
Abstract
Myotonic dystrophy type 1 (DM1) is a slowly progressive, multi-systemic disorder with clinical phenotypes that vary by age of onset and severity of symptoms. It is the most common form of muscular dystrophy occurring in adults. Abnormal regulation of alternative splicing of pre-mRNA results from a repeat expansion mutation in the dystrophia myotonica protein kinase (DMPK) gene. The resulting spliceopathy is universal across affected individuals and clinical phenotypes and drives the clinical manifestations of DM1, which include a diverse array of signs and symptoms affecting most organ systems. There is currently no disease-modifying treatment for DM1. Heterogeneity in the developmental and degenerative features and patterns of DM1 complicates the stratification, powering, and execution of interventional clinical trials in a reasonable timeframe. The use of splicing change as a surrogate endpoint in DM1 evolved from the principle that the degree of DM1-affected exons relates to the level of functional muscleblind-like (MBNL) RNA-binding protein activity in muscle cell nuclei. Surrogate endpoints based on panels of mis-splicing events reflecting the underlying DM1 molecular mechanism are reasonably likely to predict clinical benefit in a timely fashion, thus enabling accelerated clinical development of therapies that address unmet needs in DM1. Natural history data from the DM1 population support a strong correlation between dysregulated splicing and muscle function and point to the utility of a composite splicing index as a surrogate endpoint to predict future functional benefit, particularly in clinical trials of reasonable duration. Ongoing and future clinical trials will hopefully address the validity of surrogate endpoints using changes in splicing and whether the correction of spliceopathy correlates with meaningful clinical outcome assessments in individuals with DM1.
Introduction
Myotonic dystrophy type 1 (DM1) is a progressive, multi-systemic muscular dystrophy with clinical phenotypes that vary by age of onset and severity of symptoms. 1 DM1 is the most common form of muscular dystrophy occurring in adults and has an estimated prevalence of 1:8,000.2,3 In a recent meta-analysis, the pooled estimate of DM1 global prevalence was 9.27 cases (95% confidence interval [CI]: 4.73–15.21) per 100,000. 4 A cross-sectional study of dried blood spots from the New York State, United States, newborn screening program reported an incidence rate of 1 in every 2,100 births, although this does not necessarily reflect penetrant DM1 cases and may be an overestimate. 5 Individuals with DM1 incur significant clinical and economic burden. High total all-cause cost of care varies substantially between individuals underscoring the medical complexity and heterogeneity of disease.6,7 Data from a longitudinal study carried out over 47 years showed that only 18% of individuals with myotonic dystrophy survived to the age of 65 years, compared with an expected survival of 78%. 8 The socio-economic impact of DM1 is significant, as job retention is difficult and compounded by regular visits to different specialists needed for treating the array of symptoms. 9
A molecular hallmark of DM1 across clinical phenotypes is abnormal regulation of alternative splicing of pre-mRNA due to the repeat expansion mutation in the dystrophia myotonica protein kinase (DMPK) gene, resulting in mis-splicing of hundreds of genes across multiple cell types and tissues. Alternative splicing occurs in over 90% of human genes10,11 and is critical for appropriate development and tissue identify. Without accurate splicing, most protein coding genes cannot be properly expressed.
Splicing regulation is critical for the proper development and maintenance of all tissues in which the DMPK gene is highly expressed, including muscle and nervous system tissues. 12 Extensive postnatal alternative splicing in skeletal muscle is important for maturation and function of the contractile apparatus.13,14 Alternative splicing is coordinated in part by RNA-binding proteins whose function is impacted by RNA secondary structure. The muscleblind-like (MBNL) family of RNA-binding proteins are critical regulators of programmed alternative splicing, and inhibiting their function in DM1 leads to multi-organ disruption in mRNA processing and an abnormal persistence of fetal patterns of alternative splicing of hundreds of genes in adult tissues.15–22 The pathophysiology of DM1 is driven by a complex cascade of molecular events in affected tissues resulting from expansion of a cytosine, thymine, and guanine (CTG) trinucleotide repeat in the 3’ untranslated (UTR) region of the DMPK gene. Unaffected individuals typically harbor 5 to 37 trinucleotide repeats, whereas individuals with DM1 have expansions from 50 to as many as several thousand repeats. 23 The CTG expansion is unstable in somatic tissues and tends to increase in length over time, contributing to disease progression within an individual. There is also germline instability contributing to increasing repeat length and severity across successive generations of individuals with DM1. 24
Upon transcription, the expanded CUG repeats in the 3′-UTR of DMPK RNA form hairpin-loop structures that become trapped within the cell nucleus. 25 MBNL splicing factors preferentially recognize the CUG expansion abnormal structures and are sequestered to form distinct intra-nuclear aggregates, also known as foci17,26,27 (Figure 1). Sequestration and depletion of MBNL proteins reduces the levels of free MBNL proteins available for normal splicing regulation, and when combined with imbalance of other molecular switches, results in mis-splicing of hundreds of genes.15,28,29 Mis-spliced mRNAs can result in multiple outcomes, including but not limited to the inability to translate the intended protein, the production of a protein isoform that is less functional, or trafficking of the protein to an incorrect cellular compartment. The disease pathology caused by dysregulated splicing is commonly referred to as a spliceopathy. While there may be abnormal pathways in addition to mis-splicing contributing to DM1 pathology (e.g., altered polyadenylation), sequestration of MBNL and subsequent mis-splicing is necessary for disease manifestation. Assessment of free MBNL protein concentration in tibialis anterior muscle biopsies from DM1 patients suggests that in early DM1 disease stages, individuals remain pre-symptomatic as long as free MBNL levels are sufficient to maintain the transcriptional activity under its regulation. 30 As MBNL proteins are sequestered into nuclear foci, the splicing regulator CUGBP elav-like family member 1 (CELF1) protein is upregulated. 20 An imbalance in CELF and MBNL proteins results in the abnormal persistence of fetal patterns of alternative splicing in adult tissues with accompanying alterations in function.20,23 Mis-splicing of specific events has been directly linked to DM1 symptoms, including CLCN1 and myotonia, INSR and insulin resistance, SCN5A and cardiac arrhythmias, and BIN1 and muscle weakness.15,20,22 Thus, the primary DM1 molecular defect in mRNA processing establishes a spliceopathy that drives the clinical manifestations of DM1.
