
Abstract
Background
Duchenne muscular dystrophy (DMD) is a severe muscle disease with an unmet therapeutic need. Despite ongoing research efforts, only a few medicines have achieved marketing authorisation in the EU to date. We present a regulatory science overview summarising the insights obtained from the evaluation by the Paediatric Committee (PDCO) of the European Medicines Agency (EMA) of paediatric investigation plans (PIPs) for this condition, with the primary objective of providing recommendations for future developments.
Methods
We reviewed the PIPs approved by the PDCO and related regulatory procedures analysimg the available information in our own databases completing it with a search in the EU Clinical Trials Register (EudraCT) and Clinical Trials Information System (CTIS) databases to provide context.
Results
Between January 2005 and December 2024, 16 PIPs were agreed. By 2024, 1 PIP has been completed, the remaining ones are ongoing. So far, 3 medicines for the treatment of DMD have received a positive opinion by EMA's Committee for Medicinal Products for Human Use (CHMP). Main characteristics and critical factors contributing to successful developments are outlined.
Conclusions
This study presents the first in-depth evaluation of PIPs approved within the EU for DMD offering insights into potential strategic approaches for clinical development of investigational medicinal products. It highlights the accumulated experience of regulators and stakeholders, particularly regarding pivotal trials that establish clinical efficacy in key patient subgroups. Furthermore, it underscores the emerging value of innovative methodologies – such as extrapolation of efficacy and integration of real-world evidence – while acknowledging persistent challenges related to data quality.
Introduction
Duchenne muscular dystrophy
Duchenne muscular dystrophy (DMD) is a rare, progressive, degenerative muscle disease causing muscular weakness, impaired cardiac and pulmonary function, leading to a need for assisted ventilation and to premature death. 1 DMD is caused by different mutations across the gene encoding for dystrophin, located in the short arm of the X chromosome, including single-exon or multi-exon deletions (65–70%) or duplications (6–10%), small point mutations and other smaller rearrangements (in around 25–30% of cases).2,3 Whilst 70% of cases occur in patients with a known family history, approximately 30% of cases are caused by de novo mutations. 4 Approximately 7900 genetic variants have been discovered as responsible for DMD and for the less severe Becker muscular dystrophy (BMD) phenotype. 5 The worldwide estimated prevalence is 7.1 cases per 100,000 males and 2.8 cases per 100,000 in the general population. 6
Diagnosis is typically established in boys 2–4 years of age upon the onset of the first symptoms counteracting natural muscle development in children. This usually first involves weakness in the lower limbs and progresses with loss of ambulation and involvement of the upper limbs by around 12–15 years of age. Finally, during adolescence and adulthood, heart and lung function progressively decrease, with respiratory failure and cardiomyopathy leading to death around 25–30 years of age. 7
The standard of care includes coordinated multidisciplinary approach. Pharmacological therapies include the use of glucocorticoids (prednisone or deflazacort) which have shown a positive effect in preserving locomotor, heart and lung functions.8,9,10 Despite efforts made in medicines development by numerous pharmaceutical companies as well as patient organisations, academics and health authorities worldwide, the therapeutic armamentarium to treat DMD is still insufficient to fulfil the medical need in people with this condition. Even though some medicines have received regulatory approval in the USA, Europe and Japan for treatment of DMD, to date there is no curative treatment yet.
Regulatory aspects in the EU
The Paediatric Regulation (EC) No 1901/2006 came into force in the EU to support the development and clinical investigation of medicines in the paediatric populations. 11 For every new application for a marketing authorisation of a medicine, applicants are obliged, according to EU law, to submit a paediatric investigation plan (PIP), which must have been previously agreed by European Medicines Agency (EMA)'s Paediatric Committee (PDCO). A PIP includes, under a designated condition, all required studies to collect efficacy and safety data on the use of a given investigational medicinal product (IMP) (a substance or combination of substances being tested in a clinical trial) in a paediatric population in a reliable and robust manner. Conducted under agreed conditions, these studies are intended to support the inclusion of a paediatric indication in the medicine's marketing authorisation within the European Union. 12 Waivers to such requirements can be agreed for all or selected paediatric age sub-groups, based on specific scientific and legal grounds. The PDCO has been reviewing PIPs for IMPs for the treatment of DMD since the Paediatric Regulation came into force in 2007.
The PDCO opinions are the PDCO outcome documents and are published on the EMA website. However, they are made available only in an abridged form, since some parts of them are commercially confidential and cannot be published in full at the time of agreement of a PIP. In its full version such a document includes both the initial proposal of the developer (most often the pharmaceutical industry) for a paediatric development plan, the PDCO's assessment of such a proposal, as well as a final outcome section that describes all the planned (clinical) studies that are included in a PIP as finally agreed by the Committee. For each study included there are defined ‘key elements’ – which resemble a study synopsis – that need to be adhered to by the sponsors when performing the studies. Agreed PIPs can be modified and updated as needed when new data becomes available.
A scientific-regulatory guidance was published in 2015 for adult-paediatric development of medicines for DMD (Guideline on the clinical investigation of medicinal products for the treatment of Duchenne and Becker muscular dystrophy). 13 However, various stakeholders have highlighted the need for further guidance to facilitate and streamline drug development in this area since some trials were initiated and development is progressing, but others were terminated prematurely and were not completed. 14 Whilst a PIP is mandatory by law, applicants can also voluntarily request scientific advice (SA) on any aspect of paediatric development planned to be included in a PIP (or not, e.g., because paediatric requirements don’t apply) from EMA's Scientific Advice Working Party (SAWP) of the CHMP. 15
The developer of an IMP identifies questions and possible solutions: e.g., adequacy of paediatric formulation development plans, non-clinical studies to support paediatric clinical trials, design elements of clinical studies enrolling a paediatric population, or other aspects which can guide generation of robust evidence on a medicine's benefits and risks for marketing authorisation, and a Scientific Advice Letter is issued in response. These letters are commercially confidential and are free of charge if they only include questions on paediatric development but, as opposed to PIPs, would not cover the entire development of the IMP. In fact, since they are structured in a way to respond to specific questions posed by the sponsor, they would cover only specific aspects. In other words the same aspect may have already been agreed in principle in a PIP but might need further refinement (e.g., strategy to validate a pharmacodynamic end-point, dose selection strategy, etc) or would help in building a PIP to be submitted later. Submitting request for scientific advice/protocol assistance (see below) in parallel to a PIP submission/amendment review is generally discouraged, except if specifically proposed by PDCO.
In addition, IMPs for the treatment of DMD might be eligible for an orphan designation (OD). Applying for an OD is not mandatory, but most developers make use of this provision as it offers pre- and post-marketing incentives. 16 When an OD is granted by the Committee for Orphan Medicinal Products (COMP), the medicine is eligible for protocol assistance (PA), which is SA for OD products, at a fee reduction or without fee at all.
Here, we review the PIPs for treating DMD that have been agreed by the PDCO. The aim of this review is to provide an overview of these PIPs to look at experience gained by regulators in this field, identify the main challenges of development at the planning stage and discuss alternatives to facilitate and support clinical development for DMD medicines authorisation in Europe.
Registration of paediatric clinical trials
Once a paediatric study is planned to start, it needs to be recorded in the EudraCT 17 database or, for trials which are expected to continue after 30 January 2025, in the CTIS 18 database. These databases contain information on interventional clinical trials conducted in the EU or the European Economic Area (EEA). We also wanted to understand how many of these developments were going ahead observing how many PIP trials had begun.
