
Abstract
Objective
To describe the disease burden in patients with Pompe disease treated with enzyme replacement therapy (ERT) in the US as defined by comorbid conditions, supportive services, and treatment patterns.
Methods
A retrospective cohort study (01/01/2012–09/30/2022) was conducted using the Merative™ MarketScan Research Databases. Inclusion criteria were: ≥ 2 outpatient or ≥1 inpatient claims of Pompe disease, ≥ 1 claim of ERT, and continuous enrollment in medical/prescription coverage for ≥90 days before diagnosis date for patients ≥2 years and ≥1-month post-index date. Patients were stratified into infantile-onset Pompe disease (IOPD) or late-onset Pompe disease (LOPD) cohorts based on age at diagnosis and clinical presentation. Key comorbidities, supportive services, and treatment modifications were presented as cumulative incidence.
Results
A total of 105 patients were included (IOPD: n = 50; LOPD: n = 55). For IOPD and LOPD groups, the 12-month cumulative incidence was 84.5% and 79.4% for respiratory, 57.4% and 54.3% for ambulatory, 67.8% and 33.4% for gastrointestinal, and 16.9% and 28.7% for cardiovascular comorbidities, respectively; 12-month cumulative incidence was 66.9% and 31.8% for physical therapy, 59.5% and 3.9% for speech therapy, 48.6% and 1.8% for immune tolerance induction or intravenous immunoglobulin, 47.3% and 11.2% for nutritional therapy, 15.1% and 17.0% for respiratory support, 27.8% and 3.7% for occupational therapy, and 8.3% and 11.1% for ambulatory support, respectively. Fourteen (IOPD: n = 2; LOPD: n = 12) patients switched ERT therapy, and 21 (IOPD: n = 14; LOPD: n = 7) had ≥1 dose modification.
Conclusions
Patients with Pompe disease demonstrate substantial comorbidity burden and utilization of supportive services despite ERT treatment.
Keywords
Introduction
Pompe disease is a rare, severe, autosomal recessive metabolic disease with a multi-systemic and heterogeneous clinical presentation. It is characterized by progressive neuromuscular degeneration initiated by disease-causing variants in the gene that encodes the lysosomal enzyme acid alpha-glucosidase (GAA).1–3 Clinical signs of Pompe disease emerge in individuals with GAA activity below 20–30% of average normal activity.4,5 Clinical manifestations can vary between patients and throughout the disease. 6 The estimated birth prevalence of Pompe disease in the United States is between 1:10,000 and 1:25,000. 7
Pompe disease can be categorized as infantile-onset Pompe disease (IOPD) or late-onset Pompe disease (LOPD). IOPD often presents within the first few months of life and, when untreated, is a severe and rapidly progressing disorder that usually results in early childhood death if not treated.8–10 IOPD is often characterized by hypotonia, cardiomegaly, cardiomyopathy, generalized muscle weakness, feeding difficulties, failure to thrive, and respiratory distress.9–11 LOPD has a heterogeneous presentation, with patients often presenting after 12 months of age; however, it can manifest at any age.5,6,9 LOPD is characterized by progressive muscle dysfunction and damage throughout the body and is less likely to be associated with cardiac symptoms. 6 Adult patients with LOPD typically develop respiratory and skeletal muscle dysfunction, often leading to a need for assisted ventilation and loss of ambulation in those who are untreated. 12
Currently, the standard of care for Pompe disease is exogenous GAA enzyme replacement therapy (ERT), which can also be used with the enzyme stabilizer miglustat.13–16 The introduction of ERT for the management of Pompe disease has improved the prognosis for patients by reducing mortality and disease progression. 17 However, ERT is not curative, and mortality rates among patients with IOPD are estimated to be between 28–58%.18–21 Limited existing literature on the humanistic burden of Pompe disease suggests that while ERT initially stabilizes deteriorating activities of daily living, symptoms worsen in later years following ERT initiation. 14 Additionally, patients treated with ERT continue to demonstrate significantly worse health-related quality of life (HRQoL) compared to the general population, 14 and specific symptoms, such as pain, are largely unaffected by ERT. 17 Treatment with ERT requires intravenous infusions every 1–2 weeks, which can take up to 7 h22,23 and can be time-consuming for patients and caregivers. 14
Real-world data describing the disease burden in patients with Pompe disease receiving ERT are limited.14,24 We conducted a retrospective cohort study in the United States using a comprehensive and broadly nationally representative claims database to describe the disease burden of Pompe disease over time in patients treated with ERT as assessed using (1) the occurrence of comorbidities of interest, (2) the use of concomitant supportive treatments, and (3) treatment patterns of ERT use, including dose modification.
Methods
Study design
This was a retrospective cohort study of patients with Pompe disease treated with GAA ERT (hereafter referred to as ERT) identified using the Merative™ MarketScan Research Databases (Figure 1). Patients were identified with records for ERT by National Drug Code or healthcare standard procedure coding system codes.
