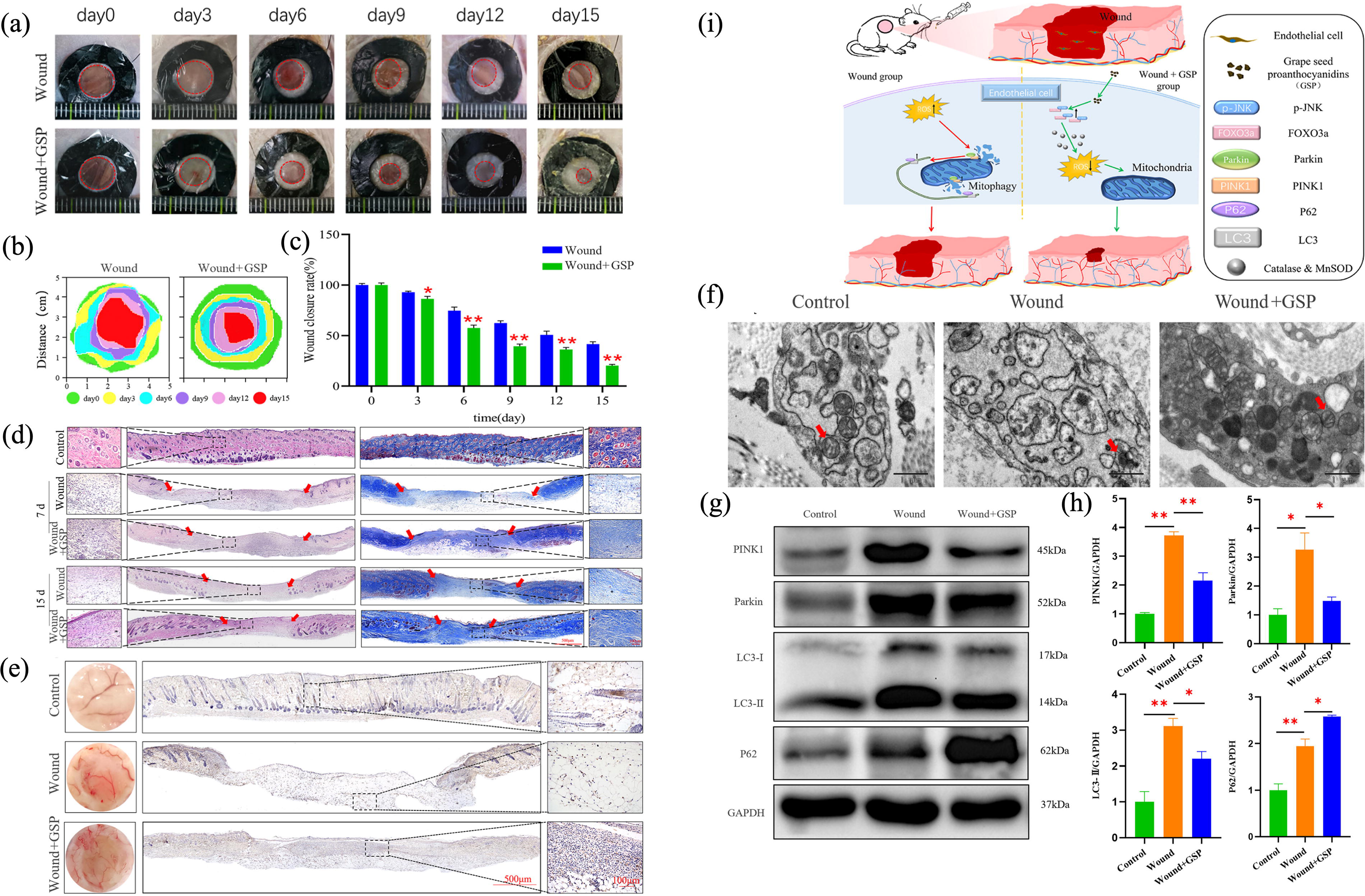
GSPs regulate mitophagy of ECs and promote wound healing in mice.
During the remodeling phase, fibroblasts release excess immature type III collagen into the matrix, which is then replaced by type I collagen. The matrix remodels, and the wound site is covered with fresh scar tissue. 219 PARK2 knockdown-mediated mitophagy suppression activated the platelet-derived growth factor receptor (PDGFR)/PI3K/AKT signaling pathway, leading to increased myofibroblast differentiation and proliferation. 168 However, pirfenidone blocks PARK2 knockdown-induced myofibroblast differentiation by lowering mtROS levels and PDGFR-PI3K-AKT activation. 169 Moreover, PDGFR signaling stimulation mediated by mitophagy inhibition further suppresses autophagy, indicating the presence of a self-amplifying loop between PDGFR activation and mitophagy inhibition. ANT2, which maintains energy homeostasis by activating glycolysis and inducing mitophagy, is upregulated in replicative senescent human diploid dermal fibroblasts and enhances cell motility and proliferation, which are key functions in wound healing. 170 Although the role of mitophagy in pulmonary, cardiac, hepatic, and renal fibrosis has been documented,220–223 its involvement in scar formation and matrix remodeling warrants further investigation.
ER-phagy serves as a potential regulatory target in wound healing
ER-phagy can be enhanced under stressful circumstances such as hypoxia, oxidative stress, and nutrient deprivation to preserve ER proteostasis.56,87 During the UPR, ER-phagy maintains cell viability by digesting misfolded/unfolded proteins and restoring ER homeostasis, which is essential for activating prosurvival mechanisms and adapting to a stressful microenvironment. In the tumor microenvironment, hypoxia triggers the UPR and ER-phagy to adjust to low oxygen availability, which accelerates tumor growth.224,225 Thus, ER-phagy may contribute to cell homeostasis in response to the hypoxic milieu of a wound. However, autophagy triggers apoptosis under ER stress. For example, inhibition of autophagy can inhibit caspase-8-mediated cell death. 226 Furthermore, antiapoptotic XIAP proteins are dramatically suppressed in response to chronic ER stress. 227 Determining the interaction between UPR-induced cellular death mechanisms and prosurvival pathways is crucial for wound healing.