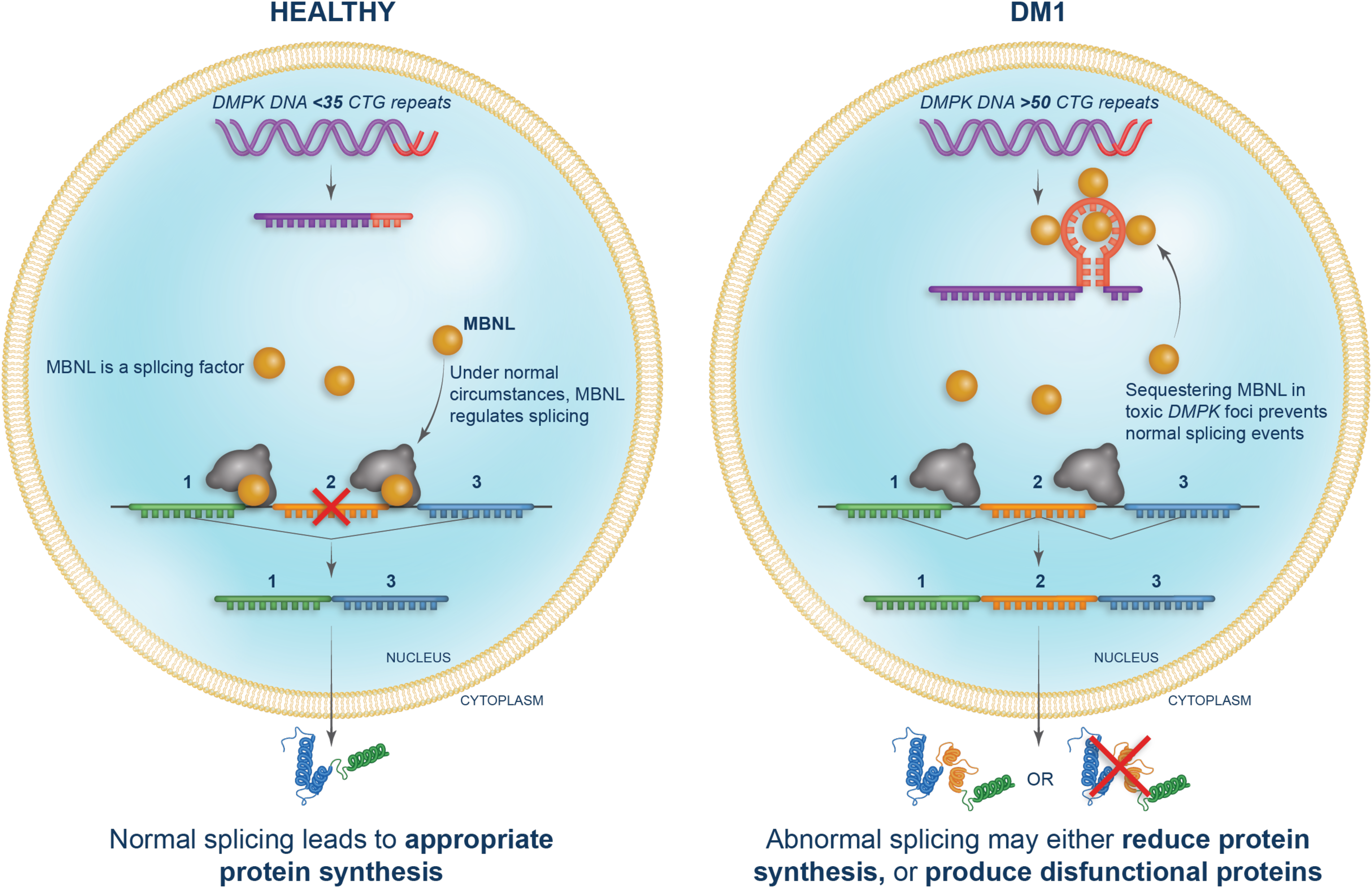
Spliceopathy in DM1. In healthy individuals, the DMPK gene contains <35 CUG repeats and normal splicing, regulated by MBNL proteins, leads to appropriate protein synthesis. The DMPK gene in individuals with DM1 contains >50 CUG repeats. RNA transcribed from DNA with expanded repeats forms hairpin loop structures that sequester MNBL splicing factors making them unavailable for splicing. This results in reduced protein synthesis or production of dysfunctional proteins.
