
Abstract
The TREAT-NMD Advisory Committee for Therapeutics (TACT) provides academic and industry applicants involved in therapy development in the neuromuscular disease (NMD) space with constructive, multidisciplinary expert advice in order to help derisk NMD drug development and avoid potential therapies entering clinical trials prematurely and to improve trial design. The ultimate goal of TACT is to reduce the risk of type 1 errors (concluding a treatment is effective, while it is not), type 2 errors (concluding a treatment is not effective, while it is), or risks of questions and regulatory rejections later on in development.
TACT celebrated its 15th birthday in 2024. From 2009–2024 TACT reviewed 90 applications, 68 from Industry and 22 from Academia. This perspective paper summarizes how TACT has evolved from its initiation. Furthermore, based on our experience, we list recurrent mistakes, such as not collecting sufficient information in pre-clinical studies, omitting wild type references in animal studies and choosing the wrong or too many outcome measures in clinical trials. By sharing common errors in preclinical and clinical trial design and providing lessons learned we aim to further improve therapy development in the NMD space.
The conception of TACT
Fifteen years ago, there was only one therapeutic specifically approved for a neuromuscular disease (NMD): enzyme replacement for Pompe disease. Clinical trials were performed for treatments for other NMDs, but most clinical trials failed to meet their clinical endpoints. In hindsight, some of these trials were initiated without sufficient understanding of mechanisms of action or lacked robust evidence of therapeutic effects in preclinical studies. Clinical trial designs suffered from poorly validated endpoints, inappropriate inclusion criteria and/or insufficient statistical power.
Identifying that therapeutic research and trial designs could be improved, the TREAT-NMD Advisory Committee for Therapeutics (TACT) was established. 1 The initiators of TACT found that for some programs there were insufficient rationale or robust data to proceed to the clinical trial phase, and key contributors (for example those with expertise in drug development) were missing from the conversation. TACT was initiated to derisk drug development in the NMD space by providing tailored expert advice to academic and industry drug developers.
TACT has recently celebrated 15 years of facilitating drug development in the NMD space by providing multidisciplinary, expert advice to academic and industry partners. From 2009–2024 TACT reviewed 90 applications, 22 from academia and 68 from industry. The majority (49/90) of applications focused on therapeutic development for Duchenne muscular dystrophy (DMD). Since the inception of TACT, the number of companies pursuing therapy development for NMDs has increased and there are now multiple regulatory approvals for novel therapeutics in this space, with e.g., vamorolone approved for DMD in the USA, the UK and the EU and givinostat receiving approval for DMD in the USA and the UK and conditional marketing authorization in the EU, and more trials reaching phase 3 for other indications, e.g., delpacibart etedesiran for myotonic dystrophy. It is challenging to quantify the succes of TACT. Some of the approaches that received TACT advice are now approved or in phase 3 clinical trials, and possibly this was facilitated by the advice provided in the report. However, the fact that some approaches were not furthered into clinical trials after TACT advice, e.g., when the scientific rationale was not sound or when preclinical results were not sufficiently robust, could also be counted as a success.
Despite the application of learnings from TACT, some of the challenges in the field that spurred the initiation of TACT in 2009, remain today and new ones have arisen related to developments in the regulatory environment. Thus, the need for such an advisory group continues in NMD and in other disciplines.2–4 Some examples of common pitfalls were discussed at the 15 years of TACT celebratory symposium, and will be elaborated on in this article including suboptimal pre-clinical data, a misunderstanding of what various types of biomarker are and what they inform on, and selection of clinical endpoints without adequate understanding of responsiveness to change or natural history. Note that this is not an exhaustive list of challenges and pitfalls and we refer the interested reader to additional papers for a more complete picture.1–3,5,6
TACT structure and process
TACT is a multidisciplinary, diverse group of stakeholders in therapeutic development for NMD including not only preclinical and clinical disease experts, but also drug developers, regulatory and toxicology experts, biostatisticians, patient representatives and more experts. 1 TACT was initially funded by the European Union Framework Program and is now supported by patient foundations and applicants, who pay a sliding scale application fee that is zero for academics, reduced for small companies and much less than a typical advisory board for established pharmaceutical companies. The review process by the committee's involves study of submitted materials, drafting an internal feedback report for the lead reviewer, discussion with the applicant and written recommendation feedback in a formal report (see box 1 for more detail).
TACT timeline from preapplication to report.
The entire TACT process is confidential. The timeline for the TACT process is shown in Figure 1. A confidentiality agreement is in place with members of the core committee and TREAT-NMD pertaining to TACT related activities. This is renewed every five years. When an applicant considers a TACT review, the first step is to put a confidentiality agreement in place between TREAT-NMD and the applicant. After this, the applicant is asked to provide a pre-application form, including a brief description of the drug-development programme along with the key questions for TACT. This pre-application is checked by the core committee to confirm eligibility and correct expectations of what TACT can and cannot offer. When the pre-application is deemed suitable, the core committee suggests names of relevant experts to add to the panel, in addition to the TACT core committee experts, who are on every panel (unless there is a conflict of interest). As there are limited spots available for online and in-person TACT meetings, it is good practise for applicants to apply early. This will also facilitate ensuring availability of key experts.
While the TACT secretariat prepares for the evaluation of the application by recruiting key experts, the applicant prepares a full application. This is a form that is more in-depth requesting information on the various aspects involved in drug development, such as the scientific rationale of the therapeutic compound, milestones and timelines of the applicant, prior preclinical and, if applicable, clinical research of the applicant for the compound, details on the preclinical models used, preclinical studies that are planned or ongoing, details on statistics and statistical models that the applicants have used and plan to use and potential collaborators and funders. Furthermore, applicants need to provide information on assays to measure drug activity, biomarkers to ensure target engagement, information on the drug (administration, distribution, metabolism and excretion (ADME), intended formulation and dose(s), PK properties, expected safety profile and therapeutic index, drug stability, the ability to produce at good manufacturing practices and the intellectual property (IP) position. Information about the planned clinical trial design needs to be provided and whether the applicant has had interactions with regulatory agencies and/or patient community advisory boards. Applicants are asked to give an overview of the therapeutic landscape for their disease and specify how their compound fits in this. Finally, the applicants are asked to outline how they will use the report and they can ask specific questions to TACT and specify in which areas they need specific guidance. In addition to this form, which can be dozens of pages long, applicants can optionally provide supporting information such as up to 5 scientific publications, and up to 5 additional documents such as posters or slides presented at conferences, an investigational brochure, a clinical trial protocol etc.
Usually the TACT committee reviews hundreds of pages of material for each application. To allow the committee sufficient time to review up to four such applications, the full application needs to be submitted 2 months prior to the meeting date. This information is then securely shared with the committee who review the application and write a report summarizing the positive aspects of the application, as well as concerns and questions they have for the applicant. Furthermore, reviewers formulate answers to the specific questions the applicants asked. As the committee members cover the various disciplines involved in drug development, the compiled report will contain feedback from all perspectives. The reports are shared with the lead reviewer no later than a week before the meeting, allowing them to collate input and prepare for the evaluation meeting.