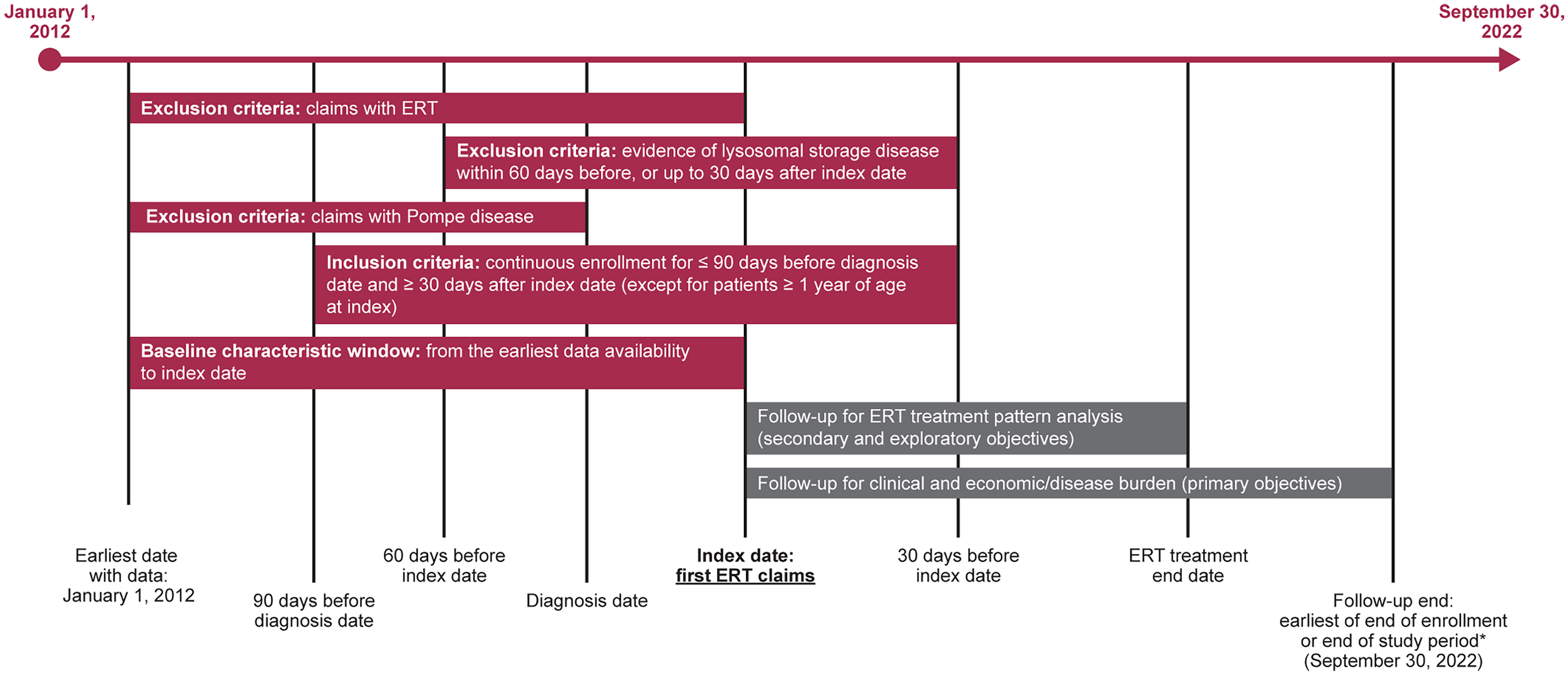
Study design. *It is possible that the follow-up and ERT treatment end dates are the same. This is not shown in the figure to avoid overcrowding. ERT, enzyme replacement therapy.
Database
The MarketScan Research Databases contain the inpatient, outpatient, and prescription drug experiences of employees and their dependents (>273 million unique patients) covered under a variety of fee-for-service and managed care health plans in the United States, including exclusive provider organizations, preferred provider organizations, point of service plans, indemnity plans, and health maintenance organizations. The medical claims are linked to outpatient prescription drug claims and person-level enrollment data through unique enrollee identifiers. Mortality data are not available in this database.
Inclusion/exclusion criteria
To be eligible for the study, patients had to meet all of the following inclusion criteria: ≥ 2 outpatient claims or ≥1 inpatient claim of Pompe disease (International Classification of Disease [ICD]-9 code 271.0 before October 1, 2015 or ICD-10 code E74.02 after October 1 2015) during the identification period (March 1, 2012 to August 31, 2022); ≥ 1 record of ERT (Myozyme, Lumizyme, or Nexviazyme); continuous enrollment in medical and prescription coverage for ≥90 days before diagnosis date for patients ≥2 years of age (waived for patients <1 year old) and ≥1-month post-index date. The diagnosis date was defined as the earliest date of claim with a diagnosis of Pompe disease during the study identification period. The index date was defined as the earliest date of ERT therapy that occurred on or after the diagnosis date.
Patients were excluded if they did not have a valid year of birth and/or sex or if they had any of the following: claims of Pompe disease during the pre-diagnosis period; claims of ERT during the baseline period; ≥ 2 outpatient or ≥1 inpatient claim of lysosomal storage disease (Gaucher disease, Fabry disease, and mucopolysaccharidoses) within 60 days before or up to 30 days after the index date.
Outcomes
This study aimed to describe the disease burden of Pompe disease in patients treated with ERT in the US. Primary outcomes included: 1) occurrence of each of the following comorbidities: cardiovascular abnormalities, respiratory manifestations, ambulatory muscle weakness, delayed motor milestones, central and peripheral nerve disorders, dry eyes/ophthalmoplegia, orthostasis, gastrointestinal dysfunction (including incontinence), sexual dysfunction, and bladder dysfunction, and 2) use of concomitant supportive treatments, including immune tolerance induction (rituximab, methotrexate or intravenous immunoglobulin), ambulatory support, respiratory support, occupational therapy, speech therapy nutritional therapy, physical therapy, and genetic counseling.
The secondary outcomes were to evaluate treatment patterns of ERT. This included: 1) frequency (weekly vs biweekly) of ERT and 2) duration of ERT (defined as the number of days from index date to date of ERT discontinuation for the first ERT treatment episode).
Exploratory outcomes were patterns of ERT dose modification, including the number of dose modifications (defined as increasing/decreasing dose per injection or increasing/decreasing injection frequency) and the days to the first dose modification.
Variables
Demographics and selected comorbidities of interest were described during the baseline period. Comorbidities in this study refer to distinct conditions that are known to be related to Pompe disease and were identified using unique diagnosis codes. The full list of diagnosis codes used in the study is included in the Supplemental Material.
