OS01.01
Phase 2/3 Study of an Investigational Anti-Inflammatory Biologic Therapy in Inclusion Body Myositis
Dr. Namita Goyal1, Dr. Pedro Machado2,3, Dr. Dana Ascherman4, Dr. Olivier Benveniste5, Dr. Said Beydoun6, Dr. Thomas Brannagan7, Dr. James Caress8, Dr. Lisa Christopher-Stine9, Dr. Michael Collins10, Dr. Jan De Bleecker11, Dr. Mazen Dimachkie12, Dr. Stacy Dixon13, Dr. Alex Dmitrienko14, Dr. David Fernandez15, Dr. Miriam Freimer16, Dr. Christopher Geiger17, Dr. Paloma Gonzalez-Perez18, Dr. Neelam Goyal19, Dr. Kelly Gwathmey20, Dr. Daragh Heitzman21, Dr. Robert Henderson22,23, Dr. Lisa Hobson-Webb24, Dr. Yessar Hussain25, Dr. Lawrence Korngut26, Dr. Christina Liang27,28,29, Dr. James Lilleker30,31, Dr. Thomas Lloyd32,33,34, Dr. Payam Mohassel32, Dr. Elie Naddaf35, Dr. Merilee Needham36,37,38,39, Dr. Ezequiel Piccione40, Dr. Colin Quinn41, Dr. Laura Rosow42, Dr. Bhaskar Roy43, Dr. Tobias Ruck44,45, Dr. Jens Schmidt46,47, Dr. Arjun Seth48, Dr. Jaimin Shah49, Dr. Aziz Shaibani50, Dr. Perry Shieh51, Dr. Zachary Simmons52, Dr. Kumaraswamy Sivakumar53, Dr. Leo Wang54, Dr. Conrad Weihl55, Dr. H. Jeffrey Wilkins56, Dr. Anthony Amato57
1
Department of Neurology, University of California Irvine, Orange, United States.
2
Department of Neuromuscular Diseases, UCL Queen Square Institute of Neurology, University College London, London, United Kingdom.
3
NIHR University College London Hospitals Biomedical Research Centre, University College London Hospitals NHS Foundation Trust, London, United Kingdom.
4
Department of Medicine, University of Pittsburgh School of Medicine, Pittsburgh, United States.
5
Department of Internal Medicine and Clinical Immunology, Pitié-Salpêtrière University Hospital, Sorbonne Université, AP-HP, Paris, France.
6
University of Southern California, Los Angeles, United States.
7
Neurological Institute, Columbia University Medical Center, New York, United States.
8
Wake Forest University School of Medicine, Winston-Salem, United States.
9
Johns Hopkins University School of Medicine, Baltimore, United States.
10
Medical College of Wisconsin, Milwaukee, United States.
11
Department of Neurology and Neuromuscular Reference Center, Ghent University Hospital, Ghent, Belgium.
12
Department of Neurology, The University of Kansas Medical Center, Kansas City, United States.
13
University of Colorado, Aurora, United States.
14
Department of Biostatistics, Mediana, San Juan, United States.
15
Department of Rheumatology, Hospital for Special Surgery, New York, United States.
16
Department of Neurology, The Ohio State Wexner Medical Center, Columbus, United States.
17
University Hospitals-Cleveland Medical Center, Case Western Reserve University, Cleveland, United States.
18
Department of Neurology, Massachusetts General Hospital, Harvard Medical School, Boston, United States.
19
Stanford Neuroscience Health Center, Palo Alto, United States.
20
Department of Neurology, Virginia Commonwealth University, Richmond, United States.
21
Texas Neurology, Dallas, United States.
22
Department of Neurology, Royal Brisbane and Women's Hospital, Brisbane, Australia.
23
Centre for Clinical Research, University of Queensland, Brisbane, Australia.
24
Neuromuscular Division/Department of Neurology, Duke University, Durham, United States.
25
Austin Neuromuscular Center, Austin, United States.
26
Hotchkiss Brain Institute, Department of Clinical Neurosciences, Cumming School of Medicine, University of Calgary, Alberta, Canada.
27
Faculty of Medicine and Health, University of Sydney, Camperdown, Australia.
28
Department of Neurology, Royal North Shore Hospital, Northern Sydney Local Health District, Sydney, Australia.
29
Neuroscience Research Austral, Sydney, Australia.
30
Manchester Centre for Clinical Neurosciences, Northern Care Alliance NHS Foundation Trust, Salford, United Kingdom.
31
The University of Manchester, Manchester, United Kingdom.
32
Department of Neurology, Johns Hopkins University School of Medicine, Baltimore, United States.
33
Department of Neurology, Baylor College of Medicine, Houston, United States.
34
Department of Clinical Research, Imricor Medical Systems, Burnsville, United States.
35
Department of Neurology, Mayo Clinic, Rochester, United States.
36
Perron Institute, QEII Medical Centre, Nedlands, Nedlands, Australia.
37
University of Notre Dame, Perth, Australia.
38
Fiona Stanley Hospital, Perth, Australia.
39
Murdoch University, Perth, Australia.
40
Department of Neurology, University of Nebraska Medical Center, Omaha, United States.
41
Neuromuscular Division, Neurology Department, University of Pennsylvania, Philadelphia, United States.
42
University of California San Francisco, San Francisco, United States.
43
Department of Neurology, Yale School of Medicine, New Haven, United States.
44
Ruhr University Bochum, BG-University Hospital Bergmannsheil Department of Neurology, Bochum, Germany.
45
Heimer Institute for Muscle Research, BG-University Hospital Bergmannsheil Department of Neurology, Bochum, Germany.
46
Department of Neurology, Neuromuscular Center, University Medical Center Göttingen, Göttingen, Germany.
47
Department of Neurology and Pain Treatment, Neuromuscular Center, Center for Translational Medicine, Immanuel University Hospital, Brandenburg Medical School, Rüdersdorf bei Berlin, Germany.
48
Department of Neurology, Northwestern Memorial Hospital, Chicago, United States.
49
Department of Neurology, Mayo Clinic, Jacksonville, United States.
50
Nerve and Muscle Center of Texas, Houston, United States.
51
Department of Pediatrics, University of California Los Angeles, Los Angeles, United States.
52
Department of Neurology, Penn State University College of Medicine, Hershey, United States.
53
The Neuromuscular Research Centre and Neuromuscular Clinic of Arizona, Phoenix, United States.
54
Department of Neurology, University of Washington, Seattle, Seattle, United States.
55
Department of Neurology, Washington University in St Louis, St. Louis, United States.
56
Abcuro, Inc., Newton, United States.
57
Brigham and Women’s Hospital, Harvard Medical School, Boston, United States
Background: Inclusion body myositis (IBM) is a rare, progressive disorder characterized by invasion of muscle by highly differentiated cytotoxic CD8+ T cells. IBM can cause difficulty walking, loss of grip strength, and dysphagia. There are no pharmacological treatments approved for IBM. Ulviprubart, a humanized monoclonal antibody, selectively depletes cytotoxic CD8+ KLRG1+ T cells by targeting KLRG1 expressed on most IBM-muscle–infiltrating T cells. Here, we describe the study design and results of a phase 2/3 study of ulviprubart for treatment of IBM.
Methods: This randomized, double-blind, placebo-controlled trial (NCT05721573) included patients aged ≥40 years with an IBM diagnosis otherwise fulfilling 2011 European Neuromuscular Center criteria. Ulviprubart (0.5 or 2.0 mg/kg) or placebo (1:1:1) was administered subcutaneously every 8 weeks for 76 weeks, with the option to receive treatment at 2.0 mg/kg in an open-label extension study. Efficacy endpoints included mean changes from baseline to week 76 in Inclusion Body Myositis Functional Rating Scale (IBMFRS; primary), manual muscle testing (MMT) of 23 muscle groups converted to a Kendall score, hand grip and quadriceps strength by dynamometry, and modified Timed Up and Go (mTUG). Safety monitoring included assessment of treatment-emergent adverse events.
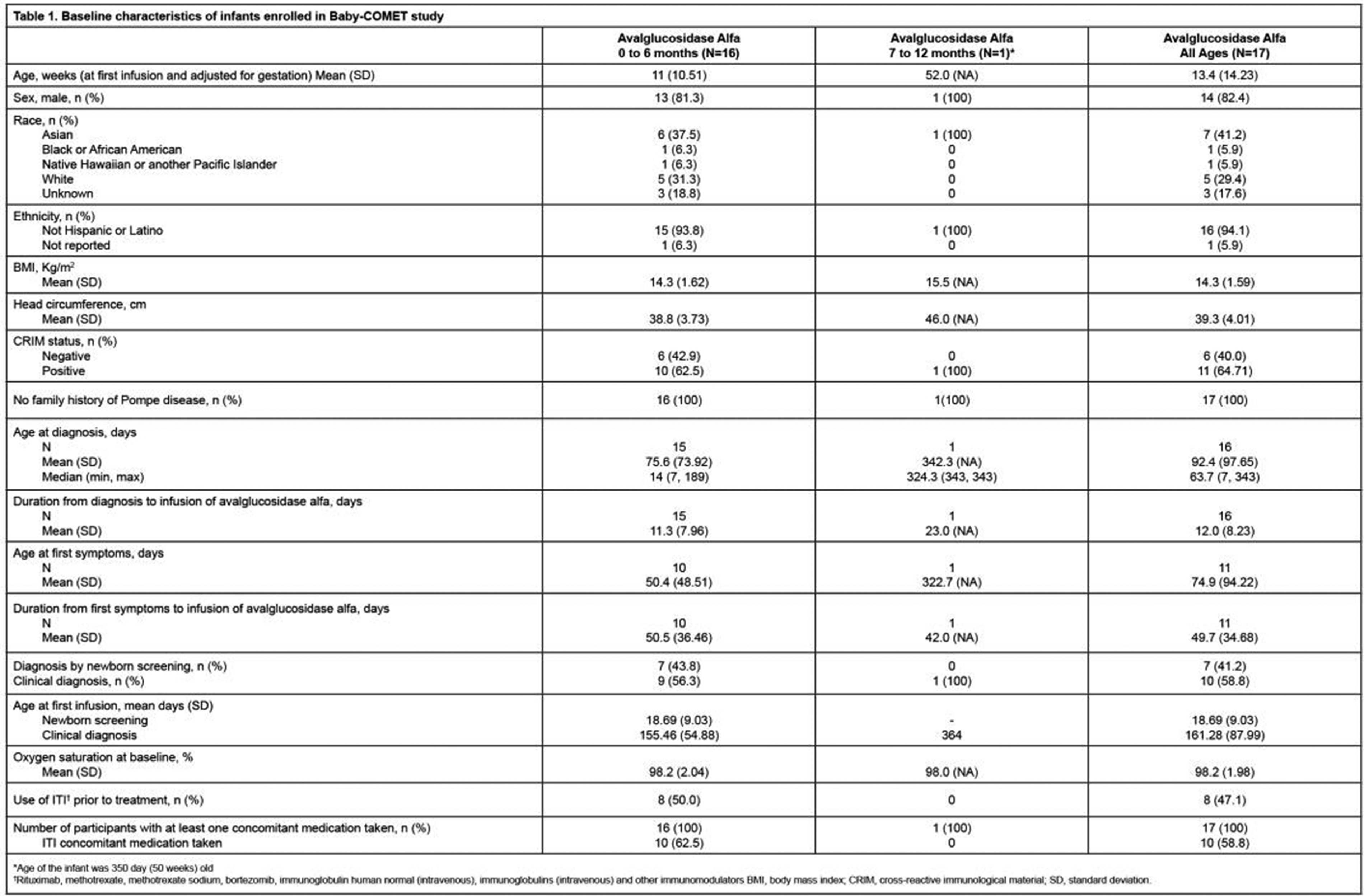
Results: Overall, 272 patients were enrolled. Mean (SD) patient age at baseline was 68.1 (7.8) years, and 186 patients were male (68%). Mean (SD) age at diagnosis was 65.1 (8.1) years. Mean (SD) baseline IBMFRS score was 27.3 (5.0); 14 patients had baseline IBMFRS scores of <20, 63 had scores of 20−24, and 195 had scores of ≥25. Mean (SD) baseline values for MMT, grip strength (dominant), quadriceps strength (dominant), and mTUG score were 7.8 (1.2), 11.7 (7.0) kg, 11.0 (7.6) kg, and 0.32 meters/second, respectively.
Conclusion: This is the largest prospective interventional study of patients with IBM with a range of clinically significant disease burden. Topline efficacy and safety results will be presented.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Inflammatory / Immune-mediated Myopathies including Inclusion Body Myositis (IBM)
OS01.02
Genetic Modifier LTBP4 Stratification of Proteomics Points to Interacting IL-23/IL-6/IL-17D Pathways in Severity of DMD
Prof. Eric Hoffman1, Assoc. Prof. Utkarsh Dang2, Asst. Prof. Yuan Fang3, Dr. Daniele Sabbatini4, Prof. Elena Pegoraro4, Assoc. Prof. Luca Bello4, Prof. Paula Clemens5, Prof. Michela Guglieri6, Prof. Johannes van den Anker7, Dr. Jesse Damsker8, Dr. Laura Hagerty8, Prof. Yetrib Hathout1, Miss Lauren Morgenroth9, Prof. Kanneboyina Nagaraju1, Prof. Jyoti Jaiswal7
1
Binghamton University - State University of New York, Binghamton, United States.
2
Carleton University, Ottawa, Canada.
3
Old Dominion University, Norfolk, United States.
4
University of Padova, Padova, Italy.
5
University of Pittsburgh School of Medicine, Pittsburgh, United States.
6
Newcastle University, Newcastle, United Kingdom.
7
Children's National Hospital, Washington, United States.
8
ReveraGen, Vienna, United States.
9
TRINDS, Pittsburgh, United States
Background: Genetic modifiers in Duchenne muscular dystrophy are gene polymorphisms, non-linked to the DMD gene, that alter disease severity or response to therapy. Studies of DMD natural history studies and patient registries have identified LTBP4, SPP1, ACTN3, CD40, ADRB2, TNFRSF10A, THBS1, DYNLT5 (TCTEX1D1) as genetic modifiers in DMD using progression (loss of ambulation) in the second decade of life as the associated phenotype. We tested associations of genetic modifiers in young (4 to <7 yr) steroid naïve association clinical trial participants. For validated modifiers, we stratified serum profiles at baseline by genotype to identify candidate cellular responses associated with clinical severity.
Methods: Participants in clinical trials (VBP15-002/003 n=48; VBP15-004 n=131) were genotyped for genetic modifier loci (n=110 genotyped). Associations of genotypes with baseline motor function were defined via an age-adjusted linear model, and p-values corrected for false discovery rate. Drug response was modeled by inheritance genotype-stratified placebo vs. steroid treatment at 12 and 24 weeks treatment (MMRM). Serum proteomic profiling was done using SomaScan aptamer panels, genotype-stratified pre-treatment serum biomarkers identified and mapped to cell types in muscle using snSeq datasets.
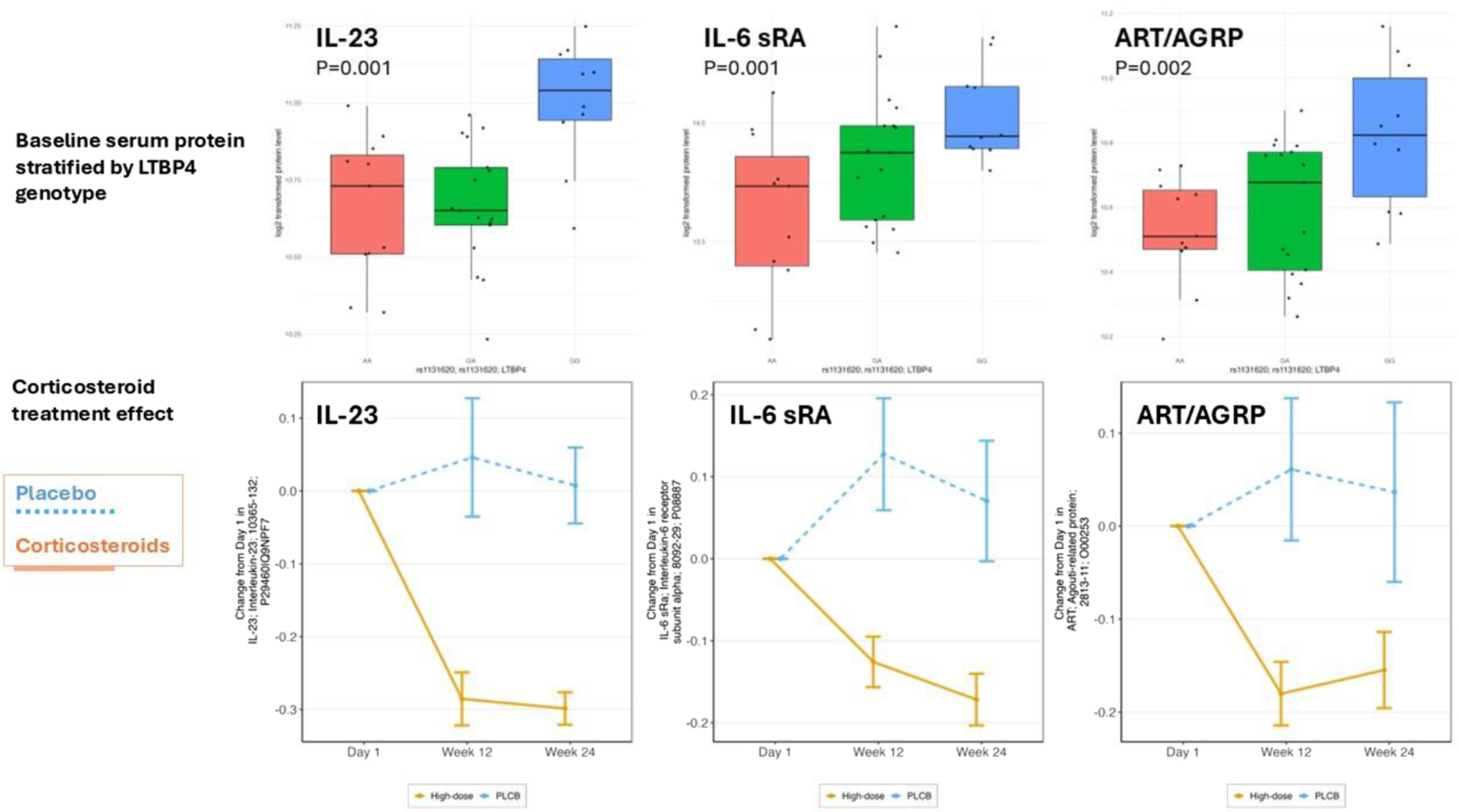
Results: LTBP4 genotype (rs1131620) was associated with baseline motor function for all five motor tests studied (time to stand from supine velocity, time to run/walk 10 m velocity, time to climb 4 stairs velocity, 6-min walk distance, and NorthStar Ambulatory Assessment). DYNLT5 genotype (rs1060575) was associated with time to stand from supine velocity, time to climb 4 stairs velocity, and time to run/walk 10 m velocity. The association of genotypes matched previous studies of loss of ambulation in older steroid-treated DMD patients. Stratification of baseline Somascan proteomic profiling data by LTBP4 genotype and mapping of genotype-associated serum proteins to specific cell types in muscle (snRNAseq) suggested that IL-23, IL-6 and IL-17D interacting pathways may be contributors to disease progression. In contrast, the dominant DYNLT5 genotype was associated with proteosome and chaperonin pathways.
Conclusion: Significant associations with improved baseline motor outcomes in young steroid naïve DMD children were found for LTBP4 and DYNLT5 loci IL-23, IL-6 and IL-17D interacting pathways may participate in disease progression.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
Images or Table (Optional)
OS01.03
Targeting Musashi-2 With Antisense Oligonucleotides Mitigates Muscle Pathology in Myotonic Dystrophy Type 1
Dr. Dulce Peris-Moreno1, Ms. NATALIA RIEDEL2,3,4, Mr. ALBERTO GARCÍA-MARTÍN3, Mr. JORGE ESPINONSA-ESPINOSA5, Dr. ARIADNA BARGIELA3, Prof. RUBÉN ARTERO2,3,4
1
Ciberer, Iscill, Madrid, Spain.
2
BIOTECMED, UNIVERSITY OF VALENCIA, VALENCIA, Spain.
3
INCLIVA, VALENCIA, Spain.
4
CIBERER, ISCIII, MADRID, Spain.
5
Universidad Particular Internacional SEK, QUITO, Ecuador
Background: Myotonic dystrophy type 1 (DM1) is a rare genetic disorder caused by an expansion of a non-coding CTG repeat in the DMPK gene, leading to the sequestration and translational repression of MBNL proteins and resulting in severe muscle dysfunction, including myotonia, atrophy and weakness. We recently identified the RNA-binding protein Musashi-2 (MSI2) as a critical contributor in DM1 pathology: MSI2 is overexpressed in the disease and represses miR-7 biogenesis, disrupts autophagy regulation and exacerbates muscle loss. Targeting MSI2 may therefore complement strategies that directly neutralize toxic CUG-expanded RNA. In this study, we explored MSI2 as a therapeutic target using antisense oligonucleotides (ASOs) to knockdown its expression in vitro and in vivo.
Methods: A total of 127 ASOs targeting MSI2 mRNA were designed in silico. Based on bioinformatics criteria and to achieve maximal coverage of transcript sequences through non-overlapping tiling, 27 candidates were selected for secondary in vitro profiling in human DM1 myotubes (hDM1-myo), from which the best candidate ASOs were chosen for detailed evaluation. In hDM1-myo, we quantified MSI2 silencing, myogenic fusion (desmin immunostaining) and transcriptomic changes. For in vivo studies, the best candidate ASOs were administered subcutaneously to HSALR mice to assess MSI2 knockdown across tissues, with MSI2 protein levels quantified by Jess simple western and ASOs levels by hybridization-based ELISA. After detecting insufficient skeletal-muscle delivery, ASO chemistry was further optimized, and the new design was re-evaluated for MSI2 silencing in skeletal muscle and other tissues, as well as for renal and hepatic tolerability.
Results: Initial in vitro screening identified 27 MSI2-directed ASOs, from which the best candidates were selected for in-depth analysis. In hDM1-myo, MSI2 silencing by these ASOs produced marked phenotype improvement, including a >75% increase in myogenic fusion capacity. Transcriptomic analyses showed restoration of pathways associated with muscle function and cytoskeletal integrity. In HSALR mice, systemic administration of ASOs in a single dose significantly reduced MSI2 levels in non-muscular tissues such as liver, but skeletal-muscle uptake and knockdown were limited. ELISA quantification confirmed that this limitation reflected poor ASO delivery to skeletal muscle. Subsequent optimization of ASO chemistry together with additional dosing frequency and concentration, substantially enhanced MSI2 silencing in skeletal muscle as well as in other tissues, without evidence of renal or hepatic toxicity at the tested doses. In a dedicated durability study, one candidate ASO achieved sustained Msi2 silencing in liver and skeletal muscle for at least 45 days post-treatment indicating a durable Msi2 knockdown in vivo.
Conclusion: MSI2 silencing with ASOs induces robust in vitro improvements in DM1 myotubes, including enhanced myogenic fusion and transcriptional recovery of muscle-structural pathways, supporting MSI2 as a relevant therapeutic target in DM1. In vivo, the studies highlight delivery to skeletal muscle as a critical bottleneck but also show that appropriate chemical optimization can markedly improve MSI2 knockdown in muscle without overt toxicity and with sustained target engagement. Together, these findings identify MSI2 as a promising therapeutic target in DM1 and support MSI2-directed ASOs, while highlighting the need to optimize skeletal-muscle delivery to enable clinical translation
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Myotonic Dystrophies
OS01.04
The Facial-Sparing FSHD Phenotype: A Distinct Entity With Implications for Diagnosis, Prognosis, and Trial Design
Dr. Andi Nuredini1, Dr. Maria Francesca Di Feo2,3, Dr. Riccardo Cuoghi Costantini4, Dr. Dario Zoppi5, Dr. Adele Barbaccia6, Dr. Noemi Albano1, Dr. Isabella Allegri7, Prof. Giovanni Antonini8, Dr. Silvia Bonanno9, Dr. Elena Canali10, Dr. Maria Grazia D'Angelo11, Prof. Massimiliano Filosto12, Prof. Marina Grandis2, Dr. Lorenzo Maggi9, Dr. Maurizio Moggio13, Prof. Elena Pegoraro14, Prof. Stefano Previtali15, Dr. Sabrina Ravaglia16, Dr. Giulia Ricci17, Prof. Gabriele Siciliano17, Dr. Maria Lucia Valentino18, Prof. Gaetano Vattemi19, Dr. Daniele Velardo13, Prof. Tiziana Enrica Mongini20, Prof. Carmelo Rodolico6, Dr. Lucia Ruggiero5, Prof. Rossella Tupler1
1
Department of Biomedical, Metabolic, and Neural Sciences, University of Modena and Reggio Emilia, Modena, Italy.
2
Department of Neuroscience, Rehabilitation, Ophthalmology, Genetics, and Maternal and Child Health (DINOGMI), University of Genoa, Genova, Italy.
3
Folkhälsan Research Center, Helsinki, Uusimaa, Finland.
4
Department of Medical and Surgical Sciences, University of Modena and Reggio Emilia, Modena, Italy.
5
Department of Neurosciences, Reproductive and Odontostomatological Sciences, University of Naples Federico II, Napoli, Italy.
6
Department of Clinical and Experimental Medicine, University of Messina, Messina, Italy.
7
Department of General and Specialized Medicine, University Hospital of Parma, Parma, Italy.
8
Neuromuscular and Rare Disease Centre, Department of Neuroscience, Mental Health and Sensory Organs (NESMOS), Sapienza University of Rome, Roma, Italy.
9
Neuroimmunology and Neuromuscular Diseases Unit, Foundation IRCCS Carlo Besta Neurological Institute, Milano, Italy.
10
Department of Neurology, IRCCS Arcispedale Santa Maria Nuova, Reggio Emilia, Italy.
11
Unit of Rehabilitation of Rare Diseases of the Central and Peripheral Nervous System, Scientific Institute IRCCS Eugenio Medea, Bosisio Parini, Italy.
12
Department of Clinical and Experimental Sciences, NeMO-Brescia Clinical Center for Neuromuscular Diseases, University of Brescia, Brescia, Italy.
13
Neuromuscular and Rare Disease Unit, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milano, Italy.
14
Neuromuscular Unit, Department of Neuroscience, University of Padova, Padova, Italy.
15
Neuromuscular Repair Unit, Institute of Experimental Neurology (INSPE) and Division of Neuroscience, Vita-Salute San Raffaele University, Milano, Italy.
16
Department of Neurology, Istituto di Ricovero e Cura a Carattere Scientifico Mondino Foundation, National Institute of Neurology, Pavia, Italy.
17
Department of Clinical and Experimental Medicine, University of Pisa, Pisa, Italy.
18
Department of Biomedical and Neuromotor Sciences, University of Bologna, Bologna, Italy.
19
Department of Neurosciences, Biomedicine and Movement Sciences, Section of Clinical Neurology, University of Verona, Verona, Italy.
20
Neuromuscular Unit, Department of Neuroscience “Rita Levi Montalcini”, University of Turin, Torino, Italy
Background: Facioscapulohumeral muscular dystrophy (FSHD) shows marked phenotypic heterogeneity. Although facial weakness is considered a disease hallmark, a facial-sparing presentation challenges diagnosis and prognosis with relevant implication for genetic counselling, and trial readiness. Using data from the Italian National Registry for FSHD, we compared the clinical, molecular, longitudinal, and family features of the facial-sparing phenotype with those of the classical phenotype.
Methods: This registry-based study included subjects evaluated between 2013 and 2025. Standardized deep phenotyping was performed using the Comprehensive Clinical Evaluation Form (CCEF). 183 patients with scapular weakness and no facial weakness (CCEF category B1) and 343 with classical phenotype (category A) were selected. Demographics, disease history, FSHD clinical score and subscores, and composite MRC strength were compared. Genotype-phenotype associations were assessed by D4Z4 allele size. Longitudinal change was analyzed in patients with follow-up. Family studies were performed when possible.
Results: In the B1 cohort (n=183), 32% were female and 58% were index cases. B1 subjects showed mean FSHD score of 2.5±1.8 with composite MRC 126.8±5.9 (out of 130). Compared with A cases, B1 had later onset (30.4±16.3 vs 25.0±15.9 years) and lower disease burden: mean FSHD score in A was 5.24±3.1 excluding the facial component, and the difference persisted after age adjustment. The distribution of D4Z4 alleles in the B1 cohort was: 1-3 RU 3.8%, 4-5 RU 13.7%, 6 RU 36.1%, 7-8 RU 18.0%, 9-10 RU 16.9%, and ≥11 RU 11.5%; in the A cohort the distribution was: 1-3 RU 16.0%, 4-5 RU 27.1%, 6 RU 30.9%, 7-8 RU 8.8%, 9-10 RU 8.5%, and ≥11 RU 8.7%. In the B1 cohort allele size did not correlate with clinical severity and B1 cases carrying a D4Z4 reduced allele were clinically indistinguishable from normal-size allele carriers. Tibialis anterior weakness was present in 45/181 (24.9%) and identified a subgroup with earlier onset (26.3±15.1 vs 39.6±15.2 years) and higher disability (mean FSHD 4.7±1.9 vs 1.8±1.1). Follow-up was available for 37 B1 patients (20.1%) over 6.28±2.90 years: 54% remained stable, while 46% progressed with a mean annual FSHD increase of 0.12 points. Family analyses showed that the proband was the only clinically affected member in 89 families out of /106 (84%). When additional affected relatives were present (17 families), an A phenotype was uncommon (3/17 families), and affected relatives were overall less severe than probands (mean FSHD 2.50±2.00 vs 3.24±2.05).
Conclusion: Facial-sparing FSHD (category B1) represents a distinct clinical entity, characterized by later onset, a milder and slowly progressive course, lower disability, and better-preserved strength than classical FSHD. In this phenotype, D4Z4 repeat size has limited prognostic value, as carriers and non-carriers are clinically indistinguishable. Clinically, the absence of facial weakness broadens the differential diagnosis and increases the risk of misclassification, particularly in single cases and in carriers of D4Z4 alleles with 4-10 RU which are frequent (3-5%) in the general population. These findings support phenotype-based stratification and request family studies. These refined diagnostic pathways should be a requisite for natural-history studies and clinical trials.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Facioscapulohumeral Muscular Dystrophies
OS01.05
A Trial of First-Line Add-on Intravenous Immune Globulin in Idiopathic Inflammatory Myopathies: TIME IS MUSCLE
Miss Pinar Özkaynar1, Mrs Sanne Evers1, Miss Renske Kamperman1, Dr. Frank Smithuis2, Dr. Floor Groepenhoff2, Dr. Filip Eftimov1, Assoc. Prof. Johannes Bogaards3, Prof. Ivo van Schaik1, Dr. Anneke van der Kooi1, Dr. Joost Raaphorst1
1
Department of Neurology, Amsterdam UMC Locatie AMC, University of Amsterdam, Amsterdam, Netherlands.
2
Department of Radiology and Nuclear Medicine, Amsterdam UMC - Locatie AMC, University of Amsterdam, Amsterdam, Netherlands.
3
Department of Epidemiology and Data Science, Amsterdam UMC - Locatie AMC, University of Amsterdam, Amsterdam, Netherlands
Background: The optimal medical treatment strategy in newly diagnosed Idiopathic Inflammatory Myopathies (IIM, excluding inclusion body myositis) is unknown. Standard first-line immunosuppressive treatment consists of high dosed glucocorticoids, often with a steroid-sparing agent. Upfront more intensive treatment may lead to faster and larger improvement of disease activity, which includes muscle strength and physical functioning. Evidence for efficacy and safety of intravenous immunoglobulin (IVIG) was shown in refractory dermatomyositis (add-on), and has been suggested in newly diagnosed IIM (monotherapy). Our hypothesis is that first-line IVIG in addition to prednisone results in a superior clinical response in treatment-naïve IIM patients, as compared to monotherapy prednisone.
Methods: We conducted a double-blind randomized clinical trial in newly diagnosed, treatment-naïve IIM patients with substantial muscle weakness. All patients started on oral prednisone 1mg/kg/day. Patients in the treatment arm received IVIG 2g/kg at baseline, 4 and 8 weeks; in the control arm intravenous placebo was administered. The primary outcome was the Total Improvement Score (TIS) at 12 weeks. Key secondary outcomes were time to improvement (time to reach TIS of ≥ 40), individual core set measures (CSM), muscle MRI and Patient Reported Outcome Measures (PROMs). Safety data were collected. Forty-two patients were required based on sample size calculations. Patients who dropped-out due to reasons unrelated to IIM or study medication were replaced. Patients with insufficient treatment response or clinical deterioration necessitating rescue medication within 12 weeks reached a premature endpoint; these data were included in the analyses of the primary outcome with last observation carried forward.
Results: Between 2021 and 2025 44 patients (50% female) with a mean age of 59 years (SD 14.9) were included. IIM subtypes were immune mediated necrotizing myopathy (n=16), dermatomyositis (n=14), overlap- or non-specific myositis (n=10) and anti-synthetase syndrome (n=4). There were two drop-outs and five patients reached a premature endpoint. One asymptomatic deep venous thrombosis was detected on muscle MRI in the IVIG group. Nine other serious adverse events were considered to be unrelated to study medication.
Conclusion: The Time Is Muscle trial provides data on efficacy and safety of first-line add-on IVIG in newly diagnosed IIM patients. The primary outcome, safety and a selection of secondary outcomes have been analyzed and will be presented at the conference.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Inflammatory/Immune-mediated Myopathies including Inclusion Body Myositis (IBM)
OS01.06
Phase 1/2 Results of an Antisense Oligonucleotide Therapy for Exon‑Skipping‑Amenable DMD
Dr. Erik Niks1, Prof. Giovanni Baranello2, Prof. Marcos Madruga-Garrido3, Prof. Marika Pane4, Prof. Valeria Sansone5, Dr. Hai Liu6, Dr. Peter Velazquez6, Dr. Orli Rosen6, Dr. Cigdem Ayanoglu7, Dr. David Neil6, Prof. Haluk Topaloglu7
1
Department of Neurology, Leiden University Medical Center, Leiden, Netherlands.
2
The Dubowitz Neuromuscular Centre, Developmental Neuroscience Research and Teaching Department, UCL Great Ormond Street Institute of Child Health, NIHR Great Ormond Street Hospital Biomedical Research Centre & Great Ormond Street Hospital NHS Foundation Trust, London, United Kingdom.
3
Neurology Department, Neurolinkia and Hospital Viamed Santa Ángela de la Cruz, Sevilla, Spain.
4
Pediatric Neurology, Università Cattolica del Sacro Cuore and Centro Clinico Nemo Pediatrico, Fondazione Policlinico Universitario Agostino Gemelli IRCCS, Roma, Italy.
5
The NEMO Clinical Center in Milan, Neurorehabilitation Unit, University of Milan, ASST Niguarda Hospital, Milan, Italy.
6
BioMarin Pharmaceutical Inc., Novato, United States.
7
Department of Pediatrics, Yeditepe University, Istanbul, Turkey
Background: Nivudirsen (BMN 351) is an investigational phosphorothioate (PS) antisense oligonucleotide (ASO) that has been chemically modified to improve specificity and stability. Nivudirsen also targets a novel binding site in the DMD pre-mRNA and induced high levels of exon 51 skipping and dystrophin expression in animal models. Here, we provide the first-in-human safety and efficacy results for nivudirsen.
Methods: BMN 351-201 (EUCT#2023-506737-30-00) is an open-label, phase 1/2, dose-escalation study of nivudirsen in ambulatory participants with Duchenne muscular dystrophy (DMD) amenable to exon 51 skipping aged 4–10 years. The primary objective of the study is to assess safety and tolerability; secondary objectives include pharmacokinetics and muscle distribution; additional objectives include exon skipping, dystrophin expression, and functional assessments. Cohort 1A (n=3) received single ascending doses of intravenous nivudirsen (0.6, 1.5, 3, and 6mg/kg). Then, cohorts 1A and 1B (n=3) received 6mg/kg once weekly, and cohort 2 (n=6) received 9mg/kg once weekly. Enrollment of cohort 3 (n=6; 12mg/kg once weekly) is ongoing. The data cutoffs for safety and efficacy assessments were November 2025 and December 2025, respectively.
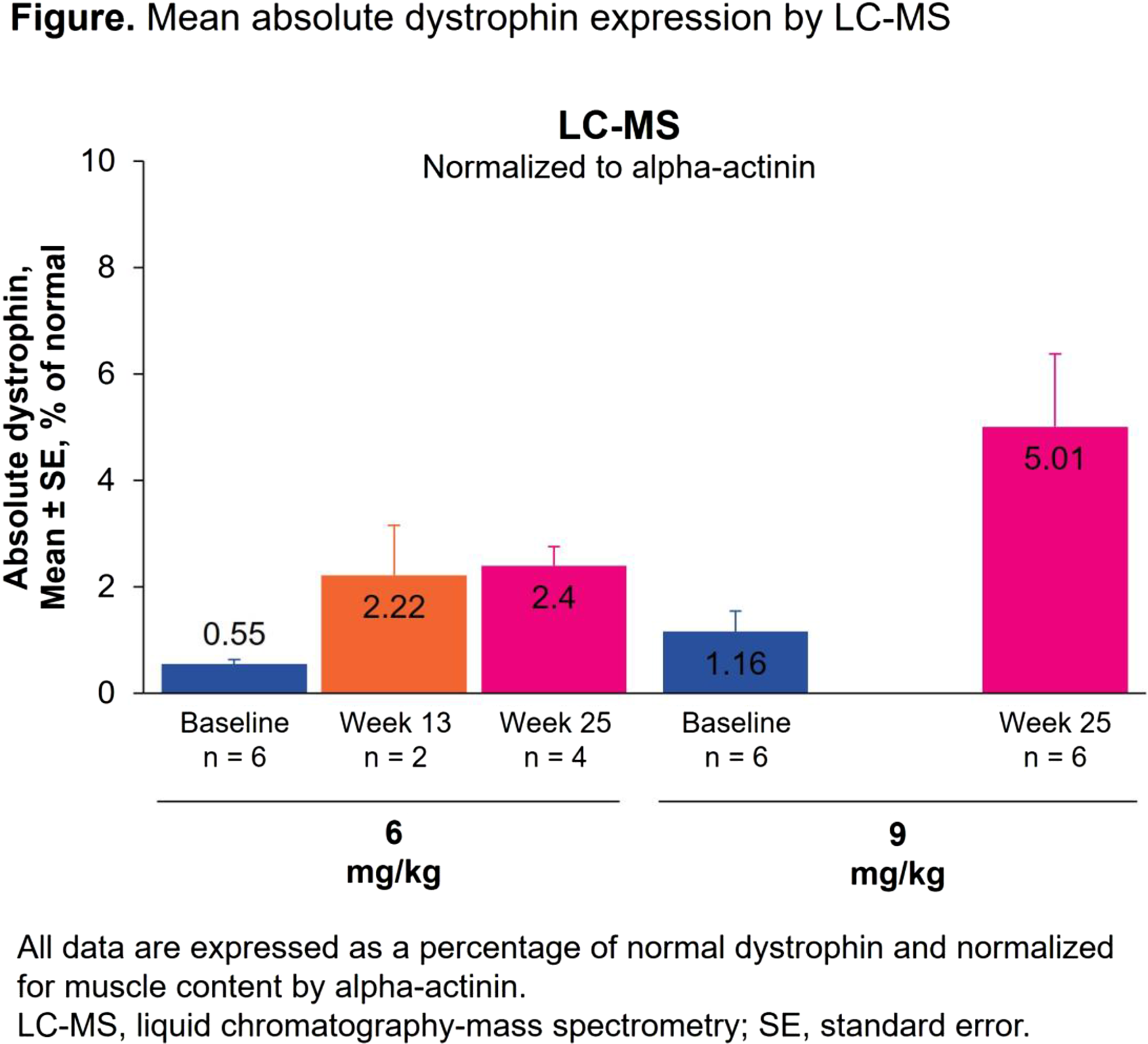
Results: In total, 14 boys were enrolled and received nivudirsen. The age range was 5.0–9.0 years. Mean duration of treatment in cohorts 1 (n=6), 2 (n=6), and 3 (n=2) was 68.4, 43.2, and 9.5 weeks. Overall, 13 participants (92.9%) experienced a treatment-emergent adverse event (AE), most of which were grade 1 or 2. The most common treatment-emergent AE was increased cystatin C that occurred in 9 participants (64.3%). No AEs led to dose reductions or permanent discontinuation. There were 2 serious AEs (grade 3 influenza and appendicitis), and both were considered unrelated to treatment. All treatment-related AEs were grade 1 or 2. As of the efficacy data cut, post-treatment muscle biopsies had been collected from all 12 participants in cohorts 1 and 2. Nivudirsen tissue concentration and exon skipping increased in a dose-dependent manner. Nivudirsen provided mean±SE exon skipping of 3.51%±2.56% (n=2; 6mg/kg) at week (W)13 and 3.17%±1.26% (n=4; 6mg/kg) and 5.03%±1.56% (n=6; 9mg/kg) at W25. By liquid chromatography-mass spectrometry, mean±SE absolute dystrophin level (% normal muscle, alpha-actinin normalized) was 2.22%±0.94% (n=2; 6mg/kg) at W13 and 2.40%±0.36% (n=4; 6mg/kg) and 5.01%±1.37% (n=6; 9mg/kg) at W25 (Figure). By western blot, mean±SE absolute dystrophin level (% normal muscle, myosin heavy chain normalized) was 1.12%±0.60% (n=2; 6mg/kg) at W13 and 1.39%±0.40% (n=4; 6mg/kg) and 3.55%±1.32% (n=6; 9mg/kg) at W25. Functional outcomes relative to natural history will be presented.
Conclusion: Interim analyses up to 25 weeks demonstrated acceptable safety and positive dystrophin changes in boys with DMD receiving nivudirsen ≤9mg/kg. Enrollment of cohort 3 at the 12mg/kg nivudirsen dose is ongoing. Pharmacokinetic and pharmacodynamic modeling of PS-based ASOs predicts that dystrophin will continue to accumulate over time and reach steady-state levels that are approximately 2-fold higher than W25.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
Images or Table (Optional)
OS02.01
Integrated Group-Based and Machine Learning Analysis Reveals Sex-Related Clinical Signatures in Autoimmune Myasthenia Gravis
Prof. Sarah Hoffmann1, Dr. Sonja Katz2, Dr. Paolo Doksani1, Dr. Frauke Stascheit1, Dr. Jan D. Lünemann3, Dr. Jana Zschuentzsch4, Dr. Maike Stein1, Dr. Lea Gerischer1, Dr. Menekse Oezturk5, Dr. Meret Herdick1, Dr. Adela Della Marina6, Dr. Amani Suboh1, Miss Carla Dusemund1, Prof. Michael Schroeter7, Prof. Andreas Meisel1, Dr. Annabel M Ruiter8, Dr. Martijn R Tannemaat8, Dr. Sophie Lehnerer1
1
Charité - Universitätsmedizin Berlin, corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Department of Neurology with Experimental Neurology, Neuroscience Clinical Research Center (NCRC) and Integrated Myasthenia Gravis Center, Berlin, Germany.
2
Laboratory of Systems and Synthetic Biology, Wageningen University & Research, Wageningen, Netherlands.
3
Department of Neurology, University and University Hospital of Münster, Münster, Germany.
4
Department of Neurology, University Medical Center Göttingen, Georg-August University, Göttingen, Germany.
5
Ruhr University Bochum, BG University Hospital Bergmannsheil, Department of Neurology, Bochum, Germany.
6
Department of Pediatric Neurology, Center for Neuromuscular Disorders in Children and Adolescents, University Hospital Essen, Essen, Germany.
7
Department of Neurology, University Hospital Cologne, Cologne, Germany.
8
Department of Neurology, Leiden University Medical Center, Leiden, Netherlands
Background: Despite epidemiological and clinical evidence for a relevant role of sex in autoimmune myasthenia gravis (MG), understanding of sex-related heterogeneity and its implications for care remains limited. This study aimed to assess sex-related differences in MG and evaluate machine learning (ML) models for sex prediction and delineation of sex-related clinical patterns using two independent registry-based populations.
Methods: We combined conventional age-adjusted group comparisons with unsupervised (PCA/UMAP) and supervised (Random Forest Classifier) ML models to conduct a cross-sectional analysis of data from the German Myasthenia Gravis Registry (MyaReg), a multicenter national cohort including 1814 patients with autoimmune MG. We validated the predictive properties of our model using external data from the Dutch MG registry (n=419). Variables included age at disease onset and diagnosis, time to diagnosis, disease severity (QMG, MG-ADL), fatigue, antibody status, electrophysiological and pharmacological test results, therapeutic strategies, and comorbidities. Model explainability was assessed using SHAP. The main outcome was biological sex (female or male).
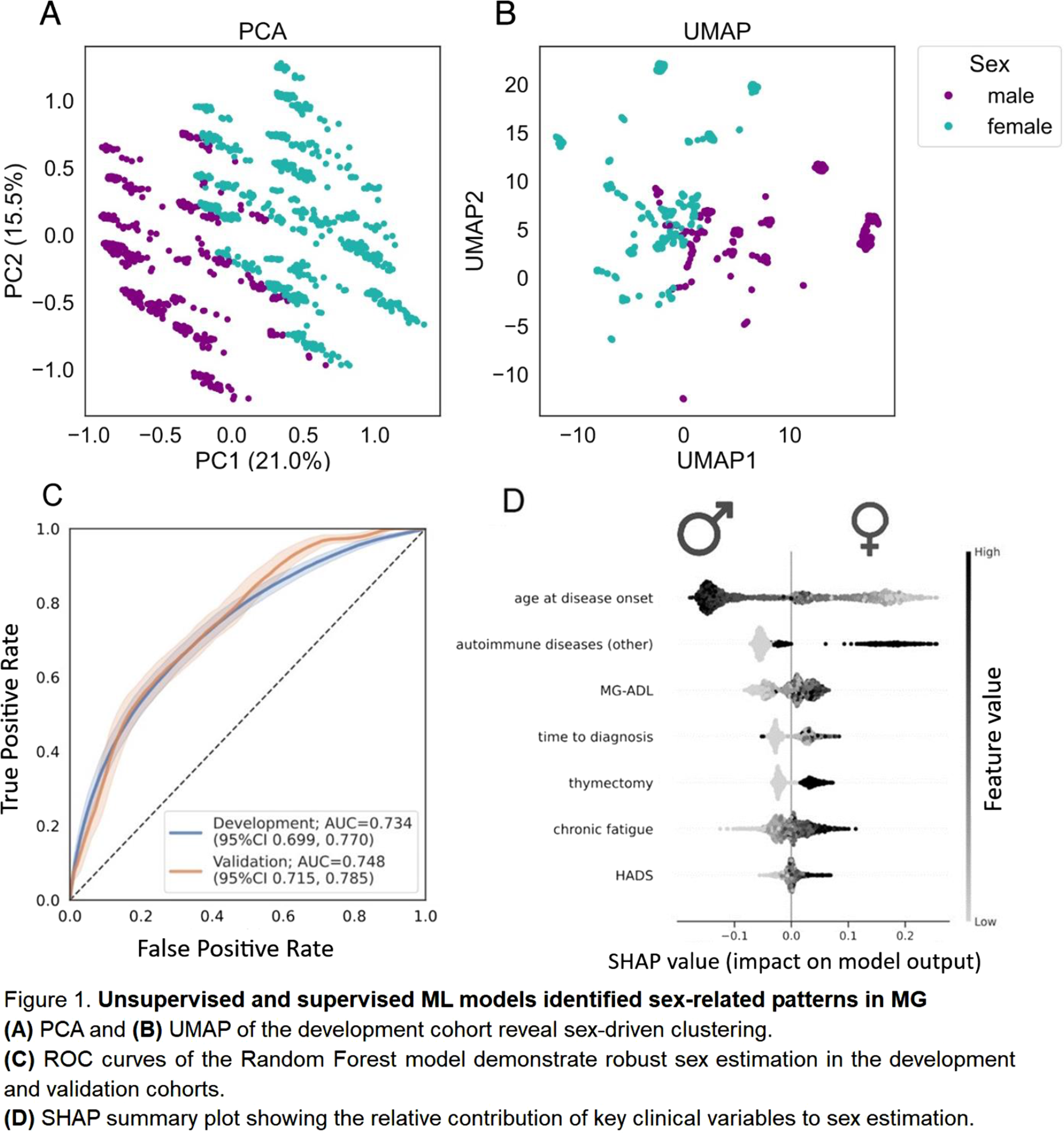
Results: In MyaReg, 56.2% (n=1019) of patients were female. Compared with men, women were significantly younger at disease onset (mean 42.9 vs. 56.9 years) and had longer time to diagnosis (mean 2.2 vs. 0.9 years). Women also showed higher disease severity (QMG mean 7.8 vs. 5.1; MG-ADL mean 4.8 vs. 3.4), a higher rate of seronegative MG (20% vs. 13%), and more autoimmune comorbidities (38% vs. 13%). Unsupervised analyses revealed sex-related structure in the data (Figure 1A & 1B). Boruta identified seven key variables as sufficient for sex estimation. A Random Forest classifier trained on these features achieved reliable discrimination (ROC–AUC 0.734 in development cohort and 0.748 in validation cohort; figure 1C). SHAP analysis showed that sex estimation was primarily driven by younger age at disease onset and autoimmune comorbidity, followed by greater functional impairment (MG-ADL), diagnostic delay, history of thymectomy, fatigue, and affective symptoms (HADS) (Figure 1D). A subset of female patients misclassified as male (24%) exhibited an intermediate clinical pattern, sharing characteristics with the male cohort consistent with a phenotypic continuum.
Conclusion: ML provides a framework to uncover sex-related signatures in MG, offering complementary insights to classical statistical comparisons. Our results reveal a reproducible, clinically meaningful pattern of sex-associated clinical features and emphasize the importance of considering both biological and clinical management related factors in understanding disease expression and access to care. ML-based methods may prove valuable in stratifying patients for personalized and equitable management in MG.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
Images or Table (Optional)
OS02.02
Muscle Transcriptome Profiling Enables In-Depth Characterization of Novel Molecular Pathways Underlying LAMA2-RD Across Disease Severity
Dr. Veronica Pini1,2, Dr. Francesco Catapano1, Dr. Rosa Bonaccorso2, Dr. Ben Weisburd3, Prof. Stefano Carlo Previtali2,4, Prof. Francesco Muntoni1,5
1
Dubowitz Neuromuscular Centre – UCL Great Ormond Street Institute of Child Health, London, United Kingdom.
2
Neuromuscular Repair Unit, Institute of Experimental Neurology, IRCCS Ospedale San Raffaele, Milan, Italy.
3
Program in Medical and Population Genetics, Broad Center for Mendelian Genomics, Broad Institute of MIT and Harvard, Cambridge (MA), United States.
4
Vita-Salute San Raffaele University, Milan, Italy.
5
NIHR Great Ormond Street Hospital Biomedical Research Centre, London, United Kingdom
Background: Merosin-deficient congenital muscular dystrophy (LAMA2-RD) is a neuromuscular disorder caused by mutations in the LAMA2 gene, coding for the α2 subunit of laminin-211. LAMA2-RD patients carrying bi-allelic LAMA2 loss-of-function mutations lack laminin-211 and have an invariably severe clinical phenotype characterized by a profound and diffuse muscle weakness that leads to the inability to acquire ambulation and respiratory insufficiency. A wider and often milder spectrum of disease severity results from missense or hypomorphic mutations mutations allowing residual protein expression. Severe muscle fibrosis and inflammation play a fundamental role in the pathogenesis of the disease, together with a severe metabolic impairment. Some of the dysregulated genes/pathways linked to LAMA2-RD muscle degeneration were previously described through microarray studies. However, a comprehensive characterization of the muscle gene expression profile of LAMA2-RD patients, as well as the identification of molecular signatures distinguishing severe from mild phenotypes, are still lacking.
Methods: RNA-sequencing was performed on muscle biopsies from patients with complete laminin-211 deficiency (n=4), partial deficiency (n=4) and age-matched healthy controls (n=4). Reads were aligned with STAR and analyzed using DESeq2. Differentially expressed genes (DEGs) enrichment was assessed using ShinyGO and MSigDB, while perturbed biological processes, canonical pathways, and upstream regulators were identified via Ingenuity Pathway Analysis. Bioinformatic predictions were complemented by curated literature review. Selected DEGs were validated by qPCR in both patient biopsies and muscle tissue from LAMA2-RD murine models of severe (Dy3K/Dy3K) and mild (Dy2J/Dy2J) disease.
Results: Transcriptome analysis revealed more extensive transcriptional dysregulation in patients with complete (4,437 DEGs) laminin-211 deficiency compared to partial deficiency (1,145 DEGs). Fibrosis and extracellular matrix remodeling emerged as shared hallmarks, with strong upregulation of COL22A1, COMP and OSTN. Metabolic dysfunction, mitochondrial impairment and oxidative stress were more pronounced in complete deficiency, with significant downregulation of PPARGC1A. Inflammasome-driven inflammation was selectively exacerbated in this group, with exclusive upregulation of pro-fibrotic and pro-inflammatory genes such as PF4 and NLRP3. Notably, the lncRNAs XIST and HOTAIR were also upregulated only in patients with complete protein deficiency, suggesting a role in amplifying inflammation and fibrotic remodelling. qPCR confirmed consistent overexpression of PF4, NLRP3, HOTAIR and XIST in patient biopsies and Dy3K/Dy3K mice, (P<0.05) but not in the milder Dy2J/Dy2J model, supporting their relevance as conserved biomarkers of severe disease.
Conclusion: This study provides the first systematic description of the main contributors to human LAMA2-RD pathology across disease severity, shedding light into novel genes and molecular pathways not previously associated with this condition that could potentially serve as targets for future therapeutic interventions.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Congenital Myopathies and congenital muscular dystrophies
OS02.03
Real-World Evaluation of Lamotrigine as an Anti-Myotonic Therapy in Myotonic Dystrophy Type 1
Dr. Barbara Risi1,2, Dr. Nesaiba Ait Allali1, Dr. Stefano Cotti Piccinelli1, Dr. Filomena Caria1, Dr. Simona Damioli1, Dr. Beatrice Labella3,4, Dr. Enrica Bertella1, Dr. Giorgia Giovanelli1, Dr. Francesca Garofali1, Dr. Giuseppina Margollicci1, Dr. Roberto Carugati1, Dr. Lucia Ferullo3,4, Dr. Emanuele Olivieri3,4, Dr. Loris Poli3, Prof. Alessandro Padovani3,4, Prof. Massimiliano Filosto1,3,4
1
NeMO-Brescia Clinical Center for Neuromuscular Diseases, Brescia, Italy.
2
Department of Molecular and Translational Medicine, University of Brescia, Brescia, Italy.
3
Unit of Neurology, ERN Euro-NMD Center ASST Spedali Civili, Brescia, Italy.
4
Department of Clinical and Experimental Sciences, University of Brescia, Brescia, Italy
Background: Myotonia, defined as delayed relaxation of skeletal muscle following voluntary contraction or electrical stimulation, is a hallmark of myotonic dystrophy type 1 (DM1) and may cause substantial functional impairment. Mexiletine, the most commonly used anti-myotonic agent, has limited availability and is often associated with adverse effects. Lamotrigine (LTG), an antiepileptic drug that inhibits voltage-gated sodium channels, has demonstrated anti-myotonic efficacy in both in vitro models and in patients with non-dystrophic myotonias (Andersen G et al. 2017; Vivekanandam V et al. 2024). The aim of this study was to evaluate the real-world use of LTG as an anti-myotonic therapy in a cohort of patients with DM1.
Methods: Fourteen consecutive adult patients with genetically confirmed DM1 and clinically relevant myotonia affecting daily activities (Myotonia Behaviour Scale, MBS >1) were enrolled. LTG was administered with a dose-escalation protocol, starting at 50 mg/day and increasing up to 200 mg/day. Treatment efficacy was analyzed using a linear mixed-effects model. Two functional timed tests, the 9-Hole Peg Test (9HPT) and a newly developed task simulating the preparation of a coffee pot (the “Coffee Task”), were performed at baseline and at each dosage level. Safety and tolerability were also assessed.
Results: The mean age of participants was 40 years, with a mean disease duration of 12 years. LTG dosage was associated with a significant improvement in 9HPT performance at the highest dose compared with baseline. Age and disease duration were significant predictors of 9HPT outcomes. No significant changes were observed in the Coffee Task performance. No serious adverse events were reported during the study.
Conclusion: This pilot open-label study provides preliminary evidence supporting the efficacy and safety of lamotrigine as an anti-myotonic treatment in patients with DM1. These results warrant further investigation in larger, randomized, placebo-controlled clinical trials to better define its therapeutic role (Risi B et al. 2025).
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Myotonic Dystrophies
OS02.04
Long‑Term Safety and Efficacy of an AAV‑Based Gene Therapy forX‑Linked Myotubular Myopathy
Dr. Nancy L. Kuntz1, Dr. Rahul Gentyala2, Dr. Julie Coats3, Dr. Atsuki Hashimoto4, Dr. Astrid Blaschek5, Dr. Wolfgang Müller-Felber5, Dr. Carsten G. Bönnemann6, Dr. Barbara K. Smith7, Dr. Andrea Seferian8, Dr. Lucy James3, Dr. Perry B. Shieh9, Dr. James J. Dowling10
1
Ann & Robert H. Lurie Children’s Hospital of Chicago, Chicago, IL, United States.
2
Astellas Pharma Global Development, Inc., Northbrook, IL, United States.
3
Astellas Gene Therapies, South San Francisco, CA, United States.
4
Astellas Pharma Europe Ltd., Addlestone, United Kingdom.
5
Department of Paediatric Neurology and Developmental Medicine, Hauner Children's Hospital, Ludwig Maximilian University of Munich, Munich, Germany.
6
National Institute of Health, Bethesda, MD, United States.
7
University of Florida, Gainesville, FL, United States.
8
Institute of Myology, French National Centre for Scientific Research, Paris, France.
9
University of California Los Angeles, Los Angeles, CA, United States.
10
Penn Medicine, Philadelphia, PA, United States
Background: X-linked myotubular myopathy (XLMTM) is a life-threatening, rare, congenital muscle disease with no approved therapies. The ASPIRO trial assessed the safety and efficacy of resamirigene bilparvovec (rAAV8-desmin-hMTM1), a gene replacement therapy that expresses human MTM1 in muscles, in children with XLMTM. With up to 4.6 years of follow-up (primary analysis), 16/24 dosed participants achieved ventilator independence and 19/24 had improved motor function compared with baseline, with 8 walking independently and 15 sitting independently. Here, results from ASPIRO with >5 years of follow-up are reported.
Methods: ASPIRO (NCT03199469) is an open-label, international, multicenter, dose-escalation trial. Male children aged <5 years with XLMTM requiring mechanical ventilator support were included. Participants received one intravenous infusion of resamirigene bilparvovec at 1.3 × 1014 vector genomes (vg)/kg bodyweight (lower-dose cohort) or 3.5 × 1014 vg/kg bodyweight (higher-dose cohort). The primary efficacy outcome was change from baseline to week 24 in hours of daily ventilator support. Other important efficacy outcomes included ventilator independence and motor milestones. ASPIRO stopped enrollment after 1 participant in the lower-dose and 3 in the higher-dose cohorts died; at time of death, all had cholestatic liver failure following gene therapy. During long-term follow-up, 1 participant in the lower-dose cohort withdrew from the study. Follow-up to 10 years of the remaining 19 participants (lower-dose cohort, n=5; higher-dose cohort, n=14) is ongoing.
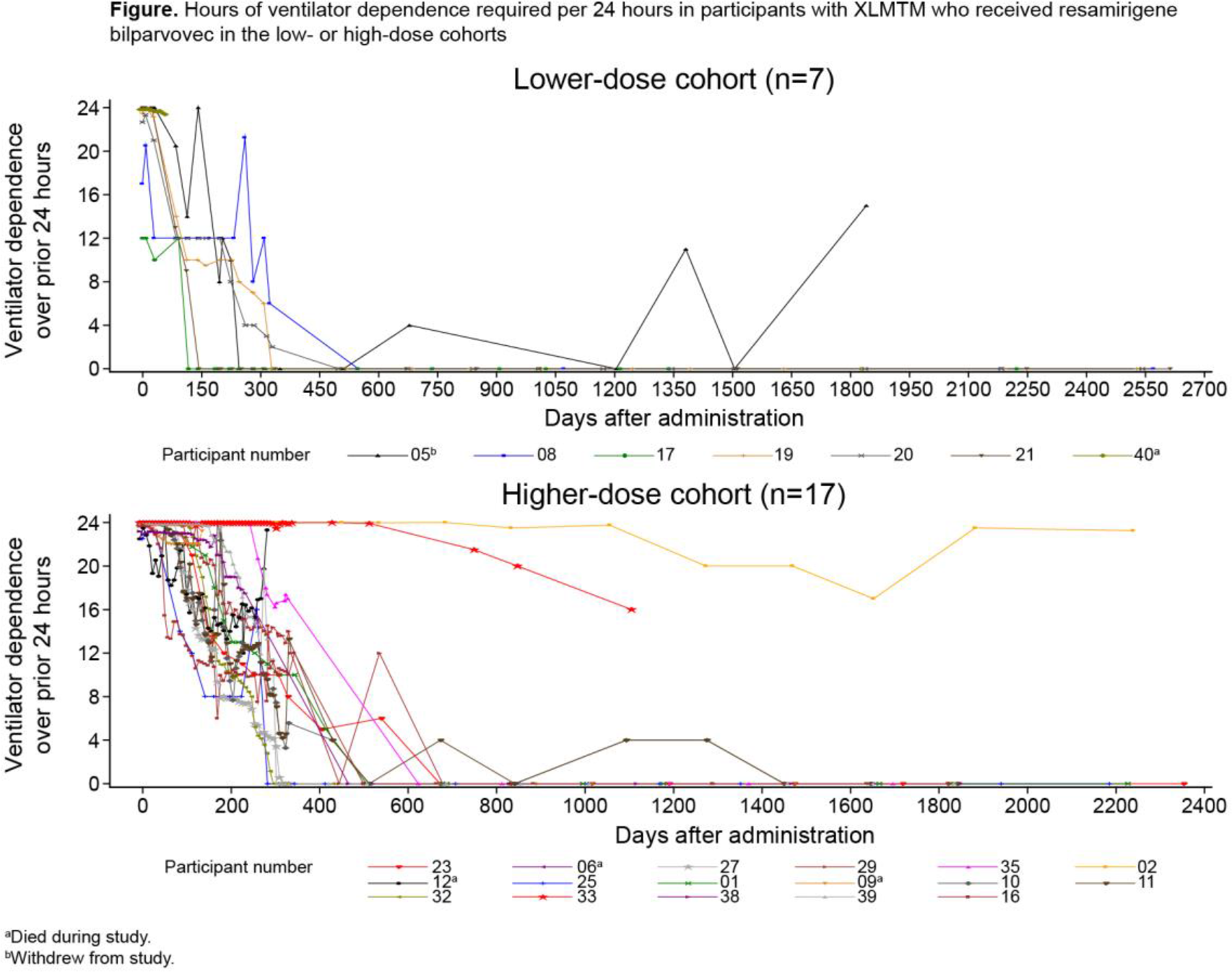
Results: As of data cutoff, June 20, 2025, median follow-up was 5.51 years (range, 0.18–7.25) for all participants (N=24; lower dose [n=7]: 6.96 years [range, 0.18–7.25]; higher dose [n=17]: 5.07 years [range, 0.28–6.48]). Among evaluable participants (n=19), 5/6 (83.3%) in the lower-dose cohort and 10/13 (76.9%) in the higher-dose cohort had ventilator independence at month 60. Most participants (n=11) achieved ventilator independence ≤1.5 years of treatment, but others (n=4) achieved independence ≥2 years after treatment. Of the 4 participants without ventilator independence, 3 required fewer hours of ventilator support daily at month 60 compared with baseline (Figure). At month 60, 5/6 (83.3%) and 12/13 (92.3%) evaluable participants demonstrated functionally independent sitting for ≥30 seconds in the lower-dose and higher-dose cohorts, respectively; 5 (83.3%) in the lower-dose and 7 (53.8%) in higher-dose cohorts were also walking independently at that time. Since the 4 deaths reported in the primary analysis, there were no additional deaths. In the overall safety analysis (N=24), 2 participants in the lower-dose cohort and 7 in the higher-dose cohort had serious treatment-related hepatobiliary adverse events (AEs), all of which had first onset 6–162 days post-dosing. Four participants had treatment-related hepatobiliary AEs that had onset ≥1 year post-dose; all had grade 1, 1 also had grade 2. Myocarditis was a treatment-related AE in 1 participant in each cohort, consistent with the primary analysis.
Conclusion: With up to 7.25 years of follow-up, most children with XLMTM who received resamirigene bilparvovec continued to show improvements in respiration and motor function. Since the primary analysis, 4 additional participants began walking independently and 1 achieved independent sitting. During long-term follow-up, no further deaths were reported, and resamirigene bilparvovec demonstrated a favorable safety profile.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Congenital Myopathies and congenital muscular dystrophies
Images or Table (Optional)
OS02.05
An HDAC Inhibitor Reduces Decline ofContractile Cross-Sectional Area andDecreases Fat Infiltration in Duchenne Muscular Dystrophy
Dr. Krista Vandenborne1, Dr. Rebecca Willcocks1, Dr. Paolo Bettica2, Dr. Sara Cazzaniga2, Ms. Federica Alessi2, Prof. Eugenio Mercuri3
1
University of Florida, Gainesville, Florida, United States.
2
Italfarmaco S.p.A., Milan, Italy.
3
Pediatric Neurology Institute, Catholic University and Nemo Pediatrico, Fondazione Policlinico Gemelli IRCCS, Rome, Italy
Background: The phase 3 EPIDYS trial (NCT02851797) was a randomized, double-blind, placebo-controlled study evaluating the safety, tolerability, and efficacy of the histone deacetylase inhibitor givinostat in Duchenne muscular dystrophy (DMD). Contractile cross-sectional area (cCSA) and fat fraction are quantitative measures of DMD disease progression, reflecting the amount of functional muscle tissue available for force generation and the extent of muscle tissue replacement by fat, respectively. The objective of analysis was to measure cCSA and fat fraction in patients from the EPIDYS trial with a baseline vastus lateralis fat fraction of >5% to ≤30% measured by magnetic resonance spectroscopy (target population) who were not expected to be at risk of a sudden loss of ambulation but could potentially experience a noticeable decline if receiving placebo.
Methods: Of the 179 patients enrolled in EPIDYS, 120 were in the target population and completed the study (givinostat n=81; placebo n=39). Quantification of cCSA and fat fraction were determined via magnetic resonance imaging (givinostat, n=76; placebo, n=37) using the Dixon technique. Data were analyzed with a mixed model for repeated measures to determine change over time with covariates including treatment group, visit, baseline value, and interaction. All P values are nominal.
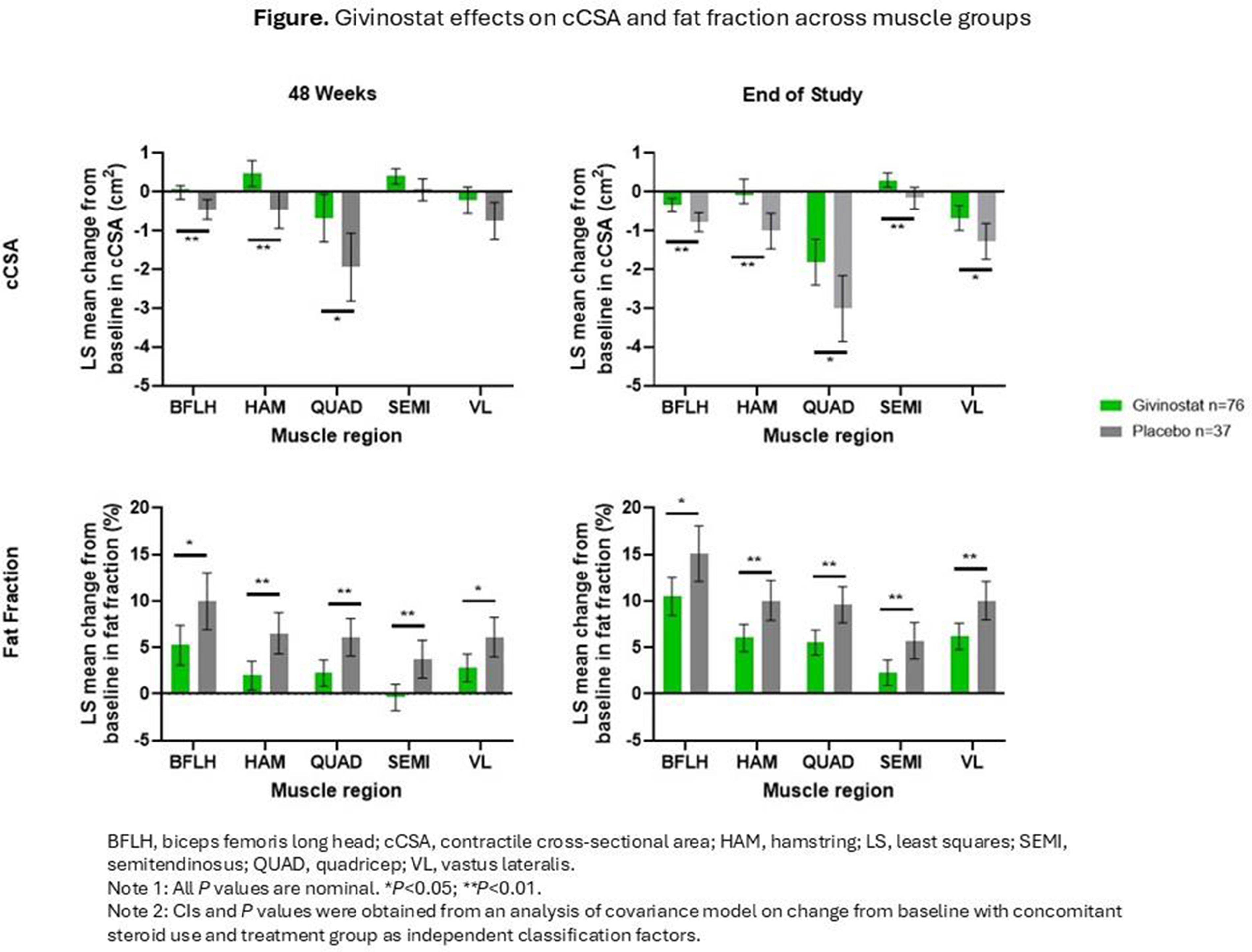
Results: At 48 weeks, differences in least squares (LS) means (95% CI) for cCSA (cm2) between givinostat and placebo groups were observed across all muscles analyzed, although not all differences reached nominal statistical significance: biceps femoris long head (0.44; 0.13, 0.74; P=0.006), hamstrings (0.94; 0.37, 1.52; P=0.002), quadriceps (1.27; 0.20, 2.34; P=0.021), semitendinosus (0.34; 0.00, 0.69; P=0.053), and vastus lateralis (0.53; -0.05, 1.12; P=0.073). In addition, differences in LS means for fat fraction (%) in the same muscles were seen at 48 weeks: -4.74 (-8.48, -1.00; P=0.013), -4.57 (-7.26, -1.88; P=0.001), -3.88 (-6.32, -1.44; P= 0.002), -4.08 (-6.54, -1.62; P=0.001), and -3.31 (-5.90, -0.71; P=0.013), respectively. At the end of the EPIDYS trial (72 weeks), the differences in LS means between the givinostat and placebo groups for cCSA in were 0.43 (0.14, 0.73; P=0.005), 1.02 (0.47, 1.58; P<0.001), 1.20 (0.16, 2.23; P=0.023), 0.47 (0.13, 0.80; P=0.006), and 0.61 (0.04, 1.17; P=0.035), respectively. Further, differences in LS means for fat fraction in the same muscles were -4.60 (-8.22, -0.99; P=0.013), -4.00 (-6.59, -1.40; P=0.003), -4.07 (-6.42, -1.72; P=0.001), -3.43 (-5.80, -1.06; P=0.005), and -3.86 (-6.36, -1.35; P=0.003), respectively (Figure).
Conclusion: Givinostat treatment appeared to be associated with a reduced decline in muscle contractile area and reduced fat infiltration compared with placebo, indicating a consistent pattern suggestive of attenuation of muscle loss in patients with DMD.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
Images or Table (Optional)
OS02.06
Characterization of Juvenile-Onset MG in a Nationwide Swedish Cohort
Dr. Oskar Sunnegardh1, Miss Wanqing Wu1, Dr. Helgi Hjartarson1,2, Prof. Thomas Sejersen1,2, Dr. Kristofer Nathorst-Böös1,2, Dr. Jing Wu1, Assoc. Prof. Susanna Brauner1,2
1
Karolinska Institutet, Stockholm, Sweden.
2
Karolinska University Hospital, Stockholm, Sweden
Background: Juvenile-onset Myasthenia Gravis (JOMG), defined as disease onset before 18 years of age, has primarily been described in Asian populations, where it accounts for up to 40% of all MG cases. In contrast, a recent Nordic population-based study reported a prevalence of only 3% in Sweden. Previous studies suggest that JOMG is associated with higher rates of spontaneous remission and seronegativity compared to adult-onset MG. However, large population-based cohorts are lacking and JOMG remains poorly characterized.
Methods: To describe the burden of disease, and treatment pattern of JOMG in Sweden.
Results: In total, 85 patients were identified in MG-reg, of which 26 (31%) incident and 59 (69%) prevalent during the study period. Twenty-seven patients were identified in the Stockholm cohort, of which 12 were not registered in MG-reg. In the combined cohort, 77% were female and 65% were diagnosed after age 12. Median diagnostic delay was 1.0 years (IQR 0.2–4.7), compared to 0.6 years (0.2–1.6) in the EOMG group. Most patients were AChR-positive (87%), while 10% were double-seronegative. Thymectomy was performed in 73 patients and 88% had hyperplasia. Of all, 97% developed generalized disease and all but one had generalized symptoms at diagnosis. However, initiation of corticosteroids and immunosuppressive therapy was substantially delayed in JOMG compared to EOMG (median 3 vs. 0.1 years and 8 vs. 0.5 years from diagnosis respectively).
Conclusion: JOMG in Sweden predominantly affects adolescents and is characterized by AChR seropositivity, thymic hyperplasia, and generalized disease—features largely comparable to the EOMG population. Notably, treatment initiation is significantly delayed, highlighting a potential gap in management strategies for this subgroup.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
OS03.01
Advances in Clinical Genetic Epidemiology and Therapy of Nlsdm/nlsdi :(Chanarin-Dorfman) as a Mediterranean Trait
Prof. Corrado Angelini1, Prof. Daniela Tavian2, Dr. Sara Missaglia2
1
University of Padova, Padova, Italy.
2
Catholic Univertsity, Milan, Italy
Background: NLSD with Ichthyosis (NLSDI) is due to the CGI activator variant ( chromosome 3 ).NLSDI or Chanarin Dorfman syndrome (CDS), is a rare AR disorder encountered in pa tients of Mediterranean and Middle Eastern origin. Patients come to medical attention for ichthyosis. Neutral Lipid Storage with Myopathy (NLSDM) is a rare lipid metabolism disorder caused by impaired Adipose Triglyceride Lipase (ATGL) activity, leading to neutral lipid accumulation in various tissues.
Methods: We did genetic epidemiological research of NLSDI/NLSDM molecularly proven cases from 1975 to 2025, investigated their genetic epidemiology, country of origin, and private mutations in 176 patients.
Results: There are two types of Neutral Lipid Storage Disease:NLSDM is a rare lipid metabolism disorder caused by impaired Adipose Triglyceride Lipase (ATGL) activity, leading to neutral lipid accumulation in various tissues. It typically manifests with progressive skeletal myopathy, with an onset of age around 35 years. An NLSDI type with Ichthyosis and liver involvement is due to the CGI activator and usually has an earlier age of involvement. We investigated the Genetic epidemiology of these two disorders and the country of origin or mutation hotspot. Patients come to medical attention for ichthyosis. We found in Mediterranean countries, 124 NLSDI cases: 10 in Italy,3 in Greece, and 48 in Turkey. Two siblings had CNS involvement, myopathy, neurosensory hearin g loss, cataracts, nystagmus, strabismus, and mental impairment; their molecular analysis revealed the presence of the N209* mutation.
Patients with ichthyosis or Jordan's anomaly are referred to the clinic for CK elevation, and muscle biopsies are consi stent with lipid storage. In Palestine,5 cases were detected (common mutation c.960+5G>A ), 9 in Israel,4 in Algeria,26 in Tunisia,3 in France,7 in Spain,4 in Egypt, and 3 in Morocco.
To date, 176 CDS patients have been reported worldwide. In India, 19 mutations were found.NLSDI patients have onset in the first/second decade. Usually, children,or people are admitted for congenital ichthyosis, Jordan's anomaly, liver steatosis; CNS changes are then detected.
Conclusion: In few NLSDI cases detected for congenital ichthyosis, Jordan's anomaly, liver steatosis; CNS changes are then detected. Myopathology is consistent with lipid storage. The myopathy can be treated with an MCT diet, and retinoids such as acitretin can be useful in skin treatment, even in the presence of liver damage.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Metabolic Myopathies: Glycogen, Lipids and Mitochondrial Myopathies
Images or Table (Optional)
OS03.02
Short Tandem Repeats as Risk Factors and Survival Modifiers of ALS
Dr. Kang-Yang Jih, Prof. Yi-Chung Lee
Taipei Veterans General Hospital, Taipei, Taiwan
Background: Amyotrophic lateral sclerosis (ALS) is a devastating neurodegenerative disorder with significant but incompletely explained heritability. Short tandem repeat (STR) expansions, established as disease mechanisms in other neurodegenerative conditions, are increasingly recognized as contributors to ALS risk and disease severity modifying factors. This study aims to investigate the role of common STRs in ALS pathogenesis.
Methods: We investigated ten STRs (C9ORF72, ATXN1, ATXN2, ATXN8/ATXN8OS, TBP, HTT, NOTCH2NLC, DMPK, CNBP and FMR1) associated with common neurological disease among 730 Taiwanese ALS patients and 1,173 controls by PCR and fragment analysis. Detail clinical information of these patients was collected. We analyzed the effect of STRs on ALS risk, survival and disease progression.
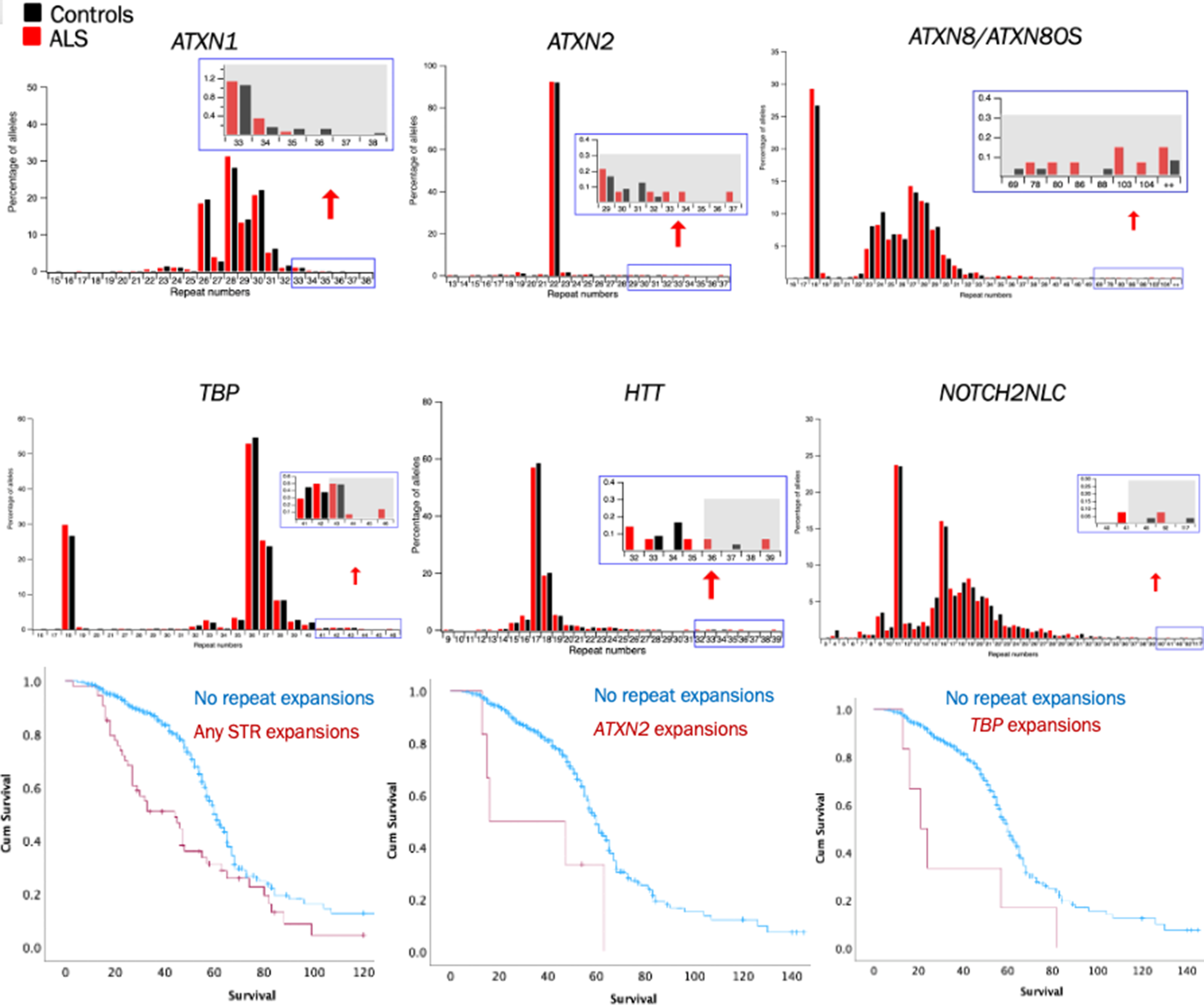
Results: STR expansions were identified in 12.7% of all ALS patients (Figure, upper). Pathogenic C9ORF72 expansions were most frequent STR among ALS patients (4.2%). High risk alleles of ATXN2, ATXN8/ATXN8OS and TBP, defining as repeat number greater than 32, 79 and 43, respectively, are significantly enriched in ALS patients over controls. In addition, ATXN1 expansions were the second most prevalent one and its pathogenicity is associated with a risk allele of UNC13ASNP, rs12608932. STR expansions, particularly in ATXN2 and ATXN8/ATXN8OS, correlated with accelerated functional decline and significantly reduced survival (Figure, lower), suggesting STR expansions are disease modifiers of ALS.
Conclusion: STR expansions in neurodegeneration-associated genes are important ALS risk and disease modifiers. The pathogenicity of STRs is likely modulated in an oligogenic basis. Recognizing the roles of STR enhances understanding of ALS genetic architecture and guides future interventions.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: ALS: Biology, Pathophysiology, Genetics
Images or Table (Optional)
OS03.03
Chronology of Subclinical Nerve Hypertrophy in Latin American Hereditary Transthyretin Amyloidosis With Polyneuropathy
Prof. Jorge Arturo Diaz-Ruiz1,2, Prof. Sandra Milena Castellar-Leones1,2,3, Prof. Edicson Ruiz-Ospina1,2, Dr. Cristian Correa-Arrieta2, Dr. Eduardo Echeverry4, Dr. Diana Ramirez-Montaño4, Dr. Juan David Lopez5, Dr. Diana Luzuriaga-Carpio6, Dr. Dario Zambrano-Vera7, Dr. Daniel César Chávez8, Dr. Edison Vasquez8, Dr. Ana Laura Castro9, Dr. Juan Pablo Muñoz10, Prof. Fernando Ortiz-Corredor1,2,3
1
Universidad Nacional de Colombia, Bogota, Colombia.
2
CIFEL, Bogota, Colombia.
3
Hospital Universitario Nacional de Colombia, Bogota, Colombia.
4
Clinica Imbanaco, Cali, Colombia.
5
Fundación Valle de Lili, Cali, Colombia.
6
Hospital General Manuel Ygnacio Monteros-IESS, Loja, Ecuador.
7
Hospital de Especialidades Carlos Andrade Marín-IESS, Quito, Ecuador.
8
Hospital Teodoro Maldonado Carbo-IESS, Guayaquil, Ecuador.
9
Hospital Universal Cartago, Cartago, Costa Rica.
10
Caja Costarricense de Seguro Social, Alajuela, Costa Rica
Background: Identifying the transition from asymptomatic carrier to symptomatic disease in hereditary transthyretin amyloidosis with polyneuropathy (hATTR-PN) remains a clinical challenge. Standard neurological examinations and clinical scales may miss early nerve involvement, whereas neuromuscular ultrasound can detect structural nerve changes at subclinical stages. This study aims to characterize the chronological pattern of nerve hypertrophy and to identify the earliest sites of structural involvement in a Latin American hATTR-PN cohort.
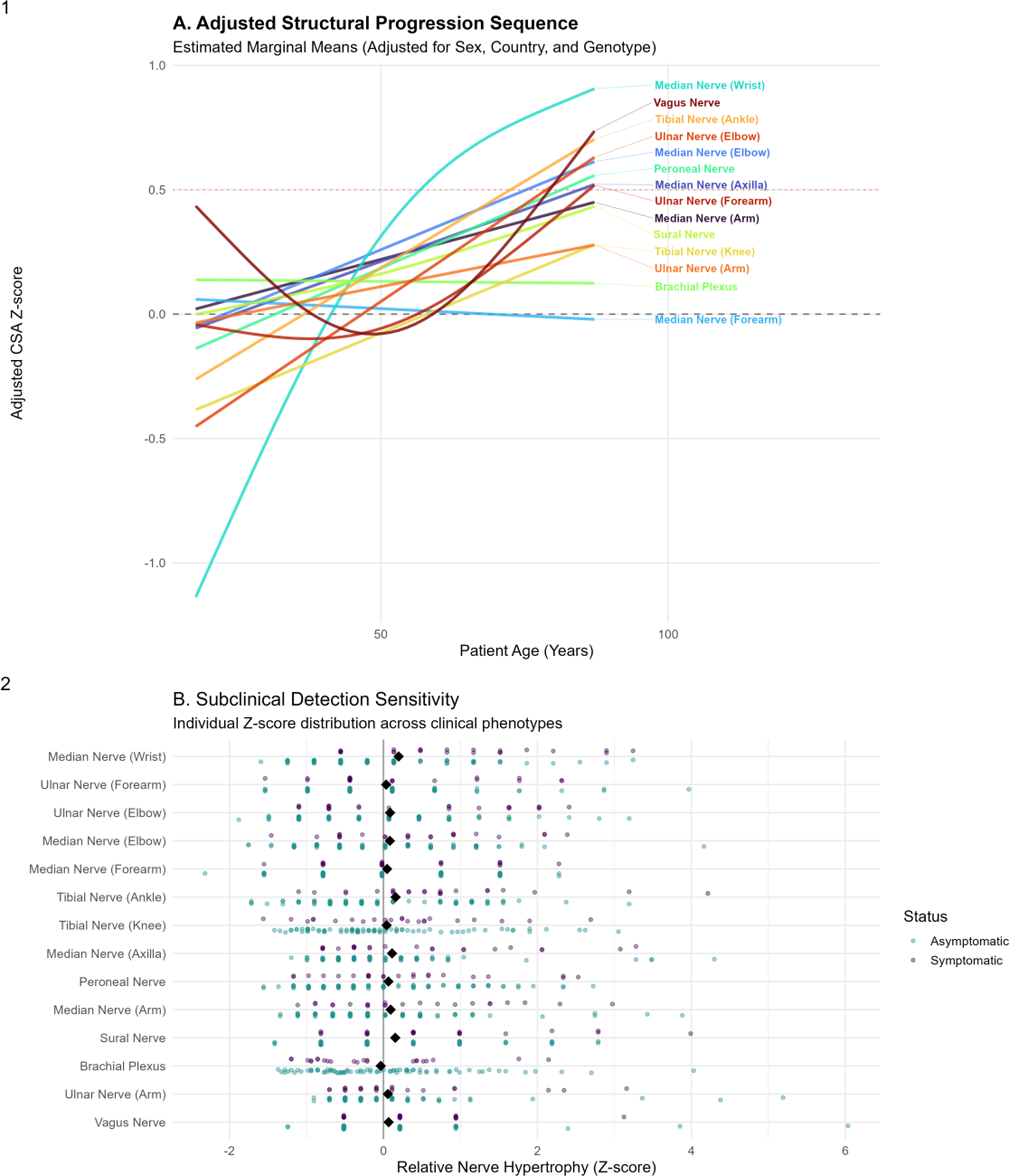
Methods: We conducted a multicenter study of 105 transthyretin (TTR) mutation carriers from Colombia, Costa Rica, and Ecuador. Clinical status was assessed using the Neuropathy Impairment Score (NIS), classifying participants as asymptomatic (NIS = 0; n = 82) or symptomatic (NIS > 0; n = 23). The cohort included 58 women (55.2%), and multiple TTR genotypes were represented, mainly Ser43Asn (33.3%), Val50Met (24.8%), and Val142Ile (22.9%). We measured the cross-sectional area (CSA) of 14 nerve sites by neuromuscular ultrasound, encompassing distal and proximal segments of the median, ulnar, tibial, peroneal, sural, brachial plexus, and vagus nerves. To account for demographic differences, CSA values were converted into adjusted Z-scores using generalized additive models controlling for age, sex, country, and mutation group. We then estimated age-related trajectories for each nerve and defined the onset of structural hypertrophy as the age at which the adjusted Z-score crossed 0.5.
Results: Ultrasound revealed a clear distal-to-proximal pattern of nerve hypertrophy. The median nerve at the wrist had the earliest modeled onset of structural involvement (57.4 years), followed by the tibial nerve at the ankle (73.0 years). Subsequent sites of involvement included the median nerve at the elbow (75.8 years), the ulnar nerve at the elbow (80.1 years), the vagus nerve (80.1 years), the peroneal nerve (81.5 years), the median nerve at the axilla (85.8 years), and the ulnar nerve at the forearm (87.2 years). In symptomatic individuals, CSA was significantly larger at key distal sites. The largest hypertrophy occurred at the median nerve at the wrist (mean Z = 0.90), followed by the tibial nerve at the ankle (Z = 0.72) and the sural nerve (Z = 0.70). Intermediate enlargement was noted at the median and ulnar nerves at the elbow, whereas proximal structures such as the brachial plexus showed minimal or no hypertrophy. By contrast, asymptomatic carriers had adjusted Z-scores near zero across all sites, despite early trajectory changes in the most susceptible nerves.
Conclusions: In this Latin American cohort, ultrasound-detected nerve hypertrophy followed a consistent distal-to-proximal pattern, with the median nerve at the wrist as the earliest site of structural involvement. Detecting enlargement at this location in carriers with normal neurological examinations may indicate early structural conversion. Focusing on these high-susceptibility distal nerves could provide a pragmatic framework for ultrasound surveillance in hATTR-PN.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Hereditary Peripheral Neuropathies
Images or Table (Optional)
OS03.04
Whole Body Imaging Reveals Stage-Dependent Imaging Markers in Calpainopathy and Dysferlinopathy
Dr. Ai Yamanaka1,2,3, Mr. Reoto Ueda4, Mr. Hiroto Azuma4, Dr. Shinichiro Hayashi2, Dr. Satoru Noguchi2, Prof. Kazuma Sugie1, Prof. Ichizo Nishino2
1
Department of Neurology, Nara Medical University, Nara, Japan.
2
Department of Neuromuscular Research, National Institute of Neuroscience, National Center of Neurology and Psychiatry, Tokyo, Japan.
3
Department of Genome Medicine Development, Medical Genome Center, National Center of Neurology and Psychiatry, Tokyo, Japan.
4
Faculty of Medicine, Nara Medical University, Nara, Japan
Background: Calpainopathy and dysferlinopathy are two major forms of autosomal recessive limb‑girdle muscular dystrophy. Although characteristic muscle involvement patterns have been described for each disorder, most previous studies have focused on limb muscles, and direct comparisons that include the trunk remain limited. This study aims to clarify stage‑related patterns of muscle involvement and to identify muscles that support differential diagnosis using whole‑body CT or MRI.
Methods: We analyzed imaging data from 69 patients with calpainopathy and 83 with dysferlinopathy. Fat replacement in 43 skeletal muscles was evaluated using the modified Mercuri score (mMS).
Results: Hierarchical clustering separated patients into mild, intermediate, and severe groups, with the mild group consisting of younger individuals. In calpainopathy, early fat infiltration was observed in the longissimus (Lo) and iliocostalis lumborum (IcL), in addition to the characteristic involvement of posterior thigh and calf muscles. Serratus anterior (SA) and gluteus medius (GMd) became involved from the intermediate stage, distinguishing mild from intermediate stage. Marked trapezius involvement appeared exclusively in severe cases, serving as an indicator of advanced progression. In dysferlinopathy, Lo and IcL showed progressive changes mainly between the mild and intermediate stages, whereas the internal abdominal oblique and rectus femoris demonstrated notable progression from intermediate to severe. Principal component analysis identified SA and GMd as major contributors to differentiating the two disorders, while Lo and IcL were key discriminators in mild cases.
Conclusion: We demonstrated that each disease exhibits a distinct set of muscles that become diagnostically informative at different stages. Lo and IcL were most informative in early stage, whereas SA and GMd provided clearer separation in more advanced stages. The differing timing of Lo and IcL involvement between the two disorders—early and severe in calpainopathy, later and milder in dysferlinopathy—may also relate to hyperlordosis risk. Importantly, the stage‑dependent changes revealed through whole‑body imaging offer a framework that may serve as a reference for future clinical studies and longitudinal evaluation.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Limb Girdle Muscular Dystrophies
OS03.05
Ultrastructural Insights
Dr. Diego Lopergolo1, Dr. Gianna Berti1, Dr. Robert Goodwin2, Dr. Francesco Michelassi3, Dr. Barbara Risi4, Prof. Marina Grandis5, Prof. Roberto Massa6, Dr. Filippo Maria Santorelli7, Dr. Gian Nicola Gallus1, Dr. Maria Lucia Valentino8, Dr. Flavia Palombo9, Dr. Dario Zoppi10, Dr. Lucia Ruggiero10, Prof. Gabriella Esposito11, Dr. Aleksandra Kawka12, Dr. Krzysztof Szczałuba12, Prof. Francesca Magri13, Prof. Stefania Corti14, Dr. Marco Savarese15, Prof. Massimiliano Filosto4, Prof. Alessandro Malandrini1, Prof. Nicola De Stefano1
1
Department of Medicine, Surgery and Neurosciences, University of Siena, Siena, Italy.
2
Medical College of Wisconsin, Milwaukee, United States.
3
Department of Neurology, Columbia University Medical Center, New York, United States.
4
NeMO-Brescia Clinical Center for Neuromuscular Diseases, University of Brescia, Brescia, Italy.
5
Department of Neuroscience, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (DiNOGMI), University of Genoa, Genoa, Italy.
6
Department of Systems Medicine, Tor Vergata University of Rome, Rome, Italy.
7
IRCCS Fondazione Stella Maris, Pisa, Italy.
8
Department of Biomedical and Neuromotor Sciences, University of Bologna, Bologna, Italy.
9
IRCCS Institute of Neurological Sciences of Bologna, Bologna, Italy.
10
Department of Neurosciences, Reproductive and Odontostomatological Sciences, University of Naples Federico II, Naples, Italy.
11
Department of Molecular Medicine and Medical Biotechnologies, University of Naples Federico II, Naples, Italy.
12
Centre of Excellence for Rare and Undiagnosed Diseases, Medical University of Warsaw, Warsaw, Poland.
13
Neuromuscular Unit, IRCCS Fondazione Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy.
14
Department of Pathophysiology and Transplantation, University of Milan, Milan, Italy.
15
Folkhälsan Research Center, Helsinki, Finland
Background: The CCDC78 gene was identified approximately ten years ago as a novel candidate gene for autosomal dominant centronuclear myopathy type 4 (CNM4) (Majczenko et al., 2012). Clinical features described in the first reported family included neonatal hypotonia, muscle weakness, myalgias, mild-to-moderate motor impairment, and mild cognitive involvement. More recently, we reported a second family harboring a nonsense CCDC78 variant, providing the first insights into CCDC78 interactors and its putative role in skeletal muscle, with localization of the protein to the sarcoplasmic reticulum (Lopergolo et al., 2024). To date, however, no systematic genotype–phenotype correlation studies have been performed, and this ultrarare muscle disease remains poorly characterized.
Methods: To establish genotype–phenotype correlations, we collected clinical, molecular, and histopathological data from 14 patients carrying CCDC78 point mutations. Patients were recruited from nine neuromuscular centers in Italy, Finland, Poland, and the United States.
Results: We identified 11 families harboring ten different likely pathogenic germline CCDC78 variants. Age at symptom onset was highly variable. Recurrent clinical features included dropped head syndrome, bilateral scapular winging, and calf hypertrophy. Common histopathological findings on light microscopy were increased internal nuclei, predominance of type I fibers, and core-like areas. Transmission electron microscopy revealed sarcoplasmic reticulum dilatation and mitochondrial abnormalities in several patients. Additional extramuscular findings included atrial structural defects and osteoarticular abnormalities.
Conclusion: This study represents the first comprehensive cohort of patients with CCDC78 mutations and significantly expands the clinical and pathological spectrum of CCDC78-related centronuclear myopathy. Our data provide robust support for the pathogenicity of germline CCDC78 variants. Despite marked phenotypic variability, consistent sarcoplasmic reticulum abnormalities on electron microscopy emerge as a potential disease hallmark, offering a valuable diagnostic clue in patients with congenital unresolved neuromuscular disorders.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Congenital Myopathies and congenital muscular dystrophies
OS03.06
Safety and Efficacy of an Enzyme Replacement Therapy in Infantile‑Onset Pompe Disease
Dr. Andreas Hahn1, Dr. Lijun Fu2, Dr. Robert J Hopkin3, Dr. Sihoun Hahn4, Dr. Peter Witters5, Dr. Katherine Denlinger6, Dr. Kristina An Haack7, Dr. Olivier Huynh-Ba7, Dr. Carlota Garcia Dominguez8, Dr. Kelly George9, Dr. Tianyue Zhou10, Dr. Madhurima Uppara Kowthalam11, Dr. Yin-Hsiu Chien12, Dr. Priya Kishnani13
1
Department of Child Neurology, United Hospital Giessen, Giessen, Germany.