The meeting spans half a day per application. The lead reviewer will introduce the application, briefly highlighting the mechanism of action, clinical development plan, positive aspects of the application, and then will facilitate the discussion to align which expert will ask which question to the applicant, to obtain further information in order to provide optimal advice. The applicant will join the meeting and will be given the opportunity to briefly introduce their approach, focusing on data that has been obtained between submitting the application and the meeting. The committee can then ask a series of clarifying questions on the various aspects, usually covering preclinical, patient, drug development, regulatory and clinical perspectives. Finally, the panel reconvenes without the applicant, where the lead reviewer coordinates the discussion, using the new learnings to collectively decide the content of the advisory report and to achieve consensus on the answers to the questions the applicant posed.
Finally, a report is drafted by the lead reviewer and circulated to the rest of the panel for input before finalising and sending to the applicant no later than six weeks after the meeting date. The report belongs to the applicant to do with as they wish and TREAT-NMD will not share the report with anyone. The applicant is asked to only share the full report, and TREAT-NMD reserves the right to share its version with any party to whom the applicant has already shared the report, provided the applicant confirms that it has been shared. Finally, a short, mutually agreed, non-confidential summary will be added to the TREAT-NMD website.
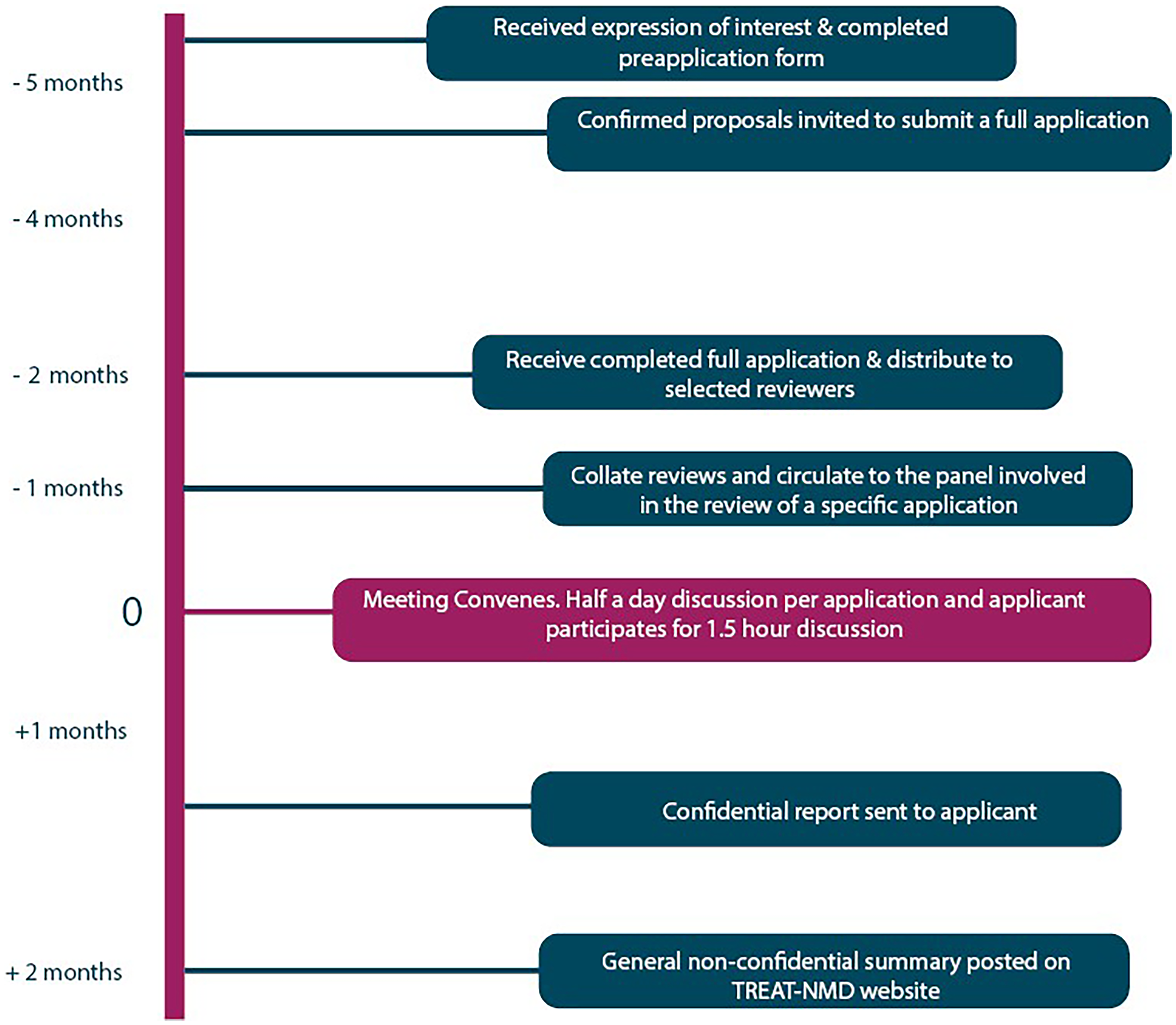
Timeline of TACT process.
The committee consists of a core group covering crucial disciplines, as well as additional members with expertise in the therapeutic approach and/or the target disease, who are invited on an as-needed basis from a growing pool of hundreds of experts in a larger pre-vetted ‘extended TACT committee’. Following a pre-meeting discussion between members, the committee meets with the applicant to ask clarifying questions, but withholds providing advice initially. The TACT team then debriefs and a subsequent confidential written report of suggestions is provided to the applicant. This report also contains answers to questions that applicants directed at TACT in the application. The applicants can use the report as they wish, with the caveat that if they choose to share the report, they are asked to share the report in its entirety.
TACT meets in person twice yearly, in spring (Europe) and autumn (North-America) from Friday to Sunday. At each meeting up to four applications can be discussed. In addition, there is an option for an online TACT review meeting that can be scheduled between the in-person meetings, thus giving drug developers extra opportunities should this be required due to e.g., strict timelines.
The importance of the patient voice
There are many ways patients can (and should) be involved in therapy development. They can be research subjects, provide samples and data for natural history studies, or participate in clinical trials to evaluate the properties, safety and efficacy of potential treatments. They can advise about trial design and on the feasibility of recruiting patients given the eligibility criteria and trial burden. Beyond this, they can also contribute by financially supporting research, and by informing representatives from academia, pharmaceutical industry, but also regulators and payers about what matters most to patients. This can result in focused translational research that aligns with the patients’ needs, and also facilitates the development of relevant outcome measures to be used in natural history studies and clinical trials. Relevant outcome measures are instrumental for benefit/risk assessments when regulators evaluate dossiers for marketing authorization.