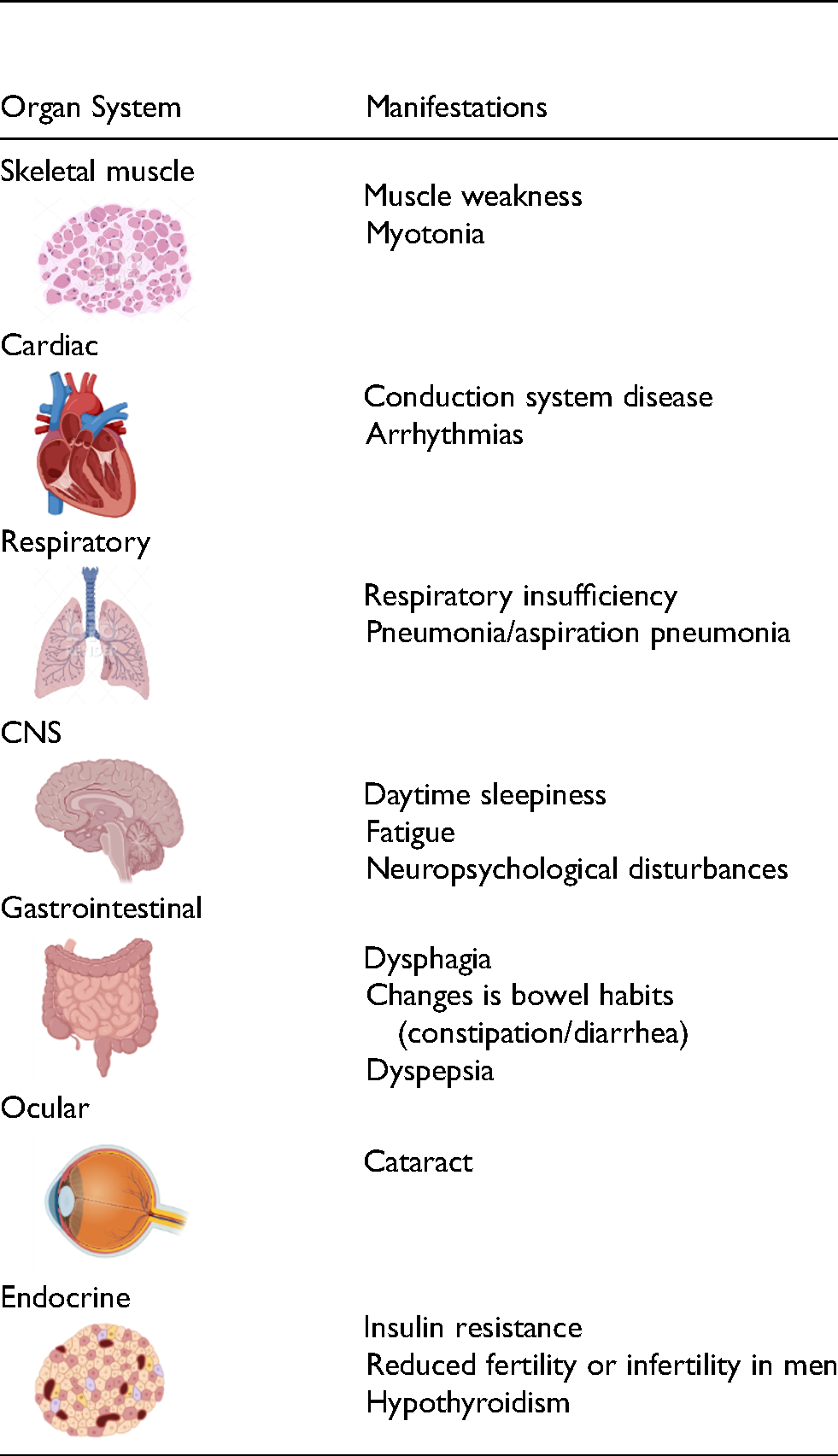
The DM1 spliceopathy is characterized by a diverse array of signs and symptoms that affects most organ systems (Table 1). While the underlying spliceopathy is the same in all individuals with DM1, four clinical phenotypes have historically been categorized: congenital, childhood, adult, and late onset, which represent the spectrum of age of onset and disease severity. 31 Congenital DM (CDM) manifests in infancy with potentially lethal outcomes. Childhood DM1 has onset between 1 and 10 years of age while adult-onset DM1 has onset after the age of 40. 23 Adult-onset DM1 is the most common clinical phenotype and is characterized by initial onset of myotonia, followed by slowly progressing weakness and atrophy predominantly of the cranial, trunk, and distal limb muscles.1,23 Other frequent clinical manifestations include cardiac conduction abnormalities, gastrointestinal and metabolic disturbances, primary hypogonadism, daytime hypersomnolence, behavioral and cognitive changes, and early-onset cataracts, 1 all of which contribute to a significantly diminished quality of life for adults living with DM1. 9 An individual's disease trajectory is a function of their unique combination of total CUG repeat load, influenced by somatic instability, and baseline MBNL levels, both of which change over a lifetime.30,32–35 A considerable portion of the variability within and across the DM1 phenotypic spectrum is likely due to variability in the extent of MBNL sequestration within different tissues, which is dependent on both repeat length and toxic DMPK RNA expression level. 36 Another key determinant of the progressive nature of DM1 is expansion of repeat length over time in somatic cells. Mechanisms that have been linked to this expansion of unstable repeats in somatic cells include DNA repair/mismatch repair mechanisms, RNA transcription-related repair, and cell turnover in tissues retaining mitotic activity, such as skeletal muscle. 37 As somatic cell CTG length increases over an individual's lifespan via repeat instability, toxic RNA accumulates and the free MBNL/expanded CUG RNA balance shifts toward decrease in regulation of alternative splicing control by MBNL.
Common clinical manifestations of DM1.
There is evidence that MBNL-dependent RNA splicing is disrupted in CDM similarly to adult-onset DM1. 38 Although disease onset and symptom presentation differ, individuals with CDM and adult-onset DM1 share a heterogenous range of alternative splicing dysregulation and a MBNL-dependent, mis-splicing signature. Infants with CDM present with severe symptoms in the first few years of life with all individuals under 2 years exhibiting low intracellular concentrations of free MBNL (determined from inferred MBNL 30 ) indicative of significant MBNL depletion and severe mis-splicing. These individuals then experience significant reversal of splicing dysregulation in early childhood with a corresponding reduction in symptoms. For a subset of these individuals, adolescence brings a return to increased splicing dysregulation and a transition from a childhood phenotype to one that is similar to adult-onset DM1 with associated myotonia and other severe symptoms. 38 These data lend support to the association between mis-splicing and DM1 symptoms that may apply to all DM1 clinical phenotypes.
In order to study mis-splicing in DM1, genes identified in the pathway between the molecular basis of disease and clinical manifestations of DM1 are used to develop panels of splicing events with reasonable likelihood to predict clinical benefit. 39 The profile of mis-spliced transcripts varies between tissues, leading to tissue-specific disease manifestations (Table 1). In skeletal muscle, a wide range of genes are impacted by mis-splicing, including genes encoding proteins responsible for sarcolemma stability (DTNA, DMD), metabolism (PKM2, INSR), ion conductance and excitability (CLCN1, CACNA1S, RYR1, SERCA1 i.e., ATP2A1), biogenesis of muscle T tubules (BIN1), sarcomere structure and stability (TNNT3, MYOM1), protease activity (CAPN3), myotube stability (NEB), myogenesis (MTMR1), and the extracellular matrix (e.g., NFIX). 20 Cardiac abnormalities in DM1 have been attributed to the mis-splicing of genes coding for proteins involved in cardiac conduction (SERCA1 i.e., ATP2A1, RBFOX2, SCN5A), maintenance of contractile properties (TNNT2), myofibril assembly and function (TTN), and cardiac fiber morphology (ZASP/LDB). A number of genes have been implicated in the pathology of DM1 in the brain, 40 including MAPT (associated with a progressive appearance of neurofibrillary tangles), NMDAR1 (required for normal long-term potentiation in the hippocampus, contributing to learning processes), 20 and GRIP1 (implicated in synaptic trafficking). 41 Abnormal splicing of INSR pre-mRNA has been associated with insulin resistance in DM1. 20 Given the widespread disruption of RNA splicing, it is likely that additional genes associated with symptoms will be implicated in the future.
