
Abstract
Introduction:
Congenital myopathies are a group of heterogenous inherited muscle diseases. With advances in genetics, newer genes with novel features are being described. Pyridine nucleotide-disulfide oxidoreductase domain 1 (PYROXD1) related myopathy is an ultrarare congenital myopathy. Only few cases have been reported worldwide till now. We report the first interesting case of PYROXD1 related myopathy from India.
Methods:
This is a retrospective study done from a quaternary neurology referral centre from southern India. All clinical, laboratory and electrophysiological data were collected from the medical records. Institutional ethics approval and informed consent from patient were obtained.
Results:
A 9 year-old-boy of non-consanguineous parentage presented with progressive fatigable proximo-distal weakness of upper and lower limbs with facial weakness from the age of 4 years. This was followed by chewing and swallowing difficulty. However speech was normal. There was profound proximal and distal joint hyperextensibility along with hip and ankle contractures. There was facial dysmorphism with high arched palate and retrognathism. Investigations showed normal serum creatine kinase levels. Nerve conduction studies showed axonal sensorimotor neuropathy. There was significant decremental response in tibialis anterior. Muscle biopsy showed both myopathic and neurogenic changes with novel findings of mitochondrial aggregates in subsarcolemmal and perinuclear regions. Next generation sequencing revealed a missense variant NM_024854.5:c.394C > T (NP_079130.2:p.Arg132Cys) of uncertain significance in exon 4 of PYROXD1 gene.
Conclusion:
This is the first report of PYROXD1 related myopathy from India. There were novel features of muscle fatigability, contractures, novel muscle biopsy features and a variant of uncertain significance expanding the phenotypic and genotypic spectrum of this rare myopathy.
Keywords
Introduction
Congenital myopathies (CM) are heterogenous group of inherited muscle disorders with usually childhood onset. CM is commonly classified based on the histopathological features into most common ones such as core, nemaline, centronuclear, myosin storage and congenital fiber type disproportion. 1 With expanding genes causing congenital myopathies, pyridinenucleotide-disulfide oxidoreductase domain 1 (PYROXD1) related myopathy is identified as an ultrarare congenital myopathy. 2 To date only 18 families have been described in literature on PYROXD1 related myopathy. Here we describe the first interesting case of PYROXD1 related myopathy from India.
Methods
This is a retrospective case report from the neuromuscular division of the quaternary neurology referral centre in south India. Detailed clinical and laboratory reports were collected from the patient records. Clinical electrophysiology studies including nerve conduction study and repetitive nerve stimulation of trapezius, anconeus, abductor digiti minimi, quadriceps and tibialis anterior muscles were performed. Cardiorespiratory evaluation such as electrocardiography, echocardiography and pulmonary function tests were done. Muscle biopsy from left biceps was carried out with histopathological analysis using stains such as hematoxylin and eosin, modified Gomori's trichrome, Nicotinamide adenine dinucleotide (NADH), Succinate dehydrogenase (SDH), ATPase, Cytochrome C oxidase (COX) stains and electron microscopy.
Genetic diagnosis: Done by Massively Parallel Sequencing (Next Generation Sequencing) - Genomic DNA from the submitted specimen was enriched for complete coding regions and splice site junctions of genes (Supplementary Table 1) using a custom bait- capture system. Paired End Sequencing was performed with 2 × 100/2 × 150 chemistry, on an Illumina platform. Reads were assembled and were aligned to reference sequences based on NCBI RefSeq transcripts and human genome build GRCh37/UCSC hg19. Data was filtered and analyzed to identify variants of interest and interpreted in the context of a single most damaging, clinically relevant transcript for the purpose of the report, indicated as a part of variant details. Enrichment and analysis focus on the coding sequence of the indicated transcripts, 5–10 bp of flanking intronic sequence, and other specific genomic regions demonstrated to be causative of disease at the time of assay design.