Given the critical role patients can and should play drug development, one or more patient representatives contribute to each TACT review. These can be academically trained individuals employed by patient organisations, patient advocates or, preferably, patients. Some patient organisations have a pool of educated patient experts, e.g., the Parent Project Muscular Dystrophy (PPMD) adult advisory committee (PAAC). Notably, the patient representative co-author on this publication has recently contributed to his tenth TACT meeting.
During the review process, patient and patient representatives are asked to provide their perspective on the mechanism of action of the approach, the clinical development plan, and whether the anticipated treatment effect justifies the burden of the trial for the individual participant. Furthermore, they can give hands-on feedback on clinical trial design as most have been involved in clinical trials themselves as subjects or caregivers.
Common recurring themes in patient representative feedback is the fact that some trial designs for childhood-onset NMDs focus on children and ignore the adult population. This means that adults are not allowed to contribute to translational research, but for the EMA often results in a limited indication only applying for the ambulatory (and thus younger) population.
Furthermore, the logistics of involving NMD patients is often underestimated, e.g., the duration to move from one area of the hospital to another or the time required for restroom breaks, but also the logistics of travel, with one or more carers.
From feedback from applicants after the review process, we have learned that having patient representatives on the panel and receiving feedback also from the patient point of view is much appreciated.
Preclinical tools and practices
Scientific research faces a significant challenge in reproducibility. A striking statistic reveals that over 70% of researchers have failed to replicate another scientist's experiments, and more than half have struggled to reproduce their own results. 7 This crisis in reproducibility extends to preclinical research in NMDs, where variability in methods, data collection, and interpretation affects the reliability of results and their translation into clinical applications. To address these challenges, international efforts have been directed toward developing standardized assessment methods for muscular dystrophy models. This initiative, spearheaded by TREAT-NMD and TACT, seeks to implement rigorous standard operating procedures (SOPs) and good laboratory practices in preclinical studies8–11(see https://www.treat-nmd.org/resources-and-support/sop-library/). This summary outlines the importance of these initiatives, their impact on NMD research, and the challenges that persist in ensuring translational success from animal models to human clinical trials.
The push for standardization began with several international workshops aimed at refining protocols for muscular dystrophy preclinical assessments.3,8,10,12
These efforts culminated in the formulation of a structured approach to testing, which included: twenty-five mdx mouse protocols, eight GRMD (Golden Retriever muscular dystrophy) dog protocols and a structured SOP template covering objectives, scope, cautions, materials, methods, interpretations, references, and appendices. An international administrative oversight group was established to maintain and update SOPs, facilitate their dissemination, and introduce new protocols as needed. This continuous process ensures the scientific community benefits from robust, well-validated methodologies. 9
TREAT-NMD's commitment to maintaining and updating SOPs has led to increased adoption within the scientific community. From June 2009 to January 2010, SOPs were downloaded 600 times. From October 2023- September 2024, they were downloaded 1930 times, reflecting growing engagement with these standardized protocols. By adhering to these guidelines, researchers can minimize variability and enhance the reproducibility of their findings. However, challenges remain, particularly in translating preclinical findings into effective human treatments.
One of the most persistent issues in NMD research is the variability in phenotype expression. Factors contributing to this variability include differences in disease onset across mouse colonies, susceptibility of different muscle groups to degeneration, histological inconsistencies between limbs of the same animal, influence of litter size on body weight and disease progression, sex differences affecting disease expression and influences of genetic background, stress, and diet. To minimize these confounding variables, good husbandry practices and controlled study designs are crucial. 10 However, even with rigorous SOPs, inter-laboratory differences persist, necessitating further refinement of experimental approaches. TACT raises awareness with applicants of the variability and the importance of good husbandry when needed. Furthermore, TACT stresses the importance of independent validation of findings (see also the next section).
To improve translational outcomes, TACT reports often contain suggestions to improve study designs that ideally should incorporate, 1) Translatable endpoints, ensuring that animal model outcomes are relevant to human disease progression, 2) Treatment timing and duration, differentiating between prophylactic and therapeutic interventions, 3) Studies to elucidate the drug's pharmacokinetic (PK) and pharmacodynamics (PD) properties, and to understand drug exposure, metabolism, and target interaction, 4) Statistical rigor, which addresses sample size considerations, biological variability, and predetermined exclusion criteria, 5) Randomization and blinding to reduce bias in data acquisition and analysis and 6) Comprehensive data reporting to ensure that all methodological details are transparent for reproducibility. A landmark study by Landis et al. emphasized the importance of these principles in enhancing study reliability. 13
In summary, reproducibility remains one of the most pressing issues in scientific research, particularly in preclinical studies for neuromuscular diseases. TREAT-NMD has taken significant steps toward standardizing methodologies, refining SOPs, and ensuring greater consistency in experimental outcomes. However, challenges persist, especially in translating preclinical successes to effective human treatments and TACT reports can raise awareness and give advice to applicants when needed.
Pre-clinical learnings over 15 years
Preclinical studies are done to collect information assess if a therapeutic approach should be tested in clinical trials and if so, to guide trial design. First, studies in model systems are done to provide proof-of-concept that a potential therapy is effective. When it is, preclinical optimization is needed to assess which dose, regimen and route of administration are optimal in the model system. Information can be collected on if, and to which extent the treatment is effective when it is started at different stages of pathology. Furthermore, information on PK and PD properties of the treatment and ADME properties of the drug can be obtained model systems, to guide clinical trial design.
In the ideal scenario these studies are done in disease models that are well studied and that match the research question. Furthermore, the use of wild type reference animals is crucial to assess the size of the treatment effect. As outlined before, data are obtained from at least two independent laboratories, rather than only generated in one laboratory or company. Finally, therapeutic effects are assessed on different levels: histology and muscle function and when available SOP as published on the TREAT-NMD website are used.
Over the past 15 years, TACT has evaluated 90 applications that all contained preclinical results. We have here pooled and anonymized recurrent mistakes and wrong assumptions, with the intention to be educational and to avoid others making the same mistakes.