2
Department of Cardiology, Shanghai Children's Medical Center, Shanghai Jiao Tong University School of Medicine, Shanghai, China.
3
Division of Human Genetics, Cincinnati Children's Hospital Medical Center and Department of Pediatrics, University of Cincinnati College of Medicine, Cincinnati, Ohio, United States.
4
Department of Pediatrics, University of Washington School of Medicine, Seattle Children’s Hospital, Seattle, Washington, United States.
5
Center for Metabolic Diseases, University Hospitals Leuven, Leuven, Belgium.
6
Children’s Hospital Colorado, Aurora, Colorado, United States.
7
Sanofi, Gentilly, France.
8
Sanofi, Madrid, Spain.
9
Sanofi, Cambridge, Massachusetts, United States.
10
Sanofi, Beijing, China.
11
Sanofi, Morristown, New Jersey, United States.
12
Department of Medical Genetics, National Taiwan University Hospital, Taipei, Taiwan.
13
Division of Medical Genetics, Department of Pediatrics, Duke University Medical Center, Durham, North Carolina, United States
Background: Infantile onset Pompe disease (IOPD) is the most severe form of Pompe disease, typically presenting shortly after birth with hypertrophic cardiomyopathy, muscle weakness, hypotonia, and respiratory compromise. If untreated, IOPD could be fatal within 6 months of birth. Early treatment is often underappreciated, yet it has been shown to improve outcomes.
In 2006, alglucosidase alfa, a recombinant human acid alpha glucosidase (rhGAA) was approved for the treatment of IOPD based on prolonged ventilator-free survival. Although alglucosidase alfa significantly improved clinical outcomes, many patients continued to exhibit declines in motor and respiratory function over time. Alglucosidase alfa uptake into muscle cells is limited due to its low content of bis-mannose-6-phosphate glycans. Avalglucosidase alfa is a next generation rhGAA designed to increase cellular uptake and target lysosomal delivery through the addition of ∼7 bis-mannose-6-phosphate glycans to the enzyme, which represents a 70-fold increase in comparison to alglucosidase alfa, hence overcoming this gap. The efficacy of avalglucosidase alfa was demonstrated in COMET study in treatment-naïve late-onset Pompe disease and Mini-COMET study in patients with IOPD previously treated with alglucosidase alfa for >6 months. In this current study, Baby-COMET, the safety and efficacy of avalglucosidase alfa is evaluated in treatment-naïve participants with IOPD aged ≤6 months, corrected for gestation, if necessary (gestational age <40 weeks will be adjusted to a full-term gestational age of 40 weeks), and not on ventilation at baseline.
Methods: Baby-COMET (NCT04910776) is a Phase 3, single-arm, open-label, international, multicenter study, assessing avalglucosidase alfa 40 mg/kg intravenous infusion every other week in treatment-naïve participants (aged ≤6 months) with IOPD. After a 4-week screening period, eligible participants received avalglucosidase alfa for 52 weeks (primary analysis period), followed by continued treatment for an additional 52 weeks (extended treatment period) and up to 104 additional weeks (extended long-term treatment period), with a 4-week follow-up. The total study duration is up to 4.08 years. An increase in dosing frequency was allowed for participants who had a suboptimal response as assessed by at least 2 consecutive assessments not less than 2 weeks apart on predefined cardiac, respiratory and motor function parameters.
The primary endpoint assessed the proportion of participants alive and free of invasive ventilation at Week 52. Secondary endpoints included proportion of participants alive and free of invasive ventilation at 12 and 18 months of age; change from baseline to Week 52 in left ventricular mass Z-score, Alberta Infant Motor Scale score, body growth Z-scores and percentiles, and urinary glucose tetrasaccharide; and safety, tolerability and immunogenicity.
Results: The study enrolled 16 IOPD participants aged ≤6 months, and 1 participant aged 7–12 months. The baseline characteristics of study participants are presented in Table 1. The efficacy and safety data are expected by May 2026 and will be presented at ICNMD.
Conclusion: The Baby-COMET study is one of the largest clinical studies in treatment-naïve patients with IOPD. The results of the Baby-COMET study have the potential to support use of avalglucosidase alfa in treatment-naïve infants living with Pompe disease.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Metabolic Myopathies: Glycogen, Lipids and Mitochondrial Myopathies
Images or Table (Optional)
OS04.01
Long-Term Remission with Anti-CD19 CAR-T Cell Therapy in Refractory Myasthenia Gravis: A Two-Year Case Series
Dr. Tobias Hegelmaier, Prof. Aiden Haghikia
Medizinische Hochschule Hannover, Hannover, Germany
Background: Myasthenia gravis (MG) is a chronic, B cell–mediated autoimmune disorder of the neuromuscular junction that remains refractory to conventional immunotherapies in a subset of patients. Anti-CD19 chimeric antigen receptor (CAR) T cell therapy has recently emerged as a promising strategy to eliminate autoreactive B cells in autoimmune diseases.
Methods: We describe the first two cases of severe, treatment-refractory, acetylcholine receptor antibody–positive MG, including one patient with concomitant rheumatoid arthritis, treated with a single infusion of anti-CD19 CAR T cells (KYV-101) following lymphodepleting chemotherapy. Clinical efficacy, immunological effects, and safety were prospectively monitored over a 24-month follow-up period. Disease activity was evaluated using the Quantitative Myasthenia Gravis (QMG) score and the MG Activities of Daily Living (MG-ADL) scale; rheumatoid arthritis activity was additionally assessed using the Disease Activity Score in 28 joints (DAS-28).
Results: Both patients achieved rapid, profound, and sustained clinical remission, with normalization of QMG and MG-ADL scores and restoration of functional capacity lasting up to two years after treatment. Clinical improvement persisted despite persistent or only modestly reduced anti–acetylcholine receptor antibody titers, indicating a dissociation between serological markers and disease activity. CAR T cell expansion and B cell depletion followed expected kinetics, with B cell reconstitution occurring after several months. Treatment was well tolerated; one patient experienced grade 1 cytokine release syndrome. No neurotoxicity, severe infections, or secondary autoimmune manifestations were observed during follow-up.
Conclusion: Anti-CD19 CAR T cell therapy induced durable clinical remission in patients with refractory MG and demonstrated an acceptable safety profile, even in the presence of persistent autoantibody levels. These findings highlight the potential of CAR T cell therapy as a transformative treatment for severe autoimmune neuromuscular disorders and support further evaluation in larger clinical studies.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
OS04.02
Comparative Effectiveness of a Conventional Immunosuppressant and a B‑Cell‑Depleting Therapy in Myasthenia Gravis
Ms. Wanqing Wu1,2, Dr. Jing Wu3, Ms. Malin Petersson1,2, Ms. Ann Eriksson-Dufva1,2,4, Prof. Fang Fang3, Prof. Fredrik Piehl1,2,4, Assoc. Prof. Susanna Brauner1,2,4
1
Department of Clinical Neuroscience, Karolinska Institutet, Stockholm, Sweden.
2
Neuroimmunology Unit, Center for Molecular Medicine, Karolinska Institutet, Stockholm, Sweden.
3
Institute of Environmental Medicine, Karolinska Institutet, Stockholm, Sweden.
4
Department of Neurology, Karolinska University Hospital, Stockholm, Sweden
Background: Myasthenia gravis (MG) is an autoimmune neuromuscular junction disorder characterized by fluctuating muscle weakness. Azathioprine has long been recommended as first-line treatment by international guidelines. However, recent randomized trials indicate a beneficial effect of several biological therapies. In Sweden, a rapid transition from azathioprine to B cell-depletion (rituximab) as preferred first-line treatment occurred around 2015. Leveraging this change, we were able to compare outcome while minimizing treatment bias. Our aim is to compare the effectiveness of azathioprine and rituximab as first-line IST in MG patients using clinical data from the Swedish MG registry (MG-reg).
Methods: We included adult MG patients from MG-reg who initiated azathioprine (n=156) or rituximab (n=91) as first-line treatment after 2005 and followed them until data withdrawal. The primary efficacy measure was relapse, identified by the use of rescue treatment. Incidence rate ratios (IRRs) were estimated using quasi-Poisson regression, adjusting for age at treatment initiation, sex, and disease duration. Analyses were performed using both intention-to-treat (ITT) and per-protocol (PP) approaches.
Results: Patients receiving rituximab were younger (57.9 vs. 62.5 years, p=0.002) and were more often Early-onset MG (24.1% vs. 9.6%, p=0.062). By year 1, 28.2% of azathioprine users and 12.1% of rituximab users had switched or added another ISTs; and after two years the proportions were 30.8% and 26.4%, respectively. By ITT, relapses occurred in 23.1% vs 14.3% of patients in year 1 and 8.9% vs 11.2% in year 2 (azathioprine vs rituximab). Adjusted IRRs for relapse in the first and second year with rituximab were 0.59 (95% CI: 0.31, 1.08) and 1.13 (95% CI: 0.48, 2.61), respectively. By PP, the relapse incidence was 298 vs. 167 per 1,000 person-years by year 1 and 194 vs. 149 per 1000 person-years by year 2. Adjusted IRRs for rescue treatment in the first and second year with rituximab were 0.55 (95% CI: 0.22, 1.23) and 0.71 (95% CI: 0.27, 1.61), respectively. Analysis of long-term follow up is ongoing.
Conclusion: Rituximab was associated with lower relapse frequency and fewer therapy switches in the first year compared with azathioprine, suggesting a faster onset of effect and greater effectiveness. Further studies are needed to identify predictors of treatment response and assess benefit–risk balance over extended follow-up.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
OS04.03
Early First-Week Changes in MG-ADL and MG-QOL-15 During C5 and FcRn-Inhibitors Treatment in Myasthenia Gravis
Dr. Claudia Vinciguerra1, Dr. Vincenzo Di Stefano2, Dr. Fiammetta Vanoli3, Dr. Nicasio Rini2, Dr. Giuseppe Di Martino2, Dr. Silvia Bonanno3, Dr. Sofia Campo2, Dr. Liliana Bevilacqua1, Dr. Eleonora Giacopuzzi Grigioli3, Dr. Rita Frangiamore3, Prof. Alvino Bisecco4, Dr. Giulia D'Alvano4, Dr. Davide Marigliano4, Dr. Marta Cheli3, Prof. Stefano Carlo Previtali5, Dr. Yuri Falzone5, Dr. Benedetta Sorrenti5, Dr. Laura Fionda6, Prof. Raffaele Iorio7,8, Dr. Silvia Falso7,8, Dr. Paolo Emilio Alboini9, Dr. Andrea Rigamonti10, Dr. Vittorio Mantero10, Dr. Dario Ricciardi11, Dr. Melania Guida12, Dr. Elena Rossini13, Prof. Marina Grandis14, Prof. Paolo Barone1, Prof. Filippo Brighina2, Dr. Marco Gemma15, Dr. Lorenzo Maggi3
1
Neurology Unit, Department of Medicine, Surgery and Dentistry “Scuola Medica Salernitana”, University of Salerno, 84131 Salerno, Italy, Salerno, Italy.
2
Department of Biomedicine, Neuroscience and Advanced Diagnostics (BiND), University of Palermo, 90127 Palermo, Italy, Palermo, Italy.
3
Neuroimmunology and Neuromuscular Diseases Unit, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy, Milano, Italy.
4
Department of Advanced Medical and Surgical Sciences (DAMSS), University of Campania “Luigi Vanvitelli”, Naples, Italy., Napoli, Italy.
5
Neurology Unit, IRCCS Ospedale San Raffaele and Vita-Salute San Raffaele University, Milan, Italy, Milano, Italy.
6
Neuromuscular and Rare Disease Centre, Neurology Unit, Sant'Andrea Hospital, Rome, Italy., Roma, Italy.
7
Department of Neuroscience, Università Cattolica del Sacro Cuore, Rome, Italy, Roma, Italy.
8
Neurology Unit, Fondazione Policlinico Universitario A. Gemelli IRCCS, Rome, Italy, Roma, Italy.
9
9Neurology Unit, Fondazione IRCCS Casa Sollievo della Sofferenza, 71013 San Giovanni Rotondo, Italy., San Giovanni Rotondo, Italy.
10
Neurology Unit, Ospedale A. Manzoni, ASST Lecco, Italy, Lecco, Italy.
11
UOC Neurophysiopathology, AORN Cardarelli, Via Antonio Cardarelli 9, Naples, taly, Napoli, Italy.
12
Department of Clinical & Experimental Medicine, Neurology Unit, University of Pisa, Italy, Pisa, Italy.
13
Department of Neuroscience, Mental Health and Sensory Organs (NESMOS), SAPIENZA University of Rome, Rome, Italy., Roma, Italy.
14
Neurology Unit, IRCCS Ospedale Policlinico San Martino, Largo R. Benzi 10, 16132 Genova, Italy, Genova, Italy.
15
Neuroanesthesia and Intensive Care Unit, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy, Milano, Italy
Background: Novel targeted therapies for generalized Myasthenia Gravis (gMG), including complement and FcRn inhibitors, are known to induce rapid clinical benefit. However, detailed daily clinical evaluations during the very early treatment stage are still limited.
The aim of this study was to assess early daily variations in functional status and quality of life during the first week of treatment with complement (C5) and neonatal Fc receptor (FcRn) inhibitors in patients with gMG.
Methods: We conducted a retrospective multicenter analysis of 95 patients with gMG receiving complement inhibitors (eculizumab, ravulizumab, zilucoplan) or FcRn inhibitors (efgartigimod, rozanolixizumab). MG Activities of Daily Living (MG-ADL) and Myasthenia Gravis Quality of Life 15-item (MG-QOL-15) scores were recorded daily from baseline to day 7. Longitudinal changes were analyzed using repeated-measures ANOVA and mixed-effects logistic regression models.
Results: MG-ADL scores showed a significant improvement over the first treatment week (F[7,736] = 7.75, p < 0.001), declining from 9.1 ± 3.1 at baseline to 6.15 ± 3.8 at day 7. A significant reduction was observed starting from day 3 (Δ = –2.04, p = 0.003), and 56.4% of patients achieved a ≥3-point improvement by day 7. The probability of clinical response increased progressively over time (day 7 vs day 1 OR = 5.5, p < 0.001). Treatment class (anti-C5 vs anti-FcRn) was not associated with differential MG-ADL outcomes (p > 0.20). MG-QOL-15 scores also improved significantly over time (F[7,480] = 2.16, p = 0.036), with a significantly higher likelihood of achieving a ≥4-point improvement by day 6 (p = 0.0002).
Conclusion: Both C5 and FcRn inhibitors are associated with rapid and clinically meaningful improvements in daily functioning and quality of life within the first week of treatment in gMG. Improvements are detectable as early as day 3 and appear comparable across therapeutic classes, underscoring the fast onset of action of these innovative immunotherapies
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
OS04.04
Repeated Cycles of an FcRn‑Targeting Therapy in Antibody‑Positive Generalised Myasthenia Gravis
Dr. Fiammetta Vanoli1,2, Dr. Artur Drużdż3, Dr. Ali A. Habib4, Dr. Dale J. Lange5, Dr. Hiroyuki Naito6, Prof. Robert Pascuzzi7, Prof. Sabrina Sacconi8, Dr. Kimiaki Utsugisawa9, Prof. John Vissing10, Dr. Tuan Vu11, Dr. Jiann-Horng Yeh12,13,14, Mr. Joel Baldwin15, Dr. Thais Tarancón16, Dr. Vera Bril17
1
Department of Neuroimmunology and Neuromuscular Diseases, Fondazione IRCCS, Instituto Nazionale Neurologico Carlo Besta, Milan, Italy.
2
Department of Human Neurosciences, Sapienza University of Rome, Rome, Italy.
3
Department of Neurology, Municipal Hospital, Poznań, Poland.
4
MDA ALS & Neuromuscular Center, Department of Neurology, University of California, Irvine, Orange, CA, United States.
5
Lange Neurology, New York, NY, United States.
6
Department of Clinical Neuroscience and Therapeutics, Hiroshima University, Hiroshima, Japan.
7
Neurology Department, Indiana University School of Medicine, Indiana University Health, Indianapolis, IN, United States.
8
Université Côte d'Azur, Peripheral Nervous System & Muscle Department, Pasteur 2 Hospital, Centre Hospitalier Universitaire de Nice, Nice, France.
9
Department of Neurology, Hanamaki General Hospital, Hanamaki, Japan.
10
Copenhagen Neuromuscular Center, Department of Neurology, Rigshospitalet, University of Copenhagen, Copenhagen, Denmark.
11
Department of Neurology, University of South Florida Morsani College of Medicine, Tampa, FL, United States.
12
Department of Neurology, Shin Kong Wu Ho-Su Memorial Hospital, Taipei, Taiwan.
13
College of Medicine, Fu Jen Catholic University, New Taipei City, Taiwan.
14
Department of Neurology, Kaohsiung Medical University, Kaohsiung, Taiwan.
15
UCB, Slough, United Kingdom.
16
UCB, Madrid, Spain.
17
Ellen and Martin Prosserman Centre for Neuromuscular Diseases, Toronto General Hospital, University of Toronto, Toronto, Ontario, Canada
Background: Anti-muscle-specific tyrosine kinase antibody-positive (MuSK Ab+) generalised myasthenia gravis (gMG) is a rare and often severe subtype of myasthenia gravis (MG). The Phase 3 MycarinG (NCT03971422) study demonstrated that six once-weekly rozanolixizumab infusions showed clinically meaningful improvements in MG-specific outcomes versus placebo in adults with gMG. Patients could then enrol in open-label extensions MG0004 (NCT04124965) then MG0007 (NCT04650854), or MG0007 directly. Here, we evaluate the efficacy and safety of repeated rozanolixizumab cycles in patients with MuSK Ab+ gMG.
Methods: In MG0004, patients received chronic, once-weekly rozanolixizumab for ≤52 weeks. In MG0007, after an initial cycle of rozanolixizumab, subsequent cycles were administered upon symptom worsening, at the investigator’s discretion. Final data (7 mg/kg or 10 mg/kg) were pooled across MycarinG, MG0004 (first 6 weeks) and MG0007 for patients with ≥2 symptom-driven cycles (efficacy pool), and across MycarinG and MG0007 for patients with ≥1 treatment cycle (safety pool). Efficacy outcomes included mean change from baseline (CFB) to Day 43 per cycle, and weighted means across Cycles 1–13 for MG Activities of Daily Living (MG-ADL) and Quantitative MG (QMG) scores.
Results: Overall, 12/129 (9.3%) patients who received ≥2 symptom-driven rozanolixizumab cycles had MuSK Ab+ gMG. In patients with MuSK Ab+ gMG, the weighted-mean MG-ADL scores across 13 cycles were 10.4 at baseline and 5.2 at Day 43. Weighted-mean CFB in MG-ADL total score across 13 cycles was −5.2, with mean (standard deviation) CFB ranging from −3.0 (3.6; n=8; Cycle 5) to −7.0 (3.5; n=12; Cycle 1). For QMG, the weighted-mean scores at baseline and Day 43 were 16.8 and 9.4, respectively. Weighted-mean CFB in QMG total score was −7.3, ranging from −5.0 (n=2; Cycle 13) to −10.6 (6.0; n=8; Cycle 3). In patients with MuSK Ab+ gMG and ≥1 treatment cycle (n=18), 14 (77.8%) reported treatment-emergent adverse events (TEAEs); most TEAEs were mild or moderate.
Conclusion: Patients with MuSK Ab+ gMG who received rozanolixizumab treatment consistently had improvements from baseline in MG-ADL and QMG scores across cycles, with an acceptable safety profile.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
OS04.05
Efficacy of Complement vs. FcRn-Inhibition in Thymoma-Associated Myasthenia Gravis: An Italian Multicentre Real-World Study
Dr. Alba Cepele1, Dr. Michelangelo Maestri Tassoni1, Dr. Paolo Emilio Alboini2, Dr. Pietro Businaro3, Assoc. Prof. Valentina Damato4, Dr. Carmen Erra5, Dr. Silvia Falso6, Dr. Laura Fionda7, Dr. Lucia Florio2, Dr. Matteo Gastaldi3, Prof. Francesco Habetswallner5, Assoc. Prof. Raffaele Iorio6, Dr. Veronica Iovino1, Dr. Aurora Lo Iacono1, Dr. Sofia Marini6, Dr. Federica Matrone5, Dr. Silvia Natoli8, Dr. Alessia Pugliese8, Dr. Elena Rossini7, Prof. Carmelo Rodolico8, Dr. Laura Tufano9, Dr. Massimiliano Ugo Verza4, Dr. Melania Guida1
1
Department of Clinical and Experimental Medicine, Neurology Unit, University of Pisa, Pisa, Italy.
2
Neurology Unit, Fondazione IRCCS Casa Sollievo della Sofferenza, San Giovanni Rotondo, Italy.
3
Neuroimmunology Research Section, IRCCS Mondino Foundation, Pavia, Italy.
4
Department of Neurosciences, Psychology, Drug Research and Child Health (NEUROFARBA), University of Florence, Firenze, Italy.
5
UOC Neurophysiopathology, AORN Cardarelli, Via Antonio Cardarelli 9, Napoli, Italy.
6
Department of Neuroscience, Università Cattolica del Sacro Cuore, Roma, Italy.
7
Department of Neuroscience, Mental Health and Sensory Organs (NESMOS), SAPIENZA University of Rome, Roma, Italy.
8
Unit of Neurology and Neuromuscular Disorders, Department of Clinical and Experimental Medicine, University of Messina, Messina, Italy.
9
Department of Neuroscience, Mental Health and Sensory Organs (NESMOS), Faculty of Medicine and Psychology, SAPIENZA University of Rome, Sant'Andrea Hospital, Roma, Italy
Background: Thymoma-associated Myasthenia Gravis (TAMG) represents a clinically challenging phenotype often underrepresented in clinical trials. While recent real-world data suggests that anti-complement therapy (C5IT) can induce significant clinical improvement and corticosteroid (CS) tapering in TAMG [1], comparative studies investigating the clinical response of this specific population to C5IT versus anti-FcRn inhibitors (aFcRn) are currently lacking.
This retrospective Italian multicentre study assessed the comparative efficacy and CS-sparing potential of C5IT vs aFcRn in TAMG patients.
Methods: We enrolled TAMG patients from eight Italian centres. Inclusion criteria were represented by Myasthenia Gravis (MG) diagnosis with positivity for antibodies anti-AChR, a histological diagnosis of thymoma, treatment with C5IT (chronic maintenance regimen) or aFcRn (individualised treatment cycles, according to Italian regulations). Patients were divided into two groups according to treatment. aFcRn patients were further stratified into “short responders” (mean intervals between cycles < 8 weeks) and “long-responder” (mean cycle intervals 8 weeks). Clinical outcomes evaluated are resumed in table n.1.
The primary endpoint included the evaluation of clinical outcomes (MG-ADL [2], QMG [3], CS dosage) from baseline over 12 months at intervals of 1, 3, 6, 9 and 12 months. The secondary endpoint was represented by the longitudinal changes in clinical outcomes in the timepoints among aFcRn long responders, short responders and anti-C5 group.
Descriptive statistics and independent t-test was computed. To assess the longitudinal evolution of clinical outcomes (MG-ADL, QMG, and CS) we approached Linear Mixed Models (LMM). Statistical significance was set at p < 0.05.
Results: 93 TAMG patient were enrolled (58 C5IT, 35 aFcRn). Demographic characteristics and baseline severity were comparable between groups for mean age at onset, age at treatment, sex, MG-ADL, QMG and corticosteroid dosage; thymoma B2 was the most represented histological type in both groups.
Both groups achieved an important improvement in all outcomes; LMM analysis demonstrated a significant treatment-by-time interaction favouring C5IT for both MG-ADL (F=8.48, p=0.004) and CS dosage (F= 6.284, p=0.013) indicating a differential response to treatment over the 12-month follow-up.
The C5IT cohort achieved greater estimated marginal mean reductions in MG-ADL (-6.43 vs -3.21), daily CS dosage (-10.8 mg vs -6-6 mg) and QMG (-6.73 vs -4.05), even if this last one was not statistically significant, compared to the aFcRn.
Subgroup analysis revealed that aFcRn “long-responders” achieved all outcomes comparable to C5IT. However, “short responders” (43% of the aFcRn cohort) showed significantly inferior improvement in MG-ADL and QMG compared to both C5IT and aFcRn long responders. Safety analysis showed no statistical differences in adverse events.
Conclusion: This real-world study suggests that C5IT in chronic maintenance regimen provides a greater overall improvement in MG-ADL scores and daily corticosteroids tapering compared to aFcRn cycle-based treatment in TAMG patients. Efficacy was heterogeneous within the aFcRn group: patients requiring frequent cycling (< 8 weeks) had suboptimal responses, whereas those with sustained durability mirrored C5IT efficacy. These founding highlight the necessity of predictive factors to identify potential “short responders”, allowing for better-personalize treatment selection and administration schedules.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
Images or Table (Optional)
OS04.06
An RNA‑Based Complement Pathway Inhibitor for gMG
Dr. Renato Mantegazza1, Dr. Saiju Jacob2, Dr. Tuan Vu3, Dr. Ali Habib4, Dr. Hiroyuki Murai5, Dr. John Vissing6, Dr. Aziz Shaibani7, Dr. Todd Levine8, Dr. Yessar Hussain9, Dr. Andreas Meisel10, Dr. Rodrigo Pavani11, Dr. Umesh Chaudhari11, Dr. Neda Jalali11, Dr. Michelle DeVeaux11, Dr. Karoline A Meagher11, Dr. Steven Sherman11, Dr. Eric Bachman11, Dr. Dipti Pawaskar11, Dr. Amal Souttou11, Dr. Lorah Perlee11, Dr. James F Howard Jr12
1
Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy.
2
University Hospitals Birmingham NHS Foundation Trust, Birmingham, United Kingdom.
3
University of South Florida Morsani College of Medicine, Tampa, FL, United States.
4
University of California Irvine, Orange, CA, United States.
5
International University of Health and Welfare, Narita, Japan.
6
University of Copenhagen, Copenhagen, Denmark.
7
Nerve and Muscle Center of Texas, Houston, TX, United States.
8
Bob Bové Neuroscience Institute, Scottsdale, AZ, United States.
9
Austin Neuromuscular Center, Austin, TX, United States.
10
Charité-Universitätsmedizin Berlin, Berlin, Germany.
11
Regeneron Pharmaceuticals, Inc., Tarrytown, NY, United States.
12
University of North Carolina, Chapel Hill, NC, United States
Background: Generalised myasthenia gravis (gMG) is characterized by auto-antibody complement activation, which causes damage to the neuromuscular junction. The phase 3 NIMBLE trial (NCT05070858) evaluated subcutaneous cemdisiran (an siRNA inhibiting hepatic C5 production) and subcutaneous pozelimab (a monoclonal antibody targeting C5) as monotherapies or in combination in adults with gMG.
Methods: NIMBLE is a double-blind, placebo-controlled trial. A total of 288 adults with gMG were randomised to receive cemdisiran (600 mg every 12 weeks), pozelimab (200 mg every 4 weeks [Q4W]), combination (pozelimab 200 mg Q4W + cemdisiran 200 mg Q4W), or placebo. The primary and key secondary efficacy endpoints (n=239, database lock August 6, 2025) were assessed using change from baseline in Myasthenia Gravis Activities of Daily Living (MG-ADL) and Quantitative Myasthenia Gravis (QMG) scores at Week 24. Other secondary efficacy endpoints included change from baseline at Week 24 in Myasthenia Gravis Composite (MGC) and Myasthenia Gravis Quality of Life (MGQoL15r) scores, and achievement of a ≥3-/≥5-point reduction in MG-ADL/QMG scores from baseline to Week 24. Safety was assessed in 281 patients in the double-blind treatment period (DBTP) (database lock December 10, 2025).
Results: NIMBLE met its primary and key secondary endpoints. Results are expressed as least-squares mean (LSM) and SE. In participants assigned to cemdisiran (n=64) and combination (n=67), the LSM (SE) change in the primary endpoint (MG-ADL) was –4.5 (0.4) and –4.0 (0.4), with placebo-adjusted differences of –2.3 (P=0.0005) and –1.7 (P=0.0086). LSM (SE) change in the key secondary endpoint (QMG) was –4.2 (0.6) and –3.3 (0.6) for cemdisiran and combination, respectively, with placebo-adjusted differences of –2.8 (P=0.0015) and –1.9 (P=0.0348). In the cemdisiran and combination arms, the LSM (SE) change from baseline at Week 24 in MGC total score was –7.8 (0.9) and –6.0 (0.9), with placebo-adjusted LSM differences of –4.3 (P=0.0009) and –2.6 (P=0.0481) (Table 1). In the cemdisiran and combination arms, the LSM (SE) change from baseline at Week 24 in MGQoL15r total score was –4.7 (0.8) and –4.3 (0.8), with placebo-adjusted LSM differences of –2.4 (P=0.0348) and –2.0 (P=0.0825) (Table 1). In MG-ADL, 76.6% (cemdisiran) and 65.7% (combination) of participants achieved ≥3-point reductions, while 48.4% (cemdisiran) and 35.8% (combination) of participants achieved ≥5-point reductions in QMG. Assessment of terminal complement inhibition was 76.6% in the cemdisiran monotherapy arm and 99.6% in the combination arm.
Cemdisiran was generally well-tolerated, with TEAEs occurring in 74.7% (59/79), 81.6% (40/49), 81.5% (66/81) and 81.9% (59/72) of participants in the cemdisiran, pozelimab, combination and placebo arms, respectively. No serious infections, meningococcal infections or deaths occurred during the DBTP; there were 2 deaths (pneumonia [cemdisiran], septic shock [combination]) in participants on concomitant immuno-suppressive therapies after the DBTP.
Conclusion: Cemdisiran, both as monotherapy and in combination with pozelimab, significantly improves clinical signs and symptoms in people with gMG and was generally well-tolerated. Quarterly subcutaneous administration of cemdisiran provides effective symptom relief and enhanced quality of life, with partial complement inhibition.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
Images or Table (Optional)
OS05.01
Prospective Study of an FcRn Inhibitor in Myasthenia Gravis Crisis
Dr. Shahar Shelly
Rambam Medical Center, Haifa, Israel. Technion Institute of Technology, Haifa, Israel
Background: We aimed to determine the safety and clinical efficacy of the FcRn blocker efgartigimod in adults presenting with impending severe myasthenic crisis.
Methods: Prospective, single-arm, open-label study conducted between October 1, 2024, and April 30, 2025, in the intensive-care and neurology units of a tertiary academic medical center. Sixteen consecutive adults (≥18 years) with generalized myasthenia gravis (AChR- or MuSK-antibody positive) who required invasive or non-invasive ventilatory support or enteral feeding were enrolled. The prespecified primary endpoint was clinically meaningful improvement (CMI) in the Myasthenia Gravis Activities of Daily Living (MG-ADL) scale—defined as a ≥2-point reduction from baseline—assessed at 4 and 8 weeks. Key secondary outcomes included a ≥3-point reduction in Quantitative Myasthenia Gravis (QMG) score, corticosteroid-sparing effect, need for rescue therapies, recurrent crisis, and safety.
Results: All 16 participants (mean [SD] age, 58.5 [17.4] years; 8 women) completed eight-weeks of follow-up. At week 8, 12 patients (75%; 95% CI, 47–91%) achieved the primary endpoint. Median (IQR) MG-ADL fell from 11 (9–13) at baseline to 6 (5–7) (median change, −5 points; p =0.003). Median QMG decreased from 21 (19–23) to 13 (11–16) (median change, −7.5 points; p < 0.001), with 9 patients (56 %) meeting QMG-CMI by week 1. Among 12 participants receiving prednisone, 6 (50 %) tapered (n = 3) or discontinued (n = 3) corticosteroids by week 8. No treatment-related serious adverse events were observed.
Conclusion: Efgartigimod produced rapid, clinically meaningful, and improvement in patients with myasthenic crisis without new safety findings. These findings suggest FcRn blockade is a practical alternative to plasma exchange or intravenous immunoglobulin in acute care and warrants confirmation in randomized controlled trials.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
OS05.02
TYMS Gene Polymorphisms and Thymic Pathology in Myasthenia Gravis
Prof. Paulo José Lorenzoni, Prof. Renata Dal-Pra Ducci, Dr. Paula Raquel do Vale Pascoal Rodrigues, Prof. Otto Jesus Hernandez Fustes, Miss Raquel Cristina Arndt, Dr. Claudia Suemi Kamoi Kay, Prof. Rosana Herminia Scola
Universidade Federal do Paraná, Curitiba, Brazil
Background: Myasthenia gravis (MG) is frequently associated with pathological alterations of the thymus, including thymoma and thymic hyperplasia. Thymidylate synthase (TYMS) is a key enzyme involved in DNA synthesis, and its expression is influenced by polymorphic variations within the thymidylate synthase enhancer region (TSER). These polymorphisms include variable numbers of tandem repeats (VNTRs), most commonly two (2R, TSER2) or three (3R, TSER3) 28-base pair repeats (rs45445694). TYMS polymorphisms have been implicated in malignancy risk and immune dysregulation; however, their potential role in thymic pathology in MG remains poorly understood. It is therefore plausible that TSER polymorphisms affecting TYMS expression may influence susceptibility to thymic abnormalities in MG.
Methods: This study aimed to determine the frequency of TYMS polymorphisms in patients with MG and to evaluate their association with thymic pathology in a real-world outpatient cohort from Southern Brazil. Additionally, the prevalence of TYMS genotypes was estimated for Brazilian MG patients. Genotyping was performed to identify the VNTR polymorphism in the TSER region of the TYMS gene (rs45445694: 2R and 3R alleles). Associations between TYMS polymorphisms and thymic pathology were assessed using univariate logistic regression, with patients presenting a normal (involuted) thymus serving as the reference group.
Results: The study included 73 patients with MG, of whom 36 underwent thymectomy. Histopathological analysis revealed a normal (involuted) thymus in 15 patients (41.7%), thymoma in 12 patients (33.3%), and follicular hyperplasia in 9 patients (25%). Genotype frequency of 2R/2R, 2R/3R and 3R/3R in the total cohort were 23.28%, 42.46% and 34.24%, respectively. Total genotype distributions were consistent with Hardy–Weinberg equilibrium in the total cohort and in the reference group with normal thymus. No significant differences were observed in allele frequencies of the 2R and 3R variants between patients with thymoma or thymic hyperplasia and those with normal thymus. The 3R allele was not associated with thymoma (OR = 0.51, 95% CI 0.17–1.55; p = 0.28) or thymic hyperplasia (OR = 0.54, 95% CI 0.16–1.83; p = 0.43). No statistically significant associations were identified between TYMS genotypes and thymic pathology. Although a trend towards reduced risk of thymic hyperplasia was observed for the 2R/3R genotype (OR = 0.095, 95% CI 0.01–1.06), this did not reach statistical significance (p = 0.08).
Conclusion: This study characterises the prevalence of TYMS 2R and 3R polymorphisms in Brazilian patients with MG. Allelic frequencies were consistent with Hardy–Weinberg expectations but differed from those reported in other Brazilian populations. No association was identified between TYMS polymorphisms and the risk of thymoma or thymic hyperplasia in MG.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
OS05.03
Subclinical Cardiomyopathy in Premanifest Carriers of Late-Onset Hereditary Transthyretin Amyloidosis with Polyneuropathy
Dr. Te-Wei Wang1,2, Asst. Prof. Mao-Yuan Su1, Assoc. Prof. Mei-Fang Cheng1, Dr. Hsueh-Wen Hsueh1, Prof. Ming-Chang Chiang3, Prof. Yen-Hung Lin1, Prof. Jyh-Ming Jimmy Juang1, Assoc. Prof. Shiou-Ru Tzeng4, Prof. Sung-Tsang Hsieh4,1, Prof. Chi-Chao Chao1
1
National Taiwan University Hospital, Taipei City, Taiwan.
2
National Taiwan University Hospital Hsin-Chu Branch, Hsinchu County, Taiwan.
3
National Yang Ming Chiao Tung University, Taipei City, Taiwan.
4
National Taiwan University, Taipei City, Taiwan
Background: Hereditary transthyretin amyloidosis (ATTRv) is a systemic disease caused by various TTR gene mutations. Recent advances in disease-modifying therapies for ATTRv have heightened the clinical significance of early diagnosis and intervention. Cardiac involvement is a common presentation of late-onset hereditary transthyretin amyloidosis with polyneuropathy (ATTRv-PN). This study explored phenotypes of cardiac amyloidosis and their clinical and neurological correlates in the premanifest stage.
Methods: Premanifest carriers with the transthyretin p.A117S variant were enrolled. Healthy controls and ATTRv-PN patients with stage 1 polyneuropathy disability (PND) were enrolled for comparison. Cardiomyopathy was evaluated by echocardiography, 99mTc-pyrophosphate (PYP) single-photon emission computed tomography (SPECT) imaging, cardiac magnetic resonance imaging (CMR), and serum N-terminal prohormone of brain natriuretic peptide (pro-BNP). Neuropathy was assessed using clinical questionnaires, skin biopsy, neurophysiology, and quantitative sensory testing. Differences between groups were compared using the Kruskal–Wallis test or Fisher’s exact test. Multivariable linear regression models were applied to adjust for age and sex. Multiple comparisons were corrected by the false discovery rate (FDR) method.
Results: Thirty-two carriers with transthyretin variants (31 p.A117S, 17 men, median age 47) were enrolled, along with 63 controls (34 men, median age 51 years) and 11 ATTRv-PN patients (9 p.A117S, 7 men, median age 64 years). None of the carriers had polyneuropathy, while 38% had carpal tunnel syndrome (CTS) and 16% had increased radiotracer uptake on 99mTc-PYP SPECT imaging. Based on the normal references from the healthy controls (the mean plus 2 standard deviations), abnormally increased extracellular volume (ECV) was noted in 25% of the carriers, prolonged myocardial native T1 in 11%, and increased left ventricular mass index (LVMi) in 6%. Compared with controls, carriers demonstrated a greater ECV (β 1.87%, 95% CI 0.76–2.98%, p=0.0025), longer native T1 (β 15.51 msec, 95% CI 1.7–29.32, p=0.0282) and greater LVMi (β 4.16 g/m²; 95% CI, 0.65–7.67; p=0.0208) on CMR, indicating subclinical cardiac amyloidosis. ATTRv-PN patients had markedly greater amyloid burden across these cardiac imaging. Among carriers, CMR myocardial interstitial and hypertrophic parameters were significantly correlated with large- and small-fiber nerve degenerations. Higher 99mTc-PYP SPECT uptake was associated with increased pro-BNP, native T1, ECV, autonomic and sensory dysfunction, and presence of CTS.
Conclusion: Subclinical cardiomyopathy, as shown by nuclear medicine imaging and CMR, is common and linked to neuropathic changes among carriers carrying predominant p.A117S variant, and may facilitate early and accurate diagnosis and intervention for ATTRv-PN.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Hereditary Peripheral Neuropathies
Images or Table (Optional)
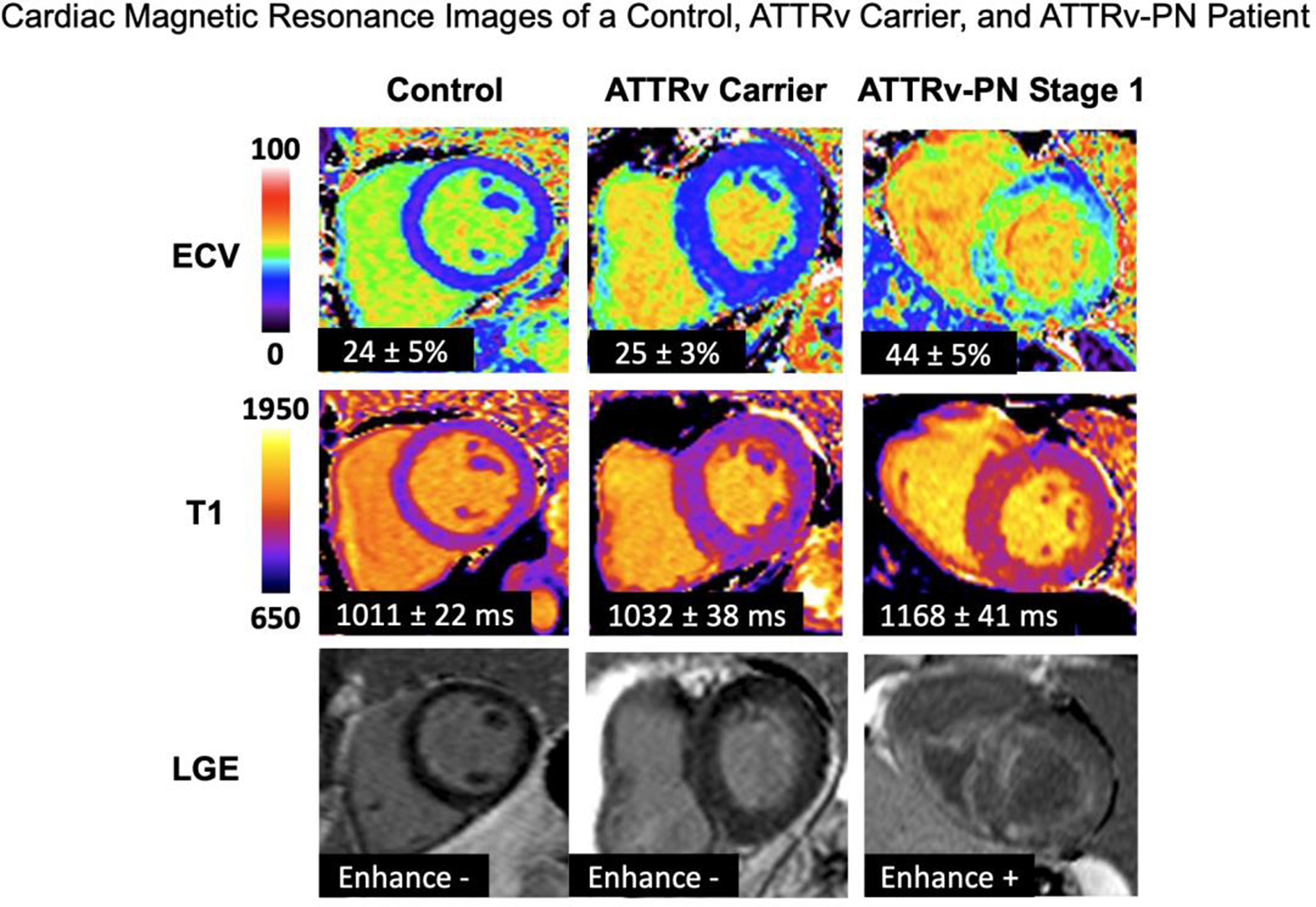
OS05.04
Serum Neurofilament Light Chain as a Biomarker of Global Axonal Burden in Immune-Mediated Demyelinating Neuropathies
Dr. Namjin Heo1, Miss Mi-Young Mi-Young2, Prof. Byoung Joon Kim1
1Department of Neurology, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea, Republic of. 2Samsung institute of future medicine, Seoul, Korea, Republic of
Background: Serum neurofilament light chain (NfL) has emerged as a sensitive biomarker of axonal injury across various neurological disorders. While elevated NfL levels are documented in chronic inflammatory demyelinating polyneuropathy (CIDP), data comparing NfL across the spectrum of immune-mediated demyelinating neuropathies—including autoimmune nodopathy and anti-myelin-associated glycoprotein (MAG) neuropathy—are sparse. We aimed to evaluate whether serum NfL reflects a common pathway of axonal burden across these distinct entities and its correlation with electrophysiological measures of motor axonal integrity.
Methods: This retrospective multicenter study analyzed patients from two tertiary academic referral centers. Serum NfL was quantified using the Ella platform (ProteinSimple, Bio-Techne). We reviewed nerve conduction studies to calculate composite compound muscle action potential (CMAP) indices, representing global motor axonal integrity. Multivariable regression analyses were adjusted for age and body mass index (BMI), both of which were predefined as key biological covariates affecting NfL distribution.
Results: A total of 72 patients were included 41 CIDP, 11 autoimmune nodopathy, and 20 anti-MAG neuropathy. Age was positively correlated (β =0.491, p < 0.001), while BMI was inversely associated (β = -0.322, p = 0.004) with serum NfL levels. After adjusting for these covariates, serum NfL levels were comparably elevated across all diagnostic groups, with no significant inter-group differences (anti-MAG vs. CIDP, p = 0.40; autoimmune nodopathy vs. CIDP, p = 0.75). Furthermore, higher NfL levels demonstrated a consistent inverse association with the composite sigma CMAP areas (standardized β = -0.167, p = 0.115), a pattern observed in both upper-limb (β = -0.122) and lower-limb distributions (β = -0.157). Notably, NfL levels did not fluctuate significantly based on clinical disease activity (p = 0.281) or treatment status (all p > 0.2), suggesting it reflects cumulative or baseline axonal strain rather than acute inflammatory fluctuations in this cohort.
Conclusion: Serum NfL functions as a convergent biological marker of axonal injury that transcends specific clinical phenotypes in immune-mediated demyelinating neuropathies. Rather than acting as a diagnostic discriminator, it provides a unified measure of global axonal burden, potentially serving as a valuable tool for long-term monitoring of neuroaxonal integrity.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Immune-mediated Neuropathies
OS05.05
Serum Peripherin as a Disease Biomarker in Hereditary Transthyretin Amyloidosis: A Multicenter Cohort Study
Dr. Guido Primiano1, Assoc. Prof. Domenico Plantone2, Dr. Delia Righi2, Dr. Angela Romano1, Dr. Luca Leonardi3, Dr. Valeria Guglielmino4, Dr. Francesca Forcina3, Asst. Prof. Marco Ceccanti5, Assoc. Prof. Maurizio Inghilleri5, Prof. Fiore Manganelli6, Asst. Prof. Stefano Tozza6, Dr. Maria Ausilia Sciarrone4, Dr. Francesca Vitali4, Dr. Andrea Sabino4, Dr. Carlo Manco2, Dr. Angela Stufano7, Dr. Maria Laura Stromillo2, Prof. Nicola De Stefano2, Prof. Paolo Calabresi4, Asst. Prof. Marco Luigetti4
1Fondazione Policlinico Universitario Agostino Gemelli IRCCS, Rome, Italy. 2University of Siena, Siena, Italy. 3Sant’Andrea Hospital, Rome, Italy. 4Università Cattolica del Sacro Cuore, Rome, Italy. 5Sapienza Università Di Roma, Rome, Italy. 6University of Naples ‘Federico II’, Naples, Italy. 7University of Bari Aldo Moro, Bari, Italy
Background: Hereditary transthyretin amyloidosis (ATTRv, v for variant) is a rare, progressive, and fatal multisystemic disease caused by pathogenic variants in the TTR gene. Peripherin, a type III intermediate filament protein predominantly expressed in the peripheral nervous system (PNS), has recently emerged as a promising biomarker for axonal damage in peripheral neuropathies. This study aims to investigate serum peripherin levels in symptomatic and presymptomatic ATTRv subjects, alongside serum neurofilament light chain (sNfL) levels, and to examine their correlations with disease progression and severity.
Methods: This multicenter, cross-sectional cohort study included 96 individuals with TTR gene variants (49 presymptomatic and 47 symptomatic ATTRv subjects) and 42 healthy controls (HCs). Serum peripherin levels were measured using a highly sensitive homebrew immunoassay developed on the Simoa SR-X platform, whereas sNfL levels were determined using a commercially available Simoa Neurology 2-Plex B assay. Statistical analyses included non-parametric Spearman correlations, Kruskal-Wallis ANOVA, ANCOVA, and binomial logistic regression.
Results: Serum peripherin levels were significantly elevated in both presymptomatic (p < 0.001) and symptomatic ATTRv groups (p < 0.001) compared to HCs, with no significant difference between the presymptomatic and symptomatic groups. Peripherin levels did not correlate with age, sNfL levels, or clinical severity (NIS scores), but showed a strong discriminative ability between ATTRv and HCs (AUC = 0.83, sensitivity = 76%, specificity = 76%, p < 0.001).
Conclusion: This study demonstrates that serum peripherin represents a promising biomarker for early detection of PNS damage in ATTRv, with elevated levels detectable even in presymptomatic stages.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Hereditary Peripheral Neuropathies
OS05.06
Static and Dynamic Stabilometric Evaluation and Rehabilitation in Peripheral Neuropathy
Dr. Edoardo Roveta1,2, Dr. Mehrnaz Hamedani3, Dr. Sara Massucco3, Dr. Chiara Gemelli4, Dr. Cristina Schenone3, Dr. Marina Grandis4, Dr. Carolina Barone3, Dr. Massimo Leandri3, Dr. Angelo Schenone4
1
University of Genoa, DINOGMI, Genoa, Italy.
2
Fondazione David Chiossone Impresa Sociale, Genoa, Italy.
3
Department of Neuroscience, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (DINOGMI), Genoa, Italy.
4
Department of Neurology, IRCCS Polyclinic Hospital San Martino, Genoa, Italy
Background: Charcot–Marie–Tooth (CMT) disease is a hereditary peripheral neuropathy characterized by progressive distal muscle weakness, sensory impairment and balance dysfunction, leading to gait instability and increased fall risk. Quantitative measures are needed to detect subtle balance deficits beyond standard clinical assessments. This study aims to evaluate the reliability and responsiveness of a static and dynamic stabilometric platform (SP) in individuals with CMT.
Methods: The full study plans to enroll 40 patients with genetically confirmed CMT and 60 healthy controls (HC), aged between 18 and 80 years. At this preliminary stage, data are available from 24 patients and 30 HC. Eligibility criteria included the absence of significant comorbidities, no recent orthopedic surgery and a pain score below 3 on the Visual Analogue Scale (VAS<3). All participants underwent balance assessment using the SP under static conditions (Eyes Open, Eyes Closed) and dynamic conditions (Eyes Open Toes Up Small Perturbation, Eyes Open Backward Stimulation). During dynamic trials, surface electromyography was used to assess activation timing of the tibialis anterior and soleus muscles. Patients with CMT also performed standardized clinical assessments, including the Berg Balance Scale (BBS), Modified Barthel Index (MBI), Tinetti Scale (TS), 10-Meter (10MWT) and 6-Minute Walking Tests (6MWT). Patients were randomized to traditional physiotherapy or platform-based rehabilitation, both consisting of 30 daily sessions of 1 hour.
Results: No significant test–retest differences were found in HC, confirming platform reliability. In patients, significant post-treatment improvements were observed in Sway Area and Sway Path during static EO and EC trials (p = 0.01). Correlation analysis revealed significant associations between Sway Area and activation times of the tibialis anterior (r = 0.65, p = 0.03) and soleus (r = 0.72, p = 0.01). Post-treatment changes in balance parameters were also significant when measured using the following clinical scales: BBS t0: 46.70 ± 2.22, t1: 51.70 ± 1.32 (p< 0.023), 10MWT time domain t0: 9.30 ± 2.65, t1: 8.17 ± 2.17 (p<0.005), TS t0: 22.50 ± 3.20, t1: 25.00 ± 1.94 (p<0.005), 6MWT t0: 362.00 ± 114.12, t1: 401.00 ± 102.61 (p<0.004).
Conclusion: Due to the current sample size, no comparison between platform-based and traditional rehabilitation was performed, and results reflect the overall rehabilitation effect in CMT patients. Combining traditional assessments with quantitative metrics improves sensitivity to small functional changes. More comprehensive results will be reported upon study completion.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Hereditary Peripheral Neuropathies
OS06.01
The Landscape of Non-Reference SINE-VNTR-Alus in Amyotrophic Lateral Sclerosis
Prof. Sulev Koks1,2, Dr. Abigail Pfaff1,2, Dr. Vivien Bubb3, Prof. John Quinn3
1
Murdoch University, Perth, Australia. 2Perron Institute, Perth, Australia. 3University of Liverpool, Liverpool, United Kingdom
Background: The fatal neurodegenerative disease, amyotrophic lateral sclerosis (ALS), causes the degeneration of motor neurons in the brain and spinal cord. Many different genetic variants are known to increase the risk of developing ALS; however, much of the disease heritability remains to be identified.
Methods: To discover novel genetic factors, we characterised SINE-VNTR-Alu (SVA) presence/absence variation in 4403 genomes from the New York Genome Centre (NYGC) ALS consortium. SVAs are a type of retrotransposon capable of mobilising within the human genome, creating new insertions that can influence gene expression and mRNA splicing. To date, 33 insertions are known to cause a range of genetic diseases.
Results: In the NYGC ALS consortium sequence data, 2831 non-reference genome SVAs were identified, and 95% of these insertions were rare, with an insertion allele frequency below 0.01. Association analysis of the common SVAs with ALS risk, age at onset, and survival did not reveal any SVAs that remained significant after correction for multiple testing. However, three distinct rare SVA insertions in the ALS-associated gene NEK1 were identified in four different individuals with ALS. The frequency of these rare NEK1 insertions was significantly higher in individuals with ALS from the NYGC ALS cohort compared to gnomAD SV non-neuro controls (p = 0.0002).
Conclusion: This study was the first to characterise non-reference SVA presence/absence variation in a large cohort of ALS patients, pinpointing insertions as potential candidates involved in disease development for future research.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: ALS: Biology, Pathophysiology, Genetics
Images or Table (Optional)
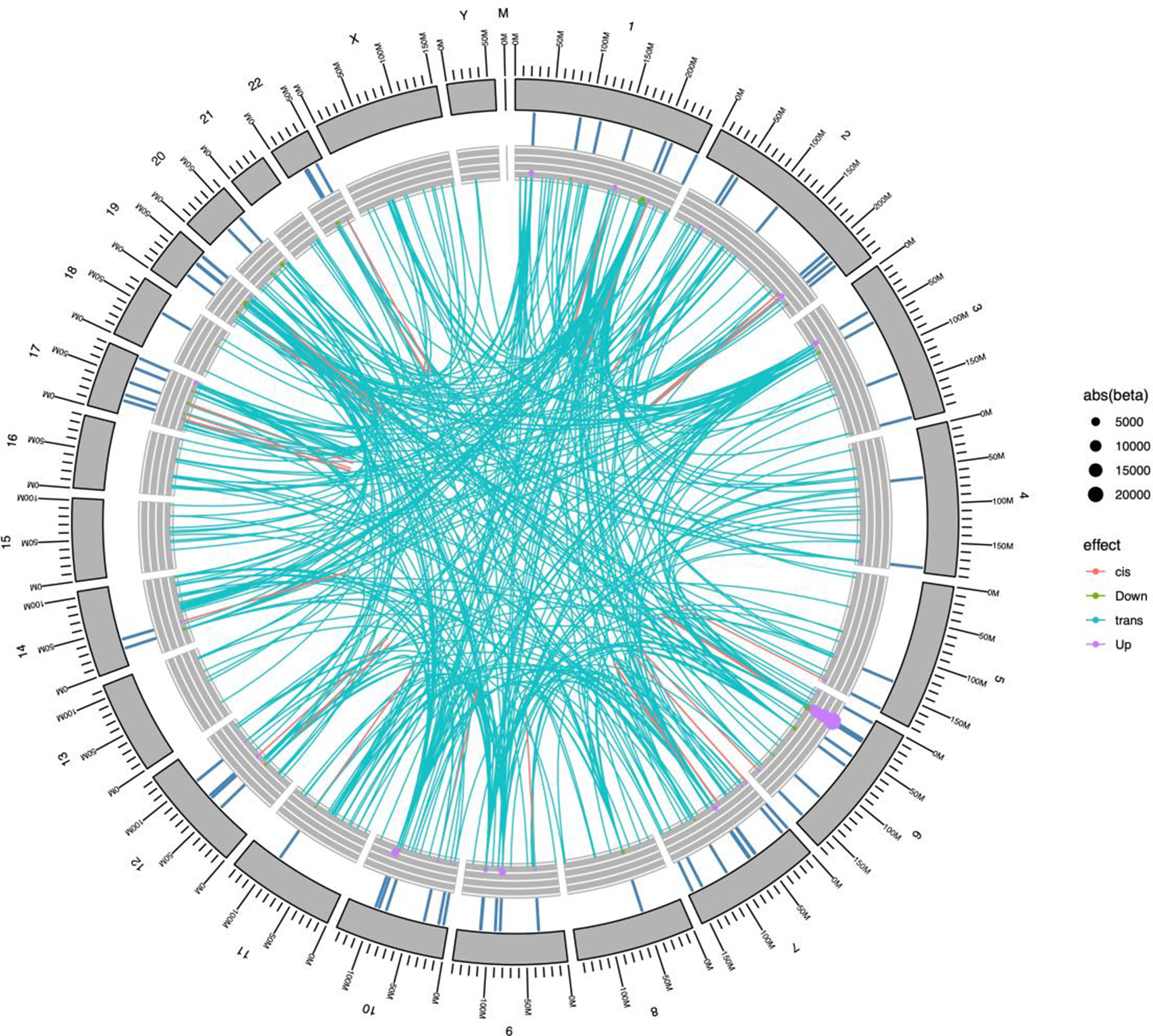
OS06.02
Prognostic Factors in Guillain–Barré Syndrome: A Retrospective Cohort Study in a Tertiary Referral Centre
Assoc. Prof. Massimo Russo, Dr. Marcella De Luca, Dr. Angelica Squillaci, Prof. Antonio Toscano, Prof. Carmelo Rodolico, Prof. Anna Mazzeo, Prof. Sonia Messina
Department of Clinical and Experimental Medicine, University of Messina, Messina, Italy
Background: Guillain–Barré syndrome (GBS) is an acute immune-mediated polyradiculoneuropathy and early identification of prognostic indicators is essential to optimize therapeutic strategies and management.
We aimed to identify the association of clinical, neurophysiological and laboratory parameters with functional outcome at 6 months. We also evaluated the response to immunomodulatory treatments among different subtypes.
Methods: We performed a single-center, retrospective observational study on patients consecutively admitted between 2015 and 2024 in a tertiary referral centre. Functional disability, muscle strength and autonomic symptoms were assessed using the Hughes Functional Grading Scale (HFGS), the Medical Research Council (MRC) sum score and the COMPASS-31 questionnaire. Neurophysiological studies allowed classification into Acute Inflammatory Demielinating Polyneuropathy (AIDP), Acute Motor Axonal Neuropathy (AMAN), Acute Motor and Sensory Axonal Neuropathy (AMSAN) or mixed forms. Laboratory parameters, including serum sodium, albumin, fibrinogen and neurofilament light chain (NfL) were analyzed. Treatment modalities included intravenous immunoglobulin and/or plasma exchange.
Results: We included 50 patients (mean age at onset of 56, SD:19 yrs, M:F, 58% vs 42%). The clinical severity at admission was high [(HFGS score of 4 (IQR 3—4)]. Twenty-eight percent of patients exhibited dysphagia and 20% required ventilatory support. COMPASS-31 score was 23,SD:12. The average time from symptom onset to specific therapy was 7.5,SD:4 days. Neurofilament light chain (NfL) levels in CSF were 470.8 pg/mL, SD:378.1.
Regarding the functional outcome at 6 months, the median Hughes score was 2 (IQR 2-3), and 62% of patients achieved autonomous walking (Hughes <2). Approximately 76% were defined as “responders” to treatment (improvement >1 point on the Hughes scale within 4 weeks). The factors found to be associated with an unfavourable outcome (HFGS >3 at 6 months) were: age >60 years (p=0.03), HFGS >4 at admission (p=0.01), time from onset to therapy >7 days (p=0.04), serum sodium <136 mmol/L (p=0.04), and NfL levels >500 pg/mL (p=0.02). The correlation between the COMPASS-31 score and HFGS at nadir was modest (p= 0.22).
Conclusion: This real-world study confirms the heterogeneity of GBS and supports the value of an integrated, multidimensional assessment combining clinical, neurophysiological, and laboratory data to improve prognostic stratification. Such an approach may facilitate earlier identification of high-risk patients, guide individualized management, and optimize follow-up strategies.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Immune-mediated Neuropathies
OS06.03
Oxidative Stress-Driven Somatic Mosaicism Marks a Distinct Molecular Subtype of Sporadic ALS
Dr. Hyunjin Kim1, Mr. Ben Thompson2, Mr. Jinhyeong Kim3, Prof. Jung-Joon Sung4, Prof. Eleanora Aronica5, Prof. Junho Kim3, Prof. Jeong Ho Lee6
1Asan Medical Cener, Seoul, Korea, Republic of. 2University College London, London, United Kingdom. 3Sungkyunkwan University, Suwon, Korea, Republic of. 4Seoul National University Hospital, Seoul, Korea, Republic of. 5Amsterdam UMC, Amsterdam, Netherlands. 6Korea Advanced Institute of Science and Technology, Daejeon, Korea, Republic of
Background: Amyotrophic lateral sclerosis (ALS) is a clinically and molecularly heterogeneous neurodegenerative disease, with most sporadic cases lacking an identified cause. While familial ALS is linked to germline mutations, the contribution of somatic mutations arising during neurodevelopment remains poorly understood in the onset of sporadic ALS. We aimed to investigate whether somatic mutations arising during brain development contribute to sporadic ALS, and to define their molecular characteristics and potential pathogenic role.
Methods: We performed whole-genome sequencing (∼60× coverage) on approximately 1,000 primary motor cortex neurons per brain from 27 sporadic ALS cases and 8 neurotypical controls. Neurons were isolated using laser capture microdissection to ensure cell-type specificity. Matched neuronal and peripheral tissue (liver) whole-genome sequencing data were compared to identify somatic variants. Variant calling was performed using Mutect2, Strelka, and MosaicForecast, and only single-nucleotide variants detected by at least two of these callers were included in the final callset. Mutational signatures and variant allele fractions were analyzed to infer timing and mechanisms of mutation accumulation.
Results: We observed a significantly increased burden of somatic single-nucleotide variants (sSNVs) in a subset of ALS cases corresponding to the ‘ALS-Oxidative stress’ molecular subtype identified in previous transcriptomic studies. Gene set enrichment analysis demonstrated that these mutations were enriched in genes involved in neurogenesis. Mutational signature analysis revealed enrichment of COSMIC SBS36-like (oxidative damage) and SBS44-like (defective mismatch repair) signatures in high-burden ALS neurons, while absent in controls and bulk motor cortex. Genes harboring these mutations were preferentially expressed during fetal neurodevelopment, suggesting a developmental window of vulnerability to oxidative stress–associated DNA damage.
Conclusion: Our findings support a model in which early neurodevelopmental oxidative stress promotes focal vulnerability in motor neurons, as reflected by associated clonal somatic mutations. This work highlights the importance of somatic mosaicism and developmental origins in ALS pathogenesis and suggests that targeting oxidative stress pathways during critical developmental periods may hold potential for prevention or early intervention in susceptible individuals.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: ALS: Biology, Pathophysiology, Genetics
OS06.04
GM1 Neuropathies, From Antibodies to Phenotype
Dr. Nicolas Dubuisson1,2, Dr. Claire Bergstrom Johnson1, Dr. Roberto Bellanti1, Dr. Mateusz Makuch1, Dr. Robert Prior3, Dr. Mariya Misheva1, Assoc. Prof. Simon Rinaldi1
1Oxford University, Oxford, Oxford, United Kingdom. 2Cliniques Universitaires St-Luc, Brussels, Belgium. 3KULeuven, Leuven, Belgium
Background: Multifocal motor neuropathy (MMN) is a chronic immune-mediated, purely motor, neuropathy characterized by the presence of GM1 antibodies. Currently, the underlying immune processes, particularly the role of B-cell clonality and GM1 antibodies, remain incompletely understood. This study aims to better characterize the origin and pathogenic potential of GM1 antibodies to inform more targeted therapies.
Methods: A cohort of 35 MMN patients was clinically characterized using MRC-SS, ONLS, and MMN-RODS. The presence of paraproteins and light-chain restriction was evaluated using serum protein electrophoresis (SPEP), ELISA and high-resolution mass spectrometry. PBMCs from MMN patients vs controls were analysed by flow cytometry, followed by bulk and single-cell sorting and in vitro culture to assess GM1-specific IgM production. Finally, the pathogenic potential of patient-derived antibodies was investigated using human iPSC-derived neuromuscular organoids.
Results: The clinical course of MMN confirmed an initial response to treatment, followed by an inevitable functional decline after approximately seven years of therapy. Serologically, GM1 antibodies were detected in 57% of patients, and evidence of B-cell clonality was observed in 35% of cases. Mass spectrometry identified additional IgM paraproteins in 30% of MMN patients and do it more frequently than in disease controls (ALS patients). Anti-GM1 reactivity was present in both IgD⁺ and IgD⁻ memory B-cell subsets, suggesting an early origin of autoreactivity involving both central and early peripheral checkpoint dysfunction, and is not dependent on germinal centre driven somatic hypermutation. Single-cell B-cell receptor sequencing data will soon be available to test this hypothesis. In human iPSC-derived neuromuscular organoids, patient-derived IgM impaired muscle contraction, supporting a direct pathogenic role.
Conclusion: MMN is characterized by a B-cell–mediated autoimmune response with evidence of clonality and GM1 antibody pathogenicity. The latter arise from memory B-cell compartments and bind motor axons while activating complement. These findings provide insight into disease induction and persistence mechanisms and support the development of targeted B-cell therapeutic strategies.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Immune-mediated Neuropathies
OS06.05
Long-Term Exposure to Air Pollution and the Risk and Prognosis of Motor Neuron Disease
Dr. Jing Wu1, Dr. Andrei Pyko1,2, Dr. Charilaos Chourpiliadis1, Dr. Yihan Hu1, Dr. Can Hou1,3,4, Assoc. Prof. Susanna Brauner5,6,7, Prof. Fredrik Piehl5,6,7, Prof. Caroline Ingre5,7, Prof. Fang Fang1
1
Department of Integrative Epidemiology, Institute of Environmental Medicine (IMM), Karolinska Institutet, Solna, Sweden.
2
Center for Occupational and Environmental Medicine, Stockholm Region, Solna, Sweden.
3
Mental Health Center and West China Biomedical Big Data Center, West China Hospital, Sichuan University, Chengdu, China.
4
Med-X Center for Informatics, Sichuan University, Chengdu, China.
5
Department of Clinical Neuroscience, Karolinska Institutet, Solna, Sweden.
6
Neuroimmunology Unit, Center for Molecular Medicine, Karolinska Institutet, Solna, Sweden.
7
Department of Neurology, Karolinska University Hospital, Solna, Sweden
Background: Air pollution exposure has been associated with an increased risk of neurodegenerative diseases; however, evidence is limited for motor neuron disease (MND), especially regarding disease progression. This study aimed to determine if long-term exposure to air pollution is associated with the risk and prognosis of MND.
Methods: This population-based, nested case-control study used Swedish health register data of incident MND cases diagnosed between 2015 and 2023 with up to 8 years of follow-up. Participants included patients with MND, 5 age- and sex-matched population controls without MND per patient with MND, and full siblings of the patients with MND. Mean yearly concentrations of particulate matters of 2.5 μm or less, 10 μm or less, or 2.5 to 10 μm in diameter (PM2.5, PM10, PM2.5-10) and nitrogen dioxide (NO2) were assessed at the residential address using a spatiotemporal model to approximate accumulated air pollution exposure.
Association between air pollution and risk of MND was assessed by comparing cases to both population and sibling controls. Flexible parametric survival models estimated the association between air pollution exposure and the risk of mortality (or use of invasive ventilation) after MND diagnosis (case-only analyses). Based on the rate of decline in the ALS Functional Rating Scale-Revised (ALSFRS-R) score and its subscores after diagnosis, patients were classified into fast (upper 25th percentile) or slow (lower 75th percentile) progression. Logistic regression was used to assess air pollution exposure and the risk of fast progression.
Results: The study included 1463 patients with MND, 7310 population controls, and 1768 sibling controls. The mean (SD) age for all patients with MND was 67.3 (11.7) years, and 814 (55.6) were male. In population comparison, air pollution was associated with an increased risk of MND; per IQR increase in the 10-year average level, odds ratio was 1.21 (95%CI, 1.09-1.34) for PM2.5, 1.30 (95%CI, 1.19-1.42) for PM2.5-10, 1.29 (95%CI, 1.18-1.42) for PM10, and 1.20 (95%CI, 1.12-1.29) for NO2. A higher level of PM10 or NO2 was associated with a higher hazard of mortality, whereas PMs were associated with faster functional decline, particularly motor and respiratory functions.
Conclusion: The findings of this case-control study suggest that air pollution, even at relatively low levels typical of Sweden, may contribute both to the risk of developing MND and disease prognosis after MND diagnosis.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: ALS: Epidemiology and Clinical Features
OS06.06
Rebalancing DNM2 Activity Ameliorates DNM2- and MTMR2-Related Charcot–Marie–Tooth Neuropathies
Dr. Marie Goret, Dr. Thomas Arbogast, Miss Gwenaelle Piccolo, Dr. jocelyn laporte
IGBMC, Inserm U1258, Strasbourg University, Illkirch, France
Background: Charcot–Marie–Tooth disease (CMT) comprises genetically heterogeneous peripheral neuropathies for which no disease-modifying therapy is currently available. Among these, recessive MTMR2 mutations cause CMT4B1, a severe demyelinating neuropathy characterized by focal myelin outfoldings and aberrant mTORC1 signaling in Schwann cells, while dominant loss-of-function mutations in DNM2 lead to intermediate or axonal CMT. Although MTMR2 and DNM2 belong to distinct molecular families, both regulate phosphoinositide-dependent membrane trafficking and cytoskeletal dynamics, suggesting convergence on shared pathogenic pathways.
Methods: Using complementary mouse models, we investigated the contribution of DNM2 dosage and activity to CMT pathophysiology and assessed gene-modulation strategies as therapeutic proof-of-concepts.
Results: In MTMR2-deficient mice, transcriptomic and biochemical analyses revealed a coordinated downregulation of DNM2 and its functional partners in peripheral nerves, concomitant with mTORC1 hyperactivation. Genetic and AAV-mediated upregulation of DNM2, either ubiquitously or selectively in Schwann cells, significantly improved neuromuscular performance, restored mechanical sensitivity, reduced paranodal myelin outfoldings, and normalized mTORC1 signaling in sciatic nerves, establishing DNM2 as a disease modifier in CMT4B1. In parallel, we addressed dominant DNM2-CMT caused by the common K562E mutation. By combining refined disease models that unmask Schwann-cell autonomous pathology with post-symptomatic AAV9-mediated DNM2 supplementation, we show that DNM2-CMT neuropathy primarily arises from early Schwann-cell dysfunction and exhibits limited reversibility once myelination defects are established. These data highlight a narrow temporal and quantitative therapeutic window for DNM2 augmentation. Importantly, genetic rebalancing of DNM2 activity—either by combining loss- and gain-of-function DNM2 alleles or by modulating BIN1, a negative regulator of DNM2—rescues both neuropathic and myopathic phenotypes of a DNM2-CMT mouse model, normalizing nerve architecture and motor function.
Conclusion: Collectively, our results support a unifying model in which precise tuning of DNM2 activity is critical for peripheral nerve homeostasis. They provide strong proof-of-concept that gene-modulation strategies targeting the DNM2 pathway, delivered through transgenesis or AAV vectors, represent a promising and broadly applicable therapeutic avenue for genetically distinct forms of CMT, while emphasizing the need for cell-type specificity, dosage control, and early intervention.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Advances in treatment of Peripheral Neuropathies
OS07.01
Can Amyotrophic Lateral Sclerosis Diagnostic Index (ALSDI) Refine ALS Diagnosis Beyond Gold Coast Criteria?
Dr. Amirul Asyraf Ab Ghapar1,2,3, Dr. Yukiko Tsuji1, Dr. Aicee Dawn Calma1,2, Dr. Nathan Pavey1,2, Dr. Mehdi A.J van den Bos1, Prof. Steve Vucic1,2, Assoc. Prof. Parvathi Menon1,2
1
Brain and Nerve Research Centre, Concord Repatriation General Hospital, NSW Sydney, Australia.
2
Department of Neurology, Concord Repatriation General Hospital, NSW Sydney, Australia.
3
Neurology Subdivision, Ministry of Health Malaysia, Putrajaya, Malaysia
Background: Diagnostic delay in ALS results from delayed recognition, further complicated by phenotypic heterogeneity and comorbidities that mimic ALS in an ageing population. Advances in therapeutic interventions have increased interest in biomarkers that enable early and accurate diagnosis of ALS. The previously described ALS Diagnostic Index (ALSDI) combines cortical and peripheral neurophysiological measures with selected demographic variables and has demonstrated good diagnostic utility in ALS. This study prospectively assessed the diagnostic utility of the ALSDI alongside the current Gold Coast Diagnostic Criteria (GCC).
Methods: Participants with a progressive upper and lower motor neuron (LMN) disorder or pure LMN disorder were prospectively recruited and underwent transcranial magnetic stimulation (TMS) using the paired-pulse threshold tracking technique to record short-interval intracortical inhibition (SICI) averaged over 1–7 ms. Examiners were blinded to participants’ history and investigation results. Clinical, peripheral neurophysiological, and demographic data were collected concurrently to score the GCC and ALSDI. Participants with an established diagnosis at progress evaluation by the census date were included in the study.
Results: One hundred and sixty-nine patients were comprehensively evaluated, with 105 participants having a final diagnosis of ALS and 64 with other neuromuscular disorders (NMD). The GCC demonstrated a diagnostic accuracy of 84% (sensitivity 87.6%, specificity 78.1%), comparable to the ALSDI accuracy of 80.5% (sensitivity 86.7%, specificity 70.3%). Combining both diagnostic indices resulted in a substantial improvement in diagnostic certainty, with excellent specificity of 96.9%, sensitivity 76.5%, and odds ratio (OR) 89.4; 95% CI 19.6–407.9. This combined approach performed equally well in participants with ≤12 months or >12 months of symptoms, in those with limited functional disability (ALSFRS-R >38), and in those with spinal onset disease, who form the majority of ALS and NMD cases (Table 1).
Conclusion: The GCC provides general neurologists with a good diagnostic tool for ALS. This study shows that combining the GCC with the ALSDI significantly increases diagnostic specificity across early and late disease and a range of functional disability, making it a potentially valuable tool for use at ALS centres to confirm ALS diagnosis.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: Biomarkers in MND
Images or Table (Optional)
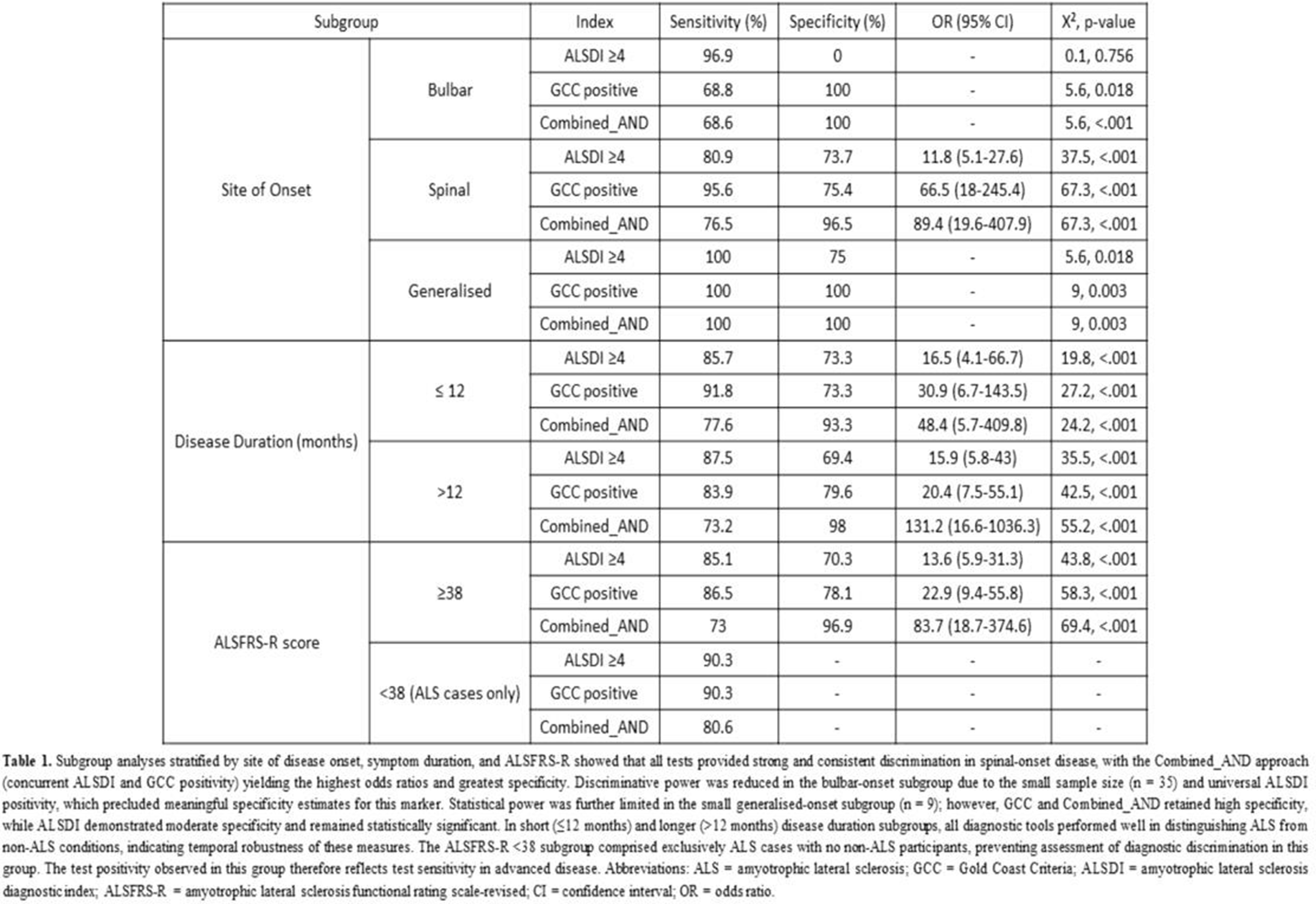
OS07.02
Diagnostic Utility of Ulnar Far Field Potentials in Amyotrophic Lateral Sclerosis
Dr. Aicee Dawn Calma1, Dr. Claudia Santos Silva1,2,3, Dr. Nathan Pavey1, Dr. Yukiko Tsuji1,4, Dr. Mehdi A.J. Van den Bos1, Prof. Michelle Farrar5,6, Assoc. Prof. Parvathi Menon1, Prof. Steve Vucic1
1
Brain and Nerve Research Centre, Concord Clinical School, University of Sydney, Concord Hospital, Sydney, Australia.
2
Department of Neurosciences and Mental Health, Unidade Local de Saúde de Santa Maria, Lisbon, Portugal.
3
Faculdade de Medicina-Instituto de Medicina Molecular, Centro de Estudos Egas Moniz, Universidade de Lisboa, Lisbon, Portugal.
4
Department of Neurology, Graduate School of Medical Science, Kyoto Prefectural University of Medicine, Kyoto, Japan.
5
Department of Neurology, Sydney Children's Hospital Network, Sydney, Australia.
6
Discipline of Paediatrics and Child Health, School of Clinical Medicine, UNSW Medicine and Health, The University of New South Wales, Sydney, Australia
Background: Far field potentials (FFP) are proposed as a neurophysiological biomarker for prognosis in amyotrophic lateral sclerosis (ALS). This study assesses the diagnostic value of ulnar nerve FFP in ALS.
Methods: A thorough peripheral neurophysiological assessment was performed on 106 prospectively recruited individuals with suspected ALS (64 ALS, 42 with ALS-mimicking disorders). The ulnar nerve was stimulated at the wrist, with the motor responses recorded over the abductor digit minimi (ADM) muscle. Measurements included conventional compound muscle action potentials (CMAP), FFP, near-field potential (NFP) amplitudes; and additional neurophysiological indices such as the split-hand index, neurophysiological index, motor unit number estimation (MScanFit-MUNE), and motor unit index (MUNIX). The diagnostic utility of ulnar FFP was analysed using receiver operating characteristic (ROC) curves.
Results: The ALS participants showed significantly lower FFP amplitudes (mean 5.07 ± 0.36 mV) compared to ALS mimics (mean 8.25 ± 0.40 mV, p < 0.001). There was a moderate to strong correlation between FFP amplitude and several neurophysiological biomarkers: CMAP amplitude (ρ = 0.77, p < 0.001), split-hand index (ρ = 0.53, p < 0.001), neurophysiological index (ρ = 0.52, p < 0.001), MUNIX (ρ = 0.69, p < 0.001), and MScanFit-MUNE (ρ = 0.66, p < 0.001). Weak to moderate correlations were also seen with clinical measures including upper limb strength, Amyotrophic Lateral Sclerosis Functional Rating Scale-Revised (ALSFRS-R) scores, and the rate of decline in the fine motor subscore of the ALSFRS-R. ROC curve analysis showed that FFP amplitude effectively differentiated ALS from mimicking disorders (AUC = 0.80, 95% CI: 0.71–0.89), with reliable diagnostic performance across various ALS phenotypes.
Conclusion: The diagnostic performance of FFP amplitude is comparable to current neurophysiological biomarkers used in ALS. It presents as a promising prognostic and diagnostic marker due to its simplicity and reproducibility – enhancing conventional neurophysiological assessments for ALS diagnosis and monitoring.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: Biomarkers in MND
OS07.03
Use of Intravenous Immunoglobulin in Participants with Post-Polio Syndrome: The Randomized, Placebo-Controlled FORCE Trial
Dr. Fran Nollet1,2, Dr. Daria A. Trojan3, Dr. Henning Andersen4, Dr. Johannes Jakobsen5, Dr. Pilar Sáiz6, Dr. Jiri Skopek7, Dr. Susanne Petri8, Dr. Karolina Piasecka–Stryczyńska9, Dr. Laura Bertolasi10, Dr. Konrad Rejdak11, Dr. Sandra Camprubi12, Dr. Beatriz Garcia12, Dr. Dermot Whyms13, Dr. Elsa Mondou14, Dr. Marinos C. Dalakas15
1
Department of Rehabilitation Medicine, Amsterdam UMC Location University of Amsterdam, Amsterdam, Netherlands.
2
Amsterdam Movement Sciences, Rehabilitation and Development, Amsterdam, Netherlands.
3
Department of Neurology and Neurosurgery, Montreal Neurological Institute-Hospital, McGill University Health Centre, McGill University, Montreal, Canada.
4
Århus Universitets Hospital, Aarhus, Denmark.
5
The Neuroscience Centre Rigshospitalet (National Hospital), Copenhagen, Denmark.
6
Hospital de Neurorehabilitación, Badalona, Spain.
7
Fakultní Thomayerova nemocnice, Praha, Czechia.
8
Hannover Medical School, Hannover, Germany.
9
Clinical Research Center Spółka z Ograniczoną Odpowiedzialnością MEDIC-R Sp. K., Poznan, Poland.
10
Azienda Ospedaliera Universitaria Integrata Verona (Ospedale Borgo Roma), Verona, Italy.
11
Department of Neurology, Medical University of Lublin, Lublin, Poland.
12
Grifols, Sant Cugat del Vallès, Spain.
13
Grifols Worldwide Operations, Dublin, Ireland.
14
Grifols, Research Triangle Park, NC, United States.
15
Thomas Jefferson University, Philadelphia, United States
Background: Post-Polio Syndrome (PPS) is a chronic, slowly progressive neuromuscular condition with no specific medical treatments. Inflammatory mechanisms are implicated in its pathophysiology, suggesting potential benefit from immunomodulatory therapies. This study assessed the use of intravenous immunoglobulin (IVIG, Flebogamma® 5% DIF) in participants with PPS.
Methods: This was a Phase II/III multicenter, randomized, double-blind, placebo-controlled trial in adult participants with PPS across Europe, Canada, and the US. Participants received monthly infusions of IVIG (1 g/kg) or placebo for 52 weeks, followed by a 24-week observation period. The primary endpoint was change from baseline in Two-Minute Walking Distance (2MWD) at Week 52. Additional endpoints included endurance measured by Six-Minute Walking Distance (6MWD), muscle strength assessed by Medical Research Council (MRC) scale in two newly weakened muscle groups (with at least one in the lower extremities) and by a dynamometer, walking activity in daily life by a pedometer, fatigue by Fatigue Severity Scale and Borg scale, and pain by Visual Analog Scale.
Results: A total of 149 participants were randomized to IVIG 1 g/kg (n=75) or placebo (n=74). Participants were mainly female (61.7%), White (95.3%), non-Hispanic (88.6%), with a mean age of 63.2 years. IVIG significantly improved 2MWD vs placebo at Week 52 (p=0.0469), meeting the primary endpoint, which was further confirmed in a sensitivity analysis (least squares mean difference [LSMD]: +6.07 m; p=0.0147). Participants in the IVIG group showed an improvement from baseline in 2MWD of 12.8 m vs 6.7 m in the placebo group. A clinically meaningful improvement in 6MWD was observed in the IVIG group vs placebo (LSMD: +15.8 m; p=0.1205); improvement from baseline in IVIG group was more than twice that seen in placebo group (+29.2 m vs +13.4 m). Muscle strength by MRC scale increased in IVIG group and decreased in placebo group (0.58 vs -0.03, p=0.0220), reflecting improvement in the selected muscle groups. No notable differences were observed for the change from baseline between both groups in muscle strength measured by a dynamometer, daily walking activity, fatigue, or pain (Table 1). IVIG 1 g/kg was well tolerated.
Conclusion: Monthly infusions of IVIG demonstrated statistically and clinically meaningful improvements in walking capacity in participants with PPS, indicating IVIG 1 g/kg monthly for one year improves physical performance and endurance. These findings support its potential as a therapeutic option in PPS.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: Post poliomyelitis Syndrome
Images or Table (Optional)
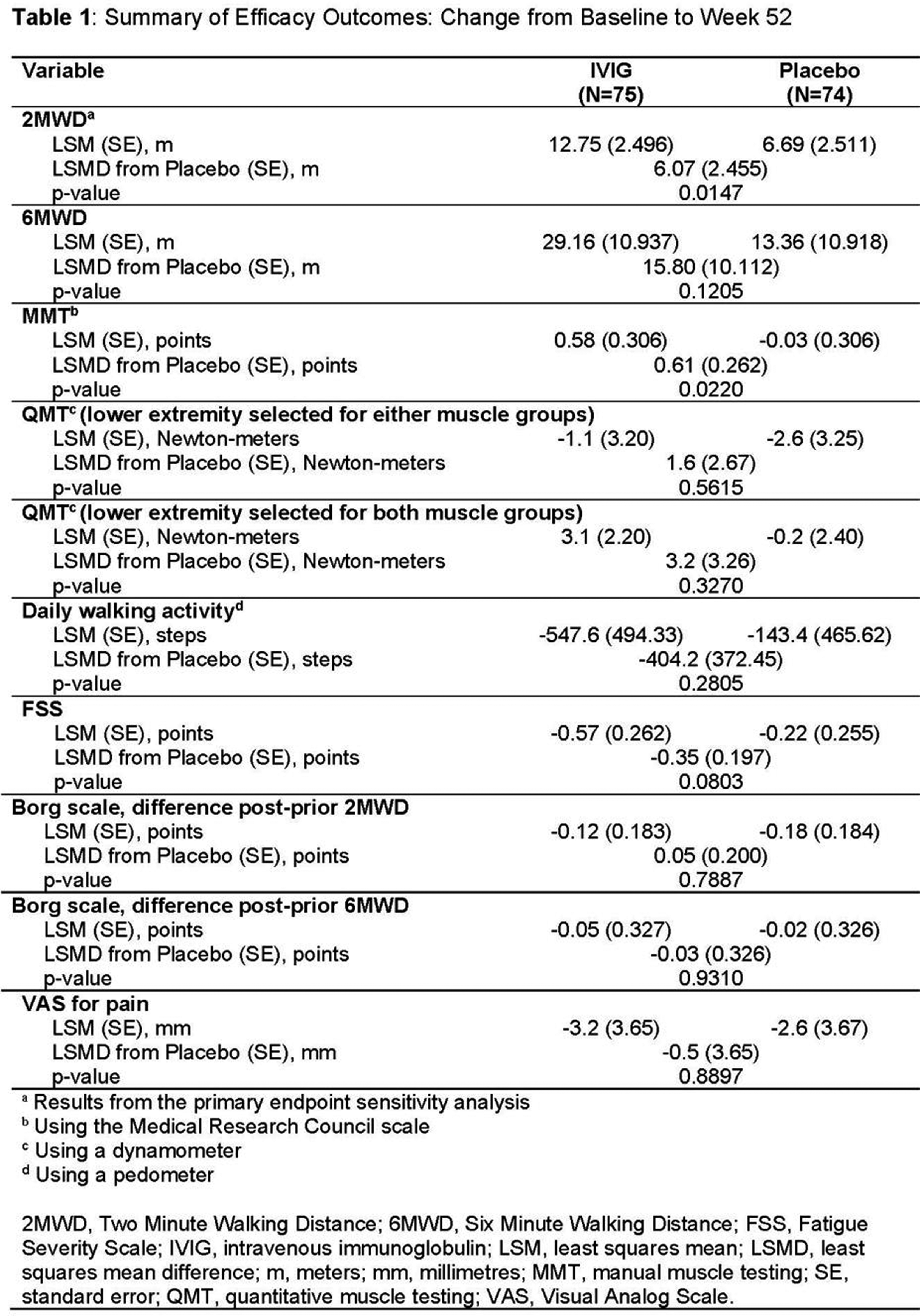
OS07.04
Respiratory Function Improvement in SMA After Long‑Term Oral SMN‑Splicing Modifier Therapy
Dr. Maria Sframeli1, Dr. Noemi Brolatti2, Asst. Prof. Federica Trucco3,4, Dr. Elena La Rosa1, Dr. Rita Lauro1, Dr. Roberto Materia1, Dr. Marisa Maniaci1, Dr. Natalia Longoni1, Dr. Ibrahim Said1, Dr. Cosimo Allegra1, Dr. Marina Pedemonte3, Dr. Claudio Bruno2,4, Prof. Sonia Messina1
1
Department of Clinical and Experimental Medicine, University of Messina, Messina, Italy.
2
Center of Translational and Experimental Myology and Maternal and Child Health, IRCCS Istituto Giannina Gaslini, Genoa, Italy.
3
Pediatric Neurology and Muscular Diseases Unit, IRCCS Istituto Giannina Gaslini, Genoa, Italy.
4
Department of Neuroscience, Rehabilitation, Ophthalmology Genetics, University of Genoa, Genoa, Italy
Background: Risdiplam is an available treatment of all patients with spinal muscular atrophy (SMA). Although the respiratory impairment is inevitably progressive also in later-onset SMA, no studies report the effect of this drug on respiratory function.
Methods: We report a case series of 14 patients, followed at two Italian tertiary referral centers with long-term disease duration and treated with risdiplam for more than 5 years. We describe changes in respiratory and motor function and the need for non-invasive ventilation (NIV).
Results: Patients (mean age: 19,7 years, range: 14-28), 11 SMA2 (78,6%) and 3 SMA3 (21,4 %), were sitter (57,1%) or non-sitter (43,9%). The Hammersmith Functional Motor Scale Expanded (HFMSE) score was ≤ 8 in 13/14 patients, and did not show significative changes after treatment (p=0.9).
Mean FVC at baseline was 1,27 l (SD:1,02). After more than 5 years (mean follow-up 5,3 years) the mean FVC improved significantly (1,41 l; SD: 1,06, p=0.04). Overall 4 patients (28%) showed minimal FVC decrease (-0,12 l; p=0.09), while the other 10 patients (72%) showed a significative improvement in FVC (+0,24; p=0.009).
None of the patients on NIV at baseline (n=8) required changes in ventilation regime at follow-up.
Conclusion: We report longer-term respiratory follow-up in treated patients and, for the first time, an improvement in commonly used pulmonary function tests, in contrast to motor functional outcome measures. Therefore, we propose to explore a high responsiveness of the FVC, mostly in severely affected SMA patients, in which commonly used motor functional outcome measures might not detect changes (PNRR-MR1-2022-12376937).
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: Spinal Muscular Atrophies: Treatment
OS07.05
NBS for 5q SMA in Minas Gerais, Brazil: 18 Months of Program Implementation
Assoc. Prof. Juliana Gurgel Giannetti, Assoc. Prof. Nelio Januario, Mrs Anna Marina, Mrs Lucina Milanez, Mrs Ana Carolina Moura, Ms. Nara Carvalho, Ms. Carolina Assis, Assoc. Prof. Keyla Cunha, Assoc. Prof. Ana Lucia Starling
Universidade Federal de Minas Gerais, BELO HORIZONTE, Brazil
Background: Newborn screening (NBS) for 5q spinal muscular atrophy (SMA) has been implemented in many countries following the availability of disease-modifying therapies (DMTs). However, implementing a statewide NBS program for 5q SMA in Minas Gerais, Brazil was challenging due to its large population of approximately 20 million inhabitants, about 20,000 monthly births, and its extensive territory covering 586,528 km² across 853 municipalities.The aim of this study is to present preliminary data from the first 18 months of the 5q SMA newborn screening program in Minas Gerais, Brazil.
Methods: NBS for 5q SMA was implemented in January/February 2024 within the public health system of Minas Gerais and the data was analyzed until July 2025. Sample collection is performed at primary health care units between the 3rd and 5th day of life. Screening is carried out using RT-PCR, and confirmatory testing is performed by MLPA both performed in a central laboratory at Federal University of Minas Gerais. Following the positive result, the first medical consultation and follow-up are conducted at the Hospital das Clínicas UFMG) including neurological evaluation, motor and bulbar scales (Chop intend, OrSAT), crying vital capacity, and Bayle scale III. The DMT is initiated on the same day of the first consultation.
Results: Over 18 months, 312,816 samples were screened, and 26 patients with 5q SMA were identified, corresponding to an estimated incidence of approximately 1:12,546. Regarding SMN2 copy number, 17 patients had 2 copies, 6 had 3 copies, 2 had 4 copies, and 1 patient had 1 copy. At the first evaluation, all patients with 3 or 4 four copies were asymptomatic and among the 17 patients with 2 SMN2 copies, 6 were classified as asymptomatic, 9 as oligosymptomatic and 2 as symptomatic. DMT was provided to 23 patients with 2 or 3 SMN2 copies, in accordance with national therapeutic protocol. The mean age at treatment initiation was 17.7 days of life, all patients started with Risdiplam. Regarding motor outcomes 11 patients completed 12 to 18 months of treatment (n=11; 8 with 2 SMN2 copies; 3 with 3 with SMN2 copies) and all achieved the ability to sit and crawl. Within this group, among those with 2 SMN2 (n=8), 50% (4/8) achieved independent ambulation and 50% (4/8) walked with support. In contrast, all patients with 3 SMN2 copies (n=3) achieved independent ambulation. Bulbar function was preserved in all treated patients (n=23) with 100% (23/23) maintaining exclusively orally feeding. Concerning respiratory function only one symptomatic patient with 2 SMN2 (1/23) has required NIV which was started at the first consultation.
Conclusion: This is the first newborn screening program for 5q SMA implemented in a large Brazilian state, and it is also the first one in South America. The organization of this program has proven to be successful and sustainable within the public health system, with excellent clinical outcomes, including motor gains, absence of bulbar and respiratory involvement (except for one symptomatic patient with 2 SMN2 copies).
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: Spinal Muscular Atrophies: Treatment
OS07.06
Treatment Switching from an Intrathecal Antisense Therapy to anOral SMN‑Modifying Therapy inSpinal Muscular Atrophy
Dr. Eshgin Maharramov1, Dr. Doruk Arslan1, Prof. Muhammed Kilinc2, Prof. Can Ebru Kurt1, Prof. Sevim Erdem-Ozdamar1, Prof. Ersin Tan1
1
Hacettepe University, Neurology Dep., Ankara, Turkey.
2
Hacettepe University, Physiotherapy and Rehabilitation Faculty, Ankara, Turkey
Background: Nusinersen was the first disease-modifying therapy approved for the treatment of adult patients with spinal muscular atrophy (SMA). In 2025, risdiplam was approved for SMA treatment in Turkey, providing an oral treatment alternative and leading to treatment switching in a subset of patients. The aim of this study is to evaluate the reasons for switching from nusinersen to risdiplam and to describe the clinical and demographic profiles of our adult SMA patients who underwent this transition.
Methods: This retrospective observational study analyzed adult patientswith spinal muscular atrophy (SMA) who were routinelyfollowed at our tertiary care center. Clinical data, treatment history, and functional outcomes were reviewed. Reasons for treatment discontinuation and switching were recorded. Functional status was assessed using the Hammersmith Functional Motor Scale Expanded (HFMSE). Patients who switched from nusinersen to risdiplam and had available follow-up data were included in the final analysis.
Results: A total of 59 patients with SMA were followed at our center. Nine patients were unable to complete the nusinersen loading phase because of administration difficulties and/or changes in treatment center. Additionally, fourteen patients discontinued nusinersen during the maintenance phase due to lack of efficacy, adverse events, or administration-related difficulties.Among the 36 patients who remained on nusinersen treatment, 13 switched to risdiplam, while two patients who had previously discontinued nusinersen subsequently initiatedrisdiplam therapy. Consequently, a total of 15 patients whoinitiated risdiplam were included in the analysis. The mean age was 32.9 (±12.4) years, and the mean age at symptom onset was 7.05 (±5.5) years. The median SMN2 copy number was 3 (2 – 4). Median HFMSE scores were 16 (0 – 62) before initiation of nusinersen and 22 (1 – 64) immediately prior to the initiation of risdiplam therapy. Patients received a median 10 (3 – 16) nusinersen doses before switching to risdiplam. In eight patients, treatment switching was undertaken after a shared decision-making process between the patient and the treating physician due to administration-related difficulties. A non-invasive therapeutic approach was preferred by five patients. Furthermore, two patients who had discontinued nusinersen owing to administration-related challenges opted to initiate risdiplam treatment.