Statistical analysis
Patients were stratified into IOPD or LOPD cohorts based on age at diagnosis and clinical presentation. Age data were only available in years (i.e., no months were recorded), therefore two alternative methods were used to identify IOPD patients. For the principal analysis, patients 0 or 1 year of age at the index date were identified as IOPD, and patients ≥2 years of age were identified as LOPD. A sensitivity analysis was conducted to confirm the accuracy of the IOPD cohort using cardiomyopathy as a variable. In the sensitivity analysis, patients 0 or 1 year of age at the index date with evidence of cardiomyopathy any time before or within 30 days of the index date were identified as IOPD; all other patients were identified as LOPD.
Key comorbidities, supportive services, and treatment modifications were presented as cumulative incidence. Kaplan–Meier analyses were used to describe time to key comorbidities, supportive services, and treatment modifications. Patients who already had a specific comorbid condition during baseline were assumed to have the condition on the index date. Cumulative incidence curves were produced for each comorbidity of interest, and the probability of having the condition was estimated at 0, 90, 180, 270, and 365 days along with 95% confidence intervals.
Frequencies and interquartile ranges were used to report the number and frequency of ERT administrations. The frequency of ERT administrations and frequency of ERT switching was described over the entire ERT treatment period, and the duration of each treatment episode was reported. All brands of alglucosidase alfa were treated as one drug when evaluating the ERT treatment pattern. Treatment duration was described on the treatment episode level and over the entire ERT treatment period. An ERT treatment episode spanned between the initiation and episode discontinuation or switching dates. Treatment discontinuation was defined as the patient having no claims for any ERT for ≥60 days. The ERT treatment period could include more than one ERT treatment episode for each patient. ERT treatment duration was defined as the time between the earliest evidence of ERT treatment and the earliest evidence of treatment discontinuation, switch to another ERT, end of enrollment or end of study period. Dose modification was defined as changes (increase or decrease) in dosing patterns, including dose per injection (e.g., 20 vs 40 mg/kg) and frequency of injections (e.g., weekly vs biweekly). The distribution of administrations that were weekly relative to the prior administration (defined as a < 12-day gap) versus biweekly (defined as a ≥ 12-day gap) were summarized.
No statistical comparisons were performed between the IOPD and LOPD groups. The sample size of this study was not based on an a priori hypothesis since all study objectives were descriptive, with no planned hypothesis testing.
Results
Baseline characteristics
A total of 2735 patients with ≥2 outpatient claims or ≥1 inpatient claim of Pompe disease between March 1, 2012, and August 31, 2022, were identified. After applying all inclusion/exclusion criteria, 105 patients were eligible and included in the analysis, 50 with IOPD and 55 with LOPD (Table 1). Baseline characteristics for the IOPD and LOPD cohorts are presented in Table 2. Overall, the mean (standard deviation [SD]) length of follow-up was 914.3 (842.9) and 987.3 (866.6) days in the IOPD and LOPD cohorts, respectively.
Patient attrition.

ERT, enzyme replacement therapy; IOPD, infantile onset Pompe disease; LOPD, late onset Pompe disease.
Baseline characteristics.

Race reported for patients on Medicaid insurance only.
Category used when brand name not clarified in the database.
ERT, enzyme replacement therapy; GI, gastrointestinal; IOPD, infantile onset Pompe disease; LOPD, late onset Pompe disease; max, maximum; min, minimum; Q1, first quartile; Q3 third quartile; SD, standard deviation.
Disease burden
Comorbidities of interests
The most common comorbidities were respiratory (IOPD: 90%; LOPD: 83.6%), cardiovascular (IOPD: 28%; LOPD: 56.4%), ambulatory (IOPD: 76%; LOPD: 80%), and gastrointestinal (IOPD: 72%; LOPD: 74.5%). Cumulative incidence curves for comorbidities of interest are shown in Figure 2. In the IOPD cohort, the 12-month cumulative incidence for the most commonly occurring comorbidities was 84.5% for respiratory, 57.4% for ambulatory, 67.8% for gastrointestinal, and 16.9% for cardiovascular comorbidities. In the LOPD cohort, the 12-month cumulative incidence for the most commonly occurring comorbidities was 79.4% for respiratory, 54.3% for ambulatory, 33.4% for gastrointestinal, and 28.7% for cardiovascular comorbidities. 12-month cumulative incidence for central and peripheral nerve disorders, orthostasis, dry eyes, sexual dysfunction, and bladder dysfunction was <20% in both cohorts.