Methods
From the EMA's internal record management system, we selected the all the regulatory procedures related to medicines developed for DMD and all PIPs submitted for the treatment of DMD since entry into force of the EU Paediatric Regulation (2007 to 2024) by applying selected filters and identified all relevant outcome documents.
From the information included in the PDCO Opinions we built a dedicated database recording quantitative information on number of studies per PIP, their type, study design, age of the subgroups, functional status of patients included, end-points, number of patients required.
In addition, for other aspects for which we considered there was no value in gathering quantitative information, as not systematically recorded in the PDCO Opinions, we performed a scoping review gathering qualitative information on the scientific rationale supporting the development chosen for each medicine as well as identified challenges in the whole dossier.
We included in this exercise also the Scientific Advice Letters provided on developments in DMD. Such review was not systematic but focused on specific aspects, including but not limited to, the role of biomarkers and the role of innovative types of evidence generating tools. These key aspects were identified through collaborative discussion performed between the paediatric team of the French National Agency for the Safety of Medicines and Health Products (ANSM), EMA's Paediatric Medicines Office, EMA's Office for Therapies for Neurological and Psychiatric Disorders, as well as members of the PDCO.
To complete the overview and to provide context and understand how many of the studies included in the PIPs were started or conducted, we performed a detailed search in publicly available clinical trials registers. These registers contain information on interventional clinical trials conducted in the EU or the EEA. This was needed since each PIP contains all the planned studies to be conducted for a given medicine. However, EMA does not have oversight of which ones are effectively started until a PIP is effectively concluded or its discontinuation is notified to the Agency.
The search of the EudraCT database to retrieve all clinical trials (CTs) authorised within and outside the EU was carried out with the key words “treatment of DMD” or “Duchenne” from 2005 (to include studies that had been started before a PIP was submitted for approval) up to 2024 to provide background to the review and understand how many PIP trials had progressed from the pure planning phase. The status of each CT was first checked in the EudraCT database and was also cross-checked with information from Clinicaltrial.gov. 19 For 2024, an additional search was also carried out in CTIS to retrieve all CTs registered in this novel platform.
Results
PIPs and related EMA procedures
Up to December 2024, 25 PIPs were submitted for evaluation to the PDCO for individual IMPs for the treatment of DMD, of which 16 were agreed and 5 are under evaluation; 1 PIP had been fully completed. Five developments so far are not progressing, after a PIP was agreed (Figure 1); it should be noted that, with the exception of two IMPs, we cannot specify if the development of the medicinal product has been discontinued or not, unless the EMA has been informed by the company accordingly. The remaining ones are expected to be ongoing (Table 1).
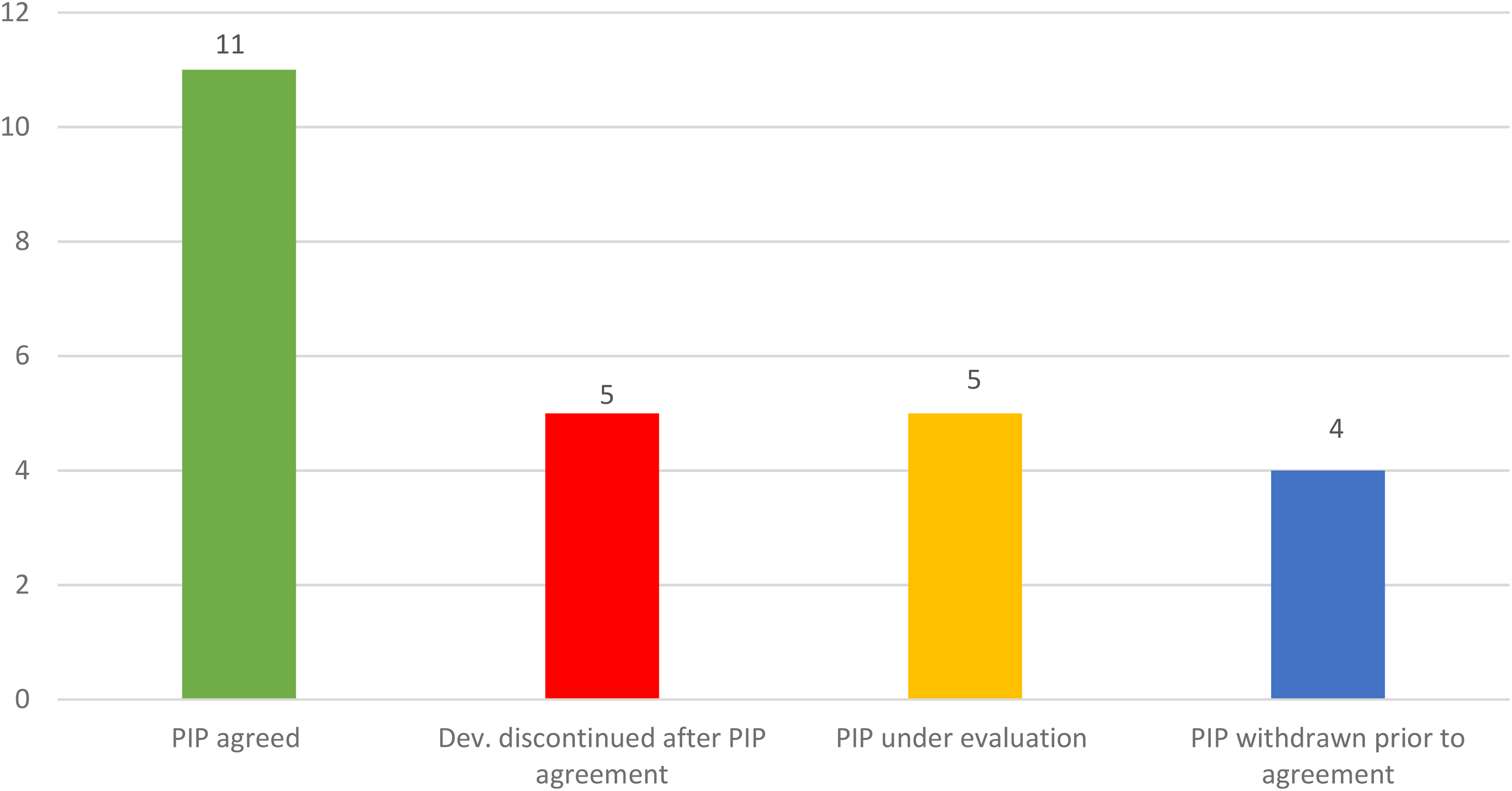
PIPs and development status. Whilst some plans are still under evaluation at the time of writing this article, many of them have been agreed by EMA and reflect an ongoing development. However, 4 submissions for PIPs were withdrawn before agreement and development seems discontinued for other 5 previoulsy agreed PIPs (see also Table 1 for details).
Details of PIPs submitted over the study period (2005–2024) and their statuses as of December 2024.