Not including wild type reference, or the wrong wild type To assess the extent of a therapeutic effect in a disease model using a wild type reference is crucial for molecular, histological and functional outcomes. If there is no difference between disease and wild type for a specific outcome, that outcome is not suitable to detect a treatment effect. However, when no wild type reference is taken along, any change can be (mis)interpreted as therapeutic, even one that moves away further from the wild type value. There are multiple mouse models available for muscular dystrophies, and these are available in different genetic backgrounds. It is critical to use a wild type of the same genetic background, rather than restoring to the commonly available C57BL/6 wild type. Muscle weight, mouse weight and function can vary for mice of different genetic backgrounds, as clearly shown in a publication comparing C57BL/10 and DBA/2J wild types.11,14 Obviously, when there are differences that are due to the strain rather than the pathology, assessing a treatment effect in the disease model becomes challenging. Planning preclinical studies without knowing the natural history To know when to intervene and which parameters to include in the analysis to assess therapeutic effects, it is important to study the natural history of the disease model, in parallel with the wild type of the same strain. The natural history for muscular dystrophy models will give insight on which skeletal muscle are more or less affected by pathology and when pathology starts and how it progresses. It will also give insight in variability between individual mice. Here the impact of sex (male vs female) is an important aspect. Especially with muscle function, a larger functional deficit has been reported for males than females for various muscular dystrophy models.11,14,15 This is crucial information, especially when preclinical studies are planned using small cohorts with an odd number of animals, as the impact of having one more poor performing male mouse in one of the groups will have an impact on the average function of the group per se. Furthermore, even with larger cohorts, the results will be noisy when the group contains both sexes. Ideally, a separate study is performed for each sex when there is such an impact of sex on functional deficits. For X-linked diseases, which generally occur primarily in males, it is recommended to assess treatment effects primarily in male animals. Developing a treatment for the animal model Natural history will identify pathological aspects in the disease model that ideally recapitulate the disease as it occurs in humans. However, there may be species specific aspects as well. Occasionally, there is a tendency to find treatments for these aspects, which occur in the animal model and not the disease. Obviously, this will likely not translate well to the human situation. Only treating the animal model before the onset of symptoms NMD patients will often be diagnosed after the onset of symptoms, when there already is significant muscle pathology present. Sometimes applicants will only show preclinical evidence that their approach can prevent pathology in model systems, rather than showing that a therapeutic approach can reduce established pathology, or prevent or slow down worsening of pathology. In these cases, additional preclinical studies are recommended. Moving to the clinical trial phase without having sufficient preclinical evidence Especially academic drug developers, but also small companies, tend to move to the clinical trial phase too quickly. The argument here is that with a progressive disease, patients do not have time for lengthy preclinical studies as every day they are at risk of irreversibly losing muscle function. While this may sound reasonable, the reality is that by moving to the clinical trial stage too quickly, e.g., without proper dose-finding studies, trial design suffers. This can result in severe toxicity of the test compound, while in hindsight a lower and more tolerable dose might have sufficed. Alternatively, this can result in no observed therapeutic effect, while in hindsight the dose or regimen was not optimal. As such, these poorly designed trials run the risk of false negative results. The impact of such errors in trial design is profound for the current and future patient community, as it is challenging to redo a clinical trial with new insights (that should have been obtained ideally before the first trial). Not taking disease aspects into account for drug repurposing Drug repurposing, where an approved drug for one disease is suitable as a treatment for another disease, allows quicker development for the second indication. TACT has evaluated many applications where repurposed drugs were proposed for the treatment of NMDs, most prominently DMD. From this we have distilled common considerations that a drug developer should go through when aiming to repurpose a treatment for a childhood-onset neuromuscular disorder: 1. Is the treatment approved for an acute disease or a chronic disease? As NMDs are chronic, likely life-long treatment is needed. Is this tolerable for the approved treatment? 2. Are data from children available? It is possible that the treatment is used only in the elderly and that there are no human PK and PD data yet for children. It is also possible that juvenile toxicity studies have not yet been done. If this is the case, there is an impact on timelines as these additional toxicity studies will first have to be done. Furthermore, a PK/PD study in patients will have to be done first as well as it is possible that this is influenced by the pathology (e.g., better or worse distribution of the treatment to the affected tissue). 3. Is the route of administration feasible? Young children will not be able to swallow large pills, and adult patients with NMDs may have swallowing problems. 4. Will the treatment be safe in the new indication? While a treatment may be safe in one disease, it may not be in another. E.g. in DMD patients there is continuous muscle turnover, which has an impact on kidney function. Therefore, DMD patients may tolerate compounds with potential mild kidney toxicity less well than others. Another example is the case of X-linked myotubular myopathy, where subclinical liver pathology became apparent after treatment with adeno associated viral vector gene therapy at a dose that was tolerated in other NMDs, but not in this disease.
16
Take the standards of care into account DMD patients use glucocorticoids (steroids) as part of the care guidelines, so ideally the drug is tested also in combination with steroids in preclinical studies. If the repurposed drug is acting to reduce inflammation, there may not be added benefit on top of steroids. When the drug is tested only by itself in animal models, this will only become apparent in patients. At the same time, taking patients off steroids, the treatment known to slow down disease progression, to test a drug that may or may not work, raises ethical concerns. Finally, there may be an unwanted drug-drug interaction with steroids and the repurposed drug. Look for experts in the disease Sometimes when a drug is repurposed for an NMD, the developers will have little or no experience with the NMD space. Ideally, they reach out to multiple key opinion leaders and patient representatives, rather than involving a single individual, who may not necessarily represent the perspective of the field or the whole patient community. Similarly, they may propose an animal model available to them for the preclinical NMD studies, while this is not the model used by the rest of the field and while this may not be the optimal model to test the treatment.
Clinical trial design and outcome measure selection
Clinical outcome assessments are measures that represent some aspect of a patient's health status. They quantify how a patient feels, functions or survives and are critical tools used to evaluate treatment safety and/or efficacy. It is important to select valid, reliable, sensitive, and meaningful measures as part of data-informed clinical trial design. Both the United States Food and Drug Administration (FDA) and European Medicines Agency (EMA) require a drug to impact patient function in a clinically meaningful way for full regulatory approval. Below we outline aspects that are regularly mentioned in reports, in part also because applicants request specific advice on these aspects.
The International Classification of Functioning, Disability and Health Model (ICF) is a useful tool to visualize the internal and external factors that contribute to a patient's overall health and wellbeing (https://www.who.int/standards/classifications/international-classification-of-functioning-disability-and-health). The ICF model can be used to contextualize how the mechanism of action of an investigational product would be expected to interact with a given health condition and ensure that individual patient and disease characteristics are captured effectively (Figure 2). Sometimes, applicants will select an outcome measure as a primary endpoint based on earlier success in another trial with another therapeutic approach. However, if the mechanism of action of the therapeutic approach is different, it is likely that this choice may not be correct. For instance, if the therapeutic approach aims to improve endurance, an outcome should be selected to capture this construct, while if it aims to reduce muscle weakness, measuring strength would be a more appropriate outcome.
