
Abstract
Objective:
Oxidative stress limits mesenchymal stem cell (MSC) efficacy in tissue repair by reducing retention and survival at injury sites. Endogenous production of trehalose may enhance MSC resilience and promote skin wound healing.
Approach:
Trehalose-6-phosphate synthase 1-expressing MSCs (TPS1-MSCs) were engineered via adenoviral transduction. Trehalose content and synthase activity were assessed. Oxidative stress models (H2O2, 0% fetal bovine serum, CoCl2) were used to evaluate reactive oxygen species (ROS), apoptosis, and cell damage. TPS1-MSCs were transplanted into mouse wounds to track retention rate via in vivo imaging. Histology and immunofluorescence were used to assess wound healing, collagen deposition, and angiogenesis. Conditioned medium (CM) was used to evaluate paracrine functions. RNA-seq identified differentially expressed genes, and mechanisms were validated using the NRF2 inhibitor ML385.
Results:
TPS1-MSCs exhibited TPS1 activity and synthesized trehalose. Under oxidative stress, these cells showed reduced ROS, enhanced viability, and decreased apoptosis. In vivo, TPS1-MSCs displayed higher retention, accelerated healing, and neovascularization. CM from TPS1-MSCs promoted keratinocyte migration, fibroblast collagen secretion, and enhanced the tube-forming capacity of endothelial cells. Transcriptome analysis revealed enrichment in the NRF2-HMOX1 pathway. TPS1-MSCs showed elevated levels of p62, nuclear NRF2, and HMOX1. ML385 treatment impaired the observed antioxidant capacity.
Innovation:
Engineering MSCs for endogenous trehalose synthesis enhanced oxidative stress resistance and retention through NRF2-HMOX1 activation, suggesting a potential novel MSC-based wound repair strategy.
Conclusion:
TPS1-MSCs improved antioxidant capacity and wound healing, potentially through the NRF2-HMOX1 pathway, and may represent a promising therapy for skin wounds.
Guangchao Xu, PhD Rongqing Pang, PhD

INTRODUCTION
Disruption of the skin barrier, caused by severe mechanical injuries, burns, vascular insufficiency, and chronic wounds, is highly prevalent and remains a major clinical challenge. Preclinical and clinical studies have demonstrated the therapeutic potential of mesenchymal stem cells (MSCs) transplantation for wound repair. 1,2 However, limited survival, reduced viability, and poor stress tolerance during healing markedly restrict the reparative efficacy of MSCs. 3 Therefore, improving their survival and resistance to apoptosis is crucial for enhancing tissue repair.
MSCs promote wound healing by secreting factors that enhance angiogenesis, proliferation, and inflammation modulation locally. 1,4 These paracrine effects depend on the persistence of viable MSCs at the injury site. In practice, transplanted MSCs rarely survive long enough to sustain complete repair. After intravenous infusion, most MSCs are retained in the lungs, liver, or spleen, with fewer than 1% reaching the wound. 5 Even MSCs that enter full-thickness skin defects disappear within 5 days. 6 Subcutaneous injection around wounds retains MSCs for about 7 days, whereas drip or smear delivery leads to disappearance within 48 h. 7,8 Thus, insufficient persistence is a major barrier to MSC-based wound therapy.
Oxidative stress is considered a key factor in the rapid loss of transplanted MSCs in wounds. Ischemia and hypoxia in injured tissues cause excessive reactive oxygen species (ROS) production and severe oxidative damage. 9 Compared with resident skin cells such as fibroblasts, MSCs are more susceptible to oxidative stress, and even low oxidative stress can impair their proliferation and survival in vitro. 10,11 Greater oxidative stress further induces dose-dependent apoptosis. 12 Importantly, exogenous interventions can attenuate oxidative stress-induced apoptosis, suggesting that strengthening antioxidant defense may improve MSC survival and function during repair. 10
Trehalose, a natural nonreducing disaccharide, has received increasing attention for its cytoprotective effects under stress conditions. 13 Previous studies showed that trehalose alleviates oxidative damage by reducing intracellular ROS, preserving mitochondrial function, and relieving endoplasmic reticulum stress. 14,15 It also improves viability and reduces apoptosis in several oxidative injury models, including hydrogen peroxide-injured keratinocytes, myofibroblasts, and bone marrow-derived MSCs. 16 –18 Its antioxidant effects are closely associated with activation of the p62(SQSTM1)-KEAP1-NRF2 pathway, in which increased p62 disrupts KEAP1-mediated repression of NRF2 and promotes NRF2 nuclear translocation, leading to antioxidant gene expression. 19 –21
Despite these protective properties, the application of trehalose to enhance MSC survival in vivo remains limited. Trehalose exhibits poor membrane permeability, and only a small fraction enters cells through SLC2A8. 22 In addition, mammals cannot synthesize trehalose endogenously. Conventional in vitro pretreatment occurs under relatively low oxidative stress and cannot mimic the sustained stress after transplantation into ischemic wounds. Once transplanted, pretreated MSCs continue to face prolonged oxidative stress but cannot replenish intracellular trehalose. These limitations raise concerns regarding whether transient supplementation can provide durable protection in vivo.
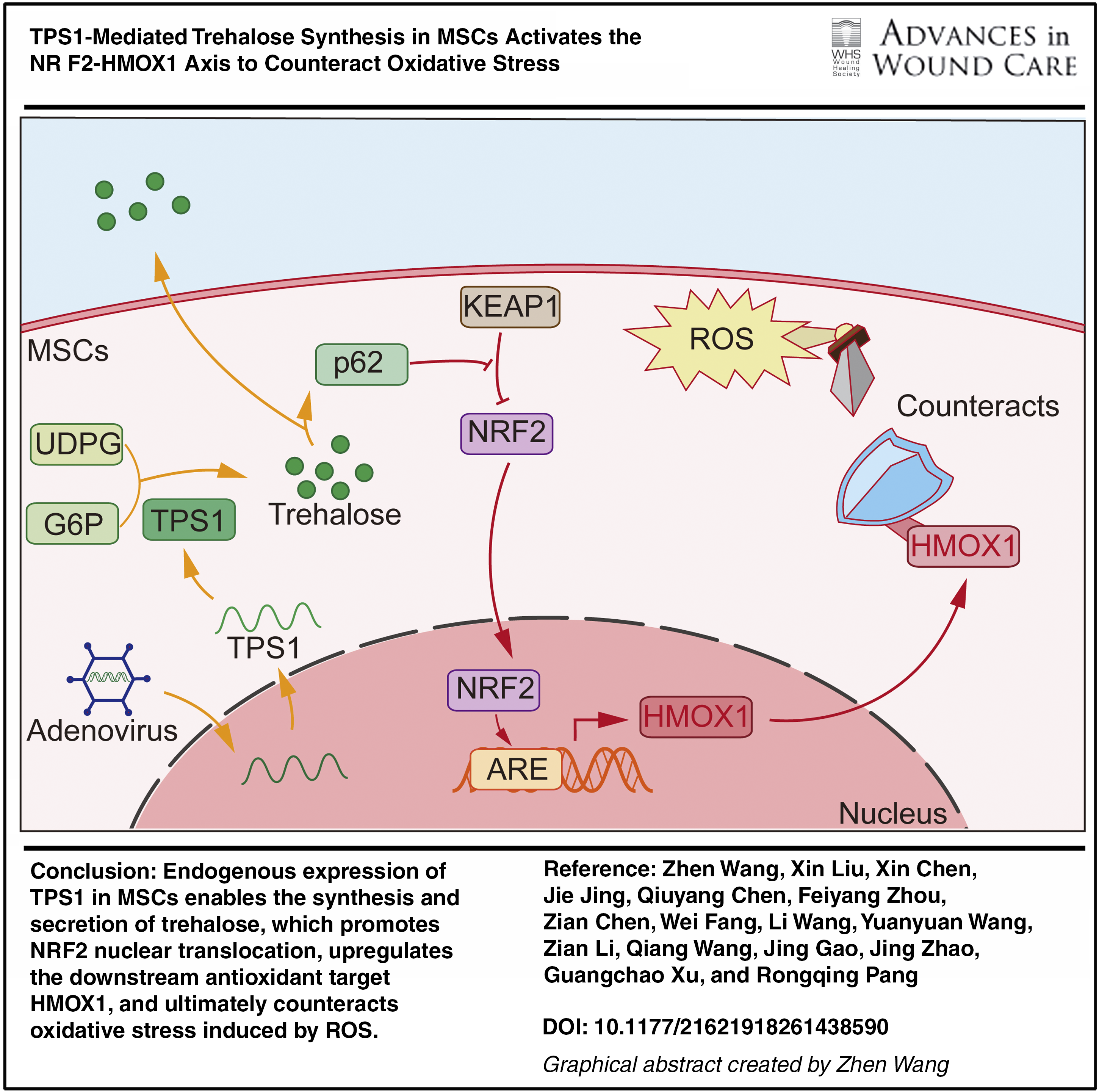
In this study, MSCs were engineered to express trehalose-6-phosphate synthase 1 (TPS1), a nonhuman enzyme, and their ability to synthesize trehalose was assessed. Using in vitro oxidative stress models and in vivo wound models, the antioxidative capacity of TPS1-MSCs, their persistence and healing efficacy in wounds, and the underlying mechanisms were investigated (Graphical abstract).