Conclusion: In this real-world cohort, switching from nusinersen to risdiplam was primarily driven by administration-related difficulties. Risdiplam represents a practical alternative for selected SMA patients, particularly those facing challenges with intrathecal therapy. Larger studies with longer follow-up and outcome data are warranted to better define functional outcomes and optimal treatment strategies after therapy switching.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: Spinal Muscular Atrophies: Epidemiology and Clinical Features
OS08.01
Human Spinal Cord Organoids Reveal Therapeutic Windows and Ciliary Pathology in Spinal Muscular Atrophy
Prof. Stefania Corti1,2, Dr. Andrea D' Angelo1, Dr. Francesca Beatrice1, Dr. Chiara Cordiglieri3, Dr. Matteo Miotto4, Prof. Simona Lodato4,5, Dr. Delia Gagliardi2, Prof. Giacomo Comi1,2, Dr. Federica Rizzo2, Prof. Linda Ottoboni1,2
1
University of Milan, Milan, Italy.
2
Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico Milano, Milan, Italy.
3
INGM, Milan, Italy.
4
Humanitas University, Pieve Emanuele, Italy.
5
Humanitas Hospital, Rozzano, Italy
Background: The advent of SMN-restoring treatments has fundamentally changed SMA prognosis, yet clinicians still face uncertainty regarding optimal intervention timing. A deeper grasp of how neurodegeneration unfolds temporally and when neurons remain responsive to therapy is crucial for improving patient outcomes. This study aims to delineate the temporal boundaries of therapeutic efficacy for SMN2-targeting compounds and to uncover novel disease mechanisms in spinal muscular atrophy (SMA) through patient-derived spinal cord organoid models.
Methods: Three-dimensional spinal cord organoids were derived from iPSCs obtained from SMA patients, employing a scaffold-free differentiation approach. Spontaneous electrical activity and network coordination were assessed through multielectrode array (MEA) technology. Transcriptomic profiling via RNA sequencing captured gene expression dynamics at sequential differentiation stages. The risdiplam-like SMN2 splicing modifier RO7021707 was applied at distinct developmental timepoints to characterize the window of therapeutic responsiveness.
Results: SMA organoids displayed marked electrophysiological abnormalities, with diminished spontaneous firing and impaired synchronous network activity relative to healthy controls. Transcriptomic analysis uncovered widespread suppression of genes encoding ciliary structures, including core architectural proteins and transition zone components. This finding points to an unexpected contribution of ciliary dysfunction to cellular signaling impairment and neuronal vulnerability in SMA. RO7021707 administration revealed a distinct pattern of temporal sensitivity: treatment at intermediate differentiation stages yielded maximal SMN protein recovery alongside normalization of network function. Intervention at early stages produced partial functional improvement, whereas treatment at advanced stages conferred limited benefit. These observations establish that a discrete developmental period determines therapeutic responsiveness, with treatment efficacy closely linked to restoration of ciliary gene expression and recovery of coordinated network activity.
Conclusion: These data reveal ciliary pathway disruption as a previously unappreciated element of SMA pathophysiology and establish specific developmental windows during which therapeutic intervention achieves optimal effect. The clinical relevance extends to treatment timing decisions, while the identification of ciliary abnormalities opens new avenues for therapeutic development. This organoid-based platform offers valuable opportunities for individualized treatment strategies in SMA and related motor neuron disorders.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: Spinal Muscular Atrophies: Treatment
OS08.02
Top Stride Velocity (SV95C) is a Validated Endpoint in DMD Below 4 Years Old
Dr. Margaux Poleur1, Mr. Guillaume Parinello2, Dr. Eva Vrščaj3, Ms. Camille Bisson2, Dr. Céline Cluzeau2, Dr. Aurore Daron1, Asst. Prof. Lena Szabo4, Prof. Damjan Osredkar3, Dr. Paul Strijbos5, Mr. Damien Eggenspieler2, Prof. Laurent Servais6,7
1
University department of neurology, Citadelle, Liege, Belgium.
2
Sysnav, Vernon, France.
3
University Children's Hospital, Ljubljana, Slovenia.
4
Pediatric Center, Semmelweis University, Budapest, Hungary.
5
F. Hoffmann-La Roche Ltd., Basel, Switzerland.
6
Neuromuscular Reference Center, Department of Pediatrics, University Hospital Liege & University of Liege, Liege, Belgium.
7
University of Oxford, Oxford, United Kingdom
Background: Early treatment of patients with Duchenne muscular dystrophy (DMD) is considered critical to maximize the effect of disease-modifying treatments. However, traditional clinical outcome measures are challenging to administer to patients younger than 4 years of age, as they have limited ability to understand and follow instructions. Wearable digital health technology (wDHT) enables passive collection of ambulation data in daily living. A digital endpoint, stride velocity 95th centile (SV95C), derived from data collected by the Syde ankle-worn sensor, was qualified as primary endpoint by the European Medicines Agency in ambulant patients with DMD from the age of 4 years. We hypothesized that this sensor could be worn and provide a robust and sensitive measure of ambulatory function in individuals between 1 and 4 years of age.
Methods: Ambulant patients and healthy volunteers below 4 years old were enrolled in an ongoing 3-year longitudinal multicentric study, ActiLiege-Next. Patients were asked to wear two ankle sensors daily for the first 3 months and subsequently for 1 month every 3 months. Controls were instructed to wear the same sensors daily for 1 month every 6 months. Two digital variables were computed: SV95C and number of strides per hour (nb strides/h). Their reliability was assessed by comparing 2 consecutive two-week periods within each recording month using intraclass correlation coefficient (ICC). Ability of the measure to differentiate patients from controls was evaluated using a Mann Whitney U test. Longitudinal changes were assessed using Wilcoxon signed-rank test. Steroids effect was tested by comparing the slope of evolution before and after steroids initiation.
Results: Twenty-eight patients were enrolled (median age [range]: 35 months [16-47]), and twenty-seven healthy boys (32 months [13-48]). Five patients were on steroids before or upon enrolment, and as of August 2025 seven initiated steroids during follow-up. Most participants were adherent to wearing the sensors, with more than 85% recording at least 50 hours of data at baseline, 6, 12 and 18 months. SV95C reliability was excellent, with intraclass correlation coefficients ≥0.97 for patients and ≥0.92 for controls at all time points, whereas reliability of nb strides/h varied between 0.60 and 0.93. Mean baseline SV95C was significantly higher in controls than in patients (p<0.001). Longitudinal analyses showed a greater increase in SV95C among controls than among patients at 12 months, with mean change from baseline of 0.51 m/s for controls (n=17), and 0.24 m/s for patients naive to steroids (n=12), respectively. For the 7 patients initiating steroids during follow-up, median SV95C increase was 0.17 m/s/year before starting steroids and 0.47 m/s/year after starting steroids.
Conclusion: These results suggest that SV95C can differentiate patients from healthy boys and quantify treatment effect even in small cohorts. Together, these findings validate SV95C as a robust measure of ambulation and support its use for evaluating treatment efficacy in clinical trials for all ambulant patients with DMD including those younger than 4 years old. All available data will be presented at the congress.
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: Outcome measures and clinimetry
OS08.03
Molecular Imaging of Muscle Involvement in Facioscapulohumeral Muscular Dystrophy Using Multispectral Optoacoustic Tomography
Dr. Mauro Monforte1,2, Dr. Sara Bortolani2, Dr. Beatrice Ravera1, Dr. Davide Marchese1, Dr. Eleonora Torchia1, Dr. Carmine Di Marco1, Prof. Marco De Spirito1, Prof. Enzo Ricci1,2, Prof. Giorgio Tasca3,4,5
1
Università Cattolica del Sacro Cuore, Rome, Italy.
2
Fondazione Policlinico Universitario A. Gemelli IRCCS, Rome, Italy.
3
John Walton Muscular Dystrophy Research Centre, Newcastle upon Tyne, United Kingdom.
4
Newcastle University Translational and Clinical Research Institute, Newcastle upon Tyne, United Kingdom.
5
Newcastle Hospitals NHS Foundation Trust, Newcastle upon Tyne, United Kingdom
Background: Facioscapulohumeral muscular dystrophy (FSHD), one of the most prevalent adult-onset muscular dystrophies, is characterized by progressive fatty replacement and degeneration of individual muscles in an asynchronous and highly variable manner. Current imaging techniques, such as magnetic resonance imaging (MRI) and ultrasonography (US), have limited ability to assess fibrosis and other disease-related molecular changes within affected muscles. Multi-spectral optoacoustic tomography (MSOT) is an innovative imaging modality that enables non-invasive characterization of muscle molecular composition using pulsed near-infrared laser light. In this study, we aimed to: (1) assess the intra- and inter-rater reproducibility of MSOT measurements; (2) evaluate the ability of MSOT to differentiate FSHD patients from healthy controls (HC); (3) investigate the relationships between MSOT-derived parameters, clinical features, and MRI findings.
Methods: Adult subjects with a genetically confirmed diagnosis of FSHD and healthy controls were recruited. Participants underwent MSOT scanning of predefined upper and lower limb muscles using a standardized protocol. FSHD patients also underwent upper and lower limb muscle MRI examinations performed close to the MSOT assessment, including T1-weighted sequences to evaluate fatty replacement and T2-weighted STIR sequences to assess edema and inflammatory changes, along with comprehensive clinical characterization. Test–retest and inter-rater reproducibility of MSOT measurements were evaluated using intraclass correlation coefficients (ICC). Optoacoustic absorption spectra (wavelength-specific signal peaks expressed in arbitrary units) were compared between FSHD and HC muscles, both globally and at the single-muscle level. Receiver operating curve (ROC) analysis was used to assess the performance of MSOT derived parameters in discriminating FSHD vs HC. Correlations between optoacoustic spectra, MRI features, and clinical measures of muscle strength and disease severity were also examined in patients.
Results: Fifteen FSHD patients and nine healthy controls were enrolled. Intra-rater (same operator, different time) and inter-rater (different operators, different times) analyses both demonstrated excellent agreement (ICC = 0.88; 95% CI: 0.87–0.89). FSHD muscles displayed distinct optoacoustic signatures, characterized by reduced signal intensity at wavelengths below 900 nm and increased signal at 930 nm. MSOT-derived lipid signals effectively discriminated FSHD patients from healthy controls (all muscles AUC = 0.83; 95% CI: 0.66–0.99; p = 0.008). Lipid content was significantly higher in FSHD muscles with severe fatty replacement on MRI (p < 0.0001). MSOT signal of normally appearing FSHD muscles on MRI overlapped with the HC. Muscles positive for T2w-STIR hyperintensity on MRI exhibited higher collagen-related MSOT signals compared with T2w-STIR–negative muscles (p = 0.0012). Oxygenated and total hemoglobin levels were reduced in fatty-replaced muscles (p = 0.006 and p < 0.0001, respectively), suggesting impaired perfusion, whereas deoxygenated and total hemoglobin levels were increased in T2w-STIR–positive muscles (p = 0.04 and p = 0.0013, respectively). MSOT parameters showed significant moderate correlations with clinical measures of disease severity and muscle strength.
Conclusion: MSOT is a reproducible non-invasive imaging technique capable of capturing disease-relevant molecular features in FSHD muscles, including lipid accumulation, alterations in muscle perfusion and collagen remodeling. MSOT appears a promising complementary tool to conventional imaging with potential applications in the identification of biomarkers of disease activity and progression.
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: Other Biomarkers
OS08.04
High Diagnostic Yield in a Large Monocentric Cohort of Patients Affected With Primary Muscle Disorders
Dr. Francesca Magri1, Dr. Daniele Velardo1, Dr. Delia Gagliardi2, Dr. Elena Abati3, Dr. Roberto Del Bo2, Dr. Daniela Piga3, Dr. Sabrina Lucchiari2, Dr. Serena Pagliarani3, Dr. Nicola Molitierno3, Dr. Claudia Alberti2, Dr. Mosè Parisi3, Dr. Ilaria Bestetti4, Dr. Monica Sciacco1, Prof. Palma Finelli4, Prof. Stefania Corti2,1, Prof. Giacomo Comi2,3, Prof. Dario Ronchi2,3
1
IRCCS Foundation Ca' Granda Ospedale Maggiore Policlinico, Neuromuscular and Rare Disease Unit, Milan, Italy.
2
Dino Ferrari Center, Department of Pathophysiology and Transplantation, University of Milan, Milan, Italy.
3
IRCCS Foundation Ca' Granda Ospedale Maggiore Policlinico, Neurology Unit, Milan, Italy.
4
IRCCS Foundation Ca' Granda Ospedale Maggiore Policlinico, Medical Genetics Laboratory, Milan, Italy
Background: Primary muscle disorders are clinically and genetically heterogeneous. The achievement of a reliable molecular diagnosis can improve clinical management, provides appropriate genetic counseling and testing of relatives, and enables potential therapeutic options. We reviewed the molecular findings obtained by using diagnostic next generation sequencing protocols at Dino Ferrari Center in Milan.
Methods: In the last 6 years, 285 patients with the suspect of primary muscle disease underwent NGS-based molecular testing consisting of i) custom targeted gene panels (n=70) or ii) exome sequencing (n=215) followed by diagnostic interpretation of variants in genes associated with matched clinical phenotypes.
Results: Overall diagnostic yield was 59%. Disease-causing variants were identified in 110 patients (38%: definitive diagnosis). Likely pathogenic variants and/or variants of uncertain significance were found in 59 additional probands (21%: probable diagnosis).
Diagnostic yield of the two approaches was similar (58.1% versus 58.5%). This rate tends to increase in patients affected with muscular dystrophies (71%) and congenital myopathies (66%) while it falls to 45% in patients with a suspect of metabolic myopathy. The simultaneous analysis of two affected members from the same family facilitated the establishment of a diagnosis (100%). More than 50% of patients who had previously performed a muscle biopsy obtained a positive genetic report. Congenital myopathies related to heterozygous RYR1 defect with or without susceptibility to malignant hyperthermia was the most recurrent clinical entity followed by recessive limb girdle muscular dystrophies due to CAPN3 and DYSF pathogenetic variants and heterogenous clinical presentations linked with heterozygous CAPN3 defects. The variants included in positive genetic reports identified >200 different molecular defects in 59 different genes. Half of them only were previously reported in patients and already classified as pathogenetic. Clinical exome sequencing allowed the analysis of additional OMIM genes leading to unexpected diagnosis or the enlargement of the clinical spectrum of known disease-related genes. Full exome sequencing contributed to identify 3 novel disease genes (GUK1, POPDC2, TNNT1) in the last years.
Conclusion: The results of our cohort study emphasize the value of an integrated, multi-faceted approach to the diagnosis of neuromuscular diseases, combining molecular genetics, muscle biopsy, functional studies, and advanced molecular techniques. This approach not only enhances diagnostic accuracy but also provides a framework for the discovery of novel molecular defects and a better understanding of the complex genetic architecture of neuromuscular diseases.
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: Genetics in Neuromuscular Diseases including Biochemical/Molecular Techniques and next generation sequencing.
OS08.05
Determining Autoimmunity in Seronegative Myasthenic Syndromes: CBA and Human Muscle Cell Assay Results in D-a-CH
Dr. Paolo Doksani1, Miss Marlene Wolfsgruber2, Prof. Fritz Zimprich2, Dr. Hakan Cetin2, Dr. Martin Krenn2, Asst. Prof. Romana Höftberger2, Asst. Prof. Lukas Weigl3, Dr. Meret Herdick1, Dr. Amani Suboh1, Miss Carla Dusemund1, Prof. Andreas Meisel1, Prof. Sarah Hoffmann1, Dr. Inga Koneczny2
1
Charité - Universitätsmedizin Berlin, corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Department of Neurology with Experimental Neurology, Neuroscience Clinical Research Center (NCRC) and Integrated Myasthenia Gravis Center, Berlin, Germany.
2
Department of Neurology, Division of Neuropathology and Neurochemistry, Medical University of Vienna, Vienna, Austria.
3
Department of Anaesthesia, Intensive Care Medicine and Pain Medicine, Medical University of Vienna, Vienna, Austria
Background: Seronegative myasthenia gravis (SNMG) represents a clinical challenge, as approximately 10–15% of MG patients remain antibody-negative on routine testing despite clinically typical myasthenic features. This diagnostic gap is associated with diagnostic uncertainty, delayed diagnosis, and limited access to targeted immunotherapies. Cell-based assays (CBAs) have demonstrated increased diagnostic yield. However, a substantial proportion of SNMG patients remains antibody-negative even with extended CBA testing. In such cases, antibody binding to NMJ-associated structures may provide evidence of autoimmunity and increase diagnostic confidence, even without antigen identification. The MYA-DACH study assesses the prevalence of autoimmune signals in SNMG patients from the D-A-CH region (Germany, Austria, Switzerland) using live (L-CBA) and fixed (F-CBA) cell-based assays for clustered AChR- and MuSK-antibody detection, complemented by a human muscle cell-based assay for autoimmunity screening.
Methods: MYA-DACH is a multicenter cross-sectional study testing samples from adult and pediatric patients with clinically characteristic myasthenic syndromes and negative routine antibody testing (AChR; MuSK and LRP4 where available). Samples were collected between November 2023 and February 2026. Using L-CBA and F-CBA, we assessed the prevalence of autoantibodies against clustered AChR and MuSK. To assess autoimmunity beyond known antigen targets, persistently seronegative sera in L-CBA testing were screened for antibody binding using an NMJ model based on primary human skeletal muscle cells with agrin-induced AChR clustering. This interim analysis focuses on baseline characteristics of 275 and antibody prevalence of 466 out of 500 collected samples. Recruitment and sample collection are complete, while final antibody analyses and comprehensive clinical correlations are ongoing, with updated results to be presented at ICNMD.
Results: As of Dezember 2025, 466 serum samples were collected. The cohort is predominantly female (69.8%) with a mean age of 51.7 years. Mean age at disease onset was 44.6 years, with a mean diagnostic delay of 2.7 years. Most patients had generalized MG (76.9%), while 23.1% presented with purely ocular symptoms. Diagnostic features frequently supported autoimmune MG, including positive repetitive nerve stimulation (34.3%), abnormal single-fiber EMG (26%), and a positive oral pyridostigmine test (85.2%). L-CBA detected antibodies against clustered AChR in 4,78% (22/466) and MuSK antibodies in 1.52% (6/466) of previously seronegative classified patients. Of the L-CBA–positive samples, 86.4% (19/22) of AChR-positive sera were confirmed by fixed CBA, whereas none of the MuSK-positive samples (0/5) showed reactivity, indicating a higher sensitivity of the L-CBA. Among samples remaining negative on L-CBA, 6.3% (24/382) demonstrated antibody binding to NMJ-associated structures on human myotubes.
Conclusion: The interim results from the MYA-DACH study demonstrate the prevalence of detectable AChR or MuSK autoantibodies identified by live and fixed cell-based assays in seronegative myasthenia gravis is relatively low compared with published international cohorts, but consistent with previous German data. Beyond antigen-specific antibodies, additional NMJ-associated antibody binding on human muscle cells supports autoimmune involvement in a subset of patients. Ongoing analyses will address clinical correlations with specific assay results, comparing patients with and without detectable signals for autoimmunity.
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: Other Biomarkers
Images or Table (Optional)
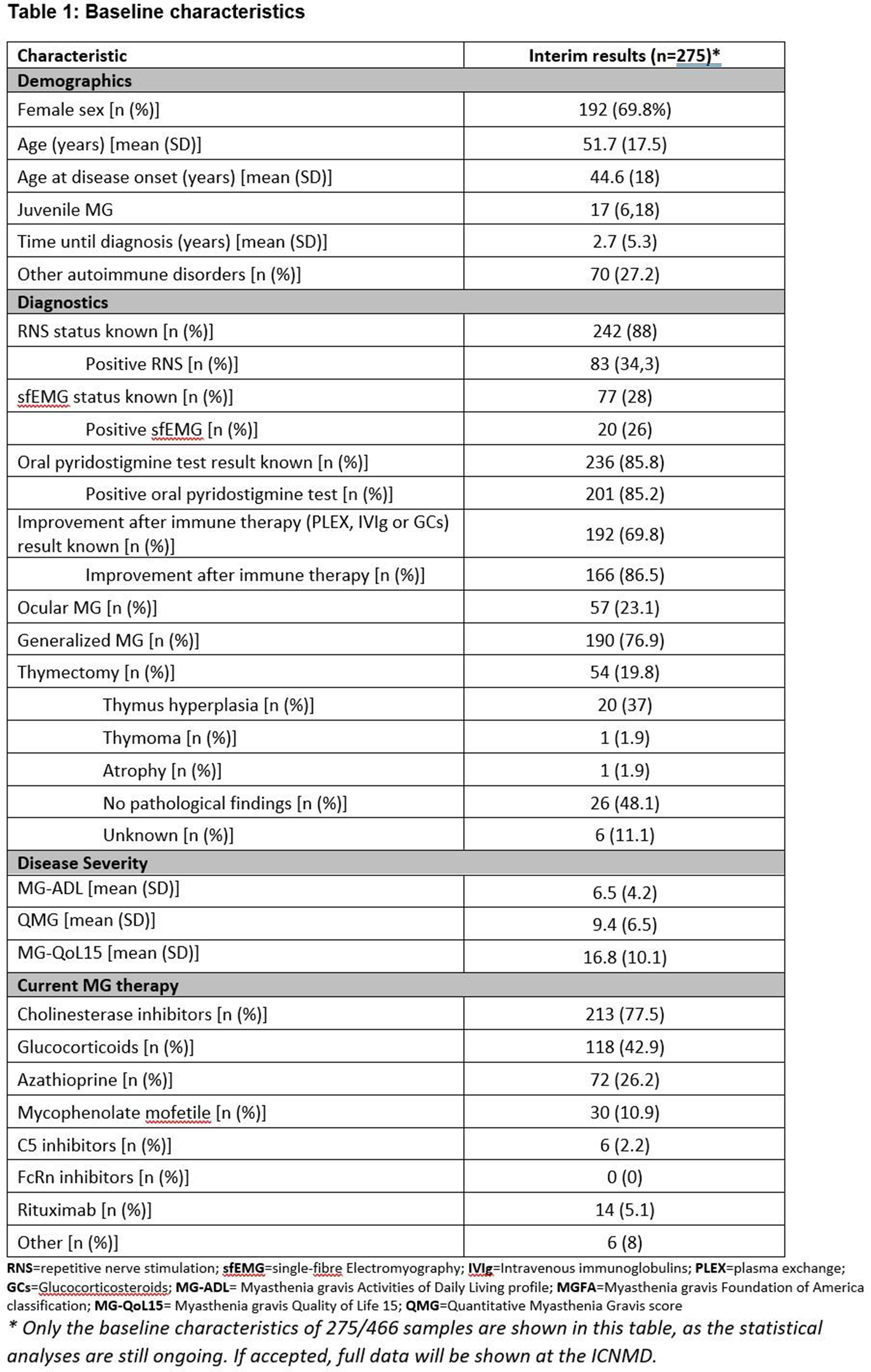
OS08.06
One-Year Longitudinal Quantitative Whole-Body Muscle MRI and Functional Outcomes in Adults With FSHD1
Mr. Matthias Opsomer1,2, Dr. Lotte Huysmans3,4, Dr. Kobe Bamps5,6, Dr. Ronald Peeters7, Dr. Veerle Goosens7, Prof. Frederik Maes3,4, Prof. Patrick Dupont8, Prof. Kristl G. Claeys1,2
1
Laboratory for Muscle Diseases and Neuropathies, Department of Neurosciences, KU Leuven, and Leuven Brain Institute (LBI), Leuven, Belgium.
2
Department of Neurology, University Hospitals Leuven, Leuven, Belgium.
3
Department ESAT/PSI, KU Leuven, Leuven, Belgium.
4
Medical Imaging Research Center, University Hospitals Leuven, Leuven, Belgium.
5
Department of Cardiovascular Sciences, KU Leuven, Leuven, Belgium.
6
Department of Cardiology, University Hospitals Leuven, Leuven, Leuven, Belgium.
7
Department of Radiology, University Hospitals Leuven, Leuven, Belgium.
8
Laboratory for Cognitive Neurology, Department of Neurosciences, KU Leuven, and Leuven Brain Institute (LBI), Leuven, Belgium
Background: Facioscapulohumeral muscular dystrophy type 1 (FSHD1) is a slowly progressive muscle disorder caused by contraction of the D4Z4 repeat on chromosome 4q35. Identifying sensitive and robust longitudinal outcome measures is essential for monitoring disease progression and clinical trial readiness.
Methods: Adults with genetically confirmed FSHD1 were assessed at baseline and after 12 months. We performed whole-body muscle MRI (T1-weighted, 6-point Dixon, and Short Tau Inversion Recovery (STIR)), with AI-driven automated whole-muscle segmentation, to quantify proton density fat fraction (PDFF) across leg, axial, and shoulder muscles. Clinical outcome measures (COMs) were the Vignos and Brooke scales, FSHD clinical severity score (Ricci Score), FSHD clinical score, FSHD composite outcome measure (FSHD-COM), Motor Function Measure 32 (MFM32), 6-Minute Walk Test (6MWT), Timed Up and Go (TUG), 30-Second Sit-To-Stand (30STS), grip, key pinch, tip pinch, Biodex® isometric lower limb strength testing. Patient-reported outcome measures (PROMs) comprised FSHD Rasch-built overall disability scale (FSHD-RODS), ActivLim, Individualized Neuromuscular Quality of Life (INQoL), the Fatigue Severity Scale (FSS), and Brief Pain Inventory (BPI).
Results: Of the 60 patients included, 57 completed 12-month follow-up assessment (mean age 52.1±15.7 years, 55.0% female, mean number of D4Z4 repeats 7.2±1.8). Quantitative MRI revealed progressive fat infiltration, with mean PDFF increasing by 0.8% and 0.3% in proximal and distal leg muscles respectively, most pronounced in the musculus biceps femoris brevis and longus, semimembranosus and gluteus minimus. A significant decline was observed in the MFM32 total score (-1.7%, p<0.001), driven by the MFM32-D1 (standing and transfers) subscore (-3.1%, p<0.001). Clinical worsening over one year was further confirmed by the FSHD-COM total score (+1.22 points; p=0.002), and the FSHD clinical score (+0.22; p=0.01). Functional measures such as the 6MWT (-1.5m; p = 0.76) and the TUG (p=0.80) remained stable. Similarly, strength testing based on Biodex®, grip, key pinch, and tip pinch strength did not show significant differences over 1 year. However, when analyzing the strongest side at baseline, a significant 1-year decline was observed in knee extension torque (-6.56Nm; p=0.008), hand grip strength (-2.07kg; p=0.0002), and key pinch strength (-0.48kg; p = 0.017), which were not significant at the weakest side. PROMs revealed a measurable worsening by a significant decrease in the FSHD-RODS (-2.02; p=0.02) and a downward trend in ActivLim (-1.06; p=0.05), while the INQoL score (p=0.67), FSS (p=0.31) and BPI (p=0.49) remained stable.
Analysis of PDFF of upper limbs and paraspinal muscles, and analysis for STIR images is ongoing and results will be presented at the congress.
Conclusion: This 12-month longitudinal study demonstrates that PDFF fat fraction of biceps femoris brevis and longus, semimembranosus and gluteus minimus muscles, MFM32 (specifically the D1 subscore), FSHD-RODS and strongest-side isometric strength provided to be sensitive markers for disease progression in FSHD1. These findings highlight the potential of these measures as sensitive, targeted endpoints for upcoming 12-month clinical trials in FSHD1.
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: Outcome measures and clinimetry
OS09.01
Enhancing the Diagnosis of Myasthenia Gravis Using Objective Oculomotor Assessment
Dr. Fatemeh Rezania1,2, Mr. Murray Worner1, Dr. Lauren Ross1,2, Ms. Linda Seiderer1, Ms. Natasha Willems1, Assoc. Prof. Patrick Waters3, Mr. Jake Canning3, Dr. Ezgi Bakircioglu-Duman3, Assoc. Prof. David Szmulewicz4,2, Assoc. Prof. Leslie Roberts1,2
1
St Vincent's Hospital, Melbourne, Australia.
2
The University of Melbourne, Melbourne, Australia.
3
University of Oxford, Oxford, United Kingdom.
4
The Royal Victorian Eye and Ear Hospital, Melbourne, Australia
Background: The prevalence of myasthenia gravis (MG) is increasing, and delayed diagnosis is associated with substantial morbidity. Early ocular involvement is common, so we investigated whether optokinetic nystagmography (OKN) could improve diagnostic accuracy.
Methods: We recruited 31 individuals from the Neurophysiology Department at St Vincent’s Hospital, Melbourne, presenting with ptosis and/or diplopia. Diagnostic assessment comprised single-fibre electromyography (SFEMG) of the orbicularis oculi, repetitive nerve stimulation (RNS) of the nerve to the nasalis muscle, ice-pack testing when ptosis was present, and serological testing using radioimmunoprecipitation assay (RIPA) and live cell-based assays (LCBA).
Definite MG was identified in 12/31 participants based on positive serology and/or electrophysiology, while 4/31 were classified as probable MG based on a clear therapeutic response to acetylcholinesterase inhibitor therapy +/- corticosteroids despite negative serological and electrophysiological testing. The remaining 15 participants were determined to have alternative diagnoses.
All participants underwent OKN testing using binocular videonystagmography (VNG), recording eye-movement velocities during a 60-second visual tracking task.
Results: Of the 16 patients with MG, 9 (56%) were positive for acetylcholine receptor (AChR) antibodies using RIPA, while 10 (63%) were positive using LCBA. Combined RIPA and LCBA testing identified AChR antibodies in 11 of 16 MG patients (69%).
SFEMG was abnormal in 57% (8/14) of patients with MG and normal in those without. Facial RNS was abnormal in 27% (4/15) of MG patients and none of those without. Notably, all individuals with abnormal neurophysiological findings were seropositive, except for one patient who was SFEMG-positive and seronegative. In participants with ptosis, ice-pack testing was positive in 40% (6/15) with MG compared with 9% (1/11) without.
OKN yielded interpretable recordings in 12/16 with MG and 10/15 without; remaining recordings were uninterpretable due to excessive blinking, anatomical factors, severe ophthalmoplegia, or difficulty complying with the task requirements. OKN testing demonstrated significantly lower velocities and greater beat-to-beat variability in MG subjects (P<0.05). Decrement in OKN response - calculated as the percentage change between maximum and minimum OKN velocities - was significantly greater in MG than in non-MG participants for both eyes (P<0.001). ROC analysis showed 92% sensitivity and 89% specificity for the left eye at a 77.5% threshold and 100% sensitivity and 89% specificity for the right eye at a 72% threshold. Using the mean decrement of both eyes yielded 100% sensitivity and 90% specificity at a 76% cut-off (P < 0.0001). Subgroup analysis demonstrated similar thresholds for definite (75.8%) and probable MG (76.6%), with identical sensitivity (100%) and specificity (90%).
Conclusion: In this small cohort, combining LCBA with RIPA improved sensitivity for AChR antibody detection from 56% to 69% in 16 patients with MG with ocular manifestations. OKN metrics demonstrated promising diagnostic performance, with 100% sensitivity and 90% specificity, although the interpretable recording rate was 71% due to participant-related factors. Single-fibre EMG showed lower sensitivity than expected, likely reflecting the small sample size. Overall, these results highlight the potential utility of OKN-based measures as a non-invasive adjunct in the evaluation and diagnosis of MG.
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: Other Biomarkers
OS09.02
Decoding the Mechanisms of Muscle Wasting for Developing a Rna-Based Multitarget Therapeutic Approach
Prof. Marco Sandri1,2, Dr. Camilla Pezzini1, Assoc. Prof. Roberta Sartori1
1
University of Padova, Padova, Italy.
2
Veneto Institute of Molecular Medicine, Padova, Italy
Background: Muscle wasting is a debilitating process that occurs in many diseases, and its onset detrimentally affects patients drug treatment, quality of life and clinical outcome. The decrease of muscle mass is consequent to a net loss of cytoplasm due to an increased turnover of contractile proteins and organelles. Among the different proteolytic systems, ubiquitin-proteasome is the main one that removes sarcomeric proteins while autophagy-lysosome controls organelles turnover. Enhancement of protein breakdown requires transcriptional dependent programs that induce the expression of a subset of genes, the atrophy-genes or atrogenes, which encode for rate limiting enzymes not only of the degradation systems (ubiquitin proteasome, autophagy-lysosome) but also of the Integrated Stress Response (IRS) related to the unfolded protein response and translation regulation. The atrophy-genes also control other cellular processes such as mitochondrial bioenergetics and dynamics, calcium homeostasis, chromatin structure and DNA damage response. Several transcription factors such as FoxOs, NFkB, STAT3, Smad2/3 have been described to regulate atrogenes expression and muscle loss during catabolic conditions and their inhibition has been proposed as therapeutic option to preserve muscle mass and strength. Despite several attempts to develop therapeutic approaches, muscle wasting is still orphan of any specific drug/treatment mainly because our understanding of the insights of this process are still largely unknown. By combining cutting edge technologies, mouse models, patients’ biopsies and organoids we dissected the mechanisms that alter the gene signature to induce an anti-neurotrophic, pro-catabolic and pro-inflammatory environment inside muscle tissue in a gender and disease specific manner. The identification of this catabolic gene network enables us to test a novel patient- and disease-specific RNA-based therapeutic approach to preserve muscle mass and prevent muscle loss.
Methods: We have performed a multiomic approach, in which snRNA seq and ATAC seq (Multiome) have be simultaneously analyzed, to determine the transcriptional and chromatin signatures of the different cell types in muscles both from rodent models and muscle biopsies of patients. Sections of the same muscles have been also processed for spatial transcriptomic. By cross-referencing the data-sets we have identified the TFs network and the changes of cellular landscape and cytokines-receptors. We have developed myotropic AAV9 vectors expressing shRNAs ad as well as vivo-Morpholinos ASO against the most critical pro-catabolic factors/TFs and tested both in murine models and in patient's derived neuromuscular organoids (NMOs).
Results: We have used tumor bearing mice and cancer patients as first catabolic condition. Our findings depicted an unexpected scenario in which almost all the cell types activated a catabolic signature and synergized to promote a pro-catabolic environment. Data analyses identified that FoxOs binding sites were enriched in the open promoter regions of differentially expressed genes in cachectic muscles. We also found that the muscle-specific ubiquitin ligase MuRF1 was upregulated in all the muscle cells. The simultaneous inhibition of FoxO1, 3 and MuRF1 resulted in anabolic and pro-neurotrophic action in mice and human NMOs.
Conclusion: These findings pave the way for an innovative RNA multi-target therapy that matches with the concept of precision medicine and could be applied to many muscle wasting diseases.
Abstract Topic Groups (Submission Categories)
Topic Group 6 - Basic Sciences in Neuromuscular Diseases: Muscle Atrophy / Degeneration
OS09.03
Characterization of AChR-Specific CD4+ T Cell Responses in Myasthenia Gravis with HLA-DRB Risk Alleles
Dr. Mingming Li1, Dr. Hiroshi Kubo2, Prof. Fredrik Piehl1,3,4, Dr. Naoshi Fukushima2, Assoc. Prof. Susanna Brauner1,4, Assoc. Prof. Nicolas Ruffin1
1
Department of Clinical Neuroscience, Karolinska Institutet, Stockholm, Sweden.
2
Denka Co., Ltd., Denka Innovation Center, Tokyo, Japan.
3
Center for Neurology, Academic Specialist Center, Stockholm, Sweden.
4
Department of Neurology, Karolinska University Hospital, Stockholm, Sweden
Background: Myasthenia gravis (MG) is an archetypal autoantibody-mediated disease, with a well-established genetic susceptibility HLA class II locus. Specific alleles, such as DRB1*03:01 and DRB1*15:01, exhibit strong associations with disease, suggesting a central role for AChR-specific CD4+ T cell-HLA interactions in the pathogenesis. However, the role of these disease-associated HLA alleles in the initiation and regulation of AChR specific T-cell responses remains incompletely characterized.
Methods: By screening healthy donor peripheral blood mononuclear cells (PBMC) carrying MG HLA-class II risk alleles, seven AChR alpha 1 subunit derived peptides (#1-7) were identified using the MHC-Associated Peptide Proteomics (MAPPs) methodology. Subsequently, 16 MG patients (DRB1*03:01+DRB3*01:01, n=5; DRB1*15:01, n=5; triple carrier, n=1; triple-negative, n=5) and 7 HLA-matched healthy controls (HCs: DRB1*03:01+DRB3*01:01, n=3; DRB1*15:01, n=3; triple carrier, n=1) were recruited. Ex vivo immunophenotyping was performed, and PBMC were subsequently stimulated with individual peptides and expanded in vitro. On day 7 of culture, cells were restimulated with peptide and subsequently phenotyped and sorted for activated IFN-γ+ CD4+ T cells, for future RNA seqencing. In parallel, supernatants were analyzed by multiplex cytokine analysis. Clinical data, including treatment status, were collected and correlated to experimental outcomes.
Results: Higher peptide-specific CD4+ T cell responses were observed in MG patients compared to HLA-matched controls, with highest responses in untreated patients (p<0.0001). Among the tested peptides, peptide #7 elicited in the most robust immunoactivation, particularly in patients carrying the DRB1*15:01 allele. The frequency of peptide #7-induced IFN-γ+ CD4+ T cells correlated strongly with IFN-γ measured in the supernatants (R²=0.75, p<0.000). Analysis of supernatants revealed that, despite low absolute concentrations, IL-17A and IL-10 appeared largely co-expressed with IFN-γ. Notably, patients receiving B-cell depleting therapy (n=8) exhibited profoundly suppressed AChR-specific T-cell responses compared to untreated patients (p<0.0001), with levels comparable to healthy donors, suggesting a significant immunomodulatory effect of this treatment.
Conclusion: This study provides ex vivo evidence that AChR-derived peptides are targets of CD4+ T cells in MG. Among the screened peptides, one peptide emerged as the most immunogenic epitope, particularly in patients carrying the DRB1*15:01 allele. Furthermore, B-cell depletion therapy seems to dampen these AChR-specific CD4+ T cell response. These findings establish a foundation for further investigation into antigen-specific T cells in the context of MG pathogenesis and possible therapeutic targets.
Abstract Topic Groups (Submission Categories)
Topic Group 6 - Basic Sciences in Neuromuscular Diseases: Immune Mechanisms in Neuromuscular Diseases
OS09.04
Diagnostic Support for Myotonic Dystrophy Type 1 (DM1, ORPHA:273) Using German Real-World Data
Assoc. Prof. Jana Zschuentzsch1, Mrs Elisabeth Nyougui1, Prof. Richard Röttger2, Miss Miriam Hübner3, Mr. Marco Schaarschmidt3, Mrs Romina Blasini4, Dr. Josef Schepers1
1
University medical center, Göttingen, Germany.
2
Syddansk Universitet, Odense, Denmark.
3
Charité – University medical Center, Berlin, Germany.
4
U, Gießen, Germany
Background: Myotonic dystrophy type 1 (DM1) exhibits striking clinical heterogeneity, often leading to delayed diagnosis. Early identification is critical, as timely management of cardiac and endocrine complications can improve both prognosis and quality of life. The advent of causal and disease‑modifying therapeutic trials in DM1 highlights the need for precise phenotyping and early disease staging to enable effective trial readiness and improved treatment.
Objectives: This study investigates the potential of real‑world data (RWD) from the German University Data Integration Centers (DIC) to support early DM1 detection and clinical trial preparedness. Specifically, we aim to: (1) define phenotype‑specific and laboratory‑based profiles in genetically confirmed DM1; (2) assess the diagnostic performance (positive predictive value, PPV) of a laboratory constellation comprising elevated CK, AST, ALT, and HbA1c with decreased IgG and TSH; (3) evaluate whether characteristic clinical features—such as frontal baldness, gallstones, early cataract, or insulin‑resistance‑related complications—enhance diagnostic accuracy; and (4) explore the feasibility of identifying undiagnosed DM1 cases through machine‑learning models applied to federated RWD.
Methods: As DM1 currently lacks a disease‑specific ICD‑10 code in Germany, feasibility analyses were conducted via the German Portal for Medical Research Data (FDPG), which provides federated access to the University DIC, using ORPHA coding - for DM1 this is ORPHA:273 or five different codes for subtypes. Between June and July 2025, confirmed DM1 cases rose from 50 across four DIC sites to 130 across six. Given an estimated prevalence of 1–5 per 10,000, approximately 3,000 DM1 cases are expected nationwide. For comparison, 5,460 individuals are currently listed under ICD‑10 G71.1 (“Myotonic disorders”) in the FDPG environment. Ongoing feasibility checks track the progressive rollout of ORPHA coding, with a comprehensive analytic query scheduled once ≥ 500 DM1 cases are confirmed. Planned analyses will extend to additional ICD‑10 groups and laboratory parameters, applying bell‑curve modeling to detect subgroup deviations. Supervised machine‑learning algorithms, optimized via the Youden index, will be used to define predictive thresholds and to identify laboratory‑clinical signatures characteristic of DM1.
Results: As of January 2026, the FDPG dashboard records 220 individuals with DM1 (Orpha: 273) across 11 sites. Preliminary analyses confirm the technical feasibility of detecting distinctive laboratory and clinical patterns consistent with DM1 within federated hospital RWD across DIC sites.
Conclusion: DM1 is a progressive, multisystemic disorder often obscured in routine care by non‑specific manifestations. Although no single biomarker is diagnostic, a composite laboratory signature—elevated CK, AST, ALT, HbA1c with reduced IgG and TSH— and integrated with neuromuscular or cardiac comorbidities appears sufficiently high predictive to warrant further diagnostics. Applying supervised machine‑learning approaches to such RWD‑derived patterns may enable digital diagnostic support and optimize trial readiness. This integrative framework could be extended to other rare neuromuscular diseases, facilitating earlier diagnosis and stratified recruitment for disease‑modifying therapies.
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: Outcome measures and clinimetry
OS09.05
a Potential Provocation: Adjusting Frontal Area Temperature How Alter Jitter in Myasthenics and Healthy Individuals
Dr. Selen Ozyurt Kose
Health Science University Umraniye Teaching and Research Hospital, Istanbul, Turkey
Background: Single-fiber electromyography (SFEMG) is the most sensitive electrophysiological method for detecting neuromuscular transmission abnormalities evaluating jitter and blocked potentials. Although temperature influences synaptic transmission, its effects on SFEMG parameters in healthy and diseased muscle are not fully defined. This study aims to investigate the impact of local temperature changes on neuromuscular transmission in patients with myasthenia gravis and healthy controls, with the ultimate goal of improving SFEMG interpretation and understanding neuromuscular junction physiology.
Methods: The study included 30 adult participants referred to our clinical neurophysiology laboratory for single-fiber electromyography (SFEMG) to investigate suspected neuromuscular junction disorders. Based on diagnostic outcomes, participants were divided into two equal groups: 15 patients with confirmed myasthenia gravis (MG) and 15 individuals in whom MG was excluded. Sample size was determined using ANOVA calculations. MG diagnosis was established according to clinical findings, response to anticholinesterase therapy, and supportive electrophysiological and/or serological evidence. Participants younger than 18 years and those with peripheral neuropathy, myopathy, diabetes mellitus, thyroid disease, or conditions or medications affecting neuromuscular transmission were excluded.
SFEMG recordings were obtained from the frontalis muscle. Local skin temperature was continuously monitored using a digital surface thermometer. Measurements were performed sequentially under three temperature conditions: normothermia (∼36.6 °C), mild cooling (∼32 °C) induced by an ice pack, and mild heating (∼40 °C) induced by a heat pack after return to baseline temperature. Seven potential pairs were recorded per condition, yielding 21 fiber pairs for per participant. Minimum, maximum and mean jitter values, blocked potentials, and abnormal jitter pair numbers were calculated and compared between groups and temperature conditions.
Results: A significant main effect of temperature (Greenhouse–Geisser–corrected p = 0.023) and a significant group × temperature interaction were observed (p < 0.001) at repeated measures ANOVA. Post-hoc analyses showed that group differences were greatest at 40 °C (p < 0.001) and remained significant at baseline (p < 0.001), but were attenuated during cooling. In healthy controls, mean jitter showed minimal change across temperature conditions, indicating relative stability, whereas heating markedly increased jitter in MG. Maximum jitter increased with heating in MG but remained stable in controls. A significant differance did not been observed in min jitter values. In MG-group, blocked potential pairs were rare at baseline and absent during cooling. A significant increase in blocked pairs was observed during heating compared with baseline (Wilcoxon signed-rank test, p = 0.038). Increased jitter pairs were rare at baseline and during cooling, precluding meaningful pairwise comparisons, while heating showed a non-significant tendency toward higher counts compared with baseline (p = 0.21).
Conclusion: This study demonstrates that neuromuscular transmission in MG is strongly influenced by local temperature changes. Heating markedly destabilizes the neuromuscular junction, leading to increased jitter and blocking, whereas cooling exerts a stabilizing effect. In contrast, neuromuscular transmission in healthy individuals remains largely temperature resistant. These findings highlight temperature as an important physiological modulator of safety factor at the neuromuscular junction and suggest that temperature control may enhance the diagnostic sensitivity and interpretation of SFEMG in myasthenia gravis.
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: Electrophysiology
OS09.06
The Uptake, Intracellular Trafficking and Recycling of FcRn-Blocking Therapeutics in Human Endothelial Cells in Vitro
Ms. Julija Sirina1, Dr. Lena D’Hooghe2, Dr. Laura Singhvi-Hanns2, Dr. Suzanne Cole2, Dr. Rocio Lledó-Garcia2, Dr. Mar Ribera Armengol2, Dr. Anthony Shock2, Dr. Nicholas Holliday1, Dr. Leigh Stoddart1
1
Excellerate Bioscience, Nottingham, United Kingdom.
2
UCB, Slough, United Kingdom
Background: The neonatal Fc receptor (FcRn) recycles immunoglobulin G (IgG) in cells and is responsible for the long half-life of IgG relative to other plasma proteins. FcRn also recycles pathogenic IgG autoantibodies and is the target for several new targeted therapeutic agents in IgG autoantibody-driven disorders. Several anti-FcRn agents have been approved for use in patients with generalized myasthenia gravis (gMG). These therapeutics include rozanolixizumab, a high-affinity monoclonal antibody (mAb) that directly blocks the IgG binding site on FcRn in a pH-independent manner.
Methods: This study explored the impact of these molecular characteristics on rozanolixizumab’s cellular uptake, endosomal trafficking and recycling. Another anti-FcRn therapeutic, the Fc fragment MST-HN IgG Fc (an analog of efgartigimod), a competitive antagonist with weaker affinity and pH-dependent binding to FcRn, was included for comparison. Using high-content imaging methods in human umbilical vein endothelial cells (HUVECs), a time- and concentration-dependent uptake of fluorescently-labelled rozanolixizumab into intracellular compartments was observed.
Results: Uptake was rapid, not pH-dependent (pEC50 8.38 ± 0.01 and8.11 ± 0.09 at pH 6.0 and pH 7.4, respectively), and competed out with unlabelled inhibitor, supporting a receptor-mediated mechanism. Conversely, uptake of MST-HN IgG Fc was slower and required higher concentrations to detect uptake (pEC50 <6) which was pH-dependent, not competed out with unlabelled inhibitor, and occurred with similar potency in cells that did not express FcRn, suggesting a receptor-independent mechanism such as passive fluid phase pinocytosis. Using Rab proteins associated with different endosomal compartments, FcRn inhibitors appeared to traffic through recycling compartments in a similar manner in HUVECs, and their return to the cell surface in FcRn-transfected cells occurred with similar kinetics.
Conclusion: These data demonstrate the impact of different structural features of FcRn inhibitors on functional outcomes on cells in vitro. Further studies are needed to understand the direct clinical implications of such data in gMG and other IgG autoantibody-driven disorders. However, these different characteristics may in part drive the observed pharmacodynamic differences in gMG patients, that result in different requirements in terms of dosing. Efgartigimod requires 10mg/kg IV (e.g. 760mg in a 76kg patient) to reach overall IgG reductions of 62% compared to rozanolixizumab that results in a greater reduction of 73% at a dose of 560 mg SC (e.g. in a 76kg patient) (gMG PKPD model simulations). Efgartigimod therefore requires 3.8-fold more moles compared to rozanolixizumab to keep the FcRn receptor occupied and reach clinically relevant levels of IgG decrease.
Abstract Topic Groups (Submission Categories)
Topic Group 6 - Basic Sciences in Neuromuscular Diseases: Neuromuscular Junction
PP01.01
Investigating Intravenous Efgartigimod in Juvenile Generalized Myasthenia Gravis: Results from the ADAPT JR Study
Dr. Sithara Ramdas1,2, Dr. Abigail N. Schwaede3, Dr. Nancy L. Kuntz3, Dr. Anna Bogatyreva4, Dr. Juliette Giacobbe4, Dr. Flavia Menezes4, Dr. Lan Lan4, Dr. Jan Noukens5, Dr. Tonke van Bragt5, Dr. Anna Kostera-Pruszczyk6, Dr. Erik H. Niks7
1
Department of Paediatric Neurology, John Radcliffe Hospital, Oxford, United Kingdom.
2
MDUK Oxford Neuromuscular Centre, Department of Paediatrics, University of Oxford, Oxford, United Kingdom.
3
Division of Neurology, Department of Pediatrics, Ann & Robert H. Lurie Children’s Hospital of Chicago, Northwestern University Feinberg School of Medicine, Chicago, United States.
4
argenx, Ghent, Belgium.
5
Curare Consulting BV, Liempde, Netherlands.
6
Department of Neurology, Medical University of Warsaw, Warsaw, Poland.
7
Department of Neurology, Leiden University Medical Center, Leiden, Netherlands
Background: Efgartigimod is a human immunoglobulin G1 (IgG1) antibody Fc fragment that blocks the neonatal Fc receptor. Previous Phase 3 trials (ADAPT/ADAPT+) demonstrated that intravenous (IV) efgartigimod is efficacious and well tolerated in adults with generalized myasthenia gravis (gMG). There is a recognized unmet need for treatments in patients with juvenile gMG. Here, we present interim results from an ongoing Phase 2/3 clinical trial assessing efgartigimod IV in juvenile participants with anti-acetylcholine receptor antibody–positive (AChR-Ab+) gMG (ADAPT JR; NCT04833894), aiming to confirm an age-adjusted dose of efgartigimod IV in a cohort of adolescent participants (aged 12-17 years).
Methods: ADAPT JR will ultimately recruit ≥12 patients with AChR-Ab+ gMG aged 2-17 years, using a staggered design, starting with the adolescent cohort (aged 12-17 years). This study comprises a single-dose confirmatory Part A (≥8 weeks) and a multiple-dose treatment-response confirmatory Part B (≥18 weeks). In Part B, efgartigimod IV was administered in cycles of 4 once-weekly infusions, and participants were retreated as needed per principal investigator discretion.
Results: As of February 2025, 11 adolescent participants were enrolled. Efgartigimod IV administration resulted in decreases of both total IgG and AChR-Ab levels. In Part B, mean (SE) changes from cycle baseline for Myasthenia Gravis Activities of Daily Living (MG-ADL) scores at Week 4 during Cycles 1 and 2 were −3.5 (0.94) and −4.0 (1.34) respectively. Minimal symptom expression (MSE; MG-ADL of 0-1) was achieved by 72.7% (n=8/11) of participants in Cycle 1 and 80.0% (n=4/5) of participants in Cycle 2. Mean (SE) Quality of Life in Neurological Disorders (Neuro-QoL) score changes from cycle baseline at Week 4 were −9.0 (3.19) and −10.0 (3.51) during Cycles 1 and 2, respectively. Efgartigimod IV was well tolerated and the most common adverse events reported by ≥3 participants in Parts A and/or B were headache (n=7/11) and oropharyngeal pain (n=3/11).
Conclusion: In adolescent participants, treatment with efgartigimod was well tolerated and resulted in decreased levels of both total IgG and AChR-Ab levels, similar to those observed in adults during ADAPT/ADAPT+. Additionally, improvements in total MG-ADL and Neuro-QoL scores were observed, with many participants achieving MSE. Efgartigimod IV may address an unmet need for the treatment of gMG in juvenile patients. Enrollment for the child cohort (aged 2-11 years) is ongoing.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Advances in the treatment of muscle diseases
PP01.02
Specific Myostatin Inhibition Ameliorates Sarcopenia and Extends Lifespan in α-Klotho-Null Mice
Assoc. Prof. Yutaka Ohsawa1, Prof. Shin-ichiro Nishimatsu1, Assoc. Prof. Masahiro Fujino2, Prof. Yoshihide Sunada1, Prof. Masahito Mihara1
1
Kawasaki Medical School, 577 Matsushima, Kurashiki, Okayama 701-0192, Japan.
2
Kyushu Women’s Junior College, Jiyugaoka 1‑1, Yahata Nishi, Kitakyushu, Fukuoka 807-858, Japan
Background: The longevity protein α-Klotho is a critical determinant of aging; its deficiency precipitates a shortened lifespan and diverse senescent phenotypes, including sarcopenia. We previously demonstrated that circulating α-Klotho functions as an endogenous inhibitor of transforming growth factor (TGF)-β superfamily members, such as myostatin, GDF11, activin, and TGF-β1 (Ohsawa, Am J Pathol 193, 2023). While a small-molecule compound suppressing these TGF-βs restored sarcopenia in α-Klotho (-/-), its clinical application is limited by potential cardiovascular risks.
Methods: In this study, we investigated whether targeted inhibition of a single ligand, myostatin, is sufficient to rescue the senescent phenotypes in the α-Klotho (-/-) mice. By crossing the α-Klotho (-/-) mice with the transgenic mice overexpressing myostatin prodomain [MSTNPro (+/-) Tg], we achieved ligand-specific suppression of myostatin activity.
Results: The resulting α-Klotho (-/-)/MSTNPro (+/-) Tg mice exhibited significantly increased muscle mass, enhanced grip strength, and greater absolute contractile force. At the molecular level, myostatin blockade attenuated Smad2 phosphorylation and downregulated the senescence marker Cdkn1a (p21) expression in skeletal muscle. Notably, myostatin inhibition significantly extended the median survival of the mice (15.2±2.2 vs. 10.5±1.9 weeks; n=17, P<0.05).
Conclusion: These results indicate that specific myostatin blockade not only alleviates sarcopenia but also promotes longevity in a systemic senescence model, thereby providing safe therapeutic potential of myostatin-targeted interventions for age-related disorders.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Advances in the treatment of muscle diseases
Images or Table (Optional)
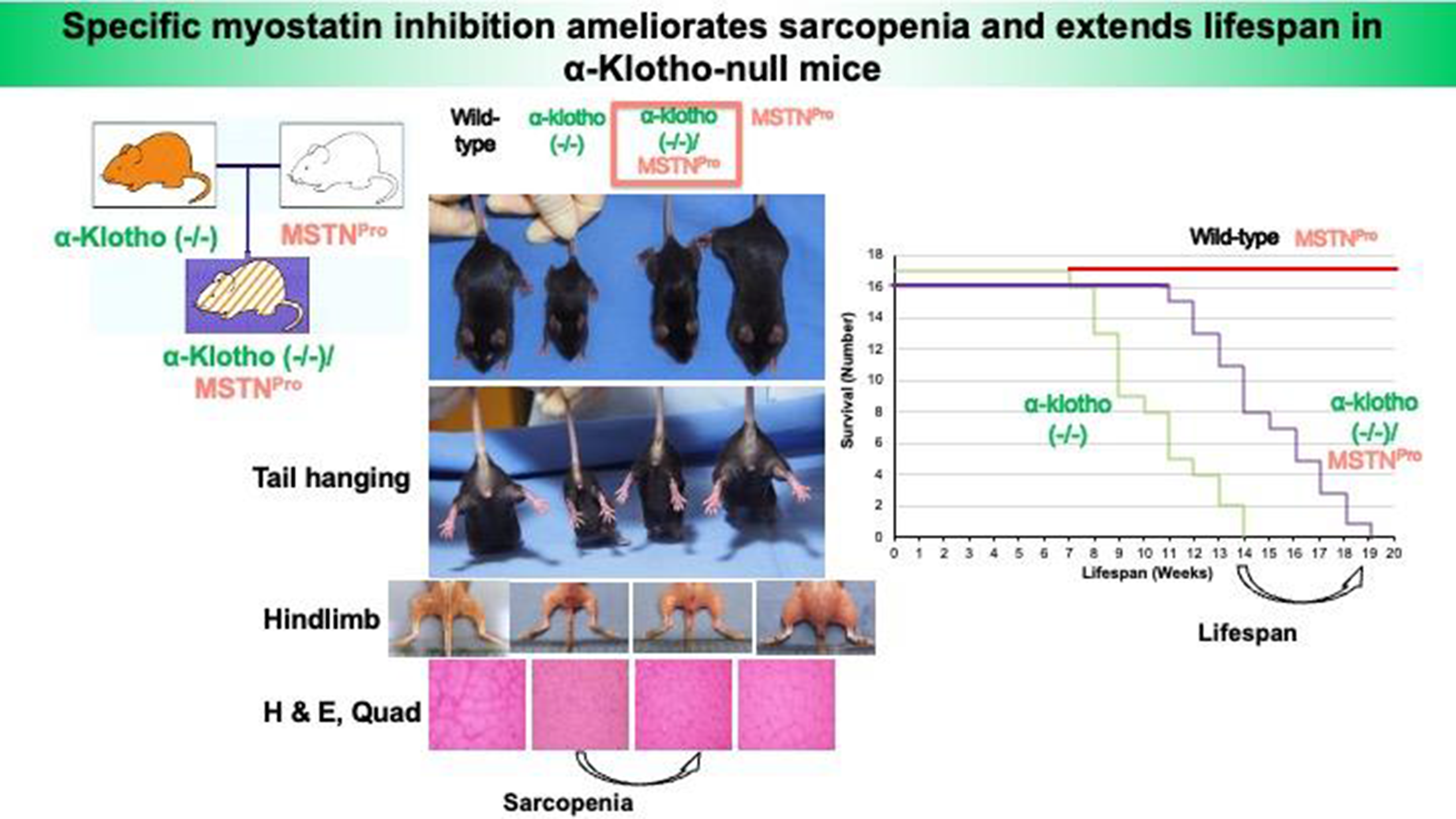
PP01.03
Clinical Correlates of Scoliosis in Spinal Muscular Atrophy Treated with Disease-Modifying Therapies: A Pilot-Observational Study
Dr. Nesaiba Ait Allali1, Dr. Barbara Risi1,2, Dr. Simona Damioli1, Dr. Giorgia Giovanelli1, Dr. Ester Franchi1, Dr. Giulia Gilberti1,3, Dr. Annamaria Sorgente1, Dr. Giorgia Garletti1, Dr. Filippo Nicolis1, Dr. Mattia Puzzi1, Dr. Francesca Garofali1, Dr. Roberto Carugati1, Dr. Samuele Pianto1, Dr. Isabella Urbano1, Dr. Chiara Colombi1, Dr. Elisa Ottelli1, Dr. Elisabetta Ferrari1, Prof. Alessandro Padovani3,4, Prof. Massimiliano Filosto1,3,4
1
NeMO-Brescia Clinical Center for Neuromuscular Diseases, Brescia, Italy, Brescia, Italy.
2
Department of Molecular and Translational Medicine, University of Brescia, Brescia, Italy, Brescia, Italy.
3
Department of Clinical and Experimental Sciences, University of Brescia, Brescia, Italy, Brescia, Italy.
4
Unit of Neurology, ERN EURO-NMD Center ASST Spedali Civili, Brescia, Italy, Brescia, Italy
Background: Spinal muscular atrophy (SMA) is a hereditary neuromuscular disorder characterized by progressive weakness and atrophy of axial and appendicular muscles. Disease-modifying therapies (DMTs) have substantially modified the clinical course of SMA, improving survival and motor outcomes. Nonetheless, scoliosis—a three-dimensional spinal deformity—remains frequently observed and may influence motor, respiratory, and daily functioning. Data on the characteristics and clinical correlates of scoliosis in treated patients are still limited. The object is to descriptively examine the onset and progression of scoliosis in patients with SMA receiving DMTs and to explore its associations with functional outcomes.
Methods: Nine patients with SMA (type 1, n = 3; type 3, n = 6; mean age 17.6 ± 13.1 years) followed at the NeMO–Brescia Center were included. All participants were receiving DMTs at baseline (T0). Scoliosis severity was assessed using Cobb angle measurements. Motor function was evaluated with CHOP-INTEND, HFMSE, RULM, and MFM-32, while respiratory function was assessed by forced vital capacity (FVC). Quality of life was evaluated using the SMA Independence Scale–Clinical Report (SMAIS-CR) in non-ambulant patients and the SF-36 in ambulant patients. Associations between Cobb angle and clinical measures were explored using Spearman’s rank correlation at baseline and after 1 year of follow-up (T1).
Results: At baseline, scoliosis was observed in 78% of patients (n = 7). Patients with SMA type 1 tended to present with more severe spinal curves (mean Cobb angle at T0: 45.0° ± 26.0°) and greater variability in progression (mean Cobb angle at T1: 48.8° ± 47.7°), with one patient undergoing a stabilization intervention. Patients with SMA type 3 generally showed milder curves (mean Cobb angle at T0: 18.5° ± 4.0°) and limited changes over time (mean Cobb angle at T1: 19.3° ± 5.1°). At T0, a negative association was observed between Cobb angle and RULM score (r = −0.832, p = 0.040). At T1, Cobb angle showed associations with FVC expressed in liters (r = 0.900, p = 0.037) and with SMAIS-CR (r = −0.900, p = 0.037). These associations should be interpreted cautiously, as increases in absolute FVC values may reflect growth or treatment-related effects rather than changes in respiratory function per se.
Conclusion: In this small, exploratory cohort, scoliosis was frequently observed in patients with SMA receiving DMTs and appeared to co-occur with measures of motor performance and daily functioning. Although these findings are preliminary and descriptive in nature, they underscore the importance of continued spinal monitoring in treated SMA populations. Larger longitudinal studies are needed to better characterize scoliosis trajectories and their clinical relevance in the context of disease-modifying therapies.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: Spinal Muscular Atrophies: Epidemiology and Clinical Features,
Images or Table (Optional)
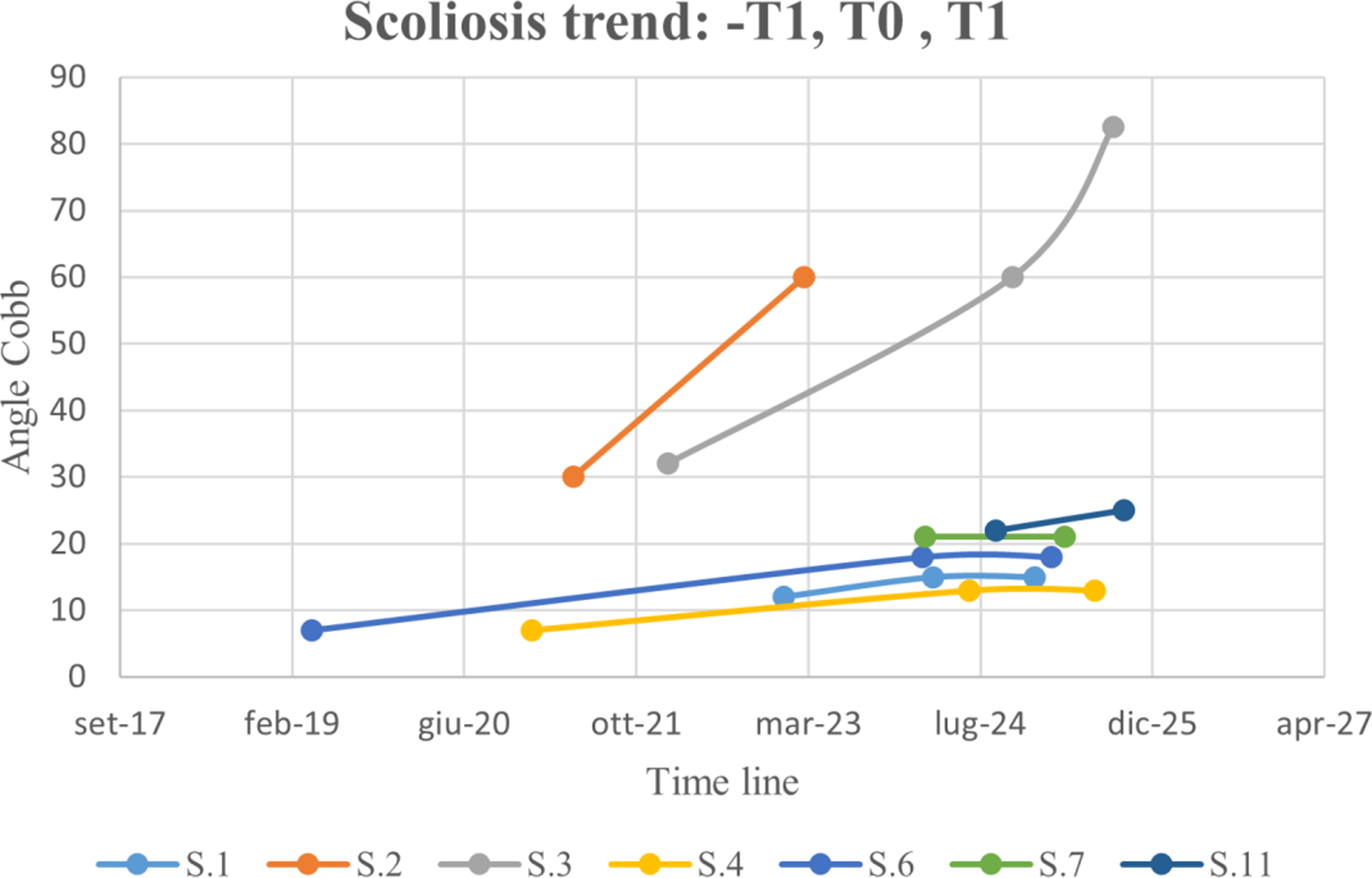
PP01.04
Diagnostic Challenges in Autosomal Dominant Multiple Acyl-CoA Dehydrogenase Deficiency: A Case Report
Dr. Barbara Risi1,2, Dr. Lucia Ferullo1, Dr. Filomena Caria1, Dr. Enrica Bertella1, Dr. Nicola Carapella3, Dr. Gaetana Lanzi4, Dr. Mattia Bugatti4, Dr. Carla Baronchelli4, Dr. Giorgia Giovanelli1, Dr. Loris Poli5, Prof. Alessandro Padovani5,6, Prof. Massimiliano Filosto1,5,6
1
NeMO-Brescia Clinical Center for Neuromuscular Diseases, Brescia, Italy.
2
Department of Molecular and Translational Medicine, University of Brescia, Brescia, Italy.
3
Department of Medical and Surgical Specialties, Radiological Sciences and Public Health, University of Brescia, ASST Spedali Civili, Brescia, Italy.
4
Unit of Pathological Anatomy, ASST Spedali Civili, Brescia, Italy.
5
Unit of Neurology, ERN Euro NMD Center ASST Spedali Civili, Brescia, Italy.
6
Department of Clinical and Experimental Sciences, University of Brescia, Brescia, Italy
Background: Multiple acyl-CoA dehydrogenase deficiency (MADD) is classically inherited in an autosomal recessive manner and is caused by defects in electron transfer flavoprotein (ETF), resulting in impaired mitochondrial fatty acid oxidation. However, rare autosomal dominant forms of MADD have been increasingly recognized. These dominant cases show variable penetrance and typically present later in life with heterogeneous clinical manifestations, including episodic exercise intolerance, myopathy, and acute metabolic decompensation.
Methods: We report the case of a 33-year-old woman who presented with a subacute onset of myalgia affecting the thighs and buttocks, exacerbated by voluntary movement and walking. Clinical, genetic and instrumental data were collected.
Results: Her past medical history was unremarkable, except for systemic leukocytoclastic vasculitis at 2 years of age and hyperhomocysteinemia. She was initially hospitalized at another center, where a diagnosis of possible post-infectious inflammatory myopathy was made based on diffuse myopathic changes on electromyography. At that time, creatine kinase (CK) levels peaked at 1,096 U/L, and myositis-associated autoantibodies, including anti-HMGCR antibodies, were negative. She was subsequently referred to our center.
Neurological examination revealed a mild waddling gait and difficulty rising from a squatting position; the patient also reported muscle pain after walking approximately 100–150 meters. A quadriceps femoris muscle biopsy demonstrated extensive vacuolization. Muscle MRI showed symmetrical muscle edema involving the gluteal muscles, with milder edema affecting the paraspinal, adductor, thigh, and calf muscles—particularly the posterior tibialis and soleus—as well as moderate atrophy of the gluteal and posterior thigh muscles. Clinical exome sequencing identified variants of uncertain significance (VUS) in three genes (LPIN1, ENO3, SGCA) not consistent with the clinical phenotype.
Over time, the patient’s symptoms progressively worsened, leading to moderate proximal (Medical Research Council grade 3/5), more than distal, weakness in all four limbs, inability to walk, distal paresthesias in the extremities, and absent deep tendon reflexes in the lower limbs. She was re-hospitalized with suspected acute inflammatory polyradiculoneuritis, metabolic acidosis with increased anion gap, and rhabdomyolysis. Subsequent analysis of urinary organic acids and plasma acylcarnitines revealed a biochemical profile consistent with MADD. A targeted reanalysis of the genetic data identified the c.1752G>C variant (VUS) in the ETFDH gene. Further mRNA analysis from muscle tissue did not reveal aberrant splicing, making the presence of a second pathogenic intronic variant unlikely.
The patient was started on riboflavin supplementation (300 mg/day) and showed gradual clinical improvement over approximately three months, with near-complete recovery. Follow-up muscle MRI demonstrated resolution of the edematous changes and improvement in thigh muscle trophism.
Conclusion: Recognition of autosomal dominant MADD broadens the phenotypic and genetic spectrum of the disease and has important implications for diagnosis, genetic counseling, and management. Notably, many dominant cases are highly responsive to riboflavin supplementation. Increased awareness of this inheritance pattern is crucial to prevent misdiagnosis and to enable timely and effective treatment.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Metabolic Myopathies: Glycogen, Lipids and Mitochondrial Myopathies
PP01.05
Late-Onset Acute Respiratory Failure Revealing SELENON-Related Myopathy: A Case Report
Dr. Barbara Risi1,2, Dr. Filomena Caria1, Dr. Simona Damioli1, Dr. Beatrice Labella3,4, Dr. Enrica Bertella1, Dr. Gaetana Lanzi5, Dr. Mattia Bugatti5, Dr. Carla Baronchelli5, Dr. Giorgia Giovanelli1, Dr. Lucia Ferullo3,4, Dr. Emanuele Oliveri3,4, Dr. Loris Poli3, Prof. Alessandro Padovani3,4, Prof. Massimiliano Filosto1,3,4
1
NeMO-Brescia Clinical Center for Neuromuscular Diseases, Brescia, Italy.
2
Department of Molecular and Translational Medicine, University of Brescia, Brescia, Italy.
3
Unit of Neurology, ERN Euro NMD Center ASST Spedali Civili, Brescia, Italy.
4
Department of Clinical and Experimental Sciences, University of Brescia, Brescia, Italy.
5
Unit of Pathological Anatomy, ASST Spedali Civili, Brescia, Italy
Background: SELENON-related myopathy (SELENON-RM) is a rare autosomal recessive congenital neuromuscular disorder caused by pathogenic variants in the SELENON gene, which encodes selenoprotein N. Disease onset usually occurs in early childhood, with axial muscle weakness, spinal rigidity, and early respiratory involvement representing characteristic clinical features (Villar-Quiles RN et al. 2020).
Methods: We describe a 44-year-old woman who required endotracheal intubation for acute respiratory failure and was successfully weaned from invasive mechanical ventilation after six months. Clinical, instrumental and genetic data were collected.
Results: Her past medical history was unremarkable; however, a detailed retrospective evaluation revealed long-standing mild motor impairment since childhood, including slow running, difficulty climbing high steps, early muscle fatigability, and reduced endurance. Neurological examination showed a waddling gait and axial and proximal limb weakness, in the absence of spinal rigidity. Muscle biopsy of the right quadriceps demonstrated non-specific myopathic changes. Clinical exome sequencing identified two heterozygous variants, c.713dupA and c.803G>A, in the SELENON gene.
Conclusion: This case underscores the marked clinical heterogeneity of SELENON-related myopathy. The absence of rigid spine and of typical core pathology on muscle biopsy should not preclude consideration of this diagnosis. Moreover, our findings emphasize that severe respiratory failure may represent a late-onset manifestation of SELENON-RM, even in middle-aged patients (Risi B et al. 2025).
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Congenital Myopathies and congenital muscular dystrophies
PP01.06
Congenital Myasthenic Syndrome: 3 Different Phenotypes and Genotypes in 8 Moroccan Families
Prof. Birouk Nazha1, Assoc. Prof. Tamaoui Leila1, Prof. Bouhouche Ahmed2
1
Department of Neurophysiology, hospital of specialities, university hospital Ibn Sina, Mohamed V university, Rabat, Morocco.
2
Laboratory of Neurogenetics, hospital of specialities, university hospital Ibn Sina, Mohamed V university, Rabat, Morocco
Background: Congenital myasthenic syndrome is due to mutations in the genes encoding the proteins of neuromuscular junction either in post-, pre-, or intra-synaptic components. The clinical presentation is usually in favour of myasthenia with ophtalmoparesis and muscle weakness exacerbated by exercise but pseudo-myopathic presentation is possible. Age at onset is usually congenital with possibly congenital respiratory distress and delayed motor millstones. Some patient can experience first symptoms later on and possibly at adult age. The aim of this study is to analyse clinical, electrophysiological, genetic and therapeutic aspects in a cohort of 13 cases belonging to 8 Moroccan Families.
Methods: All patients underwent clinical evaluation for neonatal hypotonia, respiratory and/or feeding issues at birth, ptosis and ophtalmoparesis at birth, motor millstones development and main clinical symptoms (distribution of muscle weakness in limbs, bulbar, facial and ocular muscles), functional disability and orthopaedic deformations. All patients had nerve conduction study and 3 Hz repetitive nerve stimulation for NMJ abnormalities. Genetic study consisted of exome analysis. Patients received symptomatic treatment and were followed up during 1 to 24 years.
Results: 13 patients belonging to 8 families were studied. All parents were 1st degree consanguineous except in one family. 3 families had more than one affected member. Onset of the disease was congenital in 9 cases, under 3 years in 3 cases and adult age in 1 case. We identified 3 different genotypes and phenotypes.
5 families (8 patients) had homozygous mutation in epsilon subunit of acetylcholine receptor (CHRNE) with 3 different and rare pathogenic variants identified. The p.Asn452GlufsTer4 mutation was the most frequent and concerned 3 different families. The p.Gly355AlafsTer30 mutation was diagnosed in one family (3 patients) and the NP_000071.1p? pathogenic variant in 1 family. These 5 families had similar phenotype with ptosis and ophtalmoparesis noticed at birth in most cases, all patients had moderate phenotype with walking distance limit at 100 to 500 m. All patients were noticeably improved with anticholinesterases and became asymptomatic except for ptosis and ophtalmoparesis.
One family with 3 affected members had homozygous DOK7 novel mutation (p.Leu389ProfsTer66). All had congenital onset with stridor, feeding difficulties and delayed age of walking. The phenotype was severe in 2 cases with respiratory restrictive syndrome leading to death in one case. The two remaining were dramatically improved under salbutamol with sustained effect.
One patient had heterozygous pathogenic variant in CHRNA1 (pGly173Ser), he was normal at birth and harboured first symptoms at the age of 35 years with progressive and bilateral weakness of writs and fingers extensors. Nerve conduction study showed bifid motor action potentials with postsynaptic conduction block. The patient had partial improvement with salbutamol and fluoxetine. Genetic study is still ongoing in a patient.
Conclusion: The CHRNE homozygous mutations were the most frequent with one novel homozygous mutation. One family had a novel DOK 7 homozygous pathogenic variant with typical reported phenotype. Only one patient, with a novel heterozygous CHRNA1 pathogenic variant, had a particular phenotype with late onset fingers and writs extensors which is rarely reported.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Congenital Myasthenic syndrome
PP01.07
Joint Contractures as Prominent Clinical Feature in Congenital Myopathy Related to K7del TPM2 Gene Mutation
Prof. Birouk Nazha1, Prof. Bouhouche Ahmed2
1
Neurophysiology department, Hospital of specialities, Ibn Sina University Hospital, Mohamed V University, Rabat, Morocco.
2
Laboratory of neurogenetics, Hospital of specialities, Ibn Sina University Hospital, Mohamed V University, Rabat, Morocco
Background: The TPM2 gene, encodes beta-tropomyosin an important molecule for thin filaments in skeletal muscles. TPM2 mutations are related to severe congenital myopathies and distal arthrogryposis. Some TPM2 mutations are responsible of milder phenotypes with congenital contractures and moderate muscle deficit.
Methods: We identified by, Exom sequencing, a TPM2 mutation, p.Lys7del (K7del), in 2 unrelated Moroccan families with congenital myopathy and a distinctive clinical phenotype with mostly large joint contractures during early childhood and very moderate skeletal muscle weakness.
Results: Patient 1: 65 years old male experiencing since early childhood, running difficulties with severe and progressive feet equinovarus. Clinical examination showed large joint contractures (ankles, elbows, knees with rigid spine). He was needing 1 cane for walking 500 m with difficulties in climbing stairs and slight proximal upper limbs weakness.
Patient 2: 10 years old female who had generalized congenital muscles contractures that was improved by kinesitherapy. She had normal milestones development. She had difficulties in running and climbing stairs and was able to walk unlimited distance. Her examination showed limitation in all large joints’ amplitudes with rigid spine and limited mouth opening. She was improved by kinesitherapy and was stable at 24 years. Her father had the same clinical presentation with stable signs overs years.
Both patients had normal CK and myogenic recruitment pattern with normal nerve conduction at ENMG examination. They had the same heterozygous TPM2 p.Lys7del mutation leading to the diagnosis of autosomal dominant congenital myopathy 23.
Conclusion: The TPM2 p.Lys7del (delK7) mutation leads to the loss of a highly conserved lysine residue near the N-terminus of b-tropomyosin. Mokbel et al. (2013) reported similar patients with large joint contractures as prominent clinical feature. Studies of patient muscle suggested that the mutant protein doesn't incorporate well into the thin filaments of muscle sarcomeres and likely accumulates in nemaline bodies in muscle fibers, a hallmark of nemaline myopathy. Muscle fibers become overly sensitive to calcium, leading to sustained contraction and joint stiffness. It is interesting to consider delK7 TPM2 mutation as possible diagnosis in patients with muscle stiffness and joint contractures.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Congenital Myopathies and congenital muscular dystrophies
PP01.08
Proteomic Insights into Infantile Nemaline Myopathy Reveal Dysregulated Proteins and Cellular Pathways
Assoc. Prof. Carola Hedberg-Oldfors1, Dr. Ali Zeki Bedir2, Dr. Kittichate Visuttijai1, Dr. Eva Michael3, Prof. Anders Oldfors1
1
Department of Laboratory Medicine, University of Gothenburg, Gothenburg, Sweden.
2
Department of Pediatric Neurology, İstanbul Medeniyet University / Göztepe Research and Training Hospital, Istanbul, Turkey.
3
Department of Pediatrics, University of Gothenburg, Gothenburg, Sweden
Background: Nemaline myopathy is a rare congenital neuromuscular disorder characterized by the presence of nemaline rods in skeletal muscle fibers. Pathogenic variants in several genes have been implicated in its etiology, most notably NEB and ACTA1, which encode thin filament proteins of the sarcomere. Currently, no curative treatment exists for nemaline myopathy, highlighting the urgent need to identify disease-modifying targets to support therapeutic development. Proteomic profiling of affected muscle tissue represents an emerging and promising research approach that, in combination with genomic analyses, may facilitate the identification of dysregulated proteins and molecular networks. Such insights can advance the understanding of disease pathobiology and enable the discovery of novel biomarkers and therapeutic targets in neuromuscular disorders.
The objective in this study was to identify dysregulated proteins and altered cellular pathways in infant muscle tissue across the two most common genetic forms of nemaline myopathy, which present with similar clinical phenotypes and muscle biopsy histopathology.
Methods: In this study, quantitative nanoscale liquid chromatography–tandem mass spectrometry (LC–MS³) with labeled protein analysis was performed on skeletal muscle samples obtained from five unaffected controls and seven affected patients diagnosed with nemaline myopathy caused by pathogenic variants in NEB or ACTA1. To minimize age-related effects, only infant patients were included, and all muscle samples were collected via open biopsy from living individuals. The mean age at biopsy was 5.4 months (range, 2–19 months). Among the patients, three harbored de novo dominant ACTA1 variants, while four were compound heterozygous for NEB variants.
Pathway enrichment and protein–protein interaction analyses were conducted using multiple bioinformatic tools and web-based resources, including ProteoMap and STRING.
Results: A total of 4,846 proteins were identified and quantified across all samples, of which 183 exhibited significant dysregulation. Protein–protein interaction analysis revealed nine upregulated, muscle-specific proteins: ALPK3, ANKRD2, CSRP3, FBXO40, LMOD2, NRAP, TRIM54, TRIM63 and XIRP1. Pathway analysis further demonstrated increased activity of protein synthesis and proteasomal degradation pathways, accompanied by a downregulation of glycolytic processes. Furthermore, bioinformatic analysis revealed no proteins with significantly different expression levels between the two patient groups. This finding supports the concept that mutations in different genes may be associated with very similar phenotypes, not only from a clinical and morphological perspective but also regarding protein dysregulation. This finding implies that there may be therapeutic approaches that can be efficient irrespective of the underlying genetic defect in nemaline myopathy.
Conclusion: This study identifies key dysregulated proteins and cellular pathways in the skeletal muscle of infants with nemaline myopathy. These findings underscore the value of proteomic profiling in elucidating disease pathobiology and suggest that the identified proteins merit further investigation as potential therapeutic targets for nemaline myopathy.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Congenital Myopathies and congenital muscular dystrophies
PP01.09
Biallelic SNAPC4 Variants Associatedwith Lower Motor Neuron Disease
Dr. Chiara Fiorillo1,2, Dr. Monica Traverso3, Dr. Stefania Dallorso2, Dr. Simona Baldassari3, Dr. Matteo Cataldi2, Prof. Lino Nobili1,2
1
Department of Neuroscience, University of, Genova, Italy.
2
Pediatric Neurology, Gaslini Children Hospital, Genova, Italy.
3
Medical Genetics, Gaslini Children Hospital, Genova, Italy
Background: SNAPC4 (small nuclear RNA-activating protein complex- 4) encodes the DNAbinding subunit of the snRNA-activating protein complex (SNAPc), which is required for transcription of RNA polymerases and splicing processes. Biallelic variants in SNAPC4 gene have been recently associated with a novel neurodevelopmental condition (NEDRSO, MIM 620515) featuring microcephaly, eye defects, abnormal movements, and brain atrophy. Here, we report a novel patient harboring recessive SNAPC4 variants presenting with a distinctive clinical phenotype of lower motor neuron disease.
Methods: We collected clinical, electrophysiological, imaging, and genetic evaluation of a patient followed at the IRCCS Istituto Giannina Gaslini (Genoa, Italy) for long-standing progressive distal amyotrophy of lower limb due to axonal motor neuropathy. The diagnostic assessment included nerve conduction studies, brain and spine MRI, lower limb muscle MRI, and Whole Exome Sequencing (WES) to achieve a molecular diagnosis.
Results: A 13-year-old patient was admitted to our clinical center for a diagnostic definition of gait disturbances. During infancy, he was noted to be clumsy by the parents, with a tendency to fall. At the age of 8 years, his parents noticed a slowly worsening of his walking skills requiring ambulatory support and since age 13, the use of a wheelchair for outdoor activities. Neurological examination showed wide-based gate, lower limb hyporeflexia, diffuse muscle weakness and reduced muscle mass in the lower limbs. The patient was able to walk only for few steps with unilateral support. Extended blood tests were normal except for high serum CK levels (700 U/L at rest, normal <170). Brain and spine MRI were normal. Nerve conduction studies showed axonal degeneration of motor fibers. Trio-exome sequencing identified biallelic variants in SNAPC4 in the proband: the maternal c.1436T>C, p.(Ile1479Thr) and the paternal c.1889_1899del, p.(Val630Glyfs*76) classified according to ACMG guidelines as of uncertain significance and likely pathogenetic respectively. In cultured patient’s fibroblasts using a commercially available anti-SNAPC4 antibody we observed a partially reduced protein expression in patient’s cells compared to healthy, age and sexmatched controls by western blotting.
Conclusion: SNAPC4 variants have been previously detected in association with neurodevelopmental impairment, motor regression and progressive ascending spasticity. The features observed in our case, suggestive of a distal spinal muscular atrophy (dSMA) expand the phenotype of the emerging SNAPC4-related disorders and suggest SNAPC4 playing a possible role in motorneuron disease of children with or without intellectual impairment.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: non 5q SMA/distal SMA/ Hereditary Motor Neuropathies
PP01.10
Expanding the Phenotypic Spectrum of FKTN Mutations: A Case of Generalized Epilepsy, Neurodevelopmental Delay, andHyperCKemia
Dr. Roberta Biddeci, Dr. Margherita Mancardi, Dr. Monica Traverso, Prof. Chiara Fiorillo, Prof. Lino Nobili
IRCCS Giannina Gaslini, Genova, Italy
Background: The FKTN gene (9q34.1) encodes fukutin, a ribitol-5-phosphate transferase essentialfor the functional - glycosylation of alpha-dystroglycan. This process is vital for the structural integrity of the sarcolemma in skeletal muscle and for proper neuronal migration and basement membrane assembly in the central nervous system. Mutations in FKTN lead to a spectrum of dystroglycanopathies. We report the case of a 4-year-old male who presented FKTN mutations associated with overlap phenotype, characterized by generalized epilepsy, developmental delay and muscular dystrophy.T his study describes the longitudinal clinical management of the patient, integrating multiple data.
Methods: The patient is a 4-year-old-male born via urgent C-section for cord prolapse (APGAR 4-6) requiring neonatal resuscitation. Developmental milestones were delayed (walking at 16 months, sphincter control at 3.5 years). At 4 years old, he was admitted to a regional hospital with recurrent seizures characterized by psychomotor arrest, impaired awareness, chewing automatisms, and palpebral myoclonia. Interictal EEG showed sporadic centro-posterior abnormalities and generalized spike-wave discharges during sleep. He started therapy with Levetiracetam (250 mg BID) was initiated, achieving seizure control. During a scheduled brain MRI, routine blood exam incidentally revealed a significant elevation of Creatine Kinase (>1000 U/L). This unexpected finding shifted the clinical focus toward a potential primary muscle disorder. While brain MRI was unremarkable, the EMG performed on the right deltoid suggested a myogenic process with low-amplitude, polyphasic motor unit potentials. Physical examination showed mild hypotonia and weakness of the pelvic girdle, hypotrophy of the shoulder girdle. Reflexes were hypoelicitable in the lower limbs. Gait was independent but characterized by flat-foot contact and difficulty with heel-walking.
Results: Genetic investigations via NGS panel and subsequent Trio-Exome sequencing identified biallelic compound heterozygous variants of uncertain significance in the FKTN gene (paternal c.1304A>G; Glu435Gly and maternal c.1306T>A; Phe436Ile). To establish the pathogenicity of these variants, a muscle biopsy was performed on the left thigh, showing moderate muscle damage with a marked reduction in alpha - dystroglycan expression, confirming the diagnosis of a dystroglycanopathy. Given that FKTN mutations are key drivers also of progressive cardiomyopathy and neurodevelopmental delays, the patient underwent. Echocardiography and ECG (results preserved systolic function and no structural heart disease); to characterize the patient’s cognitive and behavioural profile, the Griffiths Development Scales were administered, confirming a global developmental delay compared to chronological age.
Conclusion: This case suggests that the phenotypic spectrum of FKTN mutations may include generalized epilepsy even in the presence of paucisymptomatic muscle involvement.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Muscle Dystrophies (Non-Dystrophinopathies)
PP01.11
Clinical and Genetic Characterization of Dominant COL12A1- Related Myopathies Across European Centers
Dr. Federica Trucco1,2, Dr. Michela Catteruccia3, Dr. Monica Traverso4, Dr. Guja Astrea5, Prof. Edoardo Malfatti6, Dr. Anna Sarkozy7, Dr. Adnar Manzur7, Dr. Pinki Munot7, Dr. Daniele Galatolo5, Dr. Chiara Panicucci8, Dr. Mariafrancesca DiFeo1,9, Dr. Alexander Camizaru10, Dr. Amelia Dobrescu10, Prof. Adele D'Amico3, Prof. Enrico Bertini3, Prof. Claudio Bruno8,1, Prof. Chiara Fiorillo2,1
1
Department of Neuroscience, University of Genova, Genova, Italy.
2
Unit of Paediatric Neurology and Muscle Disorders, IRCCS Istituto G.Gaslini, Genova, Italy.
3
Unit of Neuromuscular and Neurodegenerative Disorders, Translational Paediatrics and Clinical Genetics, Bambino Gesù Children's Hospital, Rome, Italy.
4
Unit of Medical Genetic, IRCCS Istituto Giannina Gaslini, Genova, Italy.
5
Molecular Medicine, IRCCS Fondazione Stella Maris, Genova, Italy.
6
University Paris-Est Créteil, INSERM, U955 IMRB, F-94010, Paris, France.
7
The Dubowitz Neuromuscular Centre, University College London, Great Ormond Street Institute of Child Health & Great Ormond Street Hospital, London, United Kingdom.
8
Centre of Translational and Experimental Myology, IRCCS Istituto Giannina Gaslini,, Genova, Italy.
9
Folkhälsan Research Center,, Helsinki, Finland.
10
Department of Medical Genetics, University of Medicine and Pharmacy of Craiova, Cracovia, Romania
Background: Collagen 12A1 (COL12A1) is a homotrimeric extracellular matrix protein. Autosomal recessive and dominant variants in COL12A1 have been associated to Ullrich and Bethlem type 2 myopathies respectively, both characterized by muscle weakness, joint contractures and facial dysmorphism. Reports so far have been sparse and a precise genotype-phenotype correlation is lacking. Aim of the study is to describe a series of patients with COL12A1 causative variants and to correlate the clinical, histological and MRI features to patients’ genotype.
Methods: We included twelve patients from 5 European neuromuscular centers carrying causative variants in COL12A1 gene. Clinical, histological, genetic and muscle MRI characteristics have been collected in shared platform.
Results: The mean age is 8 years. 10/11 patients are reported having onset at birth or within the first 2 years. 3/11 have a relative affected with incomplete penetrance. The overall phenotype is mild and the most common clinical characteristic are joint laxity (11/11) and hypotonia at birth with delayed motor milestones (8/11). Club foot, hip dysplasia, rigid spine and pectus excavatum are also reported. In 6/11 patients muscle MRI was performed showing predominant involvement of glutei and thigh muscles. In 5/11 patients muscle biopsy was conducted displaying unspecific myopathic changes. CK is normal in 9/11. The most frequent mutations are missense (5/11) followed by splicesite (4/11) and frameshift (2/11). There is no clear genotype-phenotype correlation as either missense or null variants are related to a spectrum of manifestations.
Conclusion: This is to date the most comprehensive report of Bethlem type 2 patients. Results suggest that COL12A1 dominant mutations are associated with rather homogeneous clinical presentation of mild, early-onset myopathy without genotype-phenotype correlation.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Congenital Myopathies and congenital muscular dystrophies
PP01.12
MYBPC1-Related Congenital Myopathy With Tremor Associated With Cerebellar Atrophy and Adult-Onset Cardiac Involvement
Dr. Cristina Muntean Firanescu1, Dr. Sebastian Thams1, Dr. Kristin Samuelsson1, Dr. Malin Kvarnung2, Dr. Maria Mannila3, Dr. Rayomand Press1
1
Department of Neurology, Karolinska University Hospital, Stockholm, Sweden.
2
Department of Clinical Genetics, Karolinska University Hospital, Stockholm, Sweden.
3
Department of Cardiology and Clinical Genetics, Karolinska University Hospital, Stockholm, Sweden
Background: MYBPC1-related congenital myopathy with tremor (MYOTREM) is a rare autosomal dominant neuromuscular disorder characterized by neonatal hypotonia, muscle weakness without evidence of neuropathy, and a myogenic tremor. While primarily considered a skeletal muscle disorder, the full clinical spectrum remains incompletely defined. Only a limited number of patients with MYBPC1-related congenital myopathy have been reported, and long-term follow-up data are scarce.
Methods: We present a 26-year-old man with a previously reported heterozygous pathogenic MYBPC1 variant (p.Leu233Arg), occurring de novo, with no other affected family members. He was born at term with normal birth weight and was admitted to the neonatal unit due to involuntary movements consistent with tremor and a spontaneous pneumothorax, which resolved without intervention. Shortly after birth, hypotonia affecting both axial and limb musculature was noted.
Early motor development was delayed. Independent ambulation was achieved at 14 months and was characterized by gait instability and recurrent falls. During childhood, proximal muscle weakness predominantly involving the lower extremities developed, together with reduced muscular endurance and impaired balance. These findings persisted into adulthood. Excessive sweating developed during adolescence and has remained present.
Tremor was a prominent and persistent feature, affecting the upper and lower extremities as well as the tongue. The tremor was postural and kinetic, clinically characterized as a fine, high-frequency tremor of approximately 20 Hz.
Results: Nerve conduction studies showed no evidence of polyneuropathy. Muscle biopsy performed in childhood, including immunohistochemistry and electron microscopy, did not demonstrate any pathological abnormalities. Brain MRI demonstrated slowly progressive cerebellar vermis atrophy, first noted at 16 years of age and confirmed on follow-up imaging in adulthood. No additional structural brain abnormalities were identified. Blood workup did not reveal any alternative explanation for the cerebellar atrophy.
Cardiac evaluation performed at 26 years of age revealed mildly reduced left ventricular systolic function with an ejection fraction of approximately 45%. Long-term ECG monitoring and cardiac biomarkers were normal, and no alternative cardiac etiology was identified. Extensive evaluation of the hyperhidrosis, including assessment for endocrine, autonomic, and secondary causes, did not reveal an alternative explanation.
Conclusion: This case describes a patient with MYBPC1-related congenital myopathy and tremor, in whom cerebellar atrophy and adult-onset cardiac findings were also observed. A causal relationship between the MYBPC1 variant and the cerebellar and cardiac findings cannot be established. These findings may be coincidental or reflect an additional, as yet unidentified etiology; nevertheless, they underscore the importance of careful longitudinal neurological and cardiac follow-up. Systematic collection of long-term clinical data may help further delineate the clinical spectrum of MYBPC1-related disease.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Congenital Myopathies and congenital muscular dystrophies
PP01.13
RYR1 Variants in Pediatric Myopathies: Clinical Diversity and Diagnostic Uncertainty
Dr. Gordana Kovacevic1,2, Dr. Slavica Ostojic1,2, Assoc. Prof. Adrijan Sarajlija3,2,4, Assoc. Prof. Nina Maric5,6, Dr. Nikola Ilic3, Dr. Mihail Basa7
1
Neurology Department, Mother and Child Health Care Institute of Serbia “Dr Vukan Cupic”, Belgrade, Serbia.
2
Faculty of Medicine, University of Belgrade, Belgrade, Serbia.
3
Clinical Genetics Outpatient Clinic, Mother and Child Health Care Institute of Serbia “Dr Vukan Cupic”, Belgrade, Serbia.
4
Faculty of Medicine, University of Eastern Sarajevo, Foca, Bosnia and Herzegovina.
5
Clinic for Children Diseases, University Clinical Center of the Republic of Srpska, Banja Luka, Bosnia and Herzegovina.
6
Medical Faculty, University of Banjaluka, Banja Luka, Bosnia and Herzegovina.
7
Department of pulmology,Mother and Child Health Care Institute of Serbia “Dr Vukan Cupic”, Belgrade, Serbia
Background: Mutations in the RYR1 gene are common genetic findings in patients with congenital myopathies. The clinical symptoms of RYR1-related myopathies are very heterogenous, spanning from mild motor delay symptoms to severe neonatal hypotonia and respiratory insufficiency. The use of next-generation sequencing (NGS) has significantly contributed to accurate diagnosis. However, phenotypic variability and the frequent identification of variants of uncertain significance (VUS) continue to pose significant diagnostic challenges, especially when muscle magnetic resonance imaging (MRI) or histopathological muscle examination are unavailable. Aim of this study is to characterize the clinical and genetic variability of suspected RYR1-related myopathies in pediatric patients and to assess the diagnostic value and limitations of next-generation sequencing, particularly in cases with variants of uncertain significance.
Methods: We retrospectively analyzed clinical, genetic, and histopathological data of seven pediatric patients with suspected RYR1-related myopathy. Four patients harbored pathogenic or likely pathogenic RYR1 variants, each presenting with distinct clinical phenotypes. Three additional patients carried RYR1 VUS and exhibited clinical features compatible with congenital myopathy. Muscle biopsy was performed in two patients prior to the availability of genetic testing, while muscle MRI was not accessible. In patients with VUS, muscle biopsy was not performed due to parental refusal or recently obtained genetic results. In one family, a RYR1 variant of uncertain significance was identified in an affected sibling; however, the same variant was absent in a younger sibling with comparable hypotonia and delayed motor development. Consequently, whole genome sequencing was performed.
Results: Among patients with pathogenic RYR1 variants, clinical presentations ranged from childhood-onset motor delay with hip-girdle weakness and central core disease on biopsy, to severe neonatal hypotonia with ophthalmoplegia, bulbar and respiratory weakness requiring invasive ventilation, contractures, and centronuclear myopathy on biopsy. Other phenotypes included neonatal hypotonia with axial, and proximal weakness, and ligamentous laxity, as well as milder hypotonia with delayed motor development and proximal weakness. Patients with RYR1VUS also demonstrated variable phenotypes: congenital clubfoot with hypotonia and myopathic facies; mild axial weakness with ligamentous laxity, and delayed motor development with proximal muscle weakness. In one of these patients, extended NGS revealed a homozygous VUS in the FXR1 gene. Segregation analysis confirmed the presence of the FXR1 variant in both parents and the affected sibling, leading to a diagnosis of autosomal recessive FXR1-related congenital myopathy. In the absence of a muscle biopsy or MRI, definitive correlation between genotype and phenotype remained inconclusive in the remaining two cases.
