
Abstract
Myasthenia Gravis (MG) is a heterogeneous neuromuscular autoimmune disorder characterized by fluctuating skeletal muscle weakness and a highly variable disease course. MG subgroups are defined by antibody type, age at onset, clinical phenotype, and thymus pathology. Given the unpredictable disease course, disease-specific objective biomarkers are needed to enable personalized treatment strategies and improve clinical trial outcomes beyond conventional clinical scales. Biomarkers are measurable indicators of physiological processes, disease states, and therapy responses. Despite significant advances in MG diagnostics and therapeutics, predictive biomarkers for personalized treatment remain underdeveloped. This review explores the progress and challenges in identifying blood-based biomarkers for MG, highlighting their potential applications in diagnosis and disease monitoring. Established diagnostic blood biomarkers include autoantibodies against acetylcholine receptors (AChR) and muscle-specific tyrosine kinase (MuSK), which confirm MG diagnosis and guide initial treatment decisions. Prognostic biomarkers, such as microRNAs (miR-150-5p and miR-30e-5p), show promise in predicting disease progression. Pharmacodynamic biomarkers, including CD20+ B cell counts, may enhance treatment precision for therapies like Rituximab. Furthermore, emerging research on metabolites, T and B-cell markers, complement factors, and proteomics offer new avenues to refine MG subtyping and identify molecular signatures predictive of treatment response to novel immunosuppressants. While the journey toward clinically useful blood biomarkers in MG remains complex, ongoing collaborative efforts within the MG research community hold the potential to revolutionize disease management. Future studies integrating multi-omics approaches, large-scale longitudinal cohorts, and disease controls will be critical to translating these biomarkers from research into routine clinical practice.
Overview of myasthenia gravis and potential blood biomarkers
Myasthenia Gravis (MG) is an autoimmune disease characterised by autoantibodies against postsynaptic receptors, such as nicotinic acetylcholine receptors on the postsynaptic muscle membrane at the neuromuscular junction (NMJ), causing neuromuscular transmission failure and fluctuating fatigable weakness of skeletal muscles. One major issue in the clinical follow-up of patients with MG, as well as in clinical trials, is the disease heterogeneity due to several subgroups. These subgroups are somewhat arbitrarily defined based on clinical characteristics (clinical phenotypes and age at disease onset), antibody status, and thymus-related pathologies. 1 The clinical phenotype is typically divided into ocular or generalized, and most patients present ocular but later develop generalized fatigable muscle weakness. Based on age at onset, MG can be divided into juvenile MG (age 0–18 years), early-onset MG (EOMG; age 19–50 years); late-onset MG (LOMG; age 51–65 years) and very late-onset (VLOMG; older than 65 years). 2 The most frequent antibody subgroup consists of patients who are acetylcholine receptor antibody seropositive (AChR+), approximately 85% of patients with generalized disease, and thereafter comes the group of patients seropositive for muscle-specific tyrosine kinase antibodies (MuSK+), approximately 30–60% of patients who are seronegative for AChR antibodies. 3 The last group refers to patients with thymus pathology, mainly those with a thymoma. To further complicate the group divisions, there are also combinations of features based on pathophysiological signatures. For example, most AChR+ EOMG are young women with thymic hyperplasia, which is not common in the LOMG 4 group, where men are predominantly affected.
Given this heterogeneity, complexity, and unpredictability in MG, disease-specific objective biomarkers are an urgent need in the follow-up and in predicting disease activity to enable personalized treatment strategies and improve clinical trial outcomes beyond conventional clinical scales, as novel immunosuppressive therapies are emerging in the MG clinical trial field. Identifying specific and sensitive biomarkers in MG can also enhance our understanding of key immunopathological pathways, both in antibody-seropositive and seronegative MG, and support the validation of markers to predict and monitor treatment response. In the era of omics, such biomarkers may enable molecular stratification of MG patients and guide personalized therapies.5–7
Currently, patient-assessed scales such as the MG-Activities of Daily Living (MG-ADL) and clinician-assessed scales, including the MG Composite (MGC) or Quantitative MG (QMG) scales, are used as primary outcomes in most clinical trials. Ideally, blood biomarkers would separate MG patients from patients with other neuroimmune disorders and healthy individuals. Furthermore, those responsive to different treatment options could be utilized as pharmacodynamic biomarkers, indicating a direct or indirect link to the underlying disease mechanisms. Such blood biomarkers include antibodies, cells, proteins, metabolites, or microRNA involved in the autoimmune response, a hallmark of MG (Figure 1).