CLINICAL PROBLEM ADDRESSED
A rising number of patients needing tissue repair, especially those with wound healing requirements, places heavy burdens on society and the medical system. Current MSC-based therapies, however, are hindered by poor resistance to oxidative stress and low retention at the transplantation site, which limits their therapeutic efficacy. This project aims to engineer MSCs to express TPS1, enabling endogenous trehalose synthesis to enhance their stress resistance and retention, thereby improving tissue repair outcomes.
MATERIALS AND METHODS
Cell culture
Human umbilical cord MSCs were isolated as described previously. 23 The study was approved by the Medical Ethics Committee of the Affiliated Hospital of Zunyi Medical University (Approval Number: KLL-2024-727). The culture medium was the serum-free medium for Umbilical Cord MSCs (Yocon, China). Cells were cultured at 37°C and 5% CO2. Passaging was performed when the cell confluence reached approximately 80%.
Human fibroblasts (GNHu49) and human umbilical vein endothelial cells (GNHu39) were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China), while human keratinocytes (CL-0090) were obtained from Wuhan Procell Life Science & Technology (Wuhan, China).
Adenovirus production and cell transfection
The full-length coding sequence of Drosophila melanogaster TPS1 (Gene ID: 33642, RefSeq: NM_134983.3) was cloned into the pAdEasy-EF1-MCS-3× Flag-CMV-EGFP adenoviral shuttle vector. The TPS1 coding sequence was inserted downstream of the EF1α promoter and fused in-frame with a C-terminal 3× Flag tag to facilitate detection, while EGFP driven by the CMV promoter served as a reporter for infection efficiency. The empty vector expressing EGFP alone (HBAD-EGFP) served as the negative control. Recombinant adenoviral plasmids were generated according to the AdEasy system and transfected into HEK293 cells using Lipofiter™ transfection reagent. Viral supernatants were collected, amplified, and purified. Viral titers were determined using the TCID50 assay and converted to plaque-forming units (PFU). The titer of the HBAD-EGFP control virus was 2.51 × 1010 PFU/mL, and the titer of the HBAD-Adeasy-o-TPS1-3× Flag-EGFP virus was 1.26 × 1010 PFU/mL. MSCs were infected with recombinant adenovirus at multiplicities of infection (MOIs) of 0, 10, 30, 100, 300, and 500. Green fluorescent protein (GFP) expression was assessed by fluorescence microscopy, and cell morphology was monitored to evaluate cytotoxicity. Based on infection efficiency and cell viability, an MOI of 100 was selected for subsequent experiments. Flow cytometry was used to further confirm infection efficiency. MSCs infected with TPS1-overexpressing adenovirus were designated as TPS1-MSCs, while those infected with the empty vector served as negative controls (NC-MSCs).
MSCs triple lineage induction capacity
Osteogenic, chondrogenic, and adipogenic differentiation media were prepared following the manufacturer’s instructions provided with the Umbilical Cord MSC Directed Induced Differentiation Kit (OriCell, China). NC-MSCs and TPS1-MSCs were induced according to the instructions. Alizarin Red S, Alcian Blue, and Oil Red O were used for staining and observation at the end of induction, respectively.
Flow characterization of MSCs
Cells were harvested for flow cytometric identification 72 h postinfection for phenotypic characterization. NC-MSCs and TPS1-MSCs were resuspended in 100 μL of phosphate buffered saline (PBS) and incubated with 5 μL of each of the following surface marker antibodies for 30 min in the dark: CD29-PE, CD105-APC, CD34-PE, and CD45-PerCP (Thermo Fisher Scientific, USA), and CD73-PE, CD90-CL647, and HLA-DR-CL647 (Proteintech, China). Following three washes with PBS, cells were resuspended in 500 μL of PBS for flow cytometric analysis.
Measurement of trehalose levels
Intracellular and extracellular trehalose levels were measured using a Trehalose Content Assay Kit (Boxbio, China) according to the manufacturer’s instructions. For intracellular trehalose measurement, harvested cells were resuspended in extraction buffer at a density of 1 × 104 cells/mL and lysed by sonication on ice. The lysates were incubated at room temperature for 45 min, followed by centrifugation to remove cellular debris. The resulting supernatants were collected as cell samples. For extracellular trehalose measurement, 100 μL of culture supernatant was mixed with 900 μL of extraction buffer (1:10 dilution) and incubated at room temperature for 45 min. All samples were then centrifuged, and the supernatants were collected as the final samples for analysis. After the addition of the chromogenic reagent, the absorbance was measured at 620 nm using a microplate reader (Epoch, BioTek, USA). Trehalose concentrations were determined based on a standard curve and normalized to the cell number (104 cells) for intracellular samples or corrected for the dilution factor for culture supernatants.
Measurement of TPS1 enzymatic activity
TPS1 enzymatic activity was measured using a commercial TPS1 Activity Assay Kit (Boxbio, China) according to the manufacturer’s instructions. Briefly, harvested cells were resuspended in extraction buffer at a density of 5 × 106 cells/mL. Cell lysis was performed via sonication on ice, followed by centrifugation to obtain the supernatant. The supernatant was incubated with the reaction reagent at 30°C for 20 min. The reaction was subsequently terminated by heating at 95°C for 5 min. After cooling to room temperature, the sample was centrifuged again, and the supernatant was retained as the final detection sample. Absorbance was monitored at 340 nm at 5 min and 10 min using a microplate reader (Epoch, BioTek, USA). The change in absorbance (ΔA) was calculated, and TPS1 activity was determined according to the manufacturer’s formula and normalized to cell number (U/104 cells).
Cell Counting Kit–8 (CCK8)
Cell proliferation capacity was assessed using Cell Counting Kit-8 (UElandy, China). NC-MSCs and TPS1-MSCs were seeded into 96-well plates and incubated overnight at 37°C in 5% CO2. For proliferation assays, 10 μL of CCK8 reagent was added to each well at 12 and 24 h postseeding. The plates were then incubated at 37°C for 2 h in the dark. For inhibition assays involving ML385, TPS1-MSCs were treated with the inhibitor for 24 h prior to the addition of the CCK8 reagent. Finally, the absorbance at 450 nm was detected using a microplate reader (Epoch, BioTek, USA). Blank wells containing culture medium and CCK8 reagent without cells served as background controls. For oxidative stress experiments, cell viability was normalized to that of the corresponding untreated control group (set as 100%). The relative cell viability was calculated as the ratio of the optical density (OD) of the experimental group to that of the control group after background subtraction.
Preparation of conditioned medium
Upon completion of the adenoviral transduction of NC-MSCs and TPS1-MSCs, the complete culture medium was discarded. The cells were washed three times with PBS to eliminate residual serum and subsequently cultured in serum-free medium for 24 h. The cell culture supernatant was collected and centrifuged at 800 × g for 10 min at room temperature to remove cellular debris. The supernatant was then sterilized using a 0.22 µm filter, aliquoted, and stored at −80°C until use.
Wound healing assay
When human keratinocytes reached 90–100% confluence, a linear scratch was created across the cell monolayer using a sterile 200-μL pipette tip. Detached cells were removed by washing with PBS, and the cells were subsequently incubated with the conditioned medium (CM) prepared as described above. Images were acquired at 0, 12, and 24 h postscratching using an inverted phase-contrast microscope. The wound closure area was measured using ImageJ software to quantify cell migration.
Tube formation assay
Matrigel (Solarbio, China) was thawed slowly at 4°C. Using precooled pipette tips, 20 μL of Matrigel was evenly coated onto the bottom of precooled 96-well plates. The plates were then incubated at 37°C for 30 min to allow the matrix to polymerize. Human umbilical vein endothelial cells were resuspended in the CM at a density of 1.5 × 105 cells/mL, and 100 μL of the cell suspension was seeded onto the solidified Matrigel in each well. After 12 h of incubation, tube formation was visualized using an inverted microscope. The number of branches and total tube length were quantified using ImageJ software.
RNA-seq
Total RNA was extracted from NC-MSCs and TPS1-MSCs treated with or without 200 μM H2O2 (denoted as H-NC-MSCs and H-TPS1-MSCs, respectively) using the method described above. Sequencing library was generated using the NEBNext® Ultra™ RNA Library Prep Kit for Illumina® (NEB, USA). The libraries were subsequently sequenced on an Illumina NovaSeq PE150 platform to generate 150 bp paired-end reads. Quantification was performed using FPKM values. P < 0.05 and |log2FC|≥ 1 were recognized as significantly differentially expressed genes (DEGs). Volcano plots and heatmaps were generated to demonstrate differential genes. Venn diagrams were employed to show the cross-relationships of different sets of genes. Gene ontology (GO) analysis was performed to annotate DEGs across molecular function, biological process, and cellular component. In addition, Gene Set Enrichment Analysis (GSEA) enrichment was employed to explore the contribution of the differential genes to the phenotype. The raw RNA-Seq data generated in this study have been deposited in the Genome Sequence Archive under accession number CRA028720.