Conclusion: Our cohort demonstrates the marked clinical and genetic heterogeneity of RYR1-related myopathies in pediatric patients. Although next-generation sequencing plays a central role in identifying RYR1 variants, diagnostic uncertainty persists in a subset of patients, particularly with VUS, when comprehensive investigations cannot be completed. Limited access to MRI, biopsy, and extended genetic testing restricted definitive genotype–phenotype correlation. Nevertheless, selected cases demonstrate that extended NGS and family segregation studies can clarify the diagnosis, including non-RYR1 congenital myopathies. An integrated diagnostic strategy that combines careful clinical assessment, genetic testing, and muscle biopsy—when feasible—remains essential for accurate diagnosis in suspected RYR1-related myopathies.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Congenital Myopathies and congenital muscular dystrophies
PP01.14
One-Year Follow-Up of Muscle MRI in LAMA2-Related Muscular Dystrophy and SELENON-Related Congenital Myopathy
Ms. Ilse de Laat, Dr. Karlijn Bouman, Dr. Sanne Vincenten, Dr. Saskia Houwen - van Opstal, Dr. Corrie Erasmus, Prof. Jan Groothuis, Mr. Donnie Cameron, Prof. Nicol Voermans
Radboud university medical center, Nijmegen, Netherlands
Background: Laminin- α2-related muscular dystrophy (LAMA2-MD) and selenoprotein N-related congenital myopathy (SELENON-RM) are rare congenital muscle disorders characterized by delayed motor development, slowly progressive axial muscle weakness, early-onset spinal rigidity, and respiratory insufficiency. Currently, no curative treatment options are available, however, promising preclinical studies are being performed. Clinical and functional outcome measures often show little change over short-term follow-up. In contrast, muscle magnetic resonance imaging (MRI) has shown diagnostic and prognostic value in other congenital muscle disorders, making it a promising additional outcome measure. However, no prospective quantitative MRI studies have been performed yet in LAMA2-MD and SELENON-RM. Cross-sectional imaging studies have shown an inconsistent pattern in LAMA2-MD and more uniform involvement in SELENON-RM without correlations to clinical features. Hereby, we present the one-year follow-up muscle MRI data from the LAST STRONG study, a natural history study aiming to identify suitable outcome measures and reach trial readiness for LAMA2-MD and SELENON-RM.
Methods: Patients underwent a whole-body MRI at baseline and one-year follow-up using a 1.5 tesla MRI system. The protocol included ‘Dixon’ imaging and T2-weighted STIR. In a subset of lower extremity muscles, polygonal region of interests (ROIs) were drawn to derive fat-fraction maps and cross-sectional areas. Motor function was assessed using the Motor Function Measures-32 and graded and timed function tests.
Results: Data analysis is currently ongoing. At the congress, patterns of muscle involvement and longitudinal changes in quantitative muscle MRI, including fat-fraction and cross-sectional area, and their correlations with clinical and functional outcome measures will be presented.
Conclusion: This prospective natural history study will provide important longitudinal quantitative muscle MRI data in LAMA2-MD and SELENON-RM patients. The findings are expected to clarify muscle involvement patterns, and determine if muscle MRI is a sensitive outcome measure for future clinical trials.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Congenital Myopathies and congenital muscular dystrophies
PP01.15
MEF2A Modulation as a Therapeutic Strategy for LAMA2-Related Muscular Dystrophy
Dr. Julie Yue Yuan1, Dr. Veronica Pini2,1, Prof. Stefano Carlo Previtali2,3, Prof. Francesco Muntoni1,4
1
Dubowitz Neuromuscular Centre, UCL Great Ormond Street Institute of Child Health, London, United Kingdom.
2
Neuromuscular Repair Unit, Institute of Experimental Neurology, Ospedale San Raffaele, Milan, Italy.
3
Vita-Salute Sanraffaele University, Milan, Italy.
4
NIHR Great Ormond Street Hospital Biomedical Research Centre, London, United Kingdom
Background: LAMA2-related muscular dystrophy (LAMA2-RD) is a severe congenital neuromuscular disorder caused by mutations in the LAMA2 gene, resulting in absent or defective laminin-α2 and leading to progressive muscle weakness, fibrosis, and impaired regeneration. Disease severity correlates with residual laminin-α2 expression: patients with complete deficiency typically present with a homogeneous, severe phenotype and rarely achieve independent ambulation, whereas those with residual protein show more variable and often milder disease.
We identified a patient with an atypically mild LAMA2-RD phenotype despite complete laminin-α2 deficiency, who remains ambulant at 36 years of age. This individual carries a previously unreported genomic variant in the muscle transcription factor MEF2A, located within a poorly characterised region of the protein. MEF2A plays a central role in myogenesis and muscle homeostasis, and preliminary transcriptomic data indicate altered expression of MEF2A-dependent genes in this patient.
This study investigates how the MEF2A variant influences cellular and molecular features of LAMA2-RD and evaluates its potential role as a genetic modifier and therapeutic target.
Methods: Primary fibroblast and myoblast were established from atypical and LAMA2-RD patients and healthy controls. MEF2A subcellular localisation was analysed by immunofluorescence. Myogenic differentiation capacity will be evaluated. Expression of predicted MEF2A interactors and target genes was analysed by qPCR, with proteomic profiling underway.
Results: Immunofluorescence analyses showed preserved nuclear localisation of MEF2A in patient-derived fibroblasts, suggesting that the MEF2A variant does not impair nuclear trafficking. These findings indicate that altered disease severity is unlikely to result from MEF2A mislocalisation.
Patient-derived cellular models have been successfully established, enabling ongoing analyses of myogenic capacity, proliferation, apoptosis, and gene expression. Preliminary molecular profiling supports the presence of altered MEF2A-dependent transcriptional programmes in the mild phenotype patient. Proteomic and transcriptomic studies are in progress to identify MEF2A-interacting partners and downstream pathways associated with the variant.
Conclusion: A previously unreported genomic variant in MEF2A is associated with an atypically mild LAMA2-RD phenotype despite complete laminin-α2 deficiency. Preserved nuclear localisation of MEF2A suggests that altered transcriptional regulation, rather than mislocalisation, may underlie disease modification. Ongoing molecular aim to define MEF2A-dependent pathways that influence disease severity. These findings highlight MEF2A as a potential therapeutic target for modifying disease progression in LAMA2-related muscular dystrophy.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Congenital Myopathies and congenital muscular dystrophies
PP01.16
DNM2-Related Centronuclear Myopathy With Electrical Myotonia
Prof. Jungim Suk
Department of Neurology, Catholic University of Daegu, School of Medicine, Daegu, Korea, Republic of
Background: Centronuclear myopathy (CNM) is a rare hereditary myopathy characterized by centrally located muscle fiber nuclei. Usual EMG finding is myopathic motor unit potentials without spontaneous activity. Myotonic discharge without clinical myotonia can be seen in polymyositis, acid maltase deficiency, and so on.
Methods: We reported a case of CNM with electrical myotonia.
Results: A 30-year-old man presented with progressive limb weakness. Neurologic examination revealed mild ptosis and proximal limb weakness. Neither spontaneous or percussion myotonia nor muscle atrophy was observed. In the electrophysiologic examinations, motor and sensory conduction studies were normal; needle electromyogram showed numerous myotonic discharges. CNM was confirmed by gene test without muscle biopsy. Next generation sequencing revealed a heterozygous substitution (c.1948G>A; p.Glu650Lys) in the DNM2.
Conclusion: This case suggests that DNM2-related CNM may present electrical without clinical myotonia. The diagnosis of CNM should be considered in patients with myotonic discharges of an unknown cause.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Congenital Myopathies and congenital muscular dystrophies
PP01.17
Clinical Characteristics of Patients With Congenital Myopathies From a 2-Year Prospective Natural History Study (READYCOM)
Ms. Sanne A.J.H. van de Camp1, Mr. Jeroen L.M. van Doorn1, Ms. Elisabeth C.M. de Laat1, Prof. Jan T. Groothuis1, Prof. Nens van Alfen1, Prof. Heinz Jungbluth2,3, Dr. Corrie. E. Erasmus1, Dr. Bart Bartels4, Dr. Renske I. Wadman4, Dr. Nicoline B.M. Voet1,5, Prof. W. Ludo van der Pol4, Prof. Nicol C. Voermans1
1
Radboud university medical center, Nijmegen, Netherlands.
2
Evelina London Children’s Hospital, Guy’s and St Thomas’ Hospital NHS Foundation Trust, London, United Kingdom.
3
Randall Centre for Cell and Molecular Biophysics, King’s College London, London, United Kingdom.
4
University Medical Center Utrecht, Utrecht, Netherlands.
5
Rehabilitation Centre Klimmendaal, Arnhem, Netherlands
Background: Congenital myopathies (CMYO) are a group of rare hereditary muscle diseases defined by characteristic abnormalities on muscle biopsy. Several types – including the core myopathies Central Core Disease (CCD) and Multi-minicore Disease (MmD), Nemaline Myopathy (NM) and Centronuclear Myopathy (CNM) - have been identified based on the characteristic histopathological changes and attributed to various genetic backgrounds. The most prominent clinical features are generalized muscle weakness often pronounced axially, variable cardiorespiratory and bulbar impairment, and skeletal and joint involvement. Currently, no curative therapies are available for CMs, however, a few phase I-II trials have been performed or are expected in the near future.
To reach trial readiness, an informed understanding of the disease course and a selection of relevant and sensitive clinical and functional outcome measures, and blood and imaging biomarkers is necessary. Various outcome measures exist in practice to determine the presence and severity of CM, but it is yet unknown which outcome measures are most sensitive to detect changes in muscle strength and motor abilities over the timeframe of a clinical trial in CM.
Methods: In collaboration with patient representatives, we have designed a prospective cohort study with five 6 monthly visits over a 2-year period. Due to the clinical similarities in phenotypes between CCD/MmD, NM, and CNM, we have combined them into one basket study with 15 participants per group, for a total of 45 patients, aged 2-67 years old. In this study several assessments are performed, covering patient-reported outcomes, clinical and functional outcome measures, and blood and imaging biomarkers.
Results: We will describe the baseline characteristics of the cohort, including information on respiratory function, motor function measure (MFM), Graded and Timed functions, hand-held dynamometry and patient-reported outcomes. The questionnaires contain information on fatigue, pain, quality of life, and participation.
Conclusion: This study will provide important natural history data of CMYO patients. These data can be used to select relevant and sensitive clinical and functional outcome measures, and blood and imaging biomarkers to reach trial readiness. Furthermore, it will provide insight in the occurrence and severity of muscle fatigability in CMYO patients.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Congenital Myopathies and congenital muscular dystrophies
PP01.18
Beyond Muscle: Anterior Horn Cell Involvement in The Phenotypic Spectrum of Collagen VI - Related Disorders
Dr. Thainá Louise Rodrigues1, Dr. Enzo Pellacani1, Dr. Rodrigo Frezatti2, Dr. Pedro Tomaselli2, Dr. Wilson Marques2, Dr. Alzira Carvalho1, Dr. Ana Marina Silva1
1
Centro Universitário FMABC, Santo André, Brazil.
2
Hospital das Clínicas de Ribeirão Preto, Ribeirão Preto, Brazil
Background: Collagen VI-related disorders (COL6-RD) encompass a heterogeneous phenotypic continuum extending from severe Ullrich congenital muscular dystrophy (UCMD) to the milder Bethlem myopathy (BM), with intermediate phenotypes constituting a significant proportion of cases.1,2,3,4 Emerging evidence has documented neurological manifestations affecting both the central nervous system (CNS) and peripheral nervous system (PNS).5,6 Herein, we present two cases demonstrating anterior horn cell pathology associated with pathogenic COL6 variants
Methods: Patient 1: A 17-year-old male proband, offspring of non-consanguineous parents, exhibited delayed motor milestone acquisition. Ambulation difficulties manifested at 18 months of age, with subsequent progression to predominantly proximal myopathy; loss of independent ambulation occurred at age 12 years. Contractures of the toe flexor tendons and talipes varus deformity were documented from age 3 years. Clinical examination demonstrated proximal muscle weakness, keratosis pilaris, keloid scarring, and structural scoliosis. Serum creatine kinase (CK) levels remained within normal reference ranges. Serial electromyography (EMG) studies conducted at ages 4, 13, and 17 years revealed diffuse neurogenic changes consistent with anterior horn cell dysfunction. Magnetic resonance imaging (MRI) of the proximal and distal lower extremities demonstrated diffuse fatty infiltration and muscle replacement, compatible with myopathic processes. Histopathological analysis of muscle tissue demonstrated myofiber diameter variability, endomysial fibrosis, and adipose tissue infiltration; immunohistochemical staining for collagen VI showed normal expression patterns. Multiplex ligation-dependent probe amplification (MLPA) excluded 5q-linked spinal muscular atrophy (5q-SMA). Whole-exome sequencing (WES) identified compound heterozygous variants in COL6A3: c.6359_6364delACAAAGGinsCCAAA (classified as likely pathogenic) and c.7447A>G (classified as likely pathogenic), demonstrating biparental inheritance consistent with autosomal recessive transmission. Patient 2: A 7-year-old male proband, offspring of non-consanguineous parents, presented with delayed motor developmental milestones. Physical examination revealed generalized joint hypermobility and diffuse hypotonia, with no keratosis pilaris and a single keloid scar. CK concentrations remained within normal limits. Radiographic evaluation of the pelvis demonstrated bilateral developmental dysplasia of the hip. EMG performed at ages 4 and 7 years showed diffuse chronic denervation potentials consistent with anterior horn cell pathology. MLPA for 5q-SMA was negative. WES identified a homozygous intronic variant in COL6A2: c.1970-9G>A (classified as pathogenic).
Results: The present case series documents two patients harboring pathogenic variants in COL6 genes who exhibit diffuse neurogenic patterns on electromyography, indicative of anterior horn cell pathology. While uncommon, this genotype-phenotype correlation has been documented in the literature. These observations provide further clinical evidence supporting the critical role of collagen VI in preserving the structural and functional integrity of lower motor neurons, as demonstrated through experimental studies in animal models6
Conclusion: These cases broaden the recognized phenotypic spectrum of COL6-related disorders, specifically highlighting anterior horn cell involvement as a clinically relevant manifestation. Consequently, molecular analysis for pathogenic COL6 variants should be incorporated into the diagnostic algorithm for patients presenting with non-5q spinal muscular atrophy phenotypes
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Congenital Myopathies and congenital muscular dystrophies
PP01.19
Exploring Novel Modifier Genes for LAMA2-RD by in Vitro and in Vivo studies
Dr. Veronica Pini1,2, Dr. Yue Julie Yuan2, Dr. Emanuela Porrello1, Dr. Rosa Bonaccorso1, Prof. Jennifer Morgan2, Prof. Francesco Muntoni2,3, Prof. Stefano Carlo Previtali1,4
1
Neuromuscular Repair Unit, Institute of Experimental Neurology, Ospedale San Raffaele, Milan, Italy.
2
Dubowitz Neuromuscular Centre, UCL Great Ormond Street Institute of Child Health, London, United Kingdom.
3
NIHR Great Ormond Street Hospital Biomedical Research Centre, London, United Kingdom.
4
Vita-Salute San Raffaele University, Milan, Italy
Background: Merosin-deficient congenital muscular dystrophy (LAMA2-RD) is a neuromuscular disorder caused by mutations in LAMA2 gene, coding for the α2 subunit of laminin-211. Most LAMA2-RD patients who carry LAMA2 loss-of-function mutations that prevent laminin-211 production follow an invariably severe clinical phenotype characterised by the inability to acquire ambulation. Missense mutations in LAMA2 gene allow instead the production of a partially functional protein, resulting in a wider (usually milder) severity spectrum. To date, only one LAMA2-RD patient was reported with a very mild phenotype despite the total laminin-211 absence in muscle. This patient is still ambulant at the age of 36 with no respiratory insufficiency nor cardiomyopathy. Interestingly, this patient carries the same LAMA2 loss-of-function mutation as its severely affected sibling who also completely lacks laminin-211 in muscle. We hypothesized that not yet characterized genetic modifier(s) are acting in the atypical patient to mitigate the consequences of complete laminin-211 absence via a novel mechanism.
Methods: To identify candidate genetic modifiers uniquely altered in the atypical patient, we combined genomic data from both siblings with RNA sequencing of muscle biopsies from the atypical patient and unrelated LAMA2-RD patients. Selected modifiers are being functionally assessed by modulating their expression in vitro in patient-derived and murine muscle cells, and in vivo in LAMA2-RD mouse models using pharmacological agonists and/or CRISPR interference. Functional, histological, and molecular analyses will be performed to evaluate disease-related outcomes.
Results: This integrative analysis allowed us to identify a restricted set of candidate genetic modifiers uniquely altered in the atypical patient. These candidates are predicted to act downstream of laminin-211 loss, potentially modulating secondary pathogenic mechanisms known to contribute to LAMA2-RD progression, including inflammation, fibrosis, and metabolic dysregulation. Among them, inflammatory mediators such as CSF3 and PF4, as well as metabolic and immunomodulatory signalling pathways including LXR/RXR, were prioritized for further investigation based on their differential regulation and biological relevant role in molecular traits perturbed in LAMA2-RD pathology. Ongoing functional studies are assessing the impact of modulating these targets in vitro in patient-derived and murine muscle cells and in vivo in LAMA2-RD mouse models using pharmacological and CRISPR interference approaches. Preliminary observations indicate that modulation of selected candidates may influence secondary pathological features and disease-related molecular signatures.
Conclusion: This study identifies genetic modifiers acting on distal and secondary molecular mechanisms as key contributors to phenotypic variability in LAMA2-RD. Targeting downstream pathogenic pathways may represent an effective therapeutic strategy to ameliorate disease severity independently of direct laminin-211 restoration. These findings provide a novel framework for therapeutic development in LAMA2-RD and potentially other severe muscular dystrophies.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Congenital Myopathies and congenital muscular dystrophies
PP01.20
Expanding the Phenotype of TUBA4A-Related Myopathy: Two Unrelated Korean Families
Dr. Yunjung Choi1, Dr. Hyung Jun Park2, Dr. Young-Chul Choi2, Dr. Jeong Hee Cho3, Dr. Bum Chun Suh4, Dr. Sang Beom Kim5, Dr. Woo-Kyung Kim6, Dr. Yang-Ki Minn7
1
Department of Neurology, Sanggye Paik Hospital, Inje University College of Medicine, Seoul, Korea, Republic of.
2
Department of Neurology, Gangnam Severance Hospital, Younsei University College of Medicine, Seoul, Korea, Republic of.
3
Department of Neurology, National Health Insurance Service Ilsan Hospital, Goyang, Korea, Republic of.
4
Department of Neurology, Kangbuk Samsung Hospital, Sungkyunkwan University School of Medicine, Seoul, Korea, Republic of.
5
Department of Neurology, Kyung Hee University Hospital at Gangdong, Kyung Hee University School of Medicine, Seoul, Korea, Republic of.
6
Department of Neurology, Kangdong Sacred Heart Hospital, Hallym University College of Medicine, Seoul, Korea, Republic of.
7
Department of Neurology, Kangnam Sacred Heart Hospital, Hallym University College of Medicine,, Seoul, Korea, Republic of
Background: Congenital myopathies are genetically heterogeneous disorders characterized by early-onset muscle weakness and characteristic structural changes on muscle pathology. Although pathogenic TUBA4A variants have been predominantly associated with neurodegenerative phenotypes, recent reports have established TUBA4A as a cause of primary myopathy. Here, we report two unrelated Korean families with autosomal dominant congenital myopathy associated with heterozygous TUBA4A variants.
Methods: We reviewed clinical features, electrophysiologic studies, laboratory findings, and muscle pathology in affected individuals from two unrelated families. Genetic testing was performed using PacBio Revio long-read sequencing, whole-exome sequencing (Illumina NovaSeq 6000) and Sanger sequencing to identify TUBA4A variants.
Results: In Family I, the proband, a 57-year-old woman, had childhood-onset reduced physical performance and developed progressive proximal weakness in her 30s, complicated by recurrent hypercapnic respiratory failure requiring tracheostomy and chronic ventilator support. Repetitive nerve stimulation demonstrated a significant decrement, and transient improvement with pyridostigmine and immunotherapies initially suggested myasthenia gravis. However, acetylcholine receptor antibodies were negative and electromyography reveal myopathic changes. Muscle biopsy showed minicores, consistent with multiminicore myopathy. She also had a family history suggestive of autosomal dominant inheritance, including multiple relatives with progressive limb weakness and deaths in their 40s.
Her affected brother, 49-year-old man, showed lifelong poor exercise tolerance and low body weight. In his 30s, he developed proximal weakness and dysphagia developed, followed by chronic ventilatory dependence due to CO₂ retention. Muscle biopsy showed multiminicores, and long-read sequencing identified a heterozygous TUBA4A variant, NM_001267550.2:c.850G>A (p.Glu284Lys), in the brother. Sanger sequencing confirmed the same variant in the proband, supporting familial segregation.
In Family II, proband, a 27-year-old woman, had normal developmental milestones but remained small and underweight until 10 years of age. She developed scoliosis thereafter and experienced progression of scoliosis and proximal weakness. Examination demonstrated axial and proximal-predominant weakness, knee and ankle contractures, a myopathic facial appearance, and myopathic changes on electrophysiology. She underwent multiple scoliosis surgeries over years, and postoperative monitoring revealed nocturnal hypercapnia requiring ventilatory support. Serum creatine kinase was markedly elevated and muscle biopsy was consistent with a muscular dystrophic pattern. Whole-exome sequencing identified TUBA4A variant, NM_006000.3:c.679C>T (p.Leu227Phe).
TUBA4A encodes an α-tubulin subunit critical for microtubule assembly and cellular architecture. To date, TUBA4A has been more commonly implicated in motor neuron disease and neurodegenerative phenotypes, emerging evidence indicates that specific TUBA4A variants can cause myopathic phenotype. In Family I, the proband showed decremental responses on repetitive nerve stimulation and partial responsiveness to cholinesterase inhibitors, which may confound the differential diagnosis with myasthenia gravis. Despite prior reports linking c.850G>A to reproductive or early developmental phenotypes, the proband did not exhibit infertility or reproductive complications, suggest phenotypic variability. In Family II, the proband’s early-onset axial deformity, dystrophic muscle pathology and subsequent hypercapnic respiratory insufficiency were broadly consistent with the phenotype previously reported for c.679C>T.
Conclusion: Our cases expand the clinical and pathological spectrum of TUBA4A-related myopathy and emphasize that TUBA4A should be considered in patients with proximal myopathy and early hypercapnic respiratory failure, even when myasthenic features or dystrophic pathology complicate the initial diagnosis.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Congenital Myopathies and congenital muscular dystrophies
PP01.21
Characterization of an In Vitro C2c12 Stable Model for plin4-Related Myopathy
Dr. Alessandra Carnazzi1, Ms. Eliana Iannibelli1, Dr. Veronica Sian2, Dr. Jaakko Sarparanta2,3, Dr. Per-Harald Jonson2,3, Dr. Eleonora Giagnorio1, Dr. Alenka Čopič4, Dr. Stefania Marcuzzo1,5, Dr. Isabella Moroni6, Prof. Marco Savarese2,3, Dr. Lorenzo Maggi1, Dr. Sara Gibertini1, Dr. Alessandra Ruggieri1
1
Neuroimmunology and Neuromuscular Diseases Unit, Fondazione IRCCS Istituto Neurologico “Carlo Besta”, Milan, Italy.
2
Folkhälsan Research Center, Helsinki, Finland.
3
University of Helsinki, Helsinki, Finland.
4
Centre de Recherche en Biologie Cellulaire de Montpellier-CRBM, Université de Montpellier, Montpellier, France.
5
Brain-targeted Nanotechnologies (BraiNs) Lab, Fondazione IRCCS Istituto Neurologico “Carlo Besta”, Milan, Italy.
6
Department of Pediatric Neurosciences, Fondazione IRCCS Istituto Neurologico “Carlo Besta”, Milan, Italy
Background: PLIN4-related myopathy is a rare distal vacuolar myopathy with late onset characterized by progressive muscle weakness and atrophy. The disease was first described in 2020 and since then only a few cases have been described: 63 patients from 6 families and 2 sporadic cases were identified up to now. Genetically, the disease is caused either by 99 nucleotides repeat expansions of different length or by a 14 blocks duplication, both affecting exon 5 of the PLIN4 gene. This exon encodes for the amphipathic helix of the protein, necessary for its physiological function, and possesses a peculiar structure of 31 highly similar but not identical repeats, each of them formed by 33 amino acids. Therefore, the enlargement of the final protein due to these rearrangements presumably impairs proper folding, causing it to precipitate within the muscle fibers. A specialized form of autophagy, called aggrephagy, is activated in the attempt to remove these protein aggregates but, most likely, the mechanism is impaired in time. Thus, autophagic vacuoles keep forming and accumulating, disrupting the spatial organization and contractile capacity of the myofibers leading to atrophy. Nevertheless, the exact pathogenetic mechanisms of the disease remain unknown.
Methods: Since PLIN4 gene is not expressed in patients’ primary myoblasts, we generated an in vitro model using C2C12 cell lines overexpressing GFP-tagged wild-type or mutated perilipin 4, to unveil the molecular pathways of this rare myopathy. An in-depth characterization of the model, involving immunofluorescence staining, western blot, fractionation assays and quantitative Real-Time PCR analysis is now ongoing. Moreover, we are also working at the optimization of the protocol for the generation of organoid-like structures using C2C12 stable lines. Since perilipin 4 physiological function in the muscle is yet to be clarified, the combined use of the stable cell lines in a bi-dimensional and tri-dimensional settings will allow us to investigate its function and expression.
Results: Preliminary data reveal GFP-positive accumulations exclusively in mutated myotubes, suggesting that the cell lines exhibit a distinct phenotype, consistent with the disease state. Moreover, the development of a tri-dimensional, contractile muscle organoid model, whose generation is now being optimized, could better recapitulate the disease by more accurately reflecting myofibrillar architecture and energetic requirements. Indeed, upon proper stimulation, the structure contracts, allowing the investigation of perilipin 4 aggregate formation and its potential mechanical disturbance in muscle contraction.
Conclusion: This combined use of bi-dimensional and tri-dimensional models will provide a more comprehensive understanding of the protein's pathogenic and physiological roles. Although a complete assessment of these stable C2C12 lines is still ongoing, preliminary findings demonstrate their utility as a robust model for the investigation of this rare myopathy.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Distal Myopathies
PP01.22
Microrna Profiling in Patients With plin4-Related Myopathy
Ms. Eliana Iannibelli1, Dr. Alessandra Carnazzi1, Ms. Irene Sambruni2, Dr. Erika Salvi2, Ms. Lucia Nicolini De Gaetano1, Mr. Franco Salerno1, Dr. Isabella Moroni3, Dr. Lorenzo Maggi1, Dr. Alessandra Ruggieri1, Dr. Sara Gibertini1
1
Neuroimmunology and Neuromuscular Diseases Unit – Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy.
2
Data Science Center and Computational multi-Omics of Neurological Disorders (MIND) Lab – Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy.
3
Department of Pediatric Neurosciences – Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy
Background: PLIN4‑related distal myopathy is a rare autosomal dominant disorder caused by a PLIN4 expansion resulting in protein aggregation. Perilipin 4 accumulates at the sarcolemma and in sarcoplasmic vacuoles, disrupting myofiber organization and contractility. Protein aggregation and defective aggrephagy, traditionally associated with neurodegeneration, are widely recognized as key contributors to several inherited and sporadic muscle disorders. To elucidate underlying molecular pathways, we analysed the miRNome of PLIN4‑associated myopathy and compared it with other vacuolated myopathies with protein aggregates, including Pompe disease (PD), Inclusion Body Myositis (IBM), and Oculopharyngeal Muscular Dystrophy (OPMD). miRNAs have recognized as key post‑transcriptional regulators, and this makes them promising tools for dissecting molecular pathways and for identifying future diagnostic biomarkers.
Methods: Primary myoblasts were derived from human muscle biopsies.
Libraries were generated from total RNA using standard protocols and sequenced on a high‑throughput platform. Raw reads were quality-checked using FastQC v0.12.1 and trimmed with Trim Galore v0.6.10. The reads were aligned to miRBase (Release 22.1) using Bowtie v1.3.1 and quantified using Samtools (v1.22.1). Lowly expressed miRNAs were filtered out, and differential expression analysis was performed with DESeq2 (v1.44.0, R version 4.4.1). Comparisons were performed between PLIN4-related myopathy (N=7) and PD (N=5), IBM (N=4), OPMD (N=5), as well as non-vacuolated myopathies (N=5). Only significant miRNAs (padj < 0.05) were considered for target identification using the R package multiMiR (v1.26.0) by querying validated miRNA–target interaction databases. Interactions supported by at least two databases were retained and used for over-representation analysis with the online tool WebGestalt.
Results: Over-representation analysis was performed separately for each set of differentially expressed miRNAs identified in each comparison group. Principal Component Analysis (PCA), shows a clear clustering separation between vacuolated and non-vacuolated myopathies, supporting the presence of disease‑specific regulatory signatures.
Intriguingly, the analysis highlighted a strong separation of PLIN4-related myopathy from IBM and non-vacuolated myopathy samples with twenty miRNAs differentially expressed. However, only three were differentially expressed compared to OPMD. Remarkably, the common miRNA up-regulated across all three comparisons was miR-532-5p. The comparison with IBM and non-vacuolated samples, found miR-199b-5p as downregulated. No differentially expressed miRNAs were detected when comparing to PD. These results highlight a disease-specific miRNA profile in PLIN4-related myopathy in primary myoblasts.
Conclusion: Our findings demonstrate that unbiased miRNA profiling is a valuable tool for uncovering reliable molecular biomarkers and disease‑related regulatory pathways in rare muscle disorders.
The PLIN4‑related myopathy specific miRNA signature, in the primary myoblasts, still requires deeper analysis regarding involved pathways and their biological significance. The obtained results will be combined with miRNA profiling of muscle tissues, which is currently in progress.
Further analysis will include functional validations in vitro to assess their biological relevance and regulatory impact. In parallel, their diagnostic potential will be evaluated in patients’ sera to determine their suitability as minimally invasive biomarkers.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Distal Myopathies
PP01.23
Desmin-Related Myopathy in a Korean Patient with Distal Myopathy and Atrial Fibrillation
Dr. Hyung Jun Park1, Dr. Yunjung Choi1, Dr. Young-Chul Choi1, Dr. Jeong Hee Cho2, Dr. Bum Chun Suh3, Dr. Sang Beom Kim4, Dr. Woo-Kyung Kim5, Dr. Yang-Ki Minn6
1
Department of Neurology, Gangnam Severance Hospital, Younsei University College of Medicine, Seoul, Korea, Republic of.
2
Department of Neurology, National Health Insurance Service Ilsan Hospital, Goyang, Korea, Republic of.
3
Department of Neurology, Kangbuk Samsung Hospital, Sungkyunkwan University School of Medicine, Seoul, Korea, Republic of.
4
Department of Neurology, Kyung Hee University Hospital at Gangdong, Kyung Hee University School of Medicine, Seoul, Korea, Republic of.
5
Department of Neurology, Kangdong Sacred Heart Hospital, Hallym University College of Medicine, Seoul, Korea, Republic of.
6
Department of Neurology, Kangnam Sacred Heart Hospital, Hallym University College of Medicine, Seoul, Korea, Republic of
Background: Desmin-related myopathy (desminopathy) is a myofibrillar myopathy caused by pathogenic variants in the DES gene, typically affecting both skeletal and cardiac muscle. It often presents with progressive skeletal myopathy, cardiomyopathy, and arrhythmias, which can obscure the recognition of a unifying cardioskeletal disorder. In Korea, desminopathy has been reported mainly based on pathological findings, with few genetically confirmed cases. Here, we report a Korean patient with late-onset, distal-predominant myopathy and atrial fibrillation harboring a pathogenic DES variant.
Methods: We reviewed clinical features, serum creatine kinase (CK), electromyography, and muscle computed tomography (CT) findings. Targeted sequencing was performed using the NextSeq550Dx/2000 System (Illumina).
Results: A 65-year-old man presented with a 10-year history of progressive gait disturbance beginning in his mid-fifties. He had undergone catheter ablation for atrial fibrillation three years earlier and had been otherwise healthy and physically active. Neurological examination revealed asymmetric lower-limb weakness with distal predominance, impaired toe and heel walking, and steppage gait. There was no family history of neuromuscular disease. Serum CK was mildly elevated (549 U/L). Electromyography showed myopathic changes. Lower limb muscle CT demonstrated symmetric fatty atrophy of the peroneus longus and brevis bilaterally, with mild subcutaneous edema. Targeted sequencing identified a heterozygous missense DES variant, NM_001927.4:c.35C>T (p.Ser12Phe), located in exon 1.
DES encodes desmin, the major intermediate filament protein in cardiac, skeletal, and smooth muscle. Dysfunctional desmin disrupts cytoskeletal integrity and myofibrillar organization, contributing to both skeletal myopathy and cardiac dysfunction. Desminopathy exhibits wide phenotypic heterogeneity, influenced by inheritance patterns and the location of pathogenic variants within the desmin molecule. Most cases present in the third to fourth decade with progressive distal weakness and variable cardiac involvement. Our patient developed symptoms in his mid-fifties and had atrial fibrillation, highlighting an unusually late onset and expanding the phenotypic spectrum. The coexistence of distal myopathy and cardiac arrhythmia should raise suspicion for desminopathy and underscores the diagnostic value of targeted gene panel testing. Given that cardiac manifestations are common and may evolve over time, systematic cardiologic evaluation and longitudinal follow-up are warranted even in predominantly skeletal presentations.
Conclusion: We report a genetically confirmed Korean case of desminopathy due to a pathogenic DES variant, broadening the clinicogenetic spectrum of this disease. This case emphasizes the importance of cardiac evaluation in patients presenting with distal myopathy.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Myofibrillar Myopathies
PP01.24
Expansion of the Clinical and Genetic Spectrum of hnRNPA1-Associated Distal Myopathy in Two Korean Families
Dr. Hyung Jun Park1, Dr. Yunjung Choi2, Dr. Young-Chul Choi1, Dr. Bum Chun Suh3, Dr. Sang Beom Kim4, Dr. Jeong Hee Cho5, Dr. Woo-Kyung Kim6, Dr. Yang-Ki Minn7
1
Department of Neurology, Gangnam Severance Hospital, Younsei University College of Medicine, Seoul, Korea, Republic of.
2
Department of Neurology, Sanggye Paik Hospital, Inje University College of Medicine, Seoul, Korea, Republic of.
3
Department of Neurology, Kangbuk Samsung Hospital, Sungkyunkwan University School of Medicine, Seoul, Korea, Republic of.
4
Department of Neurology, Kyung Hee University Hospital at Gangdong, Kyung Hee University School of Medicine, Seoul, Korea, Republic of.
5
Department of Neurology, National Health Insurance Service Ilsan Hospital, Goyang, Korea, Republic of.
6
Department of Neurology, Kangdong Sacred Heart Hospital, Hallym University College of Medicine, Seoul, Korea, Republic of.
7
Department of Neurology, Kangnam Sacred Heart Hospital, Hallym University College of Medicine, Seoul, Korea, Republic of
Background: Distal myopathies are a group of clinically and genetically heterogeneous myopathies characterized by progressive weakness and atrophy of the distal muscles, with 24 causative genes identified. Distal myopathy-3 (MPD3) was first described in a large Finnish pedigree in 2003, and its causative gene, HNRNPA1, was later identified in 2021. Here, we describe two unrelated Korean families with autosomal dominant distal myopathy, in whom we identified a pathogenic exon 10 deletion in HNRNPA1.
Methods: We reviewed clinical characteristics, electromyography, laboratory findings, and muscle biopsy results of affected individuals from two unrelated families. PacBio Revio long-read sequencing and Sanger sequencing were used to detect a deletion in HNRNPA1.
Results: In family MF1594, the proband, a 42-year-old man, developed hand muscle weakness at 13 years of age. Muscle weakness and atrophy were pronounced in the intrinsic hand and forearm muscles, but only mild ankle dorsiflexor weakness was observed without leg muscle atrophy. A muscle biopsy revealed no pathological abnormalities. The other affected family members exhibited similar clinical courses. They developed hand weakness in mid-adolescence followed by lower-limb involvement, including steppage gait in the third decade. Asymmetric involvement was consistently observed and mild neck weakness and dysphagia were noted. The course was indolent; one affected individual remained ambulatory with ankle–foot orthoses at 71 years, with largely preserved proximal strength.
In family MF443, the proband, a 68-year-old woman, reported hand weakness beginning 8 years of age. The disease progressed extremely slowly; however, writing and fine motor skills became difficult after 40 years of age, and foot drop developed at 45 years. Affected relatives followed a similar trajectory; despite severe weakness, one individual retained limited ambulation into the mid-sixties. Dysarthria and dysphagia were absent. Muscle biopsies demonstrated myopathic changes, with rimmed vacuoles identified in one severely affected individual.
Long-read sequencing of probands from two unrelated families identified a 317-base pair deletion in HNRNPA1 (NM_031157.4: c.1063+74_*4+40del; NP_112420.1:p.Gly304Asnfs*4) spanning exon 10. Sanger sequencing identified breakpoint junctions and confirmed the co-segregation of the deletion with the affected members of families MF443 and MF1594.
Heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1), encoded by HNRNPA1, is an RNA-binding protein involved in RNA metabolism and interacts with several proteins implicated in a spectrum of neuromuscular and neurodegenerative disorders. Two MPD3 phenotypes have been reported: a classic adult-onset autosomal dominant distal myopathy in Finnish families featuring distal hand weakness and gait disturbance with frequent stumbling and asymmetric involvement, and a rare childhood-onset form characterized by rapidly progressive, generalized myopathy. Our findings further support a genotype–phenotype correlation whereby exon 10 deletions are associated with a slowly progressive, hand-predominant distal phenotype.
Conclusion: This is the first report of HNRNPA1-associated distal myopathy in Korea. Our findings expand the known geographic and ethnic spectrum of HNRNPA1-related myopathies, indicating that this disorder is not confined to the Finnish population but represents a globally distributed neuromuscular disease entity.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Distal Myopathies
PP01.25
Myotonia in PLIN4-Related Myopathy: Investigating the Role of Ion Channels
Dr. Paola Laghetti1, Assoc. Prof. Concetta Altamura1, Dr. Ilaria Saltarella1, Dr. Isabella Moroni2, Dr. Alessandra Ruggieri3, Dr. Sara Gibertini3, Dr. Lorenzo Maggi3, Prof. Jean-François Desaphy1
1
Section of Pharmacology, Department of Precision and Regenerative Medicine and Ionian Area, School of Medicine, University of Bari “Aldo Moro”, Bari, Italy.
2
Department of Pediatric Neurosciences, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy.
3
Neuroimmunology and Neuromuscular Disease Unit, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy
Background: PLIN4-related myopathy is a rare vacuolar myopathy characterized by distal muscle weakness, caused by a pathological expansion of a 33-aminoacids sequence within the amphipathic helix of Perilipin 4 (PLIN4). This mutation leads to protein misfolding and aggregation in the subsarcolemmal region of myofibers, resulting in vacuole formation due to activation and impairment of the aggrephagy pathway. These alterations thereby disrupt the skeletal muscle fiber structure and its contractile function. In addition to myopathic symptoms, clinical myotonia has also been observed in a subset of patients affected by PLIN4-related myopathy. Myotonic syndromes are typically associated with mutations in the CLCN1 and SCN4A genes, encoding for the ClC-1 and Nav1.4 ion channels, respectively. Therefore, this study aimed to investigate the role of these ion channels in the pathogenesis of myotonia in patients with PLIN4 mutation, assessing their gene and protein expression.
Methods: Muscle biopsies from an Italian family affected by PLIN4-related myopathy and from patients without myopathy used as controls were used for proteins and RNA extraction. Gene expression of ion channels involved in myotonic syndromes (CLCN1 and SCN4A) and selectively expressed in denervated muscle cells (SCN5A) was assessed by SYBR Green quantitative PCR. Protein expression of ClC-1 and Nav1.4 was evaluated by Western blot analysis.
Results: The expression of CLCN1 and SCN4A genes was significantly increased in patients carrying the PLIN4 mutation compared with control subjects. Similarly, when patients were stratified according to the presence or absence of myotonia, both groups showed a trend toward increased expression of these genes compared with controls. However, these changes at the transcriptional levels were not associated with similar changes in protein expression, as no significant differences in protein levels were detected between control subjects and patients, regardless of myotonia. Together, these data suggest that the pathogenesis of myotonia is more likely related to functional alteration of ion channels rather than to changes in their expression levels. In addition, SCN5A gene expression was similar between patients and controls, indicating that the altered muscle excitability is not a consequence of impaired muscle innervation.
Conclusion: This study provides a first insight into the myotonic phenomenon observed in a subset of patients affected by PLIN4-related myopathy. Our data suggest that myotonia might be driven by functional dysregulation of ion channels, secondary to structural and proteostasis abnormalities induced by PLIN4 aggregates, rather than alterations in their expression. Additional studies are needed to further elucidate the relationship between PLIN4 protein aggregates and ion channel regulation, leading to the identification of potential therapeutic targets for the management of myotonia in patients affected by PLIN4-related myopathy.
This work was funded by the Italian Ministry of Health and the European Union (Project code: PNRR-MR1-2023-12377360).
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Distal Myopathies
PP01.26
H2S-Based Strategies for Duchenne Muscular Dystrophy
Ms. Anna Nalepa1,2, Ms. Kalina Hajok1, Ms. Małgorzata Myszka1, Ms. Ewa Jakubczak1, Ms. Urszula Waśniowska1, Dr. Olga Mucha1, Dr. Urszula Florczyk-Soluch1, Assoc. Prof. Józef Dulak1, Assoc. Prof. Agnieszka Łoboda1
1
Jagiellonian University, Kraków, Poland.
2
Doctoral School of Exact and Natural Sciences, Jagiellonian University, Kraków, Poland
Background: Duchenne muscular dystrophy (DMD) is a severe genetic disorder caused by mutations in the DMD gene, leading to dystrophin deficiency and progressive skeletal and cardiac muscle damage. Hydrogen sulfide (H2S), once considered toxic, is now recognized as a cytoprotective gasotransmitter. We previously found altered expression of H2S-producing enzymes in dystrophic muscles, suggesting H2S deficiency may contribute to DMD pathogenesis1.
Methods: Using two murine DMD models (mdx and D2.mdx), we tested H2S supplementation via fast- (NaHS) and slow-releasing (GYY4137) donors, and genetic overexpression of the H2S-producing enzyme cystathionine γ-lyase (CTH).
Results: NaHS altered the expression of DMD-related genes and proteins but did not improve muscle function2. In contrast, GYY4137 significantly enhanced exercise capacity and reduced muscle damage markers, such as lactate dehydrogenase (LDH), creatine kinase (CK), and osteopontin (OPN). Histological, gene, and protein analyses of the gastrocnemius muscle and diaphragm revealed decreased inflammation and fibrosis, along with reduced necrosis, improved muscle regeneration, and enhanced angiogenesis. The upregulation of phosphorylated AMPKα and cytoprotective and antioxidant heme oxygenase-1 (HO-1) and mitochondrial superoxide dismutase (SOD2), together with anti-apoptotic effects manifested by reduced caspase-3 levels and increased AKT phosphorylation, was evident after the treatment with a slow releaser of H2S. Overall, GYY4137 ameliorated DMD progression in the skeletal muscle3 and exerted cardioprotective effects. CTH overexpression also modulated key DMD hallmarks.
Conclusion: These results demonstrate that increasing H2S levels confers protection against DMD pathology and highlight the therapeutic potential of H2S-based strategies for slowing disease progression and developing mechanism-based interventions.
Acknowledgements: This work was supported by grant #2019/35/B/NZ3/02817 (to AŁ) from the National Science Centre.
References:
1. Mucha O.; Myszka M.; Podkalicka P.; Świderska B.; Malinowska A.; Dulak J.; Łoboda A.Proteome Profiling of the Dystrophic mdx Mice Diaphragm. Biomolecules, 2023, 13, 1648.
2. Myszka M.; Mucha O.; Podkalicka P.; Waśniowska U.; Dulak J.; Łoboda A. Sodium hydrosulfide moderately alleviates the hallmark symptoms of Duchenne muscular dystrophy in mdx mice. Eur J Pharmacol, 2023, 955, 175928.
3. Myszka M.; Jakubczak E.; Mucha O.; Hajok K.; Waśniowska U.; Nalepa A.; Dulak J.; Łoboda A. GYY4137, a Slow-Releasing Hydrogen Sulfide Donor, Attenuates Skeletal Muscle Abnormalities in a Murine Model of Duchenne Muscular Dystrophy. Antioxid Redox Signal, 2025, 43, 115-137
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
P01.27
Assessment of the Impact of Modifier Genes on the Clinical Course of Ambulatory DMD Patients
Dr. Ana Kosac1,2, Dr. Dragana Vucinic1, Prof. Natasa Cerovac1,2, Assoc. Prof. Jovan Pesovic3, Assoc. Prof. Milos Brkusanin3, Dr. Jelena Mladenovic1, Ms. Jelena Martinovic1, Prof. Dusanka Savic Pavicevic3, Prof. Vedrana Milic Rasic2
1
Clinic of neurology and psychiatry for children and youth, Belgrade, Serbia.
2
School of Medicine, University of Belgrade, Belgrade, Serbia.
3
Faculty of Biology, University of Belgrade, Belgrade, Serbia
Background: To investigate the influence of SPP1, LTBP4, and CD40 gene variants on disease progression in ambulatory patients with Duchenne muscular dystrophy (DMD).
Methods: Normality of distribution was assessed using the Kolmogorov–Smirnov test. A paired t-test for dependent samples was used to compare North Star Ambulatory Assessment (NSAA) scores obtained at baseline and after 12 months in subgroups defined by genotype.
Results: Twenty ambulatory DMD patients were followed for 12 months and evaluated using the NSAA. Patients were stratified according to the rs28357094 SPP1 variant, LTBP4 haplotype, and rs1883832 CD40 genotype. The mean age was 10.15 ± 2.35 years (range 7–16 years). Mutations in the DMD gene included 65% deletions, 30% small mutations, and 5% duplications. Proximal mutations involving Dp427 and Dp260 were present in 40% of patients. All patients were receiving glucocorticoid therapy. During the one-year follow-up, three patients lost independent ambulation.
The TT genotype of the rs28357094 SPP1 variant was present in 60% of patients. At baseline, NSAA scores were similar between genotypes (23.7 vs 23.0 for TT vs TG/GG). Over 12 months, patients carrying the protective T allele showed a decline in mean NSAA from 23.7 ± 8.0 to 20.8 ± 8.7 (p = 0.007), while patients with TG/GG genotypes declined from 23.0 ± 8.5 to 18.9 ± 11.6. The mean NSAA change was −2.9 ± 3.0 in T-allele carriers and −4.1 ± 4.5 in TG/GG carriers.
The rarer T allele of the rs1883832 CD40 variant was present in 70% of patients. These patients had higher baseline NSAA scores. During follow-up, T-allele carriers showed a decline from 24.8 ± 8.0 to 22.1 ± 9.4 (p = 0.02), whereas CC genotype carriers declined from 20.3 ± 7.6 to 15.2 ± 9.3 (p = 0.008). The mean NSAA change was −2.6 ± 3.7 in T-allele carriers and −5.2 ± 2.9 in CC carriers.
Eight patients carried at least one protective IAAM haplotype of the LTBP4 gene. These patients showed a smaller NSAA decline, from 28.4 ± 5.3 to 27.0 ± 6.0, compared with patients without a protective haplotype, whose scores declined from 20.2 ± 8.0 to 15.4 ± 9.0 (p = 0.001). The mean NSAA change was −1.4 ± 2.0 in patients with protective IAAM haplotype and −4.8 ± 3.9 in patients without a single protective IAAM haplotype.
Conclusion: Over one year of follow-up, ambulatory DMD patients carrying protective variants in the SPP1 and CD40 genes and/or at least one protective LTBP4 haplotype showed a smaller decline in NSAA scores compared with patients without protective genotypes, suggesting a modifying effect on disease progression.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
PP01.28
Cell-Cell Communication Reveals Novel Mechanistic Insights into Duchenne Muscular Dystrophy
Miss Annabel Sen, Dr. Kevin Chesmore, Dr. Florian Barthelemy, Dr. Sophie Renegarajan, Assoc. Prof. Stanley Nelson, Assoc. Prof. Carrie Miceli
UCLA, Los Angeles, United States
Background: Skeletal muscle is a cellularly complex and plastic tissue that responds to local injury by recruiting and expanding immune cells, fibroblasts and other muscle resident cells to guide satellite cell-mediated repair of damaged multinucleated myofibers. In Duchenne muscular dystrophy (DMD), lack of dystrophin leads to chronic muscle damage, immune infiltration, dysregulation of repair, fibro-fatty replacement, and loss of muscle function. Our understanding of the cell and molecular mechanisms regulating dystrophy and repair remain incomplete, especially in humans. Cell-cell communication using single cell and single nucli sequencing can reveal how myofibers interact with surrounding immune cell-types leading to pathogenicity. These communication networks as well as analysis into downstream signaling targets can reveal novel insights into the mechanism of dystrophinopathies. Furthermore, the identification of unique cell populations that create autoimmune disruptions can be investigated for novel therapeutic interventions.
Methods: Muscle biopsies from the Miceli and Nelson lab BioBank were submitted for single-cell and single-nucleus sequencing. Raw count matrices were filtered to remove low-quality cells or nuclei, doublets, and ambient RNA, then normalized and subjected to dimensionality reduction and graph-based clustering. Clusters were annotated using canonical markers to identify myogenic lineages, fibroblast, endothelial and smooth muscle cells, macrophages, T and B lymphocytes. To quantify intercellular communication, curated ligand–receptor databases and a probabilistic framework, such as NicheNet and OmniPath, were applied to cluster-averaged expression profiles. For each sender–receiver pair, ligand activity scores were computed, predicted target genes were inferred in receiver cells, and interactions were ranked based on expression, specificity, and concordance with downstream transcriptional programs. Differential expression analyses between DMD and control within each major cell type were used to identify disease-associated signaling. Gene set enrichment and pathway analyses highlighted inflammatory, fibrogenic, and regenerative pathways, while transcription factor activity and regulatory network modeling linked upstream ligands to downstream effectors in myogenic and immune populations.
Results: Single-cell and single-nucleus profiling revealed a marked expansion of pro-inflammatory macrophages, cytotoxic and regulatory T cells, and fibroblasts in DMD compared with controls, consistent with chronic immune activation and fibro-fatty remodeling. Ligand–receptor analysis demonstrated an enrichment of inflammatory cytokine and chemokine interactions from macrophages and T cells to myofibers, satellite cells, and fibroblasts in DMD muscle. Fibroblasts in DMD displayed upregulation of distinct ligands and receptors, suggesting profibrotic signaling in the dystrophic niche. Dystrophic myonuclei exhibited transcriptional signatures of heightened stress, cytokine responsiveness, and impaired differentiation, including activation of pathways involved in apoptosis, interferon signaling, and aberrant regeneration. Downstream target gene analyses linked specific immune ligands to altered transcriptional programs in myogenic populations, revealing candidate axes through which the diseased niche perpetuates myofiber instability.
Conclusion: Transcriptomic mapping of human dystrophic muscle reveals a profoundly remodeled inflammatory and fibrogenic niche in which specific ligand–receptor circuits between immune cells, fibroblasts, and myogenic populations drive chronic degeneration, impaired repair, and nominate discrete signaling axes and altered cell states as tractable therapeutic targets in Duchenne muscular dystrophy.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
PP01.29
Automated Patch-Clamp Electrophysiology for Drug Discovery and Genotype-Phenotype Correlation in Skeletal Muscle and Pain Disorders
Assoc. Prof. Paola Imbrici, Dr. Alessandro Giovanni Cerchiara, Dr. Brigida Boccanegra, Assoc. Prof. Paola Mantuano, Prof. Antonella Liantonio, Assoc. Prof. Ornella Cappellari, Prof. Annamaria De Luca
Department of Pharmacy-Drug Sciences, University of Bari, Bari, Italy
Background: Ion channel dysfunction underlies a wide spectrum of neuromuscular and pain disorders, including inherited skeletal muscle channelopathies, neuropathic pain syndromes, and Duchenne muscular dystrophy (DMD) (Cerchiara Biomed Pharm 2025; Cerchiara Ann N Y Acad Sci 2024). Robust and reproducible electrophysiological platforms are essential both for preclinical drug discovery and for elucidating genotype–phenotype correlations, particularly in diseases characterized by marked genetic heterogeneity. This study aimed (i) to evaluate automated patch clamp as a reliable tool for drug screening on voltage-gated sodium channels, focusing on the Nav1.7 channel and its modulation by mexiletine (Mex), and (ii) to investigate genotype–phenotype correlations in DMD by characterizing ion channel function and expression in patient-derived myogenic cell lines.
Methods: Automated whole-cell recordings were performed using the Patchliner platform. For Nav1.7 pharmacology, sodium currents were studied in TE671 cells endogenously expressing Nav1.7 and in HEK293 cells stably expressing human Nav1.7, assessing voltage-dependent, tonic, and use-dependent block by Mex. For DMD studies, two immortalized myogenic cell lines derived from patients DMD1 (harbouring a nonsense mutation in exon 59) and DMD2 (carrying a deletion spanning exons 48–50) and a healthy control line (hWT) were analyzed during differentiation. Electrophysiological parameters were combined with real-time PCR to evaluate ion channel function and gene expression.
Results: Mex produced comparable voltage-dependent, tonic, and use-dependent block of Nav1.7 currents in both TE671 and HEK293 cells, with similar IC₅₀ values under different holding potentials and stimulation frequencies. Mex also induced a dose-dependent hyperpolarizing shift of steady-state inactivation in both models, confirming its state-dependent mechanism of action.
In DMD1/2 myogenic cells, membrane capacitance was reduced compared to control during differentiation. Inward current density increased with differentiation only in hWT, while both dystrophic cell lines showed stable inward currents over time. Outward currents increased during differentiation in all cell lines. Notably, expression of several ion channel genes (SCN4A, SCN5A, CACNA1S, KCNJ2, KCNQ4) was reduced in DMD1 myotubes, compared to DMD2 and hWT cells, indicating mutation-specific phenotypic differences.
Conclusion: Automated patch clamp is a powerful and versatile approach for both drug screening and functional phenotyping of ion channel disorders. The platform reliably captured the state-dependent block of Nav1.7 by mexiletine and validated the two cell models for analgesic drug discovery. Furthermore, Patchliner enabled the identification of mutation-specific electrophysiological and molecular phenotypes in DMD, supporting its utility for genotype–phenotype correlation studies and the development of personalized therapeutic strategies.
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: Electrophysiology
PP01.30
Dysregulation of LKB1 in Duchenne Muscular Dystrophy: Disease Specificity and Epigenetic Regulation by HDAC Inhibitors
Dr. Brigida Boccanegra1, Dr. Lisamaura Tulimiero1, Dr. Raffaella Quarta1, Assoc. Prof. Elena Conte1, Dr. Simonetta Andrea Licandro2, Dr. Alessandra Decio2, Dr. Roberta Lenti1, Dr. Alberto Ladisa1, Dr. Giorgia Dinoi1, Dr. Letizia Claudione1, Assoc. Prof. Sabata Pierno1, Assoc. Prof. Paola Mantuano1, Assoc. Prof. Ornella Cappellari1, Dr. Gianluca Fossati2, Dr. Christian Steinkühler2, Prof. Annamaria De Luca1
1
Department of Pharmacy – Drug Sciences, University of Bari “Aldo Moro”, Bari, Italy.
2
Preclinical R&D Department, Italfarmaco S.p.A., Cinisello Balsamo, Milan, Italy
Background: Efficient skeletal muscle contraction relies on tight mechano-metabolic coupling regulated by AMP-activated protein kinase (AMPK). Duchenne muscular dystrophy (DMD) is characterized by aberrant AMPK activation and altered metabolic signalling. Here, we investigated the expression and regulation of liver kinase B1 (LKB1), the main upstream activator of AMPK. LKB1 activation occurs with the formation of a heterotrimeric complex with two accessory proteins, the pseudokinase STE20-related adaptor α (STRADα) and the scaffolding mouse protein 25 (MO25). We previously provided the first evidence that LKB1 and its partners are severely downregulated, at gene and protein level, in gastrocnemius muscle (GC) of 6-months-old BL10 mdx and D2mdx mice, the new murine model with a more severe dystrophic phenotype (Boccanegra et al, DMM 2023).
Methods: With the aim to assess if the impairment of LKB1 expression is a neuromuscular disease-specific feature, we analysed the expression of LKB1-STRADA-MO25 genes in tibialis anterior (TA) muscles of 15-week-old SOD1G93A mice, the primary mouse model for amyotrophic lateral sclerosis. Then, we investigated the dynamics of LKB1-STRADA-MO25 expression in early and in chronic DMD pathology phase, by qRT-PCR analyses in GC, diaphragm (DIA) and heart of the two murine dystrophic models throughout lifespan (4,8,28,52 weeks of age). Strain-matched WT mice were used as controls. Furthermore, we evaluated the gene expression profile of the complex in patient-derived immortalized cells (hDMD, deletion in exon 48-50) compared to WT human cells, throughout myogenesis (0,4,6,11 days of differentiation). Moreover, with the aim of detecting the precise localization of the kinase, we performed immunofluorescence analyses (IF) for LKB1 in human WT and dystrophic cells (11 days of differentiation).
Results: qRT-PCR analysis showed that the expression of LKB1-STRADA-MO25 genes was similar SOD1G93A mice in comparison with WT controls. In contrast, the expression of all three genes was severely downregulated in GC and DIA of both dystrophic mouse models, at all ages, with an overall reduction of more than 50% vs. background-related WT mice. Furthermore, heart muscles of both mdx mice models, as well as D2 WT mice, displayed a downregulation of LKB1 only at a late stage of disease (52 weeks). Notably, an 8-week treatment with the pan-HDAC inhibitor vorinostat (5 mg/kg, 5 times a week, i.p.) restored LKB1 gene and protein expression in D2mdx mice (5 weeks of age), with recovery scores (RS) of 197% and 204%, respectively. This effect correlated with significant downregulation of miR-451, miR-195 and miR-17, known post-transcriptional repressors of LKB1. In contrast, the selective HDAC1/2 inhibitor Rodin-A (4 mg/kg, i.p.) increased LKB1 mRNA (RS 138%) but did not restore protein levels or modify miRNA expression (Fig.1). Studies in patient-derived myoblasts (HDMD, deletion 48-50; AB1098) showed impaired LKB1 expression during differentiation (0, 6, 11 day). Moreover, LKB1 fluorescence was widespread in healthy cells but reduced and mainly nuclear in HDMD.
Conclusion: Overall, these findings identify the LKB1–STRADα–MO25 axis as a regulatory node disrupted in DMD and responsive to epigenetic modulation, highlighting its therapeutic potential. (supported by NL_DPP number 22.004, ERC SEEDS UNIBA 2023-UNBACLE-0245439, PRIN MUR 2020 Prot. 2020ELYA32)
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
PP01.31
Patient-Derived Skeletal Muscle-on-Chip as a Platform for Pre-Clinical Studies in Duchenne Muscular Dystrophy
Assoc. Prof. Ornella Cappellari1, Dr. Raffaella Quarta1, Dr. Mitchell Han2, Dr. Enrica Cristiano1, Dr. Manuel Marinelli1, Dr. Vincent Mouly3, Dr. Nikolas Gaio2, Prof. Annamaria De Luca1
1
Department of Pharmacy – Drug Sciences, University of Bari “Aldo Moro”, Bari, Italy.
2
BIOND Solutions B.V., Delft, Netherlands.
3
Sorbonne Université, Inserm, Institut de Myologie, Centre de Recherche en Myologie, Paris, France
Background: Duchenne muscular dystrophy (DMD) is an X‑linked recessive, muscle‑wasting neuromuscular disorder caused by mutations in the dystrophin gene, leading to progressive muscle degeneration, loss of function, and premature death, with no resolutive cure currently available1,2. Preclinical research has traditionally relied on 2D cell cultures and animal models, which partially reproduce human disease complexity and raise important issues in terms of time and ethics. Three‑dimensional, patient‑derived skeletal muscle organoids and muscle‑on‑chip systems have emerged as promising platforms for disease modelling and drug screening, with the high potential in developing patient‑specific models, enabling personalized medicine, and implementing the 3Rs policy by reducing animal use.
Methods: In this study, we developed hydrogel‑based skeletal muscle‑on‑chip constructs using three immortalized human myogenic cell lines obtained from the biobank MyoLine: a healthy control line (HWT), a DMD line (HDMD1) from a patient with a stop codon mutation in exon 59, and a DMD line (HDMD2) from a patient carrying an exon 48–50 deletion. Bundles were generated in microchips (BIOND Solutions) using a Matrigel/Geltrex and fibrin hydrogel, seeded at day 0, and cultured under differentiation conditions.
Results: Within 24 hours, bundles showed evident compaction, and a progressively organized 3D structure up to day 9 of differentiation. After 9 days of differentiation, HWT, HDMD1, and HDMD2 bundles displayed organized structures capable of eliciting a contractile response to electrical stimulation. Immunostaining for actin and sarcomeric α‑actinin demonstrated the presence of aligned, multinucleated myofibers and revealed the structural complexity and maturation state of the engineered tissues. In a larger series of experiments, fibrin‑based skeletal muscle organoids has been used to independently validate the platform for phenotypic profiling and preclinical applications. This analysis highlighted clear differences in muscle calcium-competent contractility between healthy and dystrophic organoids, particularly in force generation and resistance to fatigue over time. Interestingly, these functional differences tended to attenuate with prolonged culture in a stable physiological environment and in the absence of continuous nerve‑like stimulation, suggesting that a major component of the phenotype may be related to delayed myogenic maturation rather than fixed structural defects alone. This delay in differentiation program was also supported by expression of key myogenesis genes in the three cell-lines derived organoids.
Conclusion: Future work will refine the further characterization of the role of genetic background in phenotype and differentiation status and will explore how contraction‑induced stress and/or inflammation can unmask dystrophin‑related alterations in structure and function. Overall, this patient‑derived skeletal muscle‑on‑chip platform represents a feasible tool for disease modelling and preclinical studies in DMD and other rare neuromuscular disorders (supported by PNRR-Mnesys PE_00000006 project).
REFERENCES
1Aartsma‐Rus et al. 2006
2 Mercuri et al. 2019
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
PP01.32
Altered Sphingomyelin and Phosphocholine Profiles in Duchenne Cardiac Organoids
Dr. Lorenzo Fontanelli1, Ms. Carolina Ferri2, Mr. Adrian Florentin Suman3, Dr. Elisa Ceccherini3, Prof. Fabio Anastasio Recchia1, Prof. Gabriele Siciliano2, Prof. Giovanni Signore2, Dr. Silvia Rocchiccioli3
1
Health Science Interdisciplinary Center - Scuola Superiore Sant'Anna, Pisa, Italy.
2
University of Pisa, Pisa, Italy.
3
Istituto di Fisiologia Clinica, Consiglio Nazionale delle Ricerche, Pisa, Italy
Background: Duchenne muscular dystrophy (DMD) is caused by the absence of functional dystrophin - a protein encoded by the DMD gene - leading to increased membrane fragility and subsequent muscle degeneration. However, the specific effects of dystrophin deficiency on lipid composition in human cardiac cells remain largely unexplored. The aim of this study was to evaluate differences in the lipidomic landscape of human cardiac organoids modeling DMD.
Methods: Cardiac organoids were generated from a human induced pluripotent stem cell (hiPSC) line carrying a nonsense mutation (c.4375_4379del, exon 45) in the DMD gene using a direct, stepwise differentiation protocol. Simultaneously, hiPSC-derived cardiac organoids were developed from a control cell line. Five biological replicates were obtained for each condition. Lipids extraction was performed using a modified version of the Folch method. Lipid extracts were subjected to a targeted lipidomic analysis performed by HPLC-MS/MS. Statistical significance was determined using a false discovery rate threshold of <0.05.
Results: We found that nine phosphatidylcholine species and eight sphingomyelin species were significantly upregulated in the DMD condition compared to controls. Conversely, SM (38:1) and bis(monoacylglycerol)phosphates (BMPs), specifically BMP (16:1-18:1) and BMP (18:1-18:1), were significantly downregulated in DMD samples. Moreover, compared with controls, DMD samples exhibited a reduction in polyunsaturated fatty acids and an increase in monounsaturated fatty acids, accompanied by a shift toward shorter fatty acid chain lengths.
Conclusion: Cardiac organoids replicate key in vivo features of DMD, such as increased levels of PC (34:1). The observed alterations in fatty acid chain length and the degree of unsaturation are consistent with the enhanced membrane permeability reported in vivo. Moreover, since polyunsaturated fatty acids are more susceptible to oxidative stress, the increase in monounsaturated fatty acids at the expense of polyunsaturated ones may reflect an adaptive response to an elevated oxidative environment. Finally, the reduction in BMP levels may indicate lysosomal dysfunction.
Collectively, these results indicate that cardiac organoids accurately capture DMD-associated lipid remodeling and offer a powerful human-based platform for elucidating disease mechanisms and testing therapeutic approaches to restore membrane integrity and lipid homeostasis.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
PP01.33
Use of Psychopharmacological Treatments in Children and Adolescents With Duchenne Muscular Dystrophy: A Single-Centre Experience
Dr. Chiara Brusa1,2, Dr. Kate Waters2,3, Dr. Adnan Manzur2, Dr. Stephanie Robb2, Dr. Shira Rabinowicz2, Dr. Pinki Munot2, Dr. Mariacristina Scoto1,2, Prof. Giovanni Baranello1,2,4, Prof. Francesco Muntoni1,2,4, Dr. Anna Sarkozy1,2
1
Dubowitz Neuromuscular Centre, UCL Great Ormond Street Institute of Child Health, London, United Kingdom.
2
Dubowitz Neuromuscular Centre, Great Ormond Street Hospital for Children, London, United Kingdom.
3
Psychological Services, Great Ormond Street Hospital for Children, London, United Kingdom.
4
National Institute for Health Research Great Ormond Street Hospital Biomedical Research Centre, UCL Great Ormond Street Institute of Child Health, London, United Kingdom
Background: Duchenne Muscular dystrophy (DMD) may have a variable neurocognitive and neurobehavioural phenotype, largely related to the location of the DMD gene mutation. Intellectual disability affects 1/3 of DMD patients, while other frequent neurodevelopmental comorbidities include attention deficit hyperactivity disorder (ADHD) and autism spectrum disorder (ASD). Mood (anxiety, depression) and obsessive-compulsive disorder (OCD) symptoms, and behavioural problems are also common. Despite the increasing attention to the involvement of the central nervous system (CNS) in DMD, treatment of brain comorbidities remains an unmet need in this population, especially as regards to the role of psychopharmacological therapies for severe symptoms.
We aim to provide an update on the use of psychopharmacological treatments in a paediatric cohort of DMD patients.
Methods: Retrospective review of clinical notes of DMD patients aged 0-18 years seen at the Dubowitz Neuromuscular Centre, Great Ormond Street Hospital, London. We reviewed the use and type of psychopharmacological medications that are largely prescribed by external services, in particular the Child and Adolescent Mental Health Services (CAMHS) local to the patient.
Results: At the time of the study, the GOSH neuromuscular patients’ database included 252 DMD patients aged 0-18 years and, out of these, 13/252 (5%) were receiving a psychopharmacological therapy, either a CNS stimulant (5/13) for ADHD, or a selective serotonin reuptake inhibitor (8/13) for mood disorder/OCD. Median age at initiation of treatment was 13 years (range 9-16), with median duration of treatment being 2 years 5 months (range 5 months – 7 years 9 months). No side effects were reported by patients or families, nor observed during regular neuromuscular and cardiac follow-ups. All patients and/or caregivers verbally reported a perceived benefit with improvement of symptoms throughout the duration of treatment.
Conclusion: Data from this study expand the knowledge on the use of psychopharmacological therapies for paediatric patients with DMD and show preliminary, limited evidence of a positive profile both in terms of safety and efficacy once prescribed. Multicentre prospective studies are required to collect more robust data and develop evidence-based recommendations for this population to ensure a prompt start of psychopharmacological medications when required.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
PP01.34
Expanding the Phenotypic Spectrum of SYT2-Related Disorders: A Case Report of Distal Weakness and Fatigue
Dr. Elena Faedo1, Mrs Monica Traverso2, Dr. Sara Massucco1, Mrs Serena Baratto3, Dr. Chiara Panicucci4, Dr. Federica Trucco1,4, Assoc. Prof. Claudio Bruno1,4, Assoc. Prof. Marina Grandis1,5, Assoc. Prof. Chiara Fiorillo1,6
1
Department of Neurosciences, Rehabilitation, Ophthalmology, Genetic and Maternal and Infantile Sciences (DINOGMI), University of Genoa, Genoa, Italy.
2
Unit of Clinical Genetics, IRCCS Istituto Giannina Gaslini, Genoa, Italy.
3
Center of Translational and Experimental Myology, IRCCS Istituto Giannina Gaslini, Genoa, Italy.
4
Unit of Pediatric Neurology, IRCCS Istituto Giannina Gaslini, Genoa, Italy.
5
Unit of Neurology, IRCCS Ospedale Policlinico San Martino, Genoa, Italy.
6
Unit of Child Neuropsychiatry, IRCCS Istituto Giannina Gaslini, Genoa, Italy
Background: Mutations in the Synaptotagmin II (SYT2) gene—essential for synchronous neurotransmitter release—cause a rare presynaptic congenital myasthenic syndrome (CMS7A). Most recently, a length dependent distal hereditary motor neuropathy (dHMN) phenotype has been also associated with gain of function mutations of this gene. To date, less than 30 cases have been documented worldwide.
Methods: We describe the clinical, electrophysiological, imaging, and genetic evaluation of a patient followed at the IRCCS Istituto Giannina Gaslini (Genoa, Italy) for long-standing progressive distal weakness and fatigue. The diagnostic assessment included nerve conduction studies, repetitive nerve stimulation (RNS), lower limb muscle MRI, and Whole Exome Sequencing (WES) to achieve a molecular diagnosis.
Results: A 41-year-old woman presented with a childhood-onset of progressive distal lower limb weakness, fatigue, and pes cavus. Examination revealed bilateral ptosis, intrinsic hand muscle weakness (MRC 4), and severe distal leg atrophy (tibialis anterior MRC 3-4) with steppage gait. Alongside, she exhibited proximal weakness (iliopsoas MRC 4), a positive Gowers’ sign. Clinical examination revealed generalized areflexia, accompanied by reported early-morning muscle cramps and fluctuating paresthesia affecting both feet. Nerve conduction studies showed reduced CMAP amplitudes in right peroneal nerve with normal sensory findings. RNS of facial and median nerve revealed a pathological decrement at low frequencies, confirming impaired neuromuscular transmission. Muscle MRI demonstrated selective fatty degeneration in the posteromedial compartments of the distal legs, while the pelvic girdle showed reduced trophism without corresponding fatty replacement. Genetic analysis via WES identified a known pathogenic heterozygous variant in the SYT2 gene (NM_177401.5: c.923C>T, p.Pro308Leu) expected to cause a toxic gain of function. Symptomatic treatment with pyridostigmine resulted in partial improvement of fatigability.
Conclusion: This case highlights the phenotypic overlap of SYT2-related CMS. Our patient exhibited severe progressive distal amyotrophy associated with proximal weakness and unspecific fatigue. The absence of fatty infiltration in the proximal compartments suggests that the proximal deficit could be primarily driven by a functional synaptic block rather than structural muscle loss (2). SYT2 mutations should be considered in the differential diagnosis of “mixed” phenotypes presenting with distal amyotrophy and subtle myasthenic signs to ensure early access to symptomatic treatment(3)
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Congenital Myasthenic syndrome
PP01.35
Efficacy and Safety of Neridronate for the Treatment of Fragility Fractures in Duchenne Muscular Dystrophy
Dr. Chiara Panicucci1, Dr. Agnese Repetto1, Dr. Noemi Brolatti1, Dr. Marina Pedemonte2, Dr. Federica Trucco2,3, Dr. Alessia Angelelli4, Prof. Mohamad Maghnie4,3, Prof. Natascia Di Iorgi4,3, Prof. Claudio Bruno1,3
1
Centre of Translational and Experimental Myology, IRCCS Istituto Giannina Gaslini, Genova, Italy.
2
Pediatric Neurology and Muscle Diseases Unit, IRCCS Istituto Giannina Gaslini, Genova, Italy.
3
Department of Neurosciences, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health, DiNOGMI, University of Genova, Genova, Italy.
4
Department of Pediatrics, IRCCS Istituto Giannina Gaslini, Genova, Italy
Background: Children and adolescents with Duchenne muscular dystrophy (DMD) receiving long-term glucocorticoid therapy, are at high risk of secondary osteoporosis and fragility fractures. Bisphosphonates represent the main therapeutic option in this setting, by inhibiting osteoclast mediated bone resorption, improving bone strength and reducing fracture risk. Agents as pamidronate, alendronate, and zoledronate have been previously investigated in DMD patients. However, evidence on the efficacy and safety of neridronate in DMD remains limited, despite its established use in osteogenesis imperfecta and other forms of secondary osteoporosis.
The aim of this study was to evaluate the effects of intravenous neridronate on bone health in a cohort of glucocorticoid (GC)-treated DMD patients followed at IRCCS Institute Giannina Gaslini, Genova, presenting with fragility fracture, focusing on bone mineral density, fracture incidence, and treatment tolerability.
Methods: We conducted a retrospective, single-center, longitudinal study in patients with DMD treated with intravenous neridronate after at least one radiological confirmed fragility fracture. Lumbar spine (LS) and total body less head (TB) bone mineral density (BMD) and bone mineral content (BMC), as well as LS bone mineral apparent density (BMAD), were assessed by dual-energy X-ray absorptiometry (DXA) at baseline, before initiating neridronate treatment (T0), and after 24 months (T24) and 36 months (T36) of treatment. At the same time points, lateral spine X-ray was performed to assess vertebral fractures, quantified according to the Genant score. Finally, fracture incidence during follow-up, biochemical markers, and the safety profile of neridronate were assessed.
Results: Thirty-one of the 86 (36%) patients followed at our center who presented with at least one fragility fracture were enrolled in the study. Of these, 16/31 had vertebral fractures and 15/31 had low-trauma long-bone fractures; 8/31 reported multiple fragility fractures at baseline. Fractures occurred at a median age of 12.2 years (IQR: 9.1–15.2), after a median GC exposure of 5.75 years (IQR: 3.29–8.36). At T0, the median age was 14 years (IQR: 12–15). Twenty patients completed a 24-month follow-up, and 16 completed a 36-month follow-up. A significant and progressive improvement in LS BMD, BMD Z-score, BMC, and BMAD was observed over time (all p<0.05), indicating an increase in bone density. In contrast, TB BMD Z-scores did not significantly change, although TB BMC increased significantly, reflecting skeletal mineral accrual that remained below age and sex matched normative values. Three patients presented incident fractures during the treatment period (2 at T24 and 1 at T36). Biochemical markers of bone turnover did not significantly change during the treatment period. Neridronate was well tolerated; no serious complications occurred, and only mild and transient adverse events were reported, including arthralgia, fever, and mild, asymptomatic hypocalcaemia.
Conclusion: In glucocorticoid-treated patients with DMD presenting with fragility fractures, intravenous neridronate was safe and associated with a significant improvement in lumbar spine bone mineral density both at 24 and 36 months of treatment (+ 0.49 SDS and + 0.76 SDS respectively, all p<0.05), along with a low incidence of new fragility fractures. These findings support neridronate as a potential therapeutic option for the treatment of secondary osteoporosis in this population.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
PP01.36
Prospective Evaluation of Sleep-Disordered Breathing and Functional Assessment in Duchenne Muscular Dystrophy: DystREST Study
Dr. Amanda Maccarrone1,2, Dr. Lorenzo Chiarella3,4, Dr. Noemi Brolatti5, Mr. Simone Morando5, Dr. Ramona Cordani3,6, Ms. Antonella Iadarola6, Dr. Marco Veneruso3,6, Dr. Chiara Panicucci5, Prof. Carlo Minetti6, Prof. Martino Ruggieri1, Prof. Claudio Bruno5,6, Prof. Pasquale Striano2,6, Prof. Lino Nobili3,6, Dr. Marina Pedemonte2, Dr. Federica Trucco2,6
1
Unit of Pediatric Clinic, Department of Clinical and Experimental Medicine, University of Catania, Catania, Italy.
2
Pediatric Neurology and Muscular Diseases Unit, IRCCS Istituto Giannina Gaslini, Genova, Italy.
3
Child Neuropsychiatry Unit, IRCCS Istituto Giannina Gaslini, Genova, Italy.
4
. Department of Neurosciences, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health, University of Genova, Genova, Italy.
5
Centre of Translational and Experimental Myology, IRCCS Istituto Giannina Gaslini, Genova, Italy.
6
Department of Neurosciences, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health, University of Genova, Genova, Italy
Background: Sleep-disordered breathing (SDB) is a clinically relevant complication of Duchenne muscular dystrophy (DMD), encompassing obstructive and central events and nocturnal hypoventilation. Sleep is a vulnerable period during which respiratory abnormalities may precede daytime pulmonary decline. The association between sleep abnormalities, motor and respiratory functional decline, and biological dysregulation remain poorly defined in DMD. This study aims to characterize sleep disorders in DMD, pathogenesis and their role as disease biomarkers.
Methods: DystREST is an ongoing prospective, single-center observational study.
All participants underwent a standardized assessment including pulmonary (forced vital capacity, FVC; peak cough flow, PCF) and motor function tests (NSAA, PUL), overnight cardiorespiratory polygraphy (PG) with transcutaneous monitoring of gas exchange (oxycapnography, measuring tcPCO₂, SaO₂). Validated sleepiness questionnaires (PSQI, Epworth Sleepiness Scale, MEQ, Respicheck) were administered and objective assessment of circadian rhythm was conducted via actigraphy and salivary melatonin levels.
Clinical data, including age, ambulatory status, genotype, age at loss of ambulation (LOA), age at peak FVC and PCF, initiation of nocturnal non-invasive ventilation (NIV), comorbidities, and ongoing therapies were correlated with sleep disorders.
Results: Twenty male patients with genetically confirmed DMD have been enrolled to date. Age range was 8-32 years (mean 19.1). Preliminary results showed normal PG and nocturnal gas exchange findings in 17/20; 7 of these were already on NIV, supporting its effectiveness in maintaining adequate nocturnal ventilation.
PG identified SDB in 3/20 patients, with a predominance of obstructive events (2 out of 3); none of these three patients exhibited nocturnal hypoventilation. One ambulant, non-ventilated patient aged 10.8 years, showed combined central and obstructive events. Two non-ambulant, non-ventilated patients (aged 15 and 24.5 years, respectively) presented obstructive sleep apnea. The 15-year-old patient showed a more severe respiratory pattern and was affected by severe obesity.
In the subgroup of non-ventilated patients, pulmonary function was compared between subjects with and without SDB. No statistically significant differences were observed in FVC (p = 0.33), FVC% (p = 1.00), or peak expiratory flow (p = 0.22). However, despite the lack of statistical significance, patients with SDB consistently exhibited lower values.
There was a statistically significant positive correlation between age at peak absolute FVC and age at LOA (p = 0.005), indicating parallel progression of respiratory and motor decline. In a Cox proportional hazards model, later LOA was associated with a reduced hazard of NIV initiation (hazard ratio per year = 0.94, p = 0.59), although this association did not reach statistical significance. No significant correlations were observed between SDB severity and motor impairment or pulmonary function. PG findings were not significantly associated with self-reported sleep disturbances.
Analyses of potential circadian rhythm abnormalities recorded via actigraphy and salivary melatonin are ongoing.
Conclusion: Preliminary findings from the DystREST study suggest that clinically relevant SDB may occur in patients still ambulant, with normal pulmonary function and in the absence of hypoventilation and may therefore be overlooked.
Ongoing in-depth analysis of polygraphy data using diaphragm-specific SDB metrics may further enhance phenotypic characterization and the use of sleep as stage-specific disease biomarker in DMD.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
PP01.37
Preclinical Efficacy of ENTR-601-51 for the Treatent of Exon 51 Skip-Amenable Duchenne Muscular Dystrophy
Dr. Michael St. Andre, Dr. Nick Long, Dr. Xiang Li, Dr. Mahasweta Girgenrath, Ms. Mary Lou Beermann, Dr. Jia Qi Cheng-Zhang, Dr. Mohanraj Dhanabal, Dr. Haoming Liu, Dr. Christopher M. Brennan, Dr. Pauline Tan, Dr. Yongchao Mou, Dr. Terrance A. Stadheim, Dr. Wenlong Lian
Entrada Therapeutics, Boston, United States
Background: Antisense phosphorodiamidate morpholino oligomer (PMO)-mediated exon skipping therapies for Duchenne muscular dystrophy (DMD) have the potential to restore dystrophin expression in skeletal muscle tissue. To enhance PMO delivery to muscle tissues, we developed a family of therapeutics consisting of Endosomal Escape Vehicle (EEV) cyclic cell-penetrating peptides conjugated to DMD exon skipping PMOs. These EEV-PMO therapeutics utilize a common EEV construct that increases the likelihood of producing consistent and predictable pharmacokinetics across different exon skipping therapies. Preclinical studies have established the therapeutic potential of ENTR-601-44, ENTR-601-45, and ENTR-601-50 for patients with exon 44, 45, and 50 skip-amenable DMD, respectively. In addition, ENTR-601-44 was well tolerated and demonstrated significant exon 44 skipping compared to placebo in a phase 1 clinical trial in healthy human volunteers. Herein, we assessed the preclinical efficacy of ENTR-601-51, a novel EEV-PMO construct that targets exon 51 skip-amenable mutations in DMDpre-mRNA transcripts, to support its development as a potential therapy for patients with exon 51 skip-amenable DMD.
Methods: Skeletal muscle cell lines derived from patients with various DMD mutations amenable to exon 51 skipping were treated with ENTR-601-51 or vehicle, and exon 51 skipping and dystrophin protein levels were assessed. To assess the in vivo efficacy of ENTR-601-51, we utilized an exon 51 skip-amenable mouse model of DMD (del52hDMD.mdx). This model lacks endogenous mouse dystrophin and contains a human DMD transgene harboring a deletion of exon 52, thus resulting in a mouse model lacking any dystrophin expression. Upon skipping of exon 51, an internally truncated but functional dystrophin protein is produced. In skeletal and cardiac muscle of del52hDMD.mdx mice, exon 51 skipping levels and dystrophin protein levels were measured after in vivo treatment with ENTR-601-51 or vehicle. Changes in tetanic force and force retention during eccentric injury of del52hDMD.mdx gastrocnemius muscle were also measured.
Results: In patient-derived skeletal muscle cell lines with DMD exon 51 skip-amenable mutations, treatment with ENTR-601-51 resulted in robust, dose-dependent exon 51 skipping and dystrophin protein expression. In del52hDMD.mdx mice, in vivo treatment with ENTR-601-51 produced robust, dose-dependent exon 51 skipping and dystrophin protein expression in both skeletal and cardiac muscle tissues. This was accompanied by a significant increase in dystrophin-positive fibers as well as qualitative improvements in gastrocnemius muscle pathology. Importantly, a dose-dependent rescue of both specific tetanic force loss and improvement in force retention during eccentric injury assays was observed in the gastrocnemius muscle.
Conclusion: These results demonstrate that ENTR-601-51 is efficiently delivered to skeletal and cardiac muscle in vivo, thereby producing durable exon skipping and functional dystrophin protein that are able to rescue muscle contractile function in a relevant mouse model of human DMD. These findings support further evaluation of ENTR-601-51 in patients with DMD amenable to exon 51 skipping.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
PP01.38
Psychometric Validation of the DuMAND Checklist in Boys With Duchenne Muscular Dystrophy: A Belgian Cohort
Ms. Eline Cuveele1,2, Dr. Sam Geuens1,2, Ms. Kim Wittebols1, Ms. Joanna Willen1, Prof. Céline Gillebert2, Prof. Liesbeth De Waele1,2
1
University Hospitals Leuven, Leuven, Belgium.
2
KU Leuven, Leuven, Belgium
Background: Boys with Duchenne muscular dystrophy (DMD) are at increased risk for cognitive, behavioral and emotional difficulties that can substantially affect daily functioning, social participation and family life. Currently available behavioral questionnaires are largely generic and insufficiently capture neurobehavioral characteristics specific to DMD. The DuMAND (Duchenne Muscular Dystrophy-Associated Neurobehavioral Difficulties) Checklist was developed as a disease-specific instrument to systematically measure neurobehavioral functioning in boys with DMD across five categories: (1) cognition and learning, (2) social responsiveness, (3) externalizing behavior, (4) emotion regulation and (5) eating and sleeping.
Methods: This ongoing psychometric validation study uses a longitudinal design with three measurement timepoints at six-month intervals, aligned with routine clinical follow-up visits. Boys aged between 3 and 20 years with genetically confirmed DMD are recruited at the University Hospitals Leuven in Belgium. Parents or primary caregivers complete the DuMAND Checklist together with a battery of standardized questionnaires, including the Child Behavior Checklist (CBCL), the Behavior Rating Inventory of Executive Function (BRIEF), the Social Responsiveness Scale-Second Edition (SRS-2) and a social-emotional functioning questionnaire (SEV).
Results: Planned analyses include assessment of internal consistency using Cronbach’s alpha and inter-rater reliability using Cohen’s kappa. Convergent validity will be examined through correlations between DuMAND categories and conceptually related subscales of established instruments, while divergent validity will be explored using correlations with theoretically unrelated subscales. Exploratory factor analysis (EFA) will be conducted to investigate the underlying factor structure of the DuMAND Checklist.
Conclusion: This study aims to provide a methodologically robust evaluation of the psychometric properties of the DuMAND Checklist in a Belgian DMD cohort. Data collection is currently ongoing. Results from the cross-sectional and interim longitudinal analyses are anticipated to be available for presentation at the time of the 2026 ICNMD Congress. Completion of recruitment and longitudinal follow-up is expected to allow full psychometric evaluation, and final conclusions regarding reliability, validity and clinical applicability will be drawn once data collection and analyses are complete.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
PP01.39
The DuMAND Checklist: Behavioral Patterns in Typically Developing Boys and Boys with Duchenne Muscular Dystrophy
Ms. Eline Cuveele1,2, Dr. Sam Geuens1,2, Ms. Jente Dreezen2, Ms. Kim Wittebols2, Ms. Joanna Willen1, Prof. Céline Gillebert2, Prof. Liesbeth De Waele1,2
1
University Hospitals Leuven, Leuven, Belgium.
2
KU Leuven, Leuven, Belgium
Background: Duchenne muscular dystrophy (DMD) is characterized by progressive muscle weakness due to dystrophin deficiency. In addition to motor impairment, affected boys show increased vulnerability to develop cognitive, behavioral and psychiatric difficulties. These neurobehavioral challenges place a substantial burden on daily functioning and family life. Current clinical management typically focuses on motor outcomes and provides limited tools for the systemic assessment of central nervous system-related manifestations. To address this gap, the DuMAND Checklist was developed as a disease-specific instrument to measure neurobehavioral difficulties in DMD. The instrument evaluates five behavioral categories: (1) cognition and learning, (2) social responsiveness, (3) emotion regulation, (4) externalizing behavior and (5) eating and sleeping. Establishing normative reference data in typically developing boys is essential to differentiate typical developmental patterns from DMD-related deviations. This study aimed to characterize age-related behavioral patterns in typically developing boys using the DuMAND Checklist, and to compare these trajectories with preliminary data from boys with DMD to identify disease-specific differences.
Methods: A cross-sectional study was conducted among Dutch speaking boys aged 3-20 years living in Flanders. Parents of typically developing boys (boys with no history of DMD, or any other neuromuscular disorder) completed the DuMAND Checklist and background questionnaires online via REDCap after giving electronic consent. Parents of boys with DMD in the same age range completed the same instruments during clinical consultations at University Hospitals Leuven (Belgium). Data were analyzed in SPSS. Descriptive statistics were calculated for total (ranging from 43 to 230) and category specific scores. Group and age-related effects were examined using a multivariate analysis of covariance (MANCOVA) with age included as a covariate. Effect sizes were expressed as partial η².
Results: In typically developing boys (n=359), the mean Total DuMAND score was 93.4 (SD 24.1). Category-specific mean scores were 14.8 ± 4.8 for cognition and learning, 21.8 ± 6.4 for social responsiveness, 22.3 ± 6.8 for emotion regulation, 22.9 ± 8.2 for externalizing behavior and 11.6 ± 4.0 for eating and sleeping difficulties. In boys with DMD (n=38), the mean Total DuMAND score was 123.0 (SD 31.0), with elevated mean scores across all behavioral categories: cognition and learning 21.8 ± 6.6; social responsiveness 29.8 ± 7.4; emotion regulation 29.6 ± 8.3; externalizing behavior 27.1 ± 10.7 and eating and sleeping 14.2 ± 4.5. MANCOVA controlling for age confirmed significant group differences for the Total DuMAND score (F(1,394)=74.25, p<.001, partial η²=0.159) and all category scores (F-range 21.83-70.79, all p<.001), indicating more behavioral difficulties in boys with DMD. Significant effects of age were also observed for the total score (F(1,394)=79.75, p<.001, partial η²=0.168) and all categories (F-range 9.79-169.39, p≤.002).
Conclusion: The DuMAND Checklist is sensitive to both normative age-related behavioral patterns in typically developing boys and disease-specific deviations in boys with DMD. Boys with DMD showed higher total and category-specific DuMAND scores compared to peers without DMD, with behavioral outcomes varying by age. These findings support the use of the DuMAND Checklist as a measurement tool for assessing neurobehavioral difficulties in DMD. Ongoing data collection will refine values for more robust comparisons.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
PP01.40
Mapping the Mind in Duchenne Muscular Dystrophy: The DuMAND Network
Ms. Eline Cuveele1,2, Dr. Geuens Sam1,2, Dr. Céline Gillebert2, Dr. Katy De Valle3, Dr. Kathryn Irving3, Dr. Daniella Villano3, Dr. Ian Woodcock3, Dr. Eppie Yiu3, Dr. Guenther Bernert4, Dr. Simone Mahal5, Dr. Clara Camelo6, Dr. Cristiane Moreno6, Dr. Edmar Zanoteli6, Dr. Emilie Hill-Smith7,8, Dr. Hugh McMillan7,8, Dr. Laura McAdam9, Dr. Sophelia Chan10, Dr. Patrick Yee10, Dr. Edna Julieth Bobadilla11, Dr. Andrea del Pilar Calderon Castro12, Dr. Pavla Bortolomejovava Bartolomejovava13, Dr. Katerina Bukacova13, Dr. Anezka Dolanska13, Dr. Jana Haberlova13, Dr. Monika Firholz14, Dr. Ulrike Schara-Schmidt14, Dr. Janbernd Kirschner15, Dr. Astrid Pechmann15, Dr. Eugenio Maria Mercuri16, Dr. Riccardo Masson17, Dr. Stefano Parravicini17, Dr. Claudia Dosi17, Dr. Federica Silvia Ricci18, Dr. Hirofumi Komaki19, Dr. Yuko Shimizu-Motohashi19, Dr. Erit Takeshita19, Dr. Keiko Ishigaki20, Dr. Kumiko Ishiguro20, Dr. Yuki Kihara20, Dr. Sato Takatoshi20, Dr. Andreas Dybesland Rosenberger21, Dr. Tonje Elgsås22, Dr. Heidi Elisabeth Nag22, Dr. Simen Stokke22, Dr. Diana Carvalho23, Dr. Juliana Da Silva Cardoso24, Dr. Cristina Garrido24, Dr. Dušan Šuput25, Dr. Damjan Osredkar25, Dr. Nina Demsar25, Dr. Andrej Vovk25, Dr. Barbara Verovnik25, Dr. Thomas Sejersen26, Dr. Anne-Berit Ekstrom27, Dr. Jonas Gillenstrand28, Dr. Bettina Henzi29, Dr. Andrea Klein29, Dr. Leonie Steiner29, Dr. Rossane Govaarts30, Dr. Erik Niks30, Dr. Saskia Houwen-Van Opstal31, Dr. Corrie Erasmus31, Dr. Marloes Van Lieshout31, Dr. Francesco Muntoni32, Dr. Chloe Gaegan33, Dr. Michela Guglieri33, Dr. Charlotte Ilderton33, Dr. Volker Straub33, Dr. Catherine Turner33, Dr. Liesbeth De Waele1,2
1
University Hospitals Leuven, Leuven, Belgium.
2
KU Leuven, Leuven, Belgium.
3
The Royal Children's Hospital, Melbourne, Australia.
4
University Hospital Vienna, Vienna, Austria.
5
Klinik Favoriten Vienna, Vienna, Austria.
6
Universidade de São Paulo, São Paulo, Brazil.
7
Children's Hospital of Eastern Ontario Research Institute, Ottawa, Canada.
8
University of Ottawa, Ottawa, Canada.
9
Holland Bloorview Kids Rehabilitation Hospital, Bloorview Research Institute, University of Toronto, Toronto, Canada.
10
Hong Kong Children’s Hospital and The University of Hong Kong, Hong Kong, Hong Kong.
11
Hospital Pediatrico de la Misericordia, Bogota, Colombia.
12
Hospital Universitario Nacional de Colombia, Bogota, Colombia.
13
Motol and Homolka University Hospital, Charles University,, Motol, Czechia.
14
University Children’s Hospital Essen, Essen, Germany.
15
University Hospital Freiburg, Freiburg, Germany.
16
Fondazione Policlinico Universitario Agostino Gemelli IRCCS, Centro Clinico Nemo, Rome, Italy.
17
Fondazione IRCCS Instituto Neurologico Carlo Besta, Milan, Italy.
18
University of Turin, Turin, Italy.
19
National Center of Neurology and Psychiatry Tokyo, Tokyo, Japan.
20
Tokyo Women’s Medical University, Tokyo, Japan.
21
University Hospital of North Norway, Norwegian Centre for Rare Diseases, Tromsø, Norway.
22
Norwegian Centre for Rare Diseases - Unit Frambu, Tromsø, Norway.
23
Unidade Local de Saúde de Trás-os-Montes e Alto Douro (ULSTMAD, Trás‑os‑Montes e Alto Douro, Portugal.
24
Centro Materno- Infantil de Norte, Unidade Local de Saúde de Santo António (CMIN, ULSSA), Porto, Portugal.
25
University of Ljubljana, Ljubljana, Slovenia.
26
Karolinska Institute and Karolinska University Hospital, Stockholm, Sweden.
27
Region Pediatric Rehabilitation Center, Queen Silvia Children’s Hospital, Gothenburg; University of Gothenburg, Department of Pediatrics, Institute of Clinical Sciences, Sahlgrenska Academy, Gothenburg, Sweden.
28
University of Gothenburg;Habilitation & Health, Region Västra Götaland, Gamlestaden, Gothenburg, Sweden.
29
Inselspital, Bern University Hospital, University of Bern, Bern, Switzerland.
30
Leiden University Medical Center,, Leiden, Netherlands.
31
Radboud University Medical Centre Nijmegen, Nijmegen, Netherlands.
32
UCL Great Ormond Street Institute of Child Health, University College London & Great Ormond Street Hospital Trust, London, United Kingdom.
33
John Walton Muscular Dystrophy Research Centre, Newcastle Hospitals NHS Foundation Trust and Newcastle University, Newcastle, United Kingdom
Background: Duchenne muscular dystrophy (DMD) is increasingly understood as a multisystem condition that affects not only muscle but also brain development. Many boys with DMD experience cognitive difficulties, including problems with learning and attention, as well as challenges in social interaction, emotion regulation, behavior, eating and sleeping. Although these symptoms vary widely across individuals, they place a burden on children and families and may significantly contribute to reduced quality of life. Existing research remains fragmented, often cross-sectional, based on small samples and largely derived from Western, high-income countries, limiting our understanding of how genetic, treatment-related and environmental factors jointly influence neurodevelopment in DMD.
Methods: To address these limitations, the DuMAND Network (Duchenne Muscular Dystrophy-Associated Neurobehavioral Difficulties) was established as an international collaboration among clinicians and researchers in child neurology, psychology and psychiatry. A central component of the initiative is the DuMAND Checklist, a caregiver-administered instrument designed to measure a broad spectrum of neurobehavioral domains, divided into five categories: (1) cognition and learning, (2) emotion regulation, (3) social responsiveness, (4) externalizing behavior and (5) eating and sleeping. The DuMAND Checklist supports early recognition and structured monitoring of challenges within these DuMAND-categories, and offers a first step toward a more standardized approach to neurobehavioral assessment in clinical practice.
Results: The DuMAND Network currently includes partners from more than 17 countries across five continents and works toward two complementary aims. First, the network advances research efforts to examine cultural differences in behavior, explore genotype-phenotype relationships (including dystrophin isoform involvement) and investigate the influence of treatments such as corticosteroids on cognitive and behavioral outcomes. Integrating clinical, behavioral, and genetic data will facilitate the development of culturally sensitive, longitudinal insights and help guide future brain‑related outcome measures and targeted interventions in DMD. Second, by evaluating and validating the DuMAND Checklist globally, the network aims to enhance clinical care and contribute to greater consistency in the evaluation of neurobehavioral symptoms in DMD. Through shared expertise and collaborative learning, the DuMAND Network aims to strengthen clinical neurobehavioral management and support for boys with DMD.