Surrogate endpoints are needed for DM1 clinical trials
Individuals living with DM1 have a profound unmet medical need. Treatment is limited to symptom management as there is no cure and no disease-modifying treatment that slows disease progression. People living with DM1 view slowing or stopping disease progression as a priority. However, the high inter- and intra-individual variability over time particularly among adult-onset DM1, makes this difficult to evaluate in clinical trials. 42 The natural history of adult-onset DM1 suggests that the disease progresses slowly, which would potentially require a long period of time and a large sample size in order to measure clinical benefit. The extreme clinical heterogeneity between affected individuals (i.e., time of onset, affected organ systems, and severity of symptoms)23,43 complicates stratifying and adequately powering interventional clinical trials. Thus, predictions for worsening of symptoms and disease progression are not always comparable between patients. Existing literature estimates that a detectable treatment effect based upon stabilization of disease progression in a clinically meaningful outcome measure could take up to, or over, 2 years.44–46 In addition, the instability of the expanded DMPK CTG region in somatic cells leads to tissue-specific somatic mosaicism that is biased toward further expansion during aging and contributes to the variable nature of disease progression. 32 There is an unmet need for the identification of evidence-based outcomes in individuals with DM1 as clinical trials of disease-modifying modalities are emerging. The validity and reliability of muscle strength, balance, and functional mobility outcomes have recently been established in DM1 and are recognized as important to patients. However, knowledge about the responsiveness (i.e., a measure's ability to detect change in a condition over time) of these endpoints is still emerging. Important information in this regard will be provided by interventional clinical trials for which muscle strength, mobility and function are primary or secondary endpoints (e.g., del-desiran phase 3 trial, HARBOR, NCT 06411288). Nevertheless, the molecular hallmark of MBNL-dependent mis-splicing is universal across affected individuals and DM1 clinical phenotypes and provides an opportunity for splicing biomarkers to be used as surrogate endpoints that ensure target engagement.
A surrogate endpoint is a biomarker or physical sign that is used in therapeutic trials as a substitute for a clinically meaningful endpoint and is expected to predict the effect of therapy. While not all biomarkers are surrogate endpoints, surrogate endpoints are often biomarkers. 47 Thus, splicing biomarkers are candidate surrogate endpoints in DM1. The FDA Accelerated Approval Program was initiated to address the difficulties in applying the standard drug approval paradigm to rare and other diseases with unmet medical needs. Accelerated approval is based on a surrogate endpoint which is not itself a measure of clinical benefit, but is reasonably likely to predict clinical benefit (Table 2).48,49 The precedent for using surrogate endpoints in rare neuromuscular diseases was established with conditional approval of antisense oligonucleotides to treat Duchenne muscular dystrophy (DMD) based on primary endpoints measuring internally shortened dystrophin in skeletal muscle biopsies. 50 Per FDA requirements, ongoing clinical trials are assessing the functional correlation of internally shortened dystrophin protein expression. Similarly, an AAV delivered micro-dystrophin has been granted accelerated approval by the FDA in children 4–5 years of age with DMD for delandistrogene-moxeparvovec-rokl (ELEVIDYS) gene therapy based on expression of micro-dystrophin protein as a surrogate marker. 51 Another recent drug approval using a surrogate endpoint is tofersen for amyotrophic lateral sclerosis (ALS). Tofersen is an antisense oligonucleotide that targets mutated superoxide dismutase 1 (SOD1) mRNA to reduce the synthesis of SOD1 protein. However, increased SOD1 levels do not appear to be specific to ALS as these were also detected in the CSF of control individuals with another neurological disease. 52 Thus, FDA approval was based on a reduction in plasma neurofilament light (NfL), a blood-based biomarker of axonal (nerve) injury and neurodegeneration.
Surrogate endpoints used to gain accelerated approval* in rare diseases** (excluding malignancies).

*Data from the FDA on surrogate endpoints that were the basis of drug approval or licensure
**Based on NORD rare disease database
Following the precedent set by the above examples, a surrogate endpoint that reflects the underlying molecular mechanism of DM1 and is reasonably likely to predict clinical benefit in a timely fashion, may be able to accelerate clinical development of therapies that address DM1 unmet needs.
Limitations of potential biomarkers in the DM1 causal pathway
Several molecular biomarker candidates were subsequently deemed inadequate to serve as a surrogate endpoint for various reasons. DMPK RNA containing CUG trinucleotide repeats is immediately downstream from the DM1-causing genetic mutation. However, the assay used to measure DMPK levels does not distinguish between the cytoplasmic pool of wild-type DMPK and nuclear mutant RNA and cannot, therefore, specifically assess mutant DMPK knockdown. Furthermore, measuring expanded CUG transcripts from DMPK RNA using standard RNA isolation methods is difficult from the small amount of muscle obtained from needle biopsies and would require a significant amount of muscle biopsy tissue, which is not easily attainable from human subjects. 53 Therefore, DMPK RNA is a marker of target engagement, but may not be the optimal biomarker to be used as a surrogate endpoint.