Oxidative stress modeling
When cells reached 70–80% confluence, H2O2, serum deprivation, or CoCl2 were added for oxidative stress injury, respectively. For H2O2 treatment, cells were exposed to 0, 100, 200, 500, or 1,000 μM H2O2 for 3 h. For serum starvation assays, cells were cultured in medium containing 0%, 2%, 5%, or 10% fetal bovine serum (FBS) for 24 h. For chemical hypoxia induction, cells were treated with 0, 100, and 500 μM CoCl2 for 24 h. CCK8 reagent was added at the endpoint of the intervention. The absorbance at OD 450 nm was detected.
Cellular immunofluorescence
NC-MSCs and TPS1-MSCs were seeded on glass coverslips and subjected to stress conditions (200 μM H2O2, 0% FBS, and 500 μM CoCl2). Cells were fixed with ice-cold methanol for 1 h at 4°C. Subsequently, cells were permeabilized with 0.1% Tween-20 and blocked with 5% bovine serum albumin (BSA). Samples were incubated with primary antibody overnight at 4°C, followed by incubation with a secondary antibody (1:500, HA1122, Huabio, China) for 1 h at room temperature. After washing with PBS, slides were mounted using an antifade mounting medium containing DAPI. The primary antibodies used were anti-FLAG (1:800, 8146 T, CST, USA) and anti-NRF2 (1:100, 16396-1-AP, Proteintech, China).
In vitro detection of cellular ROS, apoptosis
NC-MSCs and TPS1-MSCs were seeded in 6-well plates (5 × 105 cells/well) overnight and then exposed to 500 μM H2O2 (3 h), 0% FBS (24 h), or 500 μM CoCl2 (24 h), respectively. For ML385 experiments, TPS1-MSCs with or without ML385 treatment were processed in parallel. Subsequently, cells were treated according to the instructions provided by the Reactive Oxygen Detection Kit (Boxbio, China) and the Annexin V-Alexa Fluor647/7-AAD Apoptosis Detection Kit (4A Biotech, China). Cells were analyzed by flow cytometry on a CytoFLEX instrument (Beckman Coulter, USA) to quantify intracellular ROS and apoptosis. For ROS detection, harvested cells were resuspended in diluted DHE (5 μmol/L) at 1 × 106 cells/mL, incubated at 37°C for 20 min in the dark, washed three times with serum-free medium, and fluorescence (Ex/Em ≈ 535/610 nm) was recorded in the B585 channel. For apoptosis analysis, cells were resuspended in 1 × binding buffer (1 × 106 cells/mL), and 100 μL cell suspension was stained with 5 μL rh Annexin V-Alexa Fluor 647 for 5 min at room temperature in the dark, followed by addition of 10 μL 7AAD and 400 μL PBS immediately prior to acquisition. Alexa Fluor 647 (Ex/Em ≈ 650/668 nm) and 7AAD (Ex/Em ≈ 546/647 nm) were detected in the R660 and B690 channels, respectively. Instrument settings and gating strategies are detailed in Supplementary Tables S1 and S2. Data acquisition and analysis were performed using CytExpert software (Beckman Coulter, USA).
Mouse wound healing model and live imaging
Male C57BL/6 mice (8 weeks old, 21–24 g) were purchased from Beijing Spefo Laboratory Animal Co. Ltd (China). All mice were housed in a specific pathogen-free facility with controlled temperature and humidity under a 12 h light/12 h dark cycle. Specifically, mice were anesthetized with pentobarbital and then depilated on the back using depilatory cream (Veet, China). Next, a full-thickness skin excision wound (1 cm in diameter) was created on the dorsal skin using a biopsy punch. To evaluate the therapeutic efficacy, experimental groups received subcutaneous injections of NC-MSCs, TPS1-MSCs, or ML385-pretreated TPS1-MSCs (1 × 106 cells/mouse, 200 μL) around the wound. Wounds were left open without dressing. A total of 10 mice were randomly assigned to each group. Optical images of the wounds were captured on days 0, 3, 5, 7, and 10 postwounding. Wound closure rates were quantified using ImageJ software (NIH, USA). To track cell retention, in vivo fluorescence imaging was performed on days 0, 3, and 7. On day 7, five mice from each group were randomly selected and sacrificed for histological examination. The remaining mice were monitored macroscopically until day 10 and subsequently sacrificed on day 14 for histological analysis of complete wound healing. At the end of the experiment, all mice were euthanized by cervical dislocation following induction of deep anesthesia with isoflurane (RWD, China). All animal procedures complied with the ARRIVE guidelines and were approved by the Medical Ethics Committee of Zunyi Medical University (Approval Number: zyfy-an-2024-0712).
Histology/immunofluorescence
On days 7 and 14 postinjury, the entire wounded tissue was harvested. Samples were fixed in 10% formalin for 48 h, followed by dehydration through a graded ethanol series (Supplementary Table S3), and embedding in paraffin (HistoCore Arcadia H + C, Leica, Germany). Tissues were then sectioned at a thickness of 4 μm (HistoCore AUTOCUT, Leica, Germany). Sections were deparaffinized, rehydrated (Supplementary Table S4), and stained with hematoxylin and eosin (H&E; Baso, China) to evaluate the epithelialization process. Collagen deposition was assessed using a Modified Masson’s Trichrome Staining Kit (Solarbio, China) following the manufacturer’s protocol. For tissue immunofluorescence staining, sections were processed for two separate double-staining combinations: (1) CD31 with α-SMA, and (2) Ki67 with Cytokeratin 14 (K14). Sections underwent antigen retrieval using citrate buffer, followed by inactivation of endogenous peroxidase with 3% H2O2 for 15 min. Sections were processed without additional permeabilization and were directly blocked with 5% BSA for 1 h. Sections were incubated with the first primary antibody, either anti-CD31 (1:4,000, 28083-1-AP, Proteintech, China) or anti-Ki67 (1:500, D3B5, CST, USA), overnight at 4°C. Detection was performed using a secondary antibody (HRP-conjugated Goat anti-Rabbit IgG, 1:100, AS014, ABclonal, China) at room temperature for 45 min, followed by signal amplification with AF546 dye (1:500, K-R-3650, Kaixin, China) for 15 min. For dual staining, a second round of antigen retrieval was performed to strip the antibody complex. Sections were then incubated with the corresponding second primary antibody, anti-α-SMA (0.5 μg/mL, NBP2-33006, Novus, China) or anti-K14 (1:2,000, 60320-1-Ig, Proteintech, China) overnight at 4°C, followed by detection with the same secondary antibody and CY5 dye (1:500, K-R-3307, Kaixin, China). Finally, slides were mounted using an antifade mounting medium containing DAPI (Solarbio, China). For better visual distinction in merged images, AF546 signals were assigned a green pseudo-color and CY5 signals were assigned a red pseudo-color during image acquisition and processing. 0.01M PBS washes were performed three times for 3 min each between all steps.
Quantitative real-time PCR
Total RNA was extracted from cells using a Rapid Cell/Tissue Total RNA Isolation Kit (Vazyme, China) following the manufacturer’s protocol. RNA concentration was then measured using a NanoDrop spectrophotometer (Thermo Fisher, USA). Reverse transcription was performed using HiScript III RT Super Hybrid qPCR (+gDNA wiper) (Vazyme, China). Quantitative real-time PCR (qPCR) assays were performed on a QuantStudio Real-Time PCR System (Thermo Fisher, USA) using ChamQ Universal SYBR qPCR Master Mix (Vazyme, China) and specific primers (BioWorks, China). Relative gene expression levels were calculated using the 2−ΔΔCT method. Primer sequences are listed in Supplementary Table S5.
Western blot
Cell samples were collected and lysed using RIPA lysis buffer supplemented with phenylmethylsulfonyl fluoride (Solarbio, China). Protein concentration was quantified using a BCA Protein Assay Kit (Solarbio, China). Samples were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and electrotransferred to a polyvinylidene difluoride membrane. Subsequently, they were blocked with 5% BSA for 1 h at room temperature. Membranes were incubated with the first primary antibody overnight at 4°C, followed by incubation with a secondary antibody (HRP-conjugated Goat anti-Rabbit IgG, 1:5,000, AS014, ABclonal, China) for 1 h at room temperature. After thorough washing, immunoreactive bands were visualized using an enhanced chemiluminescence (Dalian Meilun Biological, China). The following antibodies were used in this study: β-actin (1:4,000, GB11001, Servicebio, China), heme oxygenase 1 (HMOX1) (1:2,000, 10701-1-AP, Proteintech, China), NRF2 (1:5,000, 16396-1-AP, Proteintech, China), p62 (1:15,000, ab109012, Abcam, USA), and LC3b (1:5,000, 81004-1-RR, Proteintech, China).
Statistical analysis
Statistical analysis was conducted utilizing the SPSS software package. Data normality was assessed using the Shapiro–Wilk test prior to statistical comparisons. For comparisons between two groups, unpaired Student’s t-tests were performed, whereas multigroup comparisons were assessed using one-way analysis of variance, followed by Tukey’s post hoc analysis for detailed differentiation. Data were based on findings from at least three independent experiments. The results were presented as mean ± standard deviation. A p value less than 0.05 was considered indicative of statistical significance. All image quantifications were performed under blinded conditions using identical acquisition and analysis parameters across groups. Multiple randomly selected fields per sample were analyzed and averaged to minimize bias. An electronic laboratory notebook was not used.
RESULTS
Identification and characterization of TPS1-MSCs
MSCs were transduced with nonspecific control adenoviral vectors (NC-MSCs) or TPS1 adenovirus vectors (TPS1-MSCs). GFP fluorescence identified that MOI of 100 for 72 h was optimal, with high transduction efficiency and preserved morphology (Supplementary Fig. S1A and S1B). Flow cytometry showed 93.63% transduction in NC-MSCs and 87.46% in TPS1-MSCs (Fig. 1A and B). After osteogenic, adipogenic, and chondrogenic induction, both groups formed calcium deposits, lipid droplets, and glycosaminoglycans, indicating multilineage differentiation potential (Fig. 1C). Cells retained high CD29, CD105, CD73, and CD90 expression, while lacking CD34, CD45, and HLA-DR, supporting MSC identity after transduction (Fig. 1D–G, and Supplementary Fig. S1C–S1E).
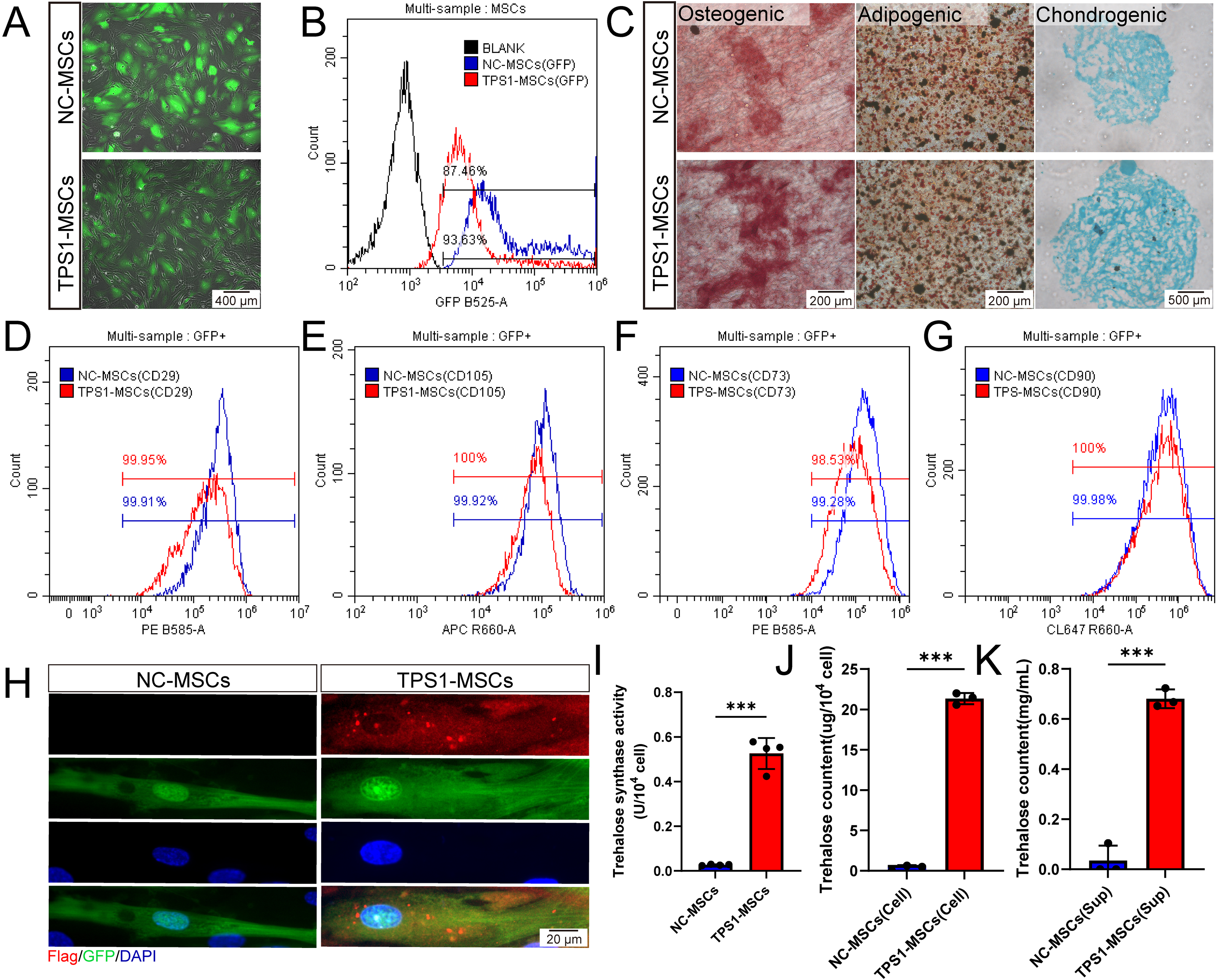
Identification and characterization of TPS1-MSCs.
To evaluate endogenous trehalose synthesis and secretion in TPS1-MSCs, TPS1 localization, activity, and trehalose distribution were examined. Immunofluorescence showed that Flag-tagged TPS1 was mainly localized in the cytoplasm (Fig. 1H). Enzymatic assays verified significant TPS1 activity, indicating functional cytoplasmic TPS1 protein (Fig. 1I). TPS1-MSCs contained 21.34 ± 0.58 μg/104 cells intracellular trehalose, while supernatant trehalose reached 0.68 ± 0.03 mg/mL (Fig. 1J and K). These data support both synthesis and secretion capacities, allowing trehalose to accumulate both intracellularly and in the extracellular microenvironment.
TPS1-MSCs exhibit enhanced antioxidant capacity under oxidative stress
After transplantation, MSCs encounter inflammatory, hypoxic, and nutrient-deprived wound conditions that induce oxidative stress. 9,24 To model this, cells were treated with H2O2 (0, 100, 200, 500, or 1,000 μM), FBS (0%, 2%, 5%, or 10%), or CoCl2 (0, 100, or 500 μM). Compared with lower doses, 200 μM H2O2 induced significant stress while preserving partial viability (Supplementary Fig. S1F). Complete serum deprivation markedly reduced viability (Supplementary Fig. S1G). Similarly, 500 μM CoCl2 caused significant cytotoxicity (Supplementary Fig. S1H). Consequently, 200 μM H2O2, 0% FBS, and 500 μM CoCl2 were selected.
Intracellular ROS levels in NC-MSCs and TPS1-MSCs were measured by DHE. Under basal conditions, ROS levels were comparable (Fig. 2A). Under 200 μM H2O2, 0% FBS, or 500 μM CoCl2, TPS1-MSCs showed significantly lower ROS accumulation than NC-MSCs (Fig. 2B–D). CCK8 assay showed significantly higher viability in TPS1-MSCs after these treatments, suggesting better functional preservation (Fig. 2E). Flow cytometry showed fewer late apoptotic cells in TPS1-MSCs under stress conditions (Fig. 2F and G). These findings demonstrate that TPS1 overexpression enhances MSC resistance to oxidative stress and apoptosis.
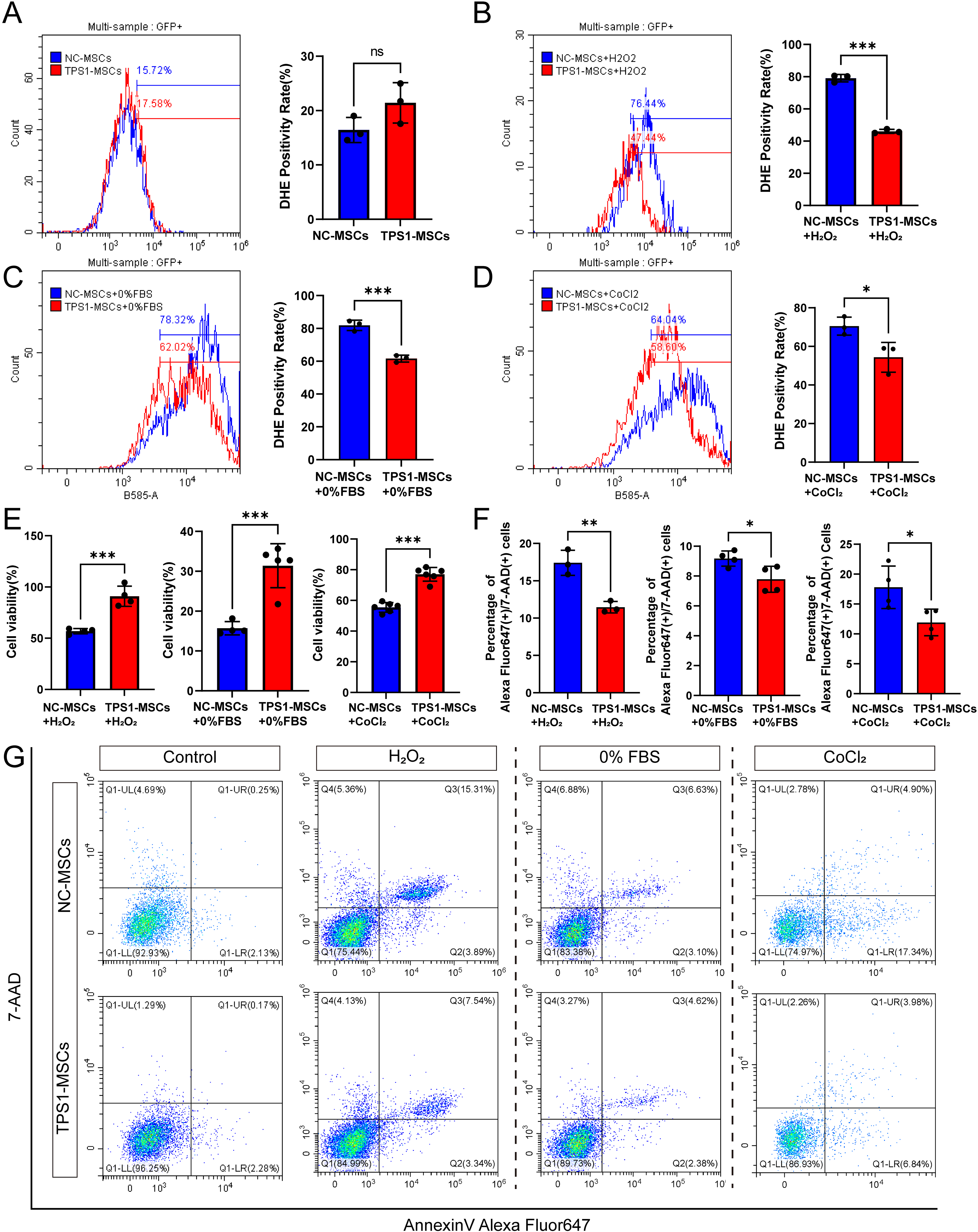
TPS1 reduces ROS accumulation and protects MSCs against multiple stress injuries.
TPS1-MSCs improve cell retention and accelerate wound closure
Based on the antioxidative capacity of TPS1-MSCs in vitro, survival in murine full-thickness excisional wounds was evaluated. In vivo fluorescence imaging on days 3 and 7 revealed that MSCs injected at wound margins migrated toward the wound center, with signal intensity decreasing over time. TPS1-MSCs nevertheless retained significantly higher fluorescence intensity than NC-MSCs on day 7, indicating greater local persistence (Fig. 3A–C). On day 7, more TPS1-MSCs were detected in wound tissue, mainly in the dermal layer (Supplementary Fig. S2A and S2B). These findings indicate improved posttransplantation survival in the wound microenvironment, possibly related to endogenous trehalose-mediated resistance to wound-associated stress.
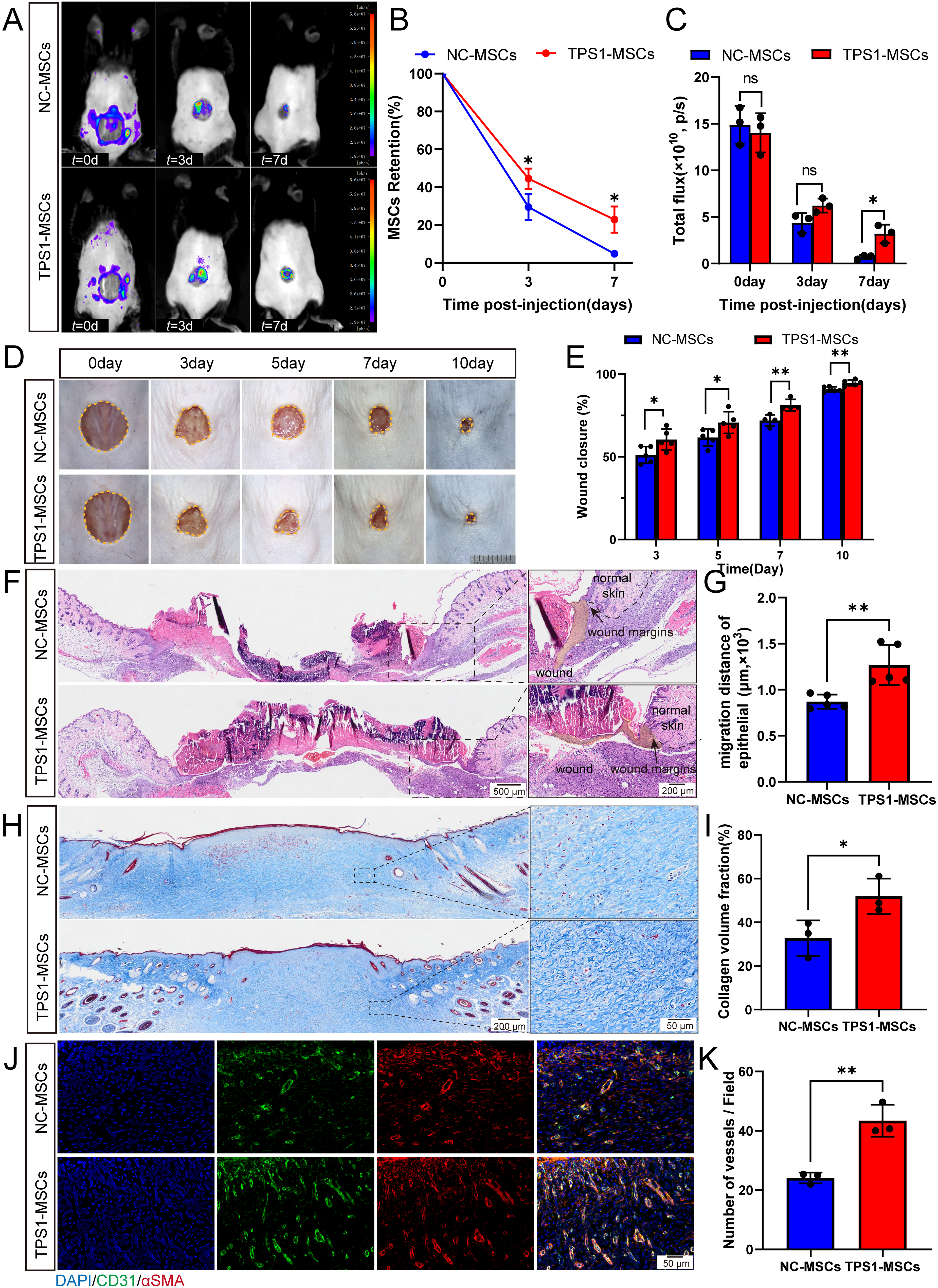
TPS1 overexpression enhances MSCs retention and accelerates wound healing.
After evaluating TPS1-MSC retention, wound healing efficacy was examined in vivo. Ten mice were initially included in each group. Wound closure was quantified from serial gross images of five mice per group monitored through day 10. TPS1-MSC treatment significantly accelerated wound closure from day 3 onward, with higher closure rates maintained through day 10 compared to the control group (Fig. 3D and E). By day 14, H&E staining demonstrated a markedly shorter intermargin distance in TPS1-MSC-treated mice (Supplementary Fig. S2C and S2D). This reduced intermargin distance indicates accelerated healing, although wound closure in this rodent model reflects both reepithelialization and wound contraction. These data demonstrate that improved antioxidative protection enhances MSC persistence and promotes wound closure.
TPS1-MSCs enhance wound reepithelialization, collagen deposition, and neovascularization
Beyond accelerated closure in TPS1-MSCs-treated wounds, repair quality was evaluated at early and late healing stages. At day 7, histology showed significantly enhanced reepithelialization with greater epithelial tongue migration at wound margins (Fig. 3F and G). Immunofluorescence showed higher density of proliferating keratinocytes (Ki67+K14+) at days 7 and 14, suggesting enhanced epidermal regeneration (Supplementary Fig. S3A–S3D). In the dermis, Masson’s trichrome staining showed increased collagen deposition and improved matrix organization at days 7 and 14, indicating more robust extracellular matrix (ECM) remodeling (Fig. 3H and I, Supplementary Fig. S3E and S3F). Immunofluorescence further showed more CD31+ and α-SMA+ vascular structures within the analyzed fields in TPS1-MSCs-treated wounds, indicating improved angiogenic remodeling (Fig. 3J and K, and Supplementary Fig. S3G and S3H).
To confirm the direct effects of TPS1-MSCs on skin constituent cells, in vitro assays were conducted. CM from TPS1-MSCs accelerated keratinocyte migration, upregulated COL1A1 and TGF-β mRNA expression in fibroblasts, and enhanced endothelial tube formation (Supplementary Fig. S4A–S4G). These findings support the in vivo results and indicate that TPS1-MSCs promote coordinated reepithelialization, dermal matrix remodeling, and angiogenesis, thereby promoting high-quality skin regeneration.
TPS1-MSCs activate NRF2-HMOX1 signaling pathway in response to oxidative stress
To investigate the mechanisms underlying the resistance of TPS1-MSCs to oxidative stress, oxidative injury was induced with 200 μM H2O2, followed by transcriptomic profiling of NC-MSCs, TPS1-MSCs, NC-MSCs+H2O2, and TPS1-MSCs+H2O2. NC-MSCs+H2O2 versus untreated NC-MSCs showed 2,055 DEGs, including 1,113 upregulated and 942 downregulated genes, enriched in oxidative phosphorylation, unfolded protein response, and oxidative stress response (Supplementary Fig. S5A and S5B). TPS1-MSCs+H2O2 versus untreated TPS1-MSCs showed 2,552 DEGs, including 2,104 upregulated and 448 downregulated genes, with similar stress-related enrichment (Supplementary Fig. S5C and S5D). These data confirm oxidative injury in both NC-MSCs+H2O2 and TPS1-MSCs+H2O2.
To identify drivers of the enhanced resilience of TPS1-MSCs, TPS1-MSCs+H2O2, and NC-MSCs+H2O2 were directly compared, identifying 2,014 DEGs, including 1,283 upregulated and 731 downregulated genes (Fig. 4A). Volcano plot analysis identified HMOX1 as one of the most significantly upregulated genes. GSEA further showed that stress-associated pathways were expressed at lower levels in TPS1-MSCs+H2O2 (Fig. 4B). Analysis of NABA_SECRETED_FACTORS showed that TPS1-MSCs retained expression of a broad panel of secreted factors under oxidative stress, supporting preserved paracrine competence despite injury (Supplementary Fig. S5E and S5F). GO analyses showed enrichment in wound healing, DNA damage repair, response to hypoxia, and heme metabolic processes, with HMOX1 recurring across categories (Supplementary Fig. S5G). Complementary KEGG analysis indicated coordinated enrichment of inflammatory and metabolic pathways (Supplementary Fig. S5H). This functional landscape, particularly the prominence of HMOX1, suggested a master upstream transcriptional regulator of antioxidant and survival programs.
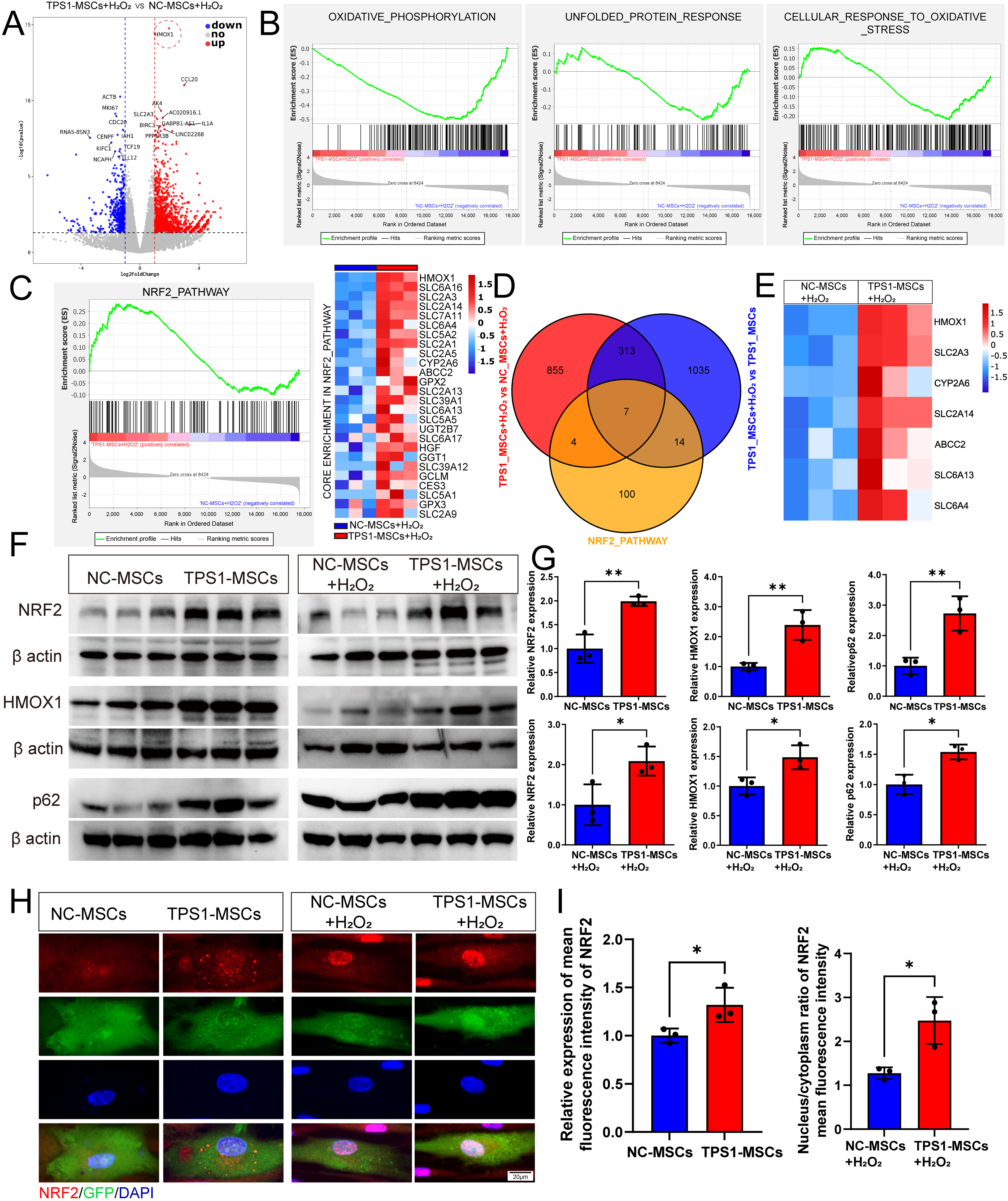
TPS1 overexpression enriches NRF2 signaling genes under oxidative stress injury.
Given the prominent differential expression of HMOX1 and its central position in enrichment analyses, NRF2 was prioritized for targeted evaluation as its canonical upstream transcriptional factor. NRF2 is a master regulator of oxidative stress responses, a direct transcriptional activator of HMOX1, and has been implicated in TNF signaling, NF kappa B signaling, and apoptosis-related processes under oxidative stress. GSEA showed NRF2 pathway enrichment in TPS1-MSCs+H2O2, with the corresponding core enrichment genes, representing the principal contributors to the enrichment score, visualized in the heatmap (Fig. 4C). To assess specificity, FOXO, SIRT1, and TFEB signaling were also analyzed. None showed significant enrichment or coordinated target-gene regulation under H2O2-induced stress, suggesting limited involvement (Supplementary Fig. S5I–S5M). Intersection analysis between the NRF2 pathway and DEGs identified seven candidate NRF2 target genes, HMOX1, SLC2A3, CYP2A6, SLC2A14, ABCC2, SLC6A13, and SLC6A4 (Fig. 4D and E). qPCR analysis under the other two oxidative stress conditions showed that only HMOX1 remained consistently more highly expressed in TPS1-MSCs (Supplementary Fig. S6A and S6B).
To corroborate the transcriptomic findings at the translational level, Western blot showed that NRF2 and HMOX1 protein levels were significantly elevated in TPS1-MSCs+H2O2 (Fig. 4F and G). Notably, this upregulation was already evident in basal TPS1-MSCs, suggesting a primed antioxidant state. To elucidate the upstream mechanism linking endogenous trehalose synthesis to NRF2-HMOX1 activation, p62 was examined. As an autophagy receptor and regulator of NRF2 through competition with KEAP1, p62 promotes NRF2 stabilization and nuclear translocation. Western blot further showed concurrent upregulation of p62 and an increased LC3-II to LC3-I ratio in TPS1-MSCs (Fig. 4F and G, and Supplementary Fig. S6C and S6D). Although increased LC3-II to LC3-I ratio suggests autophagosome formation, concurrent p62 accumulation may reflect transcriptional or autophagic modulation, consistent with reports that trehalose upregulates p62 to sequester KEAP1 and activate NRF2. 19,25
To confirm pathway activation, immunofluorescence showed significantly greater NRF2 nuclear accumulation in TPS1-MSCs under oxidative stress (Fig. 4H and I, and Supplementary Fig. S6E–S6G). Collectively, these findings suggest that TPS1 may enhance resistance to oxidative damage through p62-mediated NRF2-HMOX1 activation.
NRF2 inhibitor ML385 attenuates the antioxidant capacity of TPS1-MSCs
To investigate whether the NRF2-HMOX1 pathway mediates the antioxidant capacity of TPS1-MSCs, ML385 was employed to suppress NRF2 target gene expression. CCK8 assays showed that ML385 did not significantly impair TPS1-MSCs proliferation (Fig. 5A). Cellular activity, apoptosis, and ROS were then assessed in ML385-treated TPS1-MSCs (TPS1-MSCs+ML385) under three oxidative stress conditions. Compared with the DMSO control, ML385 significantly increased ROS levels, supporting enhanced oxidative sensitivity after NRF2 inhibition (Fig. 5B–D). Consistently, CCK8 showed reduced viability, and flow cytometry revealed a significant increase in both early and late apoptotic populations in TPS1-MSCs + ML385 (Fig. 5E–G). These findings indicate that pharmacological NRF2 inhibition weakens the antioxidant defense of TPS1-MSCs and increases susceptibility to oxidative damage.
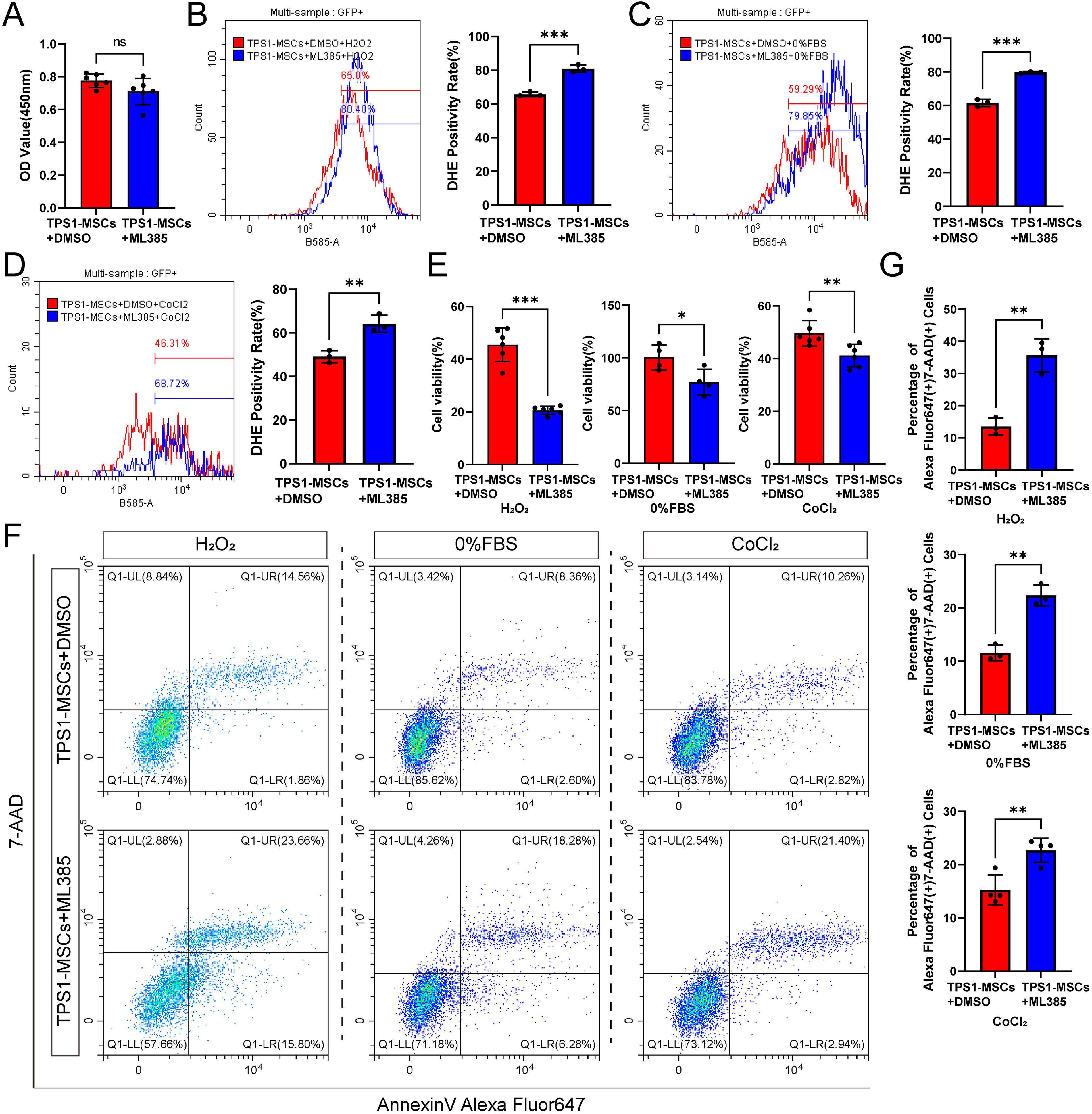
ML385 attenuates the antioxidant capacity of TPS1-MSCs.
NRF2 inhibition compromises the in vivo retention of TPS1-MSCs and delays wound healing
Following the in vitro findings, the in vivo role of NRF2 in TPS1-MSCs was further evaluated using ML385. In vivo fluorescence imaging demonstrated that the survival advantage of TPS1-MSCs was abrogated by ML385. Vehicle-treated TPS1-MSCs retained sustained fluorescence through day 7, whereas ML385 caused rapid signal decay (Fig. 6A–C). Consistently, day 7 frozen sections showed significantly fewer GFP-positive cells in the wound bed after ML385 treatment (Supplementary Fig. S7A and S7B). These findings indicate that pharmacological NRF2 inhibition reduces the survival of TPS1-MSCs in vivo.
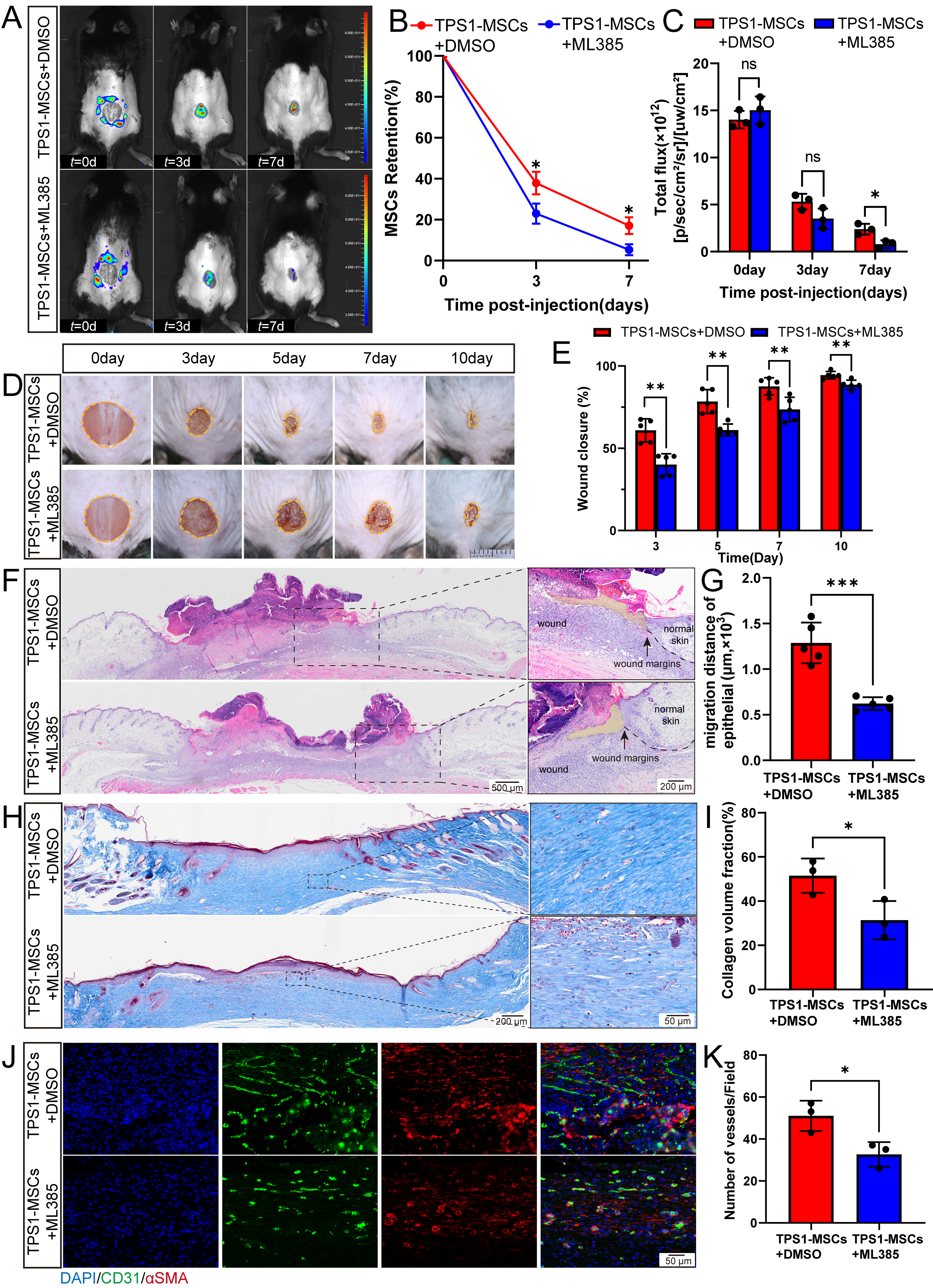
ML385 treatment attenuates TPS1-MSC-mediated retention and delays wound healing.
This impaired survival attenuated the wound healing efficacy of TPS1-MSCs. Macroscopic quantification showed that ML385 treatment of TPS1-MSCs delayed wound closure (Fig. 6D and E). Consistently, day 14 H&E staining showed a significantly wider intermargin distance in wounds treated with ML385-inhibited cells than in the TPS1-MSCs group (Supplementary Fig. S7C and S7D). Collectively, these findings suggest that therapeutic persistence of TPS1-MSCs depends on NRF2-mediated resistance to oxidative stress within the wound.
Inhibition of NRF2 attenuates TPS1-MSCs-promoted reepithelialization, collagen deposition, and angiogenesis
After wound closure was assessed, regenerated tissue structure and quality were evaluated to determine the impact of NRF2 inhibition. On day 7, histology showed that NRF2 inhibition impaired reepithelialization compared with that in the TPS1-MSC group (Fig. 6F–G). Consistently, immunofluorescence staining indicated a lower density of proliferating keratinocytes (Ki67+K14+) within the neo-epidermis of the ML385-treated group, suggesting reduced support for early epidermal continuity (Supplementary Fig. S8A–S8D). Histology also revealed that TPS1-MSCs promoted early collagen accumulation at the wound margins on day 7, and this ECM remodeling difference remained on day 14 (Fig. 6H and I, and Supplementary Fig. S8E and S8F). In parallel, CD31 and α-SMA staining showed that ML385-mediated NRF2 inhibition suppressed capillary formation, with fewer vessels at both time points than in the TPS1-MSC group (Fig. 6J–K, and Supplementary Fig. S8G and S8H). Collectively, NRF2-mediated survival supports TPS1-MSCs-driven reepithelialization, collagen deposition, and neovascularization.
DISCUSSION
Low resistance to oxidative stress and limited retention are important constraints on MSC-based tissue repair. To enhance antioxidant capacity, MSCs were engineered to express TPS1, inspired by the cytoprotective role of trehalose in nonmammalian systems. TPS1-MSCs synthesized trehalose intracellularly and released part of it extracellularly. This phenotype was associated with enhanced resistance to oxidative stress, prolonged wound retention, and improved tissue repair, supporting endogenous trehalose synthesis as a potential strategy to improve graft performance.
A major limitation of trehalose-based cytoprotection is poor membrane permeability. Consistent with prior studies, trehalose may support resistance to oxidative stress-induced damage and sustain protection within the transplant microenvironment. 26,27 Although fluid phase endocytosis and nanoparticle delivery can increase intracellular trehalose, they often fail to sustain intracellular accumulation. 28 By contrast, TPS1-MSCs continuously generated trehalose and reached approximately 21 μg/104 cells. This sustained production may help overcome low in vivo efficiency and rapid metabolic clearance while reducing dependence on repeated dosing.
Whole transcriptome sequencing indicated that the antioxidant phenotype of TPS1-MSCs under oxidative stress was associated with activation of the NRF2-HMOX1 pathway. Trehalose can induce mTOR-independent autophagy and has shown protective effects in neurodegenerative disease models. 18,29 –32 This process is accompanied by increased p62 expression and has been implicated in trehalose-associated NRF2 nuclear translocation. 19,20 Consistently, TPS1-MSCs showed NRF2 pathway enrichment under multiple oxidative stress conditions, elevated NRF2 and HMOX1 expression, increased nuclear NRF2 localization, and high p62 expression. ML385 also attenuated the antioxidant advantage of TPS1-MSCs, supporting an important contribution of NRF2 signaling. Although autophagic flux was not directly assessed, these findings suggest that trehalose may promote p62 associated signaling and thereby facilitate NRF2 nuclear translocation and downstream HMOX1 activation.
Trehalose may exert cytoprotective effects beyond antioxidant defense. As a macromolecule stabilizer, it can protect cells from environmental stress and reduce superoxide dismutase 1 misfolding and functional loss under oxidative stress. 33 Trehalose has also rescued dysfunction in H2O2-exposed keratinocytes, myofibroblasts, and bone marrow-derived MSCs. 16,17,21 Unlike single-target interventions, trehalose may act as a chemical chaperone that stabilizes protein conformation, suppresses aggregation, and supports proteostatic homeostasis under stress. 34,35 Such pleiotropic activity suggests that its protective effects may extend beyond direct antioxidant signaling. Although these processes were not directly examined, they may also contribute to the therapeutic benefits observed in TPS1-MSCs.
Because adenovirally transduced human MSCs were transplanted into immunocompetent mice without immunosuppression, the increased in vivo persistence of TPS1-MSCs may reflect not only trehalose-mediated cytoprotection but also reduced xenogeneic immune clearance. Enhanced resistance to oxidative stress may reduce oxidative stress-induced immunogenic cell death and subsequent host immune activation, thereby decreasing immune-mediated elimination of transplanted cells and partly contributing to the superior retention of TPS1-MSCs. However, this possibility requires further investigation.
TPS1 is absent from the human genome. Here, Drosophila melanogaster-derived TPS1 was heterologously expressed to confer trehalose-6-phosphate synthesis to MSCs, and the effects likely reflect trehalose-associated metabolic reprogramming rather than a native human TPS pathway. Translationally, immunogenicity of a heterologous Drosophila-derived protein may affect host recognition, inflammation, and persistence of transplanted cells, especially in immunocompetent settings. In addition, although activation of the P62-NRF2-HMOX1 axis was associated with protection against oxidative injury, sustained NRF2 activation with enhanced cell survival may raise theoretical long-term safety concerns, since persistent NRF2 signaling has been linked to aberrant survival advantages and tumor-associated phenotypes in some contexts. Therefore, the short-term wound healing model cannot rule out potential biosafety concerns following MSC transplantation, including immunogenicity, abnormal persistence, dysregulated proliferation, and tumorigenic risk, all of which warrant long-term in vivo evaluation.
Because TPS1-MSCs depend on sustained endogenous trehalose synthesis, which differs fundamentally from the transient kinetics of exogenous antioxidants, direct efficacy comparisons with antioxidant positive controls were not included. The relative contributions of metabolic reprogramming, paracrine signaling, and immune modulation, as well as the dynamics between intracellular and extracellular trehalose pools, remain unresolved. Together with differences between the murine acute wound model and human wound healing, these factors indicate a need for more human-relevant and immunologically matched systems. More broadly, trehalose supports freezing and desiccation tolerance through vitrification and water substitution. 36,37 In this context, elevated intracellular trehalose in TPS1-MSCs may indicate potential for improved cryotolerance, although this requires future investigation.
INNOVATION
Current MSC-based wound therapies are limited by poor survival under oxidative stress. Here, we engineered MSCs to express TPS1, enabling endogenous trehalose synthesis and secretion. This intrinsic capacity enhances MSCs antioxidant resistance, improves transplantation retention, and activates the NRF2-HMOX1 signaling pathway. These results advance MSC engineering by coupling metabolic reprogramming with cytoprotection, offering a clinically translatable strategy to boost wound repair efficacy.
KEY FINDINGS
TPS1-MSCs, capable of endogenously synthesizing and secreting trehalose, were successfully established.
TPS1-MSCs demonstrate a high survival and retention rate at the wound site, effectively accelerating the wound healing process and significantly improving repair outcomes.
TPS1-MSCs stably exert antioxidative stress effects and maintain their cellular activity and function, whether in the face of direct oxidative stress damage induced by H2O2 or oxidative stress caused by pathological conditions such as hypoxia and nutrient deprivation.
The molecular mechanism underlying the antioxidant capacity and wound healing-promoting effect of TPS1-MSCs may be closely related to the activation of the NRF2-HMOX1 signaling pathway.
AUTHORS’ CONFIRMATION
Z.W.: Conceptualization, data curation, formal analysis, methodology, visualization, writing—original draft, and writing—review and editing. X.L.: Conceptualization, formal analysis, methodology, writing—original draft, and writing—review and editing. X.C., J.J., Q.C., F.Z., and Z.C.: Formal analysis, methodology, and writing—review and editing. W.F., L.W., and Y.W.: Formal analysis, resources, and writing—review and editing. Z.L., Q.W., J.G., and J.Z.: Data curation, resources, and writing—review and editing. G.X. and R.P.: Conceptualization, funding acquisition, resources, supervision, and writing—review and editing.
ACKNOWLEDGMENTS AND FUNDING SOURCES
The authors thank Shanghai Tengyun Biotechnology Co., Ltd. for developing Hiplot Pro platform (https://hiplot.com.cn/) and providing technical assistance and valuable tools for data analysis and visualization. This work was supported by the
AUTHOR DISCLOSURE AND GHOSTWRITING
All authors declare that they have no conflict of interest. This thesis has been written by the named authors without the use of ghostwriters or other forms of unauthorized author assistance.
ABOUT THE AUTHORS
Footnotes
Supplemental Material
Abbreviations and Acronyms
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.