
Abstract
Spinal muscular atrophy (SMA) comprises a spectrum of clinical severities, yet the pathomechanisms of late-onset forms (Type III) remain insufficiently understood. While severe early-onset SMA has been extensively investigated using existing models, their translational relevance to adult disease is limited. Here, we recommend the 4-copy SMN2 mouse (FVB.Cg-Smn1tm1Hung Tg(SMN2)2Hung/J) as the most appropriate model for late-onset SMA. This model exhibits delayed onset, progressive motor dysfunction, and extended survival, enabling the study of chronic neurodegenerative processes, including astrocyte-mediated motor neuron pathology. Its prolonged therapeutic window makes the model suitable for mechanistic and translational investigations of late-onset SMA.
Keywords
Spinal muscular atrophy (SMA) is a clinically and genetically heterogeneous neuromuscular disorder (NMD) characterized by reduced levels of the survival motor neuron (SMN) protein, leading to the loss of spinal and bulbar motor neurons (MNs) and muscle mass.1,2 While severe early-onset forms (Type I) are mechanistically well-characterized, the pathomechanisms underlying late-onset forms, such as SMA Type III, remain poorly understood.
Spinal muscular atrophy (SMA) was historically divided into five clinical types (0–IV) based on symptom onset and the highest motor milestone achieved. This pre-therapy classification reflected the natural disease range from severe prenatal (Type 0) to mild adult-onset (Type IV) forms. With SMN-enhancing treatments and newborn screening, these categories have become less predictive, as early treated patients often surpass expected milestones.3,4 Disease severity now correlates more with SMN2 copy number, treatment timing, and achieved motor function. Similarly, the expanding therapeutic landscape and phenotypic variability call for re-evaluating how we classify and choose SMA mouse models for specific research aims.