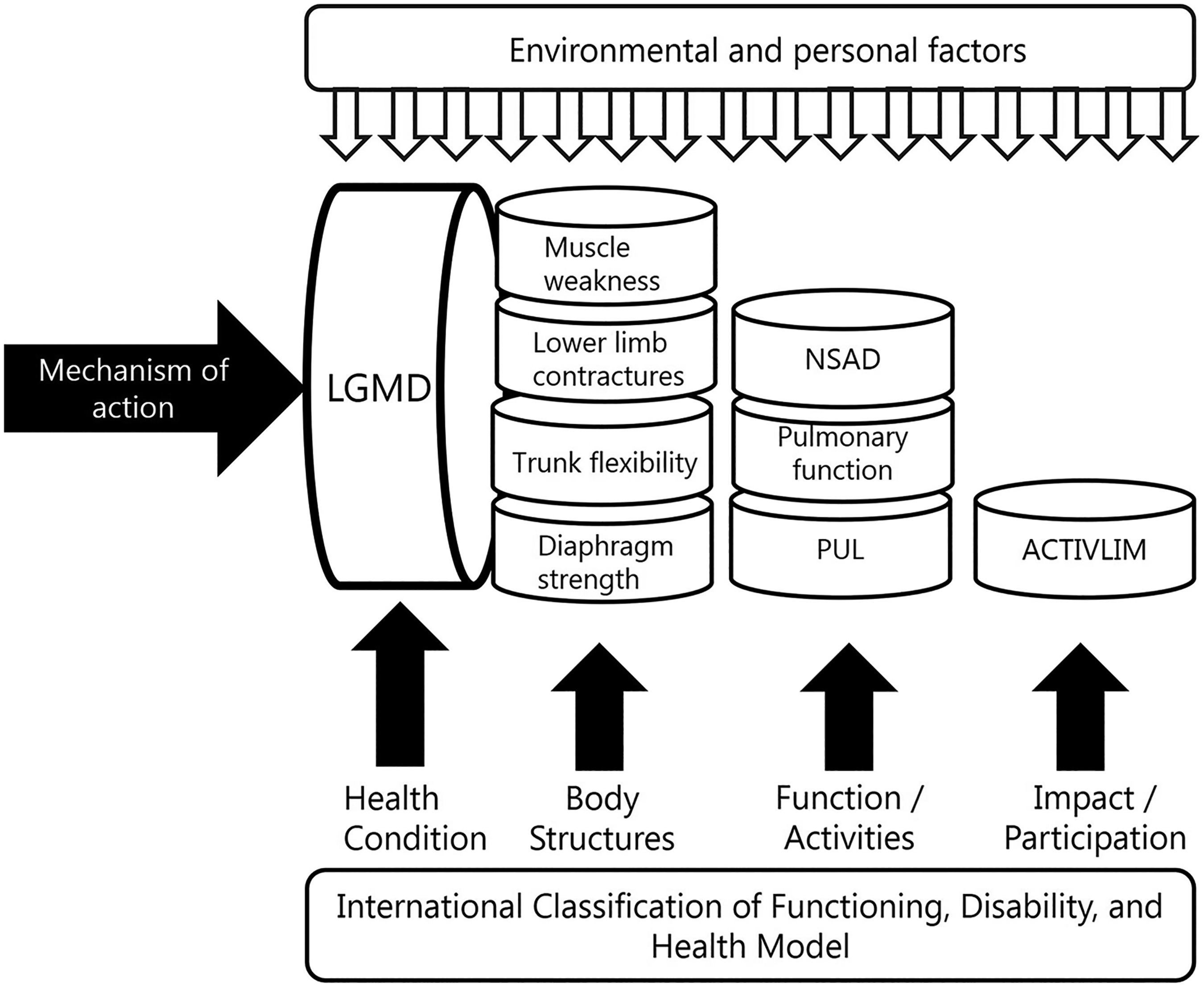
Example of use of the international classification of functioning, disability, and health model (ICF) to consider and select possible clinical outcome assessments (COA) and/or patient-reported outcomes (PRO) during clinical trial design and planning. LGMD: limb-girdle muscular dystrophy; NSAD: North-Star Ambulatory Assessment Scale for LGMD, PUL: performance upper limb.
TACT has also seen that outcomes developed for one disease are proposed for another NMD without understanding the underlying construct of an outcome. It is important to remember that existing clinical outcome assessments are often not suitable when applied to other diseases and populations and that the development of novel outcomes is an iterative (and lengthy) process. Even clinical outcome assessments that are valid and reliable in the current therapeutic landscape, may need to be refined or novel tools developed as new phenotypes emerge.
Furthermore, the selection of an appropriate clinical outcome assessment for any clinical trial should be tailored to the specific design and therapeutic context (Figure 3). Often therapeutic approaches in the NMD field aim to slow disease progression. Then it is important to select an outcome measure for which the intended trial cohort declines sufficiently during the trial duration to allow detection of a decreased decline. Natural history data are crucial for this aspect and can help with assessment construct, floor/ceiling effects, sensitivity to change, impact of covariates on reliability or validity, factors influencing data loss. Collaborative initiatives such as the Collaborative Trajectory Analysis Project (cTAP) have exemplified this approach in DMD, using pooled natural history data to refine outcome measures, reduce variability, and broaden eligibility criteria. 17
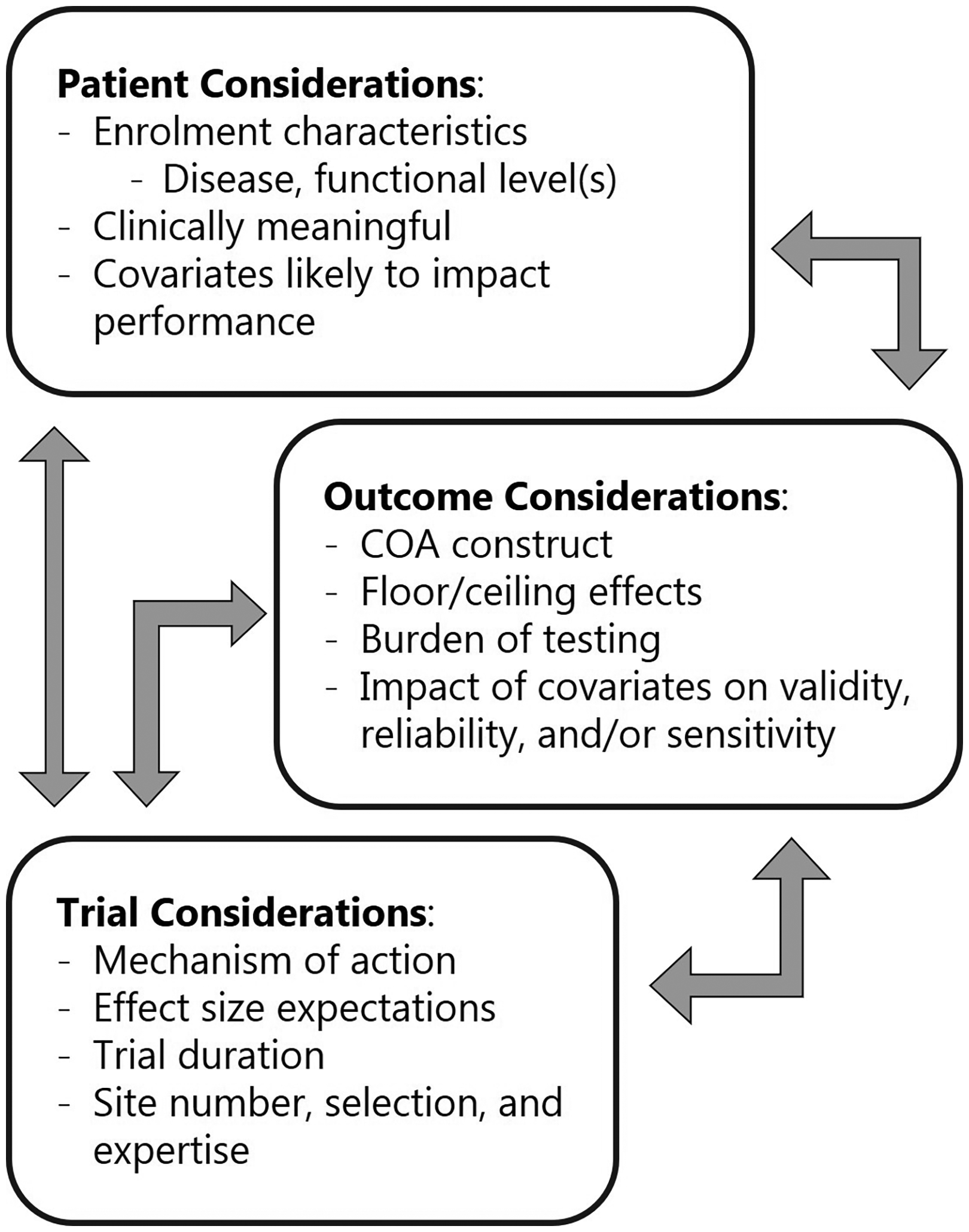
Depiction of key considerations and factors impacting the evaluation and selection of clinical trial endpoints. COA is clinical outcome assessment.