Both expanded CUG repeat length and expression levels of the toxic RNA directly impact the extent of MBNL sequestration. However, assessment of unbound MBNL from muscle biopsy tissue samples, is limited by the dynamic nature of free and sequestered MBNL pools and the inability to easily quantify the free fraction. The heterogeneity across tissues, sampling biases, and symptom severity results in poor correlation of CTG length with progressive DM1 clinical phenotypes, particularly in studies with small sample sizes,54,55 and diminishes its usefulness when limited numbers of patients are enrolled. This measure has not been associated with skeletal muscle strength, and the sizing of trinucleotide repeats is difficult. With the exception of the extremes of the pathology continuum, i.e., CDM and late-onset DM1, there is poor correlation between CTG length and severity of the DM1 clinical presentation.23,54
Measurement of expression levels of fetal protein isoforms from genes affected by DM1-related mis-splicing constitutes another candidate surrogate biomarker, but fetal and adult isoforms cannot be distinguished using simple gel electrophoresis. These isoforms typically differ by no more than a few amino acids, thereby requiring a sophisticated antibody technology.
Finally, alterations in skeletal muscle and serum levels of several micro-RNAs (miRNAs) have been reported in DM1 patients. 56 However, the lack of connection with disease mechanisms and severity is not supportive of miRNAs as biomarkers of DM1. Alterations in miRNAs in muscle do not correlate with histopathologic scoring, and results are highly inconsistent in serum. 56 Mixed and sometimes conflicting results have been reported from studies examining whether miRNAs correlate with progression of muscle atrophy.57–59 Thus, insufficient knowledge and a lack of conclusive results limit the potential of miRNAs as surrogate endpoints for therapy development in DM1. 60
In contrast to the challenges inherent with the above-mentioned biomarkers, quantifying mis-splicing events that correlate with both the underlying molecular mechanism in DM1 and with disease severity offers the potential to act as a suitable surrogate endpoint that is reasonably likely to predict clinical benefit in a timely fashion. The pathophysiology of DM1 is associated with RNA mis-splicing. While preclinical work has demonstrated that RNA splicing changes are dynamic and each event is differentially sensitive to the degree of MBNL1 sequestration, the variability posed by assessing a single splice event is being addressed by development of a composite measure of several splicing events that is representative of both DM1 disease and patient population heterogeneity and would enable detecting dose-dependent changes in the clinic.
Correction of splicing as a surrogate endpoint in DM1
The use of splicing change as a surrogate endpoint in DM1 is based on the principle that the degree of DM1-affected exons reflects the level of functional MBNL activity in muscle cell nuclei. While MBNL protein levels do not differ substantially in healthy tissue versus DM1-affected tissue, the cellular burden of CUG expansion mRNA and resulting sequestration of MBNL is the primary driver of mis-splicing (Figure 1). As stated above, the direct measurement of CUG repeat RNA length or free MBNL levels as surrogate endpoints are not feasible due to scientific or technical limitations. In contrast, the dependence of mis-splicing upon the intersection of MBNL and toxic RNA transcript levels39,61,62 supports using changes in splicing as a biomarker for the downstream effects of DMPK mutation. Furthermore, measuring combinations of splicing events within genes affected in DM1 using targeted sequencing assays has been used to infer the level of free MBNL in both human and murine cells.30,39
In order to study mis-splicing in DM1, genes identified in the pathways between the molecular basis and clinical manifestations of DM1 are used to develop panels of splicing events with reasonable likelihood to predict clinical benefit. The rationale for using a panel of splicing events and calculating a composite splicing index instead of looking at only a single event is due to the greater power that multiple events have for predicting residual MBNL activity. Previous work showed that approximately 88% of the predictive power can be captured by a minimum of 15 events. 30 The splicing events chosen for inclusion in a splicing biomarker panel should represent genes linked or implicated in pathways that cause symptoms in individuals with DM1.
Splicing outcomes (measured in muscle biopsy samples) are expressed as the percent spliced in (PSI), which is the fractional inclusion of each cassette exon expressed as the ratio of normalized events indicating inclusion of the exon to the sum of inclusion and exclusion events detected by the assay (Figure 2A).
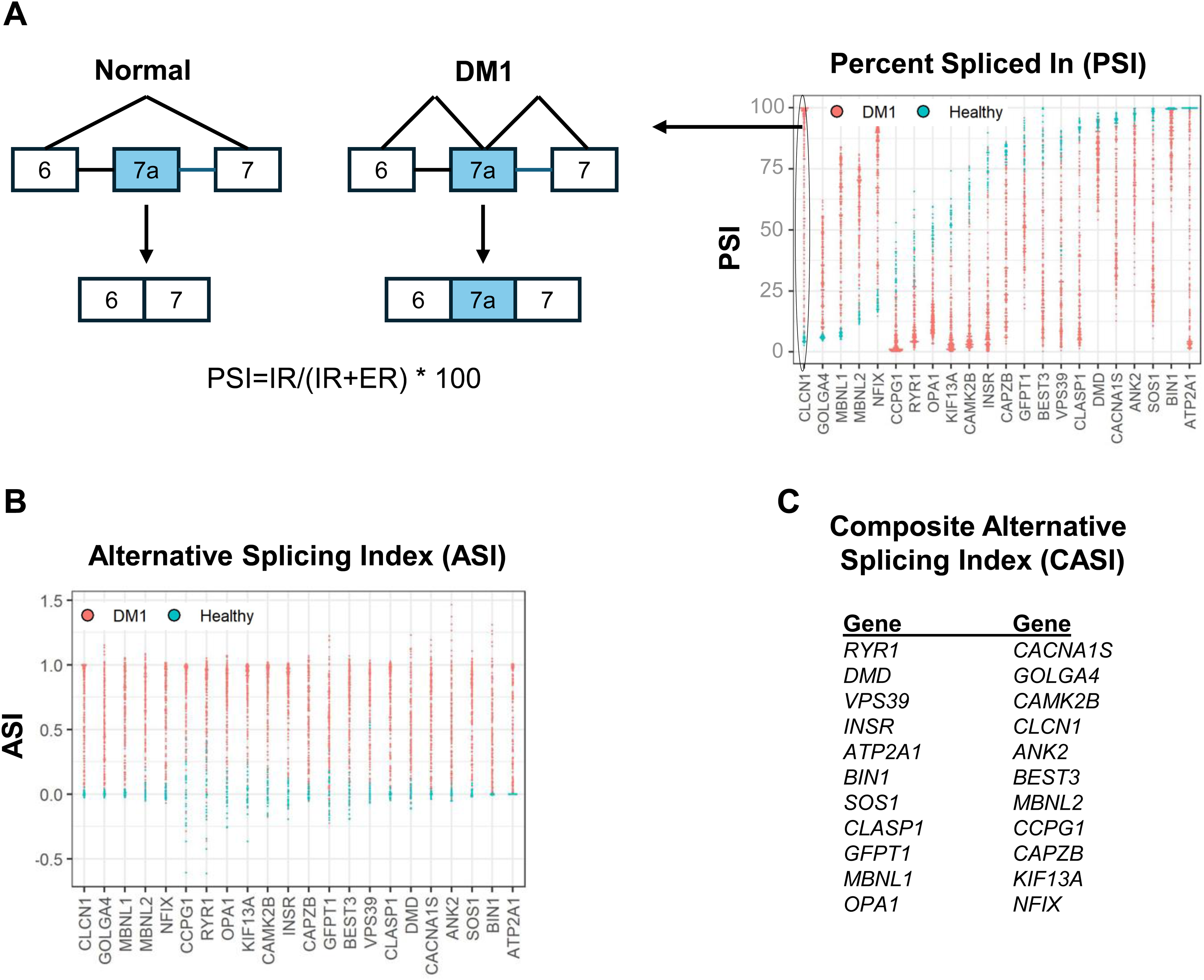