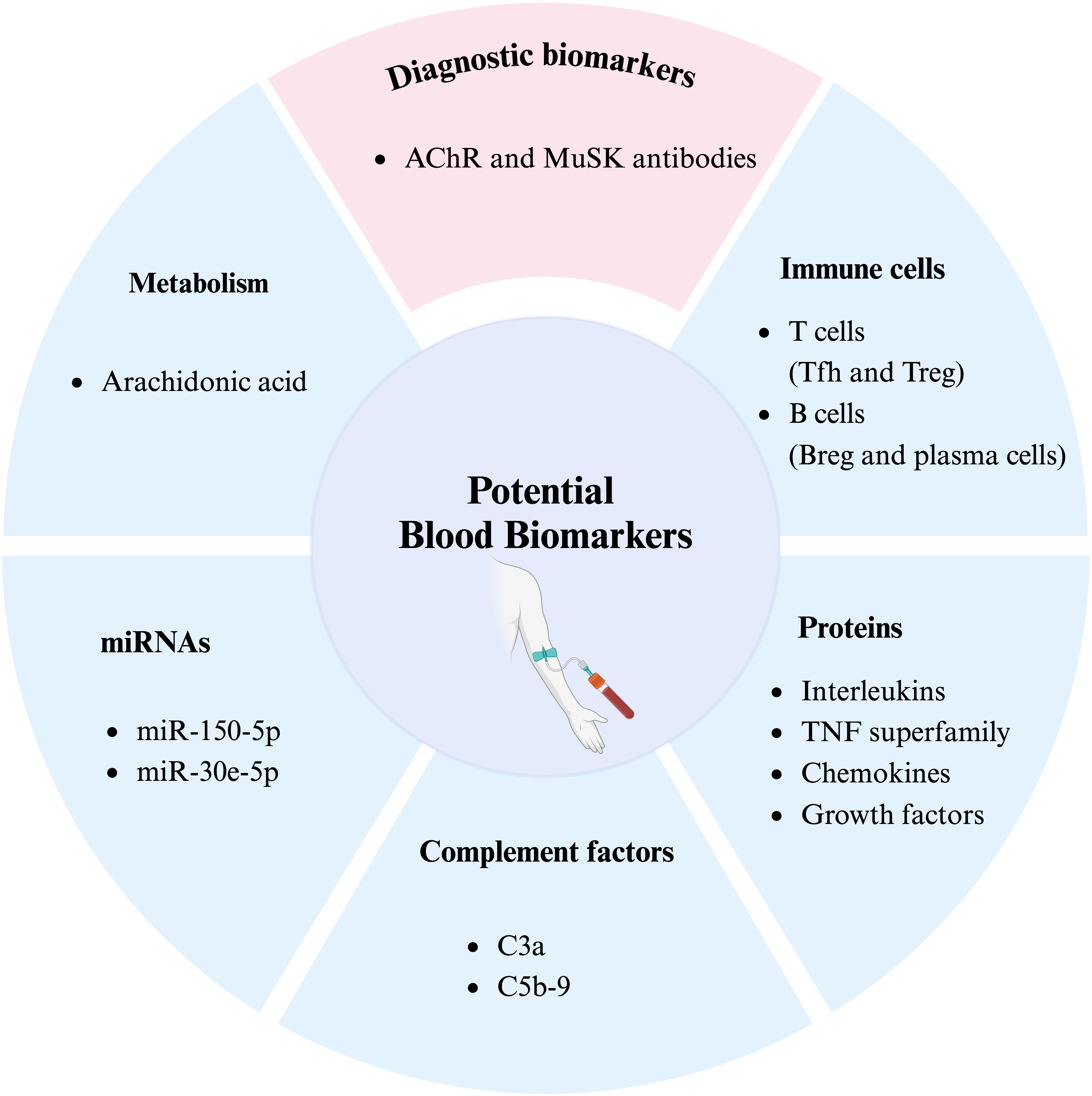
Potential future blood biomarkers in myasthenia gravis. Based on current studies, the different categories of blood biomarkers, including metabolism, immune cells, proteins, microRNAs, and complement factors, could be validated as disease-specific biomarkers MG. AChR, acetylcholine receptor; MuSK, muscle-specific tyrosine kinase; miRNA, microRNA; Tfh, T Follicular Helper cells; Treg, regulatory T cells; Breg, regulatory B cells.