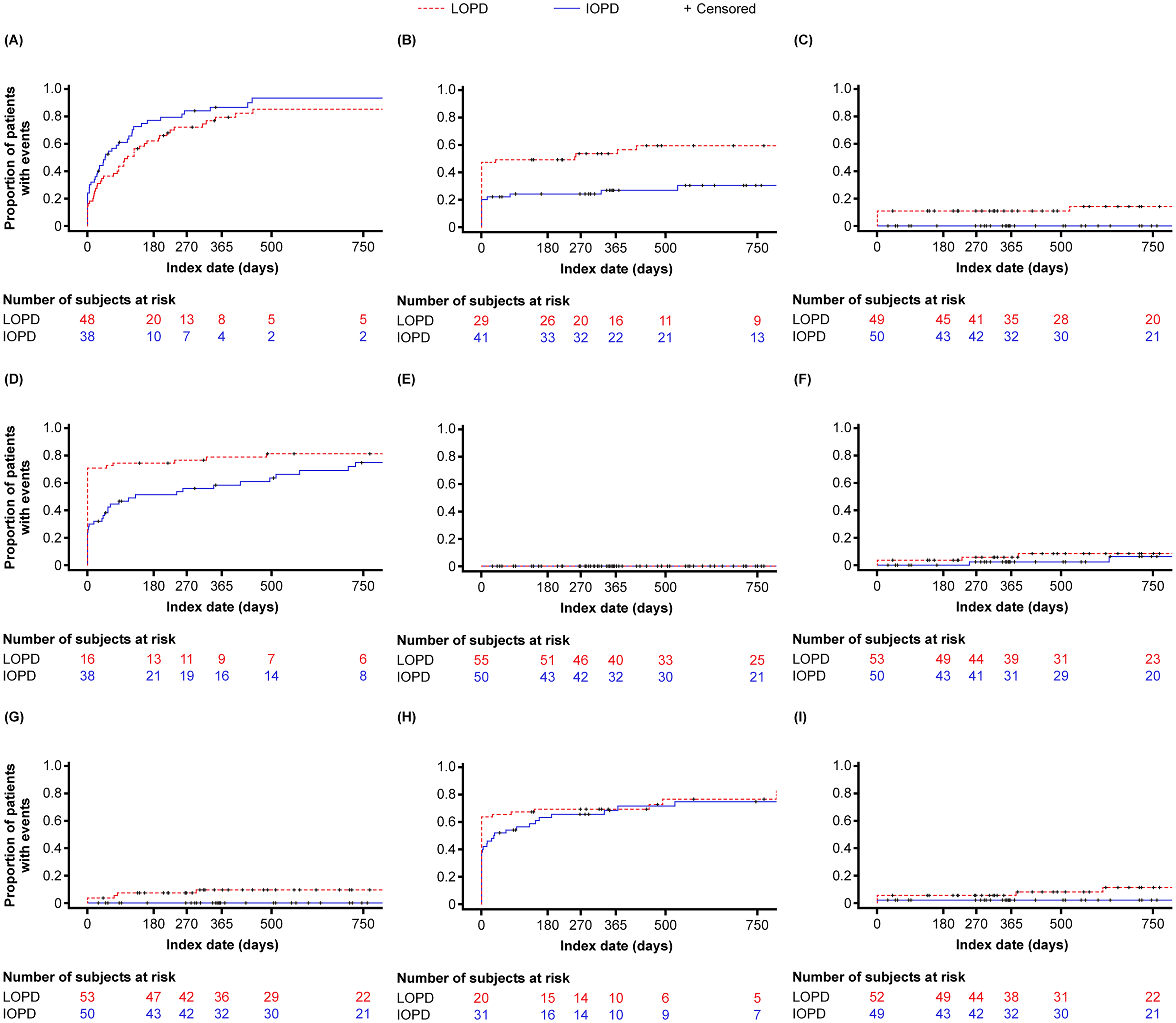
Cumulative incidence of comorbidities of interest. (A) Respiratory manifestations. (B) Cardiovascular abnormalities. (C) Central and peripheral nerve disorders. (D) Ambulatory muscle weakness or delayed motor milestones. (E) Orthostasis. (F) Dry Eyes. (G) Sexual dysfunction. (H) Gastrointestinal dysfunction. (I) Bladder dysfunction. IOPD, infantile-onset Pompe disease; LOPD, late-onset Pompe disease.
Concomitant services
Most common supportive services were immune tolerance induction or intravenous immunoglobulin (IOPD: 48%; LOPD: 1.8%), ambulatory support (IOPD: 22%; LOPD: 21.8%), respiratory support (IOPD: 22%; LOPD: 20%), occupational therapy (IOPD: 38%; LOPD: 20%), speech therapy (IOPD: 76%; LOPD: 12.7%), nutritional therapy (IOPD: 52%; LOPD: 18.2%), and physical therapy (IOPD: 80%; LOPD: 50.9%).
Cumulative incidence curves for concomitant services of interest are shown in Figure 3. In the IOPD cohort, the 12-month cumulative incidence was 48.6% for immune tolerance induction or intravenous immunoglobulin, 8.3% for ambulatory support, 0% for genetic counselling, 15.1% for respiratory support, 27.8% for occupational therapy, 59.5% for speech therapy, 47.3% nutritional therapy, and 66.9% for physical therapy. In the LOPD cohort, the 12-month cumulative incidence was 1.8% for immune tolerance induction or intravenous immunoglobulin, 11.1% for ambulatory support, 3.7% for genetic counselling, 17% for respiratory support, 3.7% for occupational therapy, 3.7% for speech therapy, 11.2% for nutritional therapy, and 31.8% for physical therapy.