The composite alternative splicing index (CASI) measures the normalized ratio of cassette exon inclusion across a panel of 22 genes involved in DM1 pathophysiology. (A) The percent spliced in (PSI) determines the fractional inclusion of each cassette exon across a panel of 22 genes in DM1 individuals and healthy controls. It is expressed as the ratio of normalized events indicating inclusion of the exon to the sum of inclusion and exclusion events detected by the assay. Example shown is CLCN1 exon 7a inclusion in DM1 but not in healthy individuals. IR = normalized read counts indicating inclusion of the exon. ER = normalized read counts indicating exclusion of the exon. (B) the alternative splicing index (ASI) is generated by normalizing the PSI of subjects with DM1 to that of healthy controls. ASI = PSI − PSIHealthy/PSIDM95 − PSIHealthy where PSIHealthy is the median PSI obtained from 25 healthy subjects and PSIDM95 is the 95th percentile for most severely affected PSI from 172 DM1 subjects. 39 (C) The composite alternative splicing index (CASI) is calculated as the average of the non-missing ASI values across the 22 genes.
Percent spliced-in (PSI) and percent spliced-out (PSO)
Mis-splicing is measured as the inclusion of a specific exon from a panel of 22 genes using a targeted RNA sequencing method similar to that described by Tanner et al.
39
For a specific transcript, the percent spliced in (PSI) is defined as the percent of inclusion-isoform reads relative to all inclusion- and exclusion-isoform reads; the percent spliced out (PSO) is defined as the percent of exclusion-isoform reads relative to all inclusion- and exclusion-isoform reads.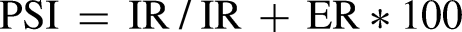
IR: Normalized read counts indicating inclusion of the exon.
ER: Normalized read counts indicating exclusion of the exon
The splice events are selected to have a large effect size (>30% PSI difference between DM1 and healthy subjects), to be contained within genes with a connection to DM1 biology, and to have a gene structure favorable for RT-PCR amplification and subsequent amplicon sequencing by RNAseq. When selecting splice events for inclusion in the composite index, preference is given to those within genes that are causally implicated in myotonic myopathy or muscle weakness, such as CLCN1 exon 7a, BIN1 exon 11, CACNA1S exon 29, INSR exon 11, and DMD exon 71/78.20,40,41 The PSI of subjects with DM1 can be normalized to that of healthy controls to generate an alternative splicing index (ASI) (Figure 2B).
Alternative splicing Index (ASI)
The alternative splicing index (ASI) is the normalized splicing value calculated as: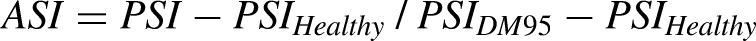
Composite alternative splicing Index (CASI)
Lastly, a composite alternative splicing index (CASI) is derived by averaging the normalized ASI across all splice events (Figure 2C). The CASI score is calculated as the average of the non-missing ASI values across the 22 genes. The CASI can be used to measure treatment outcomes by describing the extent of mis-spliced events before and after treatment. 53 In contrast to the timeframe needed to establish a clinically meaningful benefit in this slowly progressive disease, it is anticipated that a substantive change in a DM1 splice-event biomarker panel would be observed within 2–3 months due to the dynamic turnover of RNA products.
Correction of the spliceopathy central to DM1 pathophysiology has been tested in animal models that recapitulate, to varying degrees, the pathology and symptoms of DM1.29,39,61,63–69 A subset of splicing changes in DM1 mice were attributed to myotonia as a downstream consequence of MBNL loss. 39 Splice events reflected differences in the steady state levels of CUG expansion mRNA or MBNL accumulation. A composite splicing index derived from 35 splice events showed an incremental response to progressive inactivation of MBNL alleles or stepwise accumulation of CUG expansion toxic RNA. 39 Treating mice that express CUG expansion mRNA with antisense oligonucleotides caused a reduction of toxic RNA and the parallel correction of the ASI. As transgene knockdown occurred, targeted sequencing showed a parallel reduction (improvement) of the ASI, and a strong correlation between transgene knockdown and correction of mis-splicing (R2 = 0.86). Following correction of mis-splicing, myotonia in hindlimb and paraspinal muscles was reduced or eliminated. 39 Overexpression of MBNL in a mouse DM1 model not only reversed a subset of splicing events, but rescued disease-associated myotonia. 70 Similarly, antisense oligonucleotides targeting DMPK or that bind to CUG expansions and sterically block the sequestration of MBNL reverse mis-splicing of a subset of transcripts normally dysregulated in the DM1 mouse model65,69 and could improve muscle strength. 69 Similar results as well as mitigation of myotonia and muscle strength loss were obtained with antisense oligonucleotides acting via RNase H mechanisms to produce reproducible and robust reduction in skeletal muscle CUG expansion mRNA. 64 Nakamori et al. assessed the reversibility of splicing defects in transgenic mice using antisense oligonucleotides to reduce toxic RNA levels and showed that mis-splicing reflected MBNL sequestration and DM1 pathology. 61 The mis-splicing defects were fully corrected following antisense oligonucleotide injections. These preclinical proof-of-concept data support the concept that the loss of MBNL activity is a primary pathogenic event in the development of both RNA mis-splicing and skeletal muscle sequela in DM1 and created the framework for using changes in splicing as biomarkers of disease severity and therapeutic response in DM1.