Patients with severe burns often develop hypermetabolic disorders. ER stress in metabolic tissues may be a hallmark of the biological mechanism of the prolonged hypermetabolism observed in such patients. 173 Moreover, severe burns cause persistent ER stress in vivo, resulting in systemic inflammation. Prolonged ER stress releases DAMPs, which activate dendritic cells to promote the polarization of naïve T cells. 174 Burns can induce autophagy, apoptosis, and ER stress-related protein expression in human burnt fibroblasts. Chloroquine restores the migratory activity of keratinocytes and decelerates burn-induced autophagy, which may slow apoptosis-related processes. 175 Although ER stress signaling is vital for cellular defense against environmental shock, prolonged activation can disrupt protein function, accelerate metabolic disorders, and lead to cell death. To date, few studies have focused on ER stress in burn-wounded skin cells. ER-phagy restoring homeostasis of ER and cells may hold potential for future research on burn wounds.
During wound healing, the cells synthesize several proteins that require ER folding. Various cellular stress conditions in the wound microenvironment, such as hypoxia, can lead to the accumulation of unfolded or misfolded proteins in the ER, resulting in ER stress.
177
The UPR is a signaling pathway used by eukaryotic cells to control ER stress. Mild UPR activation promotes homeostasis, whereas excessive UPR activity causes cell death.
89
Cochaperones facilitate the degradation of misfolded proteins. KO and knock-in studies have demonstrated that the cochaperone ERDJ4 regulates protein synthesis and epithelial-to-mesenchymal transition, a critical step in wound healing, by degrading SREBP1c and assembling the mTORC2 protein complex.
228
Lycium barbarum polysaccharide protects skin cells from the hazardous effects of PM2.5, by modulating the oxidative stress-ER stress–autophagy–apoptosis axis.
229
Following skin injury, keratinocytes transition from a homeostatic state to a regenerative state to restore the epidermal barrier. The human skin-specific long noncoding RNA HOXC13-AS impedes molecular transit from the Golgi apparatus to the ER, causing ER stress and promoting keratinocyte differentiation.
176
KCNQ1OT1 upregulates stress-associated ER protein 1 (SERP1) expression via competitive sponging of miR-200b-3p. KCNQ1OT1 facilitates keratinocyte migration via the miR-200b-3p/SERP1 axis, playing a crucial role in wound healing.
230
Treatment with 4-phenylbutyrate enhances the migration of keratinocytes from aged leg skin, decreases the levels of ER stress indicators, and accelerates reepithelialization in a human skin wound-healing model, suggesting its potential as a therapeutic agent.
177
Furthermore, through ER stress-induced JNK activation and the UPR, N-acetyl-
Emerging roles of pexophagy in wound healing
Peroxisomes are primarily implicated in ROS metabolism and fatty acid degradation. 105 The PPAR family of nuclear receptors is activated by changes in glucose and lipid levels, thereby increasing the abundance of peroxisomes. 107 Stimulation of PPAR nuclear receptors triggers PEX11 protein production, enhancing peroxisome elongation. 234 Pseudomonas aeruginosa (PA) disrupts lipid balance by suppressing PPARδ through ceramidase expression and subsequently generates ceramide catabolic products. Downstream products of this metabolism are directly linked to PPARδ suppression. PA creates biofilms that lead to refractory wounds that disrupt the lipid balance of the skin following PPARδ downregulation. Therefore, the ability of the wound site to act as a barrier is impaired. 235 Interestingly, the mouse taste receptor type-2 member 138-N-(3-oxododecanoyl)-L-homoserine lactone (AHL-12) complex augments the degradation of LDs and releases PPARγ, thereby accelerating the clearance of AHL-12 in neutrophils to preserve homeostasis immediately during PA infection. 236 Tissue-remodeling macrophages or M2a cells are crucial for wound healing. PPARγ mediates the IL-4-dependent M2a polarization of macrophages. 237 Similarly, genipin polarizes the M2 macrophage phenotype by activating the pSTAT6-PPARγ pathway. 238 A cohort study revealed that the enhancer-region polymorphism rs10865710 increases susceptibility to traumatic sepsis by altering PPARγ expression. 239 Additionally, conditional deletion of epidermal PPARγ coactivator-1 alpha results in delayed wound healing in mice due to decreased proliferation and enhanced cell differentiation driven by UVB and inflammation. 240 Moreover, PPAR agonists enhance epidermal differentiation and suppress proliferation. PPARα or PPARγ regulates the levels of aquaporin-3, a key skin epidermal protein that affects hydration, permeability, barrier repair, and wound healing. 241 PPARs play distinct roles at various stages of wound healing; however, studies on the role of peroxisomes in wound healing are scarce. Pexophagy selectively kills excess or damaged peroxisomes, thereby maintaining the redox equilibrium and glucose and lipid metabolism in response to environmental stimuli. Managing the quality and abundance of peroxisomes via pexophagy to maintain homeostasis may offer a promising avenue for optimizing wound-healing environments.
Xenophagy potentially eradicates pathogens in wounds
Increasing evidence indicates that xenophagy represents a conserved host defense response against various intracellular pathogens via innate immunity and that ATGs play a protective role against diverse pathogens both in vivo and in vitro. 189 One of the links that connects autophagy with innate immunity is macrophages. Furthermore, various genes, including beclin1, Atg5, and Vps34, are involved in autophagy and phagocytosis in macrophages, suggesting that autophagy is a distinct phagocytic response that aids in host immunological defense in macrophages. 187 Certain PAMPs, including lipopolysaccharides, flagellin, and CpG oligodeoxynucleotides, can trigger autophagy by activating TLR4, TLR5, and TLR9. 188 By interacting with PAMPs, macrophages and other immune cells deploy PRRs to recognize invasive infections. TLRs, scavenger receptors, and C-type lectins are cell-surface receptors, whereas NLRs, peptidoglycan receptors, and IFN-γ are intracellular receptors. 189 Autophagy can be initiated in a cell-autonomous way by PRRs sensing PAMPs. In addition, macrophage autophagy deficiency alleviates cutaneous inflammation in a mouse model by upregulating CEBPB and SOCS1/3 transcription and decreasing M2 macrophage activation. 190 Thus, autophagy is necessary for M2 macrophage differentiation. Modulating autophagy may be a fantastic adjuvant tactic to support proregenerative methods. It is intriguing to leverage autophagy manipulation to promote wound healing while reducing infection. By controlling the innate immunity and immune cell conditions, selective autophagy improves the wound microenvironment and counteracts pathogenic infections.