In summary, data-driven evaluation and selection of clinical outcome assessments for clinical trials is a multistep process (Figure 4). Each clinical outcome assessment must:
Perform optimally in the cohort for which it was designed (for a given age, functional status) Take into account any expected floor and/or ceiling effects for each assessment Be sensitive to change and expected change Relate this sensitivity to establish the duration of follow up required to express efficacy
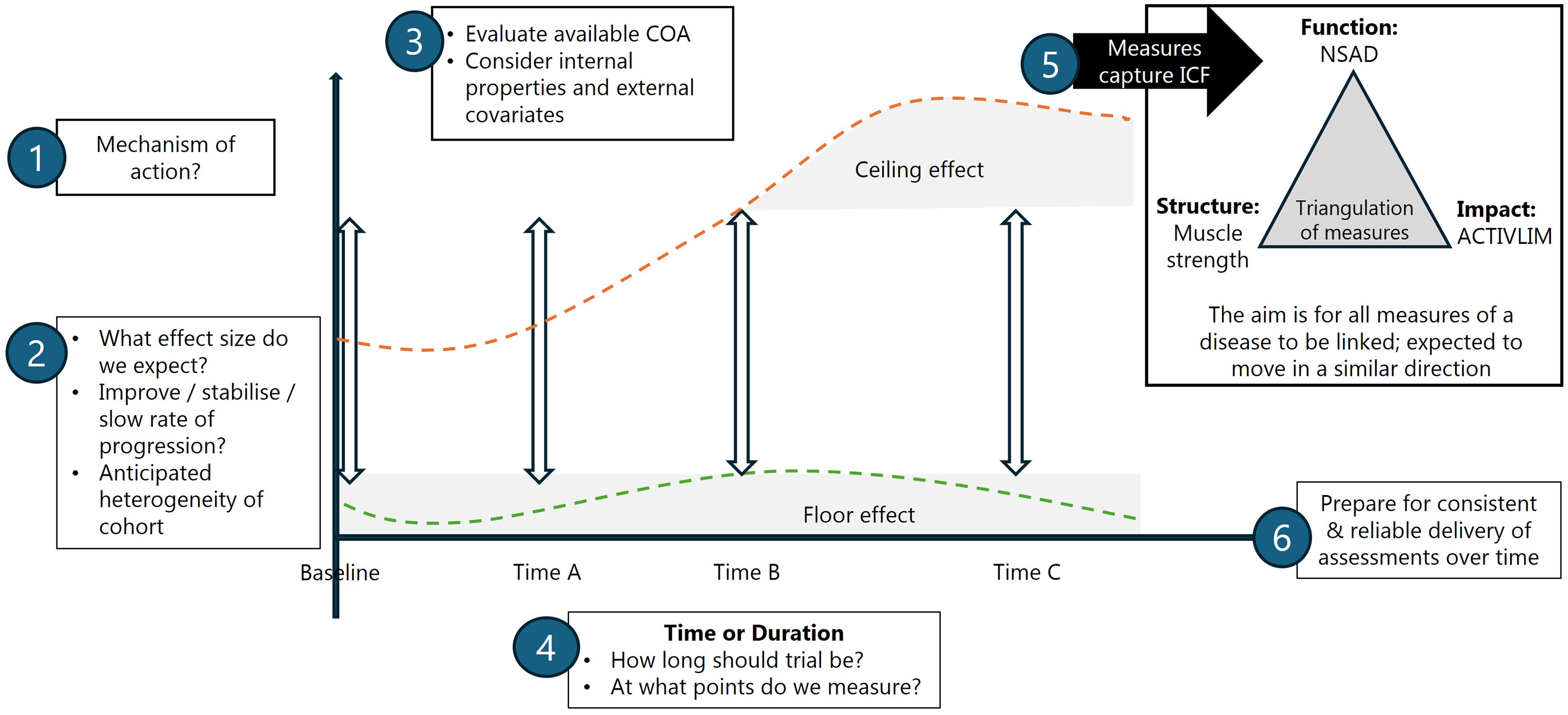
Data driven outcome selection and clinical trial design: putting all the pieces together. COA: clinical outcome assessment, NSAD: North-Star Ambulatory Assessment Scale for Limb-Girdle muscular dystrophy.
Other aspects that TACT often provides advice on are burden and feasibility. Some applicants seek to measure a significant battery of assessments, which can lead to patient fatigue, increased burden of participation, and variability in performance (over long time periods), which negatively impacts interpretability of trial efficacy. Others propose to use a study population to develop novel measures, which can confuse data interpretation. Finally, the selected clinical outcome assessment must be implemented to ensure the data are collected reliably and consistently. Many trials in rare and ultrarare disorders require multi-site international collaborations to successfully enrol and retain the number of patients needed to establish safety and efficacy. Selected clinical outcome assessments must be performed in a standardized way across sites to reduce variability which could wash out signs of treatment efficacy.