Methodology: search strategy and selection criteria
A comprehensive literature search was conducted in PubMed on January 11, 2025, and repeated on May 1, 2025, during the revision phase, using a combination of MeSH terms and free-text keywords. The search strategy included the terms “biomarker” AND “myasthenia gravis.” Although no strict lower limit was applied for the publication date, priority was given to studies published within the past 10 years that reported original data and employed any form of primary study design, including clinical trials, cohort studies, and case-control studies. Only articles published in English and available in full text were considered for inclusion.
Antibodies
Although antibodies against AChR and MuSK are promising diagnostic tools in MG, and frequently, a decrease in their levels is reported as a measure of clinical improvement in response to immunosuppressants or thymectomy, they do not accurately correlate with the severity or treatment responses. Some studies have correlated AChR antibody titers with the QMG score and MGFA class, a clinical classification according to the Myasthenia Gravis Foundation of America (MGFA) 8 , at the group level but not individually. In contrast, other studies suggest that no such correlation exists. 9
In MG patients with a more severe disease phenotype, additional antibodies against the low-density lipoprotein receptor-related protein 4 (LRP4), the potassium channel Kv1.4, the ryanodine receptor, or titin may be present alongside anti-AChR or MuSK antibodies.5,10 The co-occurrence of LRP4 and titin antibodies, particularly in conjunction with AChR or MuSK antibodies, is often associated with more complex disease mechanisms, including a thymoma, VLOMG, or overlap with myositis.11–14 These antibodies recognise antigens expressed in cardiac and skeletal muscle tissue and may serve as valuable prognostic biomarkers in selected clinical contexts.5,10,11,14
A recent systematic literature review, based on 42 eligible studies on MG antibodies, concluded that it is currently not recommended to use routine antibody-level testing to guide treatment decisions or as a biomarker, 15 due to the limited and inconsistent evidence. This review was supported by an extensive study on longitudinal serum samples and datasets from AChR+ MG, demonstrating that MG-ADL or QMG scores do not correlate with AChR antibody titers, 16 proving that antibody titers are unreliable as predictive biomarkers for severity in MG.
Immune cells
Altered immune cell frequencies and dysregulated immune functions contribute to the pathogenesis of autoimmune disorders, including MG. MG is a thymus-associated, T-cell-mediated, and B-cell-driven autoimmune disease. Table 1 provides a comprehensive overview of immune cell subset alterations across MG subtypes and their associations with clinical scores, other immune cell populations, or serum protein levels. The phenotypic markers listed in the table refer to those used in flow cytometry experiments to identify specific immune cell subtypes. Since different studies employ varying marker combinations for cell identification and quantification, this variability can lead to heterogeneous or even contradictory findings for the same immune cell subset across publications. To enhance consistency and reproducibility in MG research, it would be strongly recommended to adopt standardized flow cytometry panels, such as those established by the Human Immunology Project Consortium, for T and B cell subtype phenotyping. 17 We have excluded total RNA and/or mRNA sequencing data on immune cells from this review, as immune cell identification relies on protein expression rather than transcription alone. 18
Altered frequencies of circulating lymphocyte subtypes in human MG.