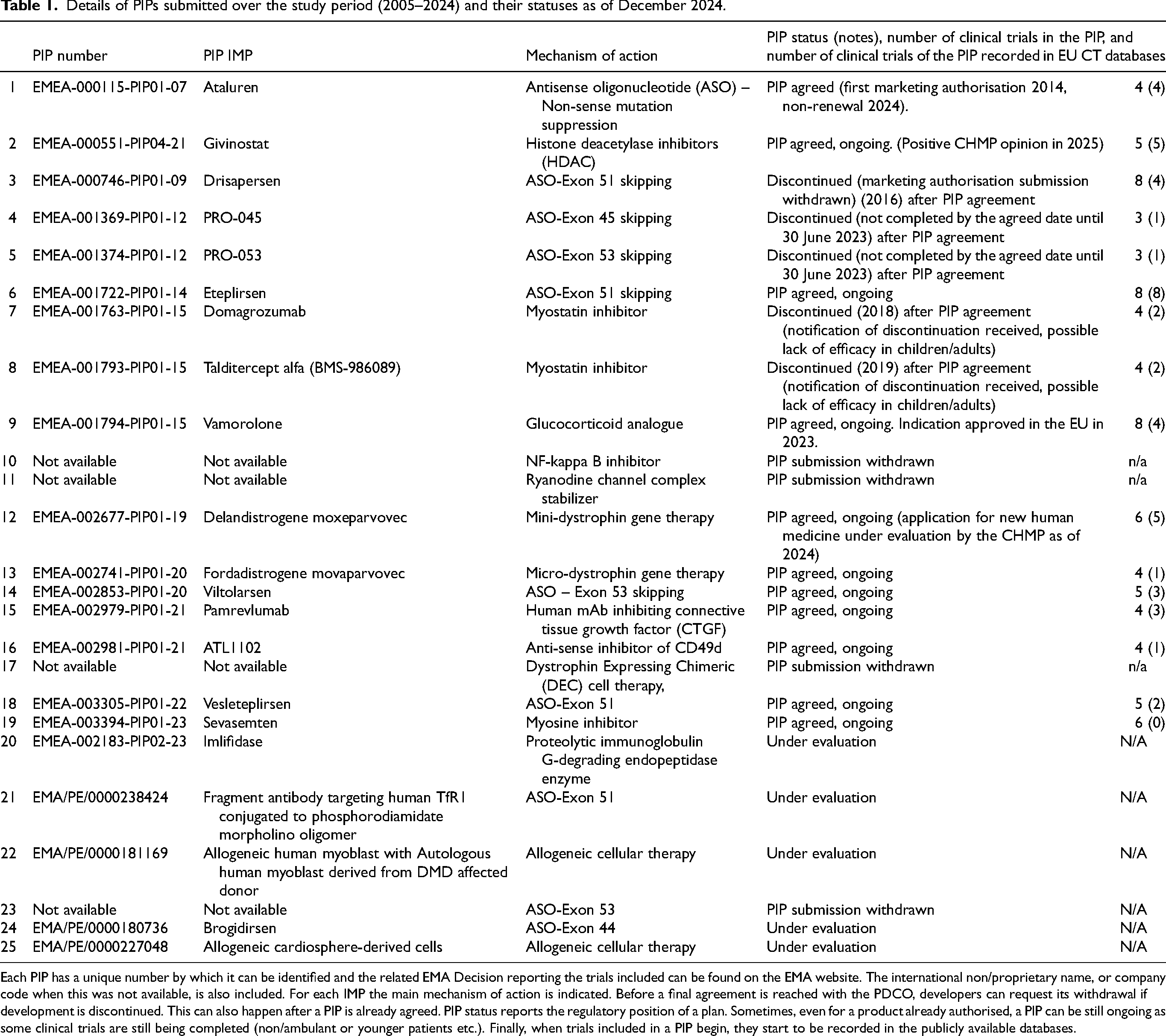
Each PIP has a unique number by which it can be identified and the related EMA Decision reporting the trials included can be found on the EMA website. The international non/proprietary name, or company code when this was not available, is also included. For each IMP the main mechanism of action is indicated. Before a final agreement is reached with the PDCO, developers can request its withdrawal if development is discontinued. This can also happen after a PIP is already agreed. PIP status reports the regulatory position of a plan. Sometimes, even for a product already authorised, a PIP can be still ongoing as some clinical trials are still being completed (non/ambulant or younger patients etc.). Finally, when trials included in a PIP begin, they start to be recorded in the publicly available databases.
The number of PIPs agreed is a much higher number compared to other neuromuscular diseases for which PIPs have been submitted in the same period. As a way of comparison, 10 PIPs were submitted for spinal muscular atrophy, 2 for myotonic dystrophy, 3 for Friederich's ataxia, 1 for facioscapulohumeral muscular dystrophy (FSHD), and 2 for limb-girdle muscular dystrophy.
So far, 3 medicines for the treatment of DMD had received a positive opinion by EMA's CHMP. These include Translarna (ataluren) which received a conditional marketing authorisation (CMA) in 2014, with a therapeutic indication restricted to DMD boys with a nonsense mutation. This CMA, nevertheless, was not renewed in 2024 after new evidence failed to confirm the efficacy of the medicine, the CHMP concluding that the benefit-risk balance was negative.
The second approved medicine, Agamree (vamorolone), was granted marketing authorisation in 2023. The pivotal study of the development was a randomised, double-blind, parallel-group, 48-week study, to evaluate the efficacy, safety, PD and population PK of vamorolone in ambulant, corticosteroid-naïve, 4-to-less-than-7-year-old boys with DMD. To support results from the pivotal study, the developer provided efficacy results from a phase 2 open-label, multicentre extension study to assess the long-term safety and efficacy of vamorolone in boys with DMD. This study used the CINRG-NHS registry as an external control arm 43 as supportive additional data.
A third medicine, Duvyzat (givinostat), was granted marketing authorisation in 2025 based on data from a subgroup of 120 patients (79 treated with givinostat and 41 with placebo) in a randomised, placebo-controlled study in ambulant DMD patients aged 6 years or older who were on concomitant steroid treatment. The primary endpoint was the change of time to complete a timed 4-stair climb (4SC) at 18 months. The differences between treatment groups were statistically significant, with the 4SC time increasing by an average of only 1.25 s in patients treated with givinostat compared to 3.03 s in those in the placebo group.
Both medicines have a therapeutic indication covering all DMD boys independent of the genotype but so far restricted to some age groups (other studies in other age groups are progressing).
Within the study period, the yearly number of DMD PIP submissions has remained relatively stable. However, the requests for SA linked to these PIPs has increased over time from a mean of 1.44 annual SA requests seen initially (period 2005–2013) compared to a mean of 5 annual requests more recently (period 2014–2024) (Figure 2 Number of PIPs, SA procedures and global Clinical Trials (CTs) over the study period (2005–9/2024).