Most chronic wounds harbor diverse bacterial species that can synergize with nonvirulent bacteria to become virulent and harm their hosts. Commonly detected bacteria include Escherichia coli, Staphylococcus, Pseudomonas, Peptoniphilus, Enterobacter, Stenotrophomonas, Finegoldia, and Serratia,242–244 often accompanied by obligate anaerobes.244,245 Studies using light and scanning electron microscopy have examined 16 acute and 50 chronic wound samples. Molecular investigations of samples from chronic wounds revealed the presence of bacteria, including strictly anaerobic bacteria and various polymicrobial communities that live as extremely durable biofilms. 246 However, owing to poor penetration of systemic antibiotics into wound biofilms and sensitization risks from topical antibiotics, antisepsis is the preferred treatment strategy. 247 Because xenophagy eradicates several harmful antibiotic-resistant bacteria, it is vital for survival. 110 Immunological equilibrium and infection outcomes may be largely determined by the housekeeping function of autophagy rather than by direct pathogen destruction. 248
Using a bacterial transposon screen, Xu et al. identified the T3SS effector SopF, which effectively inhibits Salmonella autophagy. SopF inhibits xenophagy without influencing canonical autophagy by blocking the V-ATPase-ATG16L1 axis, which is crucial for the autophagic recognition of intracellular pathogens. 191 Furthermore, SopF selectively ADP-ribosylates Gln124 on ATP6V0C, a V-ATPase component, impairing ATG16L1 recruitment to vacuoles in host cytosol-harboring bacteria. 192 To induce targeted degradation of intracellular microorganisms via xenophagy, including Streptococcus pyogenes and Escherichia coli, Lee et al. developed chemical mimics of N-degron Nt-Arg. These substances cause lysosomal degradation by inducing the synthesis and recruitment of autophagic membranes to intracellular bacteria via SQSTM1. 193 However, certain pathogens evade autophagy by preventing the formation of autophagosomes or exploiting the xenophagy system for survival. 249 Mycobacterium tuberculosis (Mtb) efficiently inhibits host-directed autophagy by utilizing virulence factors. Intracellular pathogens have evolved an extensive array of virulence factors that suppress host defenses by manipulating the host ubiquitination pathway. 194 Eukaryotic-type protein kinase G prevents autophagosome formation and disrupts autophagy by targeting the host small GTPase RAB14, supporting Mtb intracellular survival in macrophages and in vivo. 195 The TOM1L2/Rab41 pathway-mediated ESCRT is crucial for maintaining the homeostasis of xenophagolysosomes and effective elimination of microorganisms via xenophagy. 250 Collectively, innate defense mechanisms during infection rely on autophagy to identify and guide intracellular pathogens into lysosomes, whereas bacteria may release proteins to protect themselves and escape host xenophagy. Given the presence of persistent bacteria in refractory wounds and the poor efficacy of antibiotics, applying xenophagy to maintain immunological homeostasis in the wound microenvironment is a promising research frontier.
Others
Lysosomes are essential for cellular clearance, exocytosis, Ca2+ signaling, and wound healing. 251 As previously mentioned, when lysosomal membranes are damaged by extracellular stimuli, cells attempt to isolate or repair the damage to prevent hydrolytic enzyme-induced cell death. If damage is minimal, lysosomes can be repaired by the ESCRT apparatus; otherwise, they are tagged with ubiquitin to initiate lysophagy.121,252 When ESCRT activity is impaired, the cytosolic surface of damaged lysosomes undergoes metabolic conversion of sphingomyelin by neutral sphingomyelinases to facilitate lysosomal repair. 253 Moreover, annexins (ANXA1 and ANXA2) are essential for repairing damaged lysosomes independent of ESCRT. 254 Lysosomal exocytosis mediates plasma membrane healing, which relies on localized elevations in intracellular Ca2+ at damage sites.255–257 During inflammation, phagocytes produce digestive enzymes from lysosomes to degrade pathogens. Myoferlin increases cytotoxicity by stimulating the release of hydrolytic enzymes and other lysosomal contents. 258 Wu et al. delineated a newly discovered luminogen, TTTh, which selectively targets lysosomes and enhances their maturation to expedite the removal of intracellular microorganisms. TTTh eliminates methicillin-resistant Staphylococcus aureus by inducing ROS accumulation in bacteria and disrupting membrane integrity. 259 Mutations in the CXCR3 chemokine receptor enhance macrophage lysosomal function in zebrafish larvae to express vesicle trafficking and lysosomal genes. These macrophages possess large lysosomes that improve intracellular bacterial clearance. 260 Therefore, chemotaxis and lysosomal function are closely related mechanisms necessary for intracellular bacterial clearance and inflammatory responses. Furthermore, cell-intrinsic lysosomal lipolysis mediates M2 activation, which is crucial for wound healing. 261 Lysosomes and lysosome-related organelles (LROs) are dynamic organelles that maintain cellular homeostasis. Lysosomal trafficking regulator protein (LYST) controls the intracellular trafficking of lysosomes and LROs as well as membrane dynamics. The wound model demonstrated that regardless of macrophage infiltration and polarization, LYST-mutant mice exhibited poor wound healing, characterized by delayed epithelialization and collagen deposition.262,263 In summary, lysosomes perform various cellular functions such as inflammation regulation, plasma membrane repair, and pathogen destruction during wound healing. The repair or removal of damaged lysosomes potentially contributes to accelerated wound healing, making lysophagy a promising research area.
LDs consist primarily of glycerophospholipids, sphingolipids, and cholesterol. Constitutive lipids, including phospholipids, triglycerides, and sphingomyelin, are involved in metabolism, inflammation, and wound healing. 264 Stress- and inflammation-induced hypermetabolic responses are characteristics of burn recovery. The browning of white adipose tissue (WAT) is a vital component of this complex negative reaction. 265 Burn-induced WAT browning and enhanced lipolysis accelerate hepatic steatosis in mice. Lipidomic profiling revealed elevated levels of damage-inducing lipids (palmitic and stearic acids) in the plasma of burn patients and postburn rats. These lipids inhibit hepatic fat oxidation and induce ER stress. Following burn injury, hepatic ER stress enhances ER–mitochondria interactions, hepatocyte death, and oxidative stress. 266 Lipases degrade larger LDs via lipolysis, whereas smaller droplets are subjected to lipophagy, which helps mitigate the side effects caused by burn-induced WAT browning. In conclusion, selective autophagy is critical for maintaining cellular function during the physiological disruption caused by burns. Investigating the function of selective autophagy in burn wound healing holds promise for future research. Appropriate lipid metabolism is crucial for wound healing. The geographical distribution and relative abundance of cholesterol sulfate and healing time are correlated. 267 Rats lacking the lipase Pnpla5 exhibited lower levels of proteins involved in steroid metabolism and wound healing. 268 Additionally, proinflammatory signaling impedes LDs maturation in differentiated adipose-derived stem cells. The presence of adipocytes in wound tissue implies a strong anti-inflammatory milieu. 269 Moreover, effective damage repair of the EC membrane is dependent on Ca2+-dependent annexin–phospholipid interactions. 270 Keloids have a fibroblast-centered communication regulatory network, with the glycosphingolipid metabolic pathway affecting cellular communication in fibroblasts. 271 Despite the fact that increasing lipophagy decreases the formation of LDs, studies supporting the idea that lipophagy and wound healing are directly related are limited. Therefore, additional research is warranted to fully comprehend the correlation between lipophagy and wound repair.