Importantly, although SMN-enhancing therapies, including antisense oligonucleotides (nusinersen), small molecules (risdiplam), and gene addition therapy (onasemnogene abeparvovec), have markedly improved patient outcomes, individuals with late-onset SMA often experience only partial motor recovery.5–7 Nevertheless, improvements in less quantifiable aspects, such as daily functioning and quality of life, remain highly meaningful for patients.
Progressive motor deficits despite treatment suggest that SMN restoration alone does not fully reverse established pathophysiological changes, particularly in older patients where neuronal loss and maladaptive network alterations are already present.5–7
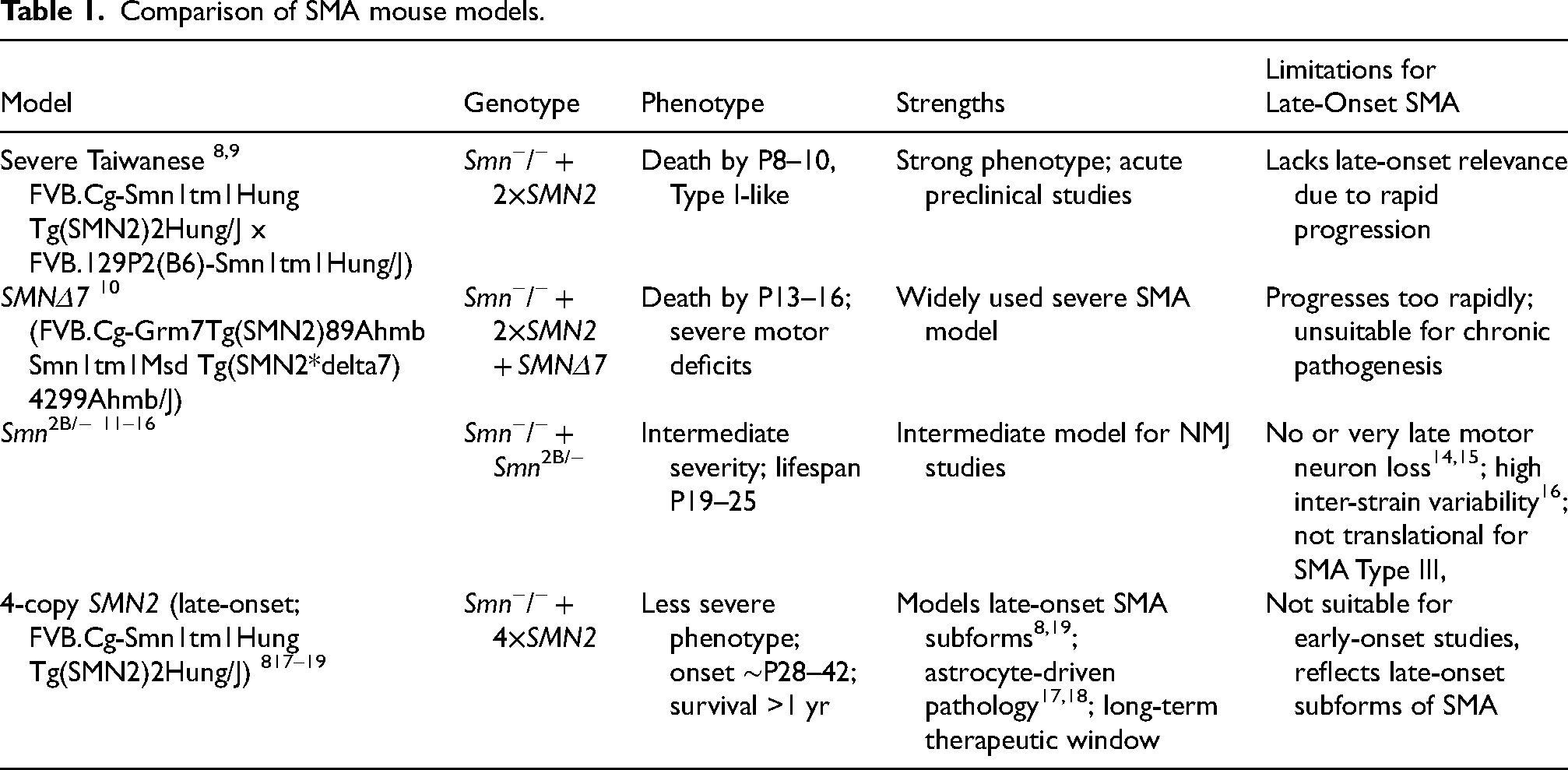
Different animal models, in particular mouse models, are used to address the diverse pathomechanisms of human SMA phenotypes. A comparative overview of the most common SMA mouse models is summarized in Table 1 and Figure 1.
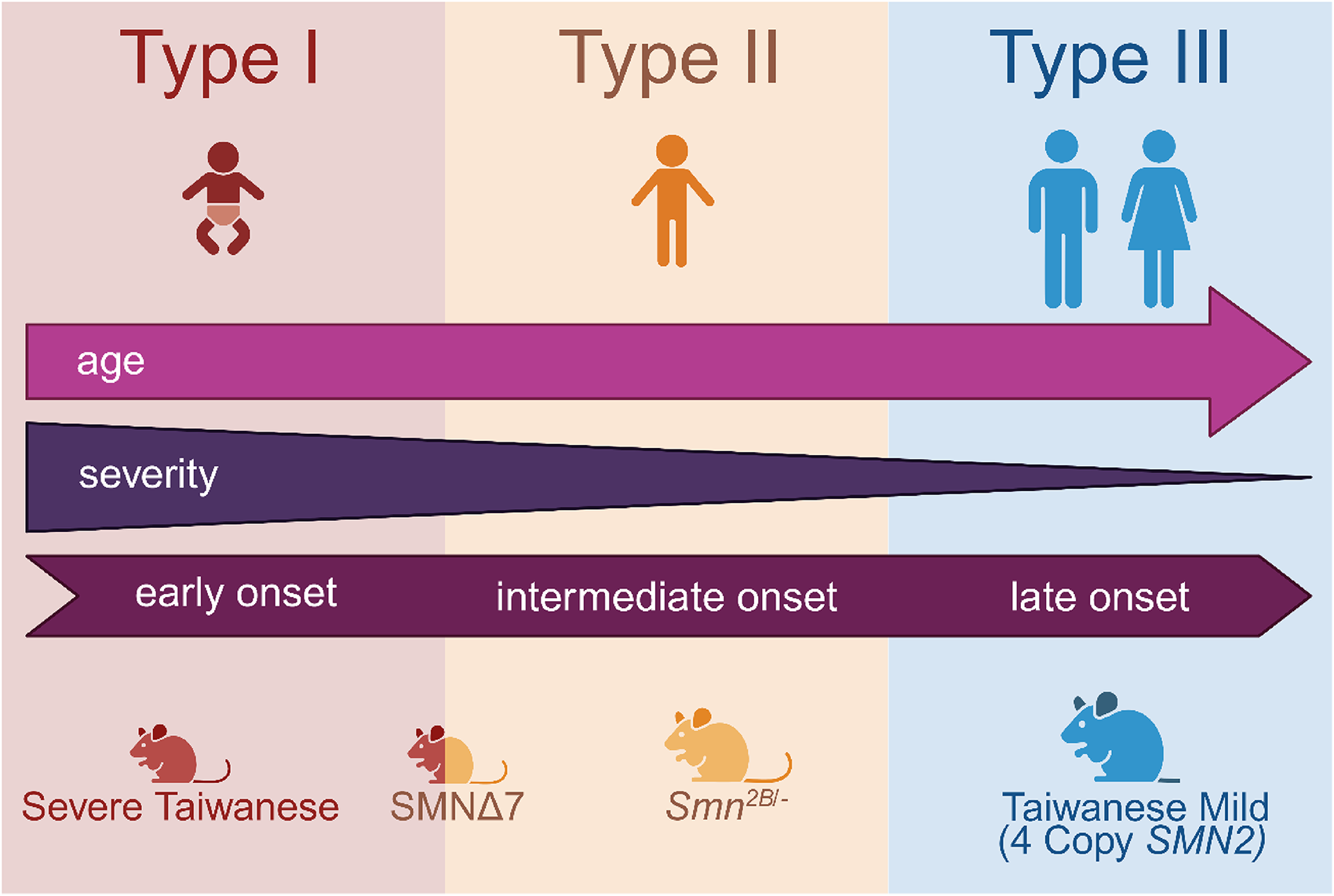
Overview of the SMA subtypes and their corresponding mouse models. SMA Type I is characterized by an early onset of symptoms and is resembled by the Severe Taiwanese mice. The SMNΔ7 model is an intermediate form set between the SMA Type I and II while the Smn2B− model is reflective of SMA Type II only. The late-onset Taiwanese Mild mouse model with four SMN2 copies is the only available model resembling SMA Type III (created with BioRender.com).
Comparison of SMA mouse models.
Proteomic studies of the Taiwanese and Smn2B/− mouse model reveal not much overlap of targets, indicating different pathomechanisms and possibly indicating diverse disease entities rather than reflecting just a gradual change. 