Correlating splicing changes and clinical outcomes in individuals with DM1
Mis-splicing has been linked to muscle weakness in several natural history studies involving tissue samples collected from individuals with DM1 (Table 3). In a detailed assessment of splicing changes and their relation to functional impairment, a group of affected splice defects were identified in a cohort of individuals with DM1 before there was evidence of muscle weakness. 61 A hierarchy of splicing changes were then identified whose number and extent were proportional to disease severity. Nakamori and colleagues concluded that mis-splicing events showed potential to function as biomarkers of severity and therapeutic response. Among splicing events examined, 20 DM1-specific events showed graded changes that correlated with weakness of ankle dorsiflexion. 61 In addition, exploratory multiple regression analyses confirmed that assaying a panel of splicing biomarkers is preferential for gauging therapeutic effects in clinical trials.
Correlation of splicing* to functional outcomes in DM1 natural history cohorts. Samples for adult cohorts across all studies were collected from tibialis anterior muscle biopsies. In the Hartman study, vastus lateralis was biopsied in children.

*CDM and adult cohorts combined. [MBNL]inferred is the concentration of MBNL as inferred by splicing changes. ADF, ankle dorsiflexion; CASI, composite alternative splice index; vHOT, video hand opening time.
Wang and colleagues characterized mis-splicing signatures in skeletal muscle in parallel with quantifying muscle performance in a cohort of 44 individuals with DM1. 71 Using a previously developed assay of mis-splicing across a large number of MBNL-dependent exons, 30 inferred MBNL activity in skeletal muscle samples were found to strongly correlate (r = 0.86) with both splicing patterns of specific transcripts and normalized ankle dorsiflexion strength. They thus concluded that: (1) individuals with DM1 exhibited a continuum of disease severity, as reflected by mis-splicing and muscle strength and (2) that either the mis-splicing pattern or measure of muscle strength might allow for quantitative assessment of the efficacy of a therapeutic intervention.
A targeted high-throughput sequencing method (multiplex alternative splice sequencing) has been used to assess splice events that were most correlated with functional impairment in DM1. The 22 splicing events selected for the panel were identified based on their connection to DM1 molecular pathology and clinical manifestations, as well as having a gene structure favorable for amplification and sequencing (Figure 2C).30,61 The panel included a muscle-specific cluster of six genes with low expression in adipose tissue, arteries, and whole blood that are potential confounding components of biopsy tissue. The remaining genes represented mis-splicing events across a range of tissues and organs, including brain, heart, kidney, gastrointestinal tract, and endocrine tissues.
Provenzano et al. 72 used this composite RNA splicing index biomarker incorporating the 22 disease-specific splice events to assess alternative splicing dysregulation for 52 individuals with DM1 in a cross-sectional cohort and longitudinally at two time points (baseline and 3 months post-baseline). The composite splicing index was determined using targeted RNA sequencing of tibialis anterior muscle biopsies and had significant associations with timepoint matched measures of muscle strength and ambulation, with ankle dorsiflexion strength, hand-grip strength, and 10-meter walk/fast run speed showing Pearson coefficients of r = −0719, r = −0.715, and r = −0.680, respectively. Importantly, not only were these timepoint-matched correlations between splicing index and function significant, but a longitudinal model assessing the prognostic ability of baseline CASI to predict future muscle function showed significant correlations, often with r values that were even more robust than those from the time-matched assessments. Baseline splicing index scores were compared to timepoint matched and 3-month outcome measures to assess temporal relationship between splicing dysregulation and functional changes. Scores exhibited stronger associations for nearly all outcomes at 3 months compared to timepoint-matched assessments, including ankle dorisiflexion strength, 10-meter walk/fast run speed, knee extension, and video hand opening time for the middle finger. A linear regression model indicated that when combined, baseline ankle dorsiflexion strength and composite splicing index were predictive of muscle strength at 3 months (adjusted R2 = 0.830). Provenzano and colleagues 72 also have shown that the variability of splicing over 3 months when assessed by different biopsy methods taken from the same side of the body or contralateral side is quite small. The variance of the composite splicing index observed between pre- and post-treatment in the placebo cohort described by Thornton et al. 53 may be accounted for by the biopsy collection method and natural progression of DM1 disease, or by the biological variability in this measurement, as demonstrated by Provenzano et.al. 72 It is worth noting that for two participants who showed apparent improvements at the highest dose of an antisense oligonucleotide targeting DMPK, 53 the shift in aggregate splicing events fell outside the biopsy-rebiopsy variance observed in a natural history study of over 100 individuals with DM1 who did not have a therapeutic intervention. These data further justify that, while splicing may have some limitations, with standardized biopsy collection procedures and development of a robust method to detect changes, splicing can serve as a molecular biomarker of DM1 disease and surrogate endpoint for therapeutic intervention.
These data by Provenzano and colleagues 72 further support the notion that the CASI is an accurate, reproducible, sensitive assessment of the relative degree of splicing dysregulation in skeletal muscle. Additionally, their data show that the CASI is sensitive to changes over time and strongly correlates with multiple measures of muscle and physical function and therefore may be predictive of future disease outcomes. Their results also suggest that changes in splicing as reflected by the CASI may be measurable before detectable phenotypic changes, and the authors suggest that this may occur as a result of a time-delay between aberrant exon inclusion patterns and the downstream biological changes that impact functional outcomes.
Hartman et al. 73 assessed the association between RNA mis-splicing and physical function in children with CDM and adults with DM1. Muscle biopsy and functional outcome data from five observational studies were normalized and correlated with an aggregate measure of alternative splicing dysregulation. Significant correlations were found between splicing dysregulation and skeletal muscle performance in both children and adults, and with myotonia in adults (there was no myotonia in the children with CDM).