It is important for sponsors to have realistic expectations for the timeline to study start up, and enrolment, each condition and the impact of chosen inclusion and exclusion criteria on recruitment. Increasing the heterogeneity of the cohort may shorten a trial but make interpretation of the results more difficult. In general, thoughtful selection of enrolment criteria to aim for the target population is recommended. Function-based enrolment criteria are often more selective and predictive of disease trajectory than age-based cutoffs.
Statistics
Statistical analysis is required at many points along the drug development process to interpret findings and shape the path forward. While it is clear to most that clinical studies involving patients require a rigorous a priori statistical plan to be carried out by a qualified statistician, the analysis of pre-clinical studies often falls short of this goal. As such, TACT advise often centres on this aspect. Many pre-clinical studies in general, but also in TACT applications, suffer from a lack of critical evaluation of study design and hypotheses, very small sample sizes with little attention to reliability or reproducibility, and analysis performed by those without a strong understanding of statistics. These weaknesses during the early drug development stage can lead to unrealistic expectations, poor decision making, and eventual failure of the program. Addressing these weaknesses early on is a cost effective and efficient method to de-risk the program.
Much has been written about guidelines on how to perform and analyse pre-clinical studies,3,10,11,19,20 however many of the same weaknesses are repeatedly encountered in TACT applications. Pseudo-replication, a phenomenon where biological and technical replicates are conflated, occurs when we measure the same outcome multiple times on the same animal or sample, but treat each assessment as if they are independent. The statistical analysis of repeated or related measurements must be done appropriately, using repeated measures methodology, to avoid an inflated sample size and artificially low P-values, which produce unreal expectations of the effect of an intervention. A lack of biological variability, where many different experiments are performed on the same biological sample, is a pervasive issue that leads to conclusions being inappropriately applied to a population while being tested on a single biological entity. Even when multiple experiments are done, when they are all performed on the same tissue sample or cell line, one cannot with certainty conclude anything about tissues or cells from another biological entity. A final common weakness is the over-interpretation of a dose response where the observation of increased means across doses is interpreted as a dose response without assessment of whether a difference between doses is present. Even if an outcome is observed to increase or decrease over increasing doses, if the outcome is not shown to be significantly different between doses, or if a significant linear trend across doses is not tested, it cannot be concluded there is a dose response.
While we have highlighted a few common issues, there is a general lack of importance given to statistical considerations when designing pre-clinical studies and in the eventual statistical analysis of the data. It is highly recommended that those performing pre-clinical studies read the available guidelines before they plan their experiments and follow SOPs where they exist. It is also recommended to consult with a statistician or someone with appropriate training early on rather than at the point where data has been collected and require statistical analysis. They can advise on experimental design to ensure that there is appropriate biological variability to allow conclusions to be generalizable beyond the individual experiment and they can implement methods to address pseudo-replication if applicable. They can perform sample size calculations to increase the likelihood of detecting a drug effect if one exists and advise on the appropriate methods for testing the hypothesis of interest. These steps yield a better understanding of the intervention effect at an early stage in development where it is much more efficient and costs much less to make changes if necessary.
Regulatory
The regulators will determine whether a new treatment receives marketing authorisation, based on a benefit/risk assessment. While the regulatory principles apply to all therapies and diseases, it is clear that each disease has its own symptoms, challenges, peculiarities and prognosis, which defines specific benefit/risk balance. The NMD disease programs that come to TACT mostly concern rare or ultra-rare diseases, which bring challenges such as limited number of patients, poorly understood diseases and the necessity to develop specific clinical outcomes which emphasizes the fundamental need to closely interact with regulatory agencies early on. TREAT-NMD has been establishing a dialogue with regulatory agencies to align on regulatory principles, and to discuss how to best develop clinical outcome assessments in NMD.21,22 TACT has had a regulatory expert on their core committee for most of its existence. This is valuable, as especially academic experts are less aware of regulatory processes, guidance documents etc.
TACT can recommend timing and type of regulatory interactions as initiatives from regulatory agencies are improved/updated to accelerate clinical development and access to innovative medicines considering rare and ultra-rare indications. This is valuable, as especially academic experts are less aware of regulatory processes and guidance documents.
Most applications deal with either the FDA (USA) and/or the EMA. Both agencies have processes to facilitate drug development for rare diseases, including orphan drug designation with fee reduction for receiving regulatory advice, and pathways to approve treatments, pending confirmatory studies.
Notably, the way the regulatory pathways are implemented vary due to legal differences. For the FDA, accelerated approval can be granted when a surrogate marker makes it reasonably likely that the treatment is effective, for example dystrophin restoration for DMD. The EMA, by contrast, can give conditional marketing authorization, but only when clinical meaningful benefit has been shown. There is no legal ground upon which EMA can grant marketing authorization based on a surrogate endpoint, unless that endpoint is validated, meaning that a clinical trial has shown that the surrogate endpoint correlates with, or is predictive off clinical meaningful results as determined with a functional outcome measure. This difference is not always clear to pharmaceutical industries based in the US and focusing on the US market. TACT has been able to point applicants to guidance documents produced by the regulators.
A topic discussed at the 15 years TACT conference and a frequent discussion topic at TACT panel meetings relates to biopsies: companies that aim to obtain accelerated approval based on a surrogate endpoint will need to obtain biopsies from clinical trial participants. It is acknowledged that muscle biopsies are an invasive procedure, especially in individuals who progressively lose muscle tissue due to their condition. Previous research has revealed that patients and families with DMD are willing to accept muscle biopsies in clinical trials that show proof-of-concept of an approach in an open label trial, but less so when a placebo-controlled trial is done to show drug efficacy. 23 Most of the applications involving biopsies relate to treatments aiming to restore dystrophin, as there is a precedent that FDA accepts this as a surrogate endpoints in exon skipping and micro-dystrophin gene therapy treatments. Typical TACT advise relates to the muscle to obtain the biopsy from, e.g., in older patients, the muscle quality of lower limb muscle will be poor and a biopsy likely not informative, so a biopsy from an upper limb muscle may be preferred. However, in young patients, the upper limb muscles are very small and an open biopsy would remove a significant part of the muscle, so this is not preferred. Magnetic resonance imaging (MRI) can help select an area of sufficient muscle quality.
Applicants will propose needle biopsies as a less invasive alternative to an open biopsy. However, this will not always work, e.g., when more material is needed for analysis than provided with a needle biopsy. Furthermore, with a needle biopsy a clinician has less influence on obtaining a piece of muscle rather than an area of adipose or fibrotic tissue that will not be informative. From the patient perspective it is clear that while biopsies are burdensome, taking a biopsy and being unable to obtain useful information from it is unacceptable.
Using biopsies for pharmacodynamic confirmation of a treatment effect for other therapeutic approaches are often proposed. However, applicants do not always realize that being able to use this as a surrogate endpoint with the FDA is not a given, and involves validation of the method to assess the treatment effects.