20 This in turn indicates the need for deep (molecular) phenotyping of the different existing SMA mouse models to warrant the selection of the appropriate model to mimic disease processes underlying the different phenotypes of SMA. Along this line, a deeper mechanistic understanding of late-onset SMA is therefore urgently needed to identify novel therapeutic targets. Complementary treatment strategies beyond SMN augmentation could substantially improve the long-term outcomes for this patient group, which remains an area of unmet clinical need. Developing such approaches critically depends on the availability of suitable preclinical models. However, currently available SMA mouse models differ considerably in severity, disease progression, and their relevance to human late-onset SMA. Careful model selection is therefore essential to accurately reflect the disease course, cellular pathology, and therapeutic windows relevant to SMA Type III.
Insights into the different mouse models
Taiwanese severe mouse model
The severe Taiwanese model is characterized by a homozygous deletion of the SMN gene and an insertion of two human SMN2 genes. This leads to low SMN protein levels and mimicking the phenotype of SMA type I patients. Onset of symptoms begins around P2-3 with signs of muscle weakness, poor motor function and reduced movement with worsened symptoms by P5-7 which is shown in clear motor impairment, reduced body weight and failure to thrive. Early lethality is one of the key-defining features of this model with most pups succumbing to the disease around P10. 8
The rapid disease progression and early death make this model an excellent representation of Type I SMA, 9 where patients typically present with severe symptoms within the first 6 months of life and have a reduced life expectancy. It makes a valuable tool for studying the mechanism of the disease and testing potential therapies aimed at increasing the SMN protein levels and the survival rate.