Promoters, untranslated regions, and other non-coding regions thought to be significant were interrogated by Sanger backfill. Sequence and copy number variants are reported according to the Human Genome Variation Society (HGVS). Clinvar, OMIM, HGMD, UCSC genome browser, Uniprot, Ensembl, dbSNP, gnomAD, ExAC, Pubmed, Dgap, icgc, Kaviar, various bioinformatics analysis, predictive tools and disease specific databases used as available and appropriate.
Sanger validation and segregation analysis: DNA was extracted from peripheral blood using Qiagen (QIAamp DNA Minikit) kit. PCR performed using primers designed to amplify the genomic region spanning the targeted variant in the exon. PCR products were confirmed by gel electrophoresis followed by treatment with ExoSAP to digest unutilized primers. The amplicons were subjected to cycle sequencing PCR using BigDye Terminator.
Informed consent was obtained from patient and his parents for publishing the clinical data and photographs with face recognition. Institutional ethics committee approval was obtained for the study (IEC no: NIMH/DO/(BS&NS) 2022).
Case report
A nine year-old-boy born of non-consanguineous parentage was evaluated in 2022. He presented with fatigable proximal limb weakness for 5 years. His perinatal period was uneventful. Child was noticed to be slow since he started walking at the age of 2 years and has never participated in sports. He initially developed difficulty in rising from the floor and climbing stairs followed by difficulty in doing overhead activities. He also developed toe walking with frequent falls. There was recurrent shoulder dislocations with routine movements. By age of 6 to 7 years he developed chewing and swallowing difficulties. There were no visual, auditory or cognitive symptoms. There was no significant family history.