Conclusion: Preparatory work has shown that the DuMAND Checklist can be feasibly implemented across diverse clinical contexts. An international validation study is planned for 2026, inviting centers with varying expertise and resources to participate. To conclude, the DuMAND Network addresses existing evidence gaps by providing a standardized international framework to advance research, clinical evaluation and ultimately management of neurobehavioral challenges in patients with DMD.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
Images or Table (Optional)
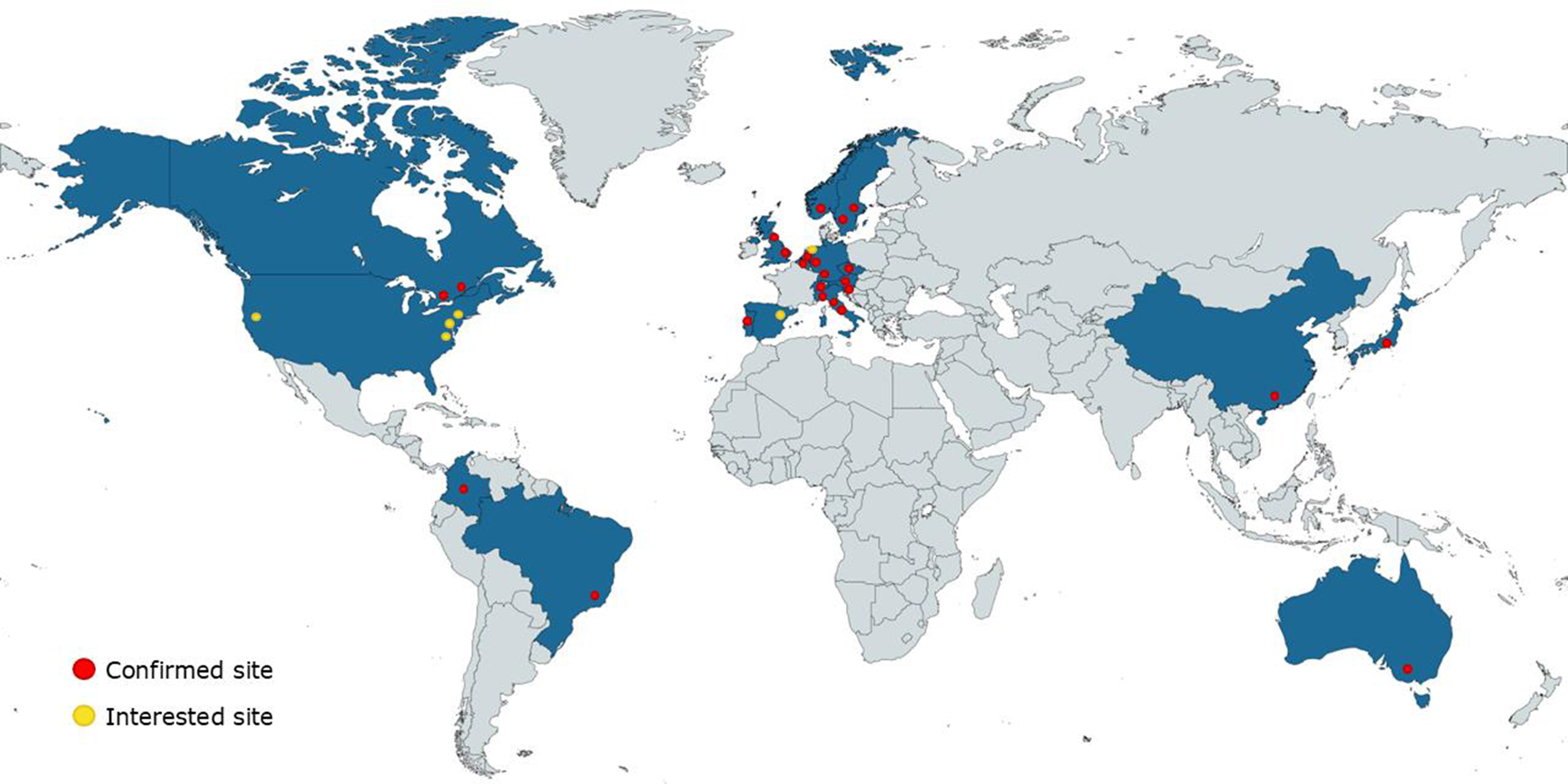
PP01.41
Efficacy and Safety of Golodirsen and Casimersen Compared With Placebo in Duchenne Muscular Dystrophy (ESSENCE)
Prof. Francesco Muntoni1,2, Prof. Emma Ciafaloni3, Dr. Fernando Chloca4, Dr. Nicolas Deconinck5, Dr. Ameneh Masud6, Dr. Ihor Sehinovych6, Dr. Kerri Drummond6, Dr. Weijian Liu6, Dr. Pamela Magistrado-Coxen6, Dr. Andrés Nascimento Osorio7, Prof. Sheffali Gulati8, Prof. Maria Judit Molnar9, Prof. Maria Mazurkiewicz-Beldzinska10, Prof. Eugenio Mercuri11, Prof. Craig McDonald12
1
Dubowitz Neuromuscular Centre, University College London, London, United Kingdom.
2
National Institute for Health Research Great Ormond Street Hospital Biomedical Research Centre, London, United Kingdom.
3
University of Rochester Medicine, Rochester, United States.
4
Favaloro Foundation - Neurosciences Institute, Buenos Aires, Argentina.
5
Centre de Référence Neuromusculaire and Paediatric Neurology Department, Hôpital Universitaire des Enfants Reine Fabiola, Université Libre de Bruxelles, Brussels, Belgium.
6
Sarepta Therapeutics, Inc., Cambridge, United States.
7
Neuromuscular Unit, Neuropaediatrics Department, Hospital Sant Joan de Déu, Fundacion Sant Joan de Déu, CIBERER - ISC III, Barcelona, Spain.
8
All India Institute of Medical Sciences, New Delhi, India.
9
Institute of Genomic Medicine and Rare Disorders, Semmelweis University, Budapest, Hungary.
10
Department of Developmental Neurology, Chair of Neurology, Medical University of Gdańsk, Gdańsk, Poland.
11
Pediatric Neurology Unit, Università Cattolica del Sacro Cuore Roma, Rome, Italy.
12
UC Davis Health, Sacramento, United States
Background: Golodirsen and casimersen are phosphorodiamidate morpholino oligomers (PMOs) used for treatment of Duchenne muscular dystrophy (DMD). The objective of this study is to report topline efficacy and safety results from the phase 3 ESSENCE trial (NCT02500381).
Methods: Eligible ambulatory males with DMD amenable to exon 53 (aged 6-13 y) or exon 45 (aged 7-13 y) skipping were randomized 2:1 to the combined treatment group (30 mg/kg golodirsen or casimersen; PMO) or placebo (PBO). A 96-wk double-blind period was followed by a 48-wk open-label period. The primary endpoint was change from baseline (CFB) in 4-step ascend velocity (step/s) at wk 96 vs PBO. Secondary functional, biological, and safety endpoints were reported. A prognostic scoring approach identified patients at high risk of 4-step ascend velocity decline (prognostically relevant subgroup [PRS]) for post hoc analysis.
Results: 225 patients were enrolled from 2016 to 2022, including through the COVID-19 pandemic, across 24 countries. Baseline characteristics were well matched. At wk 96, the primary endpoint in the PMO group (n=138) showed a numerical mean (SD) change of −0.27 (0.39) steps/s vs PBO (n=74; −0.34 [0.37]), least squares mean (LSM) difference 0.06 (95% CI, −0.05 to 0.16; P=0.309). Although not statistically significant, most secondary endpoints favored PMO. At wk 144, 4-step ascend velocity was −0.004 (−0.123 to 0.115). At wk 96, 6-minute walk test distance was 5.34 (−23.32 to 34.00), 10-meter walk/run velocity was 0.020 (−0.097 to 0.137), rise from floor velocity was −0.003 (−0.020 to 0.014), and North Star Ambulatory Assessment was 0.61 (−0.90 to 2.12). At wk 96, dystrophin expression by western blot had a mean difference of CFB of 0.69 (0.28 to 1.10; P<0.001; unadjusted) and 1.25 (0.20 to 2.31; P=0.020; muscle-content adjusted). Dystrophin fiber intensity by immunohistochemistry had a mean difference of CFB of 0.023 (0.015 to 0.031; P<0.001). Through wk 144, no new safety signals were reported. Most treatment-emergent adverse events were mild or moderate and resolved without treatment. The PRS (PMO, n=74; PBO, n=35) suggested a meaningful change in 4-step ascend velocity at wk 96 for the PMO group (LSM difference vs PBO, 0.186 steps/s; P=0.010).
Conclusion: ESSENCE confirms the established safety profile of golodirsen and casimersen. The primary endpoint was not met in ESSENCE. A post hoc analysis and the totality of PMO evidence suggest the clinical benefits of golodirsen and casimersen.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
PP01.42
Genetic Modifier Associations With Steroid Safety Outcomes in Duchenne Muscular Dystrophy
Ms. Fatemeh Ahmadiharchegani1, Prof. Eric Hoffman2, Dr. Daniele Sabbatini3, Prof. Elena Pegoraro3, Prof. Luca Bello3, Prof. Michela Guglieri4, Dr. Paula Clemens5, Prof. Utkarsh Dang1
1
Carleton University, Ottawa, Canada.
2
Binghamton University, New York, United States.
3
University of Padova, Padova, Italy.
4
Newcastle University, Newcastle, United Kingdom.
5
University of Pittsburgh, Pittsburgh, United States
Background: Duchenne muscular dystrophy (DMD) is the most common progressive, pediatric X-linked neuromuscular disorder. Traditional corticosteroids like prednisone and deflazacort can result in weight gain, adrenal suppression, growth stunting, and reduced bone biomarkers, while vamorolone, a newly approved dissociative steroid, has shown an improved safety profile, especially on bone health. Genetic modifiers are known to alter disease severity or response to treatment, but they have not been investigated in the first decade of life. Our objective was to study associations of genetic modifiers with steroid-related safety (weight gain, growth, bone biomarkers, adrenal function) in young boys with DMD.
Methods: Samples and data were obtained from the VBP15-002/003 (n=48) and VBP15-004 trials (n=121) of vamorolone. DNA samples were genotyped using the Illumina GDA-PGx platform. The analysis compared the placebo, prednisone (0.75 mg/kg/day), and vamorolone (6 mg/kg/day) treatment groups. Longitudinal analysis was used with a data mask applied to study association of pre-specified polymorphisms in UGT1A1 (steroids are glucuronidated by UGT1A1), Vitamin D Receptor, i.e., VDR (FokI, GATA, and CDX2 SNPs), and PDGFD (rs361283) genes with change in BMI z-score, height z-score, bone biomarkers of formation and turnover (Osteocalcin, P1NP, and CTXI), and cortisol levels.
Results: No PDGFD x adrenal suppression associations were seen. Certain VDR genotypes were associated with sensitivity to prednisone-related changes in bone biomarkers. Interestingly, steroid treated groups showed an association of weight gain with the pharmacogenomic/drug exposure modifier enzyme UGT1A1. We also study UGT1A1 polymorphism associations with circulating serum proteins and long-term weight gain.
Conclusion: Although based on a small sample size and needing additional studies for confirmation, we provide initial evidence of the association of genetic modifiers at an early age with common steroid safety concerns including for weight gain, which is a common reason for steroids prescribed at sub-recommended doses. These findings may be important for clinical trials and care.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
PP01.43
Real-World Ataluren Outcomes in 7 nmDMD Patients
Prof. Flávia Nardes, Prof. Jaqueline Almeida, Prof. Marcos Rebel, Prof. Aline Chacon, Dr. Clarisse Fortes, Dr. Erica Ruas, Prof. Alexandra Prufer
UFRJ, Rio de Janeiro, Brazil
Background: In our previous retrospective cohort analysis of 25 nonsense mutation DMD (nmDMD) boys with standard care (1-22 years), over the last 15 years (2007–2022), we found high rate of intelectual disability (80%), early loss of ambulation (at 9.1y ±2.1) and shortening of life expectancy (17.6y ±2.1). Now, our initial objective was to describe the clinical, motor, and cardiac parameters of seven patients receiving ataluren after five years of treatment.1,2
Methods: A descriptive, observational, and retrospective study over the last 10 years (2015–2025) was performed from our neuromuscular center of Universidade Federal do Rio de Janeiro (UFRJ), Brazil. Clinical data from 7 confirmed patients with confirmed nonsense mutation (nmDMD) were obtained from retrospective medical records. All patients are treated and followed by the current standard of care recommendations from the Brazilian consensus on Duchenne muscular dystrophy3 (corticosteroids, rehabilitation, ventilatory support, and cardiac medications) and have been received ataluren. Data sources were approved by the Ethical Committee in IPPMG/UFRJ (registered number CAAE: 01934112.0.0000.5264).
Results: Table 1 summarizes the baseline clinical and genetic characteristics of seven patients with nonsense mutation Duchenne muscular dystrophy (nmDMD) followed for up to ten years under ataluren treatment and standard care. Despite heterogeneity in mutation location, age at symptom onset, and age at diagnosis, all patients showed markedly elevated serum creatine kinase levels and typical early neuromuscular manifestations. The age at diagnosis remained relatively delayed, reflecting real-world clinical practice in a public health setting in low-income countries. There is a high prevalence of intellectual disability and learning difficulties, consistent with the known neurocognitive involvement associated with nmDMD. These findings reinforce the complexity of disease burden beyond motor impairment. Most patients initiated corticosteroids before ataluren, with ataluren introduced in mid-childhood. At the last follow-up, several patients remained ambulant into adolescence, while others experienced loss of ambulation during the second decade of life. Importantly, the majority of patients preserved cardiac function, with left ventricular ejection fraction remaining ≥55% in adolescence, suggesting relative cardiac stability during long-term follow-up. The longitudinal graphs illustrate progressive motor decline with increasing age, as expected in Duchenne muscular dystrophy, but with substantial interindividual variability. Some patients demonstrated slower deterioration of functional performance and prolonged ambulation, whereas others showed earlier functional decline. This variability likely reflects differences in baseline functional status, disease severity, timing of therapeutic intervention, and comorbidities.
Conclusion: A notable finding is the high frequency of intellectual disability (71%) of patients and learning difficulties in 85%. Cognitive impairments may influence adherence to treatment and rehabilitation, and performance in timed motor assessments, increasing variability in longitudinal functional outcomes. Overall, the data suggest that long-term ataluren treatment in routine clinical practice is feasible and may be associated with preserved cardiac function during adolescence, with some patients maintaining ambulation into their mid-teens. However, early loss of ambulation in other patients underscores that outcomes are likely influenced by baseline functional status, timing of therapy initiation, disease severity, and comorbidities. Given the very small sample size and the absence of an internal untreated comparator, conclusions must be interpreted with caution.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
Images or Table (Optional)
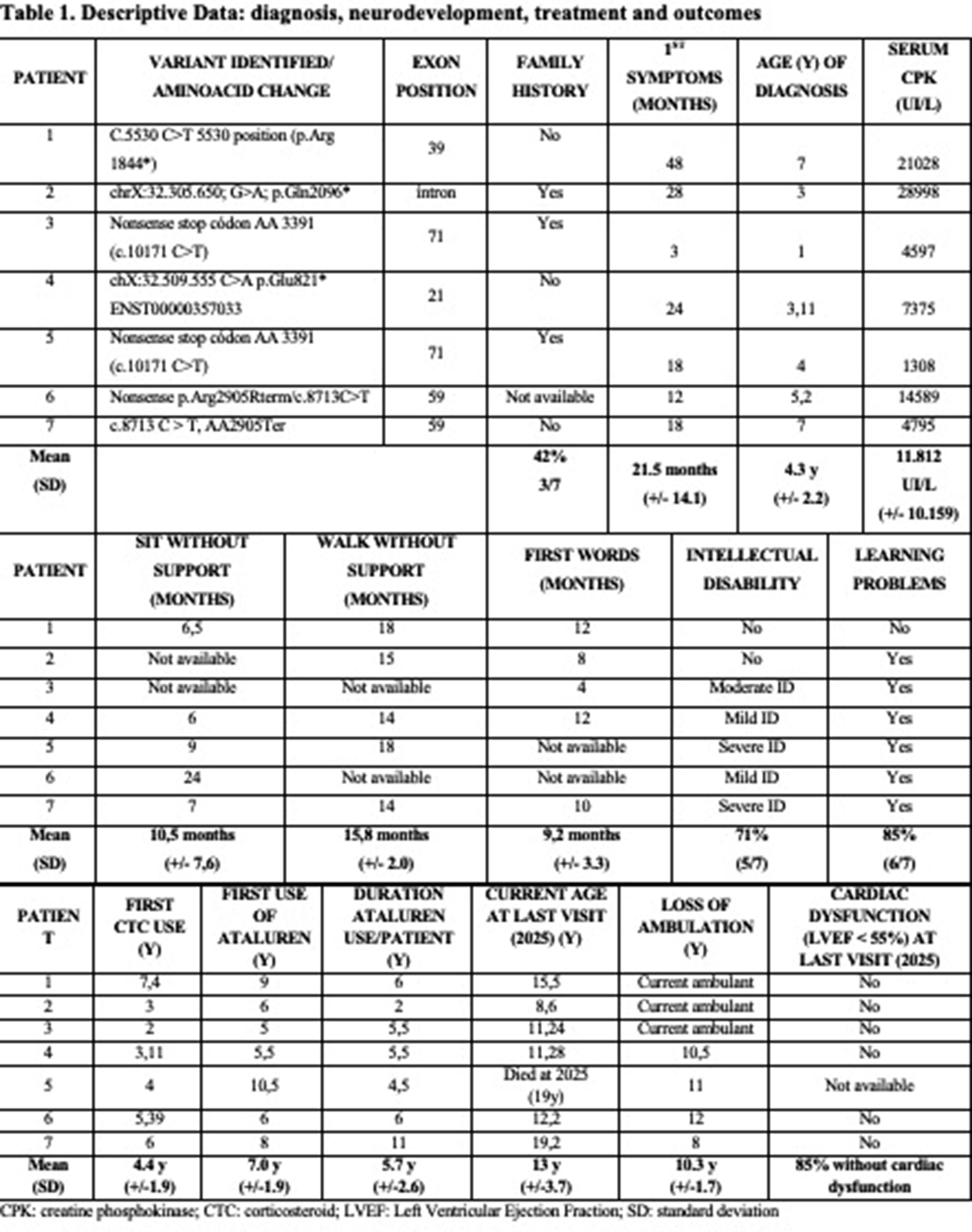
PP01.44
Characterisation of Anti-AAV9 Seropositivity in Zolgensma Treated SMA Patients
Prof. Francesco Muntoni1, Dr. Tomas Baldwin1, Mr. Ivan Doykov1, Dr. Kimberly Gilmour2, Dr. Nikki Cornell2, Dr. Mariacristina Scoto2, Dr. Pierpaolo Ala1, Prof. Haiyan Zhou1, Prof. Giovanni Baranello2, Prof. Kevin Mills1, Dr. Wendy Heywood1
1
University College London, London, United Kingdom.
2
Great Ormond Street Hospital, London, United Kingdom
Background: Gene therapy using adeno-associated virus serotype 9 (AAV9) vectors has transformed the treatment landscape for spinal muscular atrophy (SMA), offering a one-time systemic intervention. However, immune responses to AAV9 remain a significant barrier to therapy safety.
Methods: In this study, we developed a novel innovative targeted LC-MS/MS-based platform to deeply characterize subclass-specific humoral responses and systemic inflammatory profiles following AAV9 gene therapy. Paediatric Zolgensma pre and post treated patient samples ( n= 24) were collected and included patients who had moderate to severe immune response post systemic zolgensma treatment.
Results: In Zolgensma treated patients we demonstrate that the humoral response post-infusion is dominated by a robust, time-dependent increase in anti-AAV9 IgG1 antibodies, with minimal contributions from other IgG subclasses or isotypes. Importantly, IgG1 response correlate strongly with AAV9 binding of classical and terminal complement pathway components, emphasizing its mechanistic role in complement-mediated immunogenicity. Cross-reactivity analysis of treated patient serum to AAV8 and AAV2 revealed substantial IgM and IgG1 binding to AAV8, but with significantly reduced complement binding. This indicates that immunoglobulin binding alone does not predict functional immune risk - an important consideration for capsid switching strategies. IgG1 did not cross react to AAV2 capsid however IgM from the initial immune response did cross react to AAV2 with observable complement binding that was not observed for IgM against AAV9. This could have implications for patients being exposed to other AAVs after treatment. We did not observe anti-AAV9 IgG1 in pre-treated samples but we did observe positive reactions for other IgG subclasses and IgA in some patients. Additionally, targeted plasma proteomic profiling identified a panel of inflammatory proteins, including APOA1, GSTO1, IGF-2, and GSN, that stratified patients by severity of post-infusion immune responses. Notably, pre-infusion inflammatory signatures predicted severe outcomes, underscoring the potential for proteomic risk profiling to guide patient selection and immunosuppressive interventions.
Conclusion: Together, these findings highlight the critical role of IgG1-driven complement activation in AAV9 immunogenicity and establish the utility of integrating subclass-specific serology with complement and serum inflammatory profiling into precision risk stratification frameworks. Our platform advances the field by enabling safer, more individualized gene therapy delivery through mechanistically informed monitoring and intervention strategies.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: Spinal Muscular Atrophies: Treatment
PP01.45
Neuronal Micro-Dystrophin Gene Therapy Improves Emotional Behaviour and Restores Dystrophin Interactors in mdx52 mouse Model
Dr. Konstantina Tetorou1,2, Dr. Monica Rebeca Gil Garzon1, Mr. Alex Kavanagh2, Ms. Nicha Songsliph2, Mr. Shashwat Guha1, Ms. Ayana Withana2, Ms. Jenny Vartianen1, Ms. Wing Sum Chu1, Ms. Camila Vallve Maine1, Mr. Darren Chambers2,3, Dr. Riccardo Privolizzi1, Prof. Simon Waddington4,5, Dr. Simon Beggs6,7, Prof. Joanne Ng1, Prof. Francesco Muntoni1,2
1
Genetic Therapy Accelerator Centre, UCL Queen Square Institute of Neurology, London, United Kingdom.
2
Dubowitz Neuromuscular Centre, UCL Great Ormond Street Institute of Child Health, London, United Kingdom.
3
Dubowitz Neuromuscular Centre, Division of Neuropathology, UCL Queen Square Institute of Neurology, London, United Kingdom.
4
Gene Transfer Technology Group, EGA Institute for Women's Health, University College London, London, United Kingdom.
5
Wits/SAMRC Antiviral Gene Therapy Research Unit, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa.
6
Neuroscience, Physiology and Pharmacology, UCL, London, United Kingdom.
7
Developmental Neurosciences, UCL Great Ormond Street Institute of Child Health, London, United Kingdom
Background: Duchenne muscular dystrophy (DMD) is a severe neuromuscular disorder caused by mutations in X-linked DMD gene, resulting in disruption of functional dystrophin protein production. DMD patients exhibit progressive muscle weakness and 44% DMD individuals also experience intellectual disability and/or neurobehavioral complications, such as autism spectrum disorder, attention deficit hyperactivity disorder, and anxiety. These latter complications are due to deficiency of different dystrophin isoforms in the brain differentially affected by the location of the DMD gene mutation. The brain involvement is recapitulated in DMD mouse models with mutations differentially affecting Dp427, Dp140 dystrophin isoforms and displaying enhanced fear response with increased anxiety- and depressive-like behaviours.
Previous preclinical studies have shown partial restoration of dystrophin in the brain with antisense oligonucleotide (ASO) therapy delivered intracerebroventricular (ICV) resulted in partial amelioration of neurobehavioural phenotypes but low levels of Dp427 restoration.
To further address therapeutic approaches for brain dystrophin deficiency we generated Adeno-associated virus (AAV) gene therapy vector with neuronal targeted micro-dystrophin and evaluated neurobehavioural efficacy in mdx52 mice lacking both Dp427 and Dp140 isoforms.
Methods: We treated neonatal male mdx52 at P1 with ICV or intravenous (IV) 5x10vg/pup and 4x11vg/pup respectively of AAV.micro-dystrophin, controls were untreated male mdx52 and wildtype littermates n=15 per group). We performed a battery of neurobehavioural tests between 8-12 weeks including fear response, elevated zero maze, open field, light-dark box, balance beam and Catwalk XT. We also used simple western (WES) to quantify micro-dystrophin and dystrophin’s interacting proteins in the brain.
Results: We demonstrated statistically significant improvement of emotional reactivity related behaviours in treated compared to untreated mdx52 littermates. Some behaviours were restored to wildtype levels showing improved efficacy compared to previous ICV ASO studies. Micro-dystrophin protein levels showed a rostro-caudal gradient with 60% wild-type levels in hippocampus and midbrain, with 10% in the cerebellum after ICV delivery and between 10-40% in all brain areas with IV delivery.
Conclusion: This is the first preclinical therapeutic study demonstrating brain targeted micro-dystrophin improves the neurobehavioural deficits observed in DMD mouse model with ICV and IV delivery.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
PP01.46
Phase 2 Study of Vesleteplirsen in Patients With Duchenne Muscular Dystrophy: MOMENTUM Part B Results
Dr. Craig Campbell1, Dr. Eugenio Mercuri2,3,4,5, Dr. Avram Z. Traum6, Dr. Seth J. Perlman7, Dr. Hoda Z. Abdel-Hamid8, Dr. Nicolas Deconinck9,10, Dr. Giovanni Baranello11,12, Dr. Liesbeth De Waele13,14, Dr. Stefanie Mason15, Dr. Na Cai15, Ms. Joy Haugh15, Dr. Jennifer M. Thomas-Ahner15, Dr. Joseph V. Bonventre16, Dr. Andres Nascimento17, Dr. Katherine Mathews18, Dr. Han Phan19
1
Departments of Paediatrics, Clinical Neurological Sciences and Epidemiology, University of Western Ontario, London, Ontario, Canada.
2
Child Neurology and Psychiatry Unit, Fondazione Policlinico Universitario Agostino Gemelli IRCCS, Rome, Italy.
3
Clinical Psychology Unit, Fondazione Policlinico Universitario Agostino Gemelli IRCCS, Rome, Italy.
4
Pediatric Neurology, Catholic University of Sacred Heart, Rome, Italy.
5
Department of Psychiatry, Fondazione Policlinico Universitario Agostino Gemelli IRCCS, Rome, Italy.
6
Division of Nephrology, Boston Children’s Hospital, Harvard Medical School, Boston, MA, United States.
7
Seattle Children’s Hospital, University of Washington School of Medicine, Seattle, WA, United States.
8
UPMC Children's Hospital of Pittsburgh, University of Pittsburgh, Pittsburgh, PA, United States.
9
Neuromuscular Reference Center, UZ Gent, Ghent, Belgium.
10
Hôpital Universitaire des Enfants Reine Fabiola, HUB, ULB, Brussels, Belgium.
11
Dubowitz Neuromuscular Centre, UCL Great Ormond Street Institute of Child Health Great Ormond Street Hospital NHS Foundation Trust, London, United Kingdom.
12
National Institute for Health Research Great Ormond Street Hospital Biomedical Research Centre, London, United Kingdom.
13
Department of Child Neurology, University Hospitals Leuven, Leuven, Belgium.
14
Department of Development and Regeneration, KU Leuven, Leuven, Belgium.
15
Sarepta Therapeutics, Inc., Cambridge, MA, United States.
16
Division of Renal Medicine, Department of Medicine, Brigham and Women's Hospital, Boston, MA, United States.
17
Neuromuscular Unit, Neuropaediatrics Department, Hospital Sant Joan de Déu, Fundacion Sant Joan de Déu, CIBERER - ISC III, Barcelona, Spain.
18
University of Iowa Carver College of Medicine, Iowa City, IA, United States.
19
Rare Disease Research, LLC, Atlanta, GA, United States
Background: Phosphorodiamidate morpholino oligomers (PMOs) mediate exon skipping to restore the disrupted reading frames in the DMD gene that causes Duchenne muscular dystrophy (DMD), enabling production of truncated dystrophin protein that retains key functional domains. Vesleteplirsen (SRP-5051) is a next-generation, peptide-conjugated PMO (PPMO) therapy designed to skip exon 51 with increased uptake into the target tissue. MOMENTUM (NCT04004065) was a phase 2, open-label, two-part study of vesleteplirsen in patients with DMD, consisting of a dose-determination phase (Part A) and a dose-expansion phase (Part B). This abstract presents Part B results evaluating biological efficacy, safety, and pharmacokinetics of vesleteplirsen treatment at the doses determined from Part A.
Methods: MOMENTUM Part B enrolled ambulatory and non-ambulatory, corticosteroid-treated males with DMD amenable to exon 51 skipping to receive vesleteplirsen every 4 weeks. Treatment-naïve or previously treated (rolled over from Part A or an open-label extension study [NCT03675126]) patients were included. Following a 2-dose safety lead-in, patients were randomized/assigned to either a low- (750 mg if 18-49 kg; 850 mg if ≥50 kg) or a high-dose (1250 mg if 18-49 kg; 1350 mg if ≥50 kg) group. Given the hypomagnesemia risk identified in part A, patients received a twice daily oral dose of 400 mg magnesium oxide (or an equivalent formula) prophylactically starting at treatment initiation. The primary endpoint was a change from baseline to week 28 in dystrophin protein level on western blot, adjusted to muscle content, among the treatment-naïve cohort with muscle biopsy samples. Secondary endpoints included changes in exon 51 skipping from baseline to week 28, incidence of adverse events (AEs), and pharmacokinetic parameters of vesleteplirsen. Inferential statistics were prespecified for the primary endpoint only; all other results were analyzed descriptively.
Results: Sixty-two patients were enrolled (low-dose, n=32; high-dose, n=29; not treated, n=1); 21 were previously treated and 41 were treatment-naïve. Mean (range) age was 12.9 (8.0-23.0) years; 46.8% (29/62) were ambulatory. Median treatment duration was 21.7 months. Dystrophin levels increased from baseline to week 28 in both treatment groups (mean [SD] change low-dose: 1.15% [0.9], P=0.0002, n=20; high-dose: 2.94% [3.8], P=0.002, n=19). At week 28, exon 51 skipping in dystrophin mRNA for the low- and high-dose groups was 2.47% (n=20) and 11.16% (n=19), respectively (versus 0.47% and 1.07% at baseline). The most common treatment-related AEs (TRAEs) were hypomagnesemia (62/62, 100%), hypokalemia (31/62, 50.0%), and decreased glomerular filtration rate (GFR) (15/62, 24.2%). Serious TRAEs occurred in 12/62 (19.4%) patients, with hypokalemia (7/62, 11.3%) and hypomagnesemia (6/62, 9.7%) being the most common. One patient (1.6%) discontinued due to decreased GFR (Grade 3). At study end, 24/62 (38.7%) had ongoing hypomagnesemia and 39/62 (62.9%) were receiving magnesium supplementation. The plasma half-life ranged 2.15-2.62h with minimal accumulation after repeated dosing. Within 48h, 19.2% of the high-dose was recovered in urine as vesleteplirsen and 9.3% as its primary metabolite.
Conclusion: Although vesleteplirsen treatment resulted in a statistically significant increase in dystrophin expression, meeting the primary biological endpoint, MOMENTUM was terminated early due to long-term safety concerns and the evolving therapeutic landscape in DMD.
Funding: Sarepta Therapeutics, Inc.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
PP01.47
SMAD6 Heterozygous Loss-of-Function Variant as a Potential Modifier of Duchenne Muscular Dytrosphy (DMD)
Dr. Giulio Gadaleta, Dr. Giulia Maccarrone, Dr. Guido Urbano, Dr. Enrica Rolle, Dr. Eleonora Leopizzi, Dr. Silvia Boschi, Dr. Liliana Vercelli, Assoc. Prof. Tiziana Mongini
Neuromuscular Unit, Department of Neuroscience “Rita Levi Montalcini”, University of Turin, Turin, Italy
Background: Duchenne muscular dystrophy (DMD) is a severe X-linked neuromuscular disorder caused by the absence of dystrophin, leading to progressive muscle degeneration with respiratory and cardiac involvement. Despite its monogenic origin, DMD shows inter-individual variability, only partially explained by the primary dystrophin mutation. Disease progression is influenced by additional modifiers, including care-related factors and genetic modifiers that remain incompletely characterized.
Methods: Longitudinal neuromuscular, cardiac, and respiratory evaluations were collected, together with genetic analyses results.
Results: We report the case of a 23-year-old young adult with DMD, diagnosed prenatally due to a positive maternal family history with classic clinical course, confirming an exon 45 deletion of the DMD gene. Early development showed mild global motor and language delay, with independent ambulation achieved at 24 months, together with osteo-articular dysplasia.
Daily deflazacort was initiated at 4 years of age and continuously maintained, with dosage adjusted according to body weight. Independent ambulation was preserved until 23 years of age, markedly later than expected in classical DMD. Longitudinal motor assessments showed an overall slow functional decline, including a well-preserved upper limb performance even after loss of ambulation.
From a cardiac standpoint, echocardiography revealed a bicuspid aortic valve associated with hypoplasia of the tricuspid valve and right ventricle after birth, without hemodynamic compromise. Left ventricular systolic function remained preserved over time (ejection fraction 58% at 23 years), under ACE inhibitor (started at 11 yo) and beta-blocker therapy (started at 20 yo). Respiratory involvement progressed slowly, requiring nocturnal non-invasive ventilation only at 23 years, without dysphagia.
Given the atypical features and disease course, next-generation sequencing using a targeted panel for skeletal malformative disorders (>300 genes) was performed. This identified a heterozygous loss-of-function likely pathogenic variant in SMAD6 (c.1084C>T; p.Gln362*), segregating with bicuspid aortic valve disease in the patient’s father, and a de novo variant of uncertain significance in SETD5, potentially accounting for neurodevelopmental and osteo-dysmorphisms.
SMAD6 encodes an inhibitory SMAD that negatively regulates bone morphogenetic protein (BMP) signaling, a pathway involved in myogenesis, maintenance of the muscle satellite cell pool, and antagonism of transforming growth factor beta (TGF-β)–mediated fibro-adipogenic activation and extracellular matrix deposition. Nonetheless, BMP signaling exerts context- and dose-dependent effects in dystrophic muscle, as demonstrated in mdx models. Another example, instead, comes from murine LMNA−/− H2K myoblast models, in which SMAD6 overexpression inhibits satellite cell pool expansion, whereas SMAD6 inactivation restores BMP responsiveness without inducing premature or inefficient myogenic differentiation. These data may indicate that SMAD6 plays a central role in regulating the balance between satellite cell maintenance, differentiation, and regeneration. In this patient, SMAD6 heterozygous loss-of-function could therefore contribute to a milder neuromuscular phenotype by favoring timely muscle regeneration, preventing premature satellite cell exhaustion, and limiting TGF-β–mediated fibrosis.
Conclusion: This case supports the concept that additional genetic factors may modulate DMD severity. SMAD6 partial loss-of-function emerges as a plausible disease modifier and therapeutic target, although its pleiotropic role—particularly in cardiovascular development—warrants caution. Further studies are needed to validate its modifier effect in preclinical DMD models.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
PP01.48
Patient Experiences With DMD and Impacts of PMO Therapies - Interviews With Caregivers From REAL-DMD
Mrs Ivana F. Audhya1, Dr. Min Yang2, Mr. Edward Tuttle3, Dr. Bruno Martins2, Mrs Olivia Shane4, Dr. Katherine L. Gooch1
1
Sarepta Therapeutics, Inc, Cambridge, MA, United States.
2
Analysis Group, Inc, Boston, MA, United States.
3
Analysis Group, Inc, Menlo Park, CA, United States.
4
Analysis Group, Inc, San Francisco, CA, United States
Background: Duchenne muscular dystrophy (DMD) is a rare, progressively debilitating neuromuscular disease resulting from mutations in the dystrophin gene. DMD causes muscle weakness and functional decline, primarily in young males, leading to loss of ambulation during childhood. While no cures are available for DMD, several phosphorodiamidate morpholino oligomers (PMOs) have been approved in recent years. Caregivers play a crucial role in conveying patients’ health status and experiences, particularly given the early onset of the disease. However, there are limited studies describing patient experiences with PMO therapies directly from the caregivers’ perspective. This caregiver interview study provided insights into patients’ experiences with DMD and perceived impacts of PMO therapies.
Methods: REAL-DMD is an ongoing, real-world, observational, prospective, longitudinal cohort study of patients with DMD in the US, focusing on caregiver-reported patient function and experiences. Caregivers from REAL-DMD whose patients are receiving casimersen, eteplirsen, or golodirsen participated in one-on-one semi structured interviews to describe experiences following PMO treatment initiation and to assess the meaningfulness of such experiences. Transcripts were analyzed thematically.
Results: Sixteen caregivers participated in the interviews (mean ± SD age: 42.7 ± 5.9 years). Most were female (93.8%), White (87.5%), and the patient’s mother (93.8%). Patients (N=16) were 14.5 ± 5.1 years old and had been diagnosed with DMD for 10.0 ± 5.5 years. Ten (62.5%) were being treated with eteplirsen, 4 (25%) with casimersen, and 2 (12.5%) with golodirsen, with a mean ± SD treatment duration of 75.3 ± 34.5 months. Seventy-five percent of patients had been previously treated with corticosteroids. At the time of PMO treatment initiation, 88% (14/16) of patients were ambulatory. Thematic analysis revealed several key themes: i) caregivers described the immense burden of DMD given it is a progressive neuromuscular disease for which no cures currently exist; ii) caregivers report the preservation of function and stability as deeply meaningful outcomes of PMO treatments; iii) caregivers emphasize that preserving physical function through treatment is a top priority, noting that improved physical ability often enables children to maintain valuable social and recreational activities; iv) caregivers value PMO therapies because of their perceived ability to stabilize disease progression, lack of treatment side effects, and the provision of hope beyond measurable clinical outcomes; and v) children with DMD demonstrate resilience, finding meaning and joy in life by adapting and redefining participation in social and recreational activities as physical decline progresses, underscoring the importance of these therapies in helping maintain stability.
Conclusion: This interview study highlighted the profound impact of DMD on children’s lives, while also highlighting resilience families demonstrate in navigating its challenges. Caregivers’ perspectives reveal that the PMO therapies are valued for their perceived ability to preserve physical function and maintain disease stability, while also providing a sense of hope in the face of a progressive disease. Such caregiver voices are important for identifying meaningful outcomes reflecting both physical functioning and the broader quality-of-life dimensions that matter to families.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
PP01.49
Digital Transformation of Korean Articulation and Phonology Assessment: Reliability and Validity Study of New Instrument
Asst. Prof. Jee Hyun Suh1, Asst. Prof. Seo Yeon Yoon2
1
Seoul National University Bundang Hospital, Seongnam-si, Korea, Republic of.
2
Yonsei University College of Medicine, Seoul, Korea, Republic of
Background: Speech sound disorders (SSD), including articulation and phonological impairments, are prevalent in childhood and adversely affect early literacy and academic achievement. In South Korea, SSD represents a substantial proportion of pediatric communication disorders. Although telerehabilitation offers a promising solution to regional disparities in access to care, existing standardized assessments such as the Urimal Test of Articulation and Phonology (UTAP) rely on time-intensive manual scoring and face limitations in digital implementation. To address these challenges, a novel digital articulation assessment instrument, Hi-DongDong, was developed using a three-round Delphi process with multidisciplinary experts. This study aimed to evaluate the reliability and concurrent validity of Hi-DongDong in children aged 2–13 years.
Methods: This prospective multicenter study was conducted across five institutions. Children aged 2–13 years were recruited from outpatient clinics and community childcare facilities. Speech samples were recorded in a single session and assessed using Hi-DongDong and either UTAP-1 or UTAP-2 (randomized 2:1). Anonymized recordings were independently evaluated by three experienced speech-language pathologists. Inter-rater reliability was assessed using intraclass correlation coefficients (ICC). Concurrent validity and inter-instrument reliability were examined using ICCs, Pearson correlation coefficients, Wilcoxon signed-rank tests, and Bland–Altman analyses.
Results: A total of 931 children were recruited, with analyses conducted on participants with valid paired datasets. Inter-rater reliability was excellent for all variables across instruments (ICC ≥ 0.98, p<0.001). Hi-DongDong demonstrated good-to-excellent agreement with UTAP-1 and UTAP-2 for consonant- and word-level measures, particularly Total PCC and Total PCC-R. Concurrent validity showed very strong positive correlations across all outcomes (all r=1, p<0.001). Bland–Altman analyses indicated minimal mean bias for utterance-level consonant accuracy indices, while systematic differences were observed for PMLU and PVC.
Conclusion: Hi-DongDong is a reliable and valid digital articulation assessment tool, demonstrating strong agreement with established standardized instruments and supporting its clinical utility for efficient, telerehabilitation-ready evaluation of pediatric SSD.
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: Outcome measures and clinimetry
Images or Table (Optional)
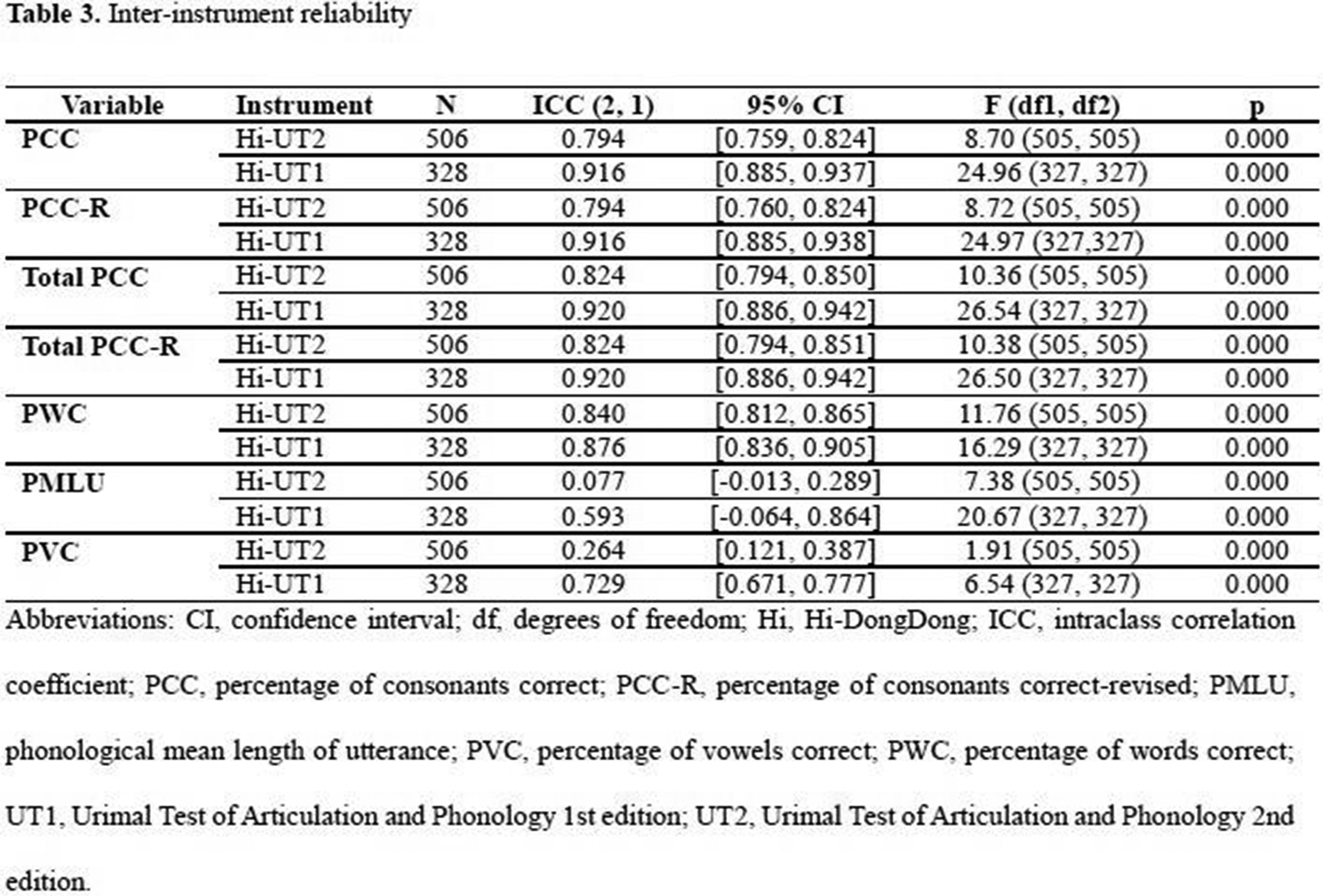
PP01.50
Management of Hip Subluxation Using Hip Bracing in Patients with Fukuyama Congenital Muscular Dystrophy
Asst. Prof. Jee Hyun Suh
Seoul National University Bundang Hospital, Seongnam-si, Korea, Republic of
Background: Fukuyama congenital muscular dystrophy (FCMD) is a rare autosomal recessive neuromuscular disorder characterized by severe muscle weakness, central nervous system abnormalities, and increased risks of joint contracture and hip dislocation. Surgically managing hip dislocation in FCMD is challenging due to cardiopulmonary risks. Although hip bracing can prevent hip displacement in cerebral palsy, its effectiveness in FCMD remains unexplored. We aimed to evaluate the potential of hip bracing to prevent hip dislocation in nonambulatory children with FCMD.
Methods: This prospective case-control study included six nonambulatory children with genetically confirmed FCMD: three wore hip braces for six months, and three were historical controls. Radiographic data, including Reimer’s migration percentage (MP), femoral head-shaft angle (HSA), and acetabular index (AI), were evaluated before and 6 months post-intervention.
Results: The mean age of the six participants was 10.83 ± 2.32 years, and each group included one male and two females. The hip brace group showed slightly improved or stabilized MP (52.40% to 50.99%), HSA (161.41° to 159.39°), and AI (15.26° to 18.41°). The control group MP deteriorated (63.85% to 78.37%); other parameters were unaffected.
Conclusion: Hip bracing may help prevent hip displacement progression in FCMD. Further large-scale studies are needed to validate these findings.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
Images or Table (Optional)
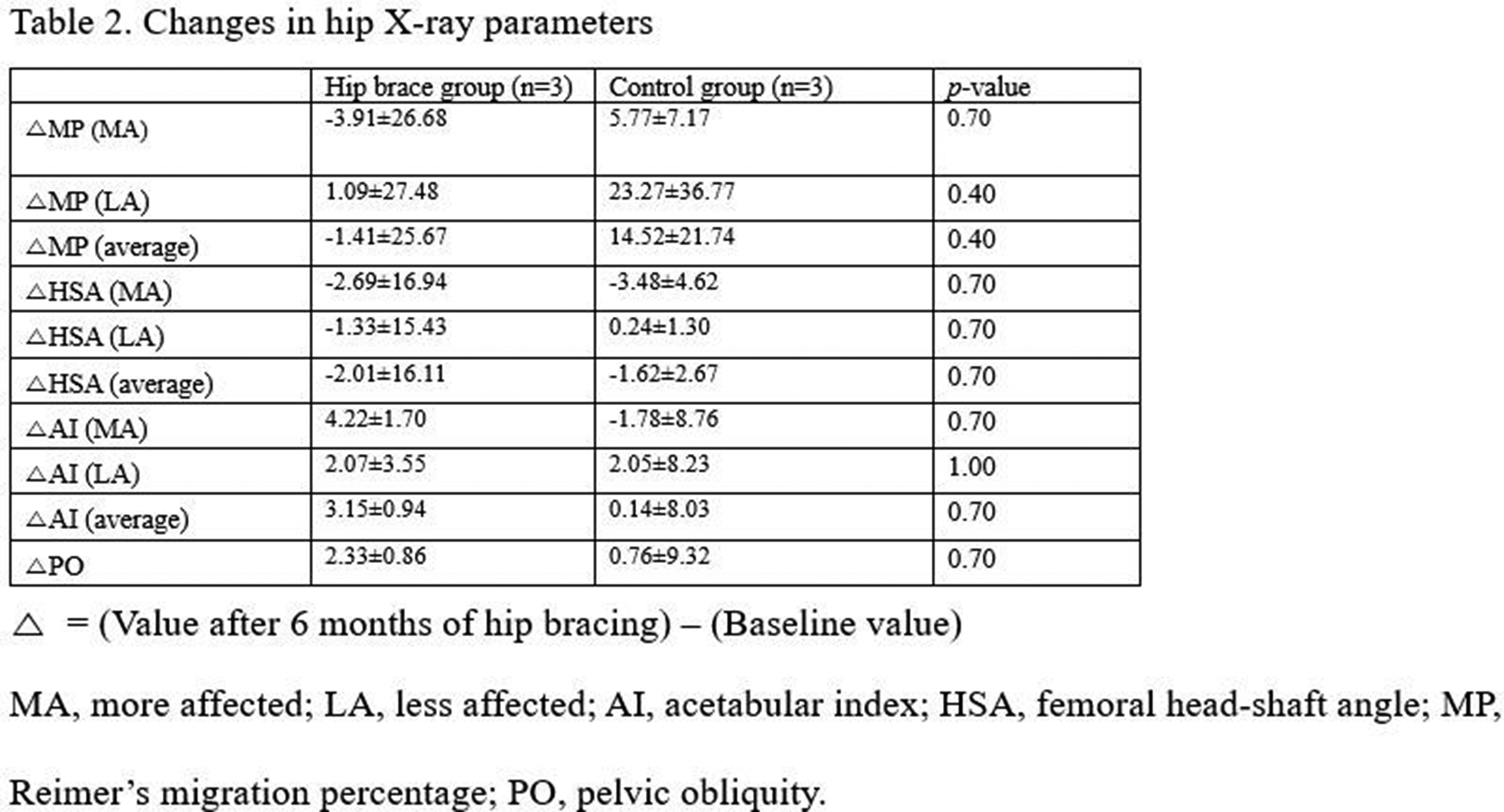
PP01.51
Considerations for Assessing Efficacy in Steroid-Naïve Interventional Trials in Duchenne Muscular Dystrophy
Ms. Kaitey Guite1, Dr. Eric P. Hoffman2, Dr. Michela Guglieri3, Dr. Paula R. Clemens4, Dr. Utkarsh J. Dang1
1
Carleton Univeristy, Ottawa, Canada.
2
Binghamton University, New York, United States.
3
Newcastle University, Newcastle Upon Tyne, United Kingdom.
4
University of Pittsburgh School of Medicine, Pittsburgh, United States
Background: Duchenne Muscular Dystrophy (DMD) is an X-linked disease characterized by progressive muscle degeneration. Its rarity, heterogenous presentation, and progressive course highlight the need for innovative trial designs to better assess drug efficacy. Strategies based on sample enrichment and/or subgroup-based primary analyses are a promising alternative to an all-comers primary analysis, while still retaining wide inclusion criteria. However, which subgroup is the most suitable for assessing efficacy is unclear. Moreover, the perception of a “honeymoon period” for the first 7 years where absolute motor function may improve and affect statistical power of comparisons in trials remains widespread. Our objective was to assess change in young 4--7-year untreated boys to understand improvement, stability, or decline in DMD in a typical clinical trial timeframe. Define subgroups that could increase statistical power compared to an all-comers analysis.
Methods: We used data from a randomized, double blinded, placebo-controlled registrational trial of vamorolone (VBP15-004), a dissociative steroid approved for DMD in boys over 2 years old, as well as data from the placebo arm of another published interventional study. A longitudinal analysis approach was employed to compare subgroup estimates to those from an all-comers analysis.
Results: The VBP15-004 trial previously showed significant steroid-related improvements in primary and multiple secondary motor outcomes. Across these outcomes (time to stand from supine [TTSTAND], time to climb 4 steps, time to run/walk 10m, six-minute walk distance [6MWD], and Northstar Ambulatory Assessment), no significant honeymoon period (improvements in motor outcomes) was identified on placebo until age 7.5 years. A literature review identified common subgroups used for DMD, typically based on age at trial baseline, specific motor outcomes, or imaging measures (fat fraction). The ability of these subgroups to improve statistical power of corticosteroid treatment effect was assessed using VBP15-004 data. The ≥ 5-second TTSTAND and 300m-400m 6MWD subgroups demonstrated the best sensitivity for detecting treatment effect of corticosteroids. Sample size and power calculations further supported the use of the ≥ 5 seconds TTSTAND subgroup demonstrating a smaller sample size required: i.e., 4 to <7 years at enrollment of steroid naïve boys, 24-wk treatment period, calculations showed 26 per group (placebo vs intervention) required for all comers analysis, whereas only 19 per group for >5 sec TTSTAND subgroup to achieve 0.9 power.
Conclusion: While some steroid-naïve boys with DMD may show a marginal improvement in some motor outcomes in early age bins, none of the motor outcomes showed consistent strength improvement until <7.5 yrs in this typical age range of interest for interventional trials. The ≥ 5-second TTSTAND and 300m-400m subgroups were found to provide greater statistical power for a corticosteroid effect than an all-comers analysis. These findings may be important both for steroid-naïve interventional trial designs and steroid add-on trials. For the former, these provide guidance for potentially capturing a range of participants with higher sensitivity to drug effects, while for the latter, importantly, underscore the large effect size for corticosteroid treatment that should be considered in clinical trials that are add-ons to corticosteroid standard of care.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
PP01.52
Long-Term Real-World Outcomes Following Onasemnogene Abeparvovec Monotherapy for Patients With SMA: Updated Findings From RESTORE
Prof. Laurent Servais1,2, Dr. John W. Day3, Dr. Darryl C. De Vivo4, Prof. Janbernd Kirschner5, Dr. Eugenio M. Mercuri6,7, Prof. Francesco Muntoni8,9, Dr. Crystal M. Proud10, Dr. Perry B. Shieh11, Dr. Eduardo Tizzano12, Prof. Susana Quijano-Roy13,14, Prof. Isabelle Desguerre15, Dr. Kayoko Saito16, Dr. Yasemin Erbaş17,18,19, Dr. Sandra P. Reyna20, Dr. Iulian Alecu20, Dr. Kamal Benguerba20, Dr. Dheeraj Raju20, Mr. David Wolff20, Dr. Richard S. Finkel21
1
Department of Paediatrics, MDUK Oxford Neuromuscular Centre & NIHR Oxford Biomedical Research Centre, University of Oxford, Oxford, United Kingdom.
2
Department of Pediatrics, Neuromuscular Reference Center, University and University Hospital of Liège, Liège, Belgium.
3
Department of Neurology, Stanford University Medical Center, Stanford, United States.
4
Departments of Neurology and Pediatrics, Columbia University Irving Medical Center, New York, United States.
5
Department for Neuropediatrics and Muscle Disease, Medical Center - University of Freiburg, Faculty of Medicine, Freiburg, Germany.
6
Department of Paediatric Neurology, Catholic University, Rome, Italy.
7
Nemo Clinical Centre, Fondazione Policlinico Universitario Agostino Gemelli IRCCS, Rome, Italy.
8
The Dubowitz Neuromuscular Centre, Developmental Neurosciences Department, University College London, Great Ormond Street Institute of Child Health, London, United Kingdom.
9
NIHR Great Ormond Street Hospital Biomedical Research Centre & Great Ormond Street Hospital, London, United Kingdom.
10
Children’s Hospital of The King’s Daughters, Norfolk, United States.
11
Department of Neurology, David Geffen School of Medicine at UCLA, University of California Los Angeles, Los Angeles, United States.
12
Department of Clinical and Molecular Genetics, Hospital Vall d’Hebron, Barcelona, Spain.
13
Garches Neuromuscular Reference Center, Child Neurology and ICU Department, APHP Raymond Poincaré University Hospital (UVSQ Paris Saclay), Garches, France.
14
Laboratoire END-ICAP - UMR 1179 (INSERM/UVSQ), Rome, Italy.
15
Reference Center for Neuromuscular Disorders, Neuropediatric Department, Hôpital Necker Enfants Malades, Paris, France.
16
Institute of Medical Genetics, Tokyo Women’s Medical University, Tokyo, Japan.
17
SMA Europe, Freiburg, Germany.
18
Spierziekten Vlaanderen, Borgerhout, Belgium.
19
Department of Developmental Psychology, Tilburg University, Tilburg, Netherlands.
20
Novartis Pharma AG, Basel, Switzerland.
21
Center for Experimental Neurotherapeutics, St. Jude Children’s Research Hospital, Memphis, United States
Background: Onasemnogene abeparvovec (OA), a one-time gene therapy for spinal muscular atrophy (SMA), has been evaluated in several clinical trials and long-term follow-up studies. However, long-term, real-world OA data are limited. RESTORE, a multinational, non-interventional SMA registry, is a successful industry/academia collaboration and robust real-world instrument. Previous RESTORE findings have been reported for OA monotherapy-treated patients (N=168; 13.7 months mean follow-up). Here, we build on long-term safety, effectiveness, and durability evidence for OA monotherapy in a larger RESTORE patient cohort with greater follow-up duration.
Methods: Motor function was assessed using the Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND), Hammersmith Functional Motor Scale – Expanded (HFMSE), and the Hammersmith Infant Neurological Examination – Section 2 (HINE-2).
Results: Of 251 patients (May 23, 2024, data cutoff), median (IQR) ages at SMA diagnosis and OA infusion were 1 (0–6) month and 3 (1–8) months, respectively. Median (IQR) time from OA infusion to last visit was 23.6 (14.3–35.7) (range, 0.5–61.6) months. Patients were diagnosed through newborn screening (n=147, 58.6%) or clinical assessment (n=104, 41.4%). Most had two (n=112, 44.6%) or three (n=106, 42.2%) survival motor neuron 2 (SMN2) gene copies and were treated in the United States (n=173, 68.9%). Of the 61 patients in safety analysis set 1 (patients for whom safety data were available from the time of OA administration), 38 (62.3%) had an adverse event (AE); 24 (39.3%) had related AEs, which were serious in four patients (6.6%). Of 227 patients in safety analysis set 2 (includes all patients with available safety data at any time after OA administration), 109 (48.0%) had an AE; 54 (23.8%) had related AEs, which were serious for seven patients (3.1%). Most common AEs of special interest were hepatotoxicity (n=52, 22.9%), cardiac AEs (n=31, 13.7%), and transient thrombocytopenia (n=28, 12.3%). Six deaths (2.4%) occurred. Overall, most patients demonstrated improvements in motor function scores, with 102 of 115 patients assessed (88.7%) achieving or maintaining CHOP INTEND scores of ≥40 points (achieved, n=33 [28.7%]; maintained, n=69 [60.0%]). Mean (SD) months between CHOP INTEND assessments was 12.9 (10.1) months. Similarly, 52 of 55 patients assessed (94.5%) achieved or maintained ≥3-point improvements in HFMSE scores (achieved, n=41 [74.5%]; maintained, n=16 [29.1%]). HINE-2 scores also demonstrated progressive increases over time, with 11 patients achieving the maximal score of 26 (SMN2 copies: two (n=3); three (n=3); ≥4 (n=5). Motor function improvements were maintained for up to 5 years.
Conclusion: These data, while limited to patients treated with OA as monotherapy, indicate therapeutic benefit for up to 5 years post-dosing, providing further evidence for OA as a durable treatment for patients with SMA.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Muscle Dystrophies (Non-Dystrophinopathies)
PP01.53
ENTR-601-44-201: First-in-Patient Results From a Phase 1/2 Study Evaluating ENTR-601-44 in Exon 44 Skip-Amenable DMD
Dr. Laurent Servais1,2, Dr. Nicolas Deconinck3,4, Dr. Liesbeth De Waele5,6, Dr. David Gómez-Andrés7, Dr. Kalina Dimova8, Dr. Jenny Li8, Ms. Nanjun Liu8, Dr. Nick Long8, Ms. Meghan MacNally8, Dr. Ridhi Parasrampuria8, Dr. Kiran Patki8, Ms. Dania M.D. Porco8, Dr. Divya Reddy8, Ms. Marie Rosenfeld8, Dr. Natarajan Sethuraman8
1
Department of Paediatrics, MDUK Oxford Neuromuscular Centre & NIHR Oxford Biomedical Research Centre, University of Oxford, Oxford, United Kingdom.
2
Department of Paediatrics, Neuromuscular Reference Centers, University Hospital of Liège, Liège, Belgium.
3
Neuromuscular Reference Center, UZ Gent, Ghent, Belgium.
4
Centre de Référence Neuromusculaire and Paediatric Neurology Department, Hôpital Universitaire des Enfants Reine Fabiola, Université Libre de Bruxelles, Brussels, Belgium.
5
Department of Paediatrics, University Hospitals Leuven, Leuven, Belgium.
6
Department of Development and Regeneration, KU Leuven, Leuven, Belgium.
7
Child Neurology, Vall d'Hebron Institut de Recerca (VHIR), Hospital Universitari Vall d'Hebron, Vall d'Hebron Barcelona Hospital Campus, Barcelona, Spain.
8
Entrada Therapeutics, Boston, United States
Background: Approximately 8% of individuals with Duchenne muscular dystrophy (DMD) have exon 44 skip-amenable dystrophin gene mutations. ENTR-601-44 is under investigation for the potential treatment of individuals with DMD who have confirmed exon 44 skip-amenable dystrophin gene mutations. ENTR-601-44 is an Endosomal Escape Vehicle peptide-phosphorodiamidate morpholino oligomer (PMO) conjugate intended to target dystrophin pre-mRNA and induce exon 44 skipping, thereby restoring expression of near full-length and functional dystrophin protein. ENTR-601-44 has demonstrated exon skipping and dystrophin restoration in skeletal muscle cells derived from individuals with DMD who have an exon 44 skip-amenable mutation. In the gastrocnemius muscle of mice with an exon 44 skip-amenable dystrophin mutation (del45hDMD.mdx), one 30-mg/kg intravenous dose of ENTR-601-44 increased dystrophin protein from <1% to 46% of wild-type levels. In nonhuman primates, 4 doses of ENTR-601-44 produced 29% DMD exon 44 skipping, indicating target engagement. Furthermore, in the first-in-human phase 1 ENTR-601-101 study in healthy adult male volunteers, ENTR-601-44 dosed at 6 mg/kg was well tolerated and showed dose-dependent increases in urinary excretion of the final metabolite (PMO-44) and exon 44 skipping. Together, these findings support further study of ENTR-601-44 in individuals with exon 44 skip-amenable DMD. Herein, we present Cohort 1 data from participants in Part A (double-blind period) of the ENTR-601-44-201 study.
Methods: ENTR-601-44-201 is a first-in-patient global 2-part phase 1/2 study of ENTR-601-44 in participants with a confirmed mutation in the dystrophin gene that is amenable to exon 44 skipping. Part A is a randomized, double-blind, placebo-controlled, multiple ascending dose design, focusing on safety/tolerability, pharmacokinetics, and pharmacodynamics (exon 44 skipping and dystrophin expression). Approximately 24 participants are planned, divided into 3 cohorts of 8 participants, each to receive ENTR-601-44 (6, ≤12, or ≤18 mg/kg) or placebo. Key study eligibility criteria include males assigned at birth aged 4–20 years, inclusive, with a confirmed genetic diagnosis of exon 44 skip-amenable DMD, and who are ambulatory with a Performance of the Upper Limb v2.0 (PUL 2.0) entry item A of at least 3 at screening. If on glucocorticoid therapy, participants must be on a stable dose and regimen for at least 3 months before dosing. No prior treatment with any exon-skipping therapy or gene therapy is allowed. All participants in Part A are eligible for continued treatment in an open-label extension period.
Results: Eight males with genetically confirmed DMD amenable to exon 44 skipping were randomized 3:1 to receive 3 doses of ENTR-601-44 (6 mg/kg) or placebo as an intravenous infusion over 6-week intervals. Participants underwent baseline muscle biopsy before the first dose of treatment during the screening period, and then again at the end of the treatment period at week 19 (6 weeks after the final dose). At baseline, mean age was 10 (range, 6–17) years and mean body mass index was 19.5 (range, 15.6–25.2) kg/m2. Mean age at DMD diagnosis was 3 (range, 0–7) years.
Conclusion: Topline data, including assessments of safety and tolerability, change in dystrophin and exon 44 skipping levels, and pharmacokinetics of ENTR-601-44 will be presented.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
PP01.54
Effects of NP12 Treatment on Skeletal and Cardiac Muscle in Thermoneutrally Housed Mdx Mice
Mr. Luc Wasilewicz1, Dr. Jessica Braun2, Dr. Sophie Hamstra1, Ms. Briana Hockey1, Dr. Rebecca MacPherson1, Dr. Val Fajardo1
1
Brock University, St.Catharines, Canada.
2
University of Copenhagen, Copenhagen, Denmark
Background: Duchenne Muscular Dystrophy (DMD) is an X-linked, genetically inheritable disease, characterized by muscle wasting and cardiorespiratory failure. To study DMD, it is important to use accurate animal models and research settings that are as closely translatable to the human population as possible, including housing temperature. Thermoneutrality is the temperature range that is most physiologically relevant to conduct research as there are no compensatory responses by the animal to alter core temperature, with this range being 28-30ºC for mice. While the mdx mouse has been widely used in the study of DMD, studies to date have housed mice at “room temperature” (22-24ºC). Although appropriate for humans, this range represents a mild cold stress in mice and imposes additional metabolic demands that may alter disease pathology and potentially conflict with the interpretation of results. Previously, we demonstrated that when DBA/2J (D2) mdx mice were housed at thermoneutrality, their muscle-wasting pathology worsened. This was demonstrated by reductions in the resistance to muscle fatigue of the extensor digitorum longus (EDL) muscle and increased cardiac impairments (i.e., reduced left ventricular internal diameter, end-diastolic volume, and cardiac output). From a mechanistic standpoint, we demonstrated that glycogen synthase kinase 3β (GSK3β) activity is higher, based on decreased inhibitory phosphorylation levels, in mice housed at thermoneutrality than at room temperature, which could contribute to the worsened pathology observed in mdx mice housed at thermoneutrality. We have previously demonstrated that GSK3β inhibition via a 4-week NP12 treatment can improve muscle strength and overall health in D2 mdx mice housed at room temperature. However, whether inhibiting GSK3β will produce similar benefits when D2 mdx mice are housed at thermoneutrality remains unknown. Thus, we sought to determine the effects of a 4-week NP12 treatment on skeletal and cardiac muscle in D2 mdx mice housed at thermoneutrality.
Methods: 20 DBA/2J mdx mice (age 5 weeks) were randomly divided into a vehicle or NP12 group (n = 10 per group). All mice were provided a vehicle (poloxamer) or NP12 solution (10 mg/kg/day) via oral gavage 5x/week for 4 weeks. Analyses; In vivo: metabolic caging, body composition scans, insulin-tolerance-test, and high-frequency ultrasound. Ex vivo: EDL muscle fatigue contractility. Biochemical: creatine kinase assay, western blotting, and histological muscle fibre-type staining.
Results: Metabolic caging revealed no differences in O2 consumption, respiratory exchange ratio, cage activity or food intake between groups. There were no differences in body composition (i.e., body mass, fat mass, lean mass), serum creatine kinase, and total GSK3β between groups. An insulin-tolerance-test demonstrated that NP12-treated mice had increased insulin sensitivity and a high-frequency ultrasound demonstrated improved cardiac function (i.e., increases in stroke volume, cardiac output, end-diastolic volume). NP12-treated mice had improved EDL fatigue resistance alongside a higher percentage of type IIA and I oxidative fibres.
Conclusion: Overall, our results demonstrate that NP12 treatment is able to improve indices of skeletal and cardiac muscle health, as well as whole-body insulin sensitivity in thermoneutrally-housed D2 mdx mice. These results help pave a path towards targeting GSK3 for DMD in the clinical setting.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
PP01.55
Development of the Duchenne Muscular Dystrophy Natural History Italian Registry: Next-Generation EU–PNRR Study
Dr. Luca Kunsztler1, Dr. Daniela Leone1, Dr. Claudia Brogna1,2, Dr. Sonia Messina3, Dr. Valeria Ada Maria Sansone4, Dr. Antonio Di Muzio5, Dr. Giulia Norcia1, Dr. Beatrice Berti1, Dr. Sophia Paolucci1, Dr. Anna Capasso1,2, Dr. Chiara Arpaia1,2, Dr. Marianna Villa1, Dr. Gianpaolo Cicala1,2, Dr. Romina Venditti1,2, Dr. Giorgia Coratti1,2, Dr. Eugenio Maria Mercuri1,2, Dr. Marika Pane1,2
1
Fondazione Policlinico Universitario Agostino Gemelli IRCCS, Rome, Italy.
2
Università Cattolica del Sacro Cuore, Rome, Italy.
3
Department of Clinical and Experimental Medicine, University of Messina, Messina, Italy.
4
The NEMO Clinical Center in Milan, Neurorehabilitation Unit, University of Milan- ERN for Neuromuscular Diseases, Milan, Italy.
5
Department of Neuroscience, Imaging and Clinical Sciences, “G. D'Annunzio” University, Chieti, Italy
Background: Duchenne muscular dystrophy (DMD) is a progressive X-linked neuromuscular disease caused by dystrophin gene mutations, affecting 1 in 3600 live male births. Typically diagnosed between ages 3-5 years with waddling gait, calf hypertrophy, and difficulty climbing stairs, the disease follows a predictable pattern of functional decline after age 7. Improved standards of care and early glucocorticoid treatment have significantly altered the natural history, affecting both survival and timing of functional milestone loss. Recent mutation-specific therapies, including antisense oligonucleotides for exon skipping and nonsense mutation treatments, show promising results. However, longitudinal data remain crucial for understanding genotype-specific progression and providing external controls for evaluating new pharmacological approaches. A 2013 three-year longitudinal study provided prospective functional data but lacked comprehensive genotype information.
Methods: Following the original 2013 Telethon-funded study, participating centers continued data collection using identical criteria and structure, accumulating over 100 additional patients with three-year follow-up data and extended observations in the original cohort. The project is funded by the Grant “Unione europea–Next Generation EU–PNRR M6C2–Investimento 2.1 Rafforzamento e potenziamento della ricerca biomedica del SSN–PNRR-MR1-2023-12377031_CUP C53C23001220007”.
This project aims to: develop a structured national electronic case report form (CRF) incorporating comprehensive functional data; migrate existing data from electronic datasets and clinical notes into the CRF; analyze three-year data from an expanded cohort exceeding 200 patients; conduct extended follow-up analysis beyond three years; evaluate non-ambulant patient outcomes; and identify prognostic indicators by analyzing functional trajectories one to three years preceding loss of ambulation.
Results: By August 31, 2025, two meetings were held among participating units to establish common terminology through a data dictionary and identify appropriate variables for CRF development. This process involved identifying ambulant and non-ambulant patients with genetically confirmed DMD who had at least three years of consecutive natural history data at each center, reviewing their scheduled follow-up appointments, and identifying deceased patients with at least three years of consecutive data. Protocols were defined for blood sample collection, storage, and shipment to Unit 1 for genetic analysis. Nine-milliliter EDTA blood samples were collected for DNA extraction and real-time PCR analysis to evaluate potential genetic modifiers (osteopontin, LTBP4, etc.) in known DMD modifier loci, specifically SNPs of LTBP4, SPP1, CD40, and THBS1 genes. During routine follow-up visits, all eligible patients with a genetic DMD diagnosis were enrolled, and new clinical, functional, respiratory, and cardiologic data were collected according to DMD care standards guidelines. The CRF includes clinical history interviews, functional assessments (6MWT, NSAA, and PUL2.0 for ambulant patients; PUL2.0 for non-ambulant patients), and laboratory and instrumental examinations (2D echocardiogram, resting electrocardiogram, 24-hour Holter ECG monitoring, spirometry, polysomnography, nocturnal oximetry) according to international DMD care standards guidelines.
As of the interim analysis, 170 patients meeting the original inclusion criteria have been consecutively enrolled following informed consent during routine clinical visits, with ongoing enrollment and data collection.
Conclusion: This initiative will advance understanding of genotype-specific disease progression, ultimately supporting improved clinical trial design and patient care in DMD.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
PP01.56
Patient Characteristics and Use of Vamorolone in Adult Patients With DMD in Germany and Austria
Dr. Marcus Erdler1, Dr. Miriam Hiebeler2, Ms. Farah J. Walter2, Dr. Kristine Tetunashvili3, Dr. Simone Thiele2, Prof. Dr. Maggie C. Walter2
1
Klinik Donaustadt, Dept. of Neurology, Vienna, Austria.
2
Friedrich-Baur-Institute, Dept. of Neurology, Klinikum LMU Munich, Munich, Germany.
3
Klinik Donaustadt, Dept. of Neurology, Wien, Austria
Background: Vamorolone is a dissociative corticosteroid approved by the FDA (patients ≥2 years) and EMA (patients ≥4 years) for treating Duchenne muscular dystrophy (DMD). We here present the first real-world experience of vamorolone use in adults.
Methods: This retrospective, real-world, early point-in-time analysis evaluated characteristics and utilization patterns of 25 adult DMD patients treated with vamorolone in one German and one Austrian centre (13 and 12 patients, respectively).
Results: The median age was 23 years (range 19-34). Mean age at diagnosis was 5,5 years, while mean symptom onset was at age 3,6 years in our cohort. All patients were non-ambulatory, since a median age of 11,6 years (range 8-19). Mean BMI was 21,4, ranging from 9 to 32. Six patients were corticosteroid (CS) naïve, 16 had prior prednisone and three prior deflazacort treatment. Of the 19 previously CS-treated subjects, eight patients directly switched from other CS; 11 were restarting treatment after previously discontinuing treatment 2-16 years prior to vamorolone, and six patients started first-line. Vamorolone was initiated to preserve upper limb motor function or due to prior CS side effects (e.g. osteoporosis, obesity, Cushing’s syndrome). Twelve patients received vamorolone at capped dose of 240 mg/day (all ≥40 kg); 13 received a lower dose of 2 mg/kg/day (if <40 kg) or capped at 80 mg/day (if ≥40 kg), following DMD guidelines for non-ambulatory patients who often remain on reduced CS dose due to prior adverse effects. In four of the latter, the dose was increased to 120 mg/day (all ≥40 kg) after 6+ month due to weight or PUL improvement. PUL was done in the Munich cohort, 4/13 patients showed PUL improvement, 4/13 stabilization, 5/13 were only recently initiated and have no follow-up data yet. All patients with prior CS-related side effects (obesity, Cushing’s syndrome, behavioural problems) showed reduced or no symptoms on vamorolone. At the last follow-up, no weight gain or new adverse events were reported.
Conclusion: This first real-world analysis demonstrates clinical efficacy of vamorolone regarding stabilization or even mild improvement in adult DMD patients who were CS-naïve, had discontinued previous CS treatment, or switched from other CS. This data further highlights the importance of continued corticosteroid treatment beyond loss of ambulation.
Conclusion: This first real-world analysis demonstrates clinical efficacy of vamorolone regarding stabilization or even mild improvement in adult DMD patients who were CS-naïve, had discontinued previous CS treatment, or switched from other CS. This data further highlights the importance of continued corticosteroid treatment beyond loss of ambulation.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
PP01.57
The DMD-NEEDS Study: Real-World Management of Duchenne Muscular Dystrophy in Spain
Dr. Carlos Ortez1, Mrs Maria Branas2, Dr. Joaquin Alejandro Fernandez3, Miss Elisa Guillen2, Miss Angeles Terrancle2, Dr. Jorge Maurino2, Ms. Marisol Montolio4, Dr. Ana Camacho5
1
Hospital Sant Joan de Deu, Barcelona, Spain.
2
Roche Farma, Madrid, Spain.
3
Hospital Reina Sofía, Cordoba, Spain.
4
Duchenne Parent Project Spain, Barcelona, Spain.
5
Hospital 12 de Octubre, Madrid, Spain
Background: Despite the existence of international clinical guidelines for Duchenne Muscular Dystrophy (DMD) management, comprehensive, updated data on their real-world implementation in Spain is limited. The DMD-NEEDS study aimed to characterize the current landscape of DMD clinical practice, validate adherence to best practices, and identify key areas for improvement in the country.
Methods: This was a non-interventional, cross-sectional, and multicenter study conducted in collaboration with the Spanish Society of Paediatric Neurology (SENEP) and Duchenne Parent Project Spain. An electronic survey containing standardized questions on the DMD patient journey, diagnostic methods, and clinical management was completed by 41 pediatric neurologists specializing in DMD care. Descriptive statistics were utilized for all reported variables.
Results: A total of 41 pediatric neurologists completed the survey, with a mean experience in DMD management of 13.3 years. The majority of participants were female (56.1%), and 41.5% had a specialization in neuromuscular diseases. Most participants (97.6%) reported following established clinical guidelines. Early diagnosis was a prominent finding for the patients under their care, with most (53.7%) receiving a diagnosis between ages 2 and 3. The majority of patients were referred from primary care (73.2%), and Multiplex Ligation-dependent Probe Amplification (MLPA) was the primary diagnostic test reported by 92.7% of participants. Corticosteroid therapy, predominantly deflazacort, was prescribed by 95.1% of neurologists and typically initiated at ages 4-5 (66.7%). Disease progression aligned with the established natural history, with loss of ambulation occurring between ages 11 and 13 (53.7%), and major complications (scoliosis, cardiac, and respiratory involvement) appearing between ages 12 and 17. While multidisciplinary care teams were reported as available in 68.3% of hospitals, a significant gap was identified in patient-centered assessment, as 58.5% of participants reported not using health-related quality of life (HRQoL) questionnaires. Furthermore, a strong proactive mindset toward novel treatments was observed, with 87.8% of pediatric neurologists indicating a willingness to recommend gene therapy for eligible patients.
Conclusion: The DMD-NEEDS study reveals an encouraging landscape in Spain, marked by high adherence to best practices like early diagnosis and widespread corticosteroid use. While these findings affirm positive trends, they also highlight critical areas for national optimization, particularly improving access to multidisciplinary care and mandating the routine application of validated HRQoL assessment tools to ensure holistic, patient-centered care.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
PP01.58
Clinical and Genetic Characterization of Duchenne Muscular Dystrophy in a Chilean Cohort
Asst. Prof. Maria de los Angeles Beytia1,2, Dr. Bernardita Suárez3,4, Dr. Javiera Jofré3,5, Asst. Prof. Daniela Ávila-Smirnov1,2, Dr. Carlos Jaque6, Dr. Claudia Castiglioni3,4,7
1
Pontificia Universidad Católica de Chile, División de Pediatría, Unidad de neurología pediátrica, Santiago, Chile.
2
Hospital Dr. Sótero del Río, Unidad de Neurología pediátrica, Santiago, Chile.
3
Clínica Meds, Unidad Neuropediatría, Departamento de Neurología, Santiago, Chile.
4
Instituto Nacional de Rehabilitación Pedro Aguirre Cerda, Santiago, Chile.
5
Hospital Dr. Luis Calvo Mackenna. Unidad de Neurología Pediátrica, Santiago, Chile.
6
Hospital Clínico Herminda Martín, Chillán, Chile.
7
Facultad de Medicina, Universidad Finis Terrae, Santiago, Chile
Background: Muscular Dystrophy (DMD) is the most prevalent pediatric neuromuscular disorder affecting approximately 1 in 3,500-5,000 male births. It is characterized by progressive muscle degeneration secondary to mutations in the DMD gene. The mean age of diagnosis globally in high-income countries is between 4 and 5 years, whereas in Latin America, it has been reported to range from 5.7 to 7 years. Internationally, the most common mutations worldwide are deletions (60-70%), duplications (20%), and point mutations (10-15%).
Objective: This study aims to evaluate the clinical characteristics, including age at symptom onset, age at diagnosis, diagnostic delay, and most frequent symptoms at onset, and the genetic profile, including type of genetic study, mutation frequency, and affected exons, in patients with DMD in a Chilean cohort.
Methods: A retrospective analysis was performed on clinical and genetic data from 130 DMD patients treated at 5 centers in Chile. 106 patients were selected, based on a confirmed diagnosis of Duchenne Muscular Dystrophy through genetic testing. Inclusion criteria were patients with a diagnosis established by multiplex ligation-dependent probe amplification (MLPA) or next-generation sequencing (NGS) panels, between January 2010 and December 2025. Exclusion criteria included patients with incomplete genetic data or those diagnosed with other forms of muscular dystrophy.
Results: The median current age of the patients was 6.4 years (range: 2.1-37.1 years). The median age of symptom onset was 2 years (range: 3 months-5 years), and the median age at diagnosis was 4.3 years (range: 4 months-32 years). In 9/106 (8.5%), the diagnosis was made before 15 months of age, predominantly due to high transaminase levels as an incidental finding with no hepatic cause identified. The most frequent reason for consultation was gait disturbance, followed by psychomotor developmental delay. Regarding genetic diagnosis, 38% (40/106) were diagnosed with MLPA as the first approach, 62% (66/106) were diagnosed using NGS panels; 19% following negative MLPA. In terms of mutation type, deletions accounted for 57% (59/106), duplications for 8.5% (9/106), including 4 complex non-contiguous, and point mutations for 36% (38/106). Of the latter, 19 (18%) were nonsense mutations. The most frequent deletions affected exons 45, 45-52, and 49-50.
Conclusion: The age at diagnosis in this Chilean cohort is comparable to that reported in high-income countries, although a diagnostic delay of approximately 2.3 years from symptom onset persists. Our population presents a higher proportion of point mutations compared to international series, but similar to some cohorts of Spanish population, suggesting a greater percentage of patients potentially eligible for “read-through” therapies targeting nonsense mutations. Potential selection bias may exist due to its multicenter, retrospective design and variations in diagnostic protocols across centers.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
PP01.59
FORZETTO, a Phase 3 Study of Z-Rostudirsen in Ambulatory Males With Exon 51 Amenable DMD
Dr. Maria Naylor, Dr. Xiaoyan Yin, Dr. Katy Meilleur, Dr. Jordan Messer, Dr. Gabriel Helmlinger, Dr. Prashant Bansal, Dr. Soma Ray, Dr. Douglas Kerr
Dyne Therapeutics, Waltham, United States
Background: Duchenne muscular dystrophy (DMD) is a progressive, X-linked neuromuscular disorder caused by pathogenic variants of the DMD gene resulting in the absence of functional dystrophin protein. DMD is a multi-system disease that manifests in progressive muscle degeneration, loss of ambulation, cardiopulmonary complications, neuropsychiatric symptoms, and premature death. Despite recent therapeutic advances, a significant unmet medical need remains for treatments that achieve durable dystrophin restoration and sustained functional benefit. Zeleciment rostudirsen (z-rostudirsen, also known as DYNE-251) leverages TfR1 to deliver an exon 51 skipping PMO to muscle, with the goal of producing near full-length, functional dystrophin. Previously disclosed data from the ongoing Phase 1/2 DELIVER trial (NCT05524883) have shown a favorable safety and tolerability profile. At the registrational dose of 20 mg/kg Q4W, z-rostudirsen demonstrated a statistically significant increase in dystrophin expression and trends in early and sustained functional improvement across clinical endpoints, supporting advancement to a Phase 3 study.
Methods: To evaluate the efficacy, safety, and tolerability of z-rostudirsen 20 mg/kg Q4W in a placebo-controlled, confirmatory study.
Results: The Phase 3 FORZETTO study will enroll ∼90 ambulatory participants aged 4-18 years with DMD caused by mutations amenable to exon 51 skipping. Participants will be randomized 1:1 to receive 20 mg/kg Q4W z-rostudirsen or placebo for 72 weeks, followed by a 96-week open-label Long-Term Extension (LTE). Endpoints include multiple measures of muscle function and PROs.
Conclusion: The Phase 3 FORZETTO study will further evaluate the clinical benefit and safety of z-rostudirsen in participants with DMD amenable to exon 51 skipping, with endpoints that measure functional improvement.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
PP01.60
RNA-Based Combinatorial Multi-Target Therapy for the Treatment of Duchenne Muscular Dystrophy
Dr. Mathieu Horeau1,2, Dr. Camilla Pezzini1,2, Assoc. Prof. Roberta Sartori1,2, Prof. Marco Sandri1,2
1
Veneto Institute of Molecular Medicine, Padova, Italy.
2
Università di Padova, Department of Biomedical Sciences, Padova, Italy
Background: Duchenne muscular edystrophy is a severe and fatal X-linked disease, characterized by the lack of dystrophin, leading to structural fragility of muscle fibers and promoting a pro-catabolic metabolic imbalance. To date, no curative treatment is available, partly due to the difficulty in restoring full dystrophin expression and developing non-immunogenic approaches. Reactivation of utrophin, a paralog of dystrophin that is naturally downregulated by microRNAs (miRNAs), has shown strong potential to compensate for dystrophin deficiency.
This project aims to address three major challenges to 1) reactivate utrophin expression to compensate for the loss of dystrophin, 2) inhibit pro-catabolic pathways to promote muscle growth, 3) develop non-immunogenic vectors for effective and tolerable combination therapy. We hypothesize that our combinatory multitarget therapy could simultaneously restore muscle structural integrity and prevent/ reverse muscle wasting and the associated loss of function.
Methods: Taking into consideration that this project only started a few months ago, we will focus solely on Aims 2 and 3.
Aim 2: To reduce the expression of MuRF1 and FOXO1/3, key players in muscle catabolism, short hairpin RNAs targeting MuRF1 and FOXO1/3, alone or in combination, were electroporated into the tibialis anterior muscle of wild-type mice.
Aim 3: We developed myotropic liposomes derived from C2C12 murine myoblast cells carrying antisense oligonucleotides against MuRF1. To date, wild-type mice were injected intramuscularly (TA) with 10¹¹ particles. After 24 and 72 hours, and 7 days, the tibialis anterior muscles were collected, and MuRF1 protein and mRNA expression were assessed.
Results: Our preliminary data for Aim 2 highlight that, in WT animals, combined electroporation of AAV-MYO-shMuRF1 and AAV-MYO-shFOXO1/3 in the tibialis anterior muscle leads to a reduction in the expression of MuRF1 (−24%; P = 0.04), FOXO1 (−36%; P = 0.002), and FOXO3 (−39%; P = 0.03) genes. This reduction is associated with muscle hypertrophy, characterized by a 30% increase in the cross-sectional area (P = 0.03) of the tibialis anterior, which is not significantly observed with non-combinatorial treatments.
For Aim 3, we observe, after only 7 days, a downregulation of the MuRF1 transcript (−46%; P = 0.038) and a slight but non-significant decrease in protein expression (−10%).
Conclusion: Our objective is now to move to pathological conditions for Aim 2. We are currently testing this approach in pre-symptomatic (3-week-old) MDX mice using adeno-associated virus vectors with a muscle-specific serotype (AAV9-MYO2/3) to deliver our AAV-MYO-shRNA against FOXO1/3 or MURF1.
Concerning Aim 3, we are now moving to a systemic approach to evaluate muscle specificity and efficiency in targeting whole-body muscle while avoiding off-target effects.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
PP01.61
Bone-Health and Fracture Risk in FSHD1: A 12-Months Longitudinal Multi-Modal Imaging and Biomarker Study
Mr. Matthias Opsomer1,2, Dr. Jilmen Quintiens3, Ms. Elena Paravisi3, Dr. Thomas Anijs3, Mr. Herman Borghs4, Mr. Walter Coudyzer5, Ms. Tine de Cuyper4, Mr. Herman Peeters4, Dr. Veerle Goosens5, Prof. Marian Dejaeger4, Prof. Patrick Dupont6, Prof. Harry van Lenthe3, Prof. Kristl G. Claeys1,2
1
Laboratory for Muscle Diseases and Neuropathies, Department of Neurosciences, KU Leuven, and Leuven Brain Institute (LBI), Leuven, Belgium.
2
Department of Neurology, University Hospitals Leuven, Leuven, Belgium.
3
Biomechanics Section, Department of Mechanical Engineering, Leuven, Belgium.
4
Metabolic Bone Center, University Hospitals Leuven, Leuven, Belgium.
5
Department of Radiology, University Hospitals Leuven, Leuven, Belgium.
6
Laboratory for Cognitive Neurology, Department of Neurosciences, KU Leuven, and Leuven Brain Institute (LBI), Leuven, Belgium
Background: Facioscapulohumeral muscular dystrophy type 1 (FSHD1) is a slowly progressive muscle disorder caused by contraction of the D4Z4 repeat on chromosome 4q35. While muscle degeneration is well-characterized, osteoporosis and fracture risk are often underestimated and conventional dual-energy X-ray absorptiometry (DXA) may not capture early microarchitectural changes. Photon-counting computed tomography (PCCT) offers a low-dose, rapid method to evaluate bone mineral density (BMD) and microarchitecture. We aim to evaluate baseline and 12-month longitudinal changes in bone health and fracture risk and identify clinical and biochemical predictors of bone loss in adults with FSHD1.
Methods: Adults with FSHD1 were prospectively examined at baseline and 12 months. Bone health was assessed via DXA (lumbar spine, bilateral hips), PCCT, vertebral fracture assessment (VFA), and serum markers (BetaCTx, P1NP, vitamin D, PTH). Clinical and functional evaluation included fall history, FRAX® scores, disease severity scales. Patient-reported outcome measures comprised ActivLim and FSHD-Rasch-built overall disability scale (FSHD-Rods) and clinical outcome measures Timed Up and Go (TUG), Motor Function Measure 32 part D1 (MFM32-D1), 4-stairs climb test, and 6-minute walk test (6MWT), and Biodex® knee extension and flexion peak isometric strength.
Results: Baseline assessment of 60 patients (mean age 52.1 ± 15.7 years, 55.0% female, mean number of D4Z4 repeats 7.2 ± 1.8) revealed that 56.7% had osteopenia and 21.7% osteoporosis. Prior fractures were reported in 50.8% of patients. Fall frequency was high with 63.3% reporting at least one fall per year). Mean DXA T-score was -0.53 ± 1.33 (spine), and -1.32 ± 0.98 (femoral neck). VFA detected mild asymptomatic vertebral fractures in three patients. Baseline 10-year FRAX® risk for major osteoporotic fractures (5.9%) and hip fractures (1.5%) increased to respectively 6.5% and 1.7%, after including DXA-derived BMD in the FRAX® score. BMD was symmetrical between left/right (p=0.45), dominant/non-dominant (p=0.59), and the strongest/weakest limbs (p=0.43).
Among patients not on anti-osteoporosis therapy, 12-month spine and hip BMD remained stable (p=0.77 and p=0.11). However, bone turnover significantly decreased, in both bone resorption (BetaCTx -66.12 ng/L, p=0.04) and bone formation markers (P1NP -6.73 µg/L, p=0.04), independent of stable vitamin D (p=0.38) and PTH (p=0.13) levels.
Regarding clinical predictors, TUG was the strongest objective predictor of 1-year hip Z-score decline (R=-0.46, p=0.003), while ActivLim was the primary patient-reported predictor (R=0.33, p=0.047). Similar trends were observed across other functional measures, including MFM32-D1 (R=0.32, p = 0.05), total FSHD Clinical Score (R=-0.31, p=0.06), FSHD-RODS (R =0.28, p=0.09), FSHD Clinical Severity Score (R =-0.29, p=0.09) and 6MWD (R=0.28, p=0.09). Conversely, bone turnover markers, number of repeat units, calcium intake and the changes in the outcome measures did not predict 1-year bone changes. Analysis of PCCT is ongoing and will be presented at the congress.
Conclusion: Adults with FSHD1 exhibit high osteoporosis prevalence, frequent falls, and suppressed bone turnover. Baseline mobility (TUG, ActivLim) predicted 12-month hip Z-score changes, while D4Z4 length and blood biomarkers did not. This suggests functional mobility drives skeletal integrity in FSHD1, highlighting the need for proactive bone screening, fall prevention, and mobility-maintaining interventions like physical therapy.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Facioscapulohumeral Muscular Dystrophies
PP01.62
Multimodal Assessment of Bone Health and Functional Status in Adults With Duchenne Muscular Dystrophy
Mr. Matthias Opsomer1,2, Dr. Jilmen Quintiens3, Ms. Elena Paravisi3, Dr. Thomas Anijs3, Mr. Herman Borghs4, Mr. Walter Coudyzer5, Ms. Tine De Cuyper4, Mr. Johan Mertens6, Dr. Veerle Goosens5, Prof. Marian Dejaeger4, Prof. Patrick Dupont7, Prof. Harry van Lenthe3, Prof. Kristl. G. Claeys1,2
1
Laboratory for Muscle Diseases and Neuropathies, Department of Neurosciences, KU Leuven, and Leuven Brain Institute (LBI), Leuven, Belgium.
2
Department of Neurology, University Hospitals LeuvenDepartment of Neurology, University Hospitals Leuven, Leuven, Belgium.
3
Biomechanics Section, Department of Mechanical Engineering, Leuven, Belgium.
4
Metabolic Bone Center, University Hospitals Leuven, Leuven, Belgium.
5
Department of Radiology, University Hospitals Leuven, Leuven, Belgium.
6
Department of Pulmonology, University Hospitals Leuven, Leuven, Belgium.
7
Laboratory for Cognitive Neurology, Department of Neurosciences, KU Leuven, and Leuven Brain Institute (LBI), Leuven, Belgium
Background: Duchenne muscular dystrophy (DMD) is a progressive neuromuscular disorder caused by defects in the DMD-gene encoding dystrophin. Despite the growing adult DMD population, validated outcome measures and comprehensive bone health assessment remain limited in clinical practice. Photon-counting computed tomography (PCCT) offers rapid, low-dose, 3D evaluation, that may provide new insights into skeletal integrity beyond conventional imaging.
Methods: Adults with genetically confirmed DMD were prospectively assessed using clinical outcome measures (COMs), comprising Motor Function Measure-32 (MFM32), Performance of Upper Limb 2.0 (PUL2.0), Egen Klassification scale version 2 (EK2), respiratory capacity, and strength test (hand grip, key, and tip pinch strength). Patient-reported outcome measures (PROMs) included ActivLim, Individualized Neuromuscular Quality of Life Questionnaire (INQoL), DMD-QoL, Fatigue Severity Scale (FSS), Brief Pain Inventory (BPI), Hospital Anxiety and Depression Scale (HADS), DMD upper limb PROM, and fracture history. Bone health was assessed in all patients via biochemistry, spinal radiographs and PCCT, supplemented by peripheral QCT (pQCT) and dual-energy X-ray absorptiometry (DXA) where physically feasible.
Results: Thirty-seven adults (median age 25.3 years, IQR 22.0–27.4) with DMD were included. Median age at corticosteroid initiation was 6 years (IQR 5–7.5); loss of ambulation 14.5 years (IQR 13–17). The majority of patients (78.4%) were on chronic corticosteroid therapy (86.2%% deflazacort and 13.8% prednisone) and 16.2% stopped treatment prior to the study due to side effects. Puberty induction using testosterone was performed in 21.6%. Functional assessments confirmed severe impairment (median MFM32 total 31.3% [IQR 22.1%–38.5%], PUL2.0 total 16.5 [IQR 10.25–19], EK2 17.5/51 [IQR 15-23], peak cough flow 165L/min [IQR 70–208], peak hand grip 3.9kg [IQR 1.7–6.1], peak key pinch 1.0kg [IQR 0.5–1.9]), and peak tip pinch strength 0.8kg [IQR 0.0–1.3]). PROMs reflected moderate to severe limitations (ActivLim median 2 [IQR 1–4], INQOL score 55.0 [IQR 48.3-66.7], DMD-QoL total 30 [IQR 27–32.5], HADS anxiety 5 (IQR 3-7), HADS depression 3 [IQR 2-5], DMD upper limb PROM 30 [22-41], FSS 32 [IQR 26–42], and BPI limitations 1.0 [0.3-2.3]). Bone involvement was profound, with 70.3% reporting prior fractures. Seven patients (21.6%) were vitamin D deficient (<20 µg/L) despite receiving supplementation. Testosterone levels were decreased (<300 ng/dL) in 27% of the patients. Bone turnover markers suggested adynamic bone disease with 37.8% exhibiting suppressed resorption (β-CTX reference value 170-704 ng/L) and 16.2% showing decreased bone formation (P1NP reference range 22-87 µg/L). Only six patients were able to undergo a DXA scan (limited by transfer and positioning challenges and metallic interference from spinal rods) revealing severe osteoporosis in all patients (median lumbar spine Z-score of -3.2). PCCT, pQCT and spinal X-ray analysis are ongoing to better characterize microarchitecture and fracture risk.
Conclusion: Adults with DMD exhibit severe functional impairment, reduced muscle strength, and a high skeletal burden. The high fracture rate and low Z-scores highlight a critical need for improved and earlier bone health diagnosis and optimized management. Advanced 3D imaging (PCCT) may be a feasible alternative to DXA and may provide essential insight into fracture risk in this population.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
PP01.63
Volumetric Brain Differences in Duchenne Muscular Dystrophy: What Happens When Accounting for Puberty?
Mr. Matthias Schelfhout1, Mr. Jayant A. Soekhradj2,3, Dr. Lara M. Wierenga4,5, Dr. Hermien E. Kan2,3, Dr. Rubén Miranda6, Dr. Liesbeth De Waele1,7, Dr. Sam Geuens1,7, Dr. Rosanne Govaarts2,3
1
Department of Development and Regeneration, KU Leuven, Leuven, Belgium.
2
C.J. Gorter MRI Center, Radiology, Leiden University Medical Center, Leiden, Netherlands.
3
Duchenne Center Netherlands, Leiden, Netherlands.
4
Institute of Psychology, Leiden University, Leiden, Netherlands.
5
Leiden Institute for Brain and Cognition, Leiden, Netherlands.
6
Department of Psychobiology and Methodology in Behavioral Sciences, Complutense University of Madrid Rector Royo Villanova s/n, Madrid, Spain.
7
Child Neurology, University Hospitals Leuven, Leuven, Belgium
Background: Duchenne muscular dystrophy (DMD) is a muscle wasting disease caused by a mutation on the X-chromosome, resulting in the absence of full-length dystrophin production. Neuroimaging studies have mainly shown smaller gray matter volumes and less structured white matter in patients with DMD compared to age- and sex-matched healthy controls. These alterations are thought to arise from dystrophin deficiency and/or long-term corticosteroid treatment, which remains a standard of care treatment in DMD. While corticosteroids are beneficial for muscle function, they are also associated with pubertal delay. Brain structure is known to change during puberty. Differences in brain volumes reported in studies using age-matched controls may thus partly reflect differences in puberal status rather than effects of dystrophin deficiency or corticosteroid exposure alone. Accounting for pubertal development, for example by using puberty matched controls, may improve the interpretation of neuroimaging findings in DMD. To address this problem, we investigated differences in grey matter (GM), white matter (WM) and cerebrospinal fluid (CSF) volumes between DMD and healthy controls while controlling for age and puberty.