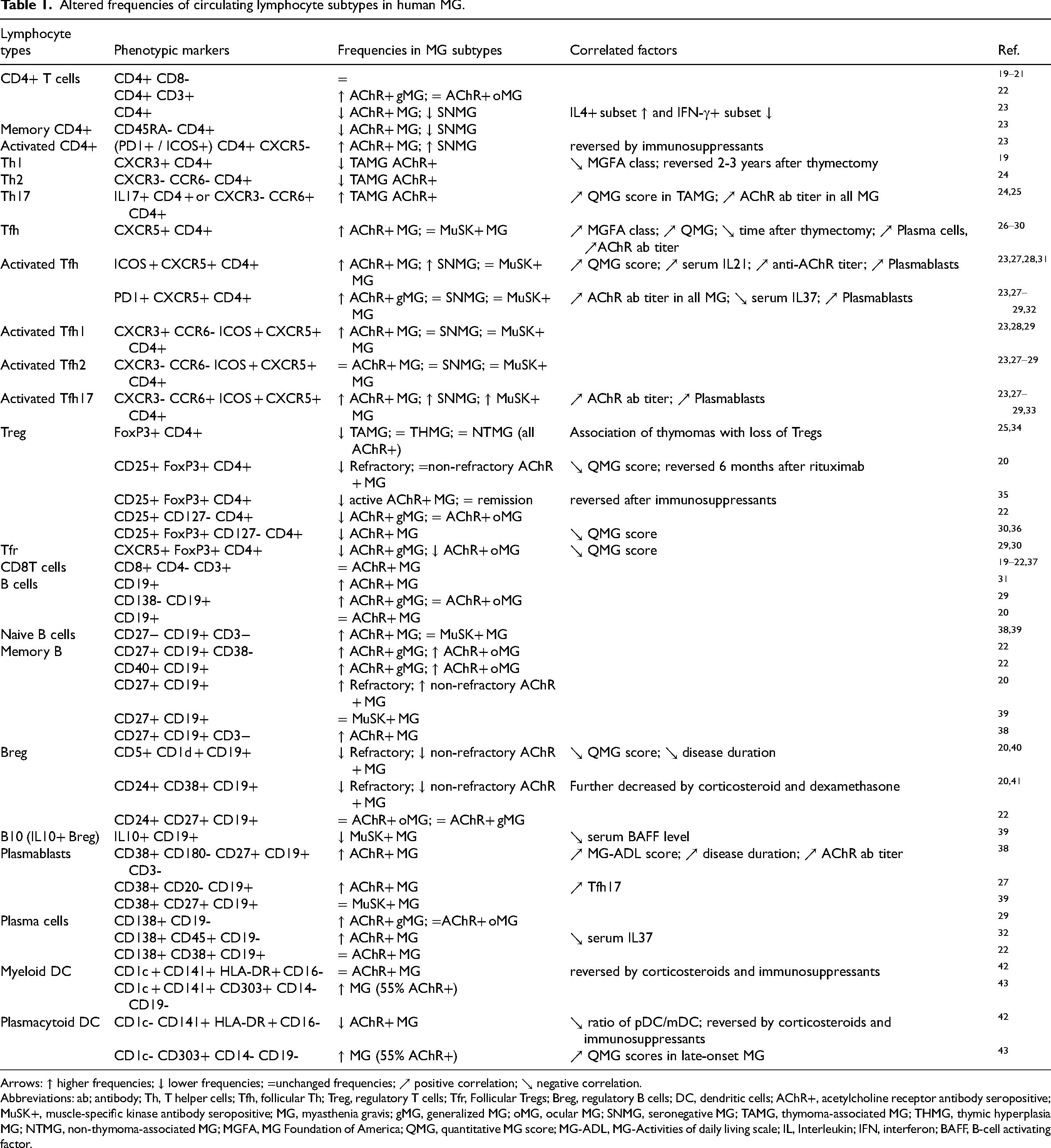
Arrows: ↑ higher frequencies; ↓ lower frequencies; =unchanged frequencies; ↗ positive correlation; ↘ negative correlation.
Abbreviations: ab; antibody; Th, T helper cells; Tfh, follicular Th; Treg, regulatory T cells; Tfr, Follicular Tregs; Breg, regulatory B cells; DC, dendritic cells; AChR+, acetylcholine receptor antibody seropositive; MuSK+, muscle-specific kinase antibody seropositive; MG, myasthenia gravis; gMG, generalized MG; oMG, ocular MG; SNMG, seronegative MG; TAMG, thymoma-associated MG; THMG, thymic hyperplasia MG; NTMG, non-thymoma-associated MG; MGFA, MG Foundation of America; QMG, quantitative MG score; MG-ADL, MG-Activities of daily living scale; IL, Interleukin; IFN, interferon; BAFF, B-cell activating factor.