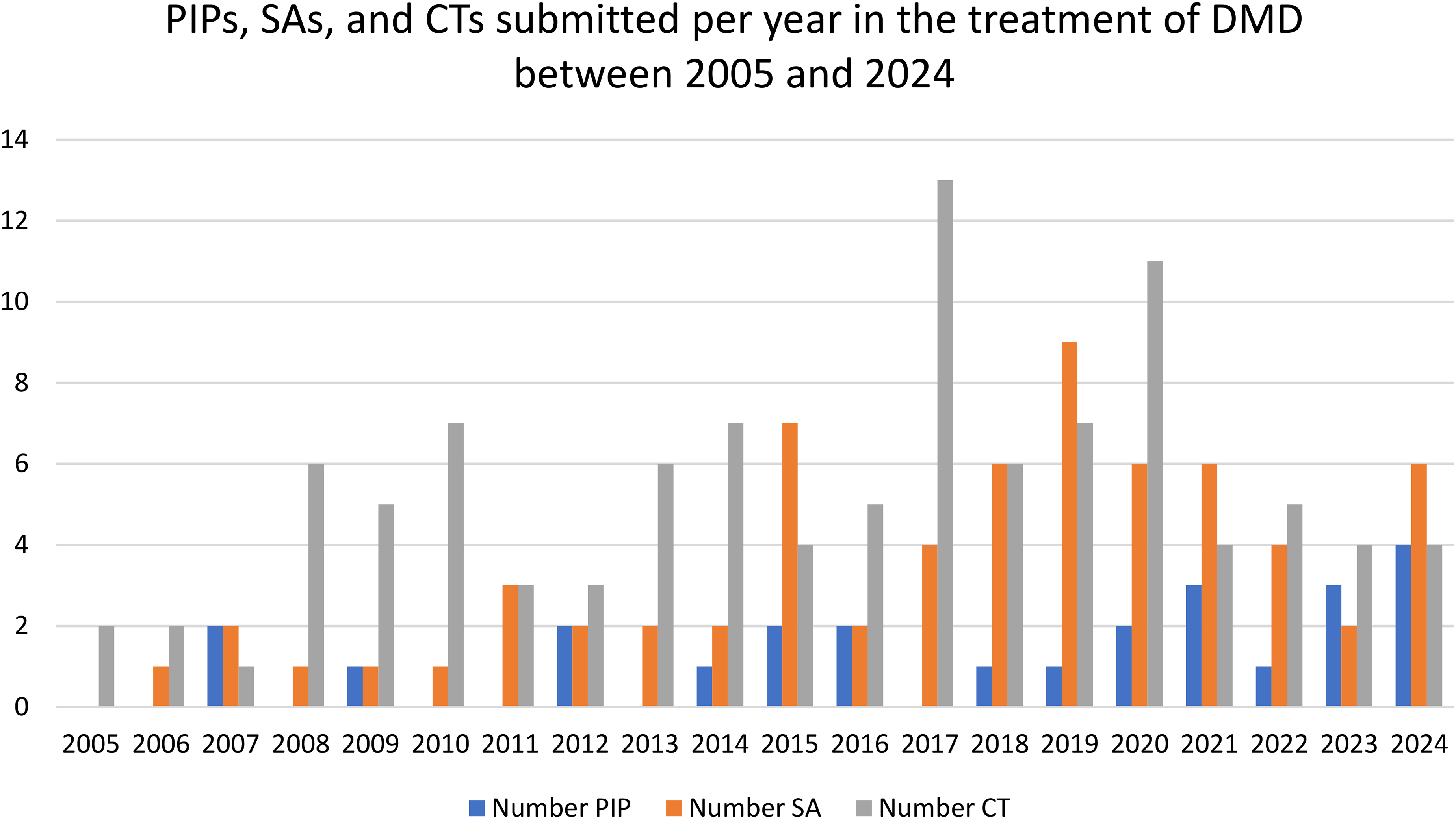
This figure shows the number of PIPs submitted, the number of scientific advice (SA) procedures requested by developers for the different IMPs under development as well as the global clinical trials (CTs) that have been gradually recorded for each year over the study period (2005–9/2024) for DMD. There seems to be a general increase in regulatory and clinical activity related to DMD over the years, with some fluctuations. CTs peaked in 2017, indicating a surge in investigational activity that year but for which no explanation can be found. SAs saw a notable rise around 2019, suggesting increased regulatory engagement, possibly reflecting a maturing pipeline or more complex development strategies. PIPs have been submitted more consistently over time, with moderate variation, reflecting ongoing efforts to ensure paediatric development is integrated early in the regulatory process.
PIPs and other procedures submitted over time
According to article 16.1 of the Paediatric Regulation, a PIP must be submitted, except in duly-justified cases, not later than upon completion of the human pharmacokinetic (PK) studies in adults. The European Commission must be informed about any lack of compliance with the Paediatric Regulation.
The EMA has recognised the uncertainty among sponsors on how to identify the date of completion of adult PK studies when these are not foreseen in a paediatric development and has clarified in the EMA Procedural advice on paediatric applications (EMA/672643/2017) that the timing of a submission should not be later than the end of healthy subject or patient PK study phase which can coincide with the initial tolerability studies, or the initiation of the adult phase-II studies (proof-of-concept studies); it also cannot be after initiation of pivotal trials or confirmatory (phase-III) trials. Submitting a first application for a new active substance during confirmatory or phase-III trials in adults, or after starting clinical trials in children, is likely to be too late to shape the development through an appropriate dialogue with regulators.
Among the 16 PIPs agreed by the PDCO, 2 were submitted before any study was started and 2 after Phase 1 studies in adults were completed. Therefore, only 25% (4 out of 16) of the agreed PIP submissions were considered on time according to the requirements of the Paediatric Regulation and the majority were submitted too late.
The challenge for developers to propose the most appropriate designs for studies covering all age subgroups of the DMD population early on, based on limited data, is well recognised. However, the timely agreement on a PIP enables a purposeful interaction between developers and regulators that increases the chances of an optimal development.
Clinical trials in the PIPs and in the European clinical trial databases (EudraCT and CTIS)
The search performed in the EU CT databases showed 113 CTs (ongoing or active but not recruiting) for treatment of DMD (Figure 2). They involved 50 different IMPs. The 16 PIPs agreed included a total of 81 CTs. A slight majority 56.84% (46/81) of the CTs included in the agreed PIPs had already been recorded in EU CT databases at the time of the analysis and were part of the 113 CTs retrieved. Ten completed CTs were included in developments (Table 1) that were discontinued, mainly due to lack of demonstrated efficacy. On average, each PIP included at least 3 CTs with a maximum of 8 CTs agreed for some of them (median = 4.5) (Table 1).
Genotypes
Five PIPs had inclusion criteria restricted to patients harbouring specific mutations that can be treated with the skipping of exon 44, 45, 51, 53, given the selective mechanism of action of the IMPs acting exclusively on those genotypes. The remaining 11 PIPs included a population without any restriction for genetic variants.
Age subgroups
Six of the 16 agreed PIPs included patients from all paediatric age subgroups, from birth to less than 18 years of age. The remaining PIPs included age-specific waivers for patients below 2 years of age (5 IMPs) or for patients below 6 months of age (5 IMPs), meaning that there was no requirement to produce data in patients below these age thresholds. Waivers for very young children were agreed by the PDCO upon submission of appropriate scientific justification, as studies were considered not feasible or could not be expected to be of significant therapeutic benefit since symptoms of the disease do not show below 2 years of age and DMD is rarely diagnosed at that age, except in families where another child had been diagnosed before.
However, in some cases including in the development patients as early as 6 months of age or from birth was considered valuable and justified based on the proposed mechanism of action. In fact, if the mechanism was related to preserving muscle function for instance and no scientifically-justified definitive age threshold could be found, it was considered appropriate to investigate early intervention in younger ages (e.g., from birth or 6 months of age) as this could potentially delay the onset of symptoms later in life.
Functional status
The functional status of the target population recruited in 113 clinical trials recorded in EU CT databases is described in Figure 3.