Methods: Retrospective data from 37 DMD patients and 21 healthy controls, collected at LUMC/UL (The Netherlands) and KUL/UZL (Belgium) were analyzed. Data variables included pubertal status (based on clinical files and expert opinion), age, corticosteroid treatment, mutation type and T1 MRI data. Volumes were standardized by the intracranial volume (ICV), which was transformed on the log-scale for small and symmetric differences. A linear model (ANCOVA) was fitted with log(GM/ICV) ratio as outcome, group (DMD vs. healthy controls) as main predictor, adjusting for age and pubertal status. Identical ANCOVA models were applied to the log(WM/ICV) and log(CSF/ICV). All model assumptions were checked and met. For interpretation purposes, estimates were transformed to the percentage scale.
Results: When adjusting for age and puberty, the log(GM/ICV) was not significantly different for group (p = 0.70) with DMD patients having a lower GM on average by -0.6% with a 95% CI [-3.8;2.7]. However, group differences were significant for the log(WM/ICV) (p<0.01) with DMD patients having a lower WM on average by -9.9% with a 95% CI [-12.6;-7.1] and log(CSF/ICV) (p<0.01) with DMD patients having a higher CSF on average by 29.8% with a 95% CI of [14.4;47.2].
Conclusion: These findings show volumetric differences between patients with DMD and healthy controls in WM and CSF, but not in GM, when adjusting for age and puberty. This is in contrast with previous research utilizing age-matched healthy controls showing volumetric differences in GM, suggesting an effect of puberty on GM volumetric differences. Future imaging research should try to account for puberty when comparing DMD with healthy controls.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
PP01.64
Outlining the Neuroimaging Landscape in Duchenne and Becker Muscular Dystrophy: A Systematic Review
Mr. Matthias Schelfhout1, Mr. Jayant A. Soekhradj2,3, Dr. Hermien E. Kan2,3, Dr. Erik H. Niks3,4, Dr. Liesbeth De Waele1,5, Dr. Sam Geuens1,5, Dr. Rosanne Govaarts2,3
1
Department of Development and Regeneration, KU Leuven, Leuven, Belgium.
2
C.J. Gorter MRI Center, Radiology, Leiden University Medical Center, Leiden, Netherlands.
3
Duchenne Center Netherlands, Leiden, Netherlands.
4
Department of Neurology, Leiden University Medical Center, Leiden, Netherlands.
5
Child Neurology, University Hospitals Leuven, Leuven, Belgium
Background: Duchenne (DMD) and Becker (BMD) muscular dystrophy are muscle wasting diseases caused by a mutation on the X-chromosome leading to absent or reduced dystrophin production. Currently, there is no cure for DMD and BMD. Symptomatic treatment consists of corticosteroids, ventilation and multidisciplinary care management. Dystrophin is well characterized in muscle tissue, but its involvement in the brain remains unclear. Imaging studies have mainly shown smaller gray matter volumes and less structured white matter in DMD patients compared to healthy controls. These brain alterations likely result from dystrophin deficiency and/or corticosteroid treatment. As no overview currently exists of this rapidly evolving field, a comprehensive and preregistered systematic review was conducted to highlight current knowledge and guide future research.
Methods: The systematic review was conducted following PICO criteria, which were applied within PUBMED, EMBASE, and Web of Science. After deduplication, studies underwent title-abstract and full text screening, supported by Rayyan, an AI-powered tool. Eligibility of studies at these stages was determined using the PICO criteria. Next, a backwards reference check was performed on the remaining articles to identify additional eligible studies not captured in the initial search. Finally, data extraction and a quality check (using the JBI critical appraisal tool for cross-sectional studies) of the included papers was conducted. The screenings, data extraction and quality check were conducted by two independent reviewers to ensure reliability, disagreements were resolved through discussion or consultation of a third independent reviewer.
Results: This process resulted in 36 included papers, with the reference check indicating no missed papers. The results show an increase in neuroimaging publications from 1977 to 2025 in the DMD and BMD population, especially in exploratory structural research. Most recent publications (<5 years) are of high quality, associates with other clinical measures and considers possible confounding factors like age, mutation and corticosteroid treatment. Despite this, some recent publications still lack the inclusion of these confounding factors. Further interpretation of the results is currently in progress and will be available for ICNMD 2026.
Conclusion: This systematic review highlights the growing interest for neuroimaging research in DMD and BMD. Exploratory structural research has provided valuable insights in understanding brain alterations of patients with DMD and BMD compared to healthy controls. Future work should try to address more specific, hypothesis-driven questions in associating neuroimaging findings to clinical symptoms, for example behavioral problems, language and cognitive impairments.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
PP01.65
A New Beginning for Duchenne: Genomic Newborn Screening and Integrated Models of Early Care
Dr. Michelle Lorentzos1,2, Prof. Kristi J Jones1,2, Ms. Sarah Shin1, Ms. Merly Babu3, Dr. Shanti Balasubramaniam3,1, Ms. Kirsten Boggs1, Dr. Julie Curtin1, Dr. Mark Davis4, Ms. Lyndal Douglas1, Prof. Michelle Farrar1,5, Ms. Rosie Junek3, Dr. Didu Kariyawasam1,5, Dr. Edwin Kirk6, Dr. Eric Law3, Ms. Jessica Lu1, Assoc. Prof. Sarah Norris7, Ms. Shelley Pirreca2, Dr. Enzo Ranieri3,8, Dr. Jacqu Russell1, Dr. Yong Kiat Wee1, Dr. Tiffany Wotton3, Dr. Gladys Ho1, Prof. Bruce Bennetts1
1
Sydney Children's Hospitals Network, Sydney, Australia.
2
The University of Sydney, Sydney, Australia.
3
NSW Newborn Screening, Sydney, Australia.
4
PathWest Laboratory Medicine Diagnostic Genomics, Perth, Australia.
5
The University of New South Wales, Sydney, Australia.
6
Randwick Genomics Laboratory, Prince of Wales Hospital, Sydney, Australia.
7
Leeder Centre for Health Policy, Economics, and Data, The University of Sydney, Sydney, Australia.
8
The University of South Australia, Adelaide, Australia
Background: Duchenne muscular dystrophy (DMD) is a severe X-linked neuromuscular disorder in which early identification can optimise clinical outcomes, prevent a protracted diagnostic journey and streamline expert care. Typical diagnosis generally occurs after symptom onset, creating delays and inequity in access to multidisciplinary care and genetic counselling. Newborn screening for DMD has historically relied on elevated creatine kinase (CK) levels, resulting in high false positives, unnecessary referrals and unwarranted family psychosocial stress. The Australian New South Wales (NSW) Newborn Screening (NBS) Scoping Study for DMD was established to assess the feasibility of population-based screening using an innovative three-tier genomic approach, while simultaneously developing and evaluating a comprehensive early-diagnosis model of care that included early DMD detection, early confirmatory testing, family-centred communication and coordinated care pathways.
Methods: The scoping study aimed to (1) evaluate the performance and operational feasibility of a novel three-tier DMD screening algorithm; (2) design and implement a structured early-diagnosis model of care to support families following a positive screen; and (3) assess the acceptability, timeliness, and clinical impact of this integrated approach. Parents of all participating newborns were offered optional DMD screening. The approach consisted of high-throughput CK-MM quantification from dried blood spots, identification of those elevated patients who were XY, followed by reflex molecular testing targeting pathogenic DMD variants. Only variants considered likely to result in a DMD phenotype were (not Becker or intermediate) were regarded as positive. Infants with screen-positive results entered a newly developed early-diagnosis pathway. This multidisciplinary model of care included: rapid family notification; streamlined confirmatory testing through a neuromuscular clinical service; structured communication protocols; early neuromuscular clinic review; and integrated genetic counselling and psychosocial support.
Results: In the first four months of the scoping study, 26,869 newborns consented to newborn screening, with an opt-in rate above 90 percent. Four newborn males had an elevated CK-MM above the threshold (>1000 ng/mL) and a variant in the DMD gene. Following a multi-disciplinary variant-calling meeting, two cases were determined likely to result in a DMD phenotype, and these underwent confirmatory testing before commencing an early diagnosis model of care. This approach demonstrated robust analytical performance and improved specificity compared with single-tier CK-based approaches, reducing unnecessary referrals and false positives. Infants identified through the pilot accessed multidisciplinary neuromuscular care substantially earlier than historically observed. The model of care proved highly feasible, and clinically impactful, enabling rapid confirmatory diagnosis and early entry into specialist services. The study is ongoing at the time of this abstract submission.
Conclusion: he NSW Newborn Screening Scoping Study for DMD uniquely combines a high-specificity, three-tier genomic approach with a comprehensive early-diagnosis model of care. Together, these advances demonstrate that population-level genomic newborn screening for DMD is feasible and impactful. This provides a blueprint for integrating advanced molecular screening with best-practice care pathways, informing future policy development and implementation of expanded newborn screening in neuromuscular care.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
PP01.66
X-Inactivation Patterns in Blood and Muscle of symptomatic Duchenne Carriers: Cross-Sectional Study of 30 Cases
Dr. Miriam Hiebeler1, Dr. Simone Thiele1, Mrs Eva Malm1, Mrs Caterina Wendel1, Prof. Angela Abicht2, Prof. Maggie Walter1
1
Friedrich-Baur-Institute, LMU, Munich, Germany.
2
MGZ München, Munich, Germany
Background: Duchenne muscular dystrophy (DMD) is an X-linked recessive neuromuscular disorder caused by mutations in the DMD gene. While mainly affecting males, female heterozygous carriers may also exhibit clinical symptoms ranging from elevated creatine kinase levels to muscle weakness, cardiomyopathy, or overt myopathy. The variability in clinical expression among female carriers is thought to be largely influenced by patterns of X-chromosome inactivation (XCI), an epigenetic mechanism that silences one of the two X chromosomes in somatic cells.
Methods: Our study aims to characterize the XCI patterns in both peripheral blood and skeletal muscle tissue of genetically confirmed symptomatic female DMD carriers and to investigate potential correlations with clinical manifestations. We will conduct a cross-sectional observational study including about 30 symptomatic female DMD carriers with a confirmed heterozygous mutation in the DMD gene. XCI analysis is performed using DNA extracted from peripheral blood and muscle biopsy samples. Skewing patterns are classified based on established thresholds. Intraindividual comparison between blood and muscle XCI patterns will be conducted, results will be correlated with clinical and cardiac data of the participants.
Results: The study will assess the degree to which blood-based XCI analysis reflects the situation in muscle tissue and how XCI skewing correlates with clinical symptoms.
Conclusion: Results are expected to improve the understanding of the molecular pathophysiology in symptomatic carriers and may contribute to more accurate diagnostic and prognostic evaluation in symptomatic female DMD carriers.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
PP01.67
Interim Safety Results From an Open-Label Extension Study of Givinostat Treatment For Duchenne Muscular Dystrophy
Dr. John F. Brandsema1, Dr. Gyula Acsadi2, Ms. Federica Alessi3, Dr. Paolo Bettica3, Prof. Odile Boespflug-Tanguy4, Dr. Michela Guglieri5, Dr. Erika L. Finanger6
1
Children’s Hospital of Philadelphia, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, Pennsylvania, United States.
2
Connecticut Children's Medical Center, Farmington, Connecticut, United States.
3
Italfarmaco S.p.A., Milan, Italy.
4
Assitance Publique Hopitaux De Paris and Université Paris Cité, Paris, France.
5
Newcastle University and Newcastle Hospital NHS Foundation Trust, Newcastle upon Tyne, United Kingdom.
6
Shriners Hospital for Children, Portland, Oregon, United States
Background: The safety and efficacy of givinostat were evaluated in the phase 3 EPIDYS study (NCT02851797). An ongoing open-label extension (OLE) is evaluating the long-term safety, tolerability, and efficacy of givinostat in patients with Duchenne muscular dystrophy who completed, or were screened but not randomized, in prior givinostat studies (NCT03373968). This interim analysis characterized safety data from the OLE as of the December 31, 2023, data cutoff date.
Methods: Based on the 2023 interim data cutoff, 207 patients have enrolled: received givinostat in previous studies (givinostat: n=119), received placebo in prior study (delayed givinostat: n=58), and not included in prior study (naive givinostat: n=30). All patients received weight-based givinostat and corticosteroids. Safety data were evaluated for patients who received ≥1 givinostat dose. Adverse events (AEs) were coded using the MedDRA version 26.1. The preferred AE terms of “thrombocytopenia” and “platelet count decreased” were assessed. The effect of givinostat on cardiac measures over time were performed via 12-lead electrocardiogram (ECG).
Results: The mean (SD) ages of patients upon study entry were 11.7 (2.1), 11.5 (2.1), and 10.5 (2.3) years in the givinostat, delayed givinostat, and naive givinostat groups, respectively. The mean (SD) duration of givinostat exposure was 1256.0 (522.9), 1134.9 (346.4), and 1073.7 (260.2) days in the givinostat, delayed givinostat, and naive givinostat groups, respectively. Overall, 98.1% of patients reported ≥1 treatment-emergent AE (TEAE; 2793 events), with similar incidence among groups. Two deaths occurred (road traffic accident and thoracic hemorrhage) in the delayed givinostat group; both were deemed unrelated to the study drug by the investigator and sponsor. Most TEAEs were mild (82.1%) or moderate (15.9%). There were no life-threatening TEAEs. Among TEAEs reported in ≥10% of the overall population, diarrhea occurred in 27.7% of patients in the givinostat, 36.2% in delayed givinostat, and 36.7% in naive givinostat groups. Falls were reported in 31.9% of patients in the givinostat, 10.3% in delayed givinostat, and 33.3% in naive givinostat groups. Thrombocytopenia occurred in 14.3% of patients in the givinostat, 24.1% in delayed givinostat, and 13.3% in naive givinostat groups. The givinostat group reported pyrexia (29.4%), increased blood triglycerides (18.5%), and decreased platelet count (12.6%); corresponding rates in delayed givinostat group were 24.1%, 19.0%, and 19.0%, and the rates in naive givinostat group were 13.3%, 40.0%, and 10.0%. Decreased platelet counts and increased triglyceride concentrations (first 2–4 weeks of treatment) were followed by stabilization in the delayed and naive givinostat groups; levels remained stable for the givinostat group (Figure 1A/1B). The mean QTcF (QT interval, Fridericia’s correction) values ranged from 383.5 to 399.9 ms for all 3 groups (Figure 1C). One patient in the givinostat group at month 40 had a QTcF interval >60 ms change from baseline with a QTcF >450 ms.
Conclusion: These results align with the known safety profile of givinostat. Early decreases in platelets and increases in triglycerides, consistent with treatment initiation, stabilized over time. One patient in the givinostat group had a single ECG over the safety threshold for QTc monitoring that subsequently resolved without intervention.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
Images or Table (Optional)
PP01.68
Update on the INSPIRE DUCHENNE Phase 1/2 Study of SGT-003 Microdystrophin Gene Therapy
Prof. Perry Shieh1, Asst. Prof. Aravindhan Veerapandiyan2, Prof. Kevin Flanigan3, Prof. Eugenio Mercuri4, Prof. Craig McDonald5, Asst. Prof. Hernan Gonorazky6, Asst. Prof. Crystal Proud7, Prof. Craig Zaidman8, Asst. Prof. Alicia Henriquez9, Dr. Patrick Gonzalez10, Dr. Matthew Harmelink10, Dr. Gabriel Brooks10
1
University of California, Los Angeles, Los Angeles, United States.
2
Arkansas Children's Hospital, Little Rock, United States.
3
Nationwide Children's Hospital, Columbus, United States.
4
Policlinico Gemelli, Rome, Italy.
5
University of California, Davis, Sacramento, United States.
6
The Hospital for Sick Children, Toronto, Canada.
7
Children's Hospital of the King's Daughters, Norfolk, United States.
8
Washington University in St. Louis, St. Louis, United States.
9
Seattle Children's Hospital, Seattle, United States.
10
Solid Biosciences, Boston, United States
Background: The INSPIRE DUCHENNE study is an actively enrolling Phase 1/2, open-label, first-in-human clinical trial designed to evaluate the safety, tolerability, and efficacy of a single intravenous infusion of SGT-003 in participants with Duchenne muscular dystrophy.
Methods: SGT-003 is a next-generation AAV-based microdystrophin gene therapy candidate that uses a novel, muscle-tropic capsid, AAV-SLB101, to deliver a uniquely functional replacement for dystrophin. AAV-SLB101 was rationally designed to improve upon the muscle-targeting ability of first-generation naturally occurring capsids by incorporating an RGD-based targeting peptide into variable loops of the capsid surface. Non-clinical studies have shown both higher biodistribution and expression in muscle tissues and lower biodistribution to the liver, compared to first-generation capsids, which could ultimately contribute positively to efficacy and safety. The microdystrophin protein encoded by SGT-003 uniquely includes spectrin-like repeats R16/R17 of full-length dystrophin, allowing for stabilization of the muscle membrane and restoration of local signaling pathways, through the anchoring of neuronal nitric oxide synthase (nNOS) that requires these repeats. The presence of this domain has been associated with protection from ischemia-induced injury and, as a result, SGT-003 microdystrophin may provide greater functionality closer to a full-length dystrophin protein compared to earlier generation constructs.
Results: As of a January 9, 2026 data cut, SGT-003 has been administered to 33 study participants ranging from 1 to 10 years of age and up to approximately 40 kg in weight. As of the data cut, SGT-003 has generally been well tolerated with the most frequent treatment-related adverse events being nausea, vomiting, and loss of appetite. Thrombocytopenia was observed at a higher frequency in early trial participants but has decreased following updates to the prophylactic steroid regimen. Muscle biopsy analyses from 10 participants reaching the Day 90 timepoint and 2 participants reaching the Day 360 timepoint demonstrated high vector genome copies with robust and durable microdystrophin expression. Evaluations of biomarkers of muscle integrity and health, including CK, ALT, AST, LDH, titin, and embryonic myosin heavy chain (eMHC) showed consistent, sustained improvements at Day 90 and out to Day 360 post-dosing.
Conclusion: Together, these results suggest an overall favorable safety profile and a positive impact on muscle integrity as a result of SGT-003 microdystrophin expression. Additional updates on this study and the SGT-003 program will be provided at the time of presentation.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
PP01.69
A Novel Tandem Quadruplication of Exons 8-40 in the DMD Gene: Phenotype and Genotype Characterization
Dr. Abdelmadjid Abdelkrim1, Asst. Prof. Ouissem Benchaabi1, Asst. Prof. Sihem Abdelhak1, Prof. Sihem Hallal1, Dr. Souheila Mousser2, Asst. Prof. Ferroudja Ramdane Cherif1, Prof. Sonia Nouioua1
1
Neurology Department, Cherchell Hospital, Chercherll, Tipaza, Algeria.
2
Neuropediatrics Medical Office, Bejaia, Algeria
Background: Dystrophinopathies result from a spectrum of pathogenic structural variants in the DMD gene, primarily deletions, duplications, and point mutations. While complex genomic rearrangements such as triplications are rare, symptomatic large-scale tandem quadruplications have not been previously reported, and their expressivity remains to be determined.
Methods: We conducted a comprehensive clinical evaluation of a family with Duchenne muscular dystrophy phenotype including neurological examination with the functional scale of the North Star Ambulatory Assessment (NSAA), biochemical analysis, and genetic testing using Next-Generation Sequencing (NGS) with sequencing-depth analysis for copy number variation.
Results: 2We identified a family consisting of three siblings (two boys and one girl). The proband, an 8-year-old boy presented with progressive proximal lower limb weakness since the age of 3. Both the proband and his 6-year-old brother exhibited a classic DMD phenotype including waddling gait, Gowers’ sign, calf muscles hypertrophy and significant pelvic girdle muscle weakness with a low North-Star Ambulatory Assessment (NSAA) score (17/34).
Biochemical tests in both boys showed markedly increased creatine-kinase (CK) levels exceeding 10,000 IU/L with associated elevated and liver enzymes (AST, ALT, LDH). The one-year-old sister is still too young for conclusive clinical examination but showed significantly elevated levels of CK consistent with possible symptomatic status. The carrier mother’s CK levels and cardiac examination were unremarkable.
NGS with sequencing-depth analysis identified an out-of-frame quadruplication of 33 contiguous exons (8-40) in the DMD gene. This pathogenic structural variant correlates with classic, severe DMD phenotype observed in the affected males.
Conclusion: This case-report documents the first detailed characterization of a large-scale tandem quadruplication in the DMD gene, a novel class of structural variant expanding the mutational spectrum of dystrophinopathies and highlighting the importance of genetic analysis in elucidating the pathophysiology, guiding therapeutic considerations and informing genetic counselling.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
PP01.70
Xp21 Contiguous Gene Deletion Syndrome Presenting as Atypical Duchenne Muscular Dystrophy With Adrenal Insufficiency
Prof. Sonia Nouioua1, Dr. Abdelmadjid Abdelkrim1, Dr. Ouissem Benchaabi1, Dr. Sihem hallal2, Dr. Ferroudja Ramdane Cherif1, Dr. Sihem Abdelhak1
1
Department of neurology EHS Cherchell, Tipaza, Algeria.
2
central laboratory CHU Mustapha, Algiers, Algeria
Background: Contiguous gene deletion syndromes (CGDS) are rare genomic disorders caused by deletions of large DNA segments involving contiguous genes, leading to the co-occurrence of apparently unrelated multi-systemic clinical features. A typical example is the Xp21 contiguous gene deletion syndrome which includes deletions of DMD, Glycerol kinase (GK), NR0B1 (DAX1) and results in a complex phenotype combining Duchenne muscular dystrophy, glycerol kinase deficiency and congenital adrenal hypoplasia.
Methods: We present the clinical, biochemical and molecular characterization of a 6-year-old male. The patient presented with a complex phenotype comprising Duchenne muscular dystrophy (DMD), profound intellectual disability and secondarily signs of adrenal insufficiency. The diagnostic workup included neurological examination, biochemical and endocrine profiling, neuroimaging, nerve conduction studies and electromyography (ENMG), muscle biopsy and genetic testing using next-generation sequencing (NGS)
Results: A 6-year-old male born to consanguineous parents at term with no significant neonatal complications apart from a neonatal infection at the age of 20 days without sequelae, presented with a global development delay (independent sitting acquired at 15 months, first ambulation at 24 months, severe language delay), frequent hypoglycemia and vomiting, proximal lower limbs weakness and failure to thrive. On admission he had poor general condition with lethargy and severe dehydration. Clinical examination revealed profound intellectual disability with autistic features; language was never acquired correctly and his vocabulary was limited to a few isolated words, with associated strabismus. He exhibited a classic DMD phenotype, including pelvic girdle muscles weakness, a waddling gait, calf pseudohypertrophy, Gowers, scapula alata signs and scoliosis. The biochemical test showed markedly increased levels of Creatine-kinase (CK) exceeding 9500 IU/L with elevated liver enzymes, adrenal insufficiency characterized by high ACTH levels (>2000 pg/mL) while cortisolemia was within the normal range. Lipid profiling revealed a pseudo hypertriglyceridemia with low HDL.
ENMG demonstrated myopathic patterns and the muscle biopsy confirmed the dystrophic process. The cardiac examination by transthoracic echocardiogram showed no dilated cardiomyopathy but revealed a patent foramen ovale, and abdominopelvic ultrasound demonstrated absent adrenal glands consistent with adrenal hypoplasia congenita. The etiological workup for cognitive impairment, including brain MRI, EEG, TSH and phenylalanine levels were unremarkable. Genetic analysis by NGS identified a large contiguous deletion of approximately 11 Mb on the short arm of the X chromosome, spanning the Xp21.1–Xp21.3 region and encompassing IL1RAPL1, NROB1 (DAX1), GK, and DMD genes. This rare pathogenic deletion correlates with the observed complex phenotype of DMD, adrenal hypoplasia congenita, glycerol kinase deficiency, and profound intellectual disability with autism spectrum features.
The patient was treated with hydrocortisone, fludrocortisone and oral salt supplementation, resulting in improvement of his general condition and metabolic stability.
Conclusion: This case highlights the considerable medical and genetic complexity of Xp21 CGDS syndrome and provides a precise molecular explanation for the co-occurrence of profound intellectual disability, adrenal agenesis and muscular dystrophy. Clinicians should consider Xp21 syndromes in male children with adrenal insufficiency and muscular or metabolic signs. Early screening and genetic testing are essential for establishing an accurate diagnosis, implementing effective management, and providing informed counselling to the family.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
PP01.71
Zeleciment Rostudirsen Increased Dystrophin and Led to Functional Improvement in Clinical Measures in DELIVER Trial
Dr. Stefano Previtali1,2, Dr. Nicolas Deconinck3,4, Dr. Liesbeth DeWaele5, Dr. Kevin Flanigan6, Dr. Chamindra Laverty7, Dr. Jeehun Lee8,9, Dr. Hugh McMillan10, Dr. Perry Shieh11, Dr. Ian Woodcock12,13, Dr. Soma Ray14, Dr. Dazhe Wang14, Dr. Douglas Kerr14, Dr. Maria Naylor14, Dr. Michaela Guglieri15
1
IRCCS San Raffaele Scientific Institute, Milan, Italy.
2
Vita-Salute San Raffaele University, Milan, Italy.
3
Neuromuscular Reference Center UZ Gent, Gent, Belgium.
4
Hôpital des Enfants Reine Fabiola (HUB), Brussels, Belgium.
5
University Hospitals Leuven,, Leuven, Belgium.
6
Nationwide Children's Hospital, Columbus, Ohio, United States.
7
UC San Diego Health, San Diego, CA, United States.
8
Samsung Medical Center, Seoul, Korea, Republic of.
9
Sungkyunkwan university School of Medicine, Seoul, Korea, Republic of.
10
Children's Hospital of Eastern Ontario, Ottawa, Ontario;, Canada.
11
University of California, Los Angeles, United States.
12
Murdoch Children’s Research Institute, Parkville, Victoria, Australia.
13
The Royal Children’s Hospital, Melbourne, Australia.
14
Dyne Therapeutics, Waltham, MA, United States.
15
Royal Victoria Infirmary, Newcastle University, Newcastle-Upon-Tyne, United Kingdom
Background: Individuals with DMD pathogenic variants amenable to exon 51 skipping have significant unmet needs, despite approved treatments. Zeleciment rostudirsen (z-rostudirsen, also known as DYNE-251) leverages the transferrin receptor, TfR1, to deliver an exon 51 skipping Phosphorodiamidate Morpholino Oligomer (PMO) to muscle with the goal of producing near-full length functional dystrophin.
Methods: In the Multiple Ascending Dose (MAD) portion of the Phase 1/2 DELIVER trial (NCT05524883), 54 participants received z-rostudirsen or placebo every 4 or 8 weeks for 6 months. Following dose selection, 32 participants were randomized 3:1 to receive 20 mg/kg Q4W z-rostudirsen or placebo for 6 months in a registrational expansion cohort (REC). After the placebo-controlled period, participants entered an open-label Long Term Extension (OLE/LTE) and transitioned to 20 mg/kg Q4W, as applicable.
Results: The REC met its primary endpoint, demonstrating a statistically significant increase in mean muscle content-adjusted dystrophin at 6 months compared to baseline (5.46% vs. 0.83% of normal, respectively, p<0.0001). Trends in functional improvement were observed across multiple clinical endpoints at 6 months including Time to Rise (TTR) velocity, 10 Min Walk Run (10MWR) velocity, North Star Ambulatory Assessment (NSAA), Stride Velocity 95 Centile (SV95C), Performance of Upper Limb (PUL) 2.0, and Forced Vital Capacity Percent Predicted (FVC%p). Sustained improvements from baseline in these measures were also observed through 18 months of treatment in participants who enrolled in the MAD at 20 mg/kg Q4W and through 24 months of treatment in participants who enrolled in the MAD at 10 mg/kg Q4W and transitioned to 20 mg/kg Q4W in the OLE/LTE. As of August 19, 2025, z-rostudirsen had a favorable safety and tolerability profile based on data from 86 participants enrolled in DELIVER and followed for up to 36 months.
Conclusion: Data from the DELIVER trial, including robust dystrophin expression, trends in functional improvement, favorable safety profile, and convenient dosing support the potential of z-rostudirsen to address the unmet needs of individuals with DMD pathogenic variants amenable to exon 51 skipping.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
PP01.72
Real-World Experience With Gene Therapy in Duchenne Muscular Dystrophy: Experience From Qatar
Prof. TAWFEG BEN OMRAN
Sidra Medicine, doha, Qatar
Background: Duchenne Muscular Dystrophy is a rare, X-linked neuromuscular disorder that leads to progressive muscle degeneration, loss of ambulation, and premature mortality due to respiratory and cardiac failure. Historically, Duchenne Muscular Dystrophy has been managed through supportive and symptomatic treatments, with limited options for disease modification. However, advancements in gene therapy have introduced promising interventions aimed at addressing the underlying dystrophin deficiency.
Delandistrogene moxeparvovec (Elevidys) received accelerated approval from the U.S. Food and Drug Administration in June 2023 for ambulatory children aged 4 to 5 years with a confirmed diagnosis of Duchenne Muscular Dystrophy. This approval represented an advancement, offering a disease-modifying therapy at an early stage when muscle function remains relatively preserved. The Food and Drug Administration expanded its approval in June 2024 to include both ambulatory and non-ambulatory children aged 4 years and older.
In this retrospective case series, we present the Qatari experience of using Delandistrogene Moxeparvovec (Elevidys)in patients with confirmed DMD, focusing on treatment outcomes, safety, and clinical implications.
Methods: This study provides a retrospective real-world analysis of eight Duchenne Muscular Dystrophy patients who received Elevidys gene therapy at our center in Qatar. Recognizing the complexities involved in treating older Duchenne Muscular Dystrophy patients, a standardized protocol for pre- and post-infusion care was implemented.
Results: Our findings highlight the safety and positive clinical outcomes of gene therapy for Duchenne Muscular Dystrophy patients in Qatar.
Both age groups show a similar pattern with a slight decrease in ALT and AST levels post-infusion. However, younger patients tend to have marginally higher ALT levels over the weeks, particularly prominent in Week 24 to 26. Older patients display greater stability in their ALT levels. GGT levels are consistently higher in the older cohort, showing a steady increase over the weeks, with a pronounced spike by Week 7 and week 14. Younger patients demonstrate a more stable hepatic response, with a less marked increase in GGT levels.
Younger patients presented markedly elevated pre-infusion CK levels, which decline substantially by Week 2 but continue to fluctuate with noticeable peaks at Weeks 5, 7 and 26. In contrast, older patients have lower initial CK levels, following a similar declining trend but with less variability. Troponin I levels remain relatively low and stable in patients aged 6 years or below, indicating minimal cardiac stress. In older patients, Troponin I levels are relatively higher with observed peaks at Weeks 1 and 3, suggesting age-related differences in cardiac response to therapy.
Our results highlight several limitations of NSAA as a sole functional outcome measure. For example, Patient 2 had no change in NSAA at two months but demonstrated measurable gains in 10MWT and get-up time, with further improvement in NSAA only observed at the four-month reassessment. These findings underscore the importance of complementary functional metrics and extended follow-up to fully capture therapeutic benefit in DMD gene therapy.
Conclusion: This study demonstrates that Elevidys is well tolerated in DMD patients aged 4–11 years. Also indicate the safety and efficacy of gene therapy in DMD when combined with a protocol.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
PP01.73
Pharmacodynamic Response of Circulating Proteins to Different Corticosteroid Types and Regimens in Duchenne Muscular Dystrophy
Assoc. Prof. Utkarsh Dang1, Assoc. Prof. Roula Tsonaka2, Ms. Chiara Degan2, Ms. Rebecca Tobin1, Prof. Cristina Al-Khalili Szigyarto3, Assoc. Prof. Yuri van der Burgt2, Prof. Jordi Diaz Manera4, Prof. Michela Guglieri4, Assoc. Prof. Pietro Spitali2, Prof. Yetrib Hathout5
1
Carleton University, Ottawa, Canada.
2
Leiden University Medical Center, Leiden, Netherlands.
3
KTH Royal Institute of Technology, Stockholm, Sweden.
4
Newcastle University, Newcastle, United Kingdom.
5
Binghamton University, Binghamton, United States
Background: Although newer drugs are gaining approval, steroids remain the most widely available standard of care for Duchenne muscular dystrophy (DMD). Moreover, most newer therapies are used in combination with steroids (prednisone, deflazacort, and vamorolone). Although steroids have proven clinical efficacy, well-known side effects (growth stunting, weight gain, adrenal suppression, bone health, puberty delay, etc.) affect adherence to recommended treatment dosage. In this context, existing literature has established some preliminary serum protein responses to steroids, but there is a paucity of large-scale, untargeted studies. Previous studies of biomarker response to steroids in DMD utilized patient samples from natural history studies, in which age range (and DMD progression) and treatment (type, regimen, lifetime exposure, etc.) may have confounded estimates. With most newer therapies used on a steroidal backbone, and the use of combination therapies likely to increase with time, it’s important to establish how biomarker levels change in response to steroids alone in a previously steroid-naïve population, to better understand response to other therapies (gene therapy, exon skipping therapy, etc.) and synergistic interaction with steroids. Our objectives were to investigate the a) longitudinal response of serum proteins, b) the dose-response between intermittent and daily prednisone, and c) the differential response to daily prednisone vs deflazacort over a shorter (1 year) and longer (up to 3 years) term using serum samples from a well-controlled clinical trial of prednisone and deflazacort in young boys with DMD.
Methods: Proteomics profiles were obtained from a Somascan panel of ∼1500 protein targets using sera from 56 boys (4 to <11 years; <8 years at steroid-naïve baseline) with DMD from the FOR-DMD (Clinicaltrials.gov: NCT01603407) study. Following pre-processing, longitudinal analysis allowing non-linear response was conducted, analysing both 1-year (appropriate for typical clinical trials) and 2/3 years (appropriate for long-term follow-up) contrasts of within-group and between-group change to establish biomarker response to drug regimens.
Results: Biosamples were from 14, 19, and 23 participants from the daily deflazacort, daily prednisone, and intermittent prednisone groups, respectively. Approximately 23%, 14%, and 5% of the protein abundances changed with daily deflazacort, prednisone, and intermittent prednisone, respectively, at year 1 (based on FDR-adjusted p<0.05), capturing proteins reflecting both efficacy and safety aspects. These proportions were all higher at the years 2/3 mark, given longer cumulative steroid exposure. Out of the subset of proteins previously identified as different between pre-treatment and healthy control samples, 22.8% responded to deflazacort at year 1, 16.6% responded to prednisone, and 6.3% responded to intermittent prednisone.
Conclusion: We studied the dose-response of serum proteins between intermittent and daily prednisone, and differential response to daily prednisone vs deflazacort in a large-scale, untargeted fashion. We validated some serum proteins previously reported as differentially responsive to prednisone vs deflazacort, but also identified newer differentially responsive biomarkers. Our work may help to better understand the mechanism of action of steroids, the predictive potential of these biomarkers and their importance to accelerated regulatory pathways, help refine therapies, define synergistic interactions between novel treatments and steroids, and potentially use as surrogate/secondary outcomes in trials.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
PP01.74
Open-Label Extension Analysis Suggests Givinostat Delays Age at Loss of Ambulation in Patients With DMD
Dr. David Gómez Andrés1, Prof. Valeria Sansone2, Dr. Andrea Parodi3, Dr. Odile Boespflug-Tanguy4, Dr. Natalie Goedeker5, Dr. Paolo Bettica3, Dr. Tracey Willis6,7, Prof. Eugenio Mercuri8
1
Hospital Universitario Vall De Hebron, Barcelona, Spain.
2
The NeMO Clinical Center in Milan, Neurorehabilitation Unit, Milan, Italy.
3
Italfarmaco S.p.A., Milan, Italy.
4
APHP and Université Paris Cité, Paris, France.
5
Washington University, St. Louis, St. Louis, Missouri, United States.
6
The Robert Jones and Agnes Hunt Orthopaedic Hospital NHS Foundation Trust, Oswestry, United Kingdom.
7
Chester University Medical School, Chester, United Kingdom.
8
Pediatric Neurology Institute, Catholic University and Nemo Pediatrico, Fondazione Policlinico Gemelli IRCCS, Rome, Italy
Background: Givinostat is an oral histone deacetylase inhibitor that was investigated in the phase 3, randomized, double-blind, placebo-controlled EPIDYS trial (NCT02851797). In 2021 interim analysis of an ongoing, single-arm, open-label extension (OLE) study of the long-term safety, tolerability, and efficacy of givinostat (NCT03373968), givinostat-treated patients showed a delay in median age at persistent loss of ambulation (LoA; ie, permanent inability to walk independently) compared with a matched, natural history corticosteroid-treated external control group. This analysis aimed to evaluate the effect of longer-term givinostat treatment, in combination with standard-of-care corticosteroids, on LoA in patients with DMD aged ≥6 years using data through the 2023 cutoff, which provided a larger patient population and an additional 2 years of follow-up since the previous interim analysis.
Methods: The ongoing OLE study includes patients who completed a prior phase 2 givinostat study (NCT01761292) or EPIDYS (NCT02851797), as well as those who were screened but not randomized in EPIDYS. All patients who received ≥1 month of givinostat in combination with corticosteroids were included in the analyses. Median age at persistent LoA was calculated using Kaplan–Meier survival analysis in this population.
Results: Among the 225 patients analyzed, 78 experienced persistent LoA. The median (95% CI) age at persistent LoA was 17.3 (15.5-18.1) years (Figure).
Conclusion: This updated interim analysis evaluated LoA in patients with DMD with 2 additional years of data from the OLE. These updated OLE data suggest that givinostat consistently delays the onset of LoA in patients with DMD. Relative to published estimates from a meta-analysis reporting a median age at LoA of 11.0-13.4 years in patients treated with corticosteroids alone, the median age at persistent LoA with givinostat was 17.3 years, suggesting a delay in the loss of ambulation in patients treated with givinostat.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
Images or Table (Optional)
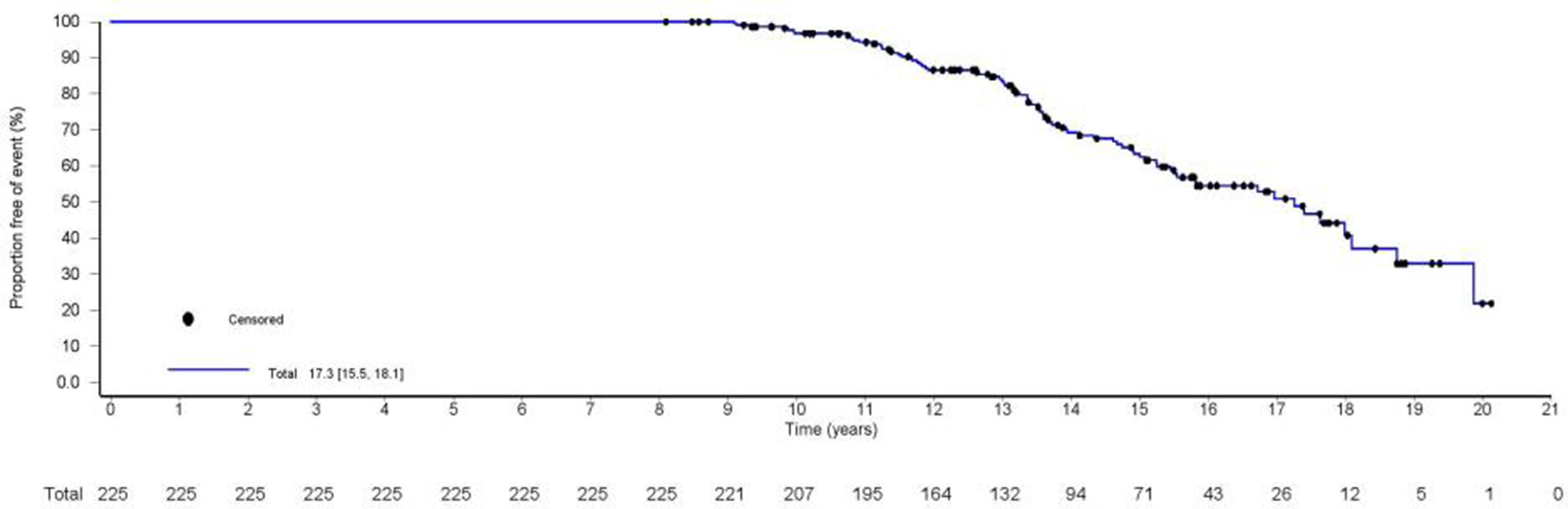
PP01.75
LT-001 a Long-Term Follow-Up Study for Participants That Completed the SAT-3247-CL-101 Study: 6-Month Update
Dr. Wildon Farwell, Dr. Phil Lambert, Dr. Ryan Mitchell, Dr. Michael Rudnicki, Mr. Frank Gleeson
Satellos Bioscience Inc., Toronto, Canada
Background: Duchenne muscular dystrophy (DMD) is caused by mutations in the dystrophin gene, leading to progressive muscle degeneration, loss of ambulation, and premature mortality. SAT-3247 is an oral small molecule that enhances muscle regeneration by modulating muscle stem cell (satellite cell) polarity and asymmetric division via regulation of adapter associated kinase 1 (AAK1). The objective of LT-001 is to assess long-term safety and efficacy of orally administered SAT-3247 in adult participants with DMD that previously completed SAT-3247-CL-101, a Phase 1 study of SAT-3247.
Methods: This open-label study will assess long-term safety, tolerability and potential efficacy of extended dosing of 60 mg of orally administered SAT-3247 in a 5-days on/2-days off (i.e. weekday dosing) regimen. Participants will receive SAT-3247 for 11 months, for a total of 12 months of treatment including the duration of the SAT-3247-CL-101 study.
Safety endpoints will include incidence, temporal profile, and severity of treatment emergent adverse events (TEAEs). Efficacy endpoints will include changes from baseline in intramuscular fat fraction in muscle quantitative magnetic resonance (qMR) in biceps brachii, changes from baseline in muscle force measurements as measured by dynamometry, and changes from baseline in the Performance of Upper Limb (PUL2.0) assessment following SAT-3247 treatment. Additionally, exploratory analyses of various biomarkers are being conducted.
Results: Summary results after 6 months of dosing will be presented.
Conclusion: Summary conclusions after 6 months of dosing will be presented
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
PP01.76
A Cyclic Peptide Targeting the Myostatin-Inhibitory Core Region Alleviates Dystrophic Pathology in D2-mdx Mice
Prof. Yoshihide Sunada1, Prof. Shin-ichiro Nishimatsu1, Assoc. Prof. Masahiro Fujino2, Assoc. Prof. Yutaka Ohsawa1
1
Kawasaki Medical School, Kurashiki, Japan.
2
Kyushu Women`s Junior College, Kitakyushu, Japan
Background: Duchenne muscular dystrophy (DMD) is a severe, X-linked recessive neuromuscular disorder caused by mutations in the DMD gene. Dystrophin deficiency results in dystrophic pathology, leading to progressive motor impairment. Myostatin, a muscle-specific TGF-β superfamily member, serves as a potent negative regulator of skeletal muscle mass; it is secreted by and acts on fast-twitch myofibers via autocrine, paracrine, and endocrine mechanisms. Previous studies have indicated that intramuscular myostatin activity is a key disease-modifying factor in DMD patients receiving corticosteroid treatment (Flanigan, Ann Neurol 73, 2013). We previously identified a 29-amino acid region within the N-terminal prodomain of the myostatin precursor protein, designated as the inhibitory core (IC), which physiologically suppresses myostatin activity (Patent: JP 6143270; Ohsawa, Sunada, J Clin Invest 116, 2006; PLoS One 10, 2015).
Methods: In this study, we developed a novel cyclic peptide that specifically targets the IC region. This peptide specifically inhibits both ligand and ligand-receptor binding, demonstrating significantly higher inhibitory potency compared to the original 29-amino acid IC peptide. Notably, the cyclic peptide showed exceptional skeletal muscle penetration, effectively suppressing autocrine and paracrine myostatin activity.
Results: Subcutaneous injection of this peptide significantly attenuated dystrophic pathology—including necrosis, impaired regeneration, fibrosis, and fatty replacement—in the D2-mdx mouse, a severe model of DMD. Furthermore, the treatment ameliorated muscle weakness and improved contractile function.
Conclusion: We are currently evaluating the therapeutic window and safety profile of this peptide in wild-type and D2-mdxmice to support its translation into first-in-human (FIH) clinical trials for patients with DMD.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Dystrophinopathies
PP01.77
Fascioscapulohumeral Dystrophy and Noonan Syndrome
Dr. Ahmet Akpınar1, Prof. Eren Gozke2
1
Kulu State Hospital, Konya, Turkey.
2
University of Health Sciences FSM Training and Research Hospital, Istanbul, Turkey
Background: Facioscapulohumeral dystrophy (FSHD) is the third most common muscular dystrophy. It is inherited as autosomal dominant (AD) and affects skeletal muscles. Noonan syndrome (NS) is a genetic disorder characterized by facial dysmorphism, cardiac conduction abnormalities, skeletal anomalies, and clinically variable cognitive deficits, which are inherited as AD.
Methods: A 30-year-old male patient was admitted to the neurology clinic with progressive weakness in both arms for several years. The patient had no known chronic disease history. In family history, her older brother had similar symptoms but had no diagnosis. There was no consanguinity between the patient’s parents, and they were completely healthy.
Results: In the neurologic examination, he could not squeeze his eyes fully; his lips were prominently protruding forward. His shoulder and arm muscles were atrophic. His bilateral shoulder abduction was 4/5, and forearm flexion was 3/5, other muscle strengths were normal. Scapula alata was seen on the left side. There was no pyramidal sign, cerebellar findings and sensory deficit. In addition, the patient had scoliosis and thoracic deformities. Creatine kinase (CK) was slightly increased (396 U/L). EMG showed widespread myopathic involvement. As a result of the genetic examination of the patient, in the PTPN11 gene, an NM_002834.5 c.781>T(p.L261F) (p.Leu261Phe) (heterozygous) change was detected and evaluated as pathological. This change is a variant associated with NS in the Human Gene Mutation Database.
Conclusion: Although neurologic involvement is rare in NS, it is associated with retinitis pigmentosa, neurofibromatosis type 1, and Charcot–Marie–Tooth disease. In addition, Chiari type 2 malformation and Dandy–Walker syndrome reported. Although myopathic involvement has been reported in NS cases, the coexistence of FSHD and NS is revealed for the first time with this case.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Facioscapulohumeral Muscular Dystrophies
PP01.78
Efficacy and Safety of Iptacopan in Patients With Generalized Myasthenia Gravis: APPRAISE Study Design
Prof. John Vissing1, Prof. Heinz Wiendl2, Prof. Vera Bril3, Prof. Srikanth Muppidi4, Prof. Stephen Reddel5, Prof. Kimiaki Utsugisawa6, Dr. Weihua Cao7, Dr. Svetlana Jevtic8, Dr. Bernd Kieseier8, Dr. Arseniy Lavrov9, Prof. James Howard10
1
Copenhagen Neuromuscular Center, Department of Neurology, Rigshospitalet, University of Copenhagen, Copenhagen, Denmark.
2
Department of Neurology and Neurophysiology, University of Freiburg, Freiburg, Germany.
3
Toronto General Hospital, Toronto, Canada.
4
Department of Neurology and Neurological Sciences, Stanford University School of Medicine, Stanford, United States.
5
University of Sydney, Departments of Neurology and Molecular Medicine, Sydney, Australia.
6
Department of Neurology, Hanamaki General Hospital, Hanamaki, Japan.
7
Novartis Pharmaceuticals Corporation, East Hanover, United States.
8
Novartis Pharma AG, Basel, Switzerland.
9
Novartis Pharmaceuticals UK Ltd., London, United Kingdom.
10
Department of Neurology, University of North Carolina School of Medicine, Chapel Hill, United States
Background: Generalized myasthenia gravis (gMG) is a rare, chronic autoimmune disorder caused by autoantibodies against neuromuscular junction components, leading to debilitating fatigable muscle weakness and other clinical consequences. Current therapies for the treatment of gMG have limitations, including infections, systemic side effects, and often require parenteral administration. Iptacopan (LNP023) is an oral, potent, and selective inhibitor of factor B in the alternative complement pathway, currently in clinical development for the treatment of gMG. The Phase 3 APPRAISE study aims to evaluate the efficacy and safety of iptacopan in patients with anti-acetylcholine receptor antibody-positive (AChR+) gMG on stable standard of care (SOC) treatment.
Methods: This ongoing randomized, double-blind, placebo-controlled, multicenter Phase 3 study (NCT06517758) is enrolling patients aged 18–75 years with Class II–IV MG (according to the Myasthenia Gravis Foundation of America) who test positive for AChR+ antibodies and are on SOC treatment. Participants must have a baseline Myasthenia Gravis Activity of Daily Living (MG-ADL) score ≥6, with ≥50% due to non-ocular symptoms, and inadequate disease control on one or more non-steroidal immunosuppressant treatments (NSIST) or frequent plasmapheresis/plasma exchange/IVIG therapy. Eligible participants will be randomized (1:1) to receive either iptacopan or placebo during the 6-month double-blind core period, followed by iptacopan administration in the open-label extension (up to 60 months). The primary endpoint is the change in MG-ADL score from baseline to Month 6. Key secondary endpoints include assessments of Quantitative MG (QMG), and MG Composite (MGC), among others.
Results: The study will enroll approximately 146 eligible participants.
Conclusion: This study will investigate the efficacy and safety of iptacopan versus placebo in adult patients with AChR+ gMG.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.79
Minimal Symptom Expression in Myasthenia Gravis: A Post Hoc Analysis of Phase 3 Rozanolixizumab Studies
Prof. John Vissing1, Dr. Ali A. Habib2, Dr. Zabeen Mahuwala3, Prof. Renato Mantegazza4, Prof. Robert Pascuzzi5, Prof. Sabrina Sacconi6, Dr. Tuan Vu7, Dr. Jana Zschüntzsch8, Mr. Joel Baldwin9, Dr. Thaïs Tarancón10, Dr. Vera Bril11
1
Copenhagen Neuromuscular Center, Department of Neurology, Rigshospitalet, University of Copenhagen, Copenhagen, Denmark.
2
MDA ALS & Neuromuscular Center, Department of Neurology, University of California, Irvine, Orange, United States.
3
Department of Neuromuscular Medicine, Epilepsy and Clinical Neurophysiology, University of Kentucky, Lexington, United States.
4
Emeritus and Past Director, Department of Neuroimmunology and Neuromuscular Diseases, Fondazione IRCCS, Istituto Nazionale Neurologico Carlo Besta, Milan, Italy.
5
Neurology Department, Indiana University School of Medicine, Indiana University Health, Indianapolis, United States.
6
Université Côte d'Azur, Peripheral Nervous System & Muscle Department, Pasteur 2 Hospital, Centre Hospitalier Universitaire de Nice, Nice, France.
7
Department of Neurology, University of South Florida Morsani College of Medicine, Tampa, United States.
8
Department of Neurology, University Medical Center Göttingen, Göttingen, Germany.
9
UCB, Slough, United Kingdom.
10
UCB, Madrid, Spain.
11
Ellen and Martin Prosserman Centre for Neuromuscular Diseases, Toronto General Hospital, University of Toronto, Toronto, Canada
Background: In the Phase 3 MycarinG study (NCT03971422) and its open-label extension studies, Myasthenia Gravis Activities of Daily Living (MG-ADL) responder rates (≥2-point improvement in MG-ADL score) of ≥63.7% were observed in Cycles 1–13 of rozanolixizumab treatment in patients with generalised myasthenia gravis (gMG). Attaining minimal symptom expression (MSE), defined as an MG-ADL score of 0 or 1, is considered a treatment goal in gMG. Achievement of MSE across rozanolixizumab treatment cycles was assessed post hoc.
Methods: In MycarinG, patients were randomised 1:1:1 to once-weekly placebo, rozanolixizumab 7 mg/kg or rozanolixizumab 10 mg/kg for 6 weeks. In MG0004 (NCT04124965), patients received once-weekly rozanolixizumab 7 mg/kg or 10 mg/kg for ≤52 weeks. In MG0007 (NCT04650854), after an initial 6-week cycle (rozanolixizumab 7 mg/kg or 10 mg/kg), further cycles were administered on symptom worsening at the investigator’s discretion. Final efficacy data were pooled across MycarinG, MG0004 (first 6 weeks) and MG0007 for patients with ≥2 symptom-driven rozanolixizumab cycles. The proportion of patients who achieved MSE (MG-ADL score: 0 or 1) at any time during treatment or observation in each symptom-driven cycle up to Cycle 13 was analysed post hoc based on MSE achievement in Cycle 1.
Results: Overall, 129 patients were assessed. In Cycle 1, 27.9% (36/129) of patients were MSE responders. Among Cycle 1 MSE responders, subsequent MSE rates were ≥60.0% in each cycle (Cycle 2–13). Among Cycle 1 MSE non-responders, 19.4% achieved MSE in a subsequent cycle (up to Cycle 13; median number of additional cycles received: 6). In every cycle (1–13), almost all patients who achieved MSE did so within the 6-week treatment period; rapid onset (1 week) was seen in some patients.
Conclusion: For patients with gMG who achieved MSE in Cycle 1, within each subsequent cycle, the majority achieved MSE, suggesting consistent efficacy with long-term rozanolixizumab treatment. Funding: UCB.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.80
Myasthenia Gravis Inebilizumab Trial (MINT): Adverse Events of Special Interest During the Randomized Controlled Period
Dr. Richard J. Nowak1, Dr. Kimiaki Utsugisawa2, Dr. Michael Benatar3, Dr. Emma Ciafaloni4, Dr. M. Isabel Leite5, Dr. John Vissing6, Dr. Fengming Tang7, Dr. Blanca Canales7, Dr. Qing Li7, Dr. Sue Cheng7, Dr. James F. Howard Jr.7
1
Yale University, New Haven, United States.
2
Hanamaki General Hospital, Hanamaki, Japan.
3
University of Miami, Miller School of Medicine, Miami, United States.
4
University of Rochester, Rochester, United States.
5
University of Oxford, Oxford, United Kingdom.
6
University of Copenhagen, Copenhagen, Denmark.
7
Amgen Inc., Thousand Oaks, United States
Background: B cells play a key upstream mechanistic role in autoimmune generalized myasthenia gravis (gMG) pathogenesis through the production of antibodies targeting the acetylcholine receptor (AChR+), muscle-specific kinase (MuSK+), or other related proteins. Inebilizumab is a monoclonal antibody targeting CD19+ B cells. B-cell-depleting therapies may impair humoral responses, increase the risk of infections and reduced response to vaccinations. Here we examine whether inebilizumab is associated with an increased risk of adverse events of special interest (AESIs) in gMG participants.
Methods: MINT (NCT04524273) was a phase 3 trial in adults with gMG. During the randomized controlled period (RCP) [52-weeks for AChR+; 26-weeks for MuSK+], participants were randomized (1:1) to receive 300mg of inebilizumab or placebo on Day-1, Day-15, and Day-183 (AChR+ only). Subsets of adverse events were identified as AESIs due to their relevance to inebilizumab’s mechanism of action.
Results: 238 patients were randomized: inebilizumab 119 (95 AChR+, 24 MuSK+), placebo 119 (95 AChR+, 24 MuSK+). Baseline demographics and disease characteristics were generally balanced between treatment groups. AESIs were infrequent, occurring in 10.1% (n=12) of the inebilizumab- and 16% (n=19) of the placebo-treated groups. Cytopenias occurred in 2.5% (n=3) of inebilzumab-treated and 9.2% (n=11) of placebo-treated participants. Serious and/or opportunistic infections were infrequent (inebilizumab: 3.4% [n=4]; placebo: 5.0% [n=6]), with COVID-19 being the most common (inebilizumab: 2/4; placebo: 3/6) and no instances of progressive multifocal leukoencephalopathy. Infusion related reactions occurred in 5.0% (n=6) of inebilizumab- and 2.5% (n=3) of placebo-treated participants (Table 1).
Conclusion: These results support a favorable safety profile of inebilizumab over a 26–52-week period.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
Images or Table (Optional)
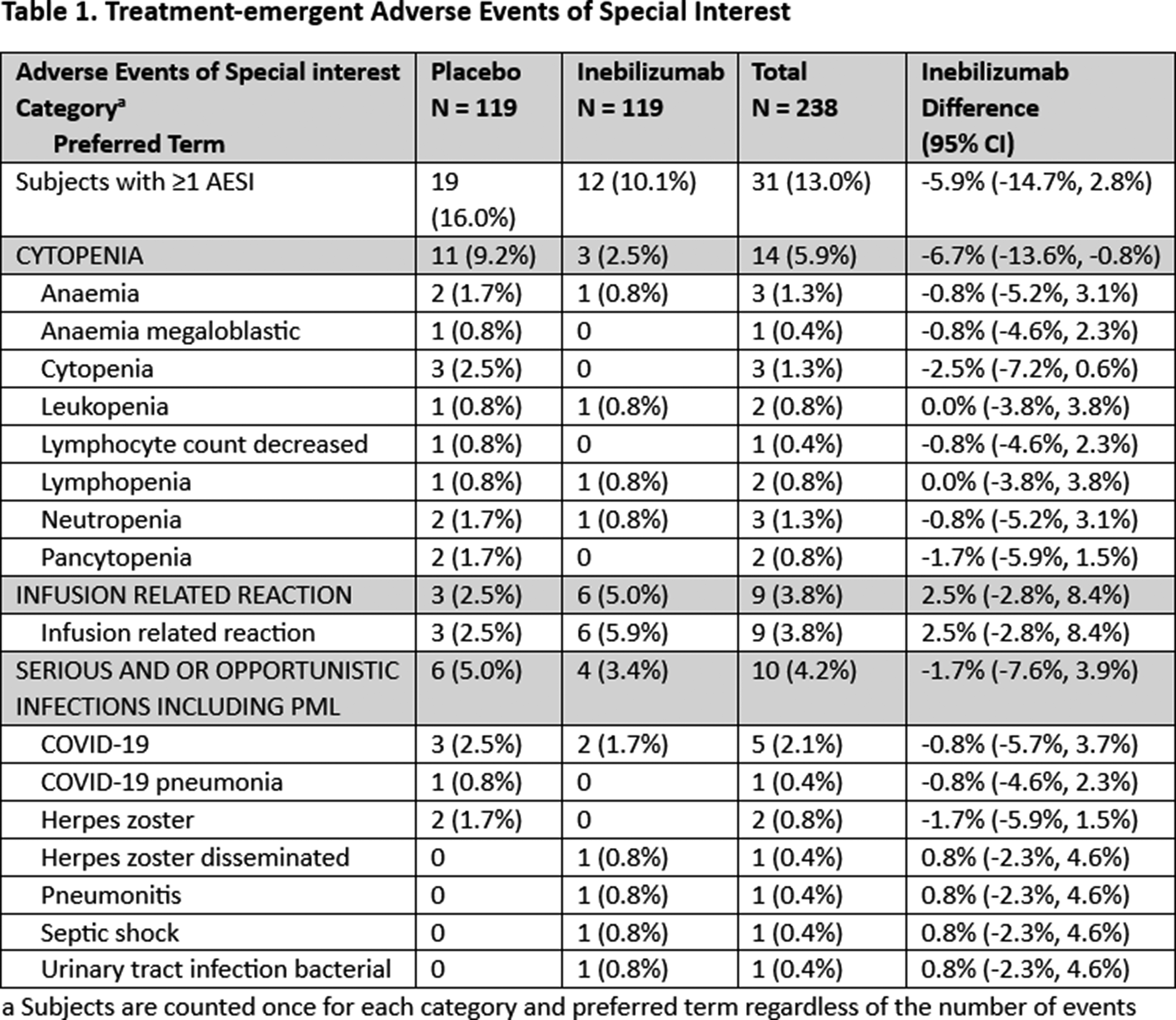
PP01.81
Myasthenia Gravis Inebilizumab Trial (MINT): Efficacy and Pharmacodynamics in AChR+ Cohort (Week 52)
Dr. Kristen A. Clarkson1, Dr. Kimiaki Utsugisawa2, Dr. Michael Benatar3, Dr. Emma Ciafaloni4, Dr. M. Isabel Leite5, Dr. John Vissing6, Dr. Fengming Tang1, Dr. Cody J. Peer1, Dr. Sue Cheng1, Dr. James F. Howard Jr.7, Dr. Richard J. Nowak8
1
Amgen Inc., Thousand Oaks, United States.
2
Hanamaki General Hospital, Hanamaki, Japan.
3
University of Miami, Miller School of Medicine, Miami, United States.
4
University of Rochester, Rochester, United States.
5
University of Oxford, Oxford, United Kingdom.
6
University of Copenhagen, Copenhagen, Denmark.
7
University of North Carolina, Chapel Hill, United States.
8
Yale University, New Haven, United States
Background: Autoreactive B-cells are central to upstream immunopathogenesis of generalized Myasthenia Gravis (gMG) through the production of autoantibodies. MINT met its primary efficacy endpoint, demonstrating a significant improvement in the Myasthenia Gravis Activities of Daily Living (MG-ADL) score at Week-26. Here, we evaluate the efficacy, pharmacodynamics, and immunogenicity of inebilizumab, a monoclonal antibody targeting CD19+ B-cells, in acetylcholine receptor antibody-positive (AChR+) gMG.
Methods: MINT (NCT04524273), a phase 3 clinical trial in adults with gMG, included a protocol-specified steroid taper. The randomized control period (RCP) was 52-Weeks for the AChR+ cohort and included additional secondary/exploratory endpoints: change from baseline (CFB) in MG-ADL and Quantitative Myasthenia Gravis (QMG) scores at Week-52, CD20+ B-cell count, and anti-drug antibodies (ADA). AChR+ participants were randomized (1:1) to receive 300mg of intravenous inebilizumab/placebo on RCP Day-1, Day-15, and Day-183.
Results: Of 238 randomized participants, 190 were AChR+ (inebilizumab: 95/placebo: 95). MG-ADL score showed improvement with inebilizumab vs. placebo at Week-52 (adjusted difference, −2.8; 95% CI, −3.9 to −1.7; nominal p<0.001). Similarly, CFB in QMG score was greater in the inebilizumab group vs. placebo at Week-52 (adjusted difference, −4.3; 95% CI, −5.9 to −2.8; nominal p<0.001).
CD20+ B-cell counts fell by 93.3% from baseline two weeks after the initial dose and remained low throughout treatment period in the AChR+ subpopulation. ADA prevalence for inebilizumab vs. placebo-treated participants was 4.2% vs. 2.1% in AChR+ subpopulation.
Conclusion: Inebilizumab leads to targeted depletion of B-cells and provides durable improvement in the AChR+ gMG subpopulation through Week-52.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.82
Trial Design for the Phase 2 FORGE Study Evaluating Apitegromab in Adults With FSHD
Dr. Jeffrey Statland1, Dr. Nicholas Johnson2, Dr. John Vissing3, Dr. Ryan Frieler4, Dr. Guochen Song4, Dr. Jing Marantz4, Dr. Tina Duong5, Dr. Leslie Nelson6, Dr. John Staropoli4
1
Department of Neurology, The University of Kansas Medical Center, Kansas City, United States.
2
Department of Neuology, Virginia Commonwealth University School of Medicine, Richmond, United States.
3
Department of Neurology, Copenhagen Neuromuscular Center, Copenhagen, Denmark.
4
Scholar Rock, Inc., Cambridge, United States.
5
Departments of Neurology and Clinical Neurosciences, Stanford University, Palo Alto, United States.
6
Department of Physical Therapy, University of Texas Southwestern Medical Center, Dallas, United States
Background: Facioscapulohumeral muscular dystrophy (FSHD) is a progressively debilitating disease characterized by progressive, facial muscle weakness that spreads to the shoulders and lower limbs. Patients with later adult-onset FSHD often present with progressive muscle weakness, accompanied by a loss of strength and function. The progressive nature of this disease presents an opportunity for therapeutic intervention with a muscle-targeted treatment to build muscle and potentially improve strength and motor function.
Apitegromab is an investigational, fully human monoclonal antibody that selectively inhibits activation of myostatin, a negative regulator of muscle growth, leading to increased muscle mass and improved motor function. In previous clinical studies for spinal muscular atrophy (Phase 2 TOPAZ, NCT03921528; Phase 3 SAPPHIRE, NCT05156320), the muscle-targeted treatment led to clinically meaningful and statistically significant improvements in motor function and was well tolerated across a broad range of patients who were also receiving a survival motor neuron-targeted treatment (nusinersen or risdiplam). Recent preclinical studies also showed that the murine analog of apitegromab (muSRK-015) significantly increased skeletal muscle mass, muscle force, and endurance in a FLExD murine model of FSHD.
Methods: FORGE, a Phase 2, randomized, double-blind, placebo-controlled, multicenter study, will assess the efficacy, pharmacokinetics, pharmacodynamics, safety, and tolerability of apitegromab in patients with FSHD.
Results: Approximately 60 ambulatory adult patients (aged 18-60 years) will be recruited to participate in the FORGE study. Patients with genetically confirmed FSHD (FSHD1 or FSHD2), a clinical severity score of 1.5-3.0 (Ricci score, range 0 to 5), and baseline 10MWRT time ≤5 seconds will be eligible for enrollment. Individuals with any medical condition, clinically significant laboratory result, or ECG value that may compromise safety, interfere with study compliance, or confound the interpretation of the results, are ineligible for the study. Exclusion criteria also include a history of hypersensitivity reactions to monoclonal antibodies or recombinant proteins, any acute or comorbid condition interfering with the well-being of the participant within 7 days prior to screening, and previous exposure to apitegromab. Eligible patients will be randomized to receive either apitegromab 10 mg/kg monotherapy or placebo by intravenous infusion every 4 weeks. The FORGE study will include a screening period of up to 4 weeks, a 52-week treatment period (13 doses), and a 20-week safety follow-up period for patients opting to not enter the open-label extension study. The primary endpoint will be percent change from baseline in total lean muscle volume by a full body MRI at week 52 of the study.
Conclusion: Supported by the pivotal outcomes from previous clinical and preclinical work, the FORGE study will investigate apitegromab, an investigational muscle-targeted treatment, in the adult FSHD patient population. FORGE is expected to be conducted across 20 sites in North America and Europe.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Facioscapulohumeral Muscular Dystrophies
PP01.83
Phenotypic Diversity and Upper Limb Function in Slovenian Patients With Facioscapulohumeral Muscular Dystrophy
Mrs Amadea Zupan1, Dr. Nataša Teran2, Mr. Matic Klopčič1, Prof. Lea Leonardis1
1
Institute of Clinical Neurophysiology, University Medical Centre Ljubljana, Ljubljana, Slovenia.
2
Clinical institute of genomic medicine, University Medical Centre Ljubljana, Ljubljana, Slovenia
Background: Facioscapulohumeral muscular dystrophy (FSHD) is a rare, phenotypically diverse hereditary muscular disorder that most commonly affects the facial muscles, the shoulder girdle, and the upper extremities. The disease progression, degree of impairment, and impact on quality of life vary significantly among individuals. The aim of this study was to retrospectively and prospectively evaluate the clinical status, functional impairment, and quality of life of Slovenian patients with FSHD.
Methods: The study included 31 patients with genetically confirmed FSHD type 1 and a control group of healthy individuals. The analysis encompassed demographic and clinical data, functional scales (Vignos-Brooke Scale, FSHD Rating Scale, Comprehensive Clinical Evaluation Scale), and questionnaires (VAS, SWLS, WHOQOL-BREF, MoCA, Zarit Burden Interview). Additionally, upper limb range of motion (ROM) was assessed using a Python-based application, which utilizes a standard 2D camera and the MediaPipe library to automatically detect body landmarks—a more accessible and less time-consuming alternative to marker-based systems or Kinect sensors.
Results: The median age of the patients was 46 years, with the disease onset at age 30. Half of the patients exhibited mild to moderate muscle impairment (FSHD Rating Scale ≤ 5), and the most frequent phenotype was myopathic D1. Regarding upper limb ROM, half of the FSHD patients achieved near-normal results (160°–180° in abduction and anteflexion), while the other half showed significant impairments, with values as low as 50° for abduction and 60° for anteflexion. Statistical analysis revealed a significant difference in ROM between FSHD patients and healthy controls. Better abduction capacity was moderately associated with higher scores in the physical health and social domains of the WHOQOL-BREF. However, upper limb function was not significantly correlated with overall life satisfaction. Furthermore, a strong correlation was observed between ROM and the Brooke Scale, confirming that greater ROM is positively associated with muscle strength scores.
Conclusion: FSHD in the Slovenian population exhibits broad phenotypic and functional diversity. While most patients maintain a satisfactory quality of life, the presence of pain, comorbidities, and physical limitations necessitates individually tailored multidisciplinary management. Objective 2D measurement of the range of motion using the MediaPipe module proves to be a feasible, accessible, and clinically useful tool for supplementing disease monitoring and tracking progress in rehabilitation or treatment.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Facioscapulohumeral Muscular Dystrophies
PP01.84
Long-Term Respiratory Follow-Up of Patients with FSHD1: An Italian Single-Center Study
Dr. Liliana Vercelli1, Dr. Giulio Gadaleta1, Dr. Fulvia Ribolla2, Dr. Guido Urbano1, Dr. Enrica Rolle1, Dr. Eleonora Leopizzi1, Dr. Silvia Boschi1, Prof. Paolo Solidoro2,3, Prof. Tiziana Mongini1
1
Neuromuscular Unit, Department of Neurosciences “Rita Levi Montalcini”, University of Turin, Turin, Italy.
2
Division of Respiratory Diseases, Cardiovascular and Thoracic Department, Città della Salute e della Scienza, University of Turin, Turin, Italy.
3
Department of Medical Sciences, University of Turin, Turin, Italy
Background: Respiratory involvement in facioscapulohumeral muscular dystrophy type 1 (FSHD1) is recognized as a meaningful clinical aspect of the disease; however, its long-term progression is still insufficiently characterized and longitudinal data describing respiratory function across multiple decades are scarce.
Methods: We performed a retrospective longitudinal study at a single center including 46 adult patients with genetically confirmed FSHD1, followed for up to 20 years. Motor and respiratory assessments were obtained at baseline and at approximately 5-year intervals. Respiratory parameters included forced vital capacity (FVC), forced expiratory volume in one second (FEV₁), maximal inspiratory pressure (MIP), and maximal expiratory pressure (MEP). Functional motor parameters included MRC scale and FSHD clinical score; we also used the Clinical Categories to classify FSHD patients. Longitudinal changes were examined using linear mixed-effects models, with exploratory analyses of potential clinical and genetic modifiers.
Results: At baseline mean age was 43.5 ± 14.2 years and mean disease duration was 20.3 ± 15.4 years; all participants were ambulatory at T0. Over the follow-up period, 17 out of 46 patients (37.0%) became non-ambulatory at a mean age of 62.0 ± 13.5 years, while 16 out of 46 (34.8%) initiated nocturnal ventilatory support at a mean age of 63.0 ± 10.4 years. Mean FVC decreased from 108.5% predicted at baseline to 91.6% after 20 years, and mean FEV₁ declined from 106.2% to 96.5%. The estimated annual rates of decline were −0.39% per year for FVC and −0.47% per year for FEV₁ (p < 0.001). Measures of respiratory muscle strength showed an earlier and more pronounced deterioration, with MIP decreasing by −0.78 cmH₂O/year and MEP by −1.54 cmH₂O/year (p < 0.001). Among the variables explored, only the CCEF FSHD score was independently associated with a faster decline in FVC (−1.42% per score point, p = 0.009) and MEP (−2.38 cmH₂O/year, p = 0.032). Sex, age at disease onset, D4Z4 fragment size, ambulatory status, and axial skeletal involvement did not significantly influence respiratory trajectories, although persistently lower respiratory values were observed in non-ambulatory individuals.
Conclusion: Over two decades of observation, patients with FSHD1 exhibited a gradual but sustained decline in FVC, FEV₁, and respiratory muscle strength, with a more pronounced reduction in MEP compared with MIP. Significant reduction of FVC and MEP values and initiation of ventilatory support were more frequently observed following loss of ambulation. Notably, respiratory impairment was primarily driven by the disease severity measured by the FSHD score, rather than by age at onset or other genetic characteristics.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Facioscapulohumeral Muscular Dystrophies
PP01.85
Clinical Characteristics and Longitudinal Changes in the Japanese FSHD Registry: Insights From 206 Patients
Prof. Tsuyoshi Matsumura1, Dr. Yuhei Hasuike1, Dr. Hotake Takizawa2, Dr. Wakako Yoshioka2, Dr. Yoshihiko Saito2, Dr. Ichizo Nishino2, Dr. Harumasa Nakamura2
1
NHO Osaka Toneyama Medical Center, Toyonaka, Japan.
2
National Center of Neurology and Psychiatry, Kodaira, Japan
Background: Facioscapulohumeral muscular dystrophy (FSHD) is caused by the disruption of mechanisms that silence the DUX4 gene. However, there is significant genetic and phenotypic variation, and epigenetic factors are implicated in its pathogenesis. The presence of gender and ethnic differences has also been noted, highlighting the importance of international comparisons and investigations into disease-modifying factors.
Methods: A nationwide patient registry for facioscapulohumeral muscular dystrophy (FSHD) was established in Japan in September 2020 and enrolled only genetically confirmed cases. It collects all core data items from the TREAT-NMD FSHD dataset version 1.0. We examined the data of 206 patients registered by the end of November 2025.
Results: Of the registered patients, 94 were male and 198 had FSHD1. Among the eight FSHD2 patients, seven carried pathogenic variants in SMCHD1 and one in DNMT3B. The mean age at registration was 43.2 ± 16.5 years. D4Z4 repeat size most commonly measured 3 units, with a median of 4. Early-onset, late-onset, and typical presentations were observed in 22, 11, and 173 patients, respectively. Early-onset cases consistently exhibited ≤4 repeats, except for one FSHD2 patient with 8 repeats, and were predominantly female (n=17). A family history was documented in 102 patients, with maternal inheritance (n=53) being more prevalent than paternal inheritance (n=23).
Regarding activities of daily living (ADLs), 148 patients remained ambulatory. Mean % forced vital capacity (%FVC) was 76.7 ± 25.1%, and 64.5% of individuals with ≤4 repeats had %FVC <80%. Mechanical ventilation was used by 22 patients. Cardiac function was largely preserved, with a mean left ventricular ejection fraction (LVEF) of 66.3 ± 6.9% and only 8 patients showing LVEF <55%.
Longitudinal data were available for 120 patients, with a mean follow-up of 2.9 ± 1.1 years. ADL deterioration occurred in 20 patients, including transitions from independent to assisted walking (n=8), independent walking to non-ambulatory status (n=2), assisted walking to non-ambulatory status (n=6), and from able to sit to unable to sit (n=4). Newly observed muscle involvement included facial (n=1), scapular girdle (n=2), lumbar girdle (n=4), and foot dorsiflexion weakness (n=3). Mechanical ventilation was newly initiated in 6 patients. Comparison of baseline and final assessments demonstrated modest but significant declines in %FVC (78.6 ± 24.9% to 76.4 ± 26.3%, p=0.028) and creatine kinase levels (log CK 2.36 ± 0.31 to 2.32 ± 0.33, p=0.012), while LVEF remained stable.
Conclusion: These findings indicate that Japanese FSHD patients tend to have shorter D4Z4 repeats and that early-onset disease is more common among women. Respiratory impairment is pronounced in individuals with shorter repeats, with a certain proportion requiring ventilatory support. Although FSHD progresses slowly, measurable declines in ADLs and respiratory function were consistently observed over approximately three years of follow-up, underscoring the value of longitudinal registry-based monitoring.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Facioscapulohumeral Muscular Dystrophies
PP01.86
Myasthenia Gravis-Myositis Overlap: A Comparative Study of Idiopathic and Immune Checkpoint Inhibitor-Related Forms
Dr. Antonio Lauletta1, Dr. Elena Rossini1, Assoc. Prof. Valentina Damato2,3, Dr. Francesca Beretta3, Dr. Simone Rossi4, Assoc. Prof. Raffaele Iorio5, Prof. Enrico Marchioni6, Dr. Antonio Petrucci7, Assoc. Prof. Alberto Vogrig8,9, Dr. Carmela Romano10, Dr. Laura Tufano1, Dr. Luca Leonardi11, Dr. stefania morino11, Dr. Francesca Forcina1, Dr. Demetrio Marando12, Dr. Valentina Vera12, Dr. Siliva Falso5, Dr. Sofia Marini5, Dr. Gregorio Spagni3, Dr. Paola Bini6, Dr. Stefano Masciocchi6, Dr. Antonio Lotti3, Assoc. Prof. Matteo Garibaldi1,11, Dr. Laura Fionda11
1
Sapienza Univesity of Rome, Nesmos Departement, Rome, Italy.
2
Department of Neurosciences, Psychology, Drug Research and Child Health (NEUROFARBA), University of Florence, Florence, Italy.
3
Neurology II Unit, Careggi University Hospital, Florence, Italy.
4
IRCCS Istituto delle Scienze Neurologiche di Bologna, bologna, Italy.
5
Department of Neuroscience, Università Cattolica del Sacro Cuore, Rome, Italy.
6
Unit of Neuroncology, Fondazione IRCCS Mondino, Pavia, Italy.
7
Center for Neuromuscular and Neurological Rare Diseases, San Camillo Forlanini Hospital, Rome, Italy.
8
Clinical Neurology, Department of Head-Neck and Neuroscience, Azienda Sanitaria Universitaria Friuli Centrale (ASU FC), udine, Italy.
9
Department of Medicine (DMED), University of Udine, udine, Italy.
10
Stroke unit, Department of Emergency, Ospedale dei Castelli-ASL6, Rome, Italy.
11
Neuromuscular and Rare Disease Centre, Neurology Unit, Sant’ Andrea Hospital, Rome, Italy.
12
Sapienza Univesity of Rome, Rome, Italy
Background: Emerging evidence highlights that a Myasthenia Gravis – Inflammatory Myopathy (MG-IM) overlap syndrome can occur as an immune-related adverse event (irAE) following immune-checkpoint inhibitor (ICI) therapy. Before the advent of ICIs, an idiopathic form of MG-IM (i-MG-IM), often thymoma-associated, had already been described.
This study compared the clinical and serological features, treatment strategies, and outcomes of i-MG-IM and ICI-related MG-IM (ir-MG-IM).
Methods: We conducted a multicenter, retrospective cohort study across eight Italian centers, including consecutive patients fulfilling established criteria for both MG and IM. Clinical features, antibody profiles, oncological history, treatments, and outcomes were systematically collected and compared. Available retrospectively collected sera were tested for reactivity against clustered adult (A) and fetal (F) AChR isoforms using live cell-based assay (L-CBA), and for complement-activating capacity using flow cytometry.
Results: Thirty-eight patients were identified (19 i-MG-IM and 19 ir-MG-IM) after excluding 13 ir-patients that did not meet MG diagnostic criteria and one i-MG-IM patient due to lack of clinical information.
Compared with i-MG-IM, the ir-MG-IM cohort was older (median 77 vs 59 years, p<0.001), predominantly male (84.2% vs 47.4%), and exclusively presented with concurrent MG/IM onset, whereas i-MG-IM often manifested sequentially. Thymoma was detected in 73.7% of i-MG-IM but in none of the ir-MG-IM patients. CK levels were higher in ir-MG-IM (median 3145 vs 1000 U/L, p=0.001). Myocarditis, orbital myositis, and severe bulbar/respiratory involvement were more frequent in ir-MG-IM (p<0.05).
Anti-AChR antibodies were found in all i-MG-IM but only in 68% of ir-MG-IM cases. Complement activation in vitro was observed only in i-MG-IM sera.
Anti-titin antibodies were identified in 85.7% of MG-IM (all thymoma-associated) and in 50% of ir-MG-IM.
Acute mortality occurred in 36.8% of ir-MG-IM and 0% in i-MG-IM patients (p<0.01). Relapses occurred in 63.2% of i-MG-IM but were absent in ir-MG-IM (p<0.001).
At last follow-up, functional outcomes were significantly worse in ir-MG-IM (median mRS 4 vs 0, p<0.001).
Conclusion: i-MG-IM and ir-MG-IM appear to represent distinct clinical and pathological entities. Although certain features overlap, our findings underscore the need for tailored diagnostic and therapeutic approaches and raise important questions regarding the pathogenic roles of antibodies and complement activation in ir-MG-IM.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Inflammatory / Immune-mediated Myopathies including Inclusion Body Myositis (IBM)
PP01.87
A Rare Autoimmune Myopathy in Later Life: Statin-Associated Anti-HMGCR IMNM Presenting with Troponin Elevation
Dr. Arkar Myo1, Dr. Ailie MCKINTY2, Dr. William Kah Howe LEE2, Dr. Alice REID3, Dr. Robert MCFARLANE4
1
Internal Medicine Trainee 2, Royal Devon and Exeter Hospital, Royal Devon University Healthcare NHS Foundation Trust, Exeter, United Kingdom.
2
Trust Resident Doctor, Royal Devon and Exeter Hospital, Royal Devon University Healthcare NHS Foundation Trust, Exeter, United Kingdom.
3
Foundation Year 2, Royal Devon and Exeter Hospital, Royal Devon University Healthcare NHS Foundation Trust, Exeter, United Kingdom.
4
Foundation Year 1, Royal Devon and Exeter Hospital, Royal Devon University Healthcare NHS Foundation Trust, Exeter, United Kingdom
Background: Immune-mediated necrotising myopathy (IMNM) is a rare autoimmune muscle disease characterised by progressive proximal weakness, markedly elevated creatine kinase (CK), and muscle fibre necrosis with minimal inflammation on biopsy. Anti-3-hydroxy-3-methylglutaryl coenzyme A reductase (anti-HMGCR) IMNM is strongly associated with statin exposure, and symptoms often persist despite statin withdrawal, requiring immunosuppressive therapy. Although cardiac involvement is uncommon, raised troponin can complicate assessment because chronic skeletal muscle regeneration may lead to re-expression of foetal troponin isoforms. We present a case of statin-associated anti-HMGCR IMNM in an elderly woman presenting with significant CK elevation and raised troponin but no evidence of myocarditis.
Methods: We reviewed the clinical course of a woman in her early 80s with a three-month history of progressive proximal weakness. Investigations included serial biochemical markers, autoimmune serology, cardiac imaging, CT and PET scans, and functional assessments. Differential diagnoses such as other inflammatory myopathies, paraneoplastic syndromes, and cardiac pathology were excluded. Treatment response was monitored through CK and troponin trends, neurological examination, and functional recovery following immunosuppressive therapy. Patient-reported experience was included to contextualise symptom progression and functional impact.
Results: The patient presented with progressive proximal weakness, CK 4,636 U/L, and troponin 258 ng/L. She had taken atorvastatin 20 mg daily for 2-3 years for dyslipidaemia, which was discontinued two weeks before admission. Despite this, CK remained markedly elevated over the following fortnight. Cardiac investigations - including ECG, echocardiography, and cardiac MRI showed no wall‑motion abnormalities or myocarditis. The extended myositis and autoimmune panels were negative. CT thorax–abdomen–pelvis and PET imaging revealed no malignancy. Two weeks after admission, anti-HMGCR antibodies sent at presentation returned strongly positive (>200 units), confirming the diagnosis. High-dose oral prednisolone (40 mg daily) was initiated immediately after antibody confirmation, with a structured taper. CK improved to 1,394 U/L after one week of steroid therapy but subsequently plateaued before rising again to 2,272 U/L by week seven of steroid therapy on recent blood. Troponin levels declined gradually without cardiac intervention. Muscle biopsy performed in week six of steroid therapy demonstrated features suggestive of denervation with possible mild necrotising myopathy, consistent with treated disease. Outpatient nerve conduction studies are pending. Clinically, the patient has shown gradual improvement in proximal strength and mobility with physiotherapy support. She remains under specialist follow-up, with plans to commence methotrexate if CK does not continue to improve.
Conclusion: This case highlights the importance of early recognition of statin-associated anti-HMGCR IMNM in older adults with persistent proximal weakness despite statin withdrawal. Strongly positive anti-HMGCR antibodies provide high diagnostic specificity and can streamline investigations by reducing reliance on early invasive testing. The combination of markedly elevated CK and raised troponin illustrates a key diagnostic pitfall, as chronic skeletal muscle injury may mimic cardiac disease; comprehensive cardiac imaging is therefore essential to avoid misinterpretation. Early immunosuppression remains central to improving outcomes, with steroid-sparing agents considered when biochemical or functional recovery plateaus. Overall, this case reinforces the value of timely antibody testing, multidisciplinary assessment, and clinical vigilance when evaluating unexplained weakness in later life.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Inflammatory / Immune-mediated Myopathies including Inclusion Body Myositis (IBM)
Images or Table (Optional)
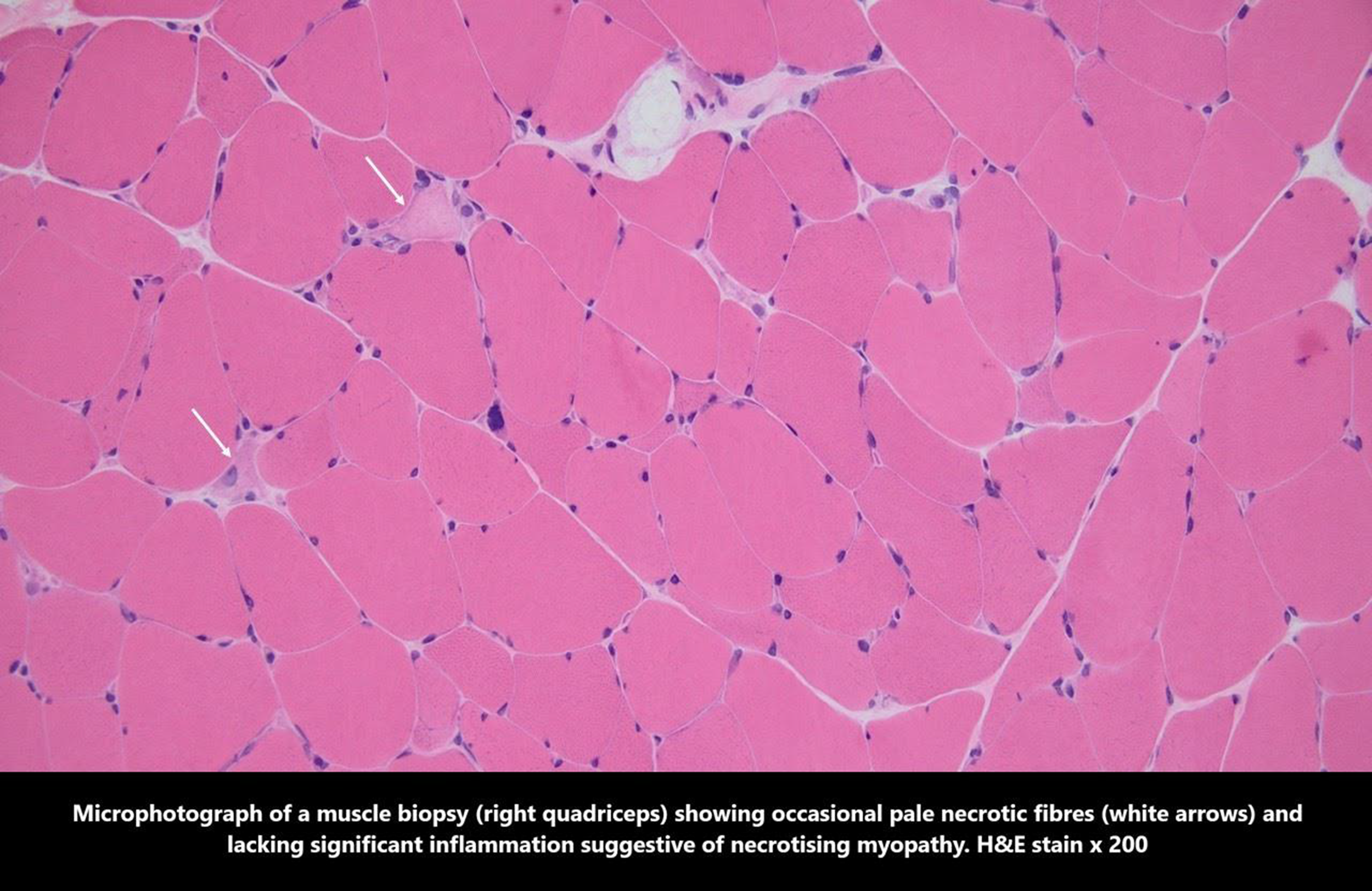
PP01.88
Clinical Characteristics and Therapeutic Response in Patients With Autoimmune Nodopathies: A Multi-Center US-Based Study
Dr. Charalampia Koutsioumpa1, Ms. Mia Weisman2, Dr. Alexander Morrison3, Dr. Waleed Khan4, Dr. Srikanth Muppidi5, Dr. Nizar Chahin6, Mr. Alexander Diefes2, Dr. Davalos Long7, Dr. Reza Sadjadi4, Dr. Karissa Gable8, Dr. Mazen Dimachkie7, Dr. Chafic Karam2, Dr. Jeffrey Allen9, Dr. Bhaskar Roy1
1
Yale University School of Medicine, New Haven, CT, United States.
2
Perelman School of Medicine, Philadelphia, United States.
3
Ohio State University School of Medicine, Columbus, United States.
4
Mass General Hospital, Boston, United States.
5
Stanford School of Medicine, Stanford, United States.
6
Oregon Health and Science University, Oregon, United States.
7
University of Kansas Medical Center, Kansas City, United States.
8
Duke University School of Medicine, Durham, United States.
9
University of Minnesota School of Medicine, Minneapolis, United States
Background: Autoimmune nodopathies (AINs) are a rare group of disorders characterized by circulating autoantibodies targeting antigens at the node of Ranvier, usually of the immunoglobulin G4 (IgG4) subclass. About 5-10% of patients with electrodiagnostic features of a demyelinating neuropathy are affected by AINs, with pathologic evidence of paranodal dissection without inflammation or demyelination. AINs may have suggestive clinical features and don’t respond to intravenous immunoglobulin (IVIg) but respond better to B-cell-depleting therapy. However, reports on the therapeutic response to AIN are limited, with most from Europe and Japan. A comprehensive review of the prevalence, characteristics, and treatment course of patients in the United States has not previously been reported.
Methods: This multi-center study, including over 12 centers in the US, is actively capturing the clinical features and therapeutic responses of patients with AINs.
Results: To date, we have included 33 patients, with 25 (76%) positive for anti-neurofascin 155 (NF155), six (18%) positive for anti-Contactin IgG4 autoantibodies, and two (6%) positive for both autoantibodies. The average age of disease onset was 50.3 ± 19.6 years, and 18 (54.5%) patients were male. 55% had distal and proximal weakness, 36% predominant distal weakness, and 24% had facial weakness. Distal sensory loss, particularly loss of vibration sensation, and ataxia were common. Areflexia was noted in more than 80% of patients. Additional clinical details will be presented. Almost 40% of patients presented acutely or sub-acutely and were initially diagnosed as Guillain-Barré syndrome. Over 90% of this cohort received IVIg, but only 33% had a partial benefit. Steroids were used in 50% of cases, neonatal Fc receptor (FcRn) inhibitors in 16% of cases, and PLEX in 20% of cases. Over 70% of patients received rituximab, and most showed objective benefit on clinical examination. Additional treatment details, including extent and durability of response will be presented.
Conclusion: This collaborative effort across the US is providing new insights into the clinical characteristics of AIN from a large US cohort, as well as critical information on management strategies.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Immune-mediated Neuropathies
PP01.89
Exercise and Functional Decline in Inclusion Body Myositis: A Study From the Yale IBM Registry
Mr. Alexander Ozonoff1, Prof. Gary Cutter2, Ms. Eesha Chakraborty1, Mr. Kurt Petschke3, Prof. A. David Paltiel3, Dr. Bhaskar Roy1, Dr. Chafic Karam4
1
Yale University School of Medicine, New Haven, CT, United States.
2
University of Alabama, School of Public Health, New Haven, CT, United States.
3
Yale School of Public Health, New Haven, CT, United States.
4
University of Pennsylvania, Perelman School of Medicine, Pennsylvania, United States
Background: Inclusion body myositis (IBM) is a rare, gradually progressive muscle disease with preferential involvement of quadriceps and long finger flexors, eventually leading to impaired ambulation and hand function. Some patients may also have dysphagia. The potential beneficial role of exercise in IBM has been implicated, but the impact of exercise in IBM, including the safety of exercise in IBM, is not well established.
Methods: We used the Yale IBM registry (IBMR) to examine the role of exercise on functional decline in IBM, based on the IBM functional rating scale score (IBM-FRS). We included patients with at least 2 longitudinal reports of IBM-FRS nine months apart, based on the IBM personalized index calculator (IBM-PIC) in the Yale IBM registry, which is a reliable remote measure of IBM-FRS.
Results: We included 401 patients (68.4 ± 8.4 years, Female 139 (34.7%)) with longitudinal data, with a baseline mean IBM-FRS score of 26.9 ± 6.8. All subjects showed a decline in IBM-FRS over time. There were no major differences in disease progression based on gender or age. The habit of exercise was associated with a slower rate of decline (p-value < 0.01), with a graded benefit for more active patients (p-value <0.001). Patients actively engaged in physical therapy and walking showed slower deterioration than those with no exercise (p-values of 0.003 and 0.04, respectively). Analyses of within-person changes emphasized the role of continuity of exercise.
Conclusion: These findings, based on patients’ self-reports, are supportive of the safety of exercise in IBM. There is also a potential benefit of remaining physically active in IBM. A longer clinical trials with a structured exercise regimen to delineate the true impact of exercise on IBM disease progression is warranted.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Inflammatory / Immune-mediated Myopathies including Inclusion Body Myositis (IBM)
PP01.90
Safety and Efficacy of Rese-cel in Severe, Refractory Idiopathic Inflammatory Myopathy
Dr. Elie Naddaf1, Dr. Erin Marie Wilfong2, Dr. Tahseen Mozaffar3, Dr. Nizar Chahin4, Dr. Huifang Lu5, Dr. Iazsmin Bauer-Ventura6, Dr. Coughi Edens6, Dr. Jason Sluzevich7, Mrs Courtney Little8, Dr. Gaurav Choudhary8, Dr. Jenell Volkov8, Mr. Thomas Furmanak8, Dr. Rohit Aggarwal9, Dr. Raj Tummala8, Dr. David J Chang8
1
Mayo Clinic, Rochester, United States.
2
Vanderbilt University Medical Center, Nashville, United States.
3
University of California, Irvine, Irvine, United States.
4
Oregon Health Sciences University, Portland, United States.
5
MD Anderson Cancer Center, Houston, United States.
6
University of Chicago, Chicago, United States.
7
Mayo Clinic, Jacksonville, United States.
8
Cabaletta Bio, Philadelphia, United States.
9
University of Pittsburgh Medical Center, Pittsburgh, United States
Background: Treatments for idiopathic inflammatory myopathy (IIM) aim to reduce inflammation and disease activity and decrease long-term morbidity and mortality. Therapies providing durable clinical responses are lacking. Rese-cel is a fully human, autologous 4-1BB CD19-CAR T cell therapy designed to deeply and transiently deplete CD19 positive cells following a single weight-based infusion with the potential to enable an “immune system reset” with durable responses without chronic immunosuppression. RESET-Myositis® (NCT06154252) is an ongoing trial evaluating rese-cel in 3 Phase 1/2 cohorts of adults with dermatomyositis (DM), antisynthetase syndrome (ASyS) and immune mediated necrotizing myopathy (IMNM) and a fourth cohort of patients with juvenile IIM (JIIM).
Methods: Eligible patients were 6 – 75 years with refractory IIM, positive myositis specific autoantibody (MSA) and at least moderate disease activity on 2 or more core set measures (CSMs). A weight-based infusion of 1x106 CAR T cells/kg was given following preconditioning with fludarabine (25 mg/m2/d on days -5, 4 and 3) and cyclophosphamide (1000 mg/m2 on day -3). All non-glucocorticoid (GC) IM agents were discontinued prior to preconditioning. Adverse events, use of IM medications and efficacy measurements to calculate the TIS were collected. Pharmacokinetic (PK) and pharmacodynamic (PD via B cell counts) profiles were determined using digital PCR and flow cytometry, respectively, in peripheral blood mononuclear cells.
Results: As of 11 September 2025, 13 patients were treated with rese-cel and completed at least 28 days of follow-up (Table 1) in the RESET-Myositis trial. Rese-cel was well-tolerated with Grade 1 cytokine release syndrome (CRS) in 4 patients and no dose-limiting toxicity, immune effector cell-associated neurotoxicity syndrome (ICANS) or serious infections. Among patients with DM or ASyS and ≥16 weeks of follow-up, 80% (3 of 3 DM and 1 of 2 ASyS) achieved at least moderate response (TIS ≥40) off IM medications on no more than low-dose steroids at Week 16. Two of 4 patients with IMNM and ≥24 weeks of follow-up achieved at least minimal response (TIS ≥20) off IM medications and on no more than low-dose steroids at Week 24. Downward trends in MSAs were observed in patients with clinical responses and sufficient follow-up. Persistence or recurrence in MSAs were seen in patients with ASyS and IMNM who did not achieve or maintain TIS response. PK and PD profiles were available for 12 adult patients. Rese-cel expanded in all patients and peaked 13 days (median; IQR: 11-14) following infusion at 25 cells/µL (median; IQR: 13-59). Peripheral B cell counts reached a minimum at 11 days (median; IQR: 8-14) post-infusion with an average depletion duration of 49 days (median; IQR: 42-66), calculated from the 11 patients who exhibited B cell repopulation at the time of the data cut. Repopulating B cells were mostly transitional naïve, supporting B cell compartment reset.