The frequency of total blood CD4+ T cells and CD8+ T cells is not affected in MG, but the frequency of CD19+ B cells is somewhat higher in MG compared to healthy individuals (Table 1). A hallmark of MG pathophysiology is the increased presence of Th17 cells and a decrease in Treg cells, resulting in a pro-inflammatory cytokine shift that promotes inflammatory responses. In the thymus, an increase in T follicular helper (Tfh, specifically, Tfh1 and Tfh17) cells plays an essential role in B cell maturation and differentiation. This may explain the observed increase in memory B cells, plasmablasts, and plasma cells, alongside a reduction in regulatory B cells (Table 1). Rituximab, a monoclonal antibody designed against the CD20 molecule on B cells, efficiently decreases all CD20+ B cell counts by a single baseline dose, highlighting CD20+ B cell count as an important pharmacodynamic marker.44,45 Although safe and effective in low doses, Rituximab's efficacy may be debated, but can be used both in newly onset AChR+ generalised (gMG) and MuSK+ MG.45,46 On the contrary, follicular Treg cells (Tfr), which regulate germinal center responses, are diminished, mirroring the peripheral T reg cell deficit. Further, alterations in dendritic cell subsets are reported with conflicting results (Table 1). Most immune cell changes have been studied in AChR+ MG, where they correlate with disease severity. In contrast, for MuSK+ MG, only Tfh17 cells have been consistently reported as elevated among the various immune cell types analyzed (Table 1).
Beyond shifts in immune cell frequencies, MG is also characterized by functional impairments that affect proliferation, cytokine secretion, chemotaxis, cytotoxicity, and antigen presentation. Alterations in cellular genetic, transcriptomic, metabolomic, and secretome profiles drive these dysfunctions. For further insights, comprehensive reviews on these mechanisms are available.44,47–50 While immune cell frequency changes in MG are currently primarily a research tool rather than a routine clinical diagnostic measure, they hold promise as predictive, diagnostic, and pharmacodynamic biomarkers. Future studies incorporating longitudinal and pharmacodynamic analyses, alongside the development of standardized, clinically feasible assays, will be essential to translating these findings into practical diagnostic and therapeutic applications.
Proteomics
As the immune cell frequencies and functions shift in MG, the quantity of secreted proteins produced by these cells also changes. These alterations are detectable in serum or plasma, providing valuable insights into disease pathophysiology. The secreted proteins include cytokines, chemokines, growth factors, ligands, ligand receptors, enzymes, proteases, and chaperone proteins. Advances in multi-omics approaches allow for the simultaneous detection of multiple proteins within the same sample, enabling robust correlations and comparisons at both the individual patient and group level. Consistent with the altered immune cell subsets (Table 1), cytokines, chemokines, their receptors, and other inflammatory mediators are dysregulated in both the periphery and the thymus of patients with AChR+ MG. Table 2 provides an overview of the reported changes in these proteins levels in AChR+ MG sera.
Altered serum inflammatory proteins in human AChR+ MG sera.

* indicates change in both AChR+ and MuSK+ MG sera.
Arrows: ↑ higher levels; ↓ lower levels; =unchanged levels.
Abbreviations: IL, interleukin; OSM, Oncostatin M (an IL-6 family member); TNF, Tumor necrosis factor; TNFSF, TNF superfamily; APRIL, A proliferation-inducing ligand; BAFF, B cell-activating factor; IFN, interferon; CCL, chemokine (C-C motif) ligand; CXCL, chemokine (C-X-C motif) ligand; MCP, Monocyte chemoattractant protein; FGF, Fibroblast growth factor; GDNF, Glial cell line-derived neurotrophic factor; HGF, Hepatocyte growth factor; VEGF, Vesicular endothelial growth factor; TGF, Transforming growth factor; MIF, Macrophage migration inhibitory factor; 4E-BP1, Eukaryotic translation initiation factor 4E-binding protein 1; EN-RAGE, extracellular newly identified receptor for advanced glycation end-products binding protein; CDCP1, CUB domain containing protein 1; MMP, Matrix metalloproteases; ITIH3, inter-alpha-trypsin inhibitor heavy chain H3; ST1A1, Sulfotransferase 1A1; SIRT2, Sirtuin 2; uPA, urokinase-type plasminogen activator; STAMBP, STAM-binding protein; sNfl, serum neurofilament light; sκ-FLC, serum kappa free light chain.