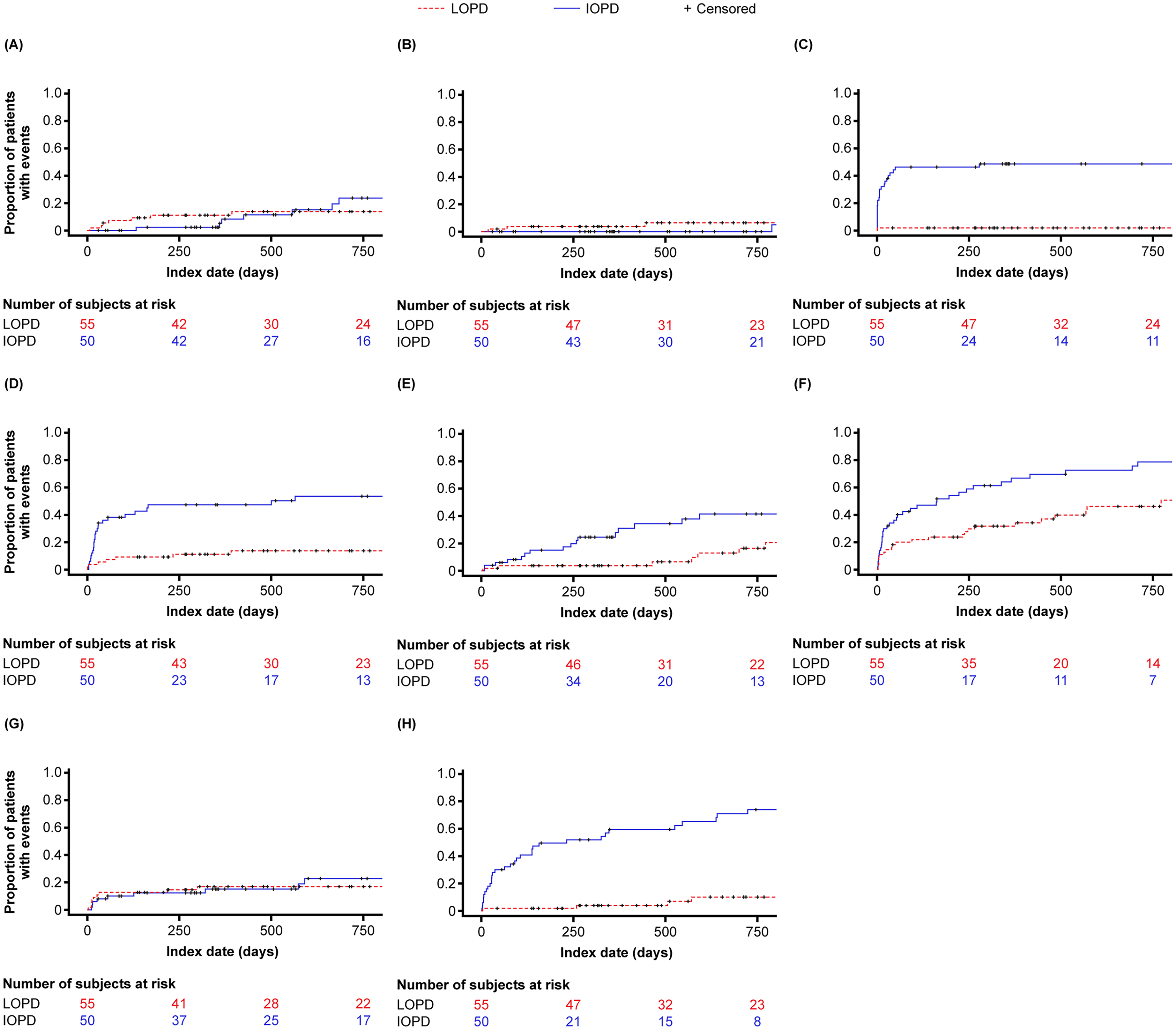
Cumulative incidence of supportive services of interest. (A) Ambulatory support. (B) Genetic counseling. (C) Immune tolerance induction and intravenous immunoglobulin. (D) Nutritional therapy. (E) Occupational therapy. (F) Physical therapy. (G) Respiratory support. (H) Speech therapy. IOPD, infantile-onset Pompe disease; LOPD, late-onset Pompe disease.
ERT treatment patterns
Frequency of ERT administrations
In the IOPD cohort, 59.3% of alglucosidase alfa administrations were biweekly, and 40.7% were weekly. Two (4%) patients in the IOPD cohort had a treatment switch, consistent with alglucosidase alfa being the only approved ERT for IOPD. In the LOPD cohort, most alglucosidase alfa administrations were biweekly (94%). The frequency of avalglucosidase alfa administrations was most commonly biweekly (87%). In the LOPD cohort, 12 (21.8%) patients had a treatment switch between alglucosidase alfa and avalglucosidase alfa.
ERT treatment duration
In the IOPD cohort, the Kaplan–Meier estimated median (interquartile range [IQR]) treatment duration for the first ERT treatment episode was 2072 days (182–not available [NA]). After 12 months, 35.3% of patients had discontinued the first ERT, and after 24 months, 48.7% of patients had discontinued the first ERT (Figure 4). In the IOPD cohort, the Kaplan–Meier estimated median (IQR) treatment duration over the entire treatment period was 2981 (2320–NA) days (i.e., 8.2 years). After 12 months, 10.3% of patients had discontinued ERT, and after 24 months, 14.1% had discontinued ERT. In the LOPD cohort, the Kaplan–Meier estimated median (IQR) treatment duration for the first ERT episode was 553 days (210–2497). After 12 months, 33% of patients discontinued their first ERT, and after 24 months, 54.5% discontinued their first ERT (Figure 4). The Kaplan–Meier estimated median (IQR) treatment duration over the entire treatment period was 1510 (553–NA) days (i.e., 4.1 years). After 12 months, 11.7% of patients discontinued ERT; after 24 months, 32% of patients discontinued ERT.