SMNΔ7 mouse model
The SMNΔ7 mouse model is characterized by the deletion of the mouse SMN gene, the insertion of two copies of the human SMN2 gene, and a transgene version of the SMN gene lacking exon 7. The resulting truncated SMN protein is unstable and partially functional. 10
The onset of symptoms occurs around P3-6, including reduced mobility and weak limb movements. By P8-12, the mice exhibit severe motor symptoms, muscle weakness, and impaired respiratory function, 21 which contribute to their eventual lethality. These mice have a limited lifespan, typically dying between P13-16. 10 This closely resembles the moderate severity seen in Type II SMA patients, who may live into childhood or adolescence but experience significant motor deficits.
The moderate severity of motor neuron degeneration and NMJ pathology, 22 with symptoms appearing early in life but with survival beyond infancy, makes the SMNΔ7 mice an ideal model for studying therapies aimed at intermediate forms of SMA. It is particularly useful for longitudinal studies of motor function and muscle strength in response to treatments and other approaches that aim to increase SMN protein levels or improve motor function. 23
Smn2B/− mouse model
The Smn2B/− model has a partially endogenous SMN gene knockout. The mice express one null allele (SMN-null) where the gene is completely inactivated and another partially functional allele, SMN2B. This allele has a C-to-T point mutation in exon 7 of the SMN gene, which is located on nucleotide position 835. This results in a splicing defect in exon 7, leading to the production of a truncated, less functional version of the SMN protein. 11
This makes the Smn2B/− mouse model distinct because it relies solely on the residual expression of SMN protein from the endogenous Smn2B allele, instead of any compensatory expression from human SMN2. Smn2B/− mice begin to show signs of motor neuron degeneration and muscle weakness around P10-15, 24 a somewhat later onset compared to the more severe models. The onset of symptoms is milder than in severe Type I models but progresses over time. Unlike the severe models, these mice do not die early, but they have reduced survival, generally living for around 4 weeks. 16
This model does not depend on the human SMN2 gene, making it a more straightforward system for investigating the effects of endogenous SMN depletion and therapeutic approaches that target the mouse SMN gene directly. Mice have a longer lifespan compared to severe models, providing an extended window for studying disease progression and the long-term efficacy of therapeutic interventions.15,16 This model closely mimics the more prevalent Type II SMA, making it highly relevant for developing therapies aimed at milder forms of SMA.
Taiwanese mild (type III SMA)/4-copy SMN2 mouse model
The late-onset SMA mouse model (Taiwanese Mild) possesses four copies of the human SMN2 gene and no murine SMN gene.8,25 This increased copy number results in higher levels of functional SMN protein being produced compared to the more severe models. The small fraction of full-length SMN protein is sufficient to allow for long-term survival and a milder phenotype. Symptoms include mild muscle weakness and reduced motor coordination.18,19 Mice retain the ability to move and function relatively normally compared to more severe SMA models. There is no impact on life expectancy.8,25 Over time, as MNs continue to degenerate and NMJs become less functional, the mice develop mild muscle atrophy.18,19,26
The higher SMN protein levels and milder symptoms are characteristic of Type III SMA patients, who typically experience a later onset of symptoms and can maintain motor function for a longer period, often into adulthood. 27 Therefore, the late-onset SMA mouse model is the only available model reflecting human Type III SMA.
Pharmacologically induced models
Pharmacologically induced SMA mouse models aim to mimic milder disease forms by treating severe models with low sub-doses of SMN-enhancing compounds. Administration of SMN2-splicing modifiers such as SMN-C1 or SMN-C3, or antisense oligonucleotides like PMO25, partially increases SMN levels, extends lifespan, and results in intermediate phenotypes.28–30 While such approaches provide useful insights into dose–response relationships and therapeutic efficacy, they do not fully capture the developmental and molecular features of genetically mild SMA models such as neurodevelopmental processes, cellular mechanisms, and cell–cell interactions. These features may manifest differently under conditions of mild SMN deficiency and cannot be faithfully reproduced by slightly increasing SMN levels in inherently severe models, because the organism's biological baseline is fundamentally different. Therefore, this model is of limited value for studying chronic or late-onset disease mechanisms.
