
Abstract
Significance:
Wound healing is an energetically demanding process that is easily disturbed by metabolic dysregulation. Metabolic dysregulation, induced by diabetes, obesity, insulin resistance, and various hormone imbalances, can disrupt each phase of wound repair, increasing the risk of post-operative complications, infection, wound dehiscence, and pathological scarring. We review the current evidence on the role of metabolic regulation in wound healing and highlight clinically relevant considerations for patient care.
Recent Advances:
Animal and human studies have advanced our understanding of how metabolic pathways influence inflammation, angiogenesis, fibroblast function, and extracellular matrix remodeling during wound healing. These insights have led to the development of novel therapies, including hormone-based topical agents and dressings that both sense and modulate metabolites during wound healing.
Critical Issues:
Despite growing recognition of the role of metabolic syndromes and hormonal regulation in wound healing, these factors are insufficiently integrated into clinical wound management. Bridging this gap requires a clear understanding of how metabolic syndromes and hormonal derangements influence healing.
Future Directions:
The continued development of treatments that modulate metabolic and hormonal pathways may enhance wound healing while minimizing systemic risk. Clinicians should also integrate local metabolic optimization with medical and lifestyle management to create the optimal wound healing environment for patients.
Keywords
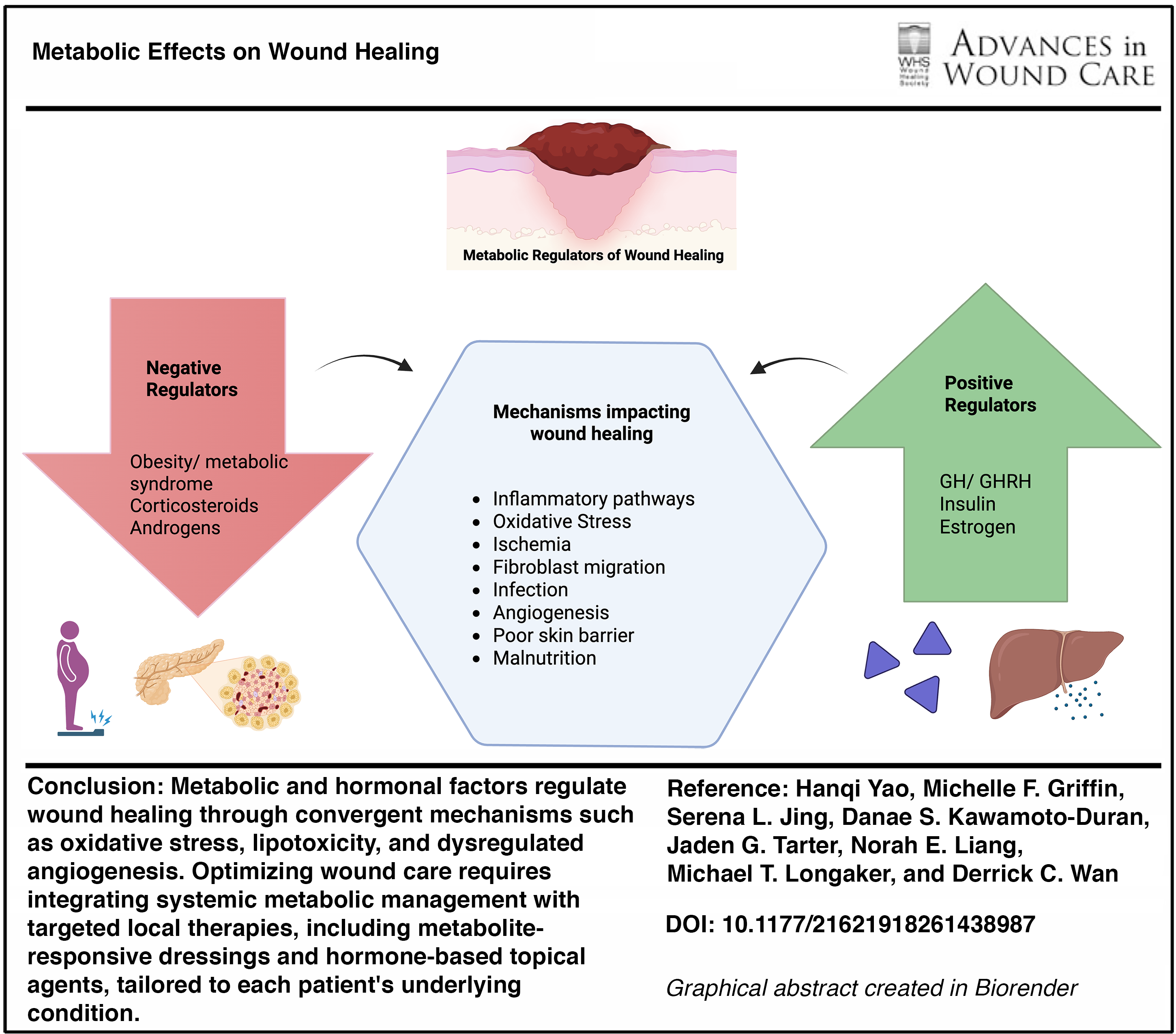
SCOPE AND SIGNIFICANCE
Wound healing is a metabolically intensive process requiring the utilization of nutrients in inflammation, cellular proliferation, and tissue remodeling. The wound healing process is greatly impacted by metabolic dysregulation from obesity, insulin resistance, corticosteroids, and various hormones. When treating wounds that coexist with specific metabolic conditions, it is important to understand these metabolic effects and mechanisms and implement interventions to optimize the wound healing process for patients. This review, therefore, aims to serve as an overview of the literature on metabolic and hormonal regulation of cutaneous wound repair, with an emphasis on clinically relevant mechanisms and considerations for patient care.
Derrick C. Wan, MD

TRANSLATIONAL RELEVANCE
Novel local treatments such as topical compound creams and hydrogels mitigate negative metabolic effects on wound healing and leverage beneficial pathways to improve outcomes in animal and human studies. Although more clinical studies are needed for widespread adaptation of these novel therapies, combining these novel treatments with medical management and lifestyle changes may provide patients with a multifaceted treatment plan to optimize the local wound-healing environment.
CLINICAL RELEVANCE
Chronic, nonhealing wounds affect 6.5 million patients in the United States and represent a multibillion-dollar industry. Not only does it present a significant health care burden in terms of resource utilization and cost but patients also often experience psychological distress and poor functional outcomes. With an ever-increasing population of patients presenting with wounds and at least one metabolic comorbidity, it is important to elucidate the differential effects and the mechanisms behind metabolic dysregulation and hormones on wound healing and take advantage of metabolic pathways beneficial to the wound-healing process.
OVERVIEW
Wound healing is a complex process that requires the tight regulation of metabolic shifts to coordinate immune activation and resolution, cell migration and proliferation, angiogenesis, and extracellular matrix (ECM) remodeling. In this review, “metabolic” refers to the cellular pathways that meet the energy demands of each of these phases in wound healing, including glucose and lipid utilization, mitochondrial function, and redox and oxidative stress regulation.
In this review, we synthesize evidence on how obesity and insulin dysregulation and other key hormone signals can alter these metabolic pathways to impact wound-healing outcomes (Graphical Abstract). We focus on corticosteroids, growth hormone (GH), growth hormone-releasing hormone (GHRH), estrogen, and androgens because their systemic and local effects have been documented clinically and directly intersect with the metabolic requirements of wound repair.
A literature search was conducted using PubMed for peer-reviewed articles published in English from database inception through December 2025. Search terms included combinations of “wound healing,” “cutaneous wound repair,” “obesity,” “insulin resistance,” “diabetes,” “glucocorticoids,” “corticosteroids,” “growth hormone,” “growth hormone-releasing hormone,” “estrogen,” “androgens,” “testosterone,” “dihydrotestosterone,” and “fibrosis.” Reference lists of reviews and key primary studies were additionally screened to capture seminal literature not retrieved by keyword searches. We included human clinical trials and observational studies when available, as well as animal and in vitro studies that elucidated mechanisms directly relevant to cutaneous wound repair. We excluded studies focused exclusively on noncutaneous tissue repair unless the wound biology was directly translatable, such as in major burn injuries. A formal meta-analysis or risk-of-bias assessment was not performed given the narrative format of this review. Throughout this review, evidence from human clinical trials is presented as the primary basis for clinical conclusions where available. Preclinical animal and in vitro findings are cited to provide mechanistic context or when human data are absent and are explicitly identified as such. Where clinical studies were small or at higher risk of bias by design, we explicitly note these limitations. Given this approach, the clinical recommendations synthesized in this review should be interpreted as expert opinion informed by the available literature rather than formal evidence-based guidelines.
DISCUSSION
Nonhealing wounds pose a large problem to individual patients and health care systems. They affect 6.5 million patients and have a health care expenditure of at least 25 billion dollars. 1 As humans age and develop diseases such as diabetes and hormonal imbalance, wound healing is significantly impacted. This review examines four metabolic scenarios—obesity, metabolic syndrome/insulin resistance, corticosteroids, and growth and sex hormones—and their clinical effect on wound healing (Fig. 1).
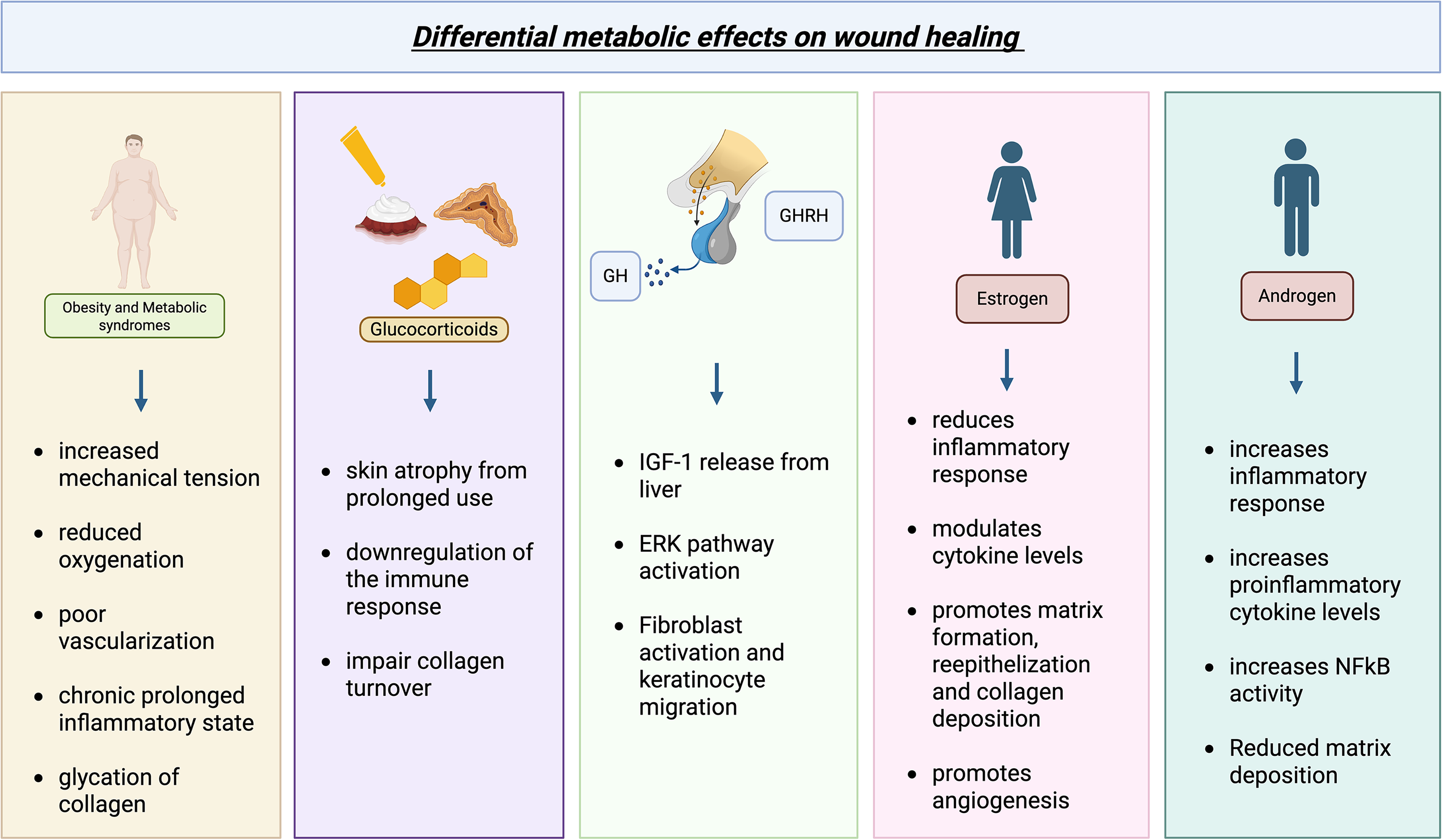
Effects of various metabolic and hormonal influences on wound healing and their mechanisms. Obesity, metabolic syndrome/insulin resistance, corticosteroids, and androgens impede wound healing while estrogen, growth hormone, and growth hormone-releasing hormone promote wound healing, each through a number of discrete mechanisms. ERK, extracellular signal-regulated kinase; GH, growth hormone; GHRH, growth hormone-releasing hormone; IGF-1, insulin-like growth factor 1; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells. Created in BioRender. Yao, H. (2025) https://BioRender.com/aeb7e5k.
Obesity and insulin dysregulation-associated clinical and pathophysiological impairments in wound healing
Approximately 42.4% of adults in the United States have a body mass index of greater than 30 kg/m2 and meet criteria for clinical obesity. 2 Among many other comorbidities, obesity is highly correlated with deficient wound healing. Patients with obesity experience an increased rate of surgical complications, including wound infection, wound dehiscence, and mortality. A 4-year prospective cohort study with National Health Service hospitals in the United Kingdom found a 1.1 to 4.4 times increase in the risk of overweight and obese patients developing surgical site infections. 3 A similar study in the United States found a 5.3-fold increase in surgical site infections for obese patients. 4 Furthermore, obese patients who underwent abdominal, sternal, hip, and upper limb surgeries all experienced a higher degree of wound dehiscence compared to patients within healthier weight limits.3,5,6 Obese patients also experience a higher rate of chronic wounds such as venous leg ulcers and are 1.27 times more likely to develop hypertrophic scars and keloids.7–10 Linked comorbid conditions such as diabetes, hypertension, and cardiovascular disease also increase the risk of aberrant scarring, chronic wounds, and impaired wound healing.11–14
Obesity affects wound healing through mechanical, physiological, and metabolic mechanisms. Mechanically, excess adipose tissue increases tension around wound edges, elevating tissue pressure which not only reduces local perfusion and predisposes patients to hematoma formation but also contributes to hypertrophic scarring (Fig. 2).14,15 Physiologically, it has been suggested that because obese patients may experience impaired diaphragmatic and chest wall relaxation, and therefore reduced vital lung capacity and overall oxygenation, this can contribute to poor oxygen delivery to wounds.14,16,17 Fibroblasts are particularly sensitive to hypoxia, requiring at least 15 mm Hg of oxygen to function effectively, such as in their differentiation into myofibroblasts, which drive wound closure through contraction.14,16,18,19
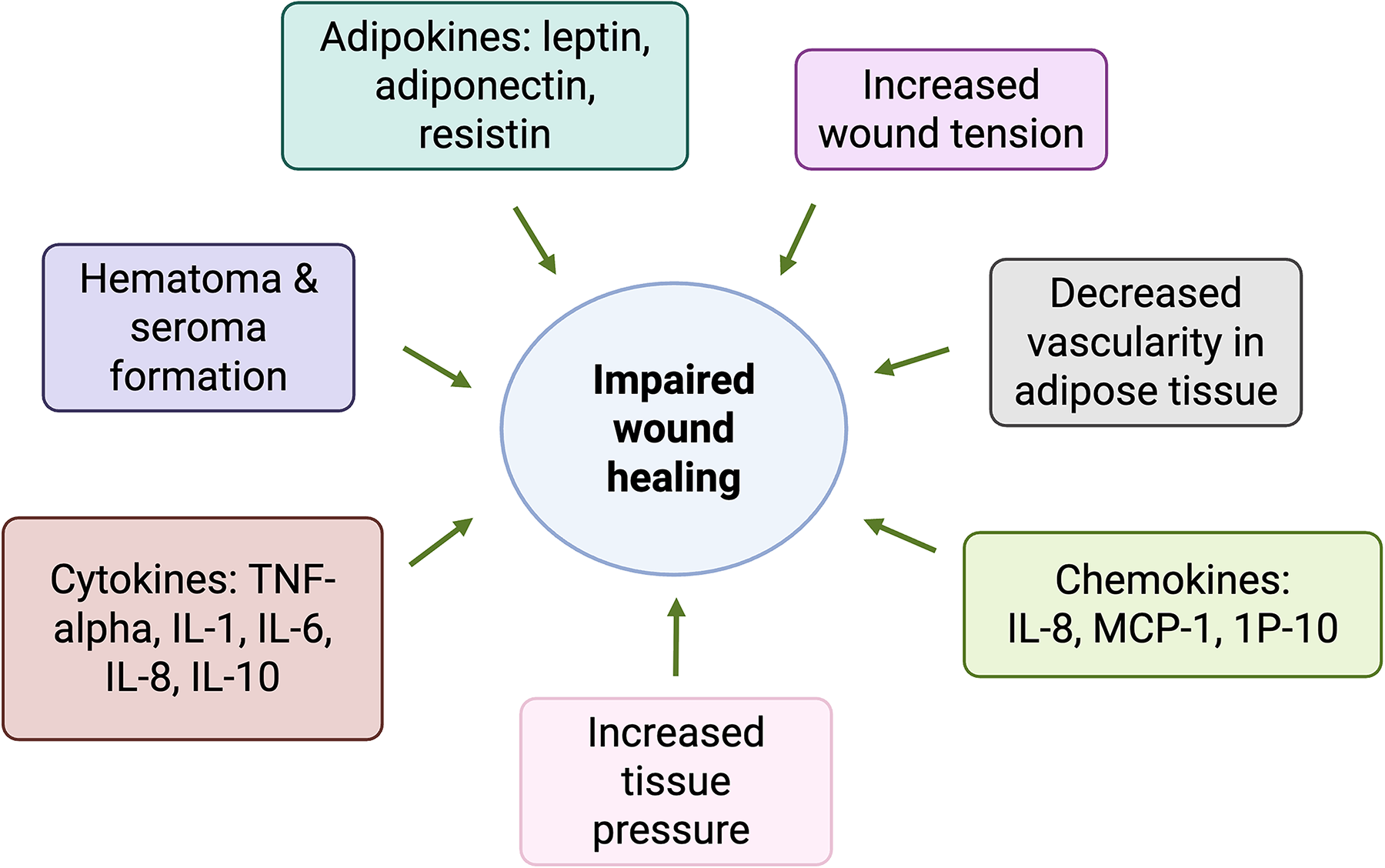
Factors in obesity impeding wound healing. Obesity is characterized by chronic inflammation, increased tissue pressure, and decreased vascularity of adipose tissue, which all deter normal wound healing. IL, interleukin; IP-10, interferon-gamma-inducible protein 10; MCP-1, monocyte chemoattractant protein-1; TNF-α, tumor necrosis factor alpha. Adapted and taken with permission from Cotterell et al. (2024). 14 Created in BioRender. Yao, H. (2025) https://BioRender.com/aeb7e5k.
Excess adipose tissue further contributes to wound hypoxia by impairing vascularization. Expansion of adipose tissue may outpace angiogenesis, and adipocytes release inhibitors of angiogenesis including endostatin and angiostatin. This chronic hypoxic environment deters collagen maturation, delays the recruitment of immune cells, hinders the delivery of nutrients to the wound, and prolongs local inflammation. 20
While mechanical stress, reduced perfusion, and hypoxia contribute to impaired wound repair in obese patients, these factors largely arise within, and are amplified by, a metabolically dysregulated, pro-inflammatory tissue environment.
Metabolic dysregulation in obesity impairs wound healing through interconnected pathways involving lipotoxicity, mitochondrial dysfunction, oxidative stress, and defective immune resolution. Disrupted lipid metabolism leads to excess lipid accumulation in non-adipose cells critical for tissue repair, including fibroblasts, immune cells, and epithelial cells.21,22 Intracellular lipid overload triggers conserved stress responses, such as endoplasmic reticulum stress, and accumulation of toxic lipid intermediates like diacylglycerol and ceramides. 22 In mitochondria, lipid overload disrupts electron transport chain activity, reducing oxidative phosphorylation efficiency.21,23 Ceramides can further alter mitochondrial membrane integrity, promoting mitochondrial depolarization and thus apoptotic signaling. 24 At the same time, excess fatty acids overwhelm mitochondrial β-oxidation capacity, causing electron leakage and excessive reactive oxygen species (ROS) generation, which further damage mitochondrial DNA, proteins, and membranes. 21 Lipotoxic stress also impairs mitophagy, allowing dysfunctional mitochondria to accumulate. 21
In macrophages, lipotoxicity impairs efferocytosis. Excess intracellular lipids disrupt phagocytic receptor signaling and skew macrophage polarization toward a pro-inflammatory M1 phenotype, reducing the clearance of apoptotic cells and delaying the resolution of inflammation and progression to the proliferative phase of wound repair.21,24 Together, these processes establish a self-sustaining cycle of cellular dysfunction, oxidative stress, and inflammation, impairing tissue repair.
Additional metabolic factors in obesity, including advanced glycation end products (AGEs) and dysregulated adipokine signaling, further exacerbate this inflamed wound environment.25,26 AGEs promote oxidative stress and inflammation while impairing matrix remodeling, whereas the adipokines leptin and adiponectin, which are elevated and reduced, respectively, in obesity, favor pro-inflammatory immune activation, and limit angiogenesis and fibroblast function.26–28
Collectively, lipotoxicity, mitochondrial dysfunction, oxidative stress, defective efferocytosis, and maladaptive metabolic signaling sustain and perpetuate obesity-associated wound-healing dysfunction. It is worth mentioning that these mechanistic insights, including lipotoxic stress response, mitochondrial dysfunction, and adipokine/AGE-linked inflammatory signaling, into obesity-associated wound impairment were largely first understood from controlled mouse models and later validated in in vitro models from human samples.25,26 These models allow researchers to isolate individual metabolic stressors and to perform tissue sampling at different phases of healing, which are rarely feasible in humans. Furthermore, human studies are often confounded by comorbidities such as diabetes and vascular disease, limiting the ability to attribute causality to obesity and metabolic dysregulation alone.
Approximately 80% of obese individuals develop insulin resistance at some point in their lives.29,30 According to the Centers for Disease Control and Prevention, approximately 38 million people live with diabetes, whereas 97 million live with prediabetes across the country. 31 Diabetic wounds, including foot ulcers, affect 25% of diabetic patients. 32 Post-surgically, regardless of surgery location or type, diabetic and insulin-dependent patients have higher rates of wound infection, dehiscence, complications, and longer post-operative hospital stay.33–36
Commonly used diabetic models, including streptozotocin-induced diabetic mice and rats and genetically diabetic db/db mice, have been instrumental in defining the immune-cell mechanisms underlying impaired wound healing, and how they contribute to altered scar formation.37,38 The hyperglycemic and chronic inflammatory state characteristic of diabetes and insulin resistance significantly alters wound repair and scar formation. Diabetic mice wounds demonstrate persistent macrophage-driven inflammation with the elevated production of pro-inflammatory cytokines, including tumor necrosis factor-alpha (TNF-α) and monocyte chemoattractant protein-1 (MCP-1). 37 At the same time, excessive neutrophil activation results in the sustained release of free radicals and cytotoxic mediators, prolonging the inflammatory phase and delaying the progression to the proliferative and remodeling phases of wound healing. 39 This dysregulated inflammatory response has been associated with a higher risk of keloids and hypertrophic scarring in both mouse models and in humans.40,41
Beyond cytokine imbalance, AGEs represent a pathophysiologic mechanism linking hyperglycemia to impaired wound healing. Chronic hyperglycemia accelerates AGE formation through increased levels of fructose, which eventually forms triose phosphate, leading to the formation of methylglyoxal, a precursor to AGEs. 38 AGEs form covalent cross-links with structural proteins, particularly collagen, resulting in increased stiffness, reduced elasticity, and diminished resistance to mechanical stress.38,42 In the dermis, AGE-mediated collagen glycation contributes to dermal elastosis, basement membrane thickening, stratum spinosum hyperplasia, and pericapillary T-cell infiltration.44,45 Intracellular AGE accumulation further activates nuclear factor-κB (NF-κB) signaling, increasing cytokine and growth factor production while amplifying oxidative stress. 38 These changes stall wounds in a prolonged inflammatory state, impair fibroblast proliferation, reduce fibroblast growth factor activity, and promote fibroblast apoptosis through caspase-3 (cysteine-dependent aspartate-directed proteases) and caspase-8 activation.38,45
Patients with glucose dysregulation also exist in a chronic state of low-grade inflammation characterized by elevated baseline levels of TNF-α, matrix metalloprotease 7 (MMP-7), and reduced metalloprotease (MMP) inhibitors. 40 Skin of patients with metabolic syndrome demonstrates increased MMP activity, whereas nonhealing diabetic foot ulcers were found to have increased TNF-α, MCP-1, and MMP-9.40,46–49 This imbalance promotes excessive ECM degradation and limits effective collagen synthesis, contributing to chronic wound persistence.
Neutrophil dysfunction and impaired clearance represent another critical aspect of diabetic wound pathology. Neutrophils act as early responders that secrete antimicrobial peptides, neutrophil extracellular traps (NETs), and inflammatory cytokines like TNF-α. 39 Their timely apoptosis initiated by macrophage efferocytosis is required for the resolution of inflammation and transition to the subsequent phases of wound healing. 50 Under hyperglycemic conditions, neutrophil metabolism shifts away from glycolysis and the pentose phosphate pathway toward the polyol pathway, resulting in sorbitol accumulation, which leads to constitutive NET formation and excess ROS generation.51,52 Constitutive NET formation is linked to impaired responses to infection as they inhibit macrophage efferocytosis through suppression of the Rac1 pathway, preventing the effective clearance of apoptotic neutrophils and prolonging inflammation. 50 This metabolic shift toward the polyol pathway also leads to the depletion of NADPH, which results in impaired activity of glutathione and other antioxidants, further increasing ROS levels. 52
Finally, diabetes disrupts wound angiogenesis through altered hypoxia signaling and microvascular rarefaction. Hyperglycemia promotes prolyl hydroxylase domain-dependent hydroxylation and degradation of hypoxia-inducible factor 1α (HIF-1α), even under hypoxic conditions. 53 This suppresses HIF-1 transcriptional activity and reduces vascular endothelial growth factor (VEGF) production, impairing neovascularization in ischemic tissues. 54 Hyperglycemia-induced HIF-1 inhibition also contributes to mitochondrial ROS overproduction, triggering endothelial apoptosis and microvascular damage. 55 This results in depletion of circulating and wound-resident endothelial progenitor cells in diabetes, further limiting angiogenic capacity and contributing to delayed and nonhealing chronic wounds.53,55
Hormonal effects on wound healing
Corticosteroid effects on wound healing
Although obesity and insulin dysregulation impair wound healing through metabolic stress and chronic inflammation, these conditions may also be characterized by alterations in stress hormone signaling, particularly glucocorticoids, which can alone significantly influence tissue repair. 56 Glucocorticoids regulate immune activation, fibroblast function, angiogenesis, and ECM remodeling—processes already disrupted in obesity and diabetes-associated wounds.
Steroids are commonly used in medicine for their anti-inflammatory and immunosuppressive effects. At the molecular level, these effects are classically mediated through binding of glucocorticoids to the intracellular glucocorticoid receptor, which translocates to the nucleus and alters gene transcription of pro-inflammatory cytokines, as well as through suppression of phospholipase A2 (PLA2) activity, an enzyme that catalyzes the release of lipid mediators of inflammation.57,58 The effect of steroids on wound healing is dependent on the dose and chronicity of use.
Acute, high-dose steroids have not been found to significantly affect wound healing. Researchers found no significant difference in the rates of post-surgical wound complications and levels of proline and collagen accumulation between patients receiving a one-time pre-operative dose of 30 mg/kg methylprednisolone and the placebo group. 59 In contrast, long-term use of systemic steroids interferes with wound healing. Studies have found a 30% reduction in wound tensile strength in animals, including rats, mice, and rabbits, receiving systemic steroid administration. 61 This is further corroborated by a retrospective human study on more than 600,000 patients that showed patients who have taken oral steroids for greater than 30 days before surgery experienced greater surgical site infections, 2- to 3-fold increase in wound dehiscence, and 4-fold increase in mortality. 60
In addition, Cushing syndrome patients with chronically high levels of adrenal corticosteroids experience a host of cutaneous problems. Most notably, these patients experience weakening of the skin barrier and skin atrophy. They also experience poor wound healing. For instance, Cushing syndrome patients have been reported to have reduced wound tensile strength, and rebound increase in wound tensile strength following adrenal cortex tumor removal has been documented. 62
At the tissue level, glucocorticoids heavily affect the turnover of the skin. Chronic topical glucocorticoid exposure on healthy skin weakens the skin barrier as it leads to atrophy of the epidermis, disturbance of the dermal–epidermal junction, and loss of keratinocytes. 62 Mechanistically, these effects are mediated in part through glucocorticoid receptor-dependent transcriptional repression of pro-inflammatory cytokines and growth factors, necessary for the initiation of the inflammatory phase of wound healing. 63 In wounded skin, topical steroid treatment decreases the expression of cytokines such as transforming growth factor beta (TGF-β), platelet-derived growth factor, TNF, keratinocyte growth factor (KGF), and interleukin (IL)-1α. 64 Corticosteroids also downregulate the expression of intercellular adhesion molecule 1 (ICAM-1) and impair macrophage recruitment to the wound.65,66 By inhibiting TGF-β and KGF, steroids reduce fibroblast and keratinocyte migration and proliferation, thereby delaying re-epithelialization. 68
At the same time, inhibition of PLA2 represents a second major mechanism by which corticosteroids impair wound healing. PLA2 enzymes play a central role in lipid metabolism by catalyzing the release of arachidonic acid and lysophospholipids from membrane phospholipids, processes that are essential for epidermal barrier integrity and eicosanoid production.68,69 Suppression of PLA2 activity by glucocorticoids limits the availability of arachidonic acid, resulting in reduced synthesis of prostaglandins and other lipid mediators that normally support inflammation resolution, vascular remodeling, and cell proliferation during wound repair.71,72 Disruption of these lipid-mediated signaling pathways further compromises cutaneous homeostasis and healing capacity.
During the remodeling phase, corticosteroids impair collagen turnover from type III to type I collagen, resulting in scars with low wound tensile strength.72–75 Collectively, these glucocorticoid receptor and PLA2-mediated mechanisms explain how chronic corticosteroid exposure disrupts multiple phases of wound healing, from inflammation and proliferation to matrix remodeling.
GH and GHRH effects on wound healing
GH and GHRH both exert positive effects on wound healing. GH is produced by the anterior pituitary gland through hypothalamic stimulation by GHRH and yields systemic effects by inducing growth in nearly every tissue and organ in the body. 76 As part of the hypothalamus–pituitary growth axis, GH induces the release of insulin-like growth factor 1 (IGF-1) from the liver, which supports proliferation in a variety of tissues, including bone, cartilage, and muscle, by promoting a pro-anabolic state. In cutaneous wounds, however, the role of circulating IGF-1 in wound healing is less clear, and emerging evidence suggests that locally produced IGF-1 in wound tissue may play a more relevant role.77,78 In mice, systemic, intraperitoneal IGF-1 administration led to significant weight gain without improvements in incisional wound strength. 78 In contrast, GH treatment that was subcutaneously injected in mice not only did not lead to significant increases in weight but also resulted in significantly increased wound strength. The failure of administered IGF-1 to improve wound strength suggests that circulating IGF-1 is not readily available to wounds and that enhanced wound integrity and collagen deposition observed with local GH administration more likely reflect autocrine and paracrine sources of IGF-1 from fibroblasts, keratinocytes, and endothelial cells.77–79
GH is also able to act directly within wound tissue. It does so by stimulating fibroblast and keratinocyte migration to the wound, facilitating the formation of granulation tissue.77,80 Mechanistically, GH activates the extracellular signal-regulated kinase (ERK) pathway to reduce oxidative stress and inflammation and promote angiogenesis. 81 This is evidenced by the increase in phosphorylated ERK1/2 proteins, epidermal growth factor (EGF), TGF-β, and VEGF. This demonstrates GH’s capacity to support repair by not only stimulating cell proliferation but also improving the wound’s inflammatory balance and pro-angiogenic environment to progress the wound from the inflammatory phase to the proliferative phase of healing. GH may also influence the quality and organization of repaired tissue. In a mouse pressure injury model, animals receiving recombinant human GH (rhGH) exhibited a greater collagen type III to type I ratio on histology, indicating a longer dermal proliferation and secretion period. 82
Separate from its role in stimulating the release of GH from the pituitary, peripheral GHRH can exert direct effects on wound healing without modulation from GH or IGF-1. GHRH is a mitogenic and antiapoptotic compound. Studies have shown that exposure to GHRH agonists induces proliferation of dermal fibroblasts through ERK1/2 and AKT pathways without increases in either GH or IGF-1, consistent with local receptor-mediated actions. 83 Importantly, fibroblasts express GHRH receptor Sv1, through which stimulation can induce alpha-smooth muscle actin (α-SMA) expression and lead to abundant fibroblasts within the wound during the inflammatory stage, resulting in a thicker dermal layer with more ECM.83,84
Clinically, GH has been used to facilitate healing from burns. In children with >40% total body surface area (BSA) and >20% total BSA full-thickness burns, 8 days of local rhGH injections facilitated accelerated healing of donor grafts on burned areas. 85 GH has also been shown to enhance donor site healing, with systemic rhGH administration associated with a 26% increase in basal lamina and a 58% increase in dermal–epidermal coverage. 86 In mice, pressure ulcers were observed to heal twice as fast following injection with rhGH. 82 The healed wounds from the rhGH group exhibited a thicker dermal ECM with greater inflammatory cell infiltration and collagen deposition. In mice, peripheral GHRH has also been observed to expedite wound healing in mice in a dose-dependent manner. 83 Histologically, the epidermis of the treated group was found to be thicker and more well-adhered compared to the control group. The treated group also harbored more myofibroblasts and α-SMA, a marker of fibroblast activation.
Estrogen effects on wound healing
There has been increasing evidence pointing to sexual dimorphism in skin properties and wound healing. Collagen content decreases by 30% in the first 5 years after menopause and drops 2% each year thereafter. The drop in collagen content results in thinner skin, a weaker skin barrier, and a loss of elasticity. 87 Skin-related changes in aging women also include a slowed re-epithelization rate following wounding, increasing the risk for the development of chronic wounds.88,89 Since many different tissues in the body are responsive to estrogen, including breast and endometrium in addition to skin, its influence on wound healing should be considered at both the systemic level and locally within wound tissue, as it also has important implications for the therapeutic approach. 90 Systemic hormone replacement therapy (HRT) can alleviate the risks of developing chronic wounds that aging women may face. In addition to strengthening the skin barrier and improving scar appearance, HRT has been shown to reduce the incidence of pressure ulcers and venous leg ulcers when compared to women receiving control, tamoxifen, and aromatase inhibitor treatments.91,92 Furthermore, a study found that young women who were in the preovulatory stage of their menstrual cycle with a higher level of estrogen had a lower risk of wound dehiscence and hypertrophic scarring following bilateral mammoplasty. 93 Locally, topical estrogen has also been associated with improvements in cutaneous wound repair. In ovariectomized mice, impaired wound healing was demonstrated to be rescued with topical estrogen application. 88 In humans, topical estrogen application before wounding and for 24 h post-wounding has also been shown to lead to a significant reduction in wound size of both men and women. 94 Systemically and locally, estrogen mediates its effects through two receptors, estrogen receptor and estrogen receptor-β, which have been described in keratinocytes, dermal fibroblasts, and adnexal structures, as well as in breast tissue, although their relative proportions and context-dependent functions across tissues remain incompletely defined.95–98
Across both systemic exposure and local delivery, estrogen’s effects on cutaneous healing appear to converge on shared actions within the wound microenvironment that modulate inflammation, angiogenesis, and matrix remodeling. First, estrogen suppresses the inflammatory response and modulates cytokine expression. More specifically, estrogen directly acts on neutrophils by downregulating L-selectin and elastase to prevent neutrophil accumulation. In addition, it also suppresses IL-1 and TNF-α and may upregulate anti-inflammatory cytokines IL-4 and IL-10. By decreasing the inflammatory response to the region, increased estrogen levels may thus promote a favorable wound-healing environment. Estrogen also decreases the level of macrophage migration inhibitory factor (MIF) and upregulates TGF-β1 produced by dermal fibroblasts.99,100 MIF is a pro-inflammatory factor secreted by macrophages, T-cells, and leukocytes, whereas TGF-β1 is an important factor for the process of cell proliferation, differentiation, and ECM production. By promoting a differential cytokine profile, estrogen may thereby facilitate wound healing. 100 Second, estrogen regulates oxidative stress by increasing superoxide dismutase and B-cell lymphoma-2, an anti-apoptotic factor, through the activation of nuclear factor 2 in fibroblasts. 101 In dermal fibroblasts and keratinocytes, 17β-estradiol, a potent form of estrogen, has been shown to protect against peroxidative injury by modulating ROS/nitric oxide (NO) release and by preserving mitochondrial integrity through the prevention of cytochrome C release, although the pathways at the basis of those effects remain to be fully understood.102,103 Estrogen also increases protease inhibitors, preventing the excessive activation of cathepsins and ensuring the synthesis of ECM. 104 Third, estrogen can stimulate the migration of endothelial cells into the wound, which promotes angiogenesis. Estradiol increases the attachment of human endothelial cells to ECM by promoting EGF and TGF-β levels. Estrogen has also been shown to induce the expression of VEGF and platelet-derived growth factor-α in endothelial cells. In contrast, estrogen’s migratory effect on endothelial cells can be blocked by an estrogen antagonist.84,88,105 However, given the wide distribution of estrogen-responsive tissues, exposure of estrogen to nontarget organs, like the breast and endometrium, introduces the risk of cancer development in these organs. As such, this therapeutic trade-off has supported the rationale for local estrogen delivery or more focused treatments that can mediate specific estrogen-responsive signaling pathways in wounded tissue. 90
Estrogen and androgens alone may not account for all the differences in wound healing between the sexes. Factors regarding skin physiology and differential gene expression in scarring also have to be considered. Men have thicker skin than women and heal more slowly from wounds. 106 This observation has been associated with greater levels of expression for collagen and elastin messenger RNA (mRNA) and lower levels of expression of FOXN1, a transcription factor crucial for the formation of hair follicles, and adipogenic genes such as PPARγ and Leptin in men. 106
Androgen effects on wound healing
Androgens have various effects on the skin and wound healing. Androgens can be synthesized from cholesterol or from steroid precursors and act on androgen receptors (ARs) systemically and locally, including in keratinocytes, fibroblasts, sebocytes, and immunoregulatory cells such as T-cells and macrophages.107–109 In general, androgens demonstrate an inhibitory effect on wound healing. Testosterone begins to decrease in men starting at age 40. Elderly men may experience a delay in excisional wound healing, while higher testosterone levels can rescue the deficiency.109,110 An early study found orchiectomy of male mice to facilitate cutaneous wound healing through a reduced inflammatory response and TNF-α levels. The castrated and wounded hairless mice also experienced hair growth, a hallmark of epithelization and cutaneous healing. 111 In another animal study, topical blockade of ARs using flutamide improved wound gap closure rate, which was faster than that of testosterone-treated mice. 112 AR levels and androgen binding capacity are elevated in keloids compared to physiologic scars and uninjured skin. 113 Furthermore, young men and women with lower testosterone levels are found to have faster mucosal healing. 114
Among the specific androgen species, dihydrotestosterone (DHT) may be the primary effector on wound healing rather than testosterone. A group of mice that received 5α-reductase inhibitor treatment that blocked the conversion of testosterone to DHT demonstrated a similar rate of healing compared to castrated mice. 109 Paradoxically, androgens have shown benefits in wound healing in specific conditions, most notably in major burns. Although the mechanism is unclear, oxandrolone, an androgen analog, has shown effectiveness in promoting the healing of cutaneous burn wounds in humans. 115 This paradox likely reflects fundamental differences between typical cutaneous wounds and major burns, which combine local tissue injury with a pronounced systemic hypermetabolic–catabolic stress response. In both murine and human models, sufficiently large burns induce sustained elevations in catecholamines, corticosteroids, and pro-inflammatory cytokines like IL-1α, IL-6, and TNF, driving increased resting energy expenditure, whole-body protein catabolism, immune dysfunction, and delayed recovery.115,116 In this setting, androgen therapies like oxandrolone have been clinically associated with increased hepatic protein synthesis, improved lean body mass, and shortened length of hospital stay, supporting the idea that androgens may benefit burn recovery in part by counteracting systemic catabolism.115,117
Mechanistically, data on burn wounds in mice also suggest that androgens may also influence immune-cell activation and inflammatory turnover locally and systemically. Following burn injury, an elevated systemic inflammatory milieu characterized by increased cytokines, like IL-1α, IL-6, and TNF, and chemokine signaling, including MCP-1, promotes monocyte recruitment into circulation and into the wound bed.115,116 These monocytes differentiate into macrophages that support bacterial clearance and, after the resolution of inflammation, contribute to matrix deposition. In this context, DHT activation of AR on neutrophils, T cells, B cells, monocytes, and macrophages has been reported to accelerate the transition through inflammatory phases locally and systemically.115,116 Collectively, these observations suggest that androgens exert predominantly local, pro-inflammatory effects that can hinder repair in non-burn cutaneous wounds, yet in the distinct physiologic state of a burn injury, they may confer systemic metabolic advantages and potentially more efficient inflammatory cycling. Whether these divergent outcomes also reflect context-dependent differences in AR expression or downstream signaling is not well defined and thus warrants further study.
In contrast to the mechanism of estrogen on wound healing, testosterone is associated with a decreased ability to clear bacteria and an increased inflammatory response. 114 Locally, androgens act on ARs on skin and immune cells and enhance local inflammation by increasing IL-6 and TNF-α levels. TNF-α further activates NF-κB, which generates more TNF-α, forming a positive feedback cycle. There is evidence to show that ARs may limit wound inflammation by modulating the activity of the TNF-α/NF-κB loop.
In addition, there are cell-type-specific cytokine profiles in response to testosterone and DHT. Both testosterone and DHT increase macrophage IL-6 gene expression and reduce fibroblast IL-6 and TGF-β1 mRNA and protein levels. This indicates that androgens may have a more potent effect in inducing inflammation in early stages of wound healing, during which macrophages are more active, compared to the later granulation and remodeling stages of wound healing. 109
Furthermore, testosterone acts on the Suppressor of Mothers against Decapentaplegic transcription factor (Smad3) signaling pathway.109,118 Castrated mice without Smad3 demonstrate slower healing compared to castrated wild-type mice. 118 Another study showed testosterone to impair keratinocyte migration by activating Wnt-β catenin signaling and by inhibiting the TGF-β/Smad pathway. 112 As previously mentioned, researchers found DHT to significantly influence wound healing. Its action on the induction of macrophage-dependent IL-6 gene expression may be dependent on mitogen-activated protein kinase (MAPK)/phosphoinositide 3-kinase (PI3K) pathways. Crosstalk between Smad3, ARs, and MAPK/PI3K pathways can modulate DNA binding and transcriptional responses in androgen-dependent diseases such as prostate cancer and kidney disease.119–122
TREATMENTS TO MITIGATE AND LEVERAGE METABOLIC EFFECT ON WOUND HEALING
Hormone-based therapies provide a clinically relevant way to modulate metabolic and endocrine pathways that influence wound repair (Table 1). Therapies, like insulin, estrogen, and growth hormone, have been explored to support wound healing in complementary ways by inducing metabolic changes that shape the wound environment and by counteracting maladaptive metabolic states, like those seen in obesity and metabolic syndrome, that hinder repair. These therapies, in addition to innovative dressings and hydrogels, have been utilized in clinical settings and show promise in improving healing outcomes in acute and chronic wounds (Table 2).
Clinically relevant metabolic and endocrine factors influencing wound healing and associated management strategies
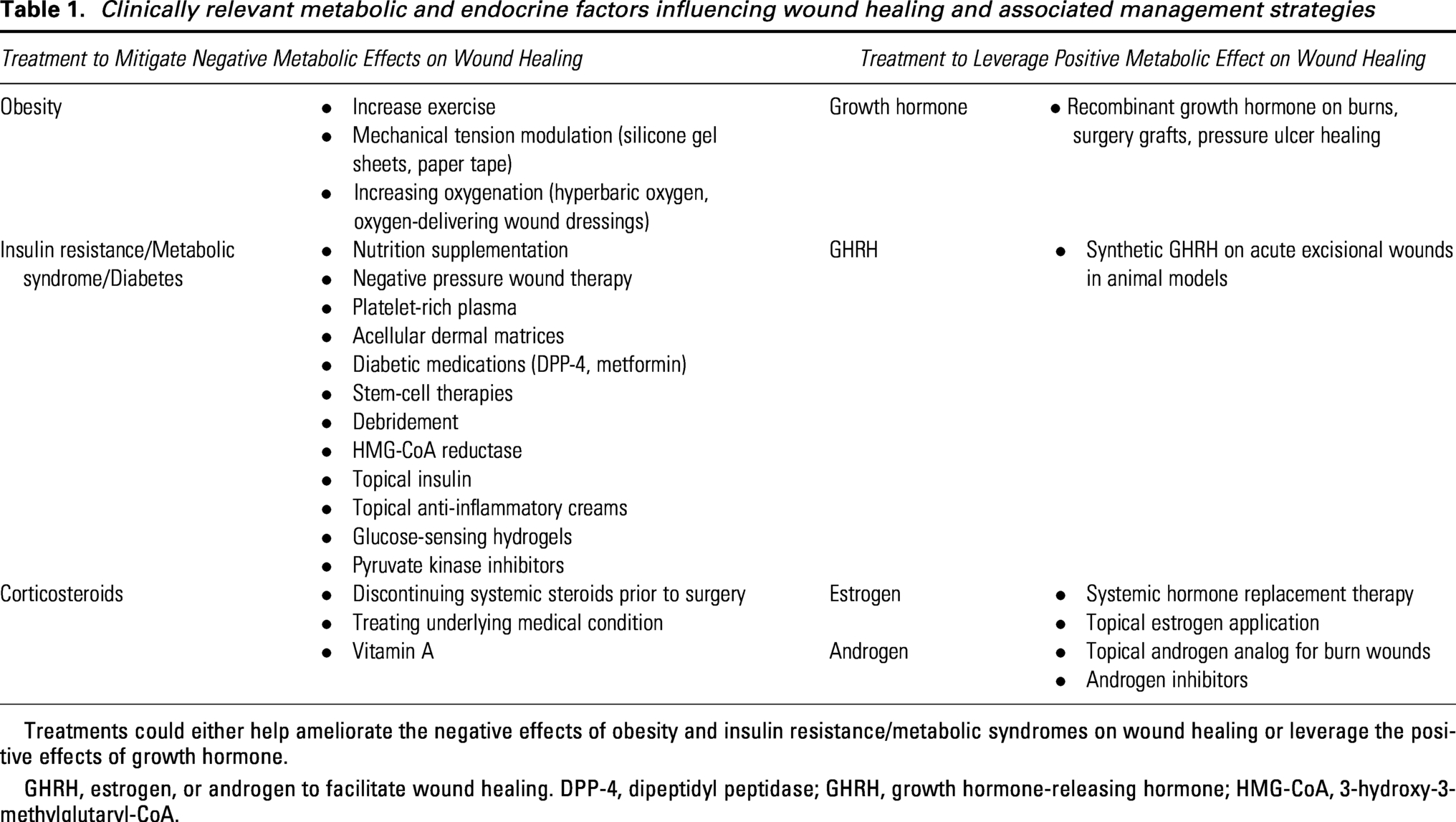
Treatments could either help ameliorate the negative effects of obesity and insulin resistance/metabolic syndromes on wound healing or leverage the positive effects of growth hormone.
GHRH, estrogen, or androgen to facilitate wound healing. DPP-4, dipeptidyl peptidase; GHRH, growth hormone-releasing hormone; HMG-CoA, 3-hydroxy-3-methylglutaryl-CoA.
Different types of wounds, possible interventions, and evidence level
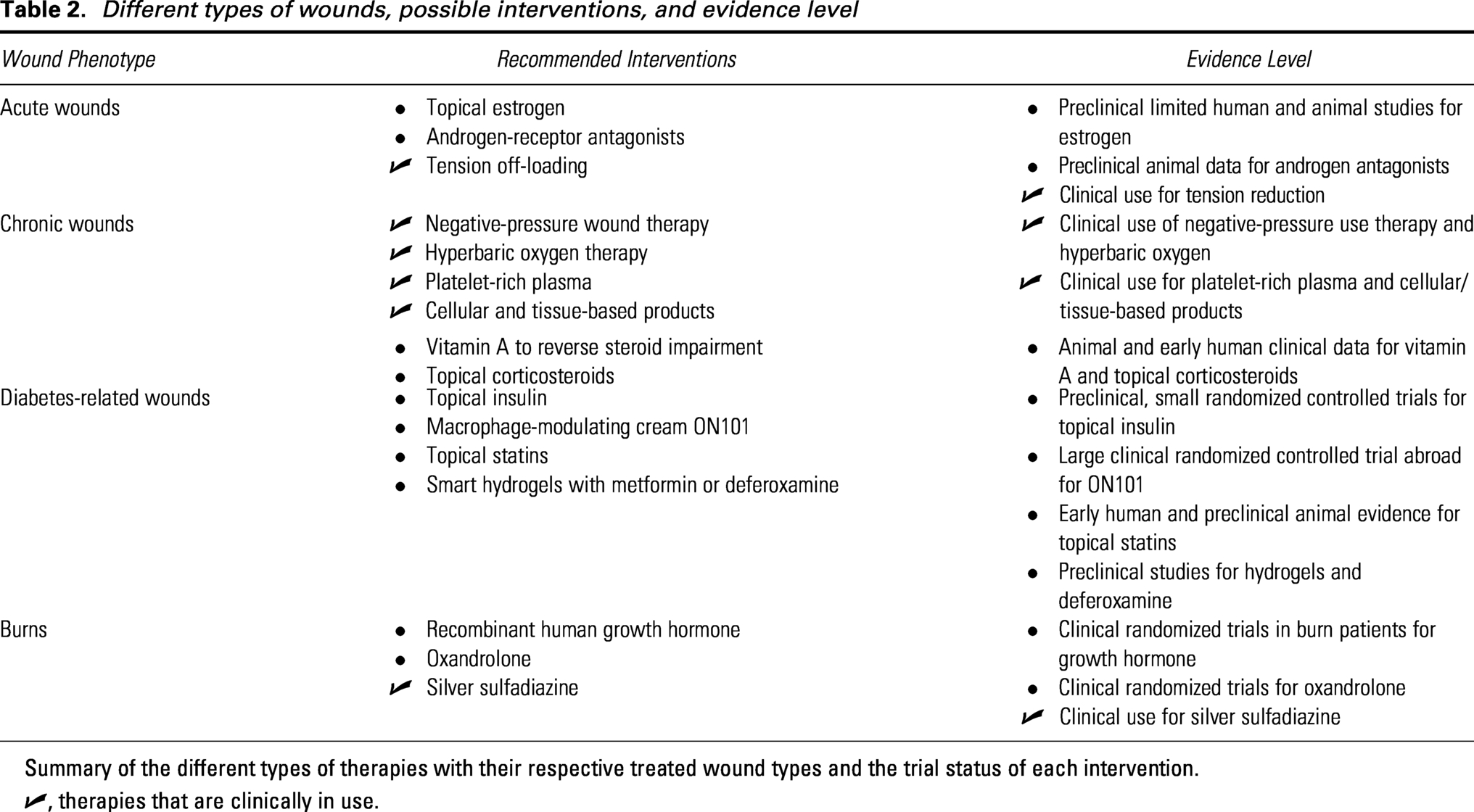
Summary of the different types of therapies with their respective treated wound types and the trial status of each intervention.
✓, therapies that are clinically in use.
Acute wounds
Topical estrogen can accelerate wound healing in acute wounds. In diabetic mice, topical estrogen was shown to induce more rapid re-epithelialization and angiogenesis in acute wounds (Fig. 3). 123 In human males and females undergoing excisional biopsies, topical estrogen treatment was similarly found to not only decrease the size of the wound but also increase collagen and fibronectin levels, as well as increase wound strength in 1 week. 94
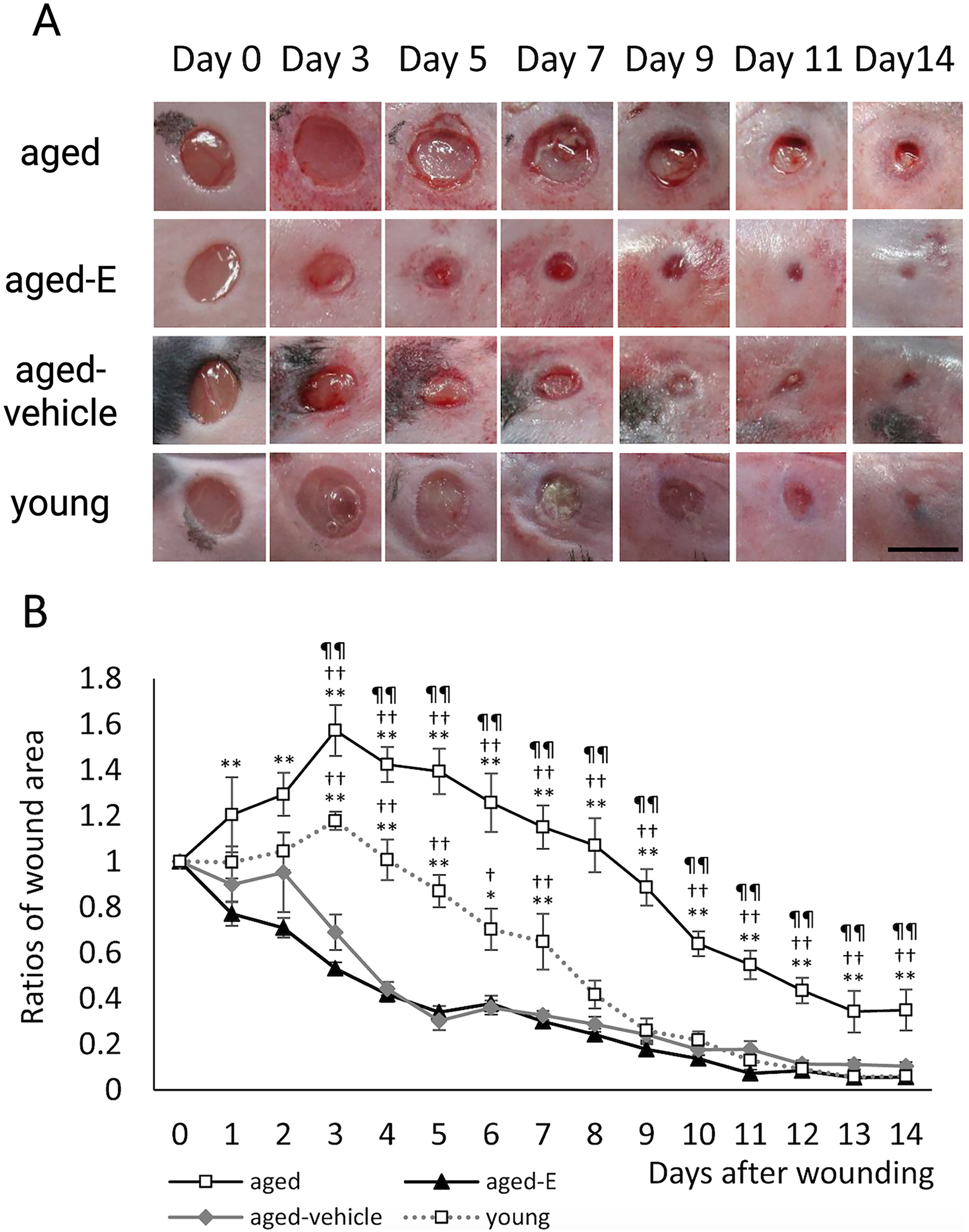
Topical estrogen accelerates excisional wound healing in mice.
Given the inhibitory effects of androgen on wound healing, it would follow that AR antagonists would improve wound healing. In mice, studies have found that after a week of flutamide application, an AR antagonist, wound re-epithelialization was significantly accelerated. 112 Another AR antagonist, ASC-19, loaded onto a dermal regenerative scaffold, facilitated the attachment of dermal fibroblasts and wound closure in mice compared to a collagen-only scaffold. 124
As previously mentioned, oxandrolone, a synthetic androgen, has interestingly demonstrated clinical utility among burn patients. Oxandrolone seems to help patients who are in hypermetabolic states to increase body mass, to synthesize protein, and to decrease the length of their hospital stay. However, more research is needed to more clearly elucidate the molecular mechanisms that may underpin the contradictory findings of topical androgens in cutaneous wounds and burns that have been observed. 117
Finally, as an anabolic hormone, recombinant growth hormone application to pressure ulcers in a human skin mouse model demonstrated twice as fast healing rate, as well as thicker skin. 82 In a burn mouse model, rhGH was found to accelerate the resolution of burn injuries in a dose-dependent manner. 81 Consistent with these findings, pooled clinical trial data on patients provided preliminary evidence that rhGH may accelerate re-epithelialization in burn wounds, reducing wound-healing time by approximately 9 days. Hospital length of stay was also found to be shortened. 125 However, it should be noted that many of these trials were composed of a small sample of participants, thereby limiting their statistical power. Interestingly, in human clinical trials where rhGH was used to treat injuries in severely burned children, rhGH was found to not increase the risk of scar formation in patients receiving daily subcutaneous injections. 126
Chronic wounds
Chronic wounds in diabetic patients are difficult to manage due to impaired vascularization and prolonged inflammation. In order to expedite healing for chronic wounds, the treatment plan should take a multidisciplinary approach. Along with the medical management of their condition, patients should incorporate nutritional supplementation and exercise to accelerate protein synthesis, improve oxygenation, decrease adipocyte storage, and decrease pro-inflammatory cytokines.127–129 Although treatments such as negative pressure wound therapy, platelet-rich plasma, and acellular dermal matrices have been used on chronic diabetic wounds, this section highlights treatments that specifically target metabolic dysregulation that underlies poor wound healing.
Several local interventions can mitigate the effects of increased mechanical tension and lack of oxygenation on wound healing from obesity. For instance, commercial silicone gel sheets and paper tape can be used to reduce mechanical tension on the surrounding skin of the wound. 130 Hyperbaric oxygen, which is Food and Drug Administration (FDA) approved, may be an option to deliver more oxygen to the wounded region. Innovative oxygen-delivering pressure dressings that use nanoenzymes to degrade hydrogen peroxide into oxygen, perfluorocarbons to directly deliver oxygen molecules, or hemoglobin-based oxygen carriers have also been developed, although their exploration has been limited to mouse models. 131
Statins have also demonstrated promise in facilitating chronic wound repair in diabetic patients. Statins inhibit the synthesis of cortisol, which, as established earlier, impairs wound healing. Statins further prevent the activation of glucocorticoid receptors, enabling transcription of pro-inflammatory cytokines and growth factors, which downstream stimulate keratinocyte migration and proliferation. 133 In particular, simvastatin promotes neovascularization by increasing VEGF and NO levels in the wound, while also reportedly possessing antibacterial properties.132,133
Insulin has demonstrated clinical benefit in promoting wound healing in both animal studies and human clinical trials. 134 In mice, topical low-dose insulin was found to shorten healing time in excisional wounds from 7 to 5 days, with insulin-treated groups demonstrating advanced infiltration and resolution of immune cells. 135 In diabetic rats, insulin cream applied for 8 days reduced wound-healing time through increased expression of VEGF and stromal cell-derived factor 1α, consistent with the paradigm that impaired inflammatory cell recruitment and impaired neovascularization drive pathology in diabetic wound healing. 136 Clinical data on humans have also yielded similar findings. In a double-blinded clinical trial of topical insulin for diabetic wound healing, 22 patients were randomly assigned to an insulin or placebo cream for 2 months, with the insulin-treated group yielding significant decreases in wound size and depth compared to patients receiving the placebo. 137 Another study examining eight chronic diabetic wound patients demonstrated greater angiogenesis and increased perfusion in the insulin-treated area of the wound. 137 Multiple studies have also reported significantly lower levels of AGEs in diabetic foot ulcers treated with topical insulin.138,139
It should be noted that these clinical studies were performed in patients with medically managed diabetes and not stratified by hemoglobin A1c. As such, the performance of topical insulin in patients with poorly controlled hyperglycemia remains uncertain, particularly because persistent systemic hyperglycemia can independently impair angiogenesis, immune function, and matrix remodeling. 140 Larger prospective studies stratified by glycemic control are needed to define effectiveness in patients with poorly controlled diabetes.
In addition to topical insulin, strategies to directly target the wound’s dysregulated glucose metabolism, either by modulating key metabolic pathways in resident cells or by sensing and responding to hyperglycemic wound conditions, are currently under development and being investigated in animal models. Shikonin, a natural compound extracted from the roots of Lithospermum erythrorhizon, has resulted in higher healing rates in rats by inhibiting the MAPK signaling pathway through reduction in phosphorylation of the ERK and c-Jun N-terminal kinases.141,142 Inhibition of the MAPK signaling pathway inhibits M1 polarization of macrophages, alleviating the inflammatory response initiated by these macrophages. 141 The higher healing rates observed in shikonin-treated rats were also accompanied by increased fibroblast and endothelial cell activity. 142 Liang et al. designed a pH and glucose-sensing, metformin-releasing hydrogel with adhesion and self-healing properties suitable for wear on the foot. 143 In a diabetic rat foot-wound model, application of the hydrogel resulted in a reduction in inflammation and improved angiogenesis, accelerating healing. Another “smart” hydrogel was engineered to detect elevated levels of glucose in the wound and, in response, release zinc ions, organic ligands, deferoxamine (DFO), and glucose oxidase. 144 Glucose oxidase degrades the excess glucose in the wound into hydrogen peroxide and glucuronic acid. DFO, an FDA-approved iron chelator, enhances HIF-1α activity by preventing iron-catalyzed degradation, promoting downstream angiogenesis (Fig. 4). 145 In fact, previous studies have documented the effectiveness of transdermal DFO patches to prevent pressure-induced diabetic wounds in mice. 145 When tested in diabetic mice, this hydrogel was observed to promote re-epithelialization, collagen deposition, and angiogenesis in their wounds. Furthermore, combining DFO with zinc ions demonstrated antibacterial activity in in vitro mouse studies. 144 While these hydrogel drug-delivery platforms have yet to be formally evaluated in humans, they represent an active area of research with translational potential as locally responsive therapies for diabetic wounds.
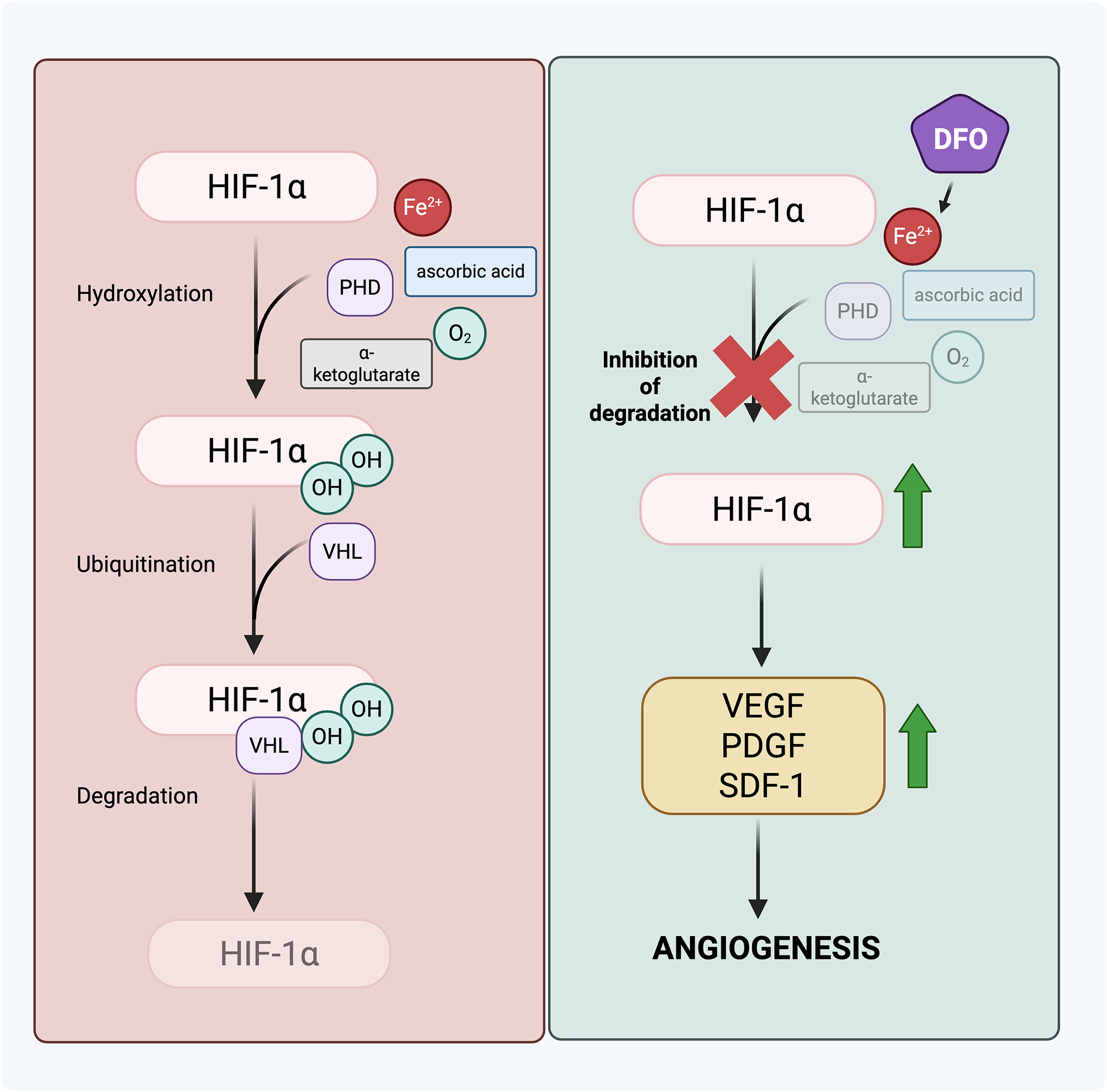
Mechanism of deferoxamine (DFO) in promoting angiogenesis. DFO binds to ferrous ions, which makes the prolyl hydroxylase domain (PHD) enzyme inactive and prevents the degradation of HIF-1α. The stabilization of the expression of HIF-1α leads to an increase in growth factors implicated in angiogenesis. HIF-1α, hypoxia inducible factor α; PDGF, platelet-derived growth factor; SDF-1, stromal cell-derived factor 1; VEGF, vascular endothelial growth factor; OH, hydroxyl group; VHL, Von Hippel-Lindau. Adapted and taken with permission from Shen et al. (2024). Created in BioRender. Yao, H. (2025) https://BioRender.com/aeb7e5k.
Other topical medications that more directly reduce inflammation during wound healing may also be effective in diabetic wounds. An international, multicenter randomized clinical trial tested ON101 cream on patients with diabetic foot ulcers. ON101 cream contains extracts derived from Plectranthus and Centella, which can suppress pro-inflammatory M1 macrophages while activating anti-inflammatory M2 macrophages. At the end of the 16-week trial, 60.7% of patients with diabetic foot ulcers in the ON101 group had complete re-epithelization of the wound compared to the 35.1% in the control group, who received only daily standard dressing changes (Fig. 5).146–148
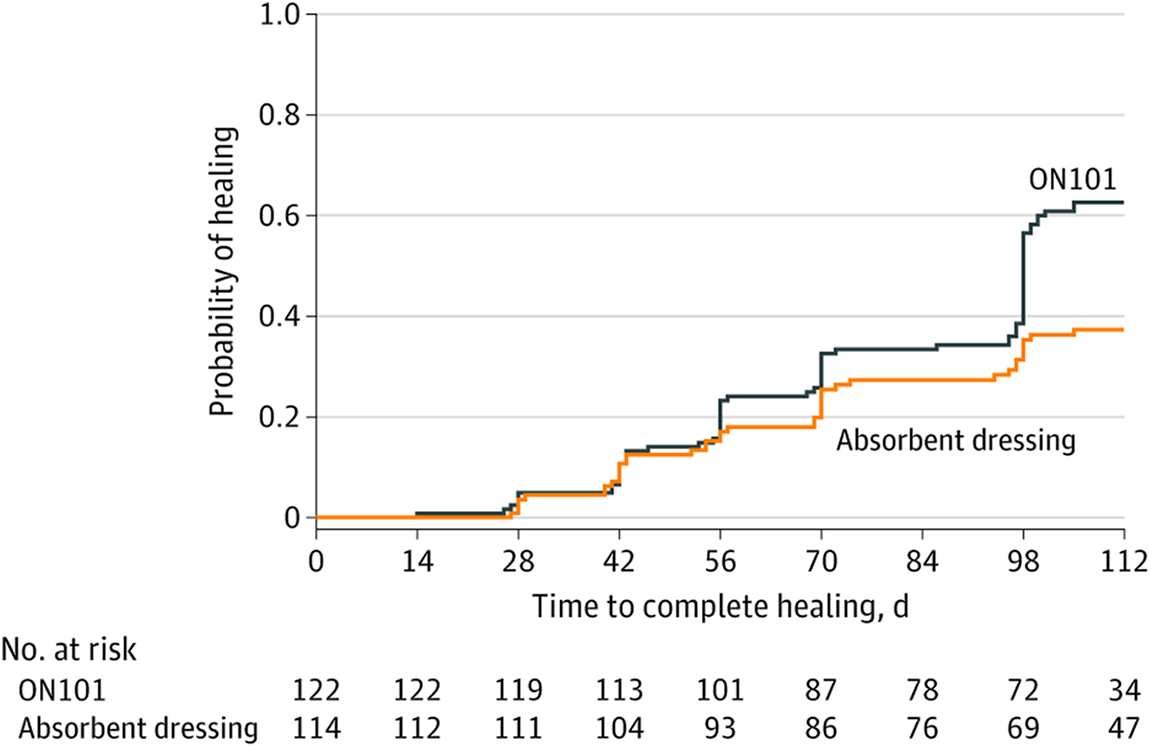
Healing rate of diabetic foot ulcers improves nearly 50% with ON101 cream. The wound healing curve shows the incidence of ulcers healed at each visit. Complete healing was defined as epithelialization without drainage observed at two consecutive visits. Patients in the ON101 group (n = 122) had a better healing rate than those in the absorbent dressing group (n = 114) (HR = 1.80 [95% CI: 1.23–2.65; P = 0.002]). CI, confidence interval. Adapted and taken with permission from Huang et al. (2021). 146
Finally, vitamin A has been shown to potentially reverse some of the inhibitory effects of glucocorticoids on wound healing. Vitamin A is crucial for healthy epithelial differentiation with its effects mediated through retinoic acid receptors. 149 Retinoids enhance TGF-β and IGF-1 expression and improve hydroxyproline content, thereby leading to increased tensile strength of healed scars.73,74,150 In fact, in a rat model of steroid-impaired wound healing, topical retinoids improved healing by restoring TGF-β and IGF-1 levels toward control levels. Furthermore, in a clinical study of human chronic leg ulcers, daily short-contact application of 0.05% retinoic acid for 4 weeks promoted visible granulation within 1 week and re-epithelialization in all patients by 4 weeks, with histologic evidence of new vessel formation and collagen deposition. 151 However, larger human studies are still needed to confirm efficacy, define optimal dosing regimens, and establish safety across different wound types.
SUMMARY
As the number of patients with metabolic dysregulation increases over time, wound healing in the setting of various metabolic conditions may become an increasingly relevant health care challenge. Metabolic changes to the body, whether due to obesity, insulin resistance, corticosteroids, or hormone excess or deficiency, can affect all stages of the wound-healing process. Notably, these conditions tend to impact local inflammation, angiogenesis, and collagen deposition. The specific mechanisms involve regulating cytokine and growth factor levels, affecting immune cell recruitment, and controlling specialized cell migration and apoptosis.
Treatments targeted for chronic wounds in general can also be useful for wound healing in patients with metabolic conditions. Treatments such as topical estrogen or recombinant growth hormone can leverage the beneficial effects of these compounds to accelerate wound healing, whereas treatments such as topical insulin and hydrogels can combat negative factors contributing to impaired wound healing. These treatments should be considered in conjunction with medical management to create the best environment for wound healing for the patient.
TAKE-HOME MESSAGES
Obesity, metabolic syndrome/insulin resistance, corticosteroids, and androgens generally impede wound healing, whereas growth hormone, growth hormone-releasing hormone, and estrogens promote wound healing. Topical androgens may promote wound healing in specific burn patients. Across these conditions, impaired wound healing reflects convergent metabolic mechanisms, such as through the production of lipotoxicity and AGEs, mitochondrial dysfunction and oxidative stress, dysregulated hypoxia signaling and angiogenesis, and impaired immune-cell clearance. In contrast, growth hormone and estrogen can promote wound repair by supporting anabolic processes and providing protection against oxidative stress. The pro-inflammatory state associated with androgens in cutaneous wounds may benefit burn injuries by countering systemic hypermetabolic catabolism. Clinicians should integrate local wound care with systemic metabolic and hormonal optimization and look at metabolite-responsive dressings or hormone-based topical therapies as adjuncts that target specific metabolic pathways contributing to impaired wound healing.
Footnotes
ACKNOWLEDGMENTS AND FUNDING SOURCES
All figures are created with BioRender.com. This work was supported by the National Institutes of Health (NIH) R01-GM136659 (M.T.L.), NIH U24DE029463 (M.T.L. and D.C.W.), Wu Tsai Human Performance Alliance (M.T.L.), NIH R01-DE032677 (M.T.L. and D.C.W.), NIH R01-AR081343 (M.T.L. and D.C.W.), and the Hagey Laboratory for Pediatric Regenerative Medicine (M.T.L. and D.C.W.).
AUTHORS’ CONTRIBUTIONS
H.Y.: Conceptualization, writing—original draft and review/editing, and visualization. S.L.J. and M.F.G.: Conceptualization and review/editing. D.S.K.-D. and J.G.T.: Review/editing (supporting). M.T.L.: Supervision (equal). N.E.L.: Conceptualization. D.C.W.: Supervision (equal), conceptualization, and review and editing.
AUTHOR DISCLOSURE AND GHOSTWRITING
M.T.L. is an inventor on patent application PCT/US2020/043717 that covers a machine learning algorithm for analysis of connective tissue networks in scarring and chronic fibroses and on patent 62/879,369 held by Stanford University that covers the use of YAP inhibition for wound healing. M.T.L. holds an equity stake in Neodyne Biosciences, Inc. and the early-stage medical device investment company TauTona. No competing financial interests exist. The content of this article was expressly written by the authors listed. No ghostwriters were used to write this article.