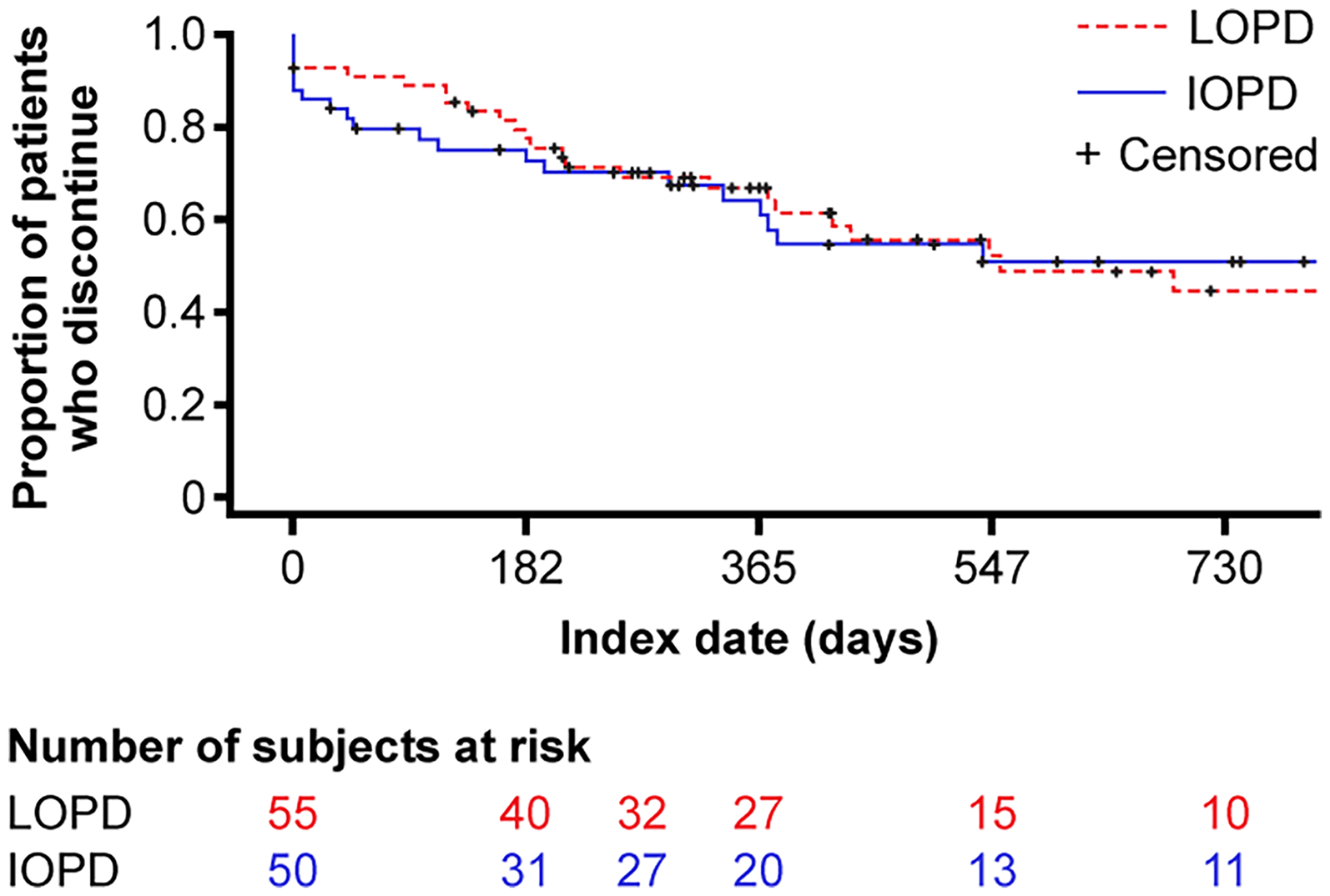
Time to discontinuation of enzyme replacement therapy. IOPD, infantile-onset Pompe disease; LOPD, late-onset Pompe disease.
Patterns of dose modification
Thirty-three and 34 patients had valid data to evaluate ERT dose in the IOPD and LOPD cohorts. In the IOPD cohort, 14 (42.4%) patients had a change in dose. A total of 12 (36.4%) patients had a dose decrease, and 14 (42.4%) had a dose increase. Kaplan–Meier median (IQR) time to first dose increase was 1491 (378–2073) days (i.e., 4.1 years), and median (IQR) time to first dose decrease was 3016 (511–3234) days (Figure 5). In the LOPD cohort, 7 (20.6%) patients had a change in dose. Five (14.7%) patients had a dose decrease, and 6 (17.6%) had a dose increase. Kaplan–Meier median (IQR) time to first dose increase was not estimable (NE) (2249–NE) days, and the median (IQR) time to first dose decrease was NE (2235–NE) days (Figure 5).
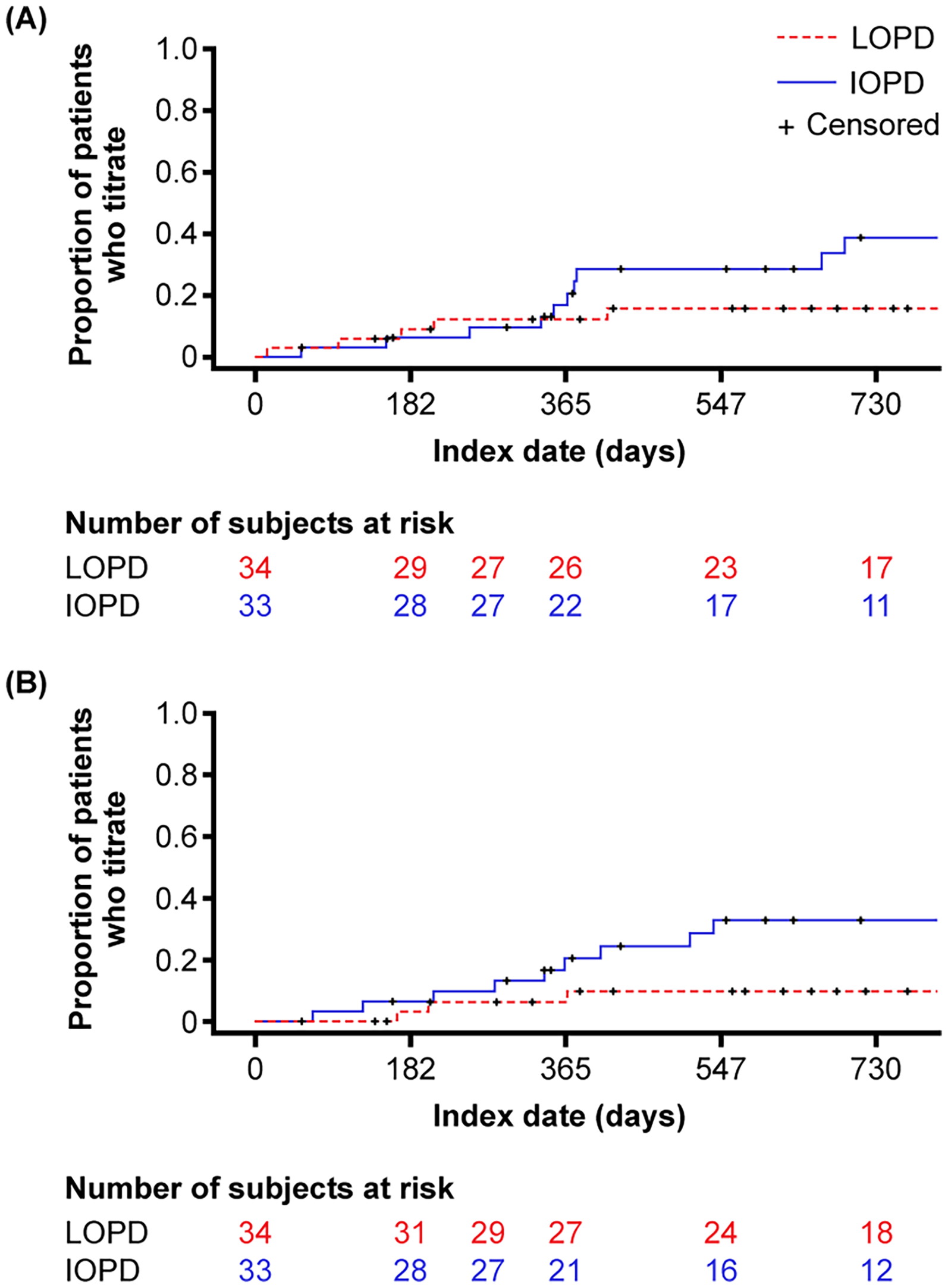
Time to (A) dose increase and (B) dose decrease of enzyme replacement therapy. IOPD, infantile-onset Pompe disease; LOPD, late-onset Pompe disease.