Limitations of current severe, milder and pharmacologically induced models
Both the Severe Taiwanese model8,9 and the SMNΔ7 model 10 have provided important insights into the pathogenesis of early-onset SMA and have been essential for testing survival-promoting therapies. However, their rapid disease progression and early lethality prevent the investigation of chronic cellular mechanisms such as long-term neuron-glia interactions, which are increasingly recognized as key contributors in late-onset SMA.
The Smn2B/− model11–13 has been proposed as a milder model placing it between the severe Taiwanese/SMNΔ7 and the late-onset model. It provides insights into systemic phenotypes, but its translational relevance regarding treatment with the existing therapeutic options remains limited. Although neuromuscular junction (NMJ) pathology is present, the model does not consistently exhibit spinal motor neuron loss14,15 and is highly sensitive to genetic background effects, resulting in substantial inter-strain variability and the absence of copies of human SMN2. 16 These limitations restrict its suitability for modeling human SMA Type III and for evaluating therapies aimed at the adult disease.
Advantages of the 4-copy SMN2 mouse model
In contrast, the 4-copy SMN2 mouse model17–19 provides a representation of both the phenotypic and mechanistic aspects of late-onset SMA. With delayed disease onset around postnatal day 28 to 42, mild but progressive motor impairment, and survival exceeding one year,17–19 which reflects more the human SMA Type III patients. These patients can exhibit a great variety of different symptoms and disease severity but have a delayed disease onset, motor neuron loss, progressive motor impairment and normal life expectancy.27,31,32
In recent years, the contribution of astrocytes to SMA pathogenesis has been revealed, both in SMNΔ7 mice 33 and Smn2B− mice, 34 as well as in human SMA astrocytes. 35 Our recent studies demonstrated that astrocyte dysfunction precedes motor neuron degeneration in the late-onset SMA mouse model.17,18,26 In particular, early reductions in astrocytic inward rectifier potassium channel 4.1 (Kir4.1) and excitatory amino acid transporter 1 (EAAT1) expression were observed, leading to impaired glutamate clearance and excitotoxicity, which ultimately contributed to the loss of MNs. Importantly, pharmacological restoration of astrocyte function with arundic acid was shown to preserve both MNs and motor performance in these mice. 18 In vitro experiments further confirmed that SMN-deficient astrocytes independently promote motor neuron hyperexcitability and degeneration, 17 supporting a central role for glial pathology in the disease process. Astrocyte-autonomous mechanisms are also conceivable, such as impaired mitochondrial function or disturbances in lactate metabolism.36,37
The 4-copy SMN2 model also provides a substantial therapeutic window for chronic treatment and supports adult CNS drug delivery paradigms,18,19,38 and muscle-directed therapies, e.g., applying myostatin inhibitor, reflecting the disease course observed in human patients with late-onset SMA. Its combination of accurate disease phenotype, glial involvement, and therapeutic responsiveness makes it the current model of choice for investigating late-onset SMA mechanisms and for testing novel interventions targeting this underserved patient population. Additionally, treatment for SMA Type III and IV is not routinely recommended, showing the importance of identifying and understanding the pathology of those patients better and possibly changing treatment policy for these late-onset individuals.
Conclusion
As SMA research increasingly focuses on addressing the unmet needs of milder and adult-onset forms of the disease, the careful selection of appropriate preclinical models is critical. While severe models remain essential for understanding early-onset SMA, they do not reflect the chronic neurodegenerative processes observed in SMA Type III. Similarly, the Smn2B/− model lacks several key translational features, including consistent motor neuron pathology. In contrast, the 4-copy SMN2 model uniquely fulfills these requirements, providing an ideal platform for both mechanistic studies and translational development in late-onset SMA. We therefore strongly recommend its preferential and increased use in preclinical research focused on late-onset SMA as the Type III phenotype, as the careful selection of an appropriate model is of particular importance in the context of translational research and for bridging the translational gap.
Footnotes
Acknowledgements
All graphics have been designed with biorender.com.
ORCID iDs
Author contributions
All authors contributed to the manuscript idea and design. ML and LIS wrote the first draft of the manuscript. All authors reviewed and revised the manuscript.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.