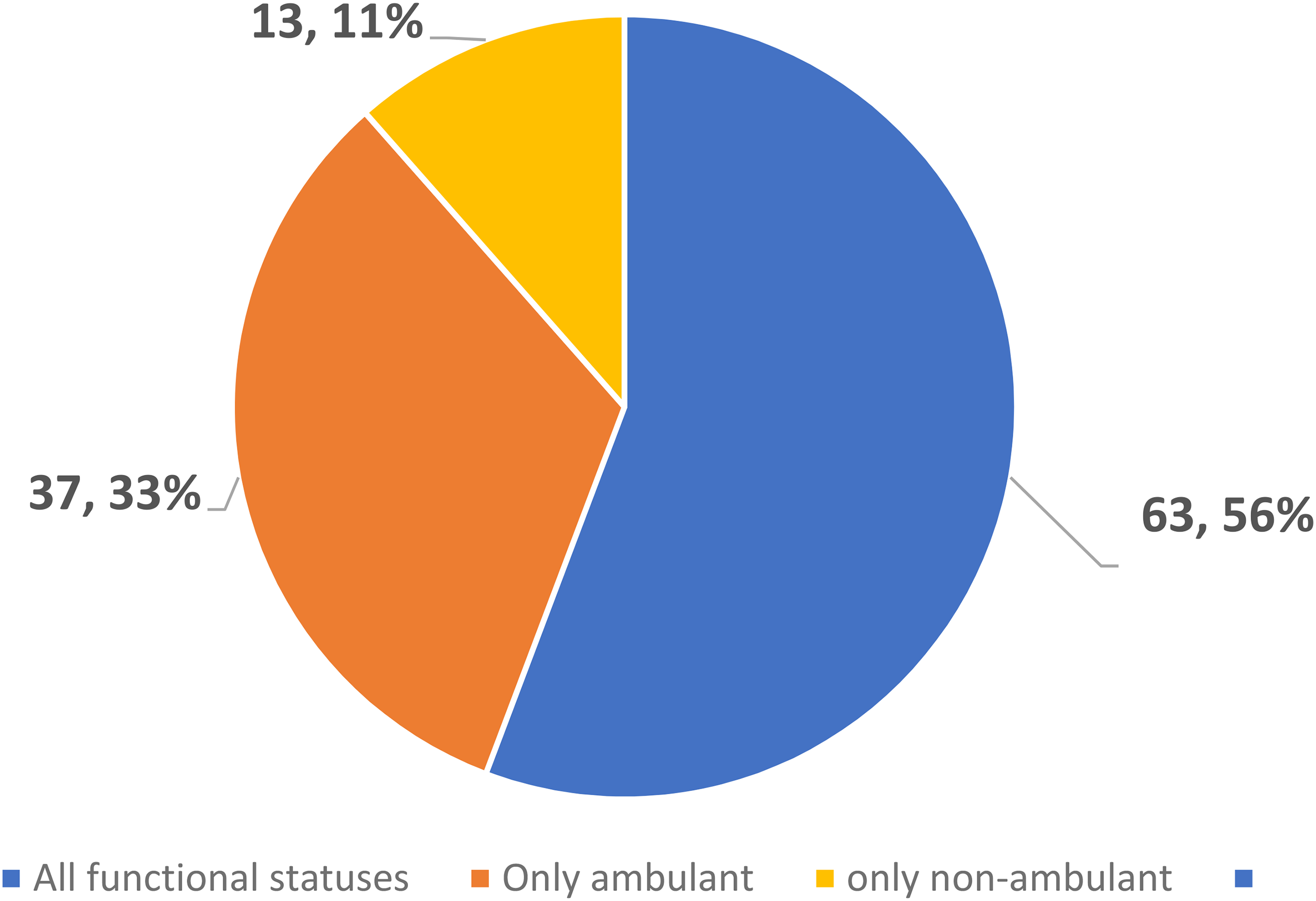
The pie chart illustrates the distribution of clinical trials or studies based on the functional status of participants – as part of the inclusion criteria – of the main DMD population that is being recruited in the all the trials retrieved from EU CT databases (number and % over the total). It is expected that as developments progresses a larger number of non-ambulant patients will be included in trials.
Similarly, among the subgroup of the 46 CTs recorded in EU CT databases which were also included in the agreed PIPs, 43.5% included all functional statuses (both ambulant and non-ambulant patients; 20 studies), 41.3% only ambulant patients (19 studies) and 15.2% only non-ambulant patients (7 studies). Regarding the subgroup of 35 studies which were part of the PIPs but were not yet recorded in EU CT databases, 42.9% included both ambulant and non-ambulant patients (15 studies), 31.4% only ambulant patients (11 studies) and 25.7% only non-ambulant patients (9 studies). This indicates that CTs involving non-ambulant patients may start later in a development programme compared to those in ambulant patients.
Study design
Among all the 113 CTs considered, 50.4% (57/113) were randomised controlled trials (RCTs), 28.3% (32/113) were single arm trials, and 19.5% (22/113) were neither single nor controlled trials (e.g., multiple doses study, ambulant vs. non-ambulant cohort). For these CTs the rate of phase 1, phase 1-2, phase 2, phase 2-3, seamless phase 1-2-3 trials, phase 3 and phase 4 trials were respectively 9% (10/113), 14% (16/113), 35% (40/113), 4% (5/113), 2% (2/113), 33% (37/113), 3% (3/113).
Looking only at the subset of the 46 CTs agreed as part of PIPs (and recorded in EU CT databases), 43.5% (20/46) were RCTs, 30.4% (14/46) were single arm trials and 19.6% (9/46) were neither single nor controlled trials (e.g., multiple doses study, ambulant vs non-ambulant cohort). The rate of phase 1, phase 1-2, phase 2, phase 2-3, seamless phase 1-2-3 trials, phase 3 and phase 4 trials were respectively 4% (2/46), 15% (7/46), 46% (21/46), 4% (2/46), 0% (0/46), 30% (14/46), 0% (0/46).
Endpoints
Among the 113 CTs considered, 83.2% specified a single primary endpoint. Of those, in 50.0% this was a primary efficacy endpoint, in 40.4% a primary safety endpoint, in 4.3% a pharmacokinetics endpoint, in 4.3% a pharmacodynamic endpoint (such as dystrophin level) and in 1.1% a quality-of-life endpoint. 16.8% of CTs had composite primary endpoints (e.g., safety and efficacy and/or dystrophin levels). Among the subgroup of 46 CTs agreed in the PIPs, 24 (52.1%) studies included primary efficacy endpoints. All of them used motor function scales (Figure 4).