Discussion
This real-world analysis showed that patients with Pompe disease in the United States experience a substantial disease burden despite treatment with standard-of-care ERT, with high rates of comorbidities, utilization of supportive services, and concomitant medications. Given the dearth of real-world data currently available, our findings, obtained using a sizable and largely representative US-based claims database, provide valuable information on clinically important aspects of patients with Pompe disease treated with ERT.
In both IOPD and LOPD subgroups, the cumulative incidence of most comorbidities increased over time, particularly for respiratory infections, and around 80% of patients in both cohorts had respiratory comorbidities at 12 months. Although published data on the disease burden of Pompe disease are limited, particularly for those with IOPD, our findings are consistent with recent real-world data in patients with LOPD. Lumgair et al. (2025) 25 found that the most frequently reported symptoms among a cohort of LOPD patients, which were also noted as being the most burdensome, were muscle weakness (98%), fatigue (95%), mobility issues (93%), pain (90%), and breathing difficulties (83%). All symptoms were experienced consistently regardless of ERT duration.
The 12-month cumulative incidence for supportive services showed that around half of IOPD patients required immune tolerance induction/intravenous immunoglobulin, speech therapy, and/or nutritional therapy, and approximately two-thirds of IOPD patients and one-third of LOPD patients required physical therapy. However, it is important to note that the management of IOPD has substantially changed during the study period, with the adoption of immunomodulation strategies to induce immune tolerance of ERT and the use of higher dose of ERT (20 mg/kg/week instead of 20 mg/kg/every other week).26–28 Additionally, the utilization of newborn screening to increase early identification of IOPD and initiation of treatment may have resulted in improved clinical outcomes during the study period. 29
The use of occupational, speech, and physical therapies increased over time for both groups. Although the substantial use of supportive services on patients’ HRQoL was not available for analysis in our study, the significant impact of LOPD on the day-to-day activities, outcomes, and HRQoL of patients has been reported in the literature. Patient-reported outcome measures have been used to demonstrate that the clinical signs and symptoms (e.g., muscle weakness and atrophy, fatigue, and pain) of LOPD adversely affected patients’ physical, emotional, and social well-being. 30 Patients with LOPD needed substantial medical care and support with daily activities, with many having to limit or cease their employment. 31 A recent study in LOPD patients found that work was the most impacted aspect of day-to-day life (73% reporting a high impact on work-life), followed by the ability to travel (59%), social life (56%), and family life (46%). 25
ERT requires frequent, persistent administration, causing significant burden for patients, caregivers, and health systems. In the present study, biweekly administrations of ERT were most common; however, 41% of administrations for IOPD were weekly. Treatment with ERT for more than a decade has been associated with additional burdensome symptoms, such as sleep issues and speaking difficulties, with 60% of participants experiencing declining LOPD status on ERT for more than 10 years. 25 The treatment burden associated with ERT could potentially explain the high rates of discontinuation seen in both the IOPD (12 months: 35%; 24 months: 49%) and LOPD (12 months: 33%; 24 months: 55%) cohorts in our study. However, it is also possible that ERT discontinuation rates were overestimated as any individual with an interruption in treatment ≥60 days were considered to have discontinued therapy regardless of whether they received treatment afterwards. Patients enrolled in clinical trials may also account for the overestimation of discontinuation rates since ERT administered as part of the clinical trial may not be coded and would therefore not be identified in the data analyzed. Since the reason for treatment discontinuation cannot be determined in a claims-based analysis, the high discontinuation rates seen in this study remain largely unexplained. It is possible that some patients may have re-started ERT, switched ERTs, or were treated elsewhere.