Chronic wounds
Diabetes often leads to vascular and nerve damage, resulting in delayed and incomplete wound healing. 272 Platelet mitophagy serves as a preventive mechanism against oxidative stress by reducing phosphorylated p53 and activating JNK, thereby preventing apoptosis. Under diabetic conditions, platelets with impaired mitophagy are more susceptible to oxidative stress, which increases the risk of thrombosis. 171 High glucose (HG) and glucometabolic toxicants disrupt mitochondrial functions, including calcium cycling and metabolism, and TRPC6, a pore subunit of transient receptor potential channels that mediate Ca2+ influx. Mesenchymal stem cell (MSC)-derived small extracellular vesicles deliver the transcription factor SP2 and the deubiquitinating enzyme USP9 to restore TRPC6 function and mitophagy to support diabetic wound healing. 172 In addition to mitophagy, mitochondrial calcium homeostasis presents a promising avenue for further research on diabetic wound healing.
The cGAS-STING signaling pathway mediates inflammatory responses to cell stress and injury. 273 Overexpression of the cGAS-STING pathway in diabetic wounds affects cellular metabolism, leading to inflammation and impaired healing. ER stress and STING are closely related, revealing novel therapeutic targets for improving healing outcomes in patients with diabetes. 183 Lower levels of miR-146a are associated with higher levels of oxidative stress and ER stress in patients with diabetic foot ulcers and in 3T3 mouse fibroblasts. 184 Exendin-4 inhibits the p38 MAPK pathway and decreases ER stress and ROS levels to promote EPC viability. 185 Neutrophil extracellular traps (NETs) are abundant in HG-induced neutrophils and diabetic wounds. High NET concentrations hamper fibroblast activity by inducing ER stress. 186 In vitro observations of HG-induced ferroptosis in human dermal fibroblasts (HDFs) revealed compromised cellular activity. Secretory autophagosomes (SAPs) produced by human umbilical vein ECs (HUVECs) inhibit ferroptosis and stimulate migration, proliferation, and tube formation to facilitate diabetic wound healing. SAPs inhibit ferroptosis by reducing ER stress-induced free Fe2+ production in HG-HDFs and increasing exosome-mediated Fe2+ removal from HG-HDFs. 180 This highlights the relationship between ER stress and iron metabolism in the HG environment, providing a new direction for exploring the mechanisms involved in diabetic wound healing. Targeting ER stress, potentially through ER-phagy, offers promise for alleviating oxidative stress and improving diabetic wound-healing outcomes.
In diabetic mice, inhibiting progestin and adipoQ receptor 3 promotes M2 macrophage polarization, HaCaT cell migration, angiogenesis, and PPARγ expression, all of which enhance cutaneous wound healing. 274 By modulating macrophages and enhancing reepithelization and collagen deposition, local administration of the PPARγ agonist GQ-11 accelerated wound closure in db/db mice. 275 Moreover, a sustained-release topical PPAR-γ agonist is effective in treating diabetic wounds with fewer systemic adverse effects and high bioavailability during the inflammatory and proliferative phases of recovery. 276 Compound Y8 promotes granulation tissue formation and ECM deposition, which in turn expedites wound healing in diabetic rats. Mechanistically, Y8 stimulates PPARβ to increase keratinocyte proliferation and decrease ROS levels in fibroblasts in vitro. 277 Because PPAR is associated with peroxisome abundance, pexophagy is a potential strategy for regulating the quantity and quality of peroxisomes, thereby preserving homeostasis of the healing environment in diabetic wounds.
The proteomic profile of chronic wound-associated fibroblasts exhibited dysregulated TGF-β signaling, lysosomal dysfunction, reduced cell division and migration, and increased ECM compression. 278 Eliminating damaged lysosomes is crucial for accelerating wound healing, highlighting the potential of lysophagy as a research focus in diabetic wound healing. In addition, diabetes is closely linked to abnormalities in iron metabolism that cause ferroptosis and ferritinophagy. NCOA4, a key regulator of ferritinophagy, was downregulated in both HG-treated senescent fibroblasts and diabetic wound tissue. Furthermore, the promotion of ferritinophagy may reverse the process of senescent fibroblasts elimination, which is hindered by resistance to ferroptosis. NCOA4 overexpression increases the susceptibility of senescent fibroblasts to ferroptosis, which is associated with the faster healing of diabetic wounds. Moreover, AMPK activation-induced ferroptosis eliminates senescent fibroblasts from diabetic wounds by inducing NCOA4-mediated ferritinophagy. In diabetic mouse model, AMPK activation decreased tissue senescence and accelerated wound healing 196 (Fig. 7). Therefore, ferroptosis and ferritinophagy are potential therapeutic targets for improving the healing of diabetic ulcers in patients with diabetes (Table 2).