For trials aiming to show clinical efficacy on a muscle function levels, the challenge is that most treatments in development for NMDs may not improve function, but slow down or stabilize functional decline. This requires long placebo-controlled clinical trials that may need to be 12, 18 or 24 months in duration. To reduce patient burden, using external controls is often proposed instead of, or to complement the placebo arm. However, this can only be done under strict criteria and using well characterized and recent natural history data. The exception would be that a treatment results in an effect that is never observed in the natural history such as was exemplified with the achievement of motor milestones and survival of type I SMA babies with disease modifying treatments. 24 However, for most therapies in development for NMDs, more subtly altering the trajectory of the disease course is the likely outcome. 5 As such, clinical trial designs using external controls may in certain circumstances be used to support regulatory-decision making justified by rarity and unmet medical need and should be discussed with regulatory agencies. In order to use external controls as pivotal evidence to establish a positive benefit-risk balance careful pre-planning is needed with matched baseline characteristics, inclusion/exclusion criteria, standard of care, endpoints and with a prespecified analysis plan. 5 Expectations for prognostic comparability and bias control are usually high. Currently the EMA is developing a reflection paper to clarify the understanding of methodological concepts and challenges and outline criteria for when the use of external controls may be acceptable and there is currently guidance available from the FDA.
Another way to reduce patient burden is to allocate more patients in the treatment arm than in the placebo arm, e.g., 2:1 or 3:1 ratios. While this may look like a way to make a clinical trial more appealing to patients, as the likelihood that they will be in the placebo arm is reduced, often this will mean that the total number of patients that need to be recruited to properly power the clinical trial will increase to the point that more patients are in the placebo arm than when a 1 to 1 allocation ratio was used.
Biomarkers
As mentioned, FDA's accelerated approval pathway enables conditional approval based on a surrogate marker deemed likely to predict clinical benefit. Furthermore, biomarkers can help provide evidence of target engagement of therapeutic approaches. These biomarkers can range from serum tests such as creatine kinase, imaging studies (i.e., lower extremity MRI) to more invasive tests such as a muscle biopsy. Biomarkers serve several purposes ranging from establishing a diagnosis, target engagement, prognosis and monitoring a therapeutic response.
Biomarkers can be used to monitor a therapeutic response. They do not need to be disease specific or related to the therapy's mechanism of action, although in some cases this may be the case (i.e dystrophin levels with eteplirsen). Instead, the ideal therapeutic response biomarker must correlate both disease progression and therapeutic response. One recent example in the neuromuscular space is the use of serum neurofilament light (NfL) chain levels in SOD1-mediated familial ALS. Intrathecal administration of an SOD1 specific antisense oligonucleotide was administered to patients with SOD1-mediated familial ALS in a 28 week phase 3 trial (VALOR). Serum NfL levels are elevated in multiple neurodegenerative disease states. In the case of ALS, an elevation of serum NfL levels coincides with disease onset and patients with higher serum NfL levels progress more rapidly than patients with lower serum NfL levels supporting its use as a therapeutic biomarker. Although, the VALOR study failed to meet its primary endpoint demonstrating a decrease in the rate ALS progression, FDA approval was given based on the compelling biomarker evidence that found a decrease in serum NfL levels following treatment with Tofersen. 18
TACT has received proposals to use a specific biomarker as a surrogate endpoint in clinical trials. Here, TACT has raised awareness about differences between different regulatory agencies and their ability to accept surrogate endpoints as primary endpoint (see previous section). Furthermore, to use a biomarker as a primary endpoint, the method to measure the biomarker must be performed according to regulatory standards. When an applicant intends to use a biomarker as a surrogate endpoint, this possibility should be discussed with the regulators early. Finally, applicants should be aware that even though FDA may accept some biomarkers as surrogate endpoints, they will also request additional phase 3 clinical trials, to confirm a clinically meaningful treatment effect. Applicants should anticipate this request, as these confirmatory trials should be planned or ideally ongoing at the time a new drug application (NDA) based on a surrogate endpoint is submitted to FDA.
The continuous need for TACT
The NMD field has made extraordinary progress over the past 15 years, from one approval to multiple approved treatments for multiple NMDs in many Western countries. However, it is also clear that there remains large unmet medical need with many of the available therapies only marginally changing the disease course and many NMDs, especially the more rare ones, still having no therapeutic options.
A steady number of new companies continue to enter the NMD field and they will be able to benefit from the TACT process and the lessons learned described in this paper. In addition, as companies move from early stage trials to late stage trials, they can still benefit from additional TACT advice. In fact, 5 applicants have returned to TACT twice, and 2 have come back up to 4 times.
The goal of TACT continues to be to derisk therapy development for NMDs and with 15 additional years of multidisciplinary expertise, advice can be based on additional lessons and success stories.
Footnotes
Author contributions
AAR, LR, LA, AM, HGD, AR, KN wrote the manuscript, all authors edited the manuscript, LR, LA and AM generated the figures. LR and AAR coordinated the writing process.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Disclosures
None of the authors have a conflict related to this publication. For full transparency, some authors disclose the following: AAR discloses being employed by LUMC which has patents on exon skipping technology, some of which has been licensed to BioMarin and subsequently sublicensed to Sarepta. As co-inventor of some of these patents AAR was entitled to a share of royalties. AAR further discloses being ad hoc consultant for PTC Therapeutics, Sarepta Therapeutics, Regenxbio, Dyne Therapeutics, Lilly, BioMarin Pharmaceuticals Inc., Eisai, Entrada, Takeda, Splicesense, Galapagos, Sapreme, Italfarmaco and Astra Zeneca. AAR also reports being a member of the scientific advisory boards of Hybridize Therapeutics (past), Silence Therapeutics, Sarepta therapeutics, Sapreme and Mitorx. Remuneration for consulting and advising activities is paid to LUMC. In the past 5 years, LUMC also received speaker honoraria from Alnylam Netherlands, Italfarmaco and Pfizer and funding for contract research from Sapreme, Eisai, BioMarin, Galapagos and Synaffix. Project funding is received from Sarepta Therapeutics and Entrada via unrestricted grants.
AGM has served on medical/scientific advisory boards for Regenxbio, Sarepta, Biogen and Roche; and has received fees for consulting and training services for Biogen, Glycomine, QED, Synox, Roche, Novartis, Biohaven, PTC, Sarepta, Italfarmaco, Dyne, Pfizer, Summit, Catabasis, Capricor, Santhera, Vision, Lysogene, Modis, Amicus, Taysha, Antisense, Analysis Group, MDUK and DUK.
KN is the co-founder of AGADA Biosciences and Reveragen Biopharma. Is is one of the board Trustees of TREAT-NMD alliance ltd.
KRW was a previous employee of Novartis and is an advisory committee member of Asklepios, MLAB, SOLVE FSHD, advisor to Cure Rare Disease, Alesta, Altay, Epi labs, Tenayathera and stock owner of Vita Therapeutics.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.