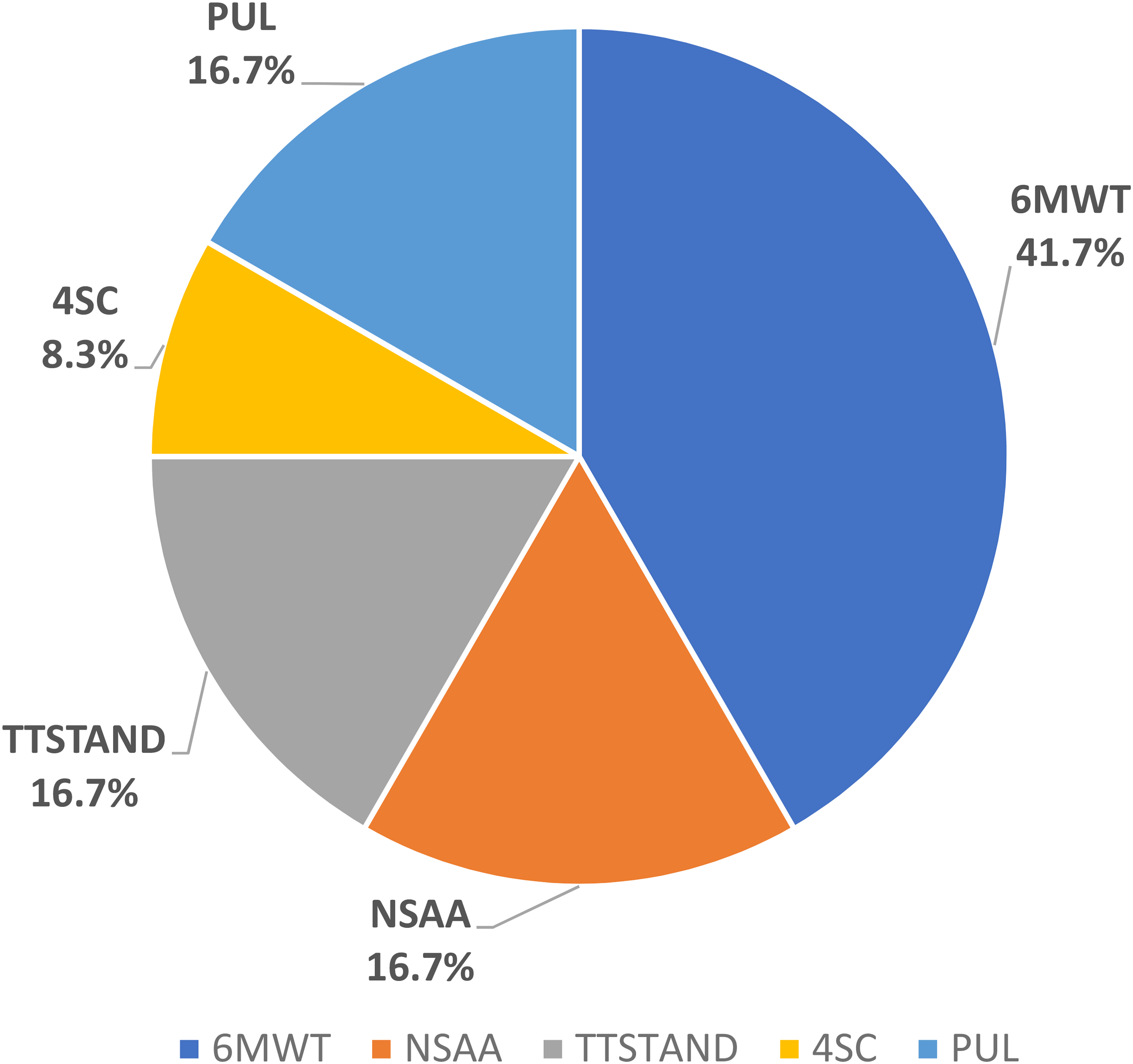
The pie chart illustrates the distribution of different motor function scales in the studies included in the PIPs with a primary efficacy end-point: 6MWT, 6 min walking test; NSAA, north start ambulatory assessment; TTSTAND, time to stand; 4SC, 4-stair climb, PUL, performance of the upper limb.
Among the timed function tests (TFTs), the 6-min walking test/distance (6MWT/6MWD) was the most frequent, probably since it is validated in paediatric patients aged 5 years and above. 20 Other TFTs, such as the 4SC or time to stand from supine velocity (TTSTAND), have also been agreed by PDCO as secondary or even primary endpoints, provided other multimodal outcomes were also measured.
Discussion
Study population
Younger ambulant patients (up to 10 years of age) in whom muscle function is rapidly decreasing are likely the most appropriate population for testing the efficacy of a new IMP (e.g. preservation or improvement in motor function), taking into consideration the residual ambulant function, the muscle damage which is mainly subclinical, and the existence of validated clinical outcome measures. Therefore, this population is, unsurprisingly, the one most often targeted for recruitment in pivotal CTs and the ones to start first. Nevertheless, according to the Paediatric Regulation, the efficacy and safety of a medicine should be studied in all relevant subsets of the paediatric population (article 15.2 of the Paediatric Regulation) to fulfil unmet therapeutic needs. This principle is still valid in treatment of DMD especially considering that non-ambulant patients are becoming the most prevalent DMD population, due to the improvements in standards of care leading to increased life expectancy. 21 Consequently the efficacy and safety of a new medicine should be studied also in the non-ambulant population, even if clinical investigation in these subpopulations with more advanced disease stage (as well as in those with still asymptomatic disease) is considered more challenging. 22
In other words, a PIP would normally have requirements to include in the various trials all population subgroups and functional status, unless otherwise scientifically justified. Patient recruitment difficulties due to the scarce number of patients available to be enrolled into clinical trials are acknowledged. Few strategies to overcome this challenge have been suggested and the role of patient organisations and specialised networks in enhancing recruitment is recognised. 23 The PDCO also discusses these challenges and can make recommendations when updates to the PIP (PIP modifications) are requested.
Outcome measures
As emphasised in the CHMP guideline on the clinical investigation of medicinal products for the treatment of DMD and BMD, 13 functional improvement (or at least delay of progression and deterioration) is considered the most relevant outcome measure. Therefore, TFTs are established as the primary endpoint of confirmatory RCTs. Additionally, measures of change in activities of daily living (ADL), respiratory and cardiac function should complement the evaluation as secondary endpoints, together with other measures of muscle function and strength, for completeness. Hence, for non-ambulant patients, specific efficacy endpoints relying on the residual motor function, such as performance of the upper limb (PUL), were included in PIPs to allow investigation of the IMPs in non-ambulant patients in clinical trials. It is important to note that these measurements have also been included in registries that have collected or are collecting long-term natural history data (such as the Cooperative International Neuromuscular Research Group DMD Natural History Study; member of the TREAT-NMD neuromuscular network). 24
Since TFTs also have limitations, such as a learning effect, the need for age- and height-normative data to account for the gained functionality due to neuro- and muscular development, high inter- and intra-personal variability due to measurement errors and age covariate, should be provided in a PIP, including a justification on how these limitations will be addressed.
The multimodal North Star Ambulatory Assessment (NSAA) 25 seems to be increasingly selected either as a primary endpoint in pivotal studies or as a key secondary endpoint in most studies assessing efficacy on motor function. This scale can be used in children from 4 years of age. The main advantage of the NSAA is that it assesses not just one, but a variety of functional motor abilities. TFTs and NSAA have a common important limitation: they are only evaluating an effect in terms of ambulation, thus restraining the continuous collection of long-term efficacy data over the disease course when the paediatric patients will lose their ambulant function.
To overcome the above limitation, the PUL was developed in 2012. 26 The PUL was designed with the aim of reflecting the proximal to distal progression of muscle weakness typically observed in DMD. The revised PUL version 2.0 was developed to remove redundancies and simplify the scoring system. 27 Further investigation is ongoing to fully validate the scale. It is not expected that these two scales are exchangeable, which limits the transition from one scale to the revised scale within the same drug development plan (or PIP).