A recent study presented a distinct biomarker profile of 23 inflammatory proteins in the sera of AChR+ MG patients from that of healthy controls 6 using proximity extension assay. Top biomarkers included ST1A1, CCL28, 4E-BP1, TNFSF14, and TGF-α. EOMG and LOMG patients vary in the levels of OPG and TGF-β1, while immunosuppressed and immunosuppressive naiive patients differed in the levels of CXCL10, CCL11, IL-17C, TNFSF14, and TGF-α. 6 It is hypothesized that such a serum profile arises from secretion by thymic follicular cells and circulating peripheral cells. The TNF superfamily is crucial in sustaining chronic inflammation in autoimmune disorders. Elevated levels of pro-inflammatory cytokines, such as IL-6 and IL-17, 47 in MG patients are likely linked to Treg dysfunction, characterized by reduced FoxP3 and IL-10 expression. IL-6, a key mediator of autoimmune pathogenesis, is implicated in diseases like MG, type 1 diabetes, multiple sclerosis, and rheumatoid arthritis and is secreted by monocytes, macrophages, B cells, muscle cells, and epithelial cells. Increased CXCL13, CCL21, and BAFF levels may recruit more B cells to the thymus, while CXCL10 and CCL21 attract T cells to inflammatory sites.48,50
Additionally, elevated IL-17 promotes the expression of inflammatory cytokines and chemokines, including IL-6, TNF-α, and IL-1β. These interactions facilitate B cell recruitment into germinal centers, stabilize their interactions with Tfh cells, and promote antibody class switching, resulting in B cell maturation, proliferation, and differentiation into memory B cells and plasma cells. 49 This process ultimately leads to AChR+ antibody production, in turn driving the disease phenotype and highlighting the role of autoreactive B cells in MG pathology.47–50
Another emerging protein family of interest in MG is the matrix metalloproteinases (MMPs). Elevated levels of MMP2, 79 MMP3,79,80 MMP979,81 and MMP106,82 have been reported in MG patients compared to healthy individuals. Together with the membrane attack complex (MAC), MMPs may contribute to the destruction of the NMJ, thereby promoting disease progression.88,89 Increased serum levels of MMPs in MG have also been associated with higher counts of and activation states of immune cells such as neutrophils, T cells, dendritic cells, and macrophages. 90 An increase of the neutrophil count is reflected by indices such as Neutrophil to Lymphocyte Ratio (NLR)91–93 and Systemic Inflammatory Response Index (SIRI)92,94 which are elevated in MG patients with severe symptoms.
A recent study that combined mass-spectrometry-based proteomics of serum samples from AChR+ MG patients with machine learning highlighted ITIH3, inter-alpha-trypsin inhibitor heavy chain H3, as a potential biomarker correlated with severity measured as change in QMG scores in AChR+ MG. 16 ITIH3 was also localized at the NMJ in AChR+ patient muscle biopsies. 16 Another masspectrometry study found high levels of serum fibrinogens (α, β, and γ) in MG patients compared to healthy individuals and patients with rheumatoid arthritis 95 ; however, this could not be reproduced in a recent follow-up study. 96 Additional proteins found elevated in sera from MG patients compared with healthy individuals include serum neurofilament light chain (sNfl, reflects abnormal neuronal activity), 83 serum kappa free light chain (sκ-FLC, reflects abnormal B cell activity),85–87 and calprotectin (a protein abundant in neutrophils), 84 mirroring the neurological and autoimmune nature of MG, together with proteins in Table 2.
Since sera are easily accessible, protein signatures for MG are promising biomarkers. However, validation against other neuroimmune disorders as well as longitudinal studies will be important in taking the next step and developing disease-specific serum protein profiles for MG.
Complement factors
Complement factors are crucial in AChR+ MG pathophysiology, as primarily by forming the MAC (C5b-9), they destroy the plasma membrane at the NMJ. There is apparent involvement of complement factors and deposition of MAC at NMJs, both in AChR+ MG 97 and antibody seronegative MG (SNMG), 98 as well as autoantibodies binding complements. Much evidence from the last decade indicates altered levels of complements in MG plasma and serum. Complement components are best assessed in plasma, as their further ex vivo activation is prevented due to the chelation of divalent ions (Ca2+, Mg2+) in the EDTA-plasma samples but not in sera. In contrast, their functional assessment is more efficient in sera. 99 The key complement component, C5b-9 (soluble C5b-9, sC5b-9), forms a pore-like structure. Elevated levels of sC5b-9,97,100,101 Ba and Bb, 101 C3 (C3a, 101 C3b 102 ), C4a, 101 C5 97 (C5a101,102), and factor I, 97 along with reduced levels of C2 and MBL 102 as well as properdin, 97 have been observed in MG plasma (Table 3). Recent data indicate the highest sensitivity and specificity for C3a and sC5b-9 in MG compared to healthy controls; however, comparisons with other disease groups are lacking. 97 Notably, the levels detected in plasma samples, and also the differences between different groups, are higher compared to those seen in serum samples. This can be due to the consumption of active complement components by lymphocytes, i.e., the difference between plasma and serum, and also due to the immunosuppressive status of MG patients. Further, almost all the changes observed in the complement components were in AChR+ MG and not in MuSK+ MG and SNMG (Table 4). This may suggest a higher tendency of MAC formation at NMJs, mainly in AChR+ MG, potentially involving the role for nicotinic AChRs on the postsynaptic muscle membrane in MAC-mediated pathophysiology in MG. 97 Hence, research highlighting these associations can further enhance the understanding of MG pathogenicity. Further, the development of serological tests to assess complement activation profiles may help determine the IgG status of the disease, thereby guiding personalized treatment choices between FcRn- and complement-targeted therapies. Although sera are considered unsuitable for reproducible and accurate measurements of complement factors, several reports show altered levels of multiple components in sera, as outlined in Table 3.