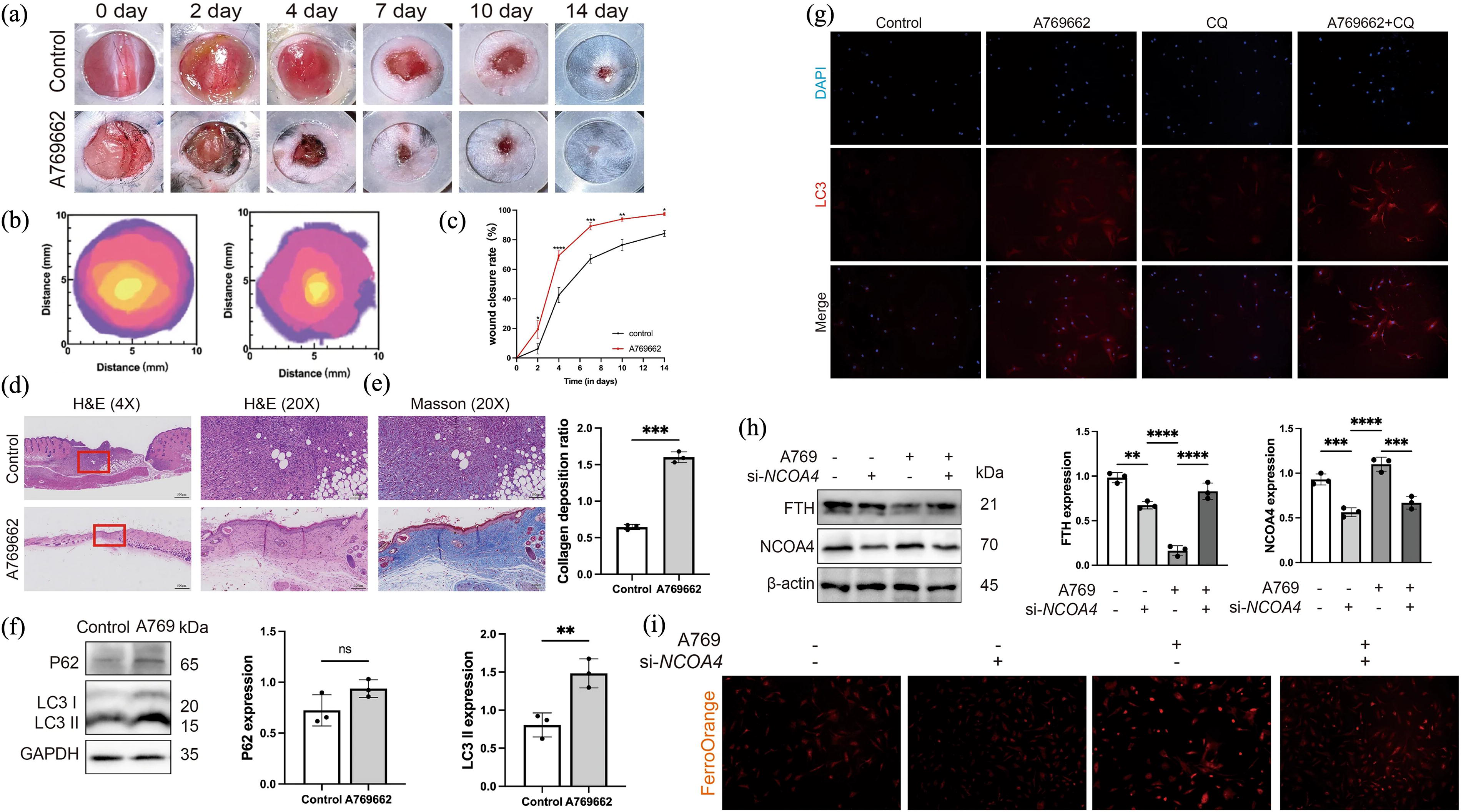
AMPK activation accelerates diabetic wound healing by inducing NCOA4-mediated ferritinophagy.
Clinical future of selective autophagy in wound healing
Therapeutic potential of selective autophagy
Given that selective autophagy is essential for wound healing, its regulation is a promising therapeutic strategy for wound care. Clinical trials of numerous autophagy activators and inhibitors that may accelerate skin wound healing are currently underway. Mitophagy is a basic process that selectively degrades damaged mitochondria to manage their quantity and quality. In response to burn injury, farnesyltransferase (FTase) and protein farnesylation levels increase; however, the administration of an FTase inhibitor mitigates mitochondrial dysfunction, elevates the HIF-1α levels, and other alterations triggered by burn injury. 279 In the healing margin of the burn sites, acute hypoxia was observed after 48 h and peaked on day 3 postinjury. 198 Because increased mitophagy significantly reduces ROS production, mTOR inhibition increases mitochondrial oxygen usage efficiency to guarantee ATP synthesis during acute hypoxia. 199 This emphasizes that targeting mTOR may offer a novel approach for enhancing acute hypoxic tolerance during wound repair. Targeting PINK1 and parkin offers additional therapeutic potential for addressing mitochondrial damage in burns. 146 Furthermore, HIF-1α mediates parkin- or BNIP3-mediated mitophagy, further emphasizing its relevance as a therapeutic target for burn wounds. Lu et al. reviewed several small-molecule modulators of mitophagy and provided valuable insights into their therapeutic applications. 50 Thus, these regulators may serve as potential targets for burn and wound healing.
Chronic hypoxia is caused by HIF-1 deficiency, and the inability to respond to hypoxic stimuli can contribute to the development of nonhealing ulcers. However, HIF-1 overexpression triggers myofibroblast differentiation, resulting in fibrotic disorders via increased matrix formation and deposition. Targeting HIF-1 offers a promising therapeutic approach for regulating wound healing.280,281 SIRT1-PINK1 signaling-mediated mitophagy attenuated reperfusion-induced oxidative stress, inflammation, apoptosis, and mitochondrial dysfunction.282,283 SIRT3 promotes mediated mitophagy and rescues cellular senescence, which can be effectively addressed as a possible candidate for aiding wound healing.284,285 Melatonin modulates mitophagy and increases the survival rate of skin flaps used in wound healing. 286 The ginsenoside, Rh2, inhibits mitophagy and restores membrane potential and mitochondrial ATP synthesis in UV-irradiated normal HDFs, which promote cell proliferation, ECM regeneration, and antioxidant capability. 287 Daiki et al. introduced an autophagy-targeting chimera (AUTAC), a unique targeted-clearance approach. In patient-derived fibroblasts, mitochondria-targeted AUTAC expedites the production of functionally normal mitochondria and eliminates defective fragmented mitochondria. 288 Zan et al. developed HCy-H2O2, a unique near-infrared fluorescence probe that consistently targets mitochondria, enabling the monitoring of H2O2 production during mitophagy. 289 Furthermore, metformin-engineered empty vector (Met-EVs) restore mitophagy to reverse senescence-induced mitochondrial dysfunction, suggesting that they provide sufficient ATP synthesis during the repair of aged tissues. Following Met-EV therapy, ATP metabolism is restored through increased OXPHOS and decreased glycolysis, thereby fixing cell dysfunction and improving healing of aged mouse skin. 290 These findings provide insights into the underlying mechanisms through which EVs ameliorate cellular senescence.