Because of the relatively slow progression of the disease and the temporal pattern of muscle involvement over the disease course, it is not possible to recommend a single instrument to capture the efficacy of an IMP over the entire disease spectrum in the typical time window of an RCT. Nevertheless, multimodal scales seem more promising for the correct evaluation of a specific function (either of lower or upper limb). Regardless of the selection of the primary efficacy endpoint, it is expected that there is consistency of the drug effect across several endpoints, if they are covering the same dimensions of motor function.
Motor Function Measure (MFM) 28 is a 32-item validated global scale for children from 6 years of age with neuromuscular disorders including DMD. A short 20-item form, the MFM-20, can be used in children as young as two years of age. 29 This scale offers a continuous assessment regardless of disease severity and ambulatory status, allowing the inclusion of children with a wide range of disease stages and facilitating a complete long-term assessment. The use of this scale has some limitations: since it was developed for all neuromuscular diseases, it is perceived to be less specific for DMD. Furthermore, completing the assessment takes substantially longer than for other tools. Nevertheless, the use of this scale, with access publicly available, 30 could be considered in planned developments.
In conclusion, key endpoints should be specifically selected based on the characteristics of the target population (according to age cohort, motor functionality), mechanism of action of the IMP and the overall study design (duration, RCT or externally controlled trial) and generic functional rating scales should also be included to support a complementary evaluation.
Wearable devices
In 2023, EMA published a qualification opinion on stride velocity 95th centile (SV95C) as a validated primary endpoint in DMD for ambulant patients 4 years of age and above, 31 to be measured by a sensitive and suitable wearable device. One of the advantages of this novel digital endpoint is that it allows the measurement of the maximal performance in a continuous and sensitive bidimensional manner and in the everyday routine environment, as opposed to the previously mentioned tests that are evaluated once per visit, in conditions that may differ from daily life of patients and might depend on caregiver and/or parents’ constraints. Simplification in outcome measures allowed by wearable devices is beneficial for the paediatric population, as they are easier to use and more suitable for daily activities’ monitoring. Therefore, the PDCO has more recently recommended inclusion of SV95C in PIPs (so far in 2 agreed PIPs). It is expected that this will allow for the gathering of further data to strengthen the long-term correlation of SV95C with functional tests, expanding normative data and further supporting the justification of the clinical relevance of the proposed Minimal Clinical Important Difference (MCID) of this measurement in patients with DMD.
Role of biomarkers
Biomarkers are critical to guide a proper dose selection or to evaluate efficacy in non-confirmatory clinical trials, by providing information on the biological effect of an IMP. Nevertheless, a therapeutic effect in a biomarker alone is unlikely to be sufficient for granting a positive opinion for a marketing authorisation in the EU. In the case of (micro)dystrophin, for instance, changes in the level of expression are currently not agreed in the EU as a surrogate marker of efficacy. According to the Muscular Dystrophy Association, “less than 5% of the normal quantity of dystrophin is related to DMD, a level 5% to 20% of normal quantity is related to intermediate disease, and more than 20% of normal level is related to Becker's muscular dystrophy (BMD)”. 32 However, the level of increased (micro)dystrophin that is needed to obtain a clinical benefit is still to be determined. Because of the uncertain value of dystrophin levels as a biomarker correlating to clinical outcomes, the performance of muscle biopsies in children are even less justified, considering also all the ethical and pragmatic constrains. Furthermore, it is unclear whether a different level of increase may be needed depending on the disease stage. Finally, it should be recognised that patients in later disease phases face not only muscle loss but also inflammatory and fibrotic changes that further negatively impact muscle function.
In the USA, the value of this biomarker information alone, based on a reasonable probability of a functional benefit, has been considered sufficient for authorisation purposes despite the lack of information on clinical improvement. However, the IMPs in question were approved only under accelerated assessment and are still subject to further data to confirm the clinical benefit. For instance, the application for a marketing authorisation for eteplirsen was approved under the provisions of accelerated approval by the FDA in 2016, based on an increased dystrophin expression, while it received a negative opinion from EMA's CHMP in 2018. No difference in 6MWD between eteplirsen and placebo was found in a short study period of 24 weeks in a limited set of patients (n = 12). 33
Type of study
The CHMP Guideline on the clinical investigation of medicinal products for the treatment of DMD and BMD states that confirmatory trials to show clinical improvement should be randomised, double-blind, parallel-group controlled trials. About 40% of studies specified in the PIPs are RCTs, as requested by the PDCO for pivotal Phase 3 trials.
In DMD, in addition, single-arm trials (SATs) are also included in all PIPs. Their objective is to gather PK and pharmacodynamic (PD) data, as well as efficacy and safety data for initial testing in some subgroups of patients that remain underrepresented (e.g., youngest age or functional status groups or patients with a specific mutation), or to gather confirmatory evidence in some age subgroups not included in RCTs. EMA has published a reflection paper on single arm trials as pivotal evidence for the authorisation of medicines in the EU, which aims to stimulate the scientific discussion around key concepts and challenges associated with SATs and improve their design and conduct. 34 Whereas some medicines for the treatment of rare diseases such as spinal muscular atrophy or metachromatic leukodystrophy were approved based solely on evidence coming from SATs (e.g., Zolgensma) 35 or registrational studies (e.g., Libmeldy: non-randomised, open-label-, prospective, non-concurrent control, single centre study), 36 the demonstration of efficacy in patients with a disease like DMD needs RCTs, for reasons including slower and less homogenous disease progression, and natural developmental growth as a confounder considering that the condition evolves clinically after infancy. However, considerations discussed in the reflection paper, as well as those provided in ICH E11A, 37 are applicable for SATs conducted in some subgroups of patients with DMD.
Due to the lack of a comparator in SATs, the role of relevant external (extra-study) information is critical for the interpretation of the results derived from a SAT. A robust characterisation and evidence on the natural course of the disease, as well as identification of the more suitable endpoints, are crucial for the use of SATs to optimise medicine development. External data sets may be used to explore the understanding of natural disease progression and variability in clinical measures over different stages of the continuum of the disease, and modelling and simulation platforms can help to optimise these data. 38 They can also be used to explore ‘meaningfulness’ of changes in functional measures that can be demonstrated. 39 In this regard, a proposal for a model-based clinical trial simulation platform (CTSP) for DMD to model disease progression, with the aim of aiding clinical trial design, has recently received a Letter of Support from EMA. 40
Considerations regarding registries
Indeed, a way to address the challenges in population subgroups where the inclusion of a control arm is challenging, might be the use of registries or longitudinal cohorts to be matched with the experimental arm, provided methodology and sufficient granularity of data collection methods are agreed at the planning stage. 41 Registries can provide supportive data added to clinical trial results, to substantiate long-term effectiveness and safety. However, the best way to generate optimal quality data from registries for regulatory use remains to be further explored. The World Duchenne Organization is currently cooperating with the Duchenne Muscular Dystrophy Data Foundation (a patient-led research foundation) to build on the related disease data platform on Duchenne (which already collects RWD) as a backbone for the development of a Patient-Derived Models Repository (PDMR). Efforts are ongoing in consolidating the network of registries (TREAT-NMD Global Registry Network). 42
Following a re-examination of available data, in 2025 EMA's CHMP confirmed its previous recommendation to not renew the conditional marketing authorisation for Translarna (ataluren). Translarna (ataluren) was granted a CMA in 2014, based on the incomplete outcome from a phase 2b randomised trial evaluating 2 doses compared to placebo in 174 ambulant patients aged 5 to 20 years with nonsense mutation DMD (nmDMD), with positive post-hoc analyses after 48 weeks on treatment in the lower dose. 43 Because of uncertainties in the robustness of efficacy data from clinical studies, including two additional randomised clinical studies in patients aged 7 to 14 years, the marketing authorisation holder (MAH) had the specific obligation to provide additional data to confirm long-term effectiveness and safety of the medicine for the renewal of the marketing authorisation. As part of this latest re-examination, the CHMP also reassessed data from a study comparing the health outcomes of patients from two registries’ studies presenting disease progression in patients receiving standard of care plus ataluren (STRIDE registry; 2015–2022) and those receiving only standard of care (CINRG-NHS; 2006–2016). The differences stemming from improvements in standard of care for DMD patients during the different study periods and locations, as well as other differences and incompleteness of some data (e.g., on mutation status), in patient populations in both registries hampered the interpretation of the 3.5-year delay in the time to loss of ambulation in STRIDE compared with CINRG-NHS, highlighting the potential difficulties in using registry data to support an application.