Clinically, as IOPD and LOPD are characterized by differing enzyme deficiency severity and disease and comorbidity profiles, it would be expected that varying disease burdens for these phenotypes will be observed, even with treatment. Specifically, IOPD patients have a severe or complete GAA deficiency (<1% residual enzyme activity) with more severe cardiac involvement with hypertrophic cardiomyopathy, hypotonia, and respiratory insufficiency. In contrast, LOPD is caused by a partial deficiency (<30% residual enzyme activity) of GAA, which presents primarily as a skeletal muscle disease and has a more insidious course.7,32 In addition, since LOPD is phenotypically heterogeneous, patients who have survived to adulthood without receiving treatment are likely to be those on the milder end of the spectrum and, as such, may require less care and medical intervention than those patients with more severe forms of the disease. 17 Furthermore, LOPD is not generally associated with cardiovascular abnormalities. The unexpectedly higher incidence of cardiovascular abnormalities in the LOPD versus IOPD cohort likely reflects an older population with common cardiovascular abnormalities related to aging rather than Pompe-associated cardiomyopathy. Notably, our study identified almost similar numbers of LOPD and IOPD patients in the database, which is not reflective of the known epidemiology evaluated in population-based studies, even accounting for the variation in prevalence between countries depending on newborn screening rates.33,34
Several limitations to this study must be addressed. Firstly, the MarketScan data may not fully represent the United States population as its payer mix differs from the broader national distribution. Furthermore, uninsured patients and those covered by military-based insurance were excluded from this study. Although the Merative™ Databases have mechanisms in place to link individuals as they change employers or health plans, the mechanisms are not perfect and as such it is possible that some individuals may re-appear in the database with a different unique ID. Since only the year (not month) of birth was available in the database, some misclassification of IOPD and LOPD may have occurred in patients who were diagnosed between 1 and 2 years of age. Additionally, it is possible that cardiac abnormalities for some infants may have been coded as Pompe disease or that early infant records may have been coded under the mother's health record. Claims databases only record diagnostic and procedure codes for reimbursement purposes; measures of severity or prognostic factors are not available; thus, detailed clinical information is not available to describe the study population using the claims data. For example, there was no specific ICD-9 diagnosis code for Pompe disease before October 1, 2015, making it more challenging to correctly identify patients diagnosed with Pompe disease before then. Accurate diagnosis of Pompe disease usually requires multiple tests over a range of time, thus making it challenging to identify a precise diagnosis date; therefore, the inclusion/exclusion criteria were designed to require both evidence of diagnosis with Pompe disease and proof of initiation of ERT to ensure the patients in the cohort were indeed patients with Pompe disease treated with ERT. However, this approach excluded patients with Pompe disease who relied only on supportive care and did not utilize ERT.
Furthermore, early diagnosis and treatment due to newborn screening may reduce disease burden in IOPD; however, this was not evaluated in our study. Mortality data are not available in the MarketScan data; as such, patients who died before the end of the study period would be considered censored due to disenrollment. Since this population's mortality rate is high, the cumulative incidence of outcomes/events of interest may be overestimated. Lastly, a new ERT plus oral enzyme stabilizer (cipaglucosidase alfa plus miglustat) was recently approved in the US after the end of the study period and was therefore excluded from this analysis. 16
In conclusion, patients with Pompe disease experience a substantial disease burden despite treatment with standard-of-care ERT, with high rates of comorbidities, utilization of supportive services and concomitant medications, and relatively high rates of ERT discontinuation. Our findings highlight the need for alternative therapies to support patients with Pompe disease and reduce their burden.
Supplemental Material
sj-xlsx-1-jnd-10.1177_22143602251387210 - Supplemental material for A retrospective cohort study describing the disease burden in patients with Pompe disease treated with enzyme replacement therapy in the United States
Supplemental material, sj-xlsx-1-jnd-10.1177_22143602251387210 for A retrospective cohort study describing the disease burden in patients with Pompe disease treated with enzyme replacement therapy in the United States by Nishitha R Pillai, Faryn Solomon, Robert D Steiner, Bin Xie, Tmirah Haselkorn, Christopher Young, Nigel Rozario, Mark Walzer and Benedikt Schoser in Journal of Neuromuscular Diseases
Footnotes
Acknowledgements
Bassaam Mulk, PharmD, of Alpha (a division of Prime, Knutsford, UK), provided medical writing and editorial support under the authors’ direction. Astellas Pharma Inc. funded the support according to Good Publication Practice guidelines (Link). The authors are responsible for all opinions, conclusions, and data interpretation.
Ethical considerations
Approval by an ethics committee was not required for this claims-based analysis.
Author contributions
Nishitha R. Pillai contributed to the conceptualization, methodology, software, validation, formal analysis, investigation, and data curation. Faryn Solomon contributed to the conceptualization, methodology, investigation, supervision, and project administration. Robert Steiner contributed to the methodology and data interpretation. Bin Xie, Christopher Young, and Nigel Rozario contributed to the conceptualization, methodology, software, validation, formal analysis, investigation, and data curation. Tmirah Haselkorn contributed to the conceptualization, methodology, validation, investigation, supervision, project administration, and funding acquisition. Benedikt Schoser contributed to the conceptualization, validation, investigation, data curation, supervision, and project administration. All authors contributed to the development of the manuscript and provided final approval of the manuscript.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was sponsored by Astellas Pharma Inc. All costs related to publication were funded by Astellas Pharma Inc.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Nishitha Pillai reports no conflicts of interest. Benedikt Schoser has participated in the following advisory boards for Amicus, Argenx, Astellas, Avidity, Pepgen, and Sanofi; holds research grants from Marigold Foundation, AMDA Foundation, EU Horizon 2022 COMPASS, EU 2022 PALADIN, and EU Horizon 2022 DM-Entry; and has received speaker's honoraria from Amicus, Alexion, Argenx, Dyne, Kedrion, and Sanofi. Robert D. Steiner has been a consultant for Exact Sciences, Mirum, PTC, and Ultragenyx, has received travel support from Astellas, and has equity in Exact Sciences. He was not compensated for the work related to writing and editing this manuscript. Faryn Solomon, Bin Xie, Tmirah Haselkorn, Christopher Young, Nigel Rozario, and Mark Walzer, are employees of Astellas Pharma Global Development, Inc.
Data availability
All data generated or analyzed during this study that support the findings of this study, are included within this article and its supplementary information files. Researchers interested in further analysis not presented in the manuscript may contact the corresponding author.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.