Elevated NET concentrations disrupt fibroblast function through the induction of ER stress. MiR-17-5p in hypoxia-pretreated MSC-derived sEVs inhibited the TLR4/ROS/MAPK pathway, thereby preventing NET formation and impairing fibroblast function. 186 These findings reveal a novel NET-related mechanism in diabetic wounds and offer a potentially effective NET-targeting approach for wound care. In the DB/db mouse model, the sustained release of SAPs from Gel-SAPs facilitated diabetic wound healing by suppressing ferroptosis and reestablishing the typical activity of skin repair cells. SAPs facilitate HUVEC function, thereby accelerating the healing of diabetic wounds. The inhibitory effect of SAPs on ferroptosis is mediated by reducing the ER stress-regulated production of free Fe2+ in HG-HDFs and stimulating exosome release 180 (Fig. 8). Although preclinical studies have focused on the role of ER stress in wound healing, research on the therapeutic potential of targeting ER stress and ER-phagy during wound healing is limited. Cytokine TNF-α was demonstrated to initiate p62-mediated xenophagy targeting intracytosolic Shigella and Listeria. 67 Further research is needed to determine the impact of TNF-α on autophagy flow, which is essential for optimal cell metabolism. Lee et al. designed and synthesized pharmacological p62 agonists that targeted the N-degron pathway and facilitate p62-mediated xenophagy during microbial invasion to investigate a novel and efficient pharmaceutical method for eliminating a variety of drug-resistant intracellular bacteria. These p62 agonists have been shown to possess anti-inflammatory properties in addition to their antimicrobial ones both in vitro and in vivo. Intriguingly, antimicrobial effectiveness works in tandem with decreased generation of inflammatory cytokines, independent of rapamycin-modulated core autophagic pathways. 193 Manual chemical stimulation of universally conserved p62-dependent xenophagy may be a potential therapeutic strategy for infectious disorders caused by different bacterial strains (Table 3).

SAPs inhibit ferroptosis by reducing ER stress-regulated free Fe2+ generation in skin repair cells to accelerate diabetic wound healing.
Compounds or modulators targeting selective autophagy for burn trauma and wound healing
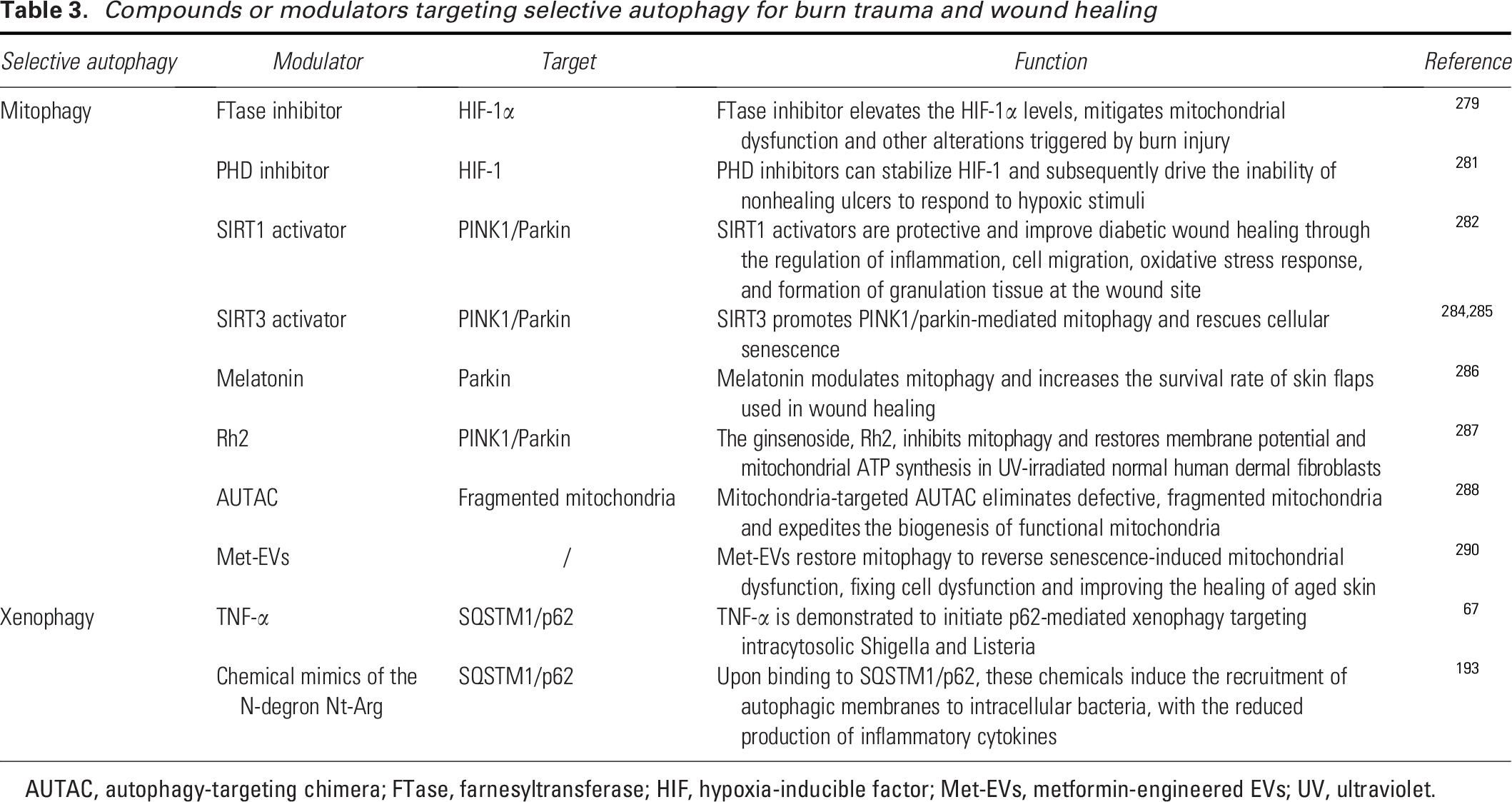
AUTAC, autophagy-targeting chimera; FTase, farnesyltransferase; HIF, hypoxia-inducible factor; Met-EVs, metformin-engineered EVs; UV, ultraviolet.