Conclusion: Acceptable safety, early IM-free clinical responses, and deep B-cell depletion were observed with rese-cel in IIM. DM and ASyS patients showed greater improvement than IMNM. These findings support rese-cel’s potential to enable an immune reset and achieve meaningful, drug-free responses in refractory myositis patients.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Inflammatory / Immune-mediated Myopathies including Inclusion Body Myositis (IBM)
Images or Table (Optional)
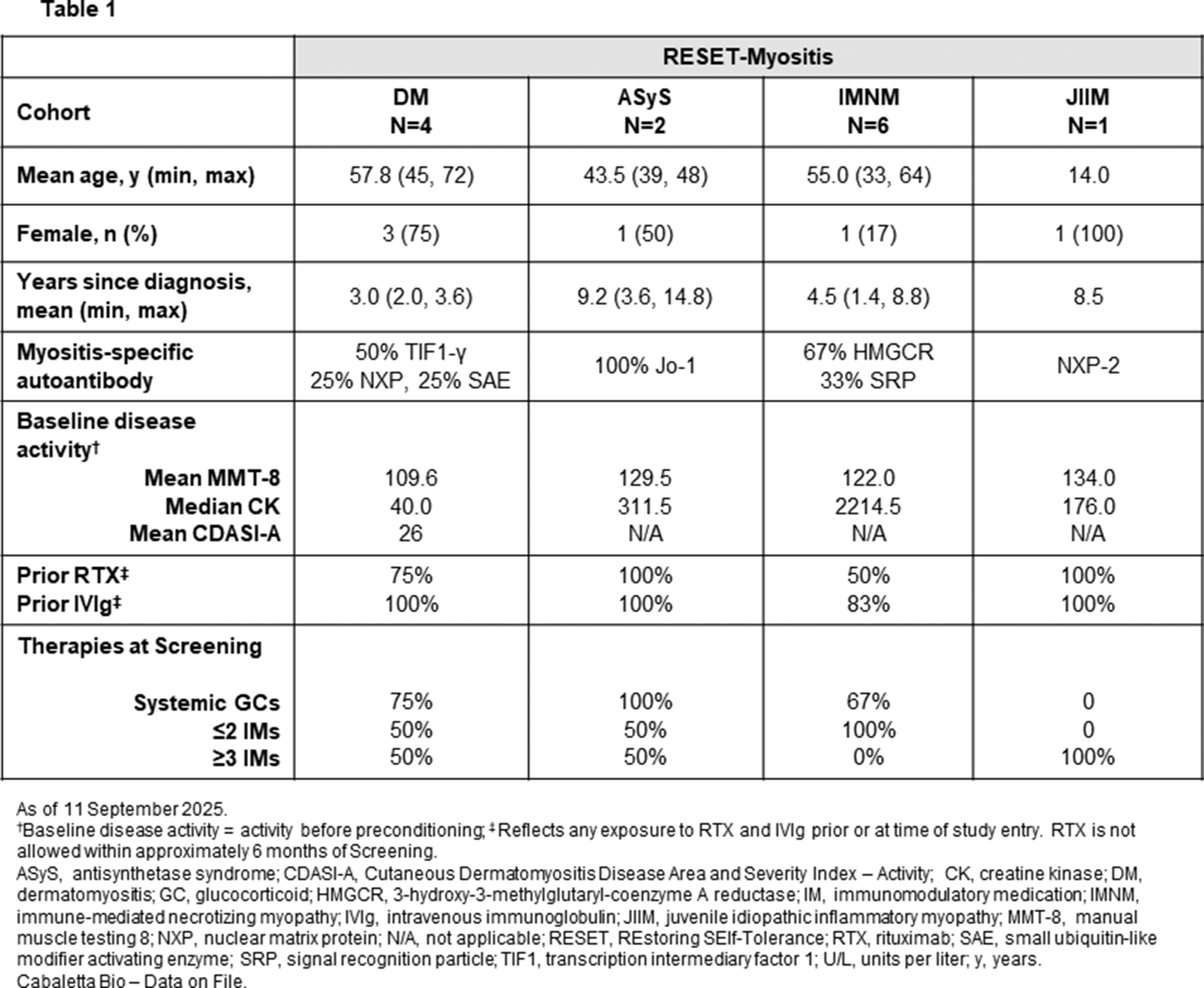
PP01.91
Tiny Molecules, Mighty Impacts: MicroRNAs in Neuromuscular Diseases
Miss Ana Moraes, Dr. Paula Fratini, Mr. Enzo Pellacani, Dr. Alzira Carvalho
Centro Universitario FMABC, Santo André, Brazil
Background: MicroRNAs (miRNAs) are small non-coding RNA molecules that regulate gene expression post-transcriptionally and play essential roles in muscle differentiation, regeneration, and metabolism. A specific subset, known as myomiRs, is predominantly expressed in muscle tissue and has been implicated in the regulation of key pathways involved in neuromuscular diseases (NMDs). Due to their stability in biological fluids, myomiRs have gained attention as potential non-invasive biomarkers for diagnosis, prognosis, and therapeutic monitoring. This review aimed to demonstrate current knowledge on the role of myomiRs in major NMDs, particullary Duchenne muscular dystrophy (DMD), myotonic dystrophy type 1 (DM1), limb-girdle muscular dystrophy (LGMD), McArdle disease, and myofibrillar myopathy (MFM), emphasizing their biological significance and clinical potential.
Methods: A narrative literature review was conducted using indexed databases to identify studies addressing myomiR expression, function, and clinical implications in neuromuscular diseases.
Results: The analysis demonstrated that specific myomiRs, such as miR-1, miR-133a/b, miR-206, miR-29b, miR-31, and miR-486, are differentially expressed among these disorders, participating in processes like muscle degeneration, regeneration, fibrosis, and inflammation. In DMD, several myomiRs are upregulated, reflecting ongoing muscle damage and regeneration, while in DM1, the downregulation of miR-1, miR-133a/b, and miR-29b contributes to defective muscle maturation and fibrosis. In contrast, miR-206 and miR-223-3p are increased as part of a compensatory regenerative response. For LGMD, miR-143-3p and miR-486 are elevated and may serve as circulating biomarkers. Only one study has evaluated the role of myomiRs in McArdle disease, underscoring the scarcity of available data and the need for further research to clarify their diagnostic and pathogenic relevance.
Conclusion: MyomiRs are central to the molecular mechanisms of NMDs and stand out as accessible biomarkers for diagnosis, monitoring, and therapeutic evaluation. Their emerging use as non-invasive endpoints in clinical trials underscores their translational relevance and the need to expand research across the different NMDs.
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: Genetics in Neuromuscular Diseases including Biochemical/Molecular Techniques and next generation sequencing.
Images or Table (Optional)
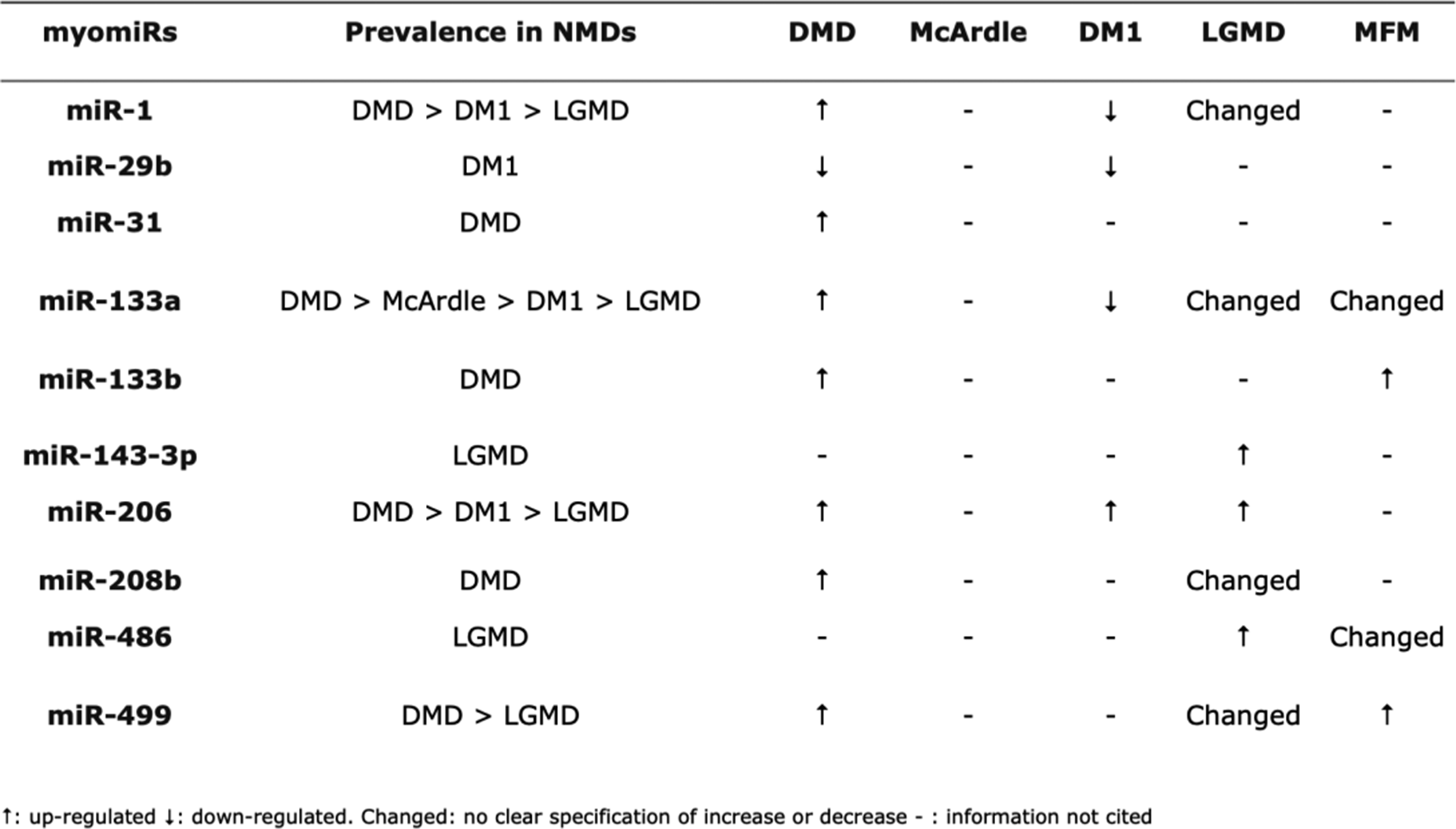
PP01.92
When Genes Fall Silent: Muscle Biopsy Guiding Lipid Storage Myopathy Management in Resource-Limited Settings
Dr. Enzo Pellacani1, Dr. Ana Marina Silva1, Dr. Alessandra Tolentino1, Dr. Roseli Corazzini1, Dr. David Feder1, Dr. Ana Luisa Hernandez2, Dr. André Macedo-Silva2, Dr. Edmar Zanoteli2, Dr. Alzira Carvalho1
1
Centro Universitário FMABC, Santo André, Brazil.
2
Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo, São Paulo, Brazil
Background: Lipid storage myopathies (LSM) comprise a heterogeneous group of metabolic disorders disrupting lipid energy metabolism (1). While Next-Generation Sequencing (NGS) has revolutionized diagnosis, access remains unequal in developing regions, and a significant proportion of histologically confirmed cases remain genetically undefined (“gene-silent”). We aim to describe the clinical-histological profile of an LSM cohort and discuss the strategic role of muscle biopsy in guiding empirical treatment in a resource-limited setting.
Methods: We conducted a multicenter, retrospective study involving patients from two tertiary centers in São Paulo, Brazil (FMABC and HCFMUSP). Inclusion criteria were defined by confirmed lipid storage myopathy on muscle biopsy (Oil Red O positive vacuoles). Genetic investigation followed a hierarchical approach based on availability and complexity (Targeted NGS Panels < WES < WGS). Peripheral blood smears were analyzed for the presence of lipid vacuoles in leukocytes (Jordan’s anomaly). Clinical phenotypes, creatine kinase (CK) levels, and therapeutic management were analyzed.
Results: Ten patients were included (mean age of symptom onset: 30.3 years; range 5–53). Weakness was the cardinal symptom (100%), predominantly proximal, but with significant overlap: distal involvement in 40%, axial in 30%, facial in 20%, and bulbar/respiratory symptoms in 30% of cases. Jordan’s anomaly was universally present in all assessed patients (8/8). Notably, this finding—classically associated with Neutral Lipid Storage Disease (NLSD)—was observed in patients with MADD (ETFDH) and genetically undefined cases. Genetic testing yielded a diagnosis in only 40% (4/10): three ETFDH and one FLAD1. The remaining 60% had inconclusive results. Crucially, histological confirmation guided the initiation of metabolic therapies (e.g., Riboflavin, L-Carnitine, CoQ10). Consistent with literature supporting riboflavin-responsive MADD, this strategy prioritizes early treatment initiation based on histopathological findings, ensuring access to disease-modifying therapy even while molecular confirmation is pending (2–6).
Conclusion: In our cohort, strictly selected by histological positivity, genetic screening failed to identify the molecular cause in the majority of patients (60%), highlighting the limitations of current accessible panels or the complexity of “gene-silent” LSMs. Although genetic confirmation is the gold standard, muscle biopsy and simple blood smears (Jordan's anomaly) proved to be robust, cost-effective tools. They serve as critical decision-points for initiating empirical metabolic therapy, bridging the gap between clinical suspicion and the often-inaccessible or inconclusive genetic diagnosis in the Brazilian public health context.
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: Neuromuscular Pathology: Muscle and Nerve Biopsy
PP01.93
Unveiling a Novel Link: Myofibrillar Myopathy Phenotype Associated with TRIM32 Gene Mutation
Dr. Enzo Pellacani, Dr. Ana Marina Silva, Dr. Alessandra Tolentino, Dr. Roseli Corazzini, Dr. David Feder, Dr. Alzira Carvalho
Centro Universitário FMABC, Santo André, Brazil
Background: The TRIM32 gene (OMIM 602290) encodes an E3 ubiquitin ligase associated with a complex phenotypic spectrum. Depending on the specific domain affected, variants may lead to the non-myopathic Bardet-Biedl syndrome or to a muscular continuum comprising LGMDR8 and Sarcotubular Myopathy (1–4). While these myopathies are classically distinguished by non-rimmed vacuoles, this morphological focus often overlooks the gene's critical role in maintaining Z-disk stability (1,3,4). We question whether TRIM32 dysfunction might also drive the structural pathology of Myofibrillar Myopathy (MFM)—a phenotype typically linked to structural Z-disk proteins rather than their regulators. MFM is strictly defined by focal myofibrillar dissolution, dark-blue trichrome inclusions, and ectopic accumulation of cytoskeletal proteins (e.g., myotilin) (5). We report a case bridging these entities, exhibiting hallmark LGMDR8 vacuolar features alongside distinct MFM pathology, supporting a potential mechanistic overlap.
Methods: A 50-year-old Brazilian male, born to consanguineous parents, presented with progressive proximal muscle weakness starting at age 15. The patient exhibited significant shoulder girdle atrophy and symmetrical lower limb weakness, with elevated CK levels and a myopathic EMG pattern; cardiac and respiratory functions were preserved.
Muscle biopsy revealed non-specific myopathic changes (fiber size variation, internalized nuclei) alongside hallmark features of both LGMDR8 and MFM. We observed slit-shaped non-rimmed vacuoles typical of LGMDR8, coexisting with evident myofibrillar disarray (NADH) and abundant intracytoplasmic protein aggregates. Immunohistochemistry confirmed the ectopic accumulation of Myotilin and αB-Crystallin, consistent with myofibrillar inclusion bodies.
Genetic testing identified a homozygous nonsense variant, c.1885C>T (p.Gln629*) in the TRIM32 gene. Electron microscopy (EM) analysis is currently in progress to further characterize the ultrastructural Z-disk disintegration expected in this phenotype.
Results: The identification of a clear MFM histophenotype in a TRIM32 patient challenges the traditional phenotypic boundaries of LGMDR8 and expands the MFM differential diagnosis beyond its classic autosomal dominant spectrum. This underscores that MFM pathology should not be disregarded in recessive pedigrees, despite the predominance of dominant forms. Given TRIM32's function as an E3 ubiquitin ligase, its failure to tag and degrade sarcomeric proteins likely leads to the accumulation of protein aggregates and subsequent Z-disk instability observed in this case. This mechanism provides biological plausibility for the MFM phenotype, suggesting that the myofibrillar disarray is not merely a secondary alteration, but a direct mechanistic consequence of the loss of TRIM32-mediated protein turnover.
Conclusion: In conclusion, we report a case of a homozygous TRIM32 nonsense variant (p.Gln629*) manifesting as a hybrid phenotype of classic vacuolar changes and distinct Myofibrillar Myopathy features. Our findings support the idea that the TRIM32-associated myopathy spectrum is broader than historically described and that TRIM32 dysfunction should be considered a potential causative mechanism when MFM features are identified on muscle biopsy.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Myofibrillar Myopathies
PP01.94
MxA Positivity Beyond Dermatomyositis: New Insights from a case of Lupus Myositis
Dr. Enzo Pellacani, Dr. Ana Marina Silva, Dr. Alessandra Tolentino, Dr. Roseli Corazzini, Dr. David Feder, Dr. Alzira Carvalho
Centro Universitário FMABC, Santo André, Brazil
Background: Myxovirus resistance protein A (MxA) is a surrogate marker for Type I Interferon (IFN1) activation. Its expression, particularly in a perifascicular pattern, is currently regarded as a highly specific hallmark of Dermatomyositis (DM) (1–3).
Systemic Lupus Erythematosus (SLE) is also driven by an IFN1 signature. However, the expression of MxA in Lupus Myositis (LM) remains controversial, with conflicting data regarding its sensitivity and staining patterns (3–5).
We report a case of Lupus Myositis (LM) presenting with patchy MxA staining. We aim to question the prevailing view that MxA is consistently negative in SLE, highlighting that Type I Interferon activation in Lupus can occasionally mirror the immunohistochemical profile of DM.
Methods: A 46-year-old man presented to the neuromuscular service, exhibiting a clinical picture of symmetrical, proximal tetraparesis, associated with ophthalmoparesis, ptosis, intense dysphagia, and severe fatigue. Laboratory tests showed elevated Creatine Phosphokinase (CK) and Aldolase levels. Electromyography (EMG) was consistent with myopathic disease, leading to a suspicion of inflammatory myopathy. The patient fulfilled the 2021 ACR/EULAR criteria for SLE, achieving 80% strength recovery under prednisone and methotrexate.
Muscle biopsy showed mild-to-moderate fiber size variation, increased internalized nuclei, and rare regenerating fibers. It featured a single fascicle with reduced fiber caliber, more evident in the periphery. Immunohistochemical (IHC) study revealed positive immunoexpression for C5b9 (sarcolemmal granular pattern) in normal-appearing fibers, along with positive and patchy MxA expression, across the whole fascicle.
Given the association of a clinical SLE diagnosis and the evident patchy MxA positivity pattern, without significant lymphomonocytic infiltrate or necrosis, and with only questionable perifascicular atrophy, a diagnosis of LM was consolidated.
Results: SLE and DM share an IFN1 signature (1–4). While Nishino et al. emphasize MxA as a highly specific marker for DM (often comparing it against SLE controls), the variable expression reported by Xing et al. in a larger LM cohort suggests a more complex landscape, raising the question of whether these discrepancies reflect distinct disease entities or variations in the intensity of a shared interferonopathy (1–4). We demonstrate MxA positivity in a patient fulfilling current ACR/EULAR criteria for SLE, lacking classic DM clinical features. Shared interferon mechanisms between DM and SLE may limit MxA specificity, suggesting that diagnosis should not rely solely on this marker's positivity.
Conclusion: MxA expression in LM raises the possibility that Type I Interferon activation is a shared feature across a broader spectrum of autoimmune myopathies, rather than a unique hallmark of DM. The patchy pattern, unlike the classic perifascicular presentation, adds complexity to the diagnostic landscape. This case invites us to reconsider the rigid categorization of autoimmune myopathies based on staining alone, suggesting that histological boundaries between these entities might be more fluid than previously thought, potentially representing variations within a common “interferonopathy” mechanism. This case suggests that finding MxA expression should not automatically default to a DM diagnosis, but rather prompt clinicians to maintain a broader differential diagnosis, ensuring that subtle SLE-specific stigmata are not overlooked, acknowledging that these diseases may exist on a shared pathophysiological spectrum.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Inflammatory / Immune-mediated Myopathies including Inclusion Body Myositis (IBM)
PP01.95
Novel TTN Variants Causing Emery-Dreifuss-Like Phenotype: Expanding the Genotypic Spectrum
Dr. Enzo Pellacani, Dr. Ana Marina Silva, Dr. Alessandra Tolentino, Dr. Roseli Corazzini, Dr. David Feder, Dr. Alzira Carvalho
Centro Universitário FMABC, Santo André, Brazil
Background: The titin gene (TTN) is the largest known gene, consisting of 363 exons, and encodes a giant sarcomeric protein essential for muscle structural integrity, elasticity, and signaling [1, 2]. Due to its size and complexity, TTN mutations are associated with significant phenotypic heterogeneity, ranging from isolated cardiomyopathies to various muscular dystrophies [2]. Recently, a rare “Emery-Dreifuss-like” phenotype without cardiomyopathy was described, characterized by the triad of early joint contractures, limb-girdle weakness, and secondary calpain-3 deficiency, distinguishing it from classic forms linked to LMNA or EMD [3]. We report a case of titinopathy with this phenotype caused by two novel compound heterozygous variants, broadening the genetic basis of this condition.
Methods: A 14-year-old female presented with hypotonia noted since infancy and delayed motor milestones, sitting unsupported at 18 months and walking at 2 years. At age 10, she developed prominent joint contractures of the elbows and ankles, as well as severe scoliosis, undergoing arthrodesis at age 12. Neurological examination revealed generalized amyotrophy, scapular winging, rigid spine, and proximal-predominant weakness (MRC 3-4/5), sparing facial and extraocular muscles. Distinctive dysmorphisms were observed, including retroverted ears, elongated face with a prominent chin, and ogival palate. Deep tendon reflexes were absent. Creatine phosphokinase (CPK) levels were normal. Two previous muscle biopsies performed at external centers showed a nonspecific dystrophic pattern (at 1.5 years) and inconclusive findings (at 11 years). Respiratory function was compromised (Forced Vital Capacity of 26%, severe restrictive pattern), contrasting with normal cardiac evaluation (echocardiogram and electrocardiogram). Thigh magnetic resonance imaging showed diffuse fatty replacement, predominantly in the posterior compartment. Next-generation sequencing (NGS) identified two novel TTN variants in compound heterozygosity: c.27607G>T (p.Glu9203*), a pathogenic nonsense variant; and c.669+1G>A, an N-terminal variant classified as pathogenic for affecting the splice site.
Results: The co-occurrence of early contractures, rigid spine, and humeroperoneal weakness mimics Emery-Dreifuss Muscular Dystrophy (EDMD) [3]. However, the absence of cardiac involvement in our patient aligns specifically with the “Emery-Dreifuss-like” titinopathy phenotype described by De Cid et al. (2015) [3]. While the literature primarily links this phenotype to M-line (C-terminal) truncations [2, 3], our patient presents an atypical combination of an N-terminal splicing variant and a central region stop codon. This suggests that compound heterozygosity affecting distinct titin domains can manifest this specific contractural phenotype. Severe respiratory restriction with preserved cardiac function distinguishes this entity from other severe early-onset titinopathies, such as Early-Onset Myopathy with Fatal Cardiomyopathy (EOMFC), where cardiac involvement is determinant [2, 3].
Conclusion: This report validates the existence of the rare “Emery-Dreifuss-like” titinopathy without cardiomyopathy and expands its mutational spectrum to include novel variants outside the classic M-line region. Titinopathy should be considered a key differential diagnosis in rigid-spine syndromes with preserved cardiac function, necessitating comprehensive TTN sequencing for definitive diagnosis.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Muscle Dystrophies (Non-Dystrophinopathies)
PP01.96
Eotaxin/CCR3-Mediated Inflammation in Inclusion Body Myositis: A Permissive Environment for Viral Entry and Muscle Degeneration
Dr. Giorgia Riolo1,2, Mr. Franco Salerno1, Dr. Alex Vicino3, Dr. Marta Cheli1, Dr. Sara Gibertini1, Dr. Alessandra Ruggieri1, Dr. Giada Giunta1, Dr. Eliana Iannibelli1, Dr. Alessandra Carnazzi1, Dr. Lucia Nicolini De Gaetano1, Dr. Massimo Costanza1, Dr. Silvia Bonanno1, Dr. Stefania Marcuzzo1,4, Dr. Paola Cavalcante1, Dr. Lorenzo Maggi1
1
Neurology 4 - Neuroimmunology and Neuromuscular Diseases Unit – Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy.
2
Ph.D. program in Pharmacological Biomolecular Sciences, Experimental and Clinical, University of Milan, Milano, Italy.
3
Nerve-Muscle Unit – Neurology Service, Department of Clinical Neurosciences, Lausanne University Hospital and University of Lausanne, Lausanne, Switzerland.
4
Brain-targeted Nanotechnologies (BraiNs) Lab, Joint Research Platform – Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy
Background: Inclusion body myositis (IBM) is the most common acquired muscle disease affecting individuals over 45 years of age. Clinically, the disease manifests as a slowly progressive and asymmetric pattern of muscle weakness, predominantly affecting the quadriceps and deep finger flexors. To date, no therapies are available, and most patients ultimately develop severe disability, becoming wheelchair-dependent.
Although IBM pathogenesis remains unclear, accumulating evidence supports a role for persistent immune activation in driving muscle fiber damage, and chronic viral infections have been suggested as potential modulators of this process. Among viruses, Hepatitis C virus (HCV) has been associated with several extrahepatic inflammatory and autoimmune manifestations. Of note, higher prevalence of HCV infection has been reported in IBM patients, although the HCV-IBM relationship remains poorly defined.
Herein, we explored the inflammatory landscape of IBM muscle, focusing on cytokine/chemokine patterns, immune cell infiltration, and the potential pathogenetic link between viral infections, anti-viral response pathways and muscle degeneration.
Methods: This study included treatment-naïve IBM (n=30), non-IBM myopathic patients (immune-mediated necrotizing myopathies, n=15; hereditary myopathies, n=22) and age- and sex-matched healthy controls (HC, n=18). Serum cytokine/chemokine profiling was performed by multiplex immunoassays. Gene expression of eotaxins, their receptor CCR3 and antiviral response mediators was assessed by qPCR in muscles, myoblasts and peripheral blood cells (PBMCs; eotaxins and CCR3 only). Immunofluorescence was used to evaluate immune cell infiltration and localize CCR3 in IBM and control tissues. HCV RNA presence in muscle biopsies from serologically HCV-positive (n=4) and -negative (n=3) IBM patients was analysed by droplet digital PCR (ddPCR) and RNAscope.
Results: We revealed a significant increase of eotaxin-1 (CCL11), eotaxin-2 (CCL24) and eotaxin-3 (CCL26) levels in serum of IBM compared to non-IBM myopathic patients and HC, thus delineating an IBM-associated inflammatory chemokine signature. Accordingly, qPCR analysis showed a significant overexpression of CCL24, CCL26 and CCR3 in muscle tissue and myoblasts, but not in PBMCs, of IBM compared to non-myopathic and non-IBM myopathic subjects. Increased CCR3 expression in IBM muscles was confirmed by immunofluorescence, and the protein mainly co-localized with macrophages and macrophage-enriched regions, thus linking the eotaxin increase with immune system cell infiltration into the muscle.
By ddPCR and RNAscope, HCV RNA was detected in 3 out of 4 serologically HCV-positive IBM patients’ muscle, where it was found to co-localize with CCR3-positive infiltrating macrophages, thus suggesting that eotaxins can mediate viral genome entry into the muscle viaCCR3. A viral contribution was further supported by qPCR data showing a significant increase in antiviral response genes, including MX1, Granzyme, TLR3, TLR7 and interferons, in IBM compared to control muscles.
Conclusion: Our findings reveal a previously unknown mechanistic framework through which HCV, or other viral components/nucleic acids, may access muscle tissue, offering a new perspective on the crosstalk between viral infections and muscle chronic inflammation in IBM. Moreover, we highlight a role for the eotaxin-CCR3 axis in mediating immune system cell infiltration and chronic inflammatory pathways contributing to muscle fiber degeneration. These insights improve our understanding of IBM pathogenesis, promising to guide the development of novel, targeted, disease-modifying therapies.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Inflammatory / Immune-mediated Myopathies including Inclusion Body Myositis (IBM)
PP01.97
Distinct Thigh Muscle MRI Patterns Across Idiopathic Inflammatory Myopathy Subtypes: A Single-Center Cohort Analysis
Dr. Hee-Jae Jung1,2, Ms. Heung-Won Kang3, Dr. Ho Cheol Kim4, Dr. Seokchan Hong5, Dr. Young Chul Kim6, Dr. Won Kim7, Dr. Eun-Jae Lee1, Dr. Young-Min Lim1, Dr. Jin-Sung Park8, Dr. Hyunjin Kim1
1
Department of Neurology, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea, Republic of.
2
Department of Neurology, Seoul, Korea, Republic of.
3
Department of Neurology, Seoul Medical Center, Seoul, Korea, Republic of.
4
Department of Pulmonary and Critical Care Medicine, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea, Republic of.
5
Division of Rheumatology, Department of Internal Medicine, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea, Republic of.
6
Department of Plastic and Reconstructive Surgery, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea, Republic of.
7
Department of Rehabilitation Medicine, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea, Republic of.
8
Department of Neurology, School of Medicine, Kyungpook National University, Kyungpook National University Chilgok Hospital, Daegu, Korea, Republic of
Background: Idiopathic inflammatory myopathies (IIMs) are a heterogeneous group of autoimmune muscle diseases, and their overlapping clinical phenotypes often complicate subtype differentiation. Muscle magnetic resonance imaging (MRI) is increasingly recognized as a valuable non-invasive diagnostic tool, yet direct comparative analyses across major IIM subtypes remain limited. We aimed to characterize and contrast thigh MRI patterns among anti-synthetase syndrome (ASyS), dermatomyositis (DM), inclusion body myositis (IBM), and immune-mediated necrotizing myopathy (IMNM).
Methods: This prospective single-center cohort included 47 patients with IIM (12 ASyS, 15 DM, 11 IBM, 9 IMNM). Thigh MRI (T1-weighted, STIR, and contrast-enhanced T1) was systematically evaluated. Muscle edema was semi-quantitatively graded across the gluteal, hamstring, quadriceps, and adductor compartments (0–3 per compartment; total edema score 0–12). Fatty infiltration was recorded dichotomously, and fascial and subcutaneous involvement, including contrast enhancement patterns, were assessed. Group comparisons were performed using non-parametric statistical tests.
Results: Distinct MRI signatures were observed across IIM subtypes. ASyS and DM demonstrated significantly greater edema, particularly in the quadriceps and gluteal compartments, compared to IMNM (quadriceps: ASyS vs. IMNM, 2.0 vs. 0.0, p=0.033; DM vs. IMNM, 2.0 vs. 0.0, p=0.023, gluteal: ASyS vs. IMNM, 3.5 vs. 1.0, p=0.003; DM vs. IMNM, 2.0 vs. 1.0, p=0.007). In contrast, IBM showed markedly higher rates of quadriceps fatty infiltration (90.9%) compared with IMNM (22.2%, p=0.008) and ASyS (41.7%, p=0.027). Fascial involvement further differentiated subtypes: hamstring fascial hyperintensity was most frequent in ASyS (75.0%), followed by DM (33.3%), whereas it was uncommon in IMNM (11.1%, p=0.008) and absent in IBM (0%, p<0.001). Furthermore, hamstring fascial enhancement was significantly more frequent in ASyS compared to DM (58.3% vs. 13.3%, p=0.04). This gradient pattern of fascial inflammation (ASyS > DM >> IMNM/IBM) served as an additional discriminator beyond muscle edema and fatty infiltration.
Conclusion: Thigh MRI demonstrates distinct, subtype-specific patterns of muscle and soft tissue involvement in IIMs. Severe anterior and gluteal muscle edema with hamstring fascial hyperintensity characterizes both ASyS and DM, while fascial enhancement further differentiates ASyS from DM. In contrast, prominent quadriceps fatty infiltration is a hallmark of IBM. Together, these radiological profiles represent clinically useful, non-invasive markers that may aid differential diagnosis in routine practice.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Inflammatory / Immune-mediated Myopathies including Inclusion Body Myositis (IBM)
PP01.98
Anti-HMGCR Myopathy in Latvia: Epidemiology and National Prevalence Data
Dr. Marija Roddate1,2, Dr. Ieva Glāzere1,2, Dr. Gundega Ķauķe1, Dr. Kristīne Ivanova3,2, Dr. Sintija Kalvāne-Brokāne3,4, Dr. Vladimirs Krutovs1,2, Dr. Inita Buliņa3,2,4, Assoc. Prof. Viktorija Ķēniņa1,2
1
Pauls Stradins Clinical University Hospital, Department of Neurology, Riga, Latvia.
2
Riga Stradins University, Riga, Latvia.
3
Pauls Stradins Clinical University Hospital, Department of Rheumatology, Riga, Latvia.
4
University of Latvia, Riga, Latvia
Background: Anti-HMGCR myopathy is a distinct subtype of immune-mediated necrotising myopathies (IMNM), characterized by proximal muscle weakness, elevated serum creatine phosphokinase (CK), and the presence of antibodies against 3-hydroxy-3-methylglutaryl–CoA reductase (HMGCR). Previously published data indicate that IMNM accounts for approximately 10% of all idiopathic inflammatory myopathies, with variable reports regarding the incidence and prevalence of specific subtypes. Although anti-HMGCR myopathy is a treatable condition, patients often exhibit poor recovery of muscle strength, particularly when diagnosis is delayed or treatment in the first two years from symptom onset is insufficient.
Methods: A retrospective cross-sectional analysis was conducted at Pauls Stradiņš Clinical University Hospital, the sole clinical center in Latvia with confirmed adult cases of IMNM. Adult patients (≥18 years) with confirmed anti-HMGCR myopathy were identified by reviewing medical records and laboratory data, with serological testing performed between 2019 and 2025. Demographic information, history of statin use, and CK levels were recorded and analyzed using descriptive statistics. Incidence and prevalence were calculated using data from the Latvian Central Statistical Bureau.
Results: A total of 14 adult patients with confirmed anti-HMGCR myopathy were identified. The cohort included 8 females (57.1%) and 6 males (42.9%), with a median age of symptom onset of 66 years (range: 3–81). Twelve of 14 patients (85.7%) had a history of statin use prior to symptom onset. All cases were confirmed between 2022 and 2026, with an annual incidence ranging from 0.56 to 2.78 cases per million population and a current point prevalence of 7.8 cases per million. All patients exhibited markedly elevated serum CK levels, with a median of 7,500 U/L (range: 2,858–20,627 U/L). Notably, 8 of 14 patients (57.1%) experienced a diagnostic delay of ≥1 year from symptom onset to initiation of treatment.
Conclusion: This study presents the first comprehensive national cohort of adult anti-HMGCR myopathy patients in Latvia. Consistent with previous reports, a slight female predominance and symptom onset primarily in the seventh decade were observed. Our cohort also demonstrated a higher prevalence of prior statin exposure compared to previous reports. The findings highlight diagnostic challenges, likely related to the limited availability of serological testing, exclusion of anti-HMGCR antibodies from standard autoimmune myopathy panels, and insufficient clinical awareness. Given the established correlation between early treatment initiation and improved clinical outcomes, these data underscore the need for heightened awareness of anti-HMGCR myopathy in patients with markedly elevated CK levels and a history of statin use, particularly among older adults.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Inflammatory / Immune-mediated Myopathies including Inclusion Body Myositis (IBM)
PP01.99
Evaluating ChatGPT’s advice and recommendations regarding exercise for people with Inclusion Body Myositis
Dr. Katherine Schutze1,2, Mr. Ruby Shehatha3, Ms. Kelly Beer3,4, Ms. Tanya Smith4, Mr. Matthew Bagg2,5, Ms. Althea Doverty3,4, Prof. Merrilee Needham2,3,4,6, Mr. Ian Cooper3,4
1
Sir Charles Gairdner Hospital, Nedlands, Australia.
2
Notre Dame University, Fremantle, Australia.
3
Perron Institute, Nedlands, Australia.
4
Murdoch University, Murdoch, Australia.
5
Neuroscience Research Australia, Rendwick, Australia.
6
Fiona Stanley Hospital, Murdoch, Australia
Background: Large language models (LLMs) have demonstrated safety and effectiveness in exercise prescription for the general population, though their advice is often less comprehensive than that of physiotherapists. Their potential value may be greater in rare disease populations, where access to specialist expertise is limited. Inclusion Body Myositis (IBM) is a rare inflammatory myopathy affecting older adults, characterised by a distinct pattern of muscle involvement and specific resistance exercise recommendations. The accuracy, safety, and appropriateness of LLM-generated exercise advice for individuals with IBM has not previously been evaluated.
Methods: This study evaluated responses generated by ChatGPT to six predefined questions addressing muscle involvement, resistance exercise prescription, and strategies to reduce falls in individuals with IBM. Twelve physiotherapists from around the world participated, with clinician experience in IBM management averaging 9.2 years. Reviewers rated each response using a five-point rubric ranging from “comprehensive and accurate” to “wrong and unsafe.”
Results: ChatGPT’s responses were judged to be safe and mostly accurate in five of the six assessed domains. Most responses demonstrated appropriate clinical reasoning and alignment with current IBM management principles, though they frequently lacked depth or specificity. Performance was strongest in general descriptions of muscle involvement and standard resistance exercise principles. Accuracy was reduced when prescribing exercise for individuals without antigravity movement, where advice was more limited and occasionally vague.
Conclusion: ChatGPT generally provided safe and mostly accurate advice related to exercise prescription and falls management in individuals with IBM, though comprehensiveness was variable. In some domains, performance was notably strong, highlighting the potential of LLMs as adjunct tools in rare disease exercise prescription where specialist access is limited. Future studies may consider whether LLMs can be used to generate individualised longitudinal regimens. Whilst LLMs may be useful in the design of a program but the responsibility for safe exercise prescription remains with the human prescriber.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Inflammatory / Immune-mediated Myopathies including Inclusion Body Myositis (IBM)
PP01.100
IBM Presenting as Early-Onset Facial Diplegia and Axial Weakness: Two Italian Cases and Literature Review
Dr. Barbara Risi1,2, Dr. Michele Giovanni Croce3, Dr. Silvia Nicolosi4, Dr. Nicola Carapella5, Dr. Daniele Velardo6, Prof. Anna Pichiecchio3,4, Prof. Giacomo Pietro Comi7,8, Prof. Stefania Corti6,7, Dr. Sabrina Ravaglia9, Prof. Massimiliano Filosto10,1
1
NeMO-Brescia Clinical Center for Neuromuscular Diseases, Brescia, Italy.
2
Department of Molecular and Translational Medicine, University of Brescia, Brescia, Italy.
3
Department of Brain and Behavioural Sciences, University of Pavia, Pavia, Italy.
4
Advanced Imaging And Artificial Intelligence Center, Neuroradiology Department, IRCCS Mondino Foundation, Pavia, Italy.
5
Department of Medical and Surgical Specialties, Radiological Sciences and Public Health, University of Brescia, ASST Spedali Civili of Brescia, Brescia, Italy.
6
Neuromuscular and Rare Disease Unit, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy.
7
Dino Ferrari Center, Department of Pathophysiology and Transplantation, University of Milan, Milan, Italy.
8
IRCCS Fondazione Ca' Granda Ospedale Maggiore Policlinico, Neurology Unit, Milan, Italy.
9
Neuromuscular Unit, IRCCS Fondazione Mondino, Pavia, Italy.
10
Department Of Clinical And Experimental Sciences, University of Brescia, Brescia, Italy
Background: Inclusion body myositis (IBM) is the most common acquired myopathy in older adults and is typically characterized at onset by slowly progressive, asymmetric weakness of the knee extensors and/or deep finger flexors. However, atypical presentations may occur, as recognized in the updated diagnostic criteria, leading to diagnostic delay or misdiagnosis. These include early involvement of facial muscles, dysphagia, axial or proximal weakness, and foot drop, which may mimic neuromuscular junction disorders, motor neuron disease, or idiopathic inflammatory myopathies. We describe two Italian female patients with an early-onset, long-lasting, atypical form of IBM.
Methods: We retrospectively analyzed two patients evaluated at two different Italian tertiary neuromuscular centers. Clinical data, laboratory results, and instrumental findings were collected. A review of the literature on atypical IBM was also performed.
Results: The clinical, laboratory, and instrumental characteristics, as well as treatment responses, of the two patients are summarized in Table 1. Neurological examination of patient 1 (P1; 53 years old) revealed slight difficulty walking on heels, inability to rise from a squatting position, severe weakness of the orbicularis oculi and oris muscles (MRC 3/5) and of the neck flexors (MRC 4−/5), inability to whistle, mild macroglossia, dysphagia requiring thickened liquids and a soft diet, dysarthria with a nasal voice, and mild left eyelid ptosis. Moderate weakness of finger extension/abduction (MRC 4−/5) and of thigh flexion and leg extension (MRC 4/5) was also noted [IBM-FRS: 34/40]. Neurological examination of patient 2 (P2; 51 years old) revealed difficulty rising from a squatting position, severe weakness of all facial muscles (MRC 3/5) and neck flexors (MRC 2/5), with dysphagia requiring a softened diet. Despite normal muscle strength, mild atrophy of the distal portion of the quadriceps femoris was evident on examination [IBM-FRS: 31/40].
Conclusion: Facial diplegia appears to be a rare presentation of IBM, accounting for approximately 4% of atypical cases (Alamr et al.). The few cases described in the literature almost exclusively involve female patients with symptom onset in their fifties or sixties. Unlike our patients, three previously reported cases required early non-invasive ventilation (Salam et al.). Data on clinical evolution are limited, except for one case in which finger flexor weakness developed within seven years of disease onset (Cummins et al.). Robust data are also lacking regarding MRI characteristics in patients with this atypical presentation—although in some reported cases a typical pattern of thigh involvement has been observed, as in both of our patients—as well as regarding the prevalence of anti-cN1A antibody positivity. The cases described here provide additional insights into symptom onset—which may occur at a younger age, as in patient P1—clinical presentation, and disease progression in IBM. Notably, neither patient developed the classical pattern of muscle weakness over an approximately ten-year disease course. In the presence of such atypical presentations, complementary investigations—particularly muscle MRI of the thigh and forearm—are essential to support the diagnosis.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Inflammatory / Immune-mediated Myopathies including Inclusion Body Myositis (IBM)
Images or Table (Optional)
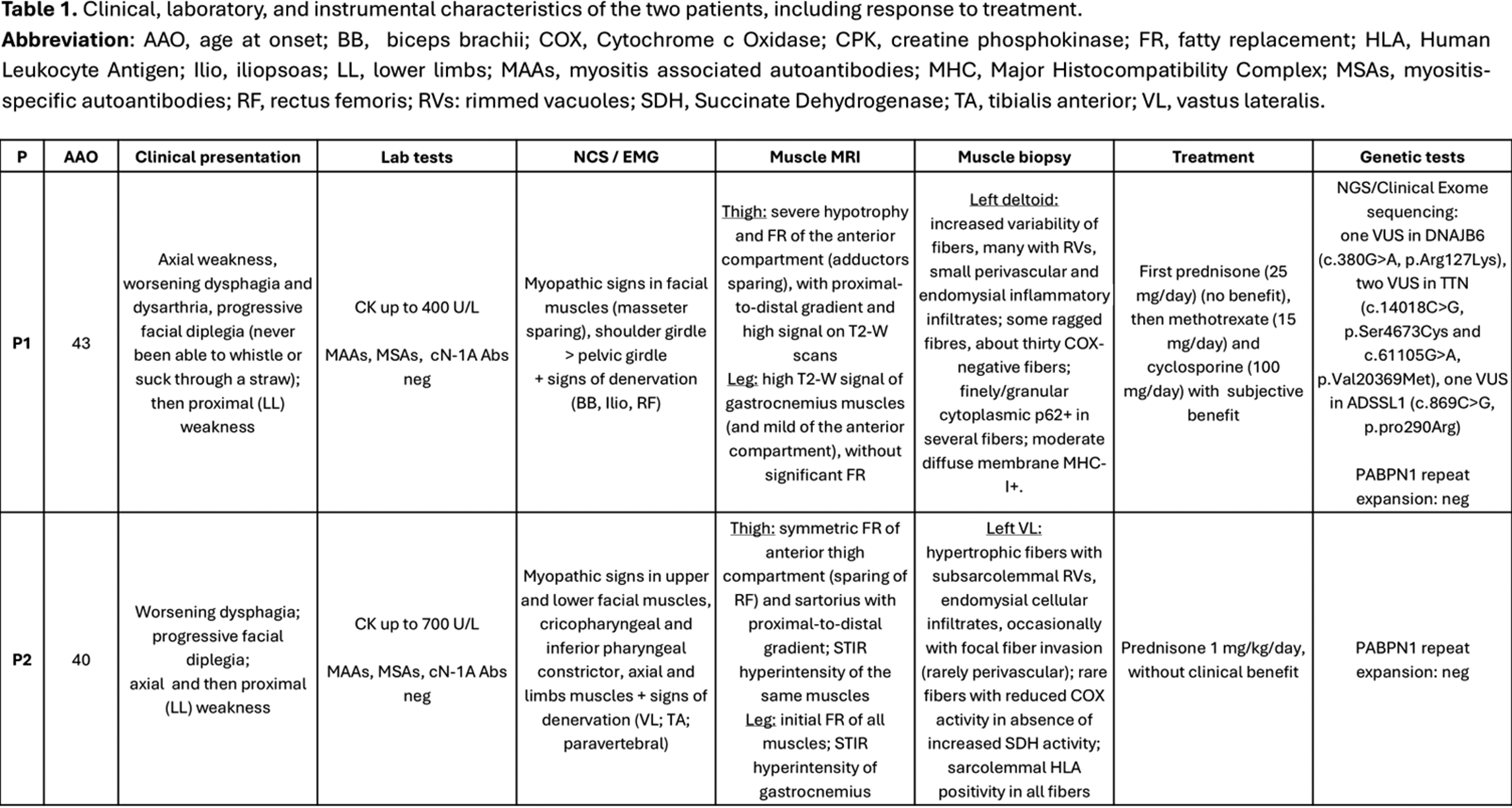
PP01.101
Systemic Anti-Neutrophil Cytoplasmic Antibody (ANCA) Vasculitis Presenting With Isolated Myositis Without Neuropathy
Dr. Nicole Rigler, Dr. Ashley Santilli
Mayo Clinic Department of Neurology, Rochester, United States
Background: ANCA-associated vasculitis (AAV) is a systemic small-vessel vasculitis leading to tissue inflammation, ischemia, and necrosis. Manifestations include constitutional symptoms such as fever, weight loss, and fatigue, though AAV may also affect specific organs including the kidneys, respiratory system, and skin. Peripheral nervous system involvement is known to occur, most classically presenting as multiple mononeuropathies (“mononeuritis multiplex”) with up to one third of patients developing neuropathy during their clinical course. Myopathy, particularly myopathy without any signs of co-occurring peripheral neuropathy, is rarer, and population-based epidemiological rates are not well established. We report the case of a patient with AAV presenting with myositis without peripheral nerve involvement.
Methods: Not applicable.
Results: A 78-year-old female presented with two months of progressive, primarily proximal, lower greater than upper limb weakness, myalgias, and fatigue. Notable medical comorbidities included hypothyroidism on appropriate hormone replacement and hyperlipidemia for which her rosuvastatin had been discontinued. The patient denied neuropathic pain, sensory loss, paresthesia, or skin rash, though endorsed weight loss with decreased appetite, dry mouth, and dysphagia. Initial neurologic exam was significant for moderate to severe axial and symmetric proximal greater than distal upper and lower limb weakness with preserved sensation and reflexes. At the time of initial neurology evaluation, the patient was unable to complete her activities of daily living secondary to weakness and required a walker for ambulation. Nerve conduction studies demonstrated reduced motor amplitudes with normal conduction velocities and preserved sensory responses without facilitation or decrement on repetitive nerve stimulation while needle electromyography revealed myopathic motor unit potentials without fibrillation potentials. Fluorodeoxyglucose positron emission tomography (FDG-PET) imaging showed diffuse, heterogenous skeletal muscle activity consistent with nonspecific myositis. Serum studies showed elevated creatinine kinase (CK) to 264 (reference range 26-192), elevated systemic inflammatory markers, and positive myeloperoxidase (MPO)/perinuclear ANCA antibodies while immune mediated necrotizing myopathy antibodies (HMGCR and SRP) and other myositis antibodies were negative. Triceps muscle biopsy was completed demonstrating necrotizing vasculitis with associated active inflammatory myopathy, denervation atrophy with reinnervation, and type 2B fiber atrophy. She was diagnosed with systemic MPO positive AAV with myositis and treated with high dose intravenous methylprednisolone and rituximab. Despite experiencing multiple complications including stress induced cardiomyopathy, acute kidney injury, and pulmonary embolism delaying her rehabilitation, she reported approximately 70% subjective improvement in her muscle weakness at follow up 3 months later. Neurologic exam at that time demonstrated mild to moderate weakness in the same distribution as prior, with preserved sensation and reflexes, and she was able to ambulate without assistance.
Conclusion: Isolated myositis without peripheral nerve involvement is a rare presentation of AAV. Early identification of this specific etiology of myopathy is important to reduce potential morbidity and mortality as patients may go on to develop additional manifestations of systemic vasculitis with other organ involvement. Additionally, patients with AAV tend to respond favorably to immunotherapy, therefore timely diagnosis and treatment initiation may reduce the risk of irreversible neurologic and systemic damage.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Inflammatory / Immune-mediated Myopathies including Inclusion Body Myositis (IBM)
PP01.102
Treatment Outcomes of Inflammatory Myopathies Stratified by Pathology and Autoantibodies: A Study of 127 Patients
Dr. Nobuyuki Eura, Dr. Yukako Nishimori, Ms. Ai Yamanaka, Dr. Tomo Shiota, Mr. Hiroaki Tanaka, Mr. Tomohito Ohashi, Mr. Hironori Shimizu, Ms. Minako Yamaoka, Mr. Nanami Yamada, Mr. Naohiko Iguchi, Mr. Akito Tanaka, Dr. Mayu Sugata, Dr. Hitoki Nanaura, Dr. Takao Kiriyama, Prof. Kazuma Sugie
Nara Medical University, Kashihara, Japan
Background: Idiopathic inflammatory myopathies (IIMs) are a heterogeneous group of autoimmune disorders primarily affecting skeletal muscle, but frequently involving extra-muscular organs such as the lung, heart, and skin. Advances in serological testing have identified a wide spectrum of myositis-specific autoantibodies (MSAs), which have improved diagnostic accuracy and enabled better disease classification. However, optimal treatment strategies and long-term outcomes remain poorly defined, particularly across different serological subtypes. Large-scale studies integrating muscle pathology, autoantibody profiles, and longitudinal clinical outcomes are still limited.
Methods: We retrospectively analyzed 127 consecutive patients with IIM who underwent muscle biopsy at our institution between 2009 and 2023. Inclusion required pathological confirmation of inflammatory myopathy. Patients with inclusion body myositis, as well as secondary myositis associated with collagen vascular diseases or immune checkpoint inhibitors, were excluded. Clinical features, laboratory findings, MSAs, organ involvement, treatment regimens, and outcomes were systematically reviewed.
Results: Dermatomyositis (DM) and immune-mediated necrotizing myopathy (IMNM) were the most prevalent subtypes (each 27.1%), followed by antisynthetase syndrome (ASyS, 14.4%) and polymyositis (PM, 2.7%). MSAs were detected in 97 patients (76.4%), supporting their diagnostic utility in routine practice. DM was mainly associated with anti-TIF1-γ, anti-MDA5, anti-Mi-2, and anti-NXP-2 antibodies, whereas IMNM was characterized by anti-SRP and anti-HMGCR antibodies. Patients positive for anti-HMGCR exhibited the highest serum creatine kinase levels. Interstitial lung disease (ILD) was frequently observed, particularly in patients with anti-MDA5 (87.5%), anti-Mi-2 (66.7%), and anti-SRP (50.0%) antibodies. Malignancies were identified across most antibody subtypes except anti-MDA5, anti-NXP-2, and anti-HMGCR, involving diverse organ systems; notably, 7 of 13 patients with traceable malignancies died within three years of IIM diagnosis. Patients with anti-AMA-M2 demonstrated a distinctive clinical phenotype, with a high prevalence of respiratory failure unrelated to ILD (40.0%) and cardiac complications, including arrhythmias and heart failure (88.9%). All patients received systemic glucocorticoids. Additional immunosuppressive therapies were administered in 80.0% of anti-MDA5, 60.0% of anti-HMGCR, and 52.6% of ASyS patients, most commonly tacrolimus, followed by methotrexate and azathioprine. Intravenous immunoglobulin was reserved for four severe cases. At three years, the mean prednisolone dose was 8.4 ± 5.5 mg/day, with only anti-SRP–positive patients achieving doses below 5 mg/day. Orthopedic complications, including fractures, occurred in 9.2% of patients.
Conclusion: This study underscores the marked clinical heterogeneity of IIM and highlights persistent treatment challenges across serological subtypes. Despite advances in diagnostic stratification using MSAs, disease control often requires prolonged immunosuppression, and outcomes remain suboptimal. These findings emphasize the urgent need for more effective, individualized therapeutic strategies tailored to immunological and pathological disease subsets.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Inflammatory / Immune-mediated Myopathies including Inclusion Body Myositis (IBM)
PP01.103
Multimodality Testing in Patients Suspected of Treatable Idiopathic Inflammatory Myopathy.A Prospective Diagnostic Accuracy Study (ADAPT-Study)
Miss Sanne Evers1, Miss Pinar Özkaynar1, Miss Renske Kamperman1, Dr. Filip Eftimov1, Prof. Eleonora Aronica2, Dr. Esther van Leeuwen3, Prof. Rohit Aggarwal4, Prof. Yves Allenbach5, Prof. Marianne de Visser1, Prof. Ivo van Schaik1, Prof. Patrick Bossuyt6, Dr. Camiel Verhamme1, Dr. Joost Raaphorst1, Dr. Anneke van der Kooi1
1
Amsterdam UMC, department of neurology, Amsterdam, Netherlands.
2
Amsterdam UMC, department of pathologydepartment of neurology, Amsterdam, Netherlands.
3
Amsterdam UMC, department of laboratory medicine, Amsterdam, Netherlands.
4
university of Pittsburgh medical centre, pittsburg, United States.
5
Pitié-Salpêtrière University Hospital, Sorbonne Université, Paris, France.
6
Amsterdam UMC, Department of Epidemiology & Data Science, Amsterdam, Netherlands
Background: Selecting a diagnostic strategy for Idiopathic Inflammatory Myopathy (IIM) is challenging due to the absence of strong evidence on the performance of potential tests. This prospective study evaluated multiple diagnostic strategies.
Methods: We conducted a prospective diagnostic accuracy study with an over-complete study design. Patients with clinical suspicion of IIM, requiring immunosuppressive treatment, underwent autoantibody testing (Ab), muscle magnetic resonance imaging (MRI), muscle ultrasound (US), electromyography (EMG), and a muscle biopsy. The reference diagnosis (IIM or IIM mimic) was assigned by an expert panel, based on the complete clinical information, including six months of follow-up. Diagnostic strategies, combining these modalities, were evaluated for sensitivity, specificity, positive/negative predictive values, and patient-reported burden.
Results: In total 100 patients were included, 67 IIM, 33 IIM mimics. Muscle MRI had the highest sensitivity 96% and the lowest specificity 42%. Muscle biopsy had the highest specificity of 97%, followed by Ab, with a specificity of 94%. Patients reported a higher burden from both EMG and muscle biopsy, compared to Ab, MRI, and US. The most optimal diagnostic strategy comprised Ab and MRI in all patients, followed by a biopsy in 34/100 patients. This yielded a sensitivity of 96% and a specificity of 94% and limited patient-reported burden. Replacing MRI by US or EMG resulted in a similar diagnostic accuracy.
Conclusion: In patients suspected of IIM, individual diagnostic tests had either limited sensitivity or specificity. Diagnostic strategies including autoantibody testing, combined with either MRI, EMG or US, and a muscle biopsy only in selected cases, were found to yield the optimal combination of highest sensitivity and specificity with a moderate burden, reported by patients.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Inflammatory / Immune-mediated Myopathies including Inclusion Body Myositis (IBM)
PP01.104
A Novel Cohort of Italian Patients With Late-Onset Pompe Disease and Extended IVS1-32-13T>G Screening
Dr. Daniele Velardo1, Dr. Alberto Lerario1, Dr. Delia Gagliardi2, Dr. Francesca Magri1, Dr. Nicola Molitierno1, Dr. Letizia Bertolasi1, Mrs Patrizia Ciscato1, Dr. Chiara Villella2, Dr. Roberto Del Bo3, Dr. Simona Zanotti1, Dr. Laura Napoli1, Dr. Michela Ripolone1, Dr. Claudia Alberti3, Dr. Mosè Parisi2, Dr. Monica Sciacco1, Prof. Stefania Corti1,3, Prof. Giacomo Pietro Comi2,3, Assoc. Prof. Dario Ronchi2,3
1
IRCCS Foundation Ca' Granda Ospedale Maggiore Policlinico, Neuromuscular and Rare Disease Unit, Milan, Italy.
2
IRCCS Foundation Ca' Granda Ospedale Maggiore Policlinico, Neurology Unit, Milan, Italy.
3
Dino Ferrari Center, Department of Pathophysiology and Transplantation, University of Milan, Milan, Italy
Background: Glycogenosis type II (GSDII, OMIM 232300), or Pompe disease (PD), is a rare autosomal recessive disorder due to biallelic pathogenic variants in GAA, resulting in the enzymatic deficiency of alpha-1,4-glucosidase. Based on whether the onset of symptoms is before or after 12 months of age, two main GSDII clinical forms are recognized, namely early onset (EOPD) and late onset (LOPD). Due to its wide clinical heterogeneity, LOPD is underrecognized, limiting the access of LOPD patients to available effective therapies.
Methods: We aim to report clinical, histological, biochemical and molecular features of a novel cohort of LOPD patients diagnosed in our Center. The cohort includes 10 males and 5 females aged between 5 and 74 years. Proximal myopathy was observed in 11 of 15 patients. 4 subjects exhibited asymptomatic hyperckemia: three of them have not developed clinical symptoms so far. Five patients currently require nocturnal Non-Invasive Ventilation (NIV): in two of them NIV was started within 2 years since the onset of myopathy. Cardiac involvement, usually described as rare in LOPD, was observed in 3 subjects. Normal serum CK levels were reported in 6 patients. Reduced alpha-1,4-glucosidase was demonstrated in 10 subjects evaluated in heterogenous tissues: lymphocytes, fibroblasts, muscle biopsy, DBS.
Results: A molecular diagnosis was established in all the patients with IVS1-32-13T>G variant identified in 14 subjects: in homozygosis in one asymptomatic patient and in compound heterozygosity with a second molecular defect in 13 patients. A single patient harboured two (non-IVS1) missense molecular defects. Muscle biopsy was available in 6 cases. Main histological findings included: vacuolated fibers, altered oxidative staining and increased intensity for Periodic Acid-Schiff and acid phosphatase staining. Histological analysis was unremarkable in a single patient reported symptomatic (myalgia) at the time of biopsy (28 years of age). Eleven patients are currently receiving enzyme replacement therapy (ERT): 6 with alglucosidase alfa biweekly and 5 with avalglucosidase alfa as replacement for alglucosidase alfa. Four patients are not currently undergoing ERT. Among treated patients, 4 tested positive for anti-GAA antibodies. To facilitate the identification of novel LOPD patients, we also performed the molecular screening (TaqMan genotyping technology) of the common IVS1-32-13T>G variant in a cohort of neuromuscular patients recruited at Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico in Milan (n = 1393). Inclusion criteria were: hyperCKemia and/or myalgia or weakness and/or myopathic non-inflammatory changes at muscle biopsy; and/or myopathic signs at EMG. 18 heterozygous carriers of the IVS1-32-13T>G variant were identified. Of these, 16 were retained for follow-up characterization (two were excluded due to an alternative diagnosis). A second GAA molecular defect was observed in 6 patients: five of them had been previously diagnosed for LOPD while one subject displayed the second molecular defect in cis with IVS1. The frequency of the IVS1-32-13T>G carriers is 0.93%, suggesting underestimated frequencies of carriers and IVS1 homozygous subjects in the general population.
Conclusion: This cohort demonstrates significant phenotypic variability with predominant IVS1 variant, underscoring the necessity of targeted molecular screening for early diagnosis and personalized therapeutic monitoring.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Metabolic Myopathies: Glycogen, Lipids and Mitochondrial Myopathies
PP01.105
Anti-HMGCR Immune-Mediated Necrotizing Myopathy: Characterization of a Monocenter Cohort and Development of a Therapeutic Algorithm
Dr. Nicola Molitierno1, Dr. Delia Gagliardi1, Dr. Mosè Parisi1, Dr. Alberto Lerario2, Dr. Patrizia Ciscato2, Dr. Letizia Bertolasi2, Dr. Simona Zanotti2, Dr. Laura Napoli2, Dr. Michela Ripolone2, Assoc. Prof. Claudia Maria Cinnante3, Prof. Stefania Corti4,5, Prof. Giacomo Pietro Comi1,5, Dr. Daniele Velardo2
1
IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Neurology Unit, Milan, Italy.
2
IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Neuromuscular and Rare Disease Unit, Milan, Italy.
3
IRCCS Istituto Auxologico Italiano, Section of Neuroradiology, Department of Radiology and Diagnostic Imaging, Milan, Italy.
4
IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Neuromuscular and Rare Disease Unit, Milan, Italy.
5
Dino Ferrari Center, Department of Pathophysiology and Transplantation, University of Milan, Milan, Italy
Background: Anti-HMGCR immune-mediated necrotizing myopathy is a rare autoimmune disease characterized by severe proximal muscle weakness, markedly elevated creatine kinase levels, and specific autoantibodies. Despite consensus recommendations, the lack of standardized criteria for therapeutic stratification represents a significant clinical challenge, particularly given the increasing recognition of phenotypic heterogeneity ranging from aggressive acute forms to chronic presentations. This study aimed to characterize a monocenter cohort and develop objective criteria for therapeutic decision-making and prognostic stratification.
Methods: We retrospectively analyzed 24 patients with anti-HMGCR myopathy diagnosed between 2014-2025. Clinical assessment included MMT-8 scoring, serum CK levels, anti-HMGCR antibody titers, and muscle MRI with semi-quantitative analysis of edema (STIR sequences) and fibro-adipose replacement (T1 sequences). Patients received either single induction therapy (high-dose corticosteroids alone) or multiple induction therapy (corticosteroids plus immunosuppressants and/or intravenous immunoglobulins). Patients were evaluated at 3, 6, and 12 months, with outcomes defined based on CK levels and muscle strength. Statistical analysis included univariate and multivariate logistic regression to identify predictors of therapeutic choices and treatment response.
Results: The cohort (median age 64.5 years, 58.3% female) showed significant demographic differences based on statin exposure: statin-exposed patients were older (66 vs 43 years) while statin-naive patients were predominantly young females. Through a retrospective evaluation of the therapeutic choices made in our cohort, we identified clinical, laboratory and imaging parameters that guided the selection between corticosteroid monotherapy and combined immunosuppressive induction therapy. Multivariate analysis of these parameters enabled development of a therapeutic algorithm integrating baseline CK levels, MMT-8 scores, and MRI edema scores (AUC=0.889, p=0.04), achieving 79.2% accuracy in predicting the need for multiple induction therapy. The application of this approach produced comparable results at 12 months between the two groups, (complete response: 50% vs 60%, p=0.697), suggesting an effective balancing between different disease severity through optimal therapeutic intensity for each patient. Prognostic analysis identified three significant baseline predictors of complete response: older age at onset, diagnostic delay (paradoxically favorable when adjusted for age and structural damage), and degree of fibro-adipose replacement on MRI (AUC=0.909, p=0.009). The unexpected finding regarding diagnostic delay supports the existence of distinct phenotypes with different biological activity and progression rates. During follow-up, persistent muscle edema on MRI and need for rescue immunoglobulin therapy predicted suboptimal response (AUC=0.898, p=0.009). Exploratory antibody kinetics analysis (n=8) revealed correlations between the percentage reduction in antibody titre and CPK values at 6 months (rho =-0.810, p=0.022) and 12 months (rho =-0.762, p=0.037). The rate of antibody reduction correlated significantly with complete clinical response (rho =0.771, p value=0.025).
Conclusion: This study supports a multiparametric approach for personalized management of anti-HMGCR myopathy, integrating easily measurable clinical, biochemical, and imaging parameters. The developed therapeutic algorithm enables objective stratification, while identification of baseline and follow-up prognostic factors facilitates early recognition of non-responders. These findings underscore the phenotypic heterogeneity of anti-HMGCR myopathy and highlight the importance of tailored therapeutic strategies in improving patient outcomes.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Inflammatory / Immune-mediated Myopathies including Inclusion Body Myositis (IBM)
PP01.106
Solid Facial and Palpebral Edema: Amyopathic Dermatomyositis, Morbihan Disease?
Dr. Zoltan Zsigmond Major1,2, Dr. Cristina Pamfil3, Dr. Elisabeta Candrea4, Mrs Kinga Andrea Major5, Dr. Ioana Crisan6
1
National Center for Spinal Disorders, Budapest, Hungary.
2
Municipal Clinical Hospital, Neurology Department, Cluj-Napoca, Romania.
3
Cluj County Clinical Emergency Hospital, Rheumatology Department, Cluj-Napoca, Romania.
4
Cluj County Clinical Emergency Hospital, Dermatology Department, Cluj-Napoca, Romania.
5
Nexus Medical Association, Cluj-Napoca, Romania.
6
Municipal Clinical Hospital, Allergology Department, Cluj-Napoca, Romania
Background: When a patient is seen with a solid, bilateral, persistent facial and eyelid edema accompanied with erythema, without definite muscle involvement, the is determined to think about rather rare diagnostic directions. Morbihan disease might be an option, at least when only signs are considered, but other diseases might also be unusual, but feasible options. One of these is amyopathic dermatomyositis.
Methods: We report a case of chronic persistent solid edema of the eyelids and face, developed gradually since 2018, with a weak response to the various applied treatments. The patient has mild associated morbidities: diabetes mellitus type II, controlled by diet to this point, with a mild axonal sensory polyneuropathy, dorsal and lumbar polysegmental discopathy and was operated for gallbladder lytiasis.
Besides swelling and erythema of the eyelids and face, the patient reports time-to-time associates paraesthesia of the lower limbs and muscular pain, without significant electrophysiological changes, and no elevated CK levels.
Imagery reveals no particular feature for atrophy or swelling of scheletal muscle, the only pathological change is related to the swelling of the soft tissues of the upper face, although local tissue biopsy reveals lymphocitic infiltrate resembling features of dermatomyositis rather than other pathology. Muscle biopsy was not characteristic. Laboratory reveals mildly increased circulating immune complexes, hypercholesterolemia, and transient positivity in the myositis profile for dermatomyositis, with a mediocre response to the common immune-suppressive drugs, but also with only a partial response to isotretinoin treatment.
Results: At this point, taking into account the responses also to the applied treatments, we consider that the above presented features are at least partially supporting the diagnosis of amyopathic dermatomyositis, although Morbihan disease remains a strong second option, and other diseases are also there as possibilities, several evaluations are still undergoing.
Conclusion: The case proves to be a diagnostic challenge, to this point without a definite diagnosis, an ongoing struggle towards a final diagnosis and proper treatment options. We are further investigating the case, along with concomitant, diverse treatment options
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Inflammatory / Immune-mediated Myopathies including Inclusion Body Myositis (IBM)
PP01.107
Interplay Infection, Locking Syndrome in Lgmd Tnpo3-Related: Sporadic Slovakian Patient With Novel Mutation
Prof. Corrado Angelini1, Prof. Giovanna Cenacchi2, Dr. Paola Tonin3, Dr. Alicia Rodriguez4, Asst. Prof. Gaetano Vattemi3
1
University of Padova, Padova, Italy.
2
University of Bologna, Bologna, Italy.
3
Hospital of Verona, Verona, Italy.
4
University of Deusto, Bilbao, Spain
Background: LGMD subtypes are highly variable in terms of age of onset, speed of disease progression, and overall severity; at a histopathological level, their common feature is progressive muscle degeneration with connective tissue substitution, CK is elevated and degenerative changes are found in muscle (biopsy,MRI) although they do not share a common pathological mechanism .The incidence for all forms is 1:100,000 and LGMD are divided into two major subgroups: autosomal dominant forms (LGMD type D) and autosomal recessive (LGMD type R).We report here a novel Slovakian patient with all these clinical features and entertain anhypthesis regarding the “locking “ symptoms that LGMD1F/D2 patients experience.
Methods: An open quadriceps muscle biopsy was done and histochemistry and electron microscopy analized. Needle EMG in muscles m. vastus lateralis l.dx., m. rectus femoris .sin., m. deltoideus .sin. shows findings of a chronic neurogenic lesion. m. biceps brachii l.dx. with a normal finding. MEP examionation In the upper and lower limbs, with bilateral cortical stimulation, total and central latencies are normal, findings are bilaterally symmetrical – so far, no convincing signs of conduction disorder in the corticospinal tract to the lower limbs. WHOLE BODY MRI: The upper limbs and legs remained outside the measured volume. Muscle structures appear to be adequately symmetrically configured, without focal atrophies, without apparent focal signal intensity changes in terms of edema or fatty transformation. Subcutaneous adipose tissue is also adequately configured, symmetrical, and without focal changes.
Results: A male patient, 31 years old, living in Slovakia, was found to be affected by a sporadic form of LGMD-TNPO3 related: a pathogenic stop codon variant c.2686 in a heterozygous state in the TNPO3 gene and a c23335 heterozygous mutation in the gene WASHC5 associated to spastic paraplegia type 8. Clinically, he developed in the third decade a weakness compatible with LGMD syndrome; he had a Gowers’ sign and difficulty walking. A detailed genetic analysis of the family showed that his parents did not carry the mutation and appeared healthy. An important clinical feature was that the patient had been frequently suffering since as a child had a lot of infections, and such an unfortunate feature persisted in adulthood. He often had sinusitis, a sore throat, and frequent headaches. His immune system is weak, but specific immune tests showed no specific disorders. He was also affected by Lyme disease and had to be treated for two years with antibiotics to eradicate the Borrelia. This treatment resulted in several gastrointestinal symptoms: for 8 years, he had daily diarrhea and/or constipation. Muscle biopsy showed COX-negative fibers on electron micoscopy abnormal nuclei (figure), fragmented cristae in mitochondria, and signs of autophagy.
Conclusion: The combination of biopsy and clinical data supports the hypothesis that LGMD-D2 is caused by several possible mechanisms likely involving nuclear transport and myofibrillar network; the interplay with infection appears to be because TNPO3 directly participates in nuclear import and infection of HIV-1.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Limb Girdle Muscular Dystrophies
Images or Table (Optional)

PP01.108
Interim Analysis from Ongoing Phase 3 FORTIFY Study of BBP-418 for LGMD2I/R9 Meets Efficacy Endpoints
Dr. Katherine Mathews1, Dr. Tahseen Mozaffar2, Dr. Volker Straub3, Dr. Robert Henderson4, Dr. Douglas Sproule5, Dr. Ada Lee5, Ms. Amy Rainey5, Dr. David Paul5, Ms. Tricia Blankenbiller5, Dr. Uma Sinha5, Ms. Thulashitha Rajasingham5, Dr. John Vissing6
1
University of Iowa, Iowa City, United States.
2
University of California Irvine, Irvine, United States.
3
Newcastle University, Newcastle upon Tyne, United Kingdom.
4
University of Queensland, Brisbane, Australia.
5
ML Bio Solutions Inc., a BridgeBio company, Palo Alto, United States.
6
University of Copenhagen, Copenhagen, Denmark
Background: Limb-girdle muscular dystrophy type 2I/R9 (LGMD2I/R9) is an autosomal recessive disorder caused by pathogenic variants in the FKRP gene that impair alpha-dystroglycan (αDG) glycosylation, leading to progressive muscle weakness and cardiopulmonary decline. No approved therapies currently exist. BBP-418 is an oral, small molecule designed to restore αDG glycosylation and stabilize muscle integrity.
Methods: FORTIFY (NCT05775848) is an ongoing randomized, double-blind, placebo-controlled, Phase 3 study evaluating the efficacy and safety of BBP-418 in individuals with LGMD2I/R9. The prespecified 1-year interim analysis assessed biochemical, functional, and safety endpoints. The primary endpoint was change from baseline in glycosylated αDG at 3 months, with key secondary endpoints including serum creatine kinase (CK), 100-meter timed test (100MTT), and forced vital capacity (FVC) at 12 months.
Results: At the interim analysis, BBP-418 treatment showed a 1.8-fold increase in glycosylated αDG from baseline at 3 months (p<0.0001), which was sustained through 12 months (p<0.0001) versus placebo. Serum CK decreased by 82% on average from baseline in the BBP-418 dosed participants at 12 months (p<0.0001 vs placebo). Statistically significant and clinically meaningful improvements were observed in 100MTT velocity (+0.14 m/s from baseline in BBP-treated participants; difference of 0.27 m/s in BBP-treated participants vs. placebo, p<0.0001) and FVC (+3% predicted from baseline in BBP-treated participants; difference of 5% predicted in BBP-treated participants vs. placebo, p=0.0071). BBP-418 was well tolerated with no new safety findings. Additional data from the interim analysis will be presented at the time of the meeting, with a focus on the Patient-Reported Outcomes Measurement Information System (PROMIS) tool.
Conclusion: The interim analysis results from the ongoing 3-year FORTIFY Phase 3 trial demonstrated that BBP-418 provides significant improvements in αDG glycosylation, decreases in CK levels and functional benefits with a favorable safety profile. These data support BBP-418 as the potential first disease-modifying therapy for LGMD2I/R9.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Limb Girdle Muscular Dystrophies
PP01.109
Childhood Limb-Girdle Muscular Dystrophies: Phenotype-to-Genotype Diagnostic Journey: Single-Center Experience
Assoc. Prof. Gulten Ozturk1, Assoc. Prof. Elif Acar Arslan1, Prof. Olcay Unver1, Dr. Bilgihan Bikmazer1, Dr. Hakki Akbeyaz1, Prof. Bilge Bilgen Geckinli2, Dr. Zeynep Yilmaz1, Dr. Kiymet Kecelioglu Binnetoglu1, Dr. Seyme Iyisenyurek1, Prof. Dilsad Turkdogan1
1
Marmara University Department of Pediatric Neurology, Istanbul, Turkey.
2
Marmara University Department of Medical Genetics, Istanbul, Turkey
Background: Limb-girdle muscular dystrophies (LGMDs) are genetically heterogeneous neuromuscular disorders characterized by progressive proximal weakness and marked clinical variability. Phenotypic overlap and variable age at onset often lead to delayed genetic confirmation. We aimed to describe the clinical features and diagnostic journey of genetically confirmed pediatric LGMD patients followed at our center
Methods: Medical records of patients with LGMD were retrospectively reviewed. Only genetically confirmed cases were included.
Results: Sixteen patients were included (9 female, 56%). The cohort’s average age was 13.4±5.5 years (median 12.5; range 6–23). Age at presentation was 50.3±41.6 months (median 60; range 0–132), and age at genetic diagnosis was 8.93±4.03 years (median 5.5; range 2.5–15). The diagnostic interval averaged 4.75±3.02 years (median 4; range 1–13). The most common initial finding was asymptomatic hyperCKemia, followed by hypotonia, neuromotor developmental delay, and progressive proximal weakness. Patients with normal creatine kinase (CK) levels experienced longer diagnostic delays. Muscle biopsy and electromyography supported myopathy but were not informative for specific genetic etiology. Pathogenic variants were identified in CAPN3 (n=3), SGCA (n=4), SGCB (n=2), ANO5 (n=2), POMT1 (n=1), HMGCR (n=1), TTN (n=2), and JAG2 (n=1). A muscular dystrophy gene panel was most commonly used (68.7%), followed by whole-exome sequencing and single-gene testing. Prematurity-related complications and normal CK were major confounding factors contributing to diagnostic delay.
Conclusion: Early recognition of LGMD in children with proximal weakness and/or hyperCKemia may reduce diagnostic delay and enable timely genetic testing.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Limb Girdle Muscular Dystrophies
PP01.110
Severe De Novo Desminopathy iwith Multisystem Involvement: A Case Report and Review of the Literature
Assoc. Prof. Marianthi Arnaoutoglou1, Dr. Korina Kaffe2, Dr. Vasiliki Poulidou2, Dr. Maria Moschou1, Dr. Maria Gavrilaki2, Assoc. Prof. Aggeliki Cheva3, Mrs Georgis Pepe4, Prof. Vasilios Kimiskidis2
1
Department of Clinical Neurophysiology , AHEPA University Hospital Aristotle University of Thessaloniki, Thessaloniki, Greece.
2
1st Department of Neurology , AHEPA University Hospital Aristotle University of Thessaloniki, Thessaloniki, Greece.
3
Department of Pathology, Aristotle University of Thessaloniki, Thessaloniki, Greece.
4
Genekor, Athens, Greece
Background: Desminopathy is a rare subtype of myofibrillar myopathy caused by pathogenic variants in the DES gene encoding desmin, a muscle-specific intermediate filament critical for cytoskeletal integrity. While desminopathies traditionally present in adulthood with skeletal and cardiac muscle symptoms, early-onset cases with multisystemic features are increasingly reported. Gastrointestinal and respiratory involvement are less common but may represent underrecognized features. Diagnosis is challenging, especially in sporadic cases without family history, making molecular diagnostics such as WES essential tools.
Methods: This is a case report presentation of a 17-year-old female presented with progressive generalized muscle weakness, low BMI, and chronic gastrointestinal symptoms, including persistent diarrhea and loss of appetite. Symptoms began subtly in early adolescence, with increasing fatigue and reduced exercise tolerance. Over time, she developed significant respiratory weakness, manifesting as dyspnoea on exertion and poor nocturnal ventilation. She experienced an acute clinical deterioration following a self-limited viral illness, with marked decline in mobility and respiratory capacity. There was no relevant family history of neuromuscular or cardiac disorders. Growth and neurodevelopment had been normal in early childhood. An extensive aboratory investigations was conducted including CK levels, Muscle biops and WES. Clinical investigation included extensive evaluation of cardiovascular function ( including cardiac MRI), evaluation of respiratory function (spirometry and sleep study) and bowel and stomach endoscopy. The differential diagnosis included: LGMD , Mitochondrial myopathies Congenital myopathies , Chronic inflammatory myopathies, GI dysmotility due to autoimmune or metabolic causes
Results: Definitive diagnosis was made by WES, which identified a heterozygous de novo pathogenic variant in DES: NM_001927.4:c.1217G>C, p.(Arg406Pro). Parental testing confirmed the variant was not inherited. Biopsy confirmed the severe and catastrophic myopathy. The patient’s condition stabilized after initial deterioration but remained significantly limited in mobility and respiratory endurance. She continued nocturnal ventilation and received ongoing multidisciplinary care. No cardiac involvement was evident at the time but will be monitored periodically due to the known cardiomyopathic risk in desminopathies.
Conclusion: Desminopathies are rare neuromuscular disorders caused by mutations in DES, which encodes desmin, an intermediate filament protein involved in muscle fiber integrity and organelle positioning. While the classic presentation involves limb-girdle weakness and cardiomyopathy in adulthood, this case highlights the early-onset, multisystem variant of desminopathy with predominant skeletal, GI, and respiratory involvement — in the absence of cardiac symptoms or family history. The patient’s c.1217G>C (p.Arg406Pro) previously reported in association with early and severe myopathy phenotypes. The de novo nature of the mutation underscores the importance of considering DES mutations even in patients without familial patterns. Respiratory muscle weakness is well-documented in desminopathy but often underrecognized. The exacerbation following viral illness further supports observations that systemic infections can unmask or accelerate muscle decompensation in genetically susceptible individuals. Gastrointestinal involvement, though less common, has been reported and may result from smooth muscle or autonomic dysfunction. Our patient’s diarrhea and appetite loss were not explained by structural abnormalities on imaging, suggesting a functional component linked to the underlying myopathy. This case adds to the growing literature on desminopathy as a phenotypically variable disorder with non-classical presentations, especially in children and adolescents.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Limb Girdle Muscular Dystrophies
PP01.111
Differential Expression of Dysferlin in Rat Fast and Slow Skeletal Muscle
Dr. Marija Meznaric1, Prof. Corrado Angelini2
1
University of Llubjana, Llubjana, Slovenia.
2
University of Padova, Padova, Italy
Background: A common form of limb girdle muscular dystrophy is dysferlinopathy, presenting as LGMDR R2 /2 B or distal Miyoshi myopathy, which shows a different clinical course. They are caused by mutations in the DYSF gene encoding the protein dysferlin, a transmembrane sarcolemmal and T-tubule protein that has a role in the repair of sarcolemma. To the best of our knowledge, dysferlin expression in different skeletal muscles has not been studied so far. We investigated by western blot dysferlin myosin ratio in human skeletal muscles, biceps brachii and tibialis anterior, and rat muscles, the slow soleus, and fast extensor digitorum longus ( EDL).
Methods: Slow and fast muscles in the man and the rat were examined for dysferlin by Western Blot. The ratio was higher in human neuromuscular controls in the biceps brachii versus the tibialis. Histochemistry was performed for Myosin heavy chain isoforms, SDH, and NADH-TR. Results show in humans a trend towards a higher amount of dysferlin in fast skeletal muscle compared to slow, which might be relevant for the different course observed in LGMDR 2 2 B and Miyoshi myopathy
Results: The ratio was higher in human neuromuscular controls in the biceps brachii than in the tibialis anterior. In rat muscles, the ratio was significantly higher in SOL than in EDL. A positive correlation was found between the area proportion of type 1 fibres and dysferlin expression, and a negative correlation for type 2, subtypes of type 2, and hybrid fibres was found. Differential dysferlin expression in human and rat fast and slow skeletal muscles is of interest and might be relevant to explain the clinical course of LGMD-R2. Our results show in humans a trend towards a higher amount of dysferlin in fast skeletal muscle compared to slow, which might be relevant for the different course observed in LGMDR R2 /2 B and Miyoshi myopathy
Conclusion: Our western blot results have discovered a different ratio of dysferlin and myosin in slow and fast muscles. This might imply a metabolic role of dysferlin,(Figure) besides the repair, on type 1 fibres that are rich in mitochondria and lipids, and in type 2 fibres with abundant T tubules
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Limb Girdle Muscular Dystrophies
Images or Table (Optional)
PP01.112
Reclassifying ISPD VUS in Siblings: The Power of Deep Phenotyping in Neuromuscular Diagnosis
Dr. Thaina Louise Rodrigues, Dr. Enzo Pellacani, Dr. Bruno Graziosi, Dr. Alessandra Tolentino, Dr. Ana Marina Silva, Dr. Roseli Corazzini, Dr. Alzira Carvalho
Centro Universitário FMABC, Santo André, Brazil
Background: Limb-girdle muscular dystrophies (LGMD) constitute a heterogeneous group of genetic myopathies characterized by progressive proximal muscle weakness. Among these, dystroglycanopathies result from defects in the glycosylation of alpha-dystroglycan 1,2. Variants in the ISPD gene acts in isoprenoid synthesis and plays a role in alpha-DG and are associated with milder phenotypes resulting from partial or complete impairment of alpha-DG, with a broad clinical spectrum ranging from severe congenital forms to late-onset limb-girdle presentations 3,4.
Methods: We describe the case of two Brazilian siblings born to non-consanguineous parents. Patient 1, male, developed symptoms at 15 months of age, presenting with frequent falls, proximal muscle weakness, toe-walking (equine gait), winged scapula, and persistent CPK elevation (1,800–27,000 U/L). Patient 2, female, at 9 years of age, presented an oligosymptomatic phenotype, with mild proximal weakness, slight scoliosis, winged scapula, and increased CPK (2,000–5,000 U/L), muscle biopsy showed a dystrophic pattern, positive immunohistochemical for dystroglycan, and thigh MRI revealed muscle atrophy and fatty replacement. Neither patient showed cardiac or respiratory involvement. Initial genetic investigation was negative for MLPA for Duchenne Muscular Dystrophy in Patient 1 and inconclusive in the whole exome analysis via next-generation sequencing (NGS), as well as in the LGMD molecular panel for Patient 2. An expanded genetic panel for neuromuscular diseases identified two compound heterozygous variants in the ISPD gene (7p21): c.636A>C (p.Glu212Asp) and an exon 10 deletion, shared by both siblings. After parents segregation was observed, each parent carried one of the variants. These were at first classified as variants of uncertain significance (VUS) and later reclassified as likely-pathogenic and pathogenic, respectively.
Results: This case highlights the phenotypic variability and intrafamilial heterogeneity associated with the ISPD gene. The heterozygous variants initially classified as VUS (c.636A>C and exon 10 deletion), later reclassified as likely-pathogenic and pathogenic, respectively, were associated for the first time with an LGMDR20 phenotype. These findings underscore the importance of accurate clinical phenotyping and systematic genetic reanalysis for establishing the correct diagnosis in hereditary myopathies.
Conclusion: This report aims to present new variants associated with LGMD.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Limb Girdle Muscular Dystrophies
PP01.113
MICU1-related Mitochondrial Myopathy Mimicking Limb-Girdle Muscular Dystrophy
Dr. Audrey Cambier1, Dr. Beatrice Labella2, Dr. Paula Panos-Basterra1, Dr. Marion Masingue1, Dr. Anthony Behin1, Dr. Juliette Nectoux3,4, Dr. Camille Verebi3,4, Dr. Pauline Gaignard5, Dr. Corinne Metay6, Dr. Tanya Stojkovic1
1
Centre de Référence des maladies Neuromusculaires Nord/Est/Ile-de-France, Institut de Myologie, Hôpital Pitié-Salpêtrière, APHP, Paris, France.
2
Functional Unit of Neuromuscular Pathology, Neuropathology Department, Pitié-Salpêtrière Hospital, APHP, Paris, France.
3
Service de Médecine Génomique Des Maladies de Système Et d'Organe, Hôpital Cochin, APHP. Centre Université de Paris Cité, Paris, France.
4
Laboratoire SeqOIA, Paris, France.
5
Service de Biochimie, UF Maladies héréditaires du métabolisme, CRMR Carammel, Filnemus, CHU Bicêtre AP-HP, Paris, France.
6
Molecular and Cellular Cardiogenetics and Myogenetics UF, Chromosomic and Molecular Genetic Center and INSERM UMRS 974, Institute of Myology, Pitié-Salpêtrière Hospital, APHP, Paris, France
Background: Biallelic pathogenic variants in the MICU1 gene, a key regulator of mitochondrial calcium uptake, cause a rare mitochondrial myopathy that often mimics limb-girdle muscular dystrophy (LGMD), resulting in diagnostic delay.
Methods: We analysed five adult patients from three unrelated consanguineous families carrying biallelic MICU1 gene variants. Clinical, biochemical, electrophysiological, imaging, histopathological and whole-genome sequencing data were collected.
Results: Five patients (two males) of Tunisian, Pakistani and Moroccan origin were included. Mean age at onset was 81 months (range 10–300), with a mean age at diagnosis of 34 years, corresponding to a mean diagnostic delay of 27 years. Weakness was predominantly proximal with a limb-girdle distribution, except for one scapuloperoneal phenotype. Scapular winging and calf hypertrophy were present in three patients each. At last follow-up, three patients were ambulant, one required unilateral support and one was wheelchair dependent. Creatine kinase levels were markedly elevated in all patients (mean peak 5,257 U/L), and muscle biopsies showed dystrophic or myopathic changes compatible with LGMD. Extra-muscular features included cognitive impairment, short stature, ganglionopathy and thyroid dysfunction. Whole-genome sequencing identified pathogenic biallelic variants in the MICU1 gene in all patients.
Conclusion: Variants in the MICU1 gene cause an LGMD-like myopathy with prominent extra-muscular features suggestive of mitochondrial disease. Systematic inclusion of the MICU1 gene in LGMD diagnostic panels may reduce diagnostic delay and improve disease classification.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Metabolic Myopathies: Glycogen, Lipids and Mitochondrial Myopathies
PP01.114
Real-world Outcomes of Very Early-treated Infantile-onset Pompe Disease in Taiwan After Switching to Avalglucosidase Alfa
Dr. Chia-Feng Yang1,2, Dr. Dau-Ming Niu3,4
1
Department of Pediatrics, Taipei Veterans General Hospital, Taipei, Taiwan.
2
School of Medicine, College of Medicine, National Yang Ming Chiao Tung University, Taipei, Taiwan.
3
Genetic Consultant Center Rare Disease Medical Research Center, Taipei Veterans General Hospital, Taipei, Taiwan.
4
Institute of Clinical medicine, National Yang Ming Chiao Tung University, Taipei, Taiwan
Background: Starting enzyme replacement therapy (ERT) before irreversible muscular damage occurs is important in infantile-onset Pompe disease (IOPD). This long-term follow-up study demonstrates our rapid diagnostic and treatment strategies for IOPD and showed better outcomes comparing to other study groups. Further, we presented our 24-month real-world experience switching all our patients to Avalglucosidase alfa and analyzed the progresses.
Methods: In this long-term follow-up study, we analyzed the outcomes of very early ERT IOPD patients who were picked up from Taiwan nationwide newborn screening programme. Out of 1,628,539 infants screened between 1 January 2010 and 31 December 2025, 43 newborns had confirmed IOPD in our group. The average age at ERT initiation was 10.06±4.17 days. The average follow-up duration was 8.18±3.14 years. We compared the long-term treatment outcomes of our patients with those of other research groups and also presented our 24-month, real-world experience of switching ERT to Avalglucosidase alfa for our 43 IOPD patients.
Results: Compared with patients in other studies, our patients had better outcomes in all aspects. The switch to new generational treatment, Avalglucosidase alfa, led to further improvements in respiratory conditions, activity endurance, muscle strength, and the levels of disease-specific biomarkers. This study highlights the benefits of very early treatment, derived from a newborn screening program combined with rapid diagnostic strategies, and furthermore, the switch to new generation ERT, which together offer the potential for substantially improved long-term outcomes in IOPD.
Conclusion: Very early initiation of enzyme replacement therapy enabled by nationwide newborn screening and rapid diagnostic strategies is associated with improved long-term outcomes in infantile-onset Pompe disease. The additional and sustained benefits observed after switching to next-generation ERT with avalglucosidase alfa further support this integrated treatment approach for long-term management of IOPD.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Metabolic Myopathies: Glycogen, Lipids and Mitochondrial Myopathies
PP01.115
Characteristics of Patients with Late-Onset Pompe Disease: Insights from the Romanian Cohort
Dr. Diana Chitimus1,2,3, Assoc. Prof. Szatmari Szabolcs4,5, Assoc. Prof. Laura Florea6, Dr. Marian Feticu7, Prof. Pascal Laforet8,9,2, Prof. Carmen Adella Sirbu3,10
1
Doctoral School, Faculty of Medicine, ‘’Carol Davila’’ University of Medicine and Pharmacy, 050474, Bucharest, Romania.
2
INSERM U 1179, Handicap Neuromusculaire, UVSQ Paris-Saclay, Paris, France.
3
Department of Neurology, “Carol Davila” Central Military Emergency University Hospital, 1010242, Bucharest, Romania.
4
2nd Clinic of Neurology, Târgu Mures County Emergency Clinical Hospital, 540136, Targu Mures, Romania.
5
George Emil Palade University of Medicine, Pharmacy, Science and Technology of Târgu Mures, 540142, Targu Mures, Romania.
6
. Department of Nephrology-Internal Medicine, Faculty of Medicine, “Grigore T. Popa” University of Medicine and Pharmacy, Iasi, Romania.
7
Department of Neurology, “Regina Maria” Military Emergency University Hospital, Brasov, Romania.
8
Neurology Department, Raymond Poincaré University Hospital, Garches, France.
9
Nord-Est-Ile-de-France Neuromuscular Reference Center, FHU PHENIX, Paris, France.
10
Department 6-Clinical neurosciences Carol Davila” University of Medicine and Pharmacy, 050474, Bucharest, Romania
Background: Late-Onset Pompe disease (LOPD) is a clinically heterogeneous neuromuscular disorder caused by the deficient activity of acid alpha-glucosidase (GAA) enzyme due to mutations in the GAA gene. Phenotypic and genotypic data from Eastern Europe remain scarce in the published literature, hence the aim of this study is to characterize the population of LOPD patients in Romania.
Methods: This is a retrospective, multi-center cohort study that included all the genetically confirmed LOPD patients in Romania. Data was extracted from three regional centers, and included demographic, clinical, functional, and molecular characteristics. Results are expressed as median and interquartile (IQR) range.
Results: We identified nine patients diagnosed with Pompe disease, in seven different families. The median age of diagnosis was 35 years-old, while the median age of symptom onset was 28. The common mutation c.-32-13T>G was identified in six patients, two siblings had the c.569G>A mutation, while one patient was heterozygous for the c.2297A>C mutation. At the time of inclusion, three patients needed walking assistance. The median of the 6-minutes-walkin-test(6MWT) was 375 meters [286.75 m-480 m], while the median FVC(%) was 81% [66.4% - 86.9%]. None of the patients are under ventilatory support. The creatine kinase (CK) median was 579 [2091-1542.5]. Eight patients received enzyme-replacing treatment (ERT) with alglucosidase alfa, and one had a positive anti-enzyme antibody titer, however without reported adverse reactions. Regarding the systemic manifestations in our cohort, two patients had history of recurrent ischemic strokes, and one patient has aortic ectasia. No history of dysphagia or macroglossia was noted in our cohort.
Conclusion: To our knowledge, this is the first study investigating the Romanian LOPD population. Our findings suggest that while the most prevalent genotype is consistent with previously reported studies in Caucasian population, the phenotypic variability remains important. In most patients the respiratory function was largely preserved, however with an important motor impairment. Despite the increasing awareness of neuromuscular disorders, LOPD remains largely underdiagnosed in the Romanian population, however with an increasing number of centers committed to monitor and treat these patients.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Metabolic Myopathies: Glycogen, Lipids and Mitochondrial Myopathies
PP01.116
Bridging the Diagnostic Gap in POLG Disorders: Phenotypic Variability and Clinical Challenges in Predominantly Brazilian-Sample
Dr. Alessandra Lima Nogueira Tolentino1, Dr. Ana Marina Silva1, Dr. Gisele Espíndola2, Dr. Euldes Júnior3, Dr. Pablo Feitoza4, Dr. Thiago Tonholo5, Dr. Thiago Oliveira6, Dr. Stephanie Delstanche7, Dr. Manon Hustinx7, Dr. Roseli Corazzini1, Prof. José Pedroso5, Prof. Alzira Carvalho1, Prof. Enzo Stinghel Pellacani8
1
Faculdade de Medicina do ABC, São Paulo, Brazil.
2
Hospital Universitário Polydoro Ernani de São Thiago - UFSC, Santa Catarina, Brazil.
3
Ambulatório de Doenças Raras, Minas Gerais, Brazil.
4
Universidade Federal do Amazonas, Amazonas, Brazil.
5
Universidade Federal de São Paulo, São Paulo, Brazil.
6
Universidade Federal de Pernambuco, Pernambuco, Brazil.
7
Centre Hospitalier Régional de la Citadelle, Liège, Belgium.
8
ABC Medical School, São Paulo, Brazil
Background
Introduction: Pathogenic variants in POLG, encoding the catalytic subunit of mitochondrial DNA polymerase gamma, produce a clinically heterogeneous spectrum of mitochondrial disorders ranging from infantile-onset Alpers-Huttenlocher syndrome to adult-onset progressive external ophthalmoplegia (PEO). Establishing genotype-phenotype correlations is critical for diagnostic precision and accurate prognostic counseling in this complex group of disorders. Objective:To delineate the genotype-phenotype correlations in a cohort of 17 predominantly Brazilian patients harboring pathogenic POLG variants, with emphasis on clinical phenotyping, natural history, and survival outcomes stratified by genotype.
Methods: A retrospective cohort study was performed utilizing comprehensive medical record abstraction. Comparative analyses focused on the two most prevalent variants: p.Ala467Thr (n=7) and p.Thr251Ile (n=4). Statistical methods included Fisher-Freeman-Halton exact test for categorical variables and Kaplan-Meier survival curves with Log-Rank testing for time-to-event analysis.
Results: Mean age at symptom onset was 34 years (SD ± 14.4), with male predominance (58.8%). Diagnostic latency averaged 15.6 years from symptom onset. Molecular analysis identified homozygous p.Ala467Thr (n=7) and homozygous p.Thr251Ile (n=4) as the most prevalent variants, with additional homozygous variants p.Val400Met and p.Trp748Ser each identified in single patients. Three patients harbored compound heterozygous variants, and one carried a single heterozygous p.Lys947Arg variant. The cardinal clinical features comprised ptosis (94.1%), ophthalmoparesis (88.2%), and sensory neuronopathy (70.6%). Epilepsy occurred in 41.2% overall, with enrichment in the p.Ala467Thr group (57.1%). The phenotypic spectrum additionally encompassed early-onset Parkinsonism, cerebellar ataxia, dystonia, chorea, and stroke-like episodes. Genotype-phenotype analysis demonstrated a significant association between p.Ala467Thr and SANDO (sensory ataxic neuropathy, dysarthria, and ophthalmoparesis) phenotype (100%; p < 0.001). Survival analysis identified p.Thr251Ile as a high-risk genotype, with 75.0% mortality (p = 0.024, Fisher’s test), significantly reduced survival probability (p = 0.040, Log-Rank test), and mean survival of 21.2 years from symptom onset.
Conclusion: This genotype-phenotype correlation study establishes p.Ala467Thr as strongly associated with the SANDO phenotype, while p.Thr251Ile confers significantly elevated mortality risk with shortened survival. The marked allelic heterogeneity and phenotypic pleiotropy observed across other variants necessitate comprehensive neuromuscular phenotyping and high clinical suspicion. The protracted diagnostic latency (mean 15.6 years) underscores the critical need for early diagnostic algorithms incorporating clinical red flags, neurophysiological studies, and molecular testing to facilitate timely therapeutic intervention and genetic counseling in POLG-related mitochondrial disorders.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Metabolic Myopathies: Glycogen, Lipids and Mitochondrial Myopathies
PP01.117
Brain Physiological Transcriptomic Architecture Underpinnings of Regional Vulnerability to Stroke-Like Lesions in MELAS
Dr. Matteo Azzimonti1, Dr. Francesca Magri2, Dr. Daniela Piga1, Dr. Guido Del Vecchio3, Dr. Marco Stroppi3, Assoc. Prof. Giorgio Conte3, Prof. Stefania Corti2,4, Assoc. Prof. Dario Ronchi1,4, Prof. Giacomo Pietro Comi1,4
1
Neurology Unit, IRCCS Fondazione Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy.