Natural history data demonstrate the relatively strong correlation between mis-splicing (CASI) and functional outcomes such as hand grip strength and 10-meter walk/run (Table 3). Other measures, such as myotonia have more modest correlation possibly due to clinical variability and the relatively short period of follow-up. Additionally, splicing in the natural history studies was assessed in tibialis anterior muscle, and given the tissue heterogeneity inherent to DM1 pathophysiology, more modest correlation of splicing measured in the lower extremity to upper extremity function is not unexpected.
In the first clinical trial of a targeted therapy to mitigate RNA toxicity in adults aged 20–55 years with DM1, 53 two of six participants treated with the highest dose level of an RNase H1-active ASO showed improvements across the CASI 22 splicing event panel assay. However, drug concentrations in skeletal muscle were generally below levels predicted to achieve substantial target reduction indicating the need to improve drug delivery efficiency.
There are currently a number of nucleic acid-based therapies or small molecules in clinical development for DM1, described in detail in recent reviews.74–77 Among these examples, evaluation of the correction of mis-splicing is being used as a surrogate endpoint in several clinical trials assessing nucleic acid-based clinical candidates. For example, measurement of mis-splicing as a biomarker outcome is studied in completed or ongoing trials assessing novel oligonucleotide conjugate platforms for treatment of DM1 in adults (NCT05027269 MARINA and NCT05481879 ACHIEVE). The CASI biomarker assay used in these studies utilizes the panel of 22 genes described in Figure 2C. These ongoing clinical trials and natural history studies will inform the scientific community on the utility and future development of splicing as a surrogate endpoint for DM1. A splicing biomarker will confirm target engagement by potential therapeutics, and effects may precede clinical benefit measurable with functional endpoints. Given the lack of a standardized method of generating the data for this biomarker, development of a universal method of biopsy collection and processing and thereafter analysis of splicing will be helpful in bridging the data across the different studies and understanding the benefit of treatment in this highly heterogeneous disease population. Once the relationship between target engagement and clinical outcomes is better understood, biopsies may not be necessary.
Overall, these data support the importance of splicing events in the pathogenesis of DM1, their direct link to functional outcomes, and their potential utility in being able to evaluate a response to therapeutic interventions. Data from ongoing and future clinical trials will help validate using measurement of mis-splicing as a surrogate biomarker endpoint and establish correlation between the extent of correction of spliceopathy with clinical outcomes in individuals with DM1.
Other considerations for early signals of efficacy
Although not in the causal pathway of DM1, quantification of myotonia as measured by video hand opening time (vHOT) could be an early signal of efficacy and may serve as an intermediate clinical endpoint in DM1 trials. vHOT has been shown to correlate with the PSI of the CLCN1 chloride channel in skeletal muscle of adults with DM1. 73 In preclinical models CLCN1 is directly linked to myotonia 78 and CLCN1missplicing is rapidly corrected following treatment.79,80
Conclusions
The slow progression of DM1, the clinical heterogeneity of the disease, and the critical unmet medical need of individuals living with DM1 warrant the use of surrogate biomarkers that are reasonably likely to predict changes in muscle function as efficacy endpoints to expedite access to a safe and effective treatment. Robust scientific evidence supports the idea that DM1 pathogenesis results from CTG trinucleotide repeat expansion mutation at the DMPK locus. This repeat expansion leads to high numbers of mutant CUG sites within the expansion mRNA that sequesters MBNL proteins and results in mis-splicing of hundreds of transcripts, including many associated with skeletal muscle structure and function, ultimately culminating in muscle weakness. Moreover, in animal models in which features of the DM1 spliceopathy recapitulate human disease, candidate therapeutics targeting mis-splicing can restore muscle strength and myotonia. Natural history data from the DM1 population support a strong correlation between dysregulated splicing and muscle function and point to the utility of the CASI to predict future functional benefit, particularly in clinical trials of reasonable duration. As a downstream outcome of mutant DMPK knockdown, measurement of correction of splicing events may represent a surrogate endpoint demonstrating that potential therapeutics have reached the intended target with the intended effect.
Footnotes
Acknowledgements
The authors would like to thank Fernanda Giupponi and Brian Newkirk of Dyne Therapeutics, Inc. and to John D. Cleary of University at Albany, SUNY, NY for providing critical feedback on the manuscript.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Dyne therapeutics supported development of this manuscript including funding for medical writing and editing support provided by Patrice C. Ferriola, PhD of KZE PharmAssociates.
Declaration of conflicting interests
AN, IIH, and SR are employees of Dyne and may hold stock in the company. JAB serves on the Scientific Advisory Committee and Board for the Myotonic Dystrophy Foundation, has consulted or currently consults for D.E. Shaw Research, Dyne Therapeutics, Entrada Therapeutics, Juvena Therapeutics, Kate Therapeutics, Syros Pharmaceuticals, Sanofi, Wayfinder Biosciences and received research funding from Agios Pharmaceuticals, Biomarin Pharmaceuticals, PepGen, Syros Pharmaceuticals and Vertex Pharmaceuticals and has received licensing royalties from the University of Florida. JAB and NEJ are co-founders and have financial interest in Repeat RNA Therapeutics Inc. JAB has received grant funding from NINDS (R01NS120485, R01NS135254, R01NS104010), NIAID (R21AI178672), USAMRAA (HT94252410190), Muscular Dystrophy Association (MDA 1363842), NSF (2244372) and NYSDEC (101274). NEJ has received grant funding from NINDS (R01NS104010, U01NS124974), NCATS (R21TR003184), CDC (U01DD001242) and the FDA (7R01FD006071). He receives royalties from the CCMDHI and the CMTHI, research funds from Novartis, Takeda, PepGen, Sanofi Genzyme, Dyne, Vertex Pharmaceuticals, Fulcrum Therapeutics, AskBio, ML Bio, and Sarepta, and has provided consultation for Arthex, Angle Therapeutics, Juvena, Rgenta, PepGen, AMO Pharma, Takeda, Design, Dyne, AskBio, Avidity, and Vertex Pharmaceuticals.