Extrapolation of efficacy
Extrapolation is a defined international agreed upon methodological approach to provide supportive evidence for effective and safe use of medicines in the paediatric population, when it can be assumed that the course of the disease and the expected response to a medicines would be sufficiently similar between the target and reference population subgroups. 37 In the case of DMD in ambulant patients, extrapolation of efficacy data from some age groups included in a RCT to older/younger ones should be considered and proposed if scientifically justified, to make the best use of evidence generated from the limited number paediatric patients available for recruitment. This is particularly important when SATs are proposed to cover a population subset.
Different modelling and simulation analyses of PK and PD are being used to optimise the data generated in appropriately-selected groups of children, to simulate and confirm the dose regimen and to support extrapolation of efficacy based on similarity of exposure, generating data for certain subgroups of patients for whom the benefit-risk cannot be directly evaluated.
In principle, extrapolation of efficacy towards an earlier phase of DMD (e.g., from older to younger patients) may be easier to be agreed in a PIP based on the general principle that any medicine aiming to halt or delay the progression of a degenerative disease may have the largest benefit when prescribed as early as possible.
Extrapolation of efficacy data from ambulant to non-ambulant DMD patients is not as straightforward, due to a more advanced stage of the disease and an expectation that effects will be more limited given that non-ambulant patients have less functional muscle tissue left. However, this would also depend on the proposed mode of action of the medicine. For vamorolone, the CHMP therefore agreed on the extrapolation of efficacy and safety data from patients aged 4 to 7 years to older-age subpopulations. The pharmacological similarity between vamorolone and glucocorticoids in terms of mode of action played a key role in this decision, as there is evidence on the benefit of the use of steroids in the non-ambulatory phase. Further encouraging safety data, although limited, supported this decision.
Overall, the experience with extrapolation gained in the evaluation of paediatric medicines development for rare neurological diseases indicates that extrapolation relies on different factors. These include the clinical course of the disease, mechanism of action and PK and PD parameters of the IMP, robustness and appropriateness of the endpoints between the source and the target population which, for extrapolation analyses, must be identical or similar. Analyses supporting an assumption of consistency in therapeutic benefit and similarity of efficacy and safety response to treatment overall need to be carefully planned.
Role of regulatory authorities
EMA's PDCO and SAWP have been working side by side to help improve paediatric medicines development, to further refine patient subgroups to be included in trials, dose approaches, endpoints and duration of the studies, and to foster streamlined global development.
For instance EMA and the US Food and Drug Administration (FDA) share information on assessment through several channels, one of them being the ‘Pediatric Cluster’, 44 a monthly forum enabling international regulators to discuss proposed paediatric development plans. Provided that PIP submissions happen early in the development phase, this can enable fruitful interaction to optimise plans and embrace global development from the outset.
In addition, EMA supports enhanced interaction frameworks between all relevant stakeholders, including regulators, with multistakeholder initiatives. Other therapeutic areas have benefited from this approach. The Accelerate 45 platform is a good example of a global space for interaction in the pre-competitive space. It is a forum for common agreement of all stakeholders in paediatric oncology (researchers and clinicians, patient representatives, medicines developers and regulators), focused on medicines in development that address unmet therapeutic needs. Such discussions and agreements take place before the initiation of paediatric clinical trials. The initiative enhances the feasibility of conducting clinical trials globally and can be considered a model for optimising paediatric development in rare and severe diseases.
Conclusion
This study presents the first in-depth evaluation of PIPs approved within the EU for DMD offering insights into potential strategic approaches for clinical development of new IMPs. It highlights the accumulated experience of regulators and stakeholders, particularly regarding pivotal trials that establish clinical efficacy in key patient subgroups. Although most IMPs have not yet reached the marketing stage, the analysis shows that there is a noticeable effort in developing new medicines for the treatment of DMD. Certainly, early agreement of PIPs by the PDCO is crucial to validate such strategies in support of a submission for a marketing authorisation in Europe. With the aim of accelerating and enhancing development of efficacious medicines for this devastating rare neuromuscular disease, all interested stakeholders – patients with DMD and their families and caregivers, the scientists, the clinicians, the industry – should consider early and continuous dialogue focussing on identifying more promptly new medicines which hold a true promising therapeutic value for DMD.
Interaction with regulators can happen by requesting an OD and/or SA as a first step, or qualification advice for an endpoint or device. While OD and SA are not mandatory regulatory procedures, these are often used by developers in early stages of development. The usual sequence would be to apply for an OD, which if granted makes the product eligible for SA (called protocol assistance in such cases) at a reduced or no fee. The SA could address any questions depending on the stage of development on specific aspects either complementing an agreed PIP or in preparation for it.
The application of real-world data and of data from disease registries is promising. Despite the known limitations of observational studies and the challenges with collecting robust and complete data through registries, their role for regulatory requirements, such as confirming long-term effectiveness and safety, can be fundamental in the regulatory decision-making, considering the regulatory recommendations. Nonetheless, robust approaches are needed to predefine, collect and analyse these longitudinal data and to explore their best use in the medicine development process based on their quality.
Extrapolation of efficacy data between a target and a reference population (e.g., across different subgroups of patients differing in age or ambulant function) has been evaluated and in some cases accepted by regulators. Acceptance of extrapolation as a complement to RCTs data to cover all relevant age groups in DMD will depend on several factors, including the specific medicine and its mechanism of action, and the associated expectations of efficacy in different stages of the disease. Because of this, extrapolation requires a well justified case-by-case discussion and decision for acceptability.
Overall, the large number of PIPs submitted and approved by the PDCO indicates that, despite numerous scientific and regulatory challenges, the regulatory science surrounding DMD development is growing, to the benefit of future developments.
Footnotes
Acknowledgments
The authors would like to thank Alix Guillot (PharmD), Anissa Bounabi (PharmD), and Louise Marsille (PharmD) for their contribution to the data analysis during their internship at the French National Agency for the Safety of Medicines (ANSM). We would like to thank Jarno Jansen for reviewing the text and for editing the English version.
ORCID iDs
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Disclaimer
The views expressed in this article are the personal views of the authors and may not be understood or quoted as being made on behalf of or reflecting the position of the regulatory agencies or organisations with which the authors are affiliated.