Altered complement components in human MG.

Arrows: ↑ higher levels; ↓ lower levels; =unchanged levels; ↗ positive correlation; ↘ negative correlation. Ba, Bb, C3a, C4a, C5a, C5b-9 were also examined in MusK+ MG and SNMG, but only C4a levels were detected higher in SNMG serum. Abbreviations: MGC, MG Composite score; MG-ADL, MG Activities of Daily Living scale; QMG, quantitative MG score.
Altered serum miRNAs in human MG sera and PBMCs.

Arrows: ↑ higher levels; ↓ lower levels. PBMCs, Peripheral blood mononuclear cells.
Circulating micrornas
MicroRNAs (miRNAs) are 22-nucleotide-long non-coding RNA sequences, highly conserved, and play a critical role in negatively regulating gene expression. They achieve this by degrading or repressing mRNA translation through miRNA-mRNA target complementarity.120–123 Despite their significance, few studies have investigated circulating miRNA levels in MG patients and compared them to those in individuals with other autoimmune or neurological diseases.
Table 4 summarizes all the miRNAs known so far to have altered levels in MG sera and PBMCs. In general, AChR+ MG sera have higher levels of miR-150-5p, miR-30e-5p, and miR-21-5p,58,105–108 but sera from gMG patients specifically have higher miR-150-5p and miR-21-5p levels than ocular MG (oMG). Further, miR-150-5p and miR-21-5p decrease in response to immunosuppressive treatment and thymectomy.58,105–108,124 Importantly, higher miR-30e-50 serum levels are observed early in the disease course in oMG patients that later generalize 125 and in MG patients who relapse with worsening of MG 108 with a hazard ratio of almost 3. Furthermore, miR-150-5p and miR-30e-5p exhibited greater short-term stability when measured weekly in sera over a month in patients with stable MG, highlighting their potential as reliable biomarkers. 126 In contrast, miR-21-5p showed significant fluctuations during the same timeframe, suggesting it may be less suitable as a biomarker. 126
Both miR-21-5p and miR-150-5p regulate autoimmune responses, particularly in the development of T- and B-cells. 126 They also correlate positively with IL-10 and inversely with IL-17 in the serum. 58 High expression levels of miR-150-5p in serum 127 and low in CD4+ T cells 128 in AChR+ MG indicate enhanced secretion by immune cells in the sera. MiR-30e-5p is upregulated in the tissues of mice with rheumatoid arthritis, negatively regulates atlastin GTPase2 (Atl2), and its knockdown suppresses the inflammatory response in TA mice. 129 Two of the top biomarkers in AChR+ MG patient sera, TNFSF14 and ST1A1, have an inverse correlation with miR-30e-5p, miR-150-5p, and miR-21-5p. 6 ST1A1, one of the top biomarkers with lower levels in MG, correlates positively with AXIN1, CASP-8, SIRT2, and STAMBP, suggesting a possible role in T-cell-mediated anti-viral immunity. 130 The serum levels of miRNAs in MG correlate with serum levels of several proteins altered in MG. Such correlations are absent in healthy controls, suggesting the role for miR-30e-5p, miR-150-5p, and miR-21-5p in direct or indirect regulation of mRNAs and proteins altered in MG. 6 Other studies have found lower serum levels of miR-15b,62,109 miR-20b,69,109,110 miR-122, miR-140-3p, miR-185, miR-192, miR-885-5p, 109 miR-27a-3p, 105 miR-146a 111 in MG patients compared to healthy individuals. In MuSK+ MG, four of the let7 family miRNAs, miR-151a-3p, miR-423-5p, let-7a-5p, and let-7f-5p are higher in the sera, 112 while the other two miRNAs, miR-210-3p, miR-324-3p, are found at lower levels compared to healthy individuals. 124
Fewer studies conducted on plasma found miR-106a-5p levels to be lower in AChR+ MG patients. 131
PBMCs from MG patients exhibit altered miRNA profiles, with overexpression of miR-146a,111,113 miR-155-5p, 114 miR-3651, miR-3654, and miR-612, 115 and downregulation of miR-181c, 57 miR-320a and miR-320b, 116 miR-150-5p, 117 miR-15a, miR-15b and miR-16, 118 and members of the let-7 family (7a, 7b, 7c, 7d). 119 The expression of mRNAs such as AKAP12 and HRH4, targets of miR-612, and CRISP3, a target of miR-3651, inversely correlates with these miRNAs. 115 Likewise, serum IL-17 levels, a target of miR-181c, show an inverse relationship with miRNA expression. 57