2
Neuromuscular and Rare Diseases Unit, IRCCS Fondazione Ca' Granda Ospedale Maggiore Policlino, Milan, Italy.
3
Neuroradiology Unit, IRCCS Fondazione Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy.
4
Dino Ferrari Center, Department of Pathophysiology and Transplantation, University of Milan, Milan, Italy
Background: Stroke-like lesions (SLLs) in mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS) show a non-random regional distribution across the brain, but the biological basis of this selective vulnerability remains unclear. Using a brain-wide transcriptomic–neuroimaging approach, we investigated how regional gene expression profiles relate to lesion susceptibility.
Methods: Five patients with MELAS due to m.3243A>G mutations and availability of at least one MRI acquired during the acute phase of a stroke-like episode were enrolled. Acute SLLs were identified and segmented on FLAIR sequences, and binarized lesion masks were averaged across patients to generate a lesion probability map (LPM). Spatial cross-correlation between the LPM and the expression of 2,802 genes related to disorders of energy metabolism (Open Targets Platform, EFO: Orphanet_79200) was evaluated using the Allen Human Brain Atlas (AHBA) and the MENGA platform (p<0.001, R²≥0.15). Cell-type-specific expression was assessed using the Darmanis human brain transcriptome database.
Results: Thirteen acute MRI scans from unique stroke-like episodes were analysed. The LPM showed a non-random spatial distribution of SLLs, with preferential involvement of occipital, parietal and temporo-polar regions. Higher lesion probability was associated with higher expression of SLBP (R²=0.725, p<0.001), a gene involved in histone mRNA processing and highly expressed in metabolically active tissues, suggesting that regions with higher intrinsic metabolic demand are more vulnerable to SLLs. In addition, higher expression of genes involved in neurovascular unit regulation, including SGCG (R²=0.667, p<0.001) and POSTN (R²=0.628, p<0.001), was associated with increased lesion probability, highlighting the role of mitochondrial microangiopathy in SLL pathogenesis. Conversely, lower expression of TMEM127 (R²=0.617, p<0.001), a negative regulator of mTOR and HIF-α signaling, characterized more vulnerable regions, suggesting that altered metabolic and hypoxic stress responses contribute to regional susceptibility.
Conclusion: In MELAS, stroke-like lesions preferentially affect brain regions with higher metabolic activity, greater dependence on neurovascular unit integrity, and reduced capacity to respond to hypoxic stress. This imaging–transcriptomic approach provides a framework for identifying molecular mechanisms and potential therapeutic targets underlying stroke-like episodes in MELAS.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Metabolic Myopathies: Glycogen, Lipids and Mitochondrial Myopathies
PP01.118
Late-Onset TK2 Deficiency in Adults: Long-Term Clinical Outcomes of Deoxynucleoside Therapy
Prof. Hacer Durmus, Prof. Asuman Gedikbası, Asst. Prof. Arman Cakar, Prof. Esen Kıyan, Prof. Yesim Parman
Istanbul Faculty of Medicine, Istanbul, Turkey
Background: TK2d is a mitochondrial myopathy causing progressive weakness, respiratory insufficiency, and early mortality. Patients typically present with ptosis, mild ophthalmoparesis, progressive limb-girdle weakness, bulbar involvement and early respiratory problems due diaphragmatic dysfunction. While nucleoside supplementation benefits pediatric cases, evidence in adults remains limited. We aimed to evaluate the long-term efficacy and safety of deoxynucleoside therapy in adult patients with late-onset thymidine kinase 2 deficiency (TK2d).
Methods: Six genetically confirmed adults with TK2d were treated under compassionate-use protocols in Turkey, receiving escalating doses of oral or PEG-administered Doxecitine and Doxribtimine up to 800 mg/kg/day. Functional and clinical outcomes included the 6-minute walk test (6MWT), Hammersmith Functional Motor Scale Expanded (HFMSE), forced vital capacity (FVC), Fatigue Severity Scale (FSS), and BMI.
Results: This cohort included six adult patients (three female) with genetically confirmed late-onset TK2 deficiency treated for a median duration of 24 months (range 6–36 months). The mean age at disease onset was 15 years (range 12–24), and the mean age at treatment initiation was 30 years (range 19–56). All patients had bilateral ptosis with preserved eye movements, nasal speech, tongue weakness, and developed neck and proximal limb weakness over the disease course. Five patients from three unrelated families harbored the NM_004614 homozygous pathogenic variant c.323C>T (p.Thr108Met), while Patient 4 carried compound heterozygous c.500G>T (p.Gly167Val) and c.248C>T (p.Ala83Val) variants. Over a median follow-up of 24 (range 6–36) months, therapy was associated with sustained functional and respiratory improvements. Mean 6MWT increased from 152 m at baseline to 468 m at 24 months and exceeded 600 m in patients with 36-month data. The mean HFMSE increased from 20.5 at baseline to 24.5 at 3 months, 29 at 6 months. Patients with the longest follow-up reached a mean HFMSE of 58 at 36 months. FVC, which had declined before treatment, stabilized or improved in all patients (mean increase from 36% to 55.6% at 36 months). The youngest patient achieved near-normal respiratory function. Fatigue severity decreased by ∼30%, and BMI improved notably in underweight individuals. Treatment was generally well tolerated; transient liver enzyme elevations and mild gastrointestinal side effects resolved spontaneously.
Conclusion: Our findings provide compelling evidence that deoxynucleoside therapy is an effective and generally well-tolerated treatment in adult-onset TK2 deficiency. While early initiation clearly maximizes functional benefit—particularly with respect to ambulation and respiratory capacity—our experience also demonstrates that treatment should not be precluded solely based on disease duration or severity. Even in advanced stages, therapy can provide stabilization of decline, improvements in fatigue, and potentially meaningful enhancements in quality of life. These results contribute to the growing clinical experience with nucleoside supplementation in this rare disorder and underscore the need for prompt diagnosis and initiation of therapy across all age groups. Future research should focus on refining dosing strategies, identifying clinical and molecular predictors of treatment response, and developing standardized protocols for monitoring long-term outcomes in diverse patient populations.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Metabolic Myopathies: Glycogen, Lipids and Mitochondrial Myopathies
PP01.119
Quantitative Gait Analysis Using a Wereable Sensor Device (Baiobit) in Patients With Metabolic Myopathies
Dr. Ignazio Giuseppe Arena, Dr. Cristian Usbergo, Dr. Selene Drago, Prof. Carmelo Rodolico, Prof. Antonio Toscano, Prof. Olimpia Musumeci
University of Messina, Messina, Italy
Background: Quantitative gait analysis provides objective and sensitive metrics capable of capturing subtle abnormalities that may not be detected by routine clinical examinations. Instrumented gait analysis using wearable sensor-based systems has emerged as a feasible and reproducible tool for the assessment of spatiotemporal parameters and gait quality in both clinical practice and research settings. In this preliminary study, we aimed to explore gait characteristics in patients with metabolic myopathies (MMs) compared with healthy controls using an instrumented gait analysis system.
Methods: 20 patients with MMs (8 Pompe Disease, 6 Mitochondrial Myopathies, 4 McArdle Disease, 1 VLCAD, 1 MADD) and 10 age-matched healthy controls underwent comprehensive gait analysis using the Baiobit system, a wearable sensor-based motion analysis platform, according to standardized acquisition protocols. Parameters measured included step velocity, cadence, symmetry, propulsion, step length and duration, stance and swing phase percentages, and pelvic kinematics (tilt, obliquity, rotation). Descriptive statistics (mean ± SD) were calculated.
Results: Compared with controls, patients exhibited reduced step velocity (1.15 ± 0.25 vs 1.21 ± 0.01 m/s) and cadence (103.5 ± 22.5 vs 134.8 ± 1.3 steps/min). Symmetry was slightly reduced in patients (93 ± 7% vs 94.3 ± 5%), and propulsion forces were lower bilaterally (left: 7.2 ± 5 vs 15 ± 1 m/s²; right: 8 ± 7 vs 14.5 ± 0.5 m/s²). Step lengths were variable but generally shorter in patients (left: 0.71 ± 0.3 vs 0.57 ± 0.03 m; right: 0.72 ± 0.3 vs 0.59 ± 0.02 m), with prolonged step durations (1.04 ± 0.2 vs 0.96 ± 0.03 s). Pelvic kinematics showed slightly increased tilt and rotation ranges in patients, likely reflecting compensatory strategies.
Conclusion: Preliminary gait analysis highlights quantifiable alterations in velocity, cadence, symmetry, propulsion, and pelvic motion in MMs patients compared to healthy controls. These findings support the feasibility of detailed gait assessment as an integration tool in evaluation of motor function in patients with MMs.
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: Outcome measures and clinimetry
PP01.120
Functional Performance, Patient-Reported Outcomes and Biomarkers in Primary Mitochondrial Myopathies During Clinical Follow-Up
Dr. Ignazio Giuseppe Arena, Dr. Selene Francesca Anna Drago, Dr. Cristian Usbergo, Prof. Vincenzo Macaione, Prof. Mhammed Aguennouz, Prof. Carmelo Rodolico, Prof. Antonio Toscano, Prof. Olimpia Musumeci
University of Messina, Messina, Italy
Background: Primary mitochondrial myopathies (PMMs) are disorders of oxidative phosphorylation characterized by predominant muscle involvement, impaired energy production, and reduced exercise tolerance. Identifying clinically meaningful outcome measures remains a major challenge in neuromuscular practice and clinical research.
Methods: To assess functional performance tests, patient-reported outcome measures (PROMs), laboratory biomarkers, and clinical scales in patients with PMMs evaluated during routine clinical follow-up, and to explore associations among these measures.
Results: Forty patients with genetically confirmed PMMs (19 with mtDNA mutations, including single deletions, and 21 with nDNA mutations) were evaluated during routine clinical follow-up over a four-year period. All patients underwent functional assessments including the Six- or Twelve-Minute Walk Test (6MWT, 12MWT), Timed Up and Go test (TUG), Five Times Sit-to-Stand test (5XSTS), spirometry, and the Newcastle Mitochondrial Disease Adult Scale (NMDAS). Patient-reported outcome measures addressing physical function and daily activity were also collected. Twenty patients additionally underwent cardiopulmonary exercise testing (CPET) according to standardized protocols, with assessment of peak oxygen uptake (VO₂peak), maximal workload (Wmax), and ventilatory and cardiovascular responses. Laboratory evaluations included serum levels of GDF15, FGF21, and neurofilament light chain (sNfl). Associations between functional, clinical, patient-reported, and laboratory variables were explored using non-parametric correlation analyses.
Conclusion: In PMM patients evaluated in a clinical setting, functional performance tests and PROMs provide complementary information on disease burden and functional limitation. The 12MWT reflected objective exercise capacity assessed by CPET, while serum neurofilament light chain was associated mostly with neurological involvement and overall clinical severity. These findings support a multidimensional approach to patient assessment in primary mitochondrial myopathies.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Metabolic Myopathies: Glycogen, Lipids and Mitochondrial Myopathies
PP01.121
Slow Channel Congenital Myasthenic Syndrome Due To c.1396G>A In CHRNA1 Favourably Responding To 3,4-DAP
Prof. Josef Finsterer
Neurology and Neurophysiology Center, Vienna, Austria
Background: Mutations in CHRNA1 are responsible for postsynaptic CMS and occur either as slow channel syndrome or fast channel syndrome. Slow channel CMS due to CHRNA1 variants responds favorably to pyridostigmine. A patient with slow-channel CMS due to a new CHRNA1 variant that responds favorably to 3,4-diaminopyridine (3,4-DAP) has not yet been reported.
Methods: Case Report
Results: The patient is a 36-year-old woman who was diagnosed with non-specific CMS at the age of one year when she presented clinically with signs of somnolence, weakness and facial dysmorphism. She later also developed limb weakness, with the upper limbs being more severely affected. Heat, low humidity, late menstruation, high fever and stress aggravated the muscle weakness. Only at the age of 17 was pyridostigmine started, which partially improved the muscle weakness. The diagnosis was genetically confirmed when the new, heterozygous variant NM_001039523:c.1396G>A in CHRNA1 p.(Gly466Arg) was detected at the age of 30. Since then, 3,4-DAP was administered, which further improved the muscle weakness.
Conclusion: CHRNA1-associated slow-channel CMS may respond favorably not only to pyridostigmine but also to the additional administration of 3,4-DAP; patients with CHRNA1-associated CMS can live for years without treatment, especially in early life; CMS should be diagnosed without delay to avoid putting people at risk of receiving medication that could potentially worsen their phenotype
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Congenital Myasthenic syndrome
PP01.122
Leigh Syndrome Due to the Compound Heterozygous Variants c.1162A > C and c.1138G > C in NDUFV1
Prof. Josef Finsterer
neurology and neurophysiology center, vienna, Austria
Background: Early-onset Leigh syndrome is usually a genetically and phenotypically heterogeneous, severe, rapidly progressive mitochondrial disorder with a fatal outcome. Leigh syndrome is genetically heterogeneous as it is based on mutations in mtDNA or nDNA genes, which mostly encode subunits of respiratory chain complexes or assembly factors. It is phenotypically heterogeneous because it is genetically heterogeneous and due to the peculiarities of mitochondrial genetics. One of the more than 100 mutated genes responsible for Leigh syndrome is NDUFV1. Here we present an infant with Leigh syndrome who suffers from a novel heterozygous variant of NDUFV1, which is phenotypically characterized by a number of previously unknown features.
Methods: Case Report
Results: The patient is a four-month-old girl with Leigh syndrome due to the compound heterozygous variants c.1162+4A>C (previously described, inherited from the mother) and c.1138G>C (novel, inherited from the father) in NDUFV1. The mutation c.1162+4A>C is a non-canonical splice site variant that has been demonstrated to result in loss of function. Bioinformatic analysis supports that the missense variant c. 1138G>C has a deleterious effect on protein structure or function. The mutations manifested phenotypically with typical cerebral lesions on imaging, developmental delay, cognitive decline, epileptiform discharges in the electroencephalography without seizures, AV block II, agenesis of a subclavian vein, right heart failure, patent foramen ovale, pulmonary hypertension, hypoaldosteronism and abdominal hernias. Within five weeks of hospitalization, the disease took a progressive course and the patient died of infectious complications despite maximum treatment.
Conclusion: This case shows that the described new heterozygous variant in NDUFV1 can occur with previously undescribed phenotypic features. It is important to diagnose mitochondrial disorders due to NDUFV1 mutations early in order not to miss the time for appropriate symptomatic treatment
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Metabolic Myopathies: Glycogen, Lipids and Mitochondrial Myopathies
PP01.123
Danon Disease in Japan: An Updated Nationwide Clinicogenetic Analysis
Prof. Kazuma Sugie1,2,3, Dr. Tomo Shiota1, Dr. Minako Yamaoka1, Dr. Ai Yamanaka1, Dr. Tomohito Ohashi1, Dr. Rui Shimazaki1, Dr. Yukako Nishimori1, Dr. Hitoki Nanaura1, Dr. Nobuyuki Eura1, Dr. Takao Kiriyama1, Prof. Eiichiro Mori4, Prof. Shuhei Nakamura2,5, Prof. Ichizo Nishino3
1
Department of Neurology, Nara Medical University, Nara, Japan.
2
Center for Autophagy and Anti-Aging Research, Nara Medical University, Nara, Japan.
3
Department of Neuromuscular Research, National Institute of Neurology, National Center of Neurology and Psychiatry, Tokyo, Japan.
4
Department of Future Basic Medicine, Nara Medical University, Nara, Japan.
5
Department of Biochemistry, Nara Medical University, Nara, Japan
Background: Danon disease is a rare X-linked dominant disorder characterized by vacuolar cardiomyopathy and skeletal myopathy, resulting from deficiency of lysosome-associated membrane protein-2 (LAMP-2). Although the disease is clinically severe, its epidemiological profile and genotype–phenotype correlations remain incompletely defined, particularly at the population level.
Methods: To refine the national landscape of Danon disease in Japan, we expanded upon a nationwide questionnaire survey conducted in 2018, which had targeted 2,617 hospitals. Newly diagnosed and subsequently reported patients were incorporated into the present analysis. Comprehensive review of clinical records, muscle pathology, and molecular genetic testing of the LAMP2 gene was undertaken.
Results: Since the previous survey, 13 additional patients (9 males and 4 females) from 9 unrelated families were identified, yielding a total cohort of 52 individuals (26 males and 26 females) from 29 families. Hypertrophic cardiomyopathy and electrocardiographic abnormalities were highly prevalent. Of the 23 deceased patients, 22 (96%) succumbed to either progressive heart failure or sudden cardiac death, underscoring the central role of cardiac involvement in disease prognosis. Heart transplantation, currently the only disease-modifying intervention, had been performed in two patients and was deemed urgently necessary in six others. Histopathological examination of skeletal muscle uniformly revealed autophagic vacuoles with sarcolemmal characteristics. Genetic analysis disclosed 23 distinct LAMP2 mutations across the 29 families, with approximately half of probands harboring de novo variants. Mutations were broadly distributed from exon 1 through exon 9; notably, three families carrying the exon 9B deletion mutation (c.1097_1098delAA), which selectively affects the LAMP-2B isoform, exhibited remarkably attenuated or absent cardiomyopathy.
Conclusion: Cardiac involvement remains the principal determinant of survival in Danon disease, accounting for nearly all disease-related mortality. However, the strikingly benign clinical course associated with the exon 9B mutation indicates that disruption of the LAMP-2B isoform alone may confer a uniquely mild phenotype. These findings provide important insights into the molecular basis of phenotypic heterogeneity in Danon disease and have direct implications for prognosis and patient management.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Metabolic Myopathies: Glycogen, Lipids and Mitochondrial Myopathies
PP01.124
Small Fiber Neuropathies: The Role of Sudoscan in Clinical Practice
Asst. Prof. BASMA MARZOUK1, Asst. Prof. Leila Tamaoui1, Asst. Prof. Fatima Ouchkat1, Dr. Bouchra Kably1, Prof. Nazha Birouk1, Dr. Leila Tamaoui2
1
Clinical Neurophysiology Department, Rabat, Morocco.
2
Clinical Neurophysiology Department Hôpital des Spécialités of Rabat / Ibn Sina University Hospital Center, Rabat, Morocco
Background: Small fiber neuropathy (SFN) is characterized by selective damage to small Aδ and C nerve fibers, which mediate thermal and nociceptive sensations as well as autonomic functions. Skin biopsy with intraepidermal nerve fiber density measurement remains the diagnostic gold standard. However, this technique is invasive, not widely accessible, and presents certain limitations. Alternative diagnostic tools, such as electrochemical skin conductance (ESC) measured by Sudoscan, offer a promising non-invasive approach. This study aimed to evaluate the diagnostic value of Sudoscan in SFN and its role within the clinical diagnostic algorithm.
Methods: All patients referred for suspected SFN were consecutively included, regardless of DN4 score or electromyography results, to encompass all possible SFN cases. Clinical and paraclinical data were collected using a standardized form. Patients with incomplete data were excluded. Assessments included Sudoscan, sympathetic skin reflex (SSR), and RR interval analysis via deep breathing test (E/I index) and orthostatic test (30/15 index). Patients were divided into two groups based on ESC results: altered vs. normal conductance. Statistical analyses were conducted with Jamovi 2.6.17; quantitative variables were compared using Mann–Whitney U test, qualitative variables using Chi-square or Fisher's exact test, and correlations assessed by Spearman's rho. A p-value < 0.05 was considered statistically significant.
Results: Twenty-nine patients (16 females, 13 males) with a median age of 57 years [range 42–65] were included. ESC was altered in 16 patients and normal in 13. Positive sensory symptoms—such as allodynia, burning, tingling, and electric shock sensations—were significantly more frequent in the altered ESC group, whereas numbness was predominant in the normal ESC group (40% vs. 16.7%, p=0.02). Associated pathologies included diabetes mellitus (n=4), drug toxicity (n=2), Sjögren’s syndrome (n=2), IgM monoclonal gammopathy of undetermined significance (MGUS) (n=1), Parkinsonian syndrome (n=1), amyloidosis (n=1), Ross syndrome (n=1), Wolfram syndrome (n=1), Thévenard disease (n=1), erythromelalgia, and vasculitis (n=1 each). Neurophysiological findings showed that the sympathetic skin reflex was abolished more frequently in the altered ESC group (76.9% vs. 23.1%, p=0.01). No significant differences were observed between groups for age, body mass index, DN4 score, sensory signs, or RR interval indices. The 30/15 index tended to be lower in the altered ESC group but without statistical significance. Correlation analysis between DN4 and Sudoscan results showed a weak, non-significant negative correlation (r = -0.28, p = 0.1).
Conclusion: SFN prevalence is estimated at approximately 53 cases per 100,000 inhabitants, with an incidence of 12 cases per 100,000 annually. It presents with positive (painful) or negative (sensory loss) symptoms, most commonly length-dependent “glove and stocking” distribution, though asymmetric or focal patterns may occur. Our findings confirm the typical clinical and etiological spectrum described in the literature. Skin biopsy limitations include variability due to age, sex, ethnicity, focal sampling, and failure to capture the pain phenotype, while primarily reflecting thermoalgesic sensory loss. Previous studies recommend combining neurophysiological tests to enhance diagnosis. Our findings support using Sudoscan with SSR and RR interval to improve SFN assessment, particularly when biopsy or extensive tests are limited.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Small fibers and painful neuropathies
PP01.125
Somatosensory Evoked Potentials (SSEP) in Phenotyping Chronic Inflammatory Demyelinating Polyradiculoneuropathy (CIDP) Variants
Asst. Prof. Leila Tamaoui1,2, Assoc. Prof. Fatima Ouchkat1,2, Asst. Prof. Basma Marzouk1,2, Dr. Bouchra Kabli1, Prof. Nazha Birouk1,2
1
Clinical Neurophysiology Department, Hôpital des Spécialités of Rabat, Ibn Sina University Hospital, Rabat, Morocco.
2
Medical school of Mohammed V University, Rabat, Morocco
Background: Diagnosis of Chronic Inflammatory Demyelinating Polyradiculoneuropathy (CIDP) remains challenging in variants that do not meet standard EAN/PNS 2021 electrodiagnostic criteria. In these cases, nerve conduction studies (NCS) may fail to capture proximal demyelination in the roots or plexuses. This study evaluates the utility of Somatosensory Evoked Potentials (SSEP) as a complementary tool to identify proximal lesions and reclassify these atypical phenotypes.
Methods: We conducted an observational study of seven patients presenting with clinical features of CIDP variants (including distal acquired demyelinating symmetric neuropathy [DADS], Lewis-Sumner syndrome, and pure motor variants) between April 2024 and October 2025. All patients underwent standard NCS and upper limb SSEP (median and/or ulnar nerve stimulation). SSEP parameters analyzed included distal latencies (N9), cervical latencies (N13), and N9-N13 interpeak intervals to assess proximal conduction.
Results: The cohort included six males and one female, aged 20 to 69 years. Phenotypes included two pure motor CIDP, two motor-predominant demyelinating polyneuropathy, and three multifocal motor-predominant forms. All patients (7/7) exhibited SSEP abnormalities despite inconclusive sensory NCS. Three patients showed complete abolition of plexic and medullary responses, while four presented with significantly prolonged latencies, indicating proximal conduction blocks. Notably, in one patient initially presenting with complete response abolition, a one-year follow-up after corticosteroid pulse therapy demonstrated the reappearance of responses, reflecting the resolution of proximal blocks and correlating with clinical recovery.
Conclusion: SSEP are essential for identifying proximal demyelination missed by distal sensory conduction studies. They provide critical evidence of sensory involvement in “pure motor” forms and help differentiate multifocal motor variants from Lewis-Sumner syndrome, which carries significant therapeutic implications. Furthermore, SSEP serve as an objective biomarker for monitoring treatment efficacy and remyelination.
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: Electrophysiology
PP01.126
Glycogen Storage Diseases in Morocco: A Clinical Series Highlighting Phenotypic Heterogeneity and Diagnostic Challenges
Asst. Prof. Leila Tamaoui1,2, Asst. Prof. Basma MARZOUK1,2, Dr. Nada Benkirane3, Prof. Nazha Birouk1,2
1
Clinical Neurophysiology Department, Hôpital des Spécialités of Rabat, Ibn Sina University Hospital, Rabat, Morocco.
2
Medical school of Mohammed V University, Rabat, Morocco.
3
private clinical practie of Neurology, Tangier, Morocco
Background: Glycogen storage diseases (GSDs) are rare inherited metabolic myopathies characterized by marked clinical heterogeneity and frequent diagnostic delay. Data from North African populations remain limited. We aim to describe the clinical, electrophysiological, and genetic features of a Moroccan series of patients with GSDs, highlighting phenotypic variability and atypical presentations.
Methods: We report a retrospective series of nine patients from six unrelated families diagnosed with GSDs. Seven patients are followed at the Clinical Neurophysiology Department, Rabat, between January 2023 and 2025. Clinical evaluation, serum creatine kinase (CK) levels, electromyography (ENMG), respiratory and cardiac assessments, enzymatic testing, and genetic analyses were reviewed.
Results: Three distinct GSD subtypes were identified.
Pompe disease (GSD II): Four patients had late-onset Pompe disease (LOPD), and one adolescent girl (13 years) had juvenile-onset disease. Clinical presentations ranged from isolated exertional fatigue to severe proximal weakness with early respiratory failure. Two patients required non-invasive ventilation, and one young female required invasive ventilation. One index case developed rapidly progressive respiratory failure and died before treatment initiation. Familial screening enabled early diagnosis in a paucisymptomatic sibling, allowing prompt initiation of enzyme replacement therapy (ERT). Enzymatic and genetic confirmation was obtained in all cases.
Tarui disease (GSD VII): The index case (37 years) presented with childhood-onset exercise intolerance followed by permanent proximal muscle weakness in adulthood. Marked intrafamilial variability was observed, with a older sister initially presenting isolated hemolytic anemia and developing pelvic girdle weakness at 69 years.
McArdle disease (GSD V): A 77-year-old male and his daughter presented with typical exercise intolerance and a clear “second-wind” phenomenon. Whole-exome sequencing identified a novel homozygous PYGM mutation.
Conclusion: This series highlights the broad phenotypic spectrum of GSDs, ranging from isolated exercise intolerance to life-threatening respiratory involvement. Diagnostic delay remains frequent, particularly in childhood-onset exertional syndromes. LOPD was the most common diagnosis, supporting systematic dried blood spot testing in suspected cases. The Tarui disease family illustrates atypical progression and striking intrafamilial variability. Consanguinity, observed in several families, likely contributed to disease occurrence and emphasizes the importance of genetic counseling and targeted family screening. Early recognition and respiratory monitoring are critical in LOPD. This series underscores the phenotypic pleiotropy of GSD VII and the diagnostic value of the “second-wind” phenomenon in GSD V. Improved access to genetic testing and ERT is essential to optimize outcomes
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Metabolic Myopathies: Glycogen, Lipids and Mitochondrial Myopathies
PP01.127
Switching From Alglucosidase Alfa to Avalglucosidase: Real-World Data From a Single-Center Experience
Prof. Olimpia Musumeci, Dr. Ignazio Arena, Dr. Selene Drago, Dr. Cristian Usbergo, Prof. Carmelo Rodolico, Prof. Antonio Toscano
University of Messina, Messina, Italy
Background: Second-generation enzyme replacement therapies (ERTs) are now available in clinical practice for the treatment of Late-Onset Pompe Disease (LOPD). Most data about their efficacy and safety came from clinical trials, while long term data on real word settings are still limited. Herein, we present real-life clinical outcomes in a cohort of eight LOPD patients treated with avalglucosidase.
Methods: We performed a longitudinal analysis of motor and respiratory functions in eleven LOPD patients followed at the Unit of Neuromuscular disorders of Messina, who received avalglucosidase for at least one year. Parameters assessed included six-minute walking test (6MWT) and forced vital capacity (FVC), which were measured at baseline (corresponding to the start of second-generation ERT) and every 12 months.
Results: The cohort mean age was 61.3 years (range 12–82) and included six males and five females. 10/11 patients were ERT-experienced, whereas one was ERT-naïve. All received avalglucosidase-alfa. The mean follow-up duration under second-generation ERT was 3.5 years (range 1–8 years). The mean 6MWT at baseline was 355 m, increasing to 370.5 m at the last follow-up. Regarding respiratory function, the mean baseline FVC% was 68.5%, increasing to 73.8% at the last follow-up, with a mean change of +5.3%. Individual patient trajectories showed that 10/11 patients stabilized or improved in at least one functional parameter.
Conclusion: To provide real-life data is critical for understanding long-term treatment effects outside controlled trial environments. About 10/11 improved or stabilized, one pt who switched from alglucosidase-alfa to avalglucosidase-alfa after a period of gradual decline, did not inverted this trend after switching, likely due to limited residual muscle function. Notably, ERT-naïve pt had stable motor and respiratory function over eight years with avalglucosidase-alfa. 5/11 patients showed improvement in both outcome measures. However, the small sample size did not allow to determine whether specific subgroups (e.g., age, ERT-naïve vs. ERT-experienced) might have differential benefit from second-generation therapies. Longer follow-up in larger cohort is warranted to confirm this positive trend.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Metabolic Myopathies: Glycogen, Lipids and Mitochondrial Myopathies
PP01.128
Sertraline-Associated Multiple Acyl-Coenzyme a Dehydrogenase Deficiency (MADD) in Sweden – a Nationwide Population-Based Study
Dr. Sofie Sunebo1,2, Dr. Emil Nilsson3, Dr. Martin Engvall3,4, Dr. Carola Oldfors5,6,7, Dr. Yan Shen8, Dr. Inger Nennesmo9, Dr. Olof Danielsson2, Prof. Anders Oldfors5,6, Dr. Hanna Appelqvist1,2, Dr. Ulrika Lindgren5,6,10
1
Department of Biomedical and Clinical Sciences, Linköping University, Linköping, Sweden.
2
Clinical Department of Neurology in Linköping, Region Östergötland, Linköping, Sweden.
3
Centre for Inherited Metabolic Diseases, Karolinska University Hospital, Stockholm, Sweden.
4
Department of Molecular Medicine and Surgery, Science for Life Laboratory, Karolinska Institutet, Stockholm, Sweden.
5
Department of Laboratory Medicine, Institute of Biomedicine, University of Gothenburg, Gothenburg, Sweden.
6
Department of Clinical Pathology, Sahlgrenska University Hospital, Gothenburg, Sweden.
7
Department of Clinical Genetics and Genomics, Sahlgrenska University Hospital, Gothenburg, Sweden.
8
Department of Clinical Chemistry, Sahlgrenska University Hospital, Gothenburg, Sweden.
9
Department of Pathology and Cancer Diagnostics, Karolinska University Hospital Solna, Stockholm, Sweden.
10
Neuromuscular Center, Department of Neurology, Sahlgrenska University Hospital, Gothenburg, Sweden
Background: Multiple acyl coenzyme A dehydrogenase deficiency (MADD) is a rare disorder of fatty acid metabolism causing a lipid storage myopathy, previously considered solely a genetic disorder, caused by recessive pathogenic variants mainly in the ETFDH gene. In 2024 we reported a strong association between MADD and the antidepressant medicine sertraline (Sunebo et al, Ann. Neurol. 2024). This association has thereafter been corroborated by other reports from different geographical areas. The number of reported patients is still low and more knowledge about sertraline-associated MADD is needed.
Methods: A nationwide population-based registry search was conducted to identify adult patients with sertraline-associated MADD in Sweden 2014-2024. Patients with muscle biopsy reports of lipid storage myopathy or biochemical investigations showing acylcarnitine profile suggesting MADD were included. Medical records were retrospectively reviewed. The aim was to increase knowledge of incidence, clinical characteristics, diagnostics and treatment effect in sertraline-associated MADD.
Results: We identified 40 (30/10 female/male) patients with sertraline-associated MADD. Median age at symptom onset was 42 years and muscle weakness the most common first symptom. All patients reported muscle symptoms during the disease course. Extra-muscular symptoms, including sensory disturbance (29 patients) and fatigue (18 patients), were common. Most patients had lipid accumulation in muscle fibers, abnormal acylcarnitine profile, myopathic electromyography pattern and elevated creatine kinase. Increased neurofilament light chain was found in six of 13 investigated patients. The length of treatment with sertraline before symptom onset varied between 0.3-20 years and the median dosage of sertraline at diagnosis was 150 mg (range 50-300 mg). Cyanocobalamin, folic acid and proton-pump inhibitors were the most frequent other medications at diagnosis. Five patients carried a heterozygous ETFDH variant. All patients were treated with riboflavin (median dose 300mg/day). Almost all (38/39) experienced clinical improvement. Acylcarnitine profile improved in all patients (31/31) with available data. The mean incidence of sertraline-associated MADD was 1.24 patients per 100 000 adults receiving sertraline treatment per year. The mean incidence of genetic adult-onset MADD was 0.0077 per 100 000 adult inhabitants per year during the same period.
Conclusion: This is the largest cohort of sertraline-associated MADD described so far, expanding the knowledge of clinical characteristics of this disease. Sertraline-associated MADD is an important differential diagnosis in patients with an initial suspicion of for example myasthenia gravis and inflammatory myopathies. New knowledge of first symptoms increases the chances of early detection and treatment, hopefully preventing severe, permanent, iatrogenic damage. No investigational method has a 100% sensitivity, and it is important that patients presenting with muscle symptoms during sertraline therapy undergo adequate evaluation to exclude MADD. Sertraline-associated MADD is much more common than genetic MADD with adult-onset in Sweden. The true incidence of sertraline-associated MADD might be higher since sertraline is a very common medication, used by 3.9% of the adult population in Sweden, and probably only the most severely affected patients undergo sufficient investigation for diagnosis.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Metabolic Myopathies: Glycogen, Lipids and Mitochondrial Myopathies
PP01.129
A Recurrent ATP2A2 p.R528Q Variant Causes Dominant Rhabdomyolysis Due to Impaired Muscle Ca²⁺ SERCA2 Pump
Dr. Kiran Polavarapu1, Dr. Romane Idoux1, Mr. Sivasankar Malaichamy1, Miss Grace Stocker1, Dr. Rachel Thompson1, Dr. Sally Spendiff1, Mr. Daniel O’Neil1, Mr. Ricardo Carmona-Martinez1, Dr. Katarina Šikic2, Dr. Elisa Holla3, Dr. Anne Schänzer4, Dr. Benno Küsters5, Dr. Andreas Hentschel6, Dr. Vera Dobelmann7, Dr. Calvin Tucht3, Dr. Meyke Schouten5, Dr. Tobias Ruck7, Dr. Ulrike Schara-Schmidt3, Dr. Erik-Jan Kamsteeg5, Dr. Danijela Petković Ramadža2, Dr. Antonia Jakovčevic2, Dr. Tamara Žigman2, Dr. Mislav Cavka2, Dr. Veronika Karcagi8, Dr. Agnes Herczegfalvi9, Dr. Steven Laurie10, Dr. Leslie Matalonga10, Dr. Sergi Beltran10, Dr. Rita Horvath11, Dr. Kevin Flanigan12, Dr. Eleni Drakou12, Dr. Christian Peña Padilla13, Dr. Nicol Voermans5, Dr. Andreas Roos3, Dr. Ivo Baric2, Dr. Hanns Lochmüller1,14,15,10
1
CHEO Research Institute, Ottawa, Canada.
2
University Hospital Center Zagreb, Zagreb, Croatia.
3
University Hospital Essen, Essen, Germany.
4
Justus Liebig University Giessen, Giessen, Germany.
5
Radboud University Medical Center, Nijmegen, Netherlands.
6
Leibniz Institut für Analytische Wissenschaften (ISAS) e.V, Dartmund, Germany.
7
Heinrich Heine University, Düsseldorf, Germany.
8
Istenhegyi Genetic Diagnostic Centre, Budapest, Hungary.
9
Semmelweis University Pediatric Center, Budapest, Hungary.
10
Centro Nacional de Análisis Genómico (CNAG), Barcelona, Spain.
11
University of Cambridge, Cambridge, United Kingdom.
12
Nationwide Children's Hospital, Columbus, United States.
13
Hospital Civil de Guadalajara, Guadalajara, Mexico.
14
The Ottawa Hospital, Ottawa, Canada.
15
University of Ottawa, Ottawa, Canada
Background: Rhabdomyolysis is a severe condition characterized by skeletal muscle breakdown, leading to the release of intracellular components into the bloodstream and causing acute myalgia and weakness. Rhabdomyolysis is typically triggered by external stressors, and it is now recognized that underlying genetic disorders, including neuromuscular diseases, can predispose individuals to recurrent episodes. ATP2A2, encoding the sarco/endoplasmic reticulum Ca²⁺-ATPase SERCA2, is essential for intracellular calcium homeostasis and is highly expressed in slow-twitch skeletal muscle. Heterozygous ATP2A2 variants are classically associated with autosomal dominant skin disorders. We identified a recurrent missense variant in ATP2A2 as a genetic cause for autosomal dominant Rhabdomyolysis in five unrelated families across the world establishing ATP2A2 as a novel Rhabdomyolysis gene.
Methods: Genomic reanalysis of unsolved neuromuscular families in RD-Connect Genome Phenome Analysis Platform (GPAP) resulted in identification of an identical heterozygous missense variant c.1583G>A; p.R528Q in ATP2A2 gene in two unrelated families with multiple affected members segregating in autosomal dominant pattern. Through further global matchmaking, three more families were identified with patients carrying identical ATP2A2 variant with matching phenotype. To investigate the underlying mechanisms, we used patient-derived myotubes and p.R528Q CRISPR/Cas9 zebrafish models. Analyses included swimming performance in zebrafish using the Zantiks tracking system, structural assessment of slow skeletal muscle fibers and their organelles using immunofluorescence and histology, and evaluation of SERCA2 function using Ca²⁺ imaging in both zebrafish and patient-derived myotubes.
Results: Across five unrelated families, we identified 17 affected individuals carrying the same heterozygous ATP2A2 variant (c.1583G>A; p.R528Q). Clinical presentation was consistent, with recurrent rhabdomyolysis episodes predominantly triggered by fever or infection, and occasionally by exercise. Serum Creatine Kinase was highly elevated during the episodes. Inter-episode periods were predominantly asymptomatic with no reports of skin and cardiac manifestations in all patients. In silico predictions were consistently damaging for the variant which is located in SERCA2 Nucleotide-binding domain. Structural modeling demonstrated loss of critical salt-bridge interactions In silico 3D modelling of wild-type and mutated SERCA2 protein using the AlphaFold structure (AF-P16615-F1) revealed loss of critical salt bridges suspected to cause loss of stability and function of the Ca²⁺-ATPase pump. Muscle biopsy revealed mild myopathic changes with core-like features, Z-band streaming, and fiber-type remodeling, while SERCA2 protein abundance was preserved. Patient-derived myotubes exhibited delayed SERCA-mediated Ca²⁺ reuptake during high KCl-induced Ca²⁺ transients. Mutant zebrafish exhibited mild phenotypes with reduced swimming performance and structural defects in slow fibers, including reduced cross-sectional area, intermyofibrillar mitochondrial abnormalities, fibrosis, and a shift toward intermediate/fast fibers. Electrically evoked Ca²⁺ transients in slow fibers of anesthetized larvae showed reduced peak amplitudes, while reuptake kinetics remained preserved.
Conclusion: Our results indicate that p.R528Q disrupts SERCA2 function, causing abnormal intracellular Ca²⁺ handling in skeletal muscle and predisposing to rhabdomyolysis. This study expands the phenotypic spectrum of ATP2A2-related disorders, previously linked only to autosomal dominant skin disorders, and established ATP2A2 variant p.R528Q as a novel but recurrent genetic cause for autosomal dominant rhabdomyolysis. These findings also highlight potential therapeutic strategies targeting SERCA function in this newly identified neuromuscular disorder.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Muscle Channelopathies and Related Disorders
PP01.130
Design of ACHILLES Phase 3 Randomised Study: Mexiletine PR vs. Mexiletine IR in Non-Dystrophic Myotonia
Prof. Kristl Claeys1,2, Prof. Remiche Gauthier3, Dr. Dipa Jayaseelan4, Dr. Guido Primiano5, Dr. Savine Vicart6, Dr. Rocío-Nur Villar-Quiles6, Dr. Jana Zschüntzsch7, Ms. Nikki Adetoro8, Mr. Russell Brierley8, Dr. Alla Zozulya-Weidenfeller9
1
Department of Neurology, University Hospitals Leuven, Leuven, Belgium.
2
Laboratory for Muscle Diseases and Neuropathies, Department of Neurosciences, KU Leuven, Leuven, Belgium.
3
Université libre de Bruxelles, Hôpital Universitaire de Bruxelles, CUB Hôpital Erasme, Service de Neurologie, Centre de Référence Neuromusculaire, Brussels, Belgium.
4
Queen Square Centre for Neuromuscular Diseases, UCL Queen Square Institute of Neurology, London, United Kingdom.
5
Dipartimento di Neuroscienze, Organi di Senso e Torace, Fondazione Policlinico Universitario Agostino Gemelli IRCCS, Rome, Italy.
6
National Reference Center for Muscle Channelopathies Rare Diseases, Service of Neuro-Myology, Assistance Publique-Hôpitaux de Paris, Sorbonne Université, INSERM, University Hospital Pitié-Salpêtrière, Paris, France.
7
Klinik für Neurologie, Universitätsmedizin Göttingen, Göttingen, Germany.
8
Lupin Research Inc., Baltimore, United States.
9
Lupin Neurosciences, Zug, Switzerland
Background: Mexiletine is indicated for myotonia in adults with non-dystrophic myotonia (NDM). Over 7000 people have received mexiletine for NDM since its approval in 2018 (Lupin Neurosciences), and its safety and efficacy in NDM are supported in scientific literature and clinical practice. Also, mexiletine has established an acceptable tolerability profile in NDM clinical-trial settings, with gastrointestinal (GI) effects being the most common adverse events (AEs) reported. ACHILLES is an exploratory, multicentre, European, open-label, crossover NDM study assessing the safety, tolerability and effectiveness of a new formulation – mexiletine prolonged release (PR) QD – versus mexiletine immediate release (IR) TID.
Methods: ACHILLES is enrolling participants aged ≥16 years with genetically confirmed symptomatic NDM and evidence of handgrip myotonia. The target study population is N=24 (n=12 mexiletine-naïve and n=12 mexiletine-experienced participants). After screening/randomisation, participants receive 12 weeks’ treatment with mexiletine PR or mexiletine IR: mexiletine PR is bioequivalent to mexiletine IR. After 7 days’ washout, participants receive the opposite treatment for 12 weeks (Figure 1). To reduce bias, ACHILLES is randomised and collects objective information on adherence and tolerability in addition to patient-reported outcomes, patient-preference, and functional-capacity data. ACHILLES also uses the clinical myotonia rating scale (CMRS) to evaluate myotonia severity and daily impact of disability, to help establish whether the CMRS is a more sensitive measure of myotonia vs. current tools. Mexiletine PR is provided as granules for oral suspension in three strengths (167 mg, 333 mg and 500 mg) for reconstitution in water. Mexiletine IR is a 167-mg hard capsule for oral administration. Dosing commences at 167 mg QD, up-titrated in Week 1 to 333 mg and in Week 2 to a maximum of 500 mg. Differences in administration and usability between PR and IR formulations are key factors in treatment preference. The open-label design permits direct assessment of these aspects in a manner relevant to real-world mexiletine use.
Results: Safety assessments include patient- and physician-reported AEs, electrocardiogram and standard clinical laboratory evaluations, physical examinations, and vital signs. The primary (safety) endpoint is the incidence of treatment emergent adverse events (TEAEs) including treatment-related TEAEs, serious AEs, AEs of special interest, and treatment discontinuations. Secondary (efficacy) endpoints: Video recording of Hand Opening Time; Individualized Neuromuscular Quality of Life questionnaire; Myotonia Behaviour Scale; Timed up-and-go; Visual-Analog Scale (myotonia/stiffness); Clinical Global Impression (efficacy and tolerability) scores. Other endpoints: 10-metre walk test; treatment preference; gastrointestinal function (Gastroesophageal Reflux Disease-Health Related Quality of Life); swallowing function (timed testing); CMRS evaluation. Cardiac safety endpoints focus on PR, QRS and QTc interval data, and average minimum heart rate. Endpoints will be assessed as mean changes from baseline to all other timepoints. Data summaries will be presented as means and confidence intervals rather than by P-value.
Conclusion: Mexiletine PR has been developed with the intention of reducing the number and impact of GI events experienced by patients. It provides a QD regimen that aims to simplify mexiletine administration, thereby improving patients’ treatment experiences and quality of life, especially for those with dysphagia. Enrolment in ACHILLES is expected to complete in summer 2026
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Muscle Channelopathies and Related Disorders
Images or Table (Optional)
PP01.131
Andersen-Tawil Syndrome without Cardiac Conduction Defect: A Genetically Confirmed Case
Dr. Mathias Maisenbacher, Prof. Pariwat Thaisetthawatkul
Department of Neurological Sciences, University of Nebraska Medical Center, Omaha, United States
Background: Andersen-Tawil Syndrome (ATS) is an extremely rare type of periodic paralysis (PP) (estimated prevalence 1:1,000,000). ATS classically presents as a triad of periodic paralysis, cardiac abnormalities, and characteristic facial and skeletal features. Approximately70% of cases are linked to autosomal dominant mutations in KCNJ2/Kir2.1 (ATS1). More frequent than dysmorphic features, cardiac manifestations of ATS1 have been found in about 90% of cases, and may include a U-wave, ventricular bigeminy, bidirectional ventricular tachycardia, and/or QT-interval prolongation. Here, we report a genetically confirmed case of ATS without cardiac conduction defect, the diagnosis of which required clinical recognition of facial and skeletal dysmorphism.
Methods: Case Report
Results: A 30-year-old male presented with a 16-year history of episodic limb weakness since adolescence. Attacks occurred nearly monthly and varied in severity, ranging from mild weakness to profound paralysis lasting several days. Common triggers included awakening after sleep and high-carbohydrate meals. There were no sensory, autonomic, or bulbar symptoms. During a prior hospitalization for severe weakness, he was found to be severely hypokalemic. The patient never suffered cardiac symptoms or syncopal episodes. There was no family history of recurrent paralysis, though a paternal relative had cardiac events. The neurologic exam was completely normal. The patient’s general appearance was notable for obesity with mild dysmorphic features, including a small chin, mild hypertelorism, low-set ears, and short fingers. Basic laboratory tests were normal. Routine nerve conduction and EMG studies were normal. The long exercise test demonstrated a post-exercise compound muscle action potential (CMAP) decrement of approximately 51%. An electrocardiogram (EKG) was normal. Genetic testing revealed a pathogenic variant of the KCNJ2 gene, confirming the diagnosis of ATS. The patient reported a reduction in attack frequency following lifestyle modifications and pharmacologic therapy with carbonic anhydrase inhibitors and potassium supplementation. Cardiology evaluation did not reveal any cardiac or rhythm abnormalities. The patient remains stable under multidisciplinary follow-up.
Conclusion: ATS is an exceedingly rare condition that occurs along a spectrum of episodic weakness, dysmorphic features, and cardiac abnormalities. However, these cardiac abnormalities may not be present at initial presentation. Therefore, clinical recognition of the facial and finger dysmorphia is essential in case of a normal EKG and absence of cardiac symptoms. Mild facial dysmorphisms such as micrognathia are commonly idiopathic phenomena in adults and may easily be overlooked. Reduced penetrance, de novo mutations, and phenotypic ATS without KCNJ2 abnormalities (ATS2) further complicate the diagnosis, making attentive clinical and electrodiagnostic examination paired with a high index of suspicion essential in all cases of PP. Early diagnosis enables targeted management, improves quality of life, and reduces potential cardiac risks.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Muscle Channelopathies and Related Disorders
PP01.132
Monitoring Generalized Myasthenia Gravis Symptoms With ME&MGTM Digital Biomarkers: DOMYA, a Validation Study
Dr. Edouard Berling1, Dr. Ima Ebong2, Dr. Céline Tard3, Dr. Pascal Cintas4, Dr. Gaurav K Guliani5, Dr. Emmeline Lagrange6, Dr. Lindsay Malatesta7, Dr. Maud Michaud8, Dr. Aleksandra Nadaj-Pakleza9, Prof. Guillaume Nicolas10, Dr. Denis Ostrovskiy11, Dr. Yann Péréon12, Prof. Hélène Prigent10, Dr. Michael T Pulley13, Dr. Shruti M Raja14, Dr. Thomas Ragole15, Dr. Sabrina Sacconi16, Dr. Sara Takacs17, Dr. Clarissa Gorin18, Miss Aurore Balique-Laplanche18, Miss Noura Sellami18, Dr. Saad Zinaï18, Dr. Benjamin Yungher19, Dr. Bhaskar Dutta19, Dr. Loïc Carment18, Dr. Christophe A Scheiner20, Prof. Pascal Laforêt10
1
APHP, Service de Neurologie, Hôpital Raymond Poincaré, Centre de Référence Nord‐Est‐Ile‐de‐France, FHU PHENIX, Garches, France.
2
Department of Neurology, University of Kentucky College of Medicine, Lexington, United States.
3
U1172, Centre de référence des maladies neuromusculaires Nord/Est/Île-de-France, service de Neurologie A, CHU de Lille, Lille, France.
4
Service de Neurologie, Centre de référence des Maladies Neuromusculaires, Toulouse University Hospital, Toulouse, France.
5
HealthPartners Institute, Bloomington. Department of Neurology, University of Minnesota, Minneapolis, MN HealthPartners Institute Neuroscience Research Center, St. Paul. Regions Hospital, Saint-Paul, United States.
6
Department of Neurology, Grenoble University Hospital, Grenoble, France.
7
Department of Neurology, Vanderbilt University Medical Center, Nashville, United States.
8
Centre de Référence des Maladies Neuromusculaires Nord-Est-Ile de France, Department of Neurology, University Hospital Centre, Nancy, France.
9
Centre de Référence des Maladies Neuromusculaires Nord-Est-Ile de France, Department of Neurology, University Hospital Centre, Strasbourg, France.
10
Nord-Est/Ile-de-France Neuromuscular Reference Center, UMR 1179, Neurology Department, Raymond-Poincaré Hospital, Garches, FHU PHENIX, UVSQ Paris-Saclay University, Garches, France.
11
Department of Neurology, Hofstra University, North shore, Mount Sinai School of Medicine. Neurological Associates of Long Island, Long Island, United States.
12
CHU Nantes, Reference Centre for Neuromuscular Disorders AOC, FILNEMUS, Euro-NMD, Hôtel-Dieu, Nantes, France.
13
College of Medicine-Jacksonville, Department of Neurology, University of Florida, Jacksonville, United States.
14
Department of Neurology, Box 3403, Duke University Medical Center, Durham, United States.
15
Department of Neurology, University of Colorado Anschutz Medical Campus, Aurora, United States.
16
Referral Centre for Neuromuscular Diseases, CHU Nice, Nice, France.
17
Department of Neurology. Indiana University School of Medicine, Indianapolis, United States.
18
Ad Scientiam, Paris, France.
19
Alexion, AstraZeneca Rare Disease, Boston, United States.
20
University of Tennessee Medical Center, Knoxville, United States
Background: Generalized Myasthenia Gravis (gMG) is an autoimmune disease characterized by fluctuating and fatigable muscle weakness, making it essential to regularly assess symptom fluctuations outside of clinic visits. ME&MGTM is a software as medical device allowing remote and objective self-assessment of gMG symptoms through its five digital biomarkers My Eyelid, My Breathing, My Voice, My Arms and My Legs respectively measuring ptosis, respiratory function, dysarthria, upper and lower limb muscle weakness.In the ME&MGopen decentralized study, proof-of-concept was established with digital biomarkers’ scores correlating with the sub-scores of the MG Activities of daily living (MG-ADL) and MG quality of life 15-item scale-revised (MG-QoL15r) questionnaires. The ongoing DOMYA (NCT05564936) study aims to validate ME&MG™ clinical performance by comparing individual’s Quantitative MG (QMG) sub-scores to home-measured ME&MG™ digital biomarkers’ scores in people with gMG and healthy controls.
Methods: DOMYA recruited adult patients with anti-acetylcholine receptor antibody-positive gMG and healthy controls in France and the USA. Patients are monitored for one year with in-clinic visits at days 0, 90 and 365, along with monthly home-performed ME&MGTM active tests in the mornings and evenings. Healthy controls performed active tests in-clinic on day 0 and at home on day 1. Correlation coefficients between dBMKs and ptosis, speech, lung function, limb stretch sub-items of QMG, will be calculated to determine the clinical significance of dBMKs and, safety related to the medical device will be reported.
Results: 94 people with gMG (France: 46; USA: 48) and 50 healthy controls (France: 22; USA: 28) have been recruited from 21 centers (France: 10; USA: 11). 23 patients (France: 10; USA: 13) completed the study with the remaining anticipated by October 2026.
Conclusion: ME&MGTM digital biomarkers reliability, consistency and quality have been established in the ME&MGopen study. Validation of their clinical performance and safety in DOMYA will further support ME&MGTM adoption for objective tracking of gMG symptoms in routine practice supporting HCPs in monitoring the symptoms and in their decision-making processes.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.133
HARMONY360 Study for Comprehensive Monitoring of Generalized Myasthenia Gravis With Passive and Active Digital Biomarkers
Dr. Sophie Lehnerer1, Dr. Raffaele Iorio2,3, Dr. Francisco JR de Rivera Garrido4, Dr. Mazen M Dimachkie5, Dr. Lea Gerischer6,7, Dr. Séverine Bieuvelet8, Dr. Loïc Carment8, Miss Blandine Landrieu8, Miss Natacha Pesic-Heuvrard8, Miss Noura Sellami8, Dr. Saad Zinaï8, Dr. Elina Järvinen9, Dr. John Vissing10, Prof. Pascal Laforêt11
1
Charité – Universitätsmedizin Berlin, corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Department of Neurology with Experimental Neurology, Berlin, Germany.
2
Department of Neuroscience, Università Cattolica del Sacro Cuore, Rome, Italy; Neurology Unit, Fondazione Policlinico Universitario A. Gemelli IRCCS, Rome, Italy.
3
Neurology Unit, Fondazione Policlinico Universitario A. Gemelli IRCCS, Rome, Italy.
4
Reference Unit of Hereditary Ataxias and Paraplegias, Department of Neurology, IdiPAZ, Hospital Universitario La Paz, Madrid, Spain.
5
Department of Neurology, University of Kansas Medical Center, Kansas City, United States.
6
Department of Neurology With Experimental Neurology, Charité-Universitätsmedizin Berlin, Corporate Member of Freie Universität Berlin and Humboldt-Universität zu Berlin,, Berlin, Germany.
7
Neuroscience Clinical Research Center, Charité-Universitätsmedizin Berlin, Corporate Member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Berlin, Germany.
8
Ad Scientiam, Paris, France.
9
Merck OY, Espoo Finland,an affiliate of Merck KGaA, Darmstadt, Germany.
10
Department of Neurology, Copenhagen Neuromuscular Center, Rigshospitalet, University of Copenhagen, Copenhagen, Denmark.
11
Nord-Est/Ile-de-France Neuromuscular Reference Center, UMR 1179, Neurology Department, Raymond-Poincaré Hospital, Garches, FHU PHENIX, UVSQ Paris-Saclay University, Garches, France
Background: Generalized Myasthenia Gravis (gMG) is a heterogeneous antibody-mediated disease characterized by a fluctuating and fatigable muscle weakness, making it difficult for people with gMG to predict or recall day-to-day fluctuations. Digital health technologies could enhance disease monitoring by integrating wearable sensors, electronic patient-reported outcomes (ePROs) and active assessments to provide objective, longitudinal data. We have developed an innovative investigational medical device combining passive monitoring via the ActiGraph LEAP (physical activity, sleep, heart and respiratory rate), smartphone-based active functional tests (ptosis, dysarthria, upper, lower limb and respiratory muscle weakness) and ePROs (MG-ADL; MG-QoL-15r; NeuroQoL fatigue and infection tracking). By combining the passive day-to-day, active digital tests, and standardized clinical questionnaires, we aim to depict a comprehensive portrayal of gMG. In this regard, the international Harmony 360 study (NCT07323316) is designed to assess the feasibility of using this tool to detect changes in disease trajectory over 12 months.
Methods: HARMONY 360 is a prospective, single-arm, multi-center, exploratory study enrolling 100 adults with gMG in five European countries (Denmark, France, Germany, Italy and Spain) over 12 months. Participants will attend in-clinic visits at Day 0 (inclusion visit), Day 180 and Day 360 (exit visit), during which socio-demographic, clinical data, and Quantitative MG (QMG) scores will be collected. Between visits, participants will wear the ActiGraph LEAP continuously, complete monthly active test sessions (morning and evening), and ePROs twice a month (Figure).
Results: Recruitment is scheduled to begin in April 2026, with a planned study duration of 12 months.
Conclusion: HARMONY 360 study will describe the feasibility of our investigational tool in capturing and predicting changes in disease trajectory in people with gMG using continuous passive and active data. Findings will inform optimization of the device and a subsequent validation study toward more personalized generalized Myasthenia Gravis care.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Muscle Channelopathies and Related Disorders
Images or Table (Optional)
PP01.134
When One Diagnosis Was Not Enough: A Rare Neuromuscular Case Evolving Into Triple Trouble
Assoc. Prof. Anahit Mehrabyan, Asst. Prof. Hailey Segall
University Of Norh Carolina, Chapel Hill, United States
Background: Coexisting genetic and acquired neuromuscular disorders have been increasingly recognized and can pose significant diagnostic and management challenges. Such overlap may delay treatment of potentially reversible acquired conditions and complicate eligibility for clinical trials targeting inherited muscle diseases. Myotonic dystrophy type 1 (DM1) has a characteristic phenotype, including clinical myotonia, early-onset cataracts, cardiac involvement, associated with distal muscle weakness, with diagnosis confirmed by genetic testing. We report a complex case of a patient with a longstanding clinical diagnosis of DM1 that was later disproven, revealing limb-girdle muscular dystrophy due to a pathogenic ANO5 mutation, compounded by vitamin B12 deficiency and POEMS syndrome.
Methods: A 60-year-old Caucasian woman with a presumed diagnosis of DM1 since adolescence, non-ischemic cardiomyopathy status post pacemaker implantation, depression, and anxiety was admitted for progressive bilateral leg numbness and weakness. Her original DM1 diagnosis was based on clinical features and a family history of similar symptoms in her father and siblings; genetic testing had never been performed. Notably, her parents were second cousins. At baseline, she was ambulatory but had required a chair lift since her 30s. Over three months, she developed worsening lower extremity weakness, sustained a fall with left ankle injury, and progressed to using a walker. Neurologic examination revealed proximal and distal lower extremity weakness (4/5 and 2/5, respectively), preserved upper extremity strength, absent deep tendon reflexes, decreased vibration and proprioception in the distal legs, and no clinical myotonia. Over the subsequent two months, she developed worsening distal greater than proximal weakness in both lower and upper extremities, accompanied by progressive sensory loss to pinprick, vibration, and proprioception, more pronounced in the feet than the hands.
Results: Initial laboratory evaluation revealed vitamin B12 deficiency (167 pg/mL; 211–911 pg/mL), which was treated. Limited electrodiagnostic testing showed normal sural sensory response, low peroneal motor amplitude, and mild myopathic changes on needle EMG. Comprehensive neuromuscular genetic testing identified a homozygous pathogenic mutation in ANO5 (c.692G>T (p.Gly231Val) (homozygous), and a variant of uncertain significance (c.5762G>C (p.Arg1921Pro), VUS) in MYH7; testing for DM1 and 2 was negative. On reevaluation two months later, repeat electrodiagnostic studies (performed with mild sedation) demonstrated a severe demyelinating sensorimotor polyneuropathy concerning for POEMS syndrome. Serum studies revealed a monoclonal IgG lambda paraprotein (0.7 g/dL), elevated lambda (87 mg/dL) and kappa (3.58 mg/dL) light chains with a normal kappa/lambda ratio, and normal VEGF levels (47.1–49.8 pg/mL). FDG-PET imaging showed extensive lytic lesions of the bilateral pubic rami. Bone marrow biopsy demonstrated low-level plasma cell neoplasm involvement, and biopsy of a lytic lesion confirmed a plasmacytoma.
Conclusion: This case highlights several critical points: (1) the longstanding misdiagnosis of DM1 in the absence of genetic confirmation; and (2) the diagnostic complexity introduced by the coexistence of acquired neuromuscular conditions. The additional MYH7 VUS remains under investigation and may explain the patient’s cardiac comorbidity, rendering this an ultra-rare “quadruple-trouble” presentation involving double-trouble genetic myopathy, POEMS and vitamin B12 deficiency.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Muscle Dystrophies (Non-Dystrophinopathies)
Images or Table (Optional)
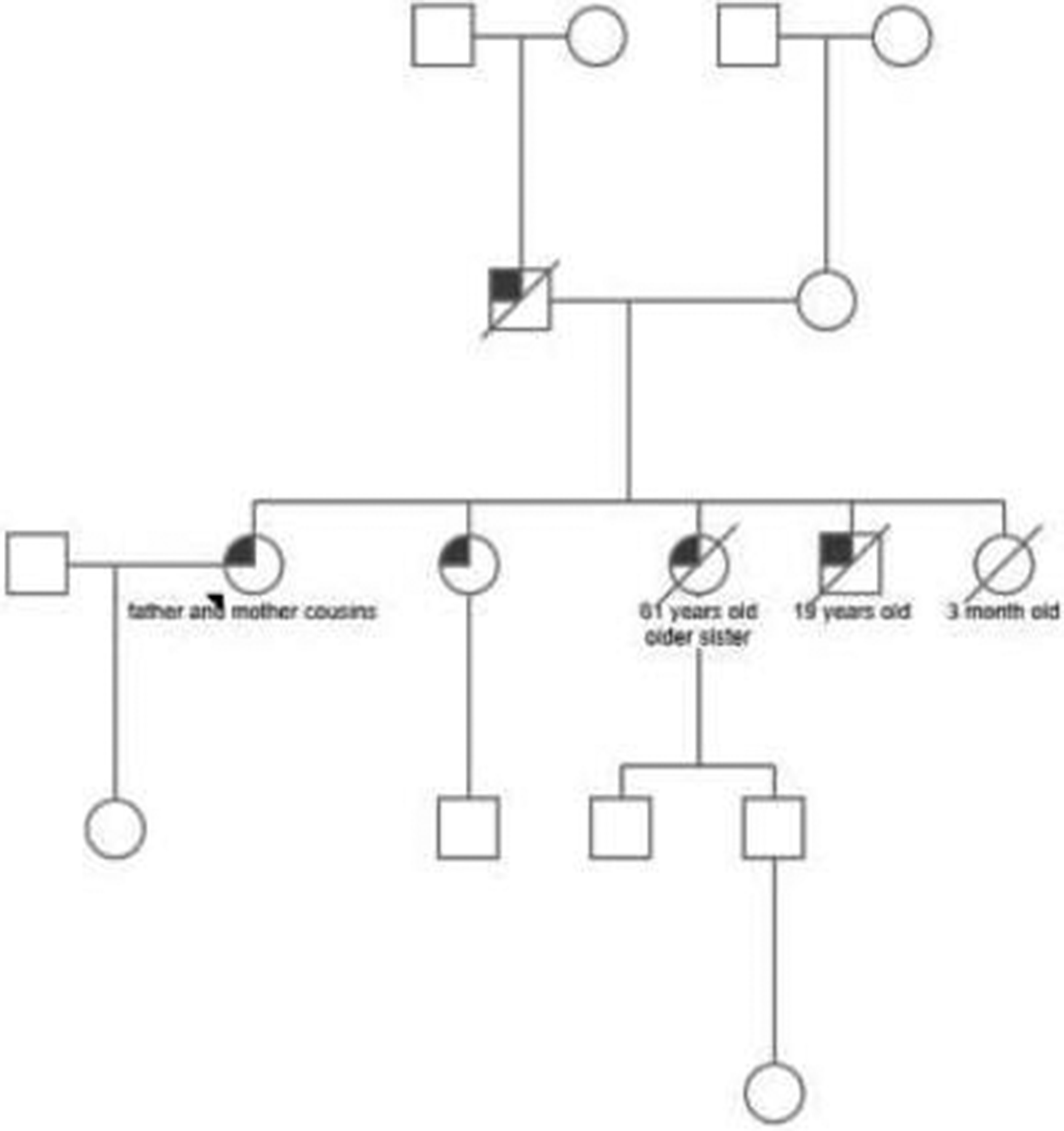
PP01.135
Precision Nutrition in Neuromuscular Diseases: Influencing Factors and Feasibility of a Protein-Optimized Intervention
Miss Farah J. Walter1, Miss Caterina M. Wendel1, Miss Daniela Grote2, Dr. Simone Thiele1, Mrs Eva Malm1, Mrs Monika Bischoff2, Dr. Gert Bischoff2, Asst. Prof. Stephan Wenninger1, Prof. Maggie C. Walter1
1
Friedrich Baur Institute at the Department of Neurology, LMU University Hospital, Munich, Germany.
2
Center for Nutritional Medicine and Prevention, Barmherzige Bruder Hospital, Munich, Germany
Background: A multidisciplinary approach is the key element in the management of patients with neuromuscular diseases. Individuals are frequently at risk of overweight, with an increased risk of undernutrition or malnutrition along with ongoing loss of muscle mass and function. Despite the essential role of nutrition in metabolic health and preserving muscle mass, there is only insufficient evidence regarding the specific nutritional requirements of neuromuscular patients and the efficacy of targeted interventions.
Methods: Nutritional status and body composition of patients with spinal muscular atrophy (SMA), facioscapulohumeral muscular dystrophy (FSHD), and Becker muscular dystrophy (BMD) will be assessed at baseline and longitudinally, patients will be individually educated and counselled on nutrition and nutritional interventions, and will regularly have BIA measurements, fill specific nutrition protocols followed by a high-protein diet, in case of overweight with a calorie-reduced high-protein diet, in case of underweight or malnutrition by adding high-caloric drink food, along with regular SMA-assessments following disease-specific protocols. The aim is to evaluate the impact of the nutritional intervention on general health, quality of life, lung function and motor function, along with correlation of current disease-modifying therapy (DMT), gastrointestinal side effects, and nutrition.
Results: Preliminary analyses demonstrate significant variability in nutritional status within the cohort. The data suggest that adherence to the nutritional intervention is significantly influenced by individual habits and disease-specific functional impairments. Furthermore, initial correlations have been identified between specific nutritional patterns and clinical-functional parameters.
Conclusion: Nutritional factors are a critical yet often overlooked component in the management of neuromuscular diseases. The findings underscore the importance of personalized approaches over generic recommendations. Further research is needed to refine these correlations, evaluate long-term effects, and establish a foundation for evidence-based clinical guidelines.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Muscle Dystrophies (Non-Dystrophinopathies)
PP01.136
CPET Validity and Aerobic Training Feasibility in Oculopharyngeal Muscular Dystrophy
Prof. Elise Duchesne1,2,3,4, Mr. Marc-Olivier Dugas1, Prof. Cynthia Gagnon1,5, Mr. Elie Fiogbé1,5, Prof. Eric Lukas Voorn6
1
Groupe de recherche interdisciplinaire sur les maladies neuromusculaires (GRIMN), Centre intégré universitaire de santé et de services sociaux du Saguenay–Lac-Saint-Jean, Saguenay, Quebec, Canada.
2
École des sciences de la réadaptation, Faculté de médecine, Université Laval, Quebec City, Quebec, Canada.
3
CHU de Québec - Centre de Recherche de l'Université Laval, Quebec City, Quebec, Canada.
4
Centre Interdisciplinaire de Recherche en Réadaptation Et Intégration Sociale (CIRRIS), Institut de Réadaptation en Déficience Physique de Québec, Quebec City, Quebec, Canada.
5
Faculté de médecine et des sciences de la santé, Université de Sherbrooke, Sherbrooke, Quebec, Canada.
6
Amsterdam UMC location University of Amsterdam, Department of Rehabilitation Medicine, Amsterdam, Netherlands
Background: Oculopharyngeal muscular dystrophy (OPMD) is a late-onset autosomal dominant neuromuscular disease caused by an expansion of a (GCN) trinucleotide repeat (11-18 repeats) in the PABPN1 gene, encoding the poly(A)-binding protein nuclear 1. Although research has traditionally focused on the cardinal symptom of dysphagia, emerging evidence, including work from our group and others, demonstrates substantial skeletal muscle impairment and mobility limitations in OPMD. As exercise-based rehabilitation gains momentum in neuromuscular diseases, objective characterization of cardiorespiratory fitness (CRF) and evaluation of aerobic training feasibility in OPMD are urgently needed. However, the validity of cardiopulmonary exercise testing (CPET) and the potential benefits of structured aerobic exercise remain unknown in this population. Objectives: To (1) establish CPET validity in individuals with OPMD; (2) characterize CRF and mobility-related limitations relative to normative values; and (3) examine the feasibility, adherence, safety, and preliminary functional effects of a 12-week individualized home-based aerobic training program.
Methods: Ambulatory adults with genetically confirmed OPMD underwent CPET and standardized mobility performance tests, including the 30-Second Chair Stand Test (30CST), 6-Minute Walk Test (6MWT), and maximal 10-Step Stair Tests for ascent and descent. CPET validity was determined using predefined physiological criteria. Participants electing to train completed a 12-week individualized home-based aerobic program following a polarized training model, consisting of two weekly low-intensity sessions and one weekly high-intensity session. Exercise intensity zones were individualized based on the first ventilatory threshold, determined from baseline CPET, and prescribed and monitored using heart-rate targets. Outcomes were assessed pre- and post-intervention. Feasibility metrics included adherence, retention, adverse events, and participant satisfaction.
Results: Sixteen participants were enrolled; CPET was valid in 14 (88%), demonstrating that maximal exercise testing is achievable in most individuals with OPMD. Mean VO₂peak and maximal heart rate reached 99.2% and 103.2% of predicted values, respectively, indicating preserved central aerobic capacity. In contrast, peak power output (66.7% of predicted), 30CST (50.6%), and 6MWT performance (65.8%) were markedly reduced, revealing a pronounced dissociation between aerobic capacity and lower-limb functional performance. Of participants initiating the training program, 60% completed the intervention with excellent adherence (98.6% of prescribed sessions). Withdrawals were due to time constraints (20%) or unrelated medical issues (20%). Peak power output improved by 24.7% following training. No adverse events occurred, and participant satisfaction was high.
Conclusion: To the best of our knowledge, this study is the first to establish CPET validity and comprehensively characterize CRF in OPMD, shifting the clinical focus beyond dysphagia to include systemic and functional limitations. Our findings provide initial evidence that individualized home-based aerobic training is feasible, safe, and capable of improving functional performance in OPMD. These results support the integration of aerobic exercise into clinical management and inform the design of future interventional trials in this underrepresented neuromuscular disease.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Muscle Dystrophies (Non-Dystrophinopathies)
Images or Table (Optional)
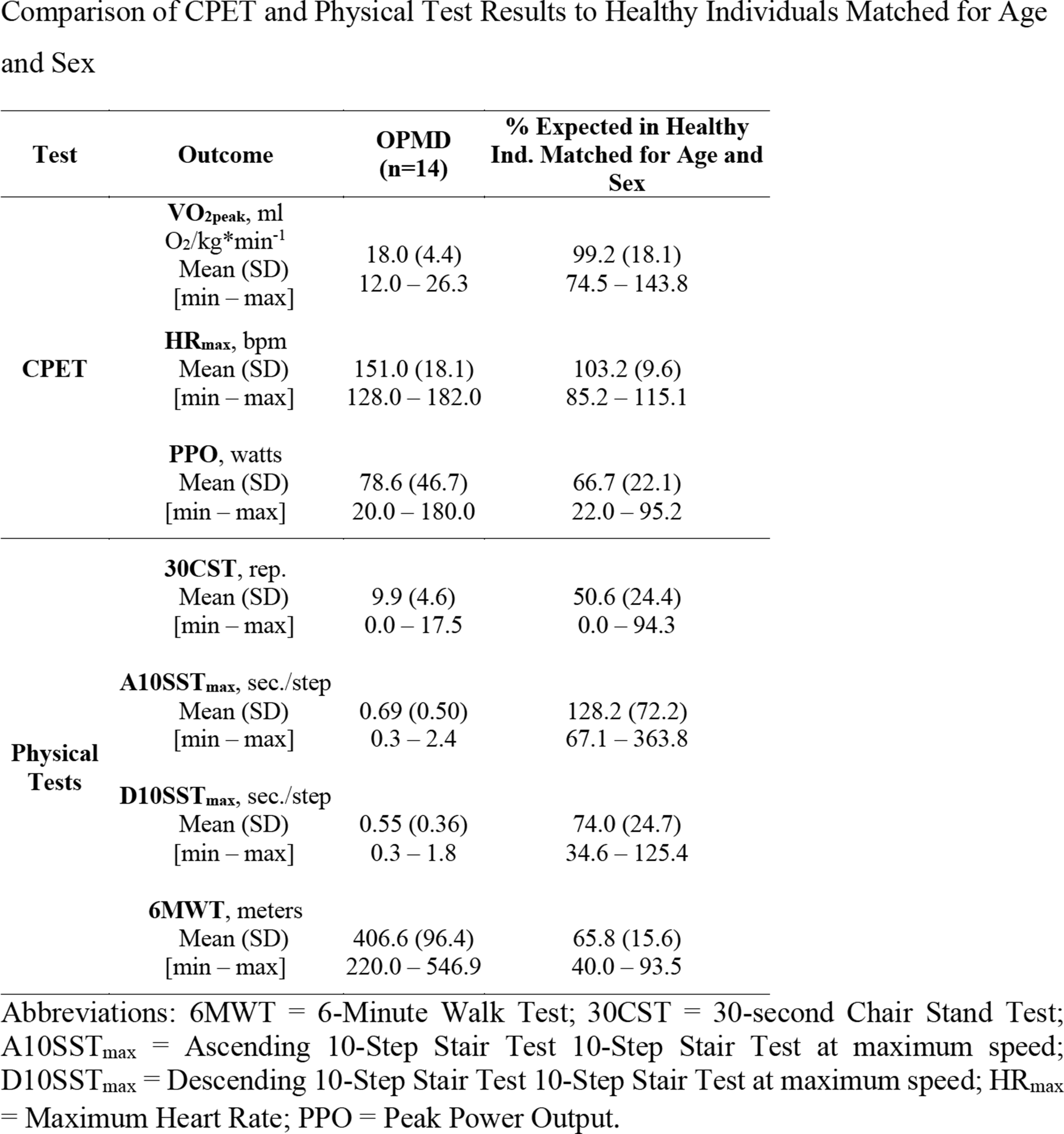
PP01.137
A Transcriptomics-Based Biomarker for Muscle Quality Measures BMN 351 Treatment Response in DMD Patient Biopsies
Dr. Kristin Obrochta Moss, Dr. Katja Hebestreit, Dr. Kevin Larimore, Dr. Daniel Gaffney, Ms. Maria Abbasi, Ms. Lexa Staskus, Dr. Ishnoor Sidhu, Dr. Janine Ilagan, Dr. Evan Witt, Dr. Ling Liu, Dr. Chris Rayner, Dr. Robert Power
BioMarin, San Rafael, CA, United States
Background: BMN 351 is an investigational antisense oligonucleotide (ASO) that is being assessed in a phase 1/2 clinical trial (NCT06280209) in patients with Duchenne Muscular Dystrophy (DMD) amenable to exon 51 skipping. BMN 351 is designed to induce exon skipping and generate near full-length dystrophin at levels sufficient to improve muscle quality and function. Dystrophin expression in skeletal muscle is widely used as a surrogate endpoint for evaluating efficacy of candidate DMD therapies. To supplement the use of dystrophin expression as a measure of response to BMN 351 treatment, we developed an exploratory biomarker that assesses muscle quality and composition using transcriptomic data. These transcriptomics data aim to provide a more comprehensive molecular view of DMD biology and the pharmacodynamic response to BMN 351 in target tissue.
Methods: Transcriptional changes were robustly assessed with limited muscle biopsy material using RNA-seq and targeted droplet digital polymerase chain reaction (ddPCR) assays focused on relevant cell-types and pathways.
Results: To develop the transcriptomics biomarker, we generated bulk RNA-seq data from skeletal muscle in the hDMDdel52/mdx mouse model of DMD and identified a comprehensive transcriptome-wide signature of disease. Genes from three biologically relevant pathways stood out as differentially expressed in the disease model compared with wildtype (WT) controls: muscle development, extracellular matrix, and adaptive immune response. Most notably, all three pathways reverted towards WT with BMN 351 treatment in this mouse model. Quantitative ddPCR assays were developed for select targets, and a composite score was implemented for the ratio of predominantly myoblast-expressed to myofiber-expressed genes. The composite expression ratio was elevated in untreated DMD mice and was reduced toward WT controls with BMN 351 treatment. These results correlated with improved muscle function in the same mice, as measured by gait score. In a set of human muscle samples from patients with DMD and controls, a similar set of disease-associated transcripts were confirmed to be differentially expressed. Reanalysis of publicly available RNA-seq data showed that transcriptomic measures of disease severity correlated with functional outcomes. Together, these findings support the utility of this transcriptomics-based biomarker as an informative measure of muscle quality in patients with DMD. Muscle biopsy samples were then collected at baseline and during treatment from participants in the ongoing BMN 351 phase 1/2 clinical trial. A composite ratio of myoblast-expressed to myofiber-expressed transcripts was elevated in patients with DMD compared to controls. The composite expression ratio was reduced in most trial participants after 13 to 25 weeks of treatment with BMN 351. In addition to interrogating target gene expression, we recently generated RNA-seq data from clinical trial biopsies to evaluate genome-wide expression changes. We hypothesize that these data will identify additional molecular pathways associated with DMD that shift with BMN 351 treatment.
Conclusion: The observed shift in transcriptome expression of muscle maturation and disease markers toward levels observed in healthy controls suggests that treatment with BMN 351 may be improving muscle quality. We anticipate that transcriptomics-based measures of treatment response will compliment dystrophin expression and functional assessments in future BMN 351 clinical trials.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Muscle Dystrophies (Non-Dystrophinopathies)
PP01.138
Personalised Neuromuscular Models Driving New CMS Therapeutics
Prof. Nalini Atchayaram, Miss Suravi Dash, Mr. Prashanth Vashishta, Assoc. Prof. Gautham Arunachal, Assoc. Prof. Seena Vengalil, Mr. Chetan Ghati, Prof. Yogananda Markandeya
National Institute of mental Health and Neurosciences, Bangalore, India
Background: Congenital myasthenic syndromes (CMS) represent a heterogeneous group of inherited neuromuscular disorders characterized by impaired signal transmission at the neuromuscular junction (NMJ). These conditions arise from pathogenic variants in single or multiple proteins critical for synaptic function. The advent of next-generation sequencing technologies has markedly advanced the discovery of novel CMS-associated genes, surpassing the limitations of earlier candidate gene and linkage-based approaches. Importantly, the genetic diversity underlying CMS translates into distinct pathogenic mechanisms, variable clinical phenotypes, and differing prognostic outcomes, thereby necessitating individualized therapeutic strategies. A treatment effective for one CMS subtype may be ineffective or even deleterious in another, underscoring the need for precision medicine. Although certain clinical features may appear gene-specific, considerable phenotypic overlap exists across mutations, and intrafamilial variability suggests the influence of genetic modifiers. Consequently, accurate molecular diagnosis is essential to guide tailored interventions.
Methods: In this study, we investigated an Indian cohort of CMS patients, clinically examined 128 individuals, and sequenced 75 patient samples to date. Analysis using GNOMAD and Varsome databases revealed 35 novel variants spanning 10 genes. Here, we present detailed findings from a single Indian-origin patient exhibiting fluctuating limb-girdle muscle weakness, ptosis, and fatigability.
Results: Family history excluded consanguinity, though her elder brother displayed similar symptoms. Electrophysiological evaluation through repetitive nerve stimulation demonstrated a decremental response in the deltoid muscle. Whole-exome sequencing identified a homozygous MUSK variant c.1742T>A (p.Ile581Asn), consistent with autosomal recessive inheritance and associated acetylcholine receptor deficiency. In silico analysis further revealed that variant p.Ile581Asn leads to substitution of a conserved hydrophobic isoleucine with a larger, polar asparagine and is predicted to disrupt hydrophobic interactions. Conservation analysis confirmed Ile581 as highly conserved across species, and structural modeling classified the variant as likely pathogenic with high confidence (score 0.957). Protein stability predictions using DDMut and MUPro indicated destabilization (ΔΔG −0.34 kcal/mol and −1.7 kcal/mol, respectively). Molecular dynamics simulations performed with GROMACS v2023.3 demonstrated increased RMSD in the MUSK p.Ile581Asn variant compared to wild type, suggesting significant structural perturbation. To establish a personalized NMJ model, patient-specific induced pluripotent stem cells (iPSCs) were generated and validated. These iPSCs exhibited compact morphology, expression of pluripotency markers (OCT4, NANOG, TRA-1-60), normal karyotype, and trilineage differentiation potential confirmed by germ layer markers (Nestin, Troponin-I, Alpha-fetoprotein). Differentiation yielded mature motor neurons expressing OLIG2, ChAT, and β3-tubulin, with patch-clamp recordings confirming neuronal maturity. Parallel differentiation produced myotubes expressing MYOD and MYHC, indicative of successful myogenesis. Future work will involve co-culturing patient-derived motor neurons and myotubes in NMJ microfluidic chambers to further dissect disease mechanisms and therapeutic responses.
Conclusion: Overall, this integrative approach—combining clinical evaluation, genetic analysis, computational modeling, and patient-specific iPSC-derived NMJ models—demonstrates the feasibility of personalized in vitro platforms for mechanistic studies and therapeutic development in CMS.
Abstract Topic Groups (Submission Categories)
Topic Group 6 - Basic Sciences in Neuromuscular Diseases: Neuromuscular Junction
PP01.139
Recurrent Homozygous SGCA Splice-Site Variant c.37+6T>C in Indian Patients with Limb-Girdle Muscular Dystrophy Type 3
Prof. Nalini Atchayaram1, Dr. Dipti Baskar1, Dr. seena vengalil1, Dr. Kiran Polavarapu2, Dr. Muddassu Keerthipriya1, Dr. Saraswati Nashi1, Dr. S G Thenral3, Dr. V L Ramprasad3, Dr. sanjana Angadi1
1
National Institute of Mental Health and Neurosciences, Bangalore, India.
2
CHEO-RI, ottawa, Canada.
3
MEDGENOME, Bangalore, India
Background: Limb-girdle-muscular-dystrophy type 3(LGMD3; OMIM #608099) is a rare autosomal recessive disorder caused by pathogenic variants in the SGCA gene (OMIM *600119), which encodes α-sarcoglycan, which is a critical component of the dystrophin–glycoprotein complex. Disruption of this leads to progressive muscle fiber damage and weakness. While numerous SGCA variants are reported, data on recurrent or founder mutations in the Indian population remain sparse.
Methods: We analyzed the clinical, biochemical, and molecular characteristics of seven unrelated Indian patients diagnosed with LGMD3. Detailed clinical evaluation included age at onset, distribution of muscle weakness, disease progression, family history, and ambulatory status. Next-generation sequencing(n=8) was performed to identify disease-causing variants. Approximately 200ng of DNA was fragmented (250bp inserts), exome-capture enriched, and sequenced on a NovaSeq (Illumina, CA) with paired-end reads to an average depth of 80-100x. SNVs/Indels were annotated using VEP (Ensembl human gene model) and incorporating allele frequencies from population databases(1000 Genomes, GnomAD, TOPmed), in silico predictions (PolyPhen‐2, SIFT, Mutation Taster2, LRT, REVEL, SpliceAI, etc.), and disease databases (OMIM, ClinVar, HGMD). Copy number variants were called using the Exomedepth method. Clinically significant variants was prioritized based on minor allele frequency, published disease associations, support from ≥ 2 in silico prediction tools, proband clinical features, and classified according to ACMG guidelines. RNA extracted from muscle tissue was processed for mRNA PolyA capture and sequenced on the Illumina NovaSeq X Plus platform(2×150 bp). Data was analyzed using RNA-Seq pipeline integrating GATK-based variant calling, differential expression, and aberrant splicing analysis.
Results: The M:F=4:3, ages ranging from 18-58 years. The median age onset and duration at presentation are 23 years(range: 16–38), and 5 years(range: 2–20). All presented with progressive limb girdle weakness. A consistent and notable finding across patients was weakness of the latissimus dorsi and rhomboideus muscles. None demonstrated cardiac or respiratory involvement. Additional clinical features included scapular winging, calf hypertrophy, ankle contractures, macroglossia, and sensorineural hearing loss, highlighting phenotypic variability. Serum CK ranged from 1111-3654 IU/L. Disease progression was slow, and all patients remained ambulant at presentation. All eight patients were homozygous for the shared intronic SGCA variant c.37+6T>C [NC_000017.11(NM_000023.4): c.37+6T>C], located near the 5' splice site of intron 1. This rare variant, affecting a highly conserved nucleotide was absent in population databases (1000 Genomes, TOPMed, gnomAD), segregated with the disease in one family, showing homozygosity in affected siblings and heterozygosity in an unaffected parent, consistent with autosomal recessive inheritance. This variant, predicted to cause aberrant splicing (SpliceAI = 0.5). mRNA testing to confirm the variant pathogenicity is being conducted. The variant's recurrence in unrelated families suggests a founder effect or population-specific origin.
Conclusion: This study identifies a recurrent homozygous SGCA splice-site variant, c.37+6T>C, as a cause of LGMD3 in Indian patients. This expand the genetic and phenotypic spectrum of SGCA-related muscular dystrophy and underscores the importance of considering population-specific variants. Recognition of this recurrent mutation has significant implications for molecular diagnosis, genetic counselling and development of targeted screening approaches in the Indian population.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Muscle Dystrophies (Non-Dystrophinopathies)
PP01.140
Chronological and Spatial Patterns of Muscle Fat Replacement in FHL1-Related Myopathies
Dr. Rui Shimazaki1,2, Dr. Satoru Noguchi2, Dr. Shinichiro Hayashi2, Prof. Kazuma Sugie1, Prof. Ichizo Nishino2
1
Nara Medical University, Kashihara, Japan.
2
National Center of Neurology and Psychiatry, Kodaira, Japan
Background: Variants in the FHL1 gene cause FHL1-related myopathies (FHL1-RMs), a group of neuromuscular disorders with diverse clinical presentations. The pattern of fatty replacement in leg muscle of patients with FHL-RMs have been previously described. This study aimed to comprehensively characterize the spatial and temporal patterns of skeletal muscle fat replacement throughout the whole body in FHL1-RMs, to examine disease progression over time, and to evaluate the relationship between imaging findings and clinical symptoms.
Methods: We retrospectively analyzed 21 whole-body imaging studies from 10 patients with genetically confirmed FHL1-RMs. Fatty replacement was scored in 47 muscles using the modified Mercuri score (mMS). Longitudinal data were used to stratify patients into slow, moderate, and rapid progression groups. K-means clustering was applied to classify muscles based on their chronological patterns of fatty infiltration. Hierarchical clustering and violin plots were used to explore inter-muscle and inter-patient variations.
Results: Despite notable variability in the rate of disease progression, a consistent pattern of muscle involvement was observed across patients. Muscles were classified into three progression clusters: early-onset and early attainment of the maximal mMS (e.g., paraspinal and posterior thigh muscles), steadily progressive (e.g., trunk and lower leg muscles), and late-onset with slow changes (e.g., shoulder and anterior thigh muscles). Initial symptoms appeared in the legs or trunk, followed later by symptoms in the arms in almost all patients. This order appeared to be consistent with the pattern of muscle fatty replacement. Additionally, joint contractures, spinal rigidity, and winged scapula were observed in one to three out of ten individuals, which would be related to fat replacement in the paraspinal spinal muscles and trapezius. These patterns paralleled the clinical symptom progression. In early-stage patients, STIR imaging revealed muscle signal abnormalities preceding fat replacement detectable on T1-weighted images.
Conclusion: This study demonstrates that longitudinal muscle imaging assessments in FHL1-RMs reveal consistent spatial patterns of muscle fat replacement despite marked inter-individual variability in age of onset and progression rates. Fat replacement typically begins in the posterior thigh, lower leg, and paraspinal muscles, later involving the arms and shoulders, with continuous progression in trunk muscles. STIR imaging detected early muscle involvement before overt fat replacement. These findings support longitudinal imaging as a sensitive biomarker for disease staging, early diagnosis, and evaluation of therapeutic efficacy in future clinical trials.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Myofibrillar Myopathies
PP01.141
Progression in Myotonic Dystrophy Type 1; Following Up of Patients With Myotonic Dystrophy Type 1
Dr. Andrea Sipos1, Dr. Endre Pál1, Dr. Dávid Varga1, Dr. Nándor Hajdú2, Miss Réka Héjas1, Mrs Brigitta Ruszin-Perecz1
1
UPMS, Pécs, Hungary.
2
ELTE, Budapest, Hungary
Background: Myotonic dystrophy type 1 (DM1) is the most common adult-onset muscular dystrophy and is characterized by progressive muscle weakness and impaired muscle relaxation (myotonia). The disease typically presents with early distal muscle involvement followed by a gradual distal-to-proximal progression. Myotonia contributes substantially to functional impairment.
Methods: Patients with myotonic dystrophy type 1 underwent a standardized, multimodal evaluation encompassing neuromuscular function, functional performance, and patient-reported outcomes. Muscle strength was assessed using Manual Muscle Testing (MMT) with Medical Research Council–based scoring and complemented by quantitative hand-held dynamometry. Myotonia was evaluated using clinical assessments and functional relaxation tests. Upper limb dexterity was measured with the Nine-Hole Peg Test (9HPT), and lower limb function and walking capacity with the Six-Minute Walk Test (6MWT). Activity limitations and disability were captured using the DM1-Activ scale and the DM Disability Scale. Health-related quality of life was assessed with the SF-36, mood with the Beck Depression Inventory (BDI), and cognitive function with the Montreal Cognitive Assessment (MoCA) and Addenbrooke’s Cognitive Examination.
Results: Longitudinal linear mixed-effects models revealed clear progression of muscular impairment. After correction for multiple comparisons, 35 of 62 outcomes (56.5%) demonstrated significant annual change. Distal lower limb muscles were most affected: ankle dorsiflexion declined ∼0.25 points/year bilaterally (padj < .001). Upper limb strength also deteriorated, with shoulder elevation decreasing 0.17–0.20 points/year (padj < .001), and the composite MIRS score increased over time. Hand grip strength declined 0.38–0.40 kg/year (p< 0.001). In contrast, 9HPT, 6MWT, and myotonia relaxation tests showed no significant longitudinal change. Cognitive function, assessed by MoCA and Addenbrooke’s Cognitive Examination, remained largely stable (MoCA β = −0.37/year), as did SF-36 subscales and the DM1-Activ scale (β = −0.06/year). Only the activities of daily living subscale of the DM1 Disability Scale showed modest but significant worsening (β = 0.19/year). Other self-reported outcomes, including fatigue, social participation, and depressive symptoms remained stable.
Conclusion: These results confirm that DM1 is characterized by progressive muscular weakness, particularly in distal lower limb and proximal upper limb muscles, while functional performance, cognitive function, quality of life, and most patient-reported outcomes remain relatively preserved. Objective strength measures, including manual muscle testing (MMT) parameters and hand grip dynamometry, were the most sensitive indicators of disease progression. Integrating quantitative neuromuscular assessments with patient-reported outcomes provides a comprehensive understanding of disease trajectory and is essential for monitoring progression and evaluating therapeutic interventions in myotoniy dystrophy type 1.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Myotonic Dystrophies
PP01.142
HARMONIA: A Global Phase 3 Trial Assessing the Efficacy and Safety of Z-basivarsen in DM1
Dr. Douglas Kerr, Dr. Soma Ray, Dr. Shuli Yu, Dr. Sowmya Chary
Dyne Therapeutics, Waltham, United States
Background: Myotonic dystrophy type 1 (DM1) is a spliceopathy that results in multi-system clinical manifestations. Zeleciment basivarsen (z-basivarsen, also known as DYNE-101) is an investigational therapeutic that consists of a TfR1-binding Fab conjugated to an ASO designed to target mutant nuclear DMPK RNA in both muscle and CNS with the goal of correcting the underlying spliceopathy. Data from the ongoing Phase 1/2 ACHIEVE trial showed that z-basivarsen had a favorable safety profile. At the selected dose of 6.8 mg/kg Q8W, z-basivarsen led to substantial knockdown of DMPKRNA levels and improvement in splicing, functional improvement across diverse clinical measures, and improvement in patient reported outcomes, including CNS-related measures.
Methods: ACHIEVE informed the initial safety and efficacy of z-basivarsen, which will continue to be evaluated in HARMONIA, a global Phase 3, placebo-controlled, randomized, double-blind study designed to assess the efficacy, safety, and tolerability of 6.8 mg/kg Q8W z-basivarsen administered intravenously in participants with DM1.
Results: HARMONIA plans to enroll ∼150 participants aged ≥16 years, expanding upon ACHIEVE by including a broader DM1 population. Participants will be randomized 1:1 to receive either z-basivarsen 6.8 mg/kg Q8W or placebo during the 48-week placebo-controlled period, after which they will be eligible to enter an LTE portion. The primary objective is to evaluate the effect of z-basivarsen compared with placebo on improvement in muscle function as measured by the change from baseline in 5 Times Sit-to-Stand (5xSTS). 5xSTS is a validated and reliable measure of lower-extremity strength and functional mobility. It measures the ability to perform activities of daily living and has been shown to be an independent predictor of fall risk, of particular importance in DM1 due to the intrinsically high risk of falls and subsequent injury. Thus, 5xSTS is a clinically relevant endpoint to capture functional improvement in DM1. Key secondary objectives include measures of myotonia, muscle strength and function, as well as patient and clinician-reported outcomes. Moreover, given DM1’s significant CNS component, the study will evaluate potential effects on CNS-related disease burden, including both cognitive and non-cognitive symptoms (e.g., fatigue, sleep), using a combination of actigraphy, clinical, and patient-reported outcomes measures.
Conclusion: HARMONIA is designed to rigorously evaluate the multi-systemic impact of spliceopathy correction using functional endpoints related to activities of daily living.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Myotonic Dystrophies
PP01.143
Somatic Repeat Expansion Rate Is Higher in DM1 Patients With a Faster Muscle Weakness Progression
Prof. Dusanka Savic-Pavicevic1, Asst. Prof. Jovan Pesovic1, Dr. Lana Radenkovic1, Asst. Prof. Milos Brkusanin1, Prof. Goran Brajuskovic1, Dr. Vladimir Jovanovic2, Prof. Vidosava Rakocevic-Stojanovic3,4, Asst. Prof. Stojan Peric3,4, Mr. Nemanja Radovanovic1
1
University of Belgrade-Faculty of Biology, Center for Human Molecular Genetics, Belgrade, Serbia.
2
Human Biology and Primate Evolution, Department of Biology, Chemistry, and Pharmacy, Freie Universität Berlin, Berlin, Germany.
3
University of Belgrade-Faculty of Medicine, Belgrade, Serbia.
4
Neurology Clinic, University Clinical Center of Serbia, Belgrade, Serbia
Background: Myotonic dystrophy type 1 (DM1) is caused by an unstable expansion of CTG repeats in the 3' untranslated region of the DMPK gene within a range from 50 to 6000 repeats. Ongoing somatic repeat instability is expansion-biased, contributes to individual patient variability, confounds genotype-phenotype correlation and is assumed to underlie the progressive nature of DM1. Here, we aimed to examine somatic expansion dynamics and their relation to the progression of skeletal muscle weakness.
Methods: We performed a longitudinal study on 39 adult DM1 patients with pure CTG repeats, followed up for 7.6±1.3 years (range 5-9 years). Patients were blood-genotyped at baseline and follow-up using single-cell small-pool PCR. We used baseline 10th percentile allele length as the best estimate of progenitor allele length (ePAL), modal allele length (MAL) as a measure of somatic expansions, and MAL increment as a measure of longitudinal increase in somatic expansion. Muscle strength was manually assessed using the MRC scale with a maximal score of 20 points. Patients were classified as progressive based on a decrease of ≥2 points during follow-up. Statistical analysis included linear mixed-effect (LME) models and logistic regression.
Results: According to an LME model, the MAL increased over time with larger ePAL (p=1.22e–15), older baseline age (p=2.83e–4) and elapsed time (p=6.55e–13), with a slight decrease due to the interaction between ePAL and elapsed time (p=0.037). A representative cohort individual with a mean ePAL of 425 repeats and a mean baseline age of 40 years was characterized by an average annual rate of MAL increase of approximately 20 repeats. The model explained 79.1% of the variance in MAL increase (marginal R²=0.791) with an additional 15.5% when accounting for baseline individual differences (conditional R²=0.946). To examine the annual rate of MAL increase in progressive versus non-progressive patients, we incorporated the interaction between elapsed time and progression status in the above described LME model while accounting for baseline MRC score. We observed a higher annual rate of MAL increase in the progressive group compared to the non-progressive (21.8 vs. 13.8 repeats) (p=0.030). To explore the risk of MRC score-defined muscle weakness progression, we performed logistic regression using progression status as the outcome variable and MAL increment and elapsed time as the predictors. When adjusting for baseline age, each 10-repeat gain in MAL increment corresponded to a 15% increase in the odds of progression (p=0.019), while each additional year corresponded to an increase in odds by a factor of 2.39 (p=0.020).
Conclusion: Our results demonstrate that the rate of somatic expansions is higher in blood cells of patients experiencing a more rapid decline in muscle strength and emphasize the usefulness of MAL increment as a biomarker for disease progression. Collectively, our findings suggest that therapeutic interventions aimed at stabilizing ongoing somatic expansions could significantly alter the natural history of DM1 by slowing the functional skeletal muscle decline. This research was supported by the Science Fund of the Republic of Serbia, #7754217, READ-DM1.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Myotonic Dystrophies
PP01.144
Cognitive and Behavioral Impairment in Myotonic Dystrophy Type 2: Evidence From ECAS and Plasma Biomarkers
Dr. Erica