Clinical research on the application of selective autophagy
Few clinical studies have investigated the potential therapeutic benefits of autophagy in wound healing. Lipidomic analysis revealed that propranolol treatment induced an anti-inflammatory lipidomic profile after burn injury (p < 0.05), which could be partially attributed to reduced ER stress, as evidenced by a decrease in p-JNK levels (p < 0.05). The considerable improvement in stress responses was attributed to the ability of propranolol to ameliorate pathological alterations in critical metabolic pathways. 291 However, relevant research on the relationship between lipidomic metabolism and ER-phagy is lacking. Microparticles (MPs) derived from thrombin-activated platelets of four obese women showed reduced expression of proteins involved in mitophagy and antioxidant defense. MPs produce cyclooxygenase-2 in human cardiac microvascular ECs, potentially increasing the risk of thrombosis (National Clinical Trial [NCT]01581801). 292
Several clinical trials have investigated the roles of autophagy in various human diseases. Hydroxychloroquine, an autophagy inhibitor, has been clinically approved for use in clinical studies in cancer (NCT05953350, NCT01266057, and NCT04760080). 39 Another trial explored the function of autophagy in systemic lupus erythematosus (NCT03583853). Moreover, a study from Duke University identified a novel signaling mechanism that regulates autophagy and apoptosis in human satellite cells, with the hope of discovering novel pharmaceutical targets in satellite cells (NCT03473288). Additionally, clinical investigations focused on the therapeutic relevance of mitophagy and pexophagy in human diseases (NCT05708547 and NCT03856866). However, the clinical translational applications of these medications remain in their infancy. These challenges include selective drug development and technical difficulties in dynamically assessing autophagy in vivo. The ability to monitor autophagy in real-time is critical for diagnosing and evaluating the effectiveness of autophagy-based therapies, which has been extensively studied. For example, a monomeric tandem mCherry-GFP-LC3 construct has been established in peripheral and central nervous system neurons, enabling dynamic assessment of autophagic activity under disease-related and pharmacological conditions. 293 Elena et al. described several screening systems using autophagy reporters, which are mainly based on autophagy markers, such as LC3, or autophagy substrates, such as aggregation-prone proteins or p62/SQSTM1. The sensitivity of autophagy reporters and assays has improved over time, allowing high-throughput drug discovery applications to measure autophagic flux. 294 Rieko et al. developed a CRISPR/Cas9 system-based mouse genome-wide screening to identify autophagic flux regulators in mice. Using the pHluorin-LC3-mCherry reporter in mice, researchers developed an effective strategy for evaluating genes influencing autophagic flux. 295
Challenges and future perspectives
Autophagy facilitates the migration, proliferation, and survival of cells involved in wound healing. However, excessive autophagy contributes to the formation of hypertrophic scars. Therefore, it is critical to define the precise function of autophagy at each stage of wound healing and develop autophagy-targeting medications that accelerate wound healing. Selective autophagy facilitates organelle turnover and provides an accurate and energy-efficient method for eliminating undesirable components. However, when exposed to the same stimulus, multiple organelles may undergo structural disruptions or functional failures. For example, hypoxia-induced suppression of glucose oxidation disrupts mitochondrial calcium flux, resulting in mitochondrial dysfunction due to refractory ER stress. 296 In addition to mitophagy, mitochondrial self-digestion (MSD), another pathway for mitochondrial degradation, has been proposed. Hypoxia induces extensive intermitochondrial contacts and eventual fusion, which transforms regular tubular mitochondria into megamitochondria. Crucially, hypoxia promotes mitochondria–lysosome interactions and the engulfment of some lysosomes by megamitochondria, termed megamitochondria engulfing lysosome (MMEL). MSD, an MMEL-mediated form of mitochondrial degradation, increases ROS generation in mitochondria 297 (Fig. 9). However, to date, no studies have focused on the role of MSD in wound healing. Further research into the precise molecular processes and clinical implications of organelle quality control and the corresponding selective autophagy or self-digestion is warranted as it may represent a viable therapeutic target for wound healing. Few studies have focused on the role of selective autophagy in endogenous mucosal injury, which may be a promising avenue for future research.
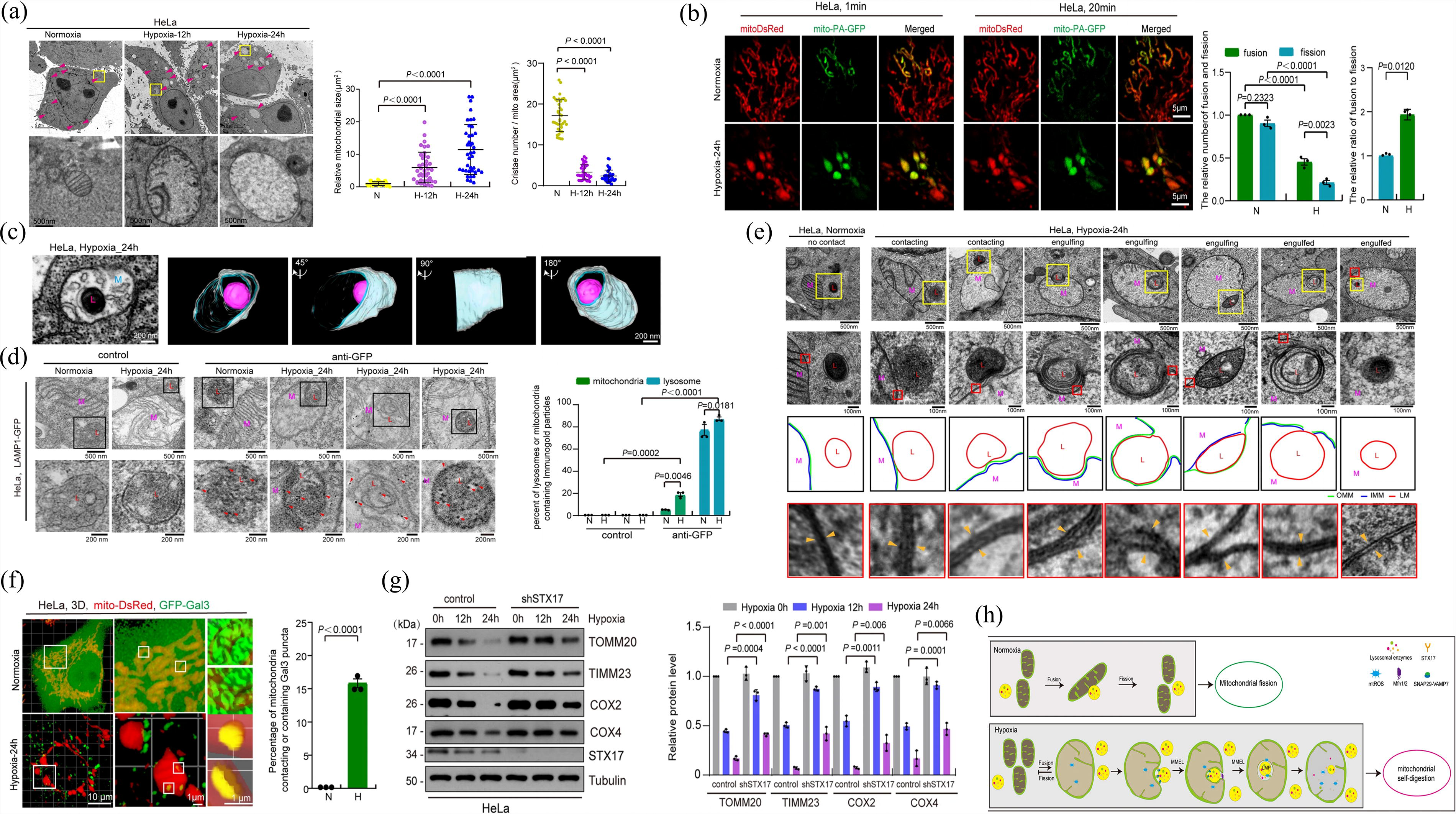
Hypoxia-reprogramed MMEL mediates mitochondrial self-digestion.
Selective autophagy plays a critical role in managing the quality of intracellular elements and regulating biological behaviors, thereby potentially contributing to wound healing. However, the current understanding of selective autophagy in wound healing is primarily based on preclinical animal investigations, with no published clinical data. Clinical assessment of toxicity, safety, specificity, off-target effects, pharmacokinetics, and resistance mechanisms of relevant modulators is lacking. Therefore, determining the mechanisms underlying selective autophagy in humans is imperative. As previously mentioned, dynamic assessment of autophagy in vivo remains challenging. Overcoming this limitation is crucial for establishing a diagnosis and monitoring the efficacy of autophagy-based therapies. Furthermore, crosstalk between various forms of selective autophagy plays a critical role in homeostasis and cellular functions. A thorough understanding of selective autophagy could advance further research into intracellular quality control and foster a general awareness of the intracellular self-protective mechanisms involved in wound healing. Comprehending the fundamental processes underlying selective autophagy and selective autophagy-associated crosstalk may facilitate the development of more comprehensive selective agents and therapeutic strategies. To fully define the clinical efficacy of selective autophagy modulators, new pharmacological methods can be formulated to develop drugs that uniquely manipulate a single form of autophagy without interfering with other organelles. Rational medication design and screening initiatives can provide more selective agents that target the core autophagy machinery. Exploring natural products, derivatives, small-molecule compounds, complexes, combination drugs, and the functional reorientation of drugs may further develop more specific and effective therapies targeting selective autophagy in wound healing. Contemporary assays must be developed to enhance future techniques for identifying and evaluating selective autophagy modulators. Modern assay technologies, including artificial intelligence technology, machine learning, and quantitative structure–activity relationships, can be leveraged to accelerate drug development. 50 With the help of new detection, the process of discovering novel modulators can be based on phenotypic screening, rational drug design on the basis of particular target proteins, or unintentional discovery on the basis of natural products. However, the absence of specific pharmacological reagents and biomarkers to precisely profile the effects of compounds on different types of autophagy and cellular responses has significantly impeded the preclinical and clinical development of selective autophagy modulators. Translational biomarkers should contain predictive biomarkers that stratify and select the appropriate patient cohort and pharmacodynamic biomarkers that demonstrate a given drug’s target engagement. Both are now severely lacking in autophagy-centric drug development. 298 Despite advancements in our understanding of these processes, the limited absorption and bioavailability of existing medications targeting selective autophagy—a very complex process—make it difficult to translate mechanistic research into clinically effective drugs. 298 Nanotechnology-based delivery systems hold considerable potential for improving the low bioavailability of selective autophagy modulators by utilizing the superior drug delivery capabilities of nanocapsules and reducing the off-target effects of some small-molecule modulators. However, the development of nanocarriers and their clinical applications are still far off, since they have limited clinical translation owing to inadequate therapeutic impact and off-target toxicity to vital organs.
TAKE-HOME MESSAGES
As a housekeeping and self-renewal process, selective autophagy leverages lysosomal machinery to degrade and recycle specific cellular components, enabling cells to adapt and survive under stressful conditions.
Selective autophagy, including mitophagy, ER-phagy, pexophagy, xenophagy, lysophagy, ferritinophagy, and lipophagy, can more accurately regulate the biological behavior involved in wound healing.
The therapeutic potential and clinical value of selective autophagy in wound healing provide novel insights into the management of refractory wounds.
Challenges to clinical translation include recognition of the limitations of the role of selective autophagy in wound healing, difficulties in identifying and evaluating selective autophagy modulators, and the low bioavailability of targeted selective autophagy medications.
SUMMARY
Herein, we comprehensively reviewed the fundamental mechanisms of selective autophagy and its functions in burn and wound healing. Various stressors induce physiologically selective autophagy to preserve cellular homeostasis. To date, many studies have focused on the processes behind selective autophagy; however, some forms of autophagy, such as pexophagy, lysophagy, and lipophagy, remain poorly understood. We focused on the role of ferritinophagy, which may be a novel therapeutic target for patients with delayed wound healing. In addition to the mechanisms underlying selective autophagy, we summarize the therapeutic potential, clinical value and challenges in the clinical translation of dedicated therapies for wound healing. Regulation of selective autophagy may be an effective and viable technique for improving wound healing.
AUTHORS’ CONTRIBUTIONS
J.W.X. wrote the article and drew diagrams. G.S.X. provided ideas for the text and reviewed and revised it. R.D.Y., Y.M.R., L.Y., and Z.W. reviewed the literature and approved the article before submission. All authors reviewed the article.
Footnotes
ACKNOWLEDGMENTS AND FUNDING SOURCES
The authors thank Chunchun Li, Yanshan Liu, and Lihong Chen from Center for Basic and Translational Research, Second Affiliated Hospital Zhejiang University School of Medicine for their support and encouragement of this article. The authors would like to thank Editage (![]() ) for English language editing. This work was supported by the following grants: National Natural Science Foundation of China (NSFC) (grant no. 82272259) and National Key Research Project of China (grant no. 2022YFC2403100), Zhejiang Provincial Natural Science Foundation of China (grant no. Q23H150010), and Zhejiang Provincial Medical and Health Science and Technology Project (no. 2023574234).
) for English language editing. This work was supported by the following grants: National Natural Science Foundation of China (NSFC) (grant no. 82272259) and National Key Research Project of China (grant no. 2022YFC2403100), Zhejiang Provincial Natural Science Foundation of China (grant no. Q23H150010), and Zhejiang Provincial Medical and Health Science and Technology Project (no. 2023574234).
AUTHOR CONFIRMATION
The authors confirm that they have the written consent of all contributing authors and that all authors accept complete responsibility for the contents of the article.
AUTHOR DISCLOSURE AND GHOSTWRITING
The authors have no conflicts of interest or ghostwriters to declare.