Metabolomics
Metabolomics can distinguish AChR+ MG patients from rheumatoid arthritis patients and healthy controls.132–134 While studies have varied in identifying specific metabolites that define MG, protein and fatty acid metabolism alterations have been consistently observed. Notably, arachidonic acid metabolism is disrupted in MG, aligning with the activation of pro-inflammatory pathways. This pathway was also highlighted in a metabolomic study evaluating changes before and after prednisone treatment. 135 Additionally, a discovery study identified metabolites such as free fatty acid (13:0), histidine, guanosine, and gamma-cholestenol as strong predictors of a treatment response to prednisone. 136 Pathway analysis reveals that alterations in xenobiotic metabolism might drive treatment resistance, suggesting that the metabolism of prednisone plays a critical role in patient response. 136
Conclusions
An ideal blood biomarker for MG should be biologically relevant, specific, sensitive, quantifiable, reproducible, accessible, and cost-efficient. It should also reflect the underlying neuroimmune pathophysiology and predict treatment responses across different subgroups. Although AChR and MuSK antibodies are essential in the MG diagnosis, they don’t present the potential to predict disease progression or activity and treatment responsiveness and, thus, cannot be used as longitudinal or pharmacodynamic biomarkers. Identifying and validating novel, promising, and specific biomarkers with the potential to reflect on distinct neuroimmune pathological mechanisms in MG will offer the development of molecular phenotype-based subgroups and, thus, personalized treatment options in MG precision medicine and may replace the need for traditional MG subgrouping. Certain biomarkers, for example, a combination of serum protein, complement, and microRNA markers, show early but promising evidence that could support clinical evaluation in specific MG subtypes, enabling individualized treatment strategies.
For many biomarkers, large-scale, longitudinal, and multicenter validation studies are still lacking, which limits their generalizability and implementation in clinical practice. Furthermore, while several biomarker candidates appear promising, such as microRNA, their clinical utility remains largely unproven in longitudinal studies due to a lack of methodological standardization and limited external validation. Achieving this requires extensive collaborative multicenter studies to evaluate potential protein, complement, microRNA, and metabolomics markers, not only in MG patients and healthy controls but also in diseases with overlapping neuroimmune features, such as multiple sclerosis, autoimmune neuropathies, and myopathies.
The MG community should prioritize these studies, especially with emerging biological therapies that depend on subjective and objective clinical scales. Nevertheless, biomarkers should be viewed as complementary tools to refine and enhance clinical assessment, rather than replace it. Validating and integrating biomarker research with clinical phenotyping and treatment response data in future studies will enable early diagnosis, prognosis prediction, and personalized treatment. Overcoming the challenges of disease heterogeneity, small sample sizes, and limited longitudinal data requires collaborative efforts to drive progress in this critical area of research.
Footnotes
Acknowledgements
The figure was created with Biorender. The authors are grateful for the participation of MG patients in research studies and acknowledge the support from funding bodies (MGFA, Myasthenia Gravis Foundation of America, and Erling Persson Foundation).
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Erling-Perssons Stiftelse, Myasthenia Gravis Foundation of America (grant number 2022_0030, N/A).
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: ARP has received consulting fees from Dianthus Therapeutics, Argenx, UCB Pharma, AMGEN, Alexion, and Toleranzia. The other authors report no competing interests.