On examination the boy had a slender body habitus along with facial dysmorphism (Figure 1(a),(b)) including high arched palate and retrognathism. He also had hyperextensible proximal and distal joints. At the shoulder joints the head of humerus could be easily dislocated with minimal movements (Figure 1(e)). There was bilateral scapular winging (Figure 1(f)) with hip flexors and tendo-achilles contractures. He had bi-facial weakness. The palatal and pharyngeal movements to gag reflex was reduced. The muscle power assessment according to modified Medical Research Council (MRC) grading showed upper limb (proximal and distal): 4, iliopsoas: 3, gluteus maximus:4, hip abductors: 3, hip adductors: 4, quadriceps: 3, hamstrings: 4+, tibialis anterior: 2, gastrocsoleus: 4. There was significant fatigability in arm abduction and foot drop. All tendon reflexes were hypoactive. Clinically the possibilities of congenital myopathy or limb girdle muscular dystrophies were considered. Investigations showed normal serum creatine kinase level of 100 IU/L. Nerve conduction studies showed reduced compound muscle potential amplitude of common peroneal nerve and absent sensory potential in superficial peroneal nerve suggestive of sensorimotor axonal neuropathy. Repetitive nerve stimulation showed a decremental response of 25% in tibialis anterior muscle (Figure 1(g)). Echocardiography was normal and pulmonary function tests showed moderate restrictive pattern. Muscle biopsy done from left biceps brachii showed myopathic features with desmin condensation and myofibrillar disarray. There was also thin subsarcolemmal accumulation of red granular material in modified Gomori's trichrome stain. Electron microcopy showed variable mitochondrial size in subsarcolemmal and perinuclear regions with myelinic figures in subsarcolemmal areas (Figure 2). Next generation sequencing showed a homozygous variation of uncertain significance in exon 4, c.394C > T (p. Arg132Cys) in PYROXD1 gene (chr12:g.21602605C > T, NM_024854.5) which was further confirmed by Sanger sequencing. The missense variant NC_000012.12 (NM_024854.5): c.394C > T (Genomic position: NC_000012.12:g.21602605C > T) located at position 109 of 129 (coding) has been reported as a variant of uncertain significance in ClinVar (Accession ID: VCV001934646.2, LOVD Submission: 00448529). The p.Arg132Cys variant is novel (not in any individuals) in 1kG All. The p.Arg132Cys variant is observed in 4 / 1459992 (Allele Frequency - 0.00000273974) alleles in gnomAD v4.0 Exomes and 1 / 152052 (Allele Frequency - 0.0000065767) alleles in gnomAD Genomes only in heterozygous state. Further, the variant was not found in GenomeAsia or Indigene databases. There is a large physicochemical difference between arginine and cysteine, which is likely to impact secondary protein structure as these residues differ in polarity, charge, size and/or other properties (Figure 3). The p.Arg132Cys missense variant is predicted to be damaging by both SIFT and PolyPhen2. The arginine residue at codon 132 of PYROXD1 is conserved in all mammalian species (Figure 4(c)). The nucleotide c.394 in PYROXD1 is predicted conserved by GERP++ and PhyloP across 100 vertebrates. The phenotype in the proband matches with that of the disorder (with additional novel features) caused by pathogenic variants in the gene PYROXD1. For these reasons, this variant has been classified as a variant of uncertain significance (ACMG Criteria - PM2_Supporting PP2 PP3 PP4_Supporting) - PM2_Supporting - The p.Arg132Cys variant is observed in 4 / 1459992 (AF - 0.000002740) alleles in gnomAD v4.0 Exomes and 1 / 152052 (AF - 0.000006577) alleles in gnomAD Genomes only in heterozygous state;
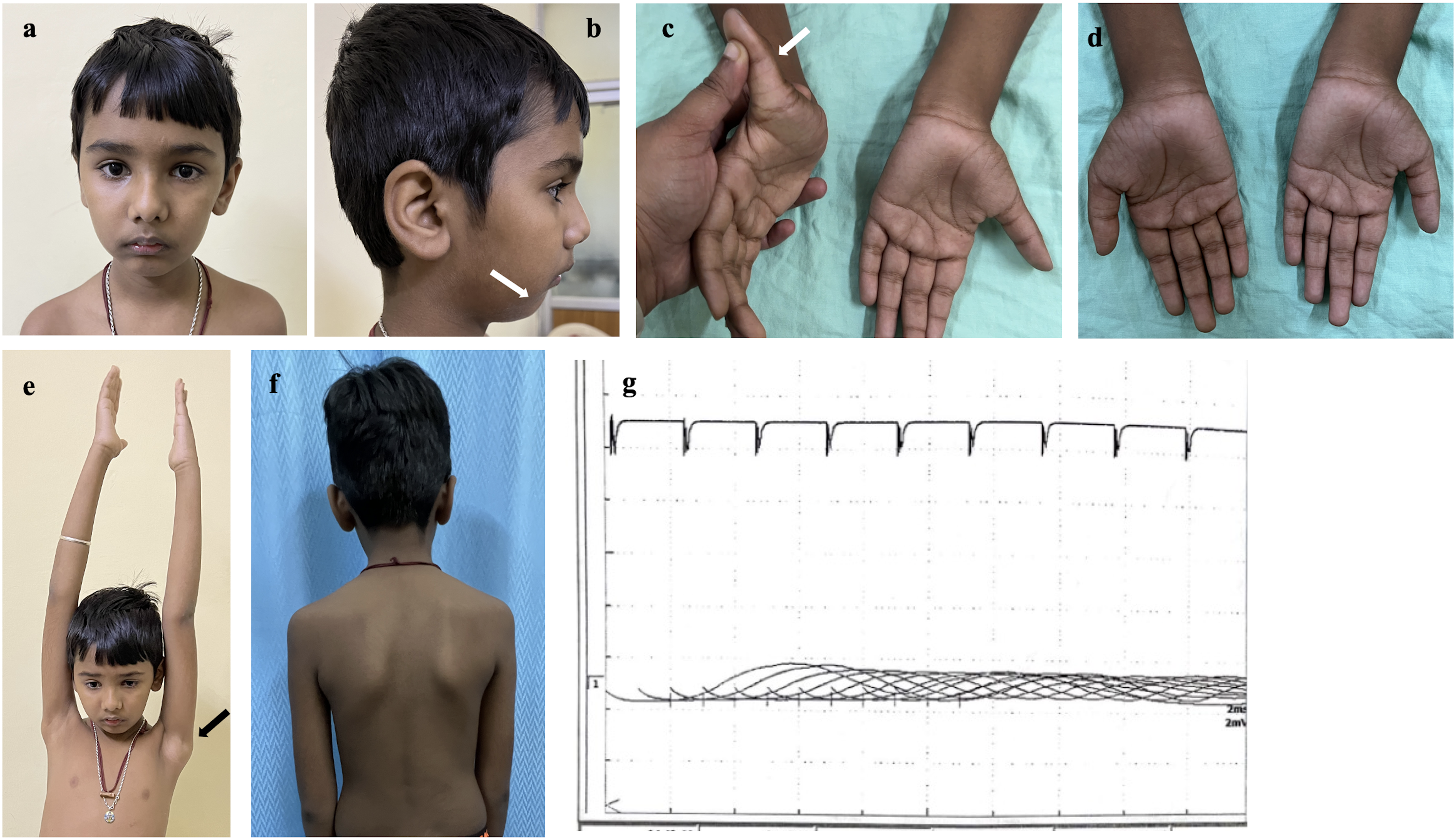
Clinical images and repetitive nerve stimulation of the patient. (a and b) retrognathism (arrow in b). (c) hyperextensile finger joints (arrow). (d) wasting of small muscles of the hand. (e) shoulder joint dislocation (black arrow). (f) bilateral scapular winging. (g) repetitive nerve stimulation at tibialis anterior showing significant decremental response of about 25%.
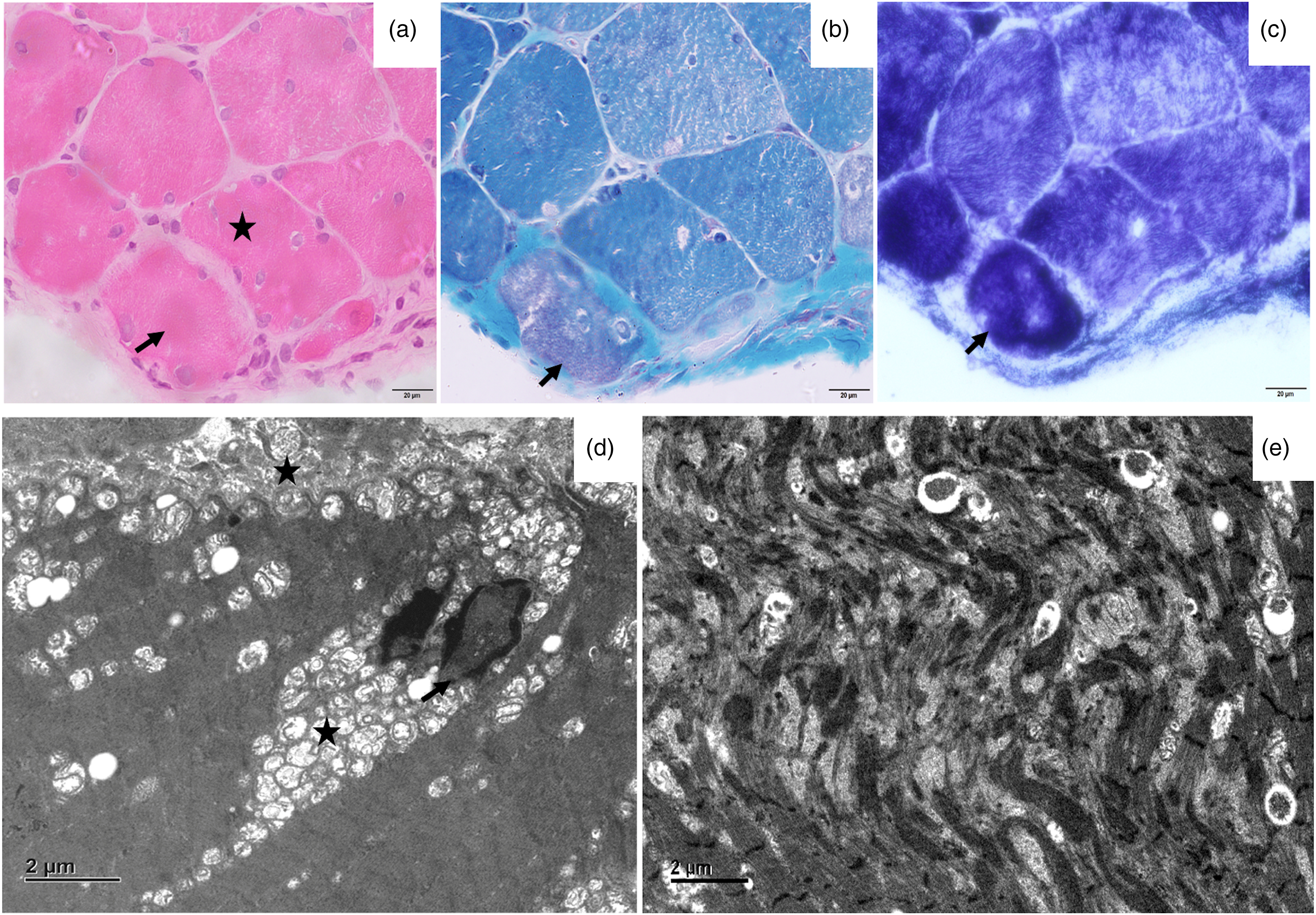
Histopathology images of open muscle biopsy (biceps brachii): a–c, Transverse section of skeletal muscle shows marked variation in fiber size, multiple internalized myonuclei (*). Few fibres show eosinophilic sarcoplasmic inclusions (arrow, a) which appear reddish on MGT stain (arrow, b). These fibers are intensely stained on NADH-Tr (c, arrow). d: Electron micrographs show abnormal nuclei (arrow) and numerous small mitochondria in aggregates (*) in the perinuclear and subsarcolemmal region (x1500). e: Large areas of disorganization of the myofibrils with formation of Z-band aggregates seen (x1500).

Representation of the 3D structure of the PYROXD1 protein (UniProt ID: Q8WU10) along with p.Arg132Cys amino acid substitution. The structures were analysed by Missense3D tool using the avaible X-ray structure of the protein (PDB ID: 6ZK7). The bottom panel represents expanded view of the location of the substitution in the protein.

(a) Chromatograms of mother, father and proband showing the variant in Exon 4 of PYROXD1 gene. (b) Spectrum and location of mutations reported in PYROXD1 gene in literature.The exons are represented as rectangular boxes with curved edges with respective exonic numbers with non-coding regions shaded in black and grey at the ends. The variant observed in this study is shaded in green. Triangles represent missense, hexagons represent intronic, square represent inframe deletion and circles represent frameshift variations. Size of exons /introns is not represented at scale. (c) Multiple sequence alignment shows the p.R132 amino acid residue to be conserved across species.
Of the 40 cases of congenital myopathy cases genetically diagnosed at our centre between 2016–2022, only one is of PYROXD1 related myopathy.
Discussion
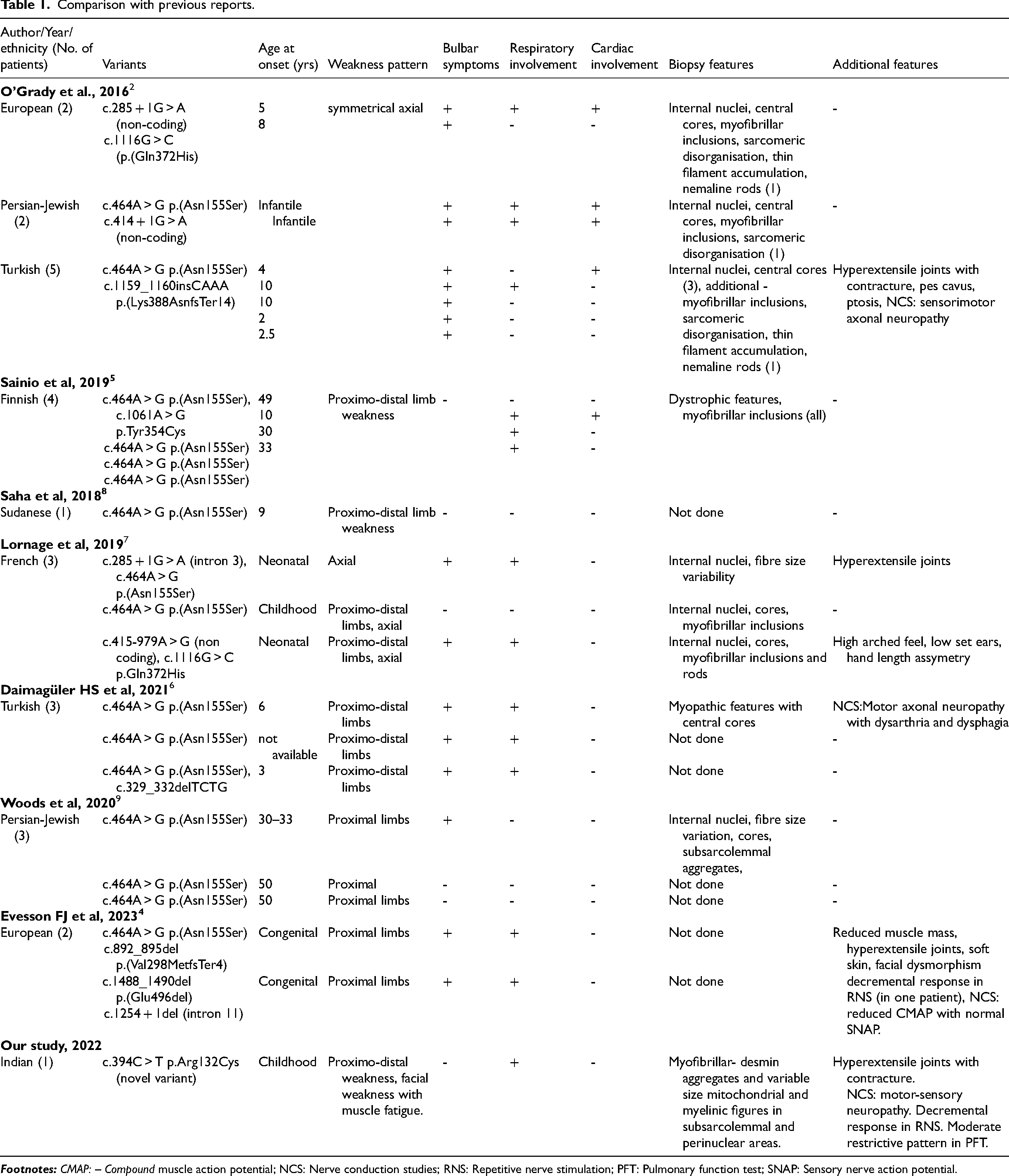
PYROXD1 (pyridine nucleotide-disulfide oxidoreductase domain containing protein 1) is expressed in many tissues including skeletal muscle with nuclear and cytosolic localization. It is encoded by PYROXD1 which is located in chromosome 12p21.1. 2 PYROXD1 acts as an oxidoreductase, potentially implicated in energy metabolism. Pyridine- nucleotide-disulphide-reductases (PNDRs) are flavoproteins (FAD binding) and catalyze the pyridine nucleotide (NAD/NADH)-dependent reduction of cysteine residues in their substrates. These PNDRs are involved in various complex reduction reactions involving electron transfers using FAD and NAD thus, playing an important role in energy metabolism. 3 The previously described phenotypes in PYROXD1 related muscle diseases include early onset myopathy with multiple internalised nuclei, cores and rods 2 and limb-girdle muscular dystrophy.4,5 We report the first case of PYROXD1 related myopathy from India. Our patient presented with typical features of this rare myopathy as previously described with childhood onset limb girdle weakness with hyperextensible joints.6,7 Significant contractures as noted in our patient is not described in previous studies.2,3 There was no ocular involvement but dysphagia was seen in our patient in contrast to previous reports were ophthalmoparesis and bulbar involvement were noted.2,3,7 Recently, in 2023, Evesson FJ et al., reported two European patients with congenital onset myopathy with bulbar symptoms. These patients had prominent connective tissue features like blue sclera, osteopenia, hyperextensible joints and soft skin. Significant descremental response to RNS in abductor digiti minimi was noted in one patient though without any response to Pyridostigmine and Salbutamol. The second patient had partial response to Pyridostigmine. 4 Our patient had fatigability of proximal upper limbs and distal lower limbs with significant decremental response in tibialis anterior on RNS. This signifies the possible involvement of neuromuscular junction involvement in addition to myopathy and connective tissue in PYROXD-1 related myopathy. The proposed pathophysiology is likely due to reduced oxido-reductase activity that may result in increased sensitivity of muscle cell to oxidative stress resulting in mitochondrial dysfunction and impaired energy metabolism. 2 Respiratory involvement by reduced vital capacity is reported by Lornage et al. 7 However, our patient presented with moderately restrictive pattern in pulmonary function tests but was asymptomatic. Muscle biopsy findings showed additional findings apart from myopathic features and myofibrillar aggregates that are described previously.5,7 However, there were no common features of central nuclei, cores, rods and myofibrillar disarray.2,3 Our patient's biopsy also showed subsarcolemmal and perinuclear accumulation of mitochondria with myelinic figures which is a novel finding. The most common variant reported is c.464A > G which is the Turkish founder mutation but also reported in other ethnicities. 3 Previous reports have shown that patients with pathogenic variant c.464A > G in homozygous or compound heterozygous state usually present with limb-girdle muscular dystrophy like phenotype.2,5,8 In contrast, patients with splice site pathogenic variations tend to have onset in infancy with joint hypermobility, contractures, scoliosis and hand-feet anomalies.2,7 However, our patient showed a novel missense variant of c.394C > T in exon 4 of PYROXD-1 gene showing intermediate features between Turkish founder missense variation and splice site variation with early onset, contractures and joint hypermobility along with myasthenic muscle weakness. Comparison of this case with previous reports is presented in Table 1.
Comparison with previous reports.
Conclusion
This is the first case of PYROXD-1 related myopathy from India with novel clinical features of significant muscle fatigability, joint contractures, joint hypermobility with missense mutation expanding the phenotypic and genotypic spectrum. Thus, its prudent to consider neuromuscular junction involvement testing in patients with suspected PYROXD-1 related myopathy with prominent connective tissue features.
Supplemental Material
sj-docx-1-jnd-10.1177_22143602241301635 - Supplemental material for A rare case of myopathy with fatigability due to PYROXD1 variation
Supplemental material, sj-docx-1-jnd-10.1177_22143602241301635 for A rare case of myopathy with fatigability due to PYROXD1 variation by Dipti Baskar, Aneesha Thomas, Vijay Kumar Boddu, Rashmi Santhoshkumar, Ram Murthy Anjanappa, Saraswati Nashi, Kosha Srivastava, Kiran Polavarapu, Gautham Arunachal, Ananthapadmanabha Kotambail, Bhoomika Rao, Anita Mahadevan, Atchayaram Nalini and Seena Vengalil in Journal of Neuromuscular Diseases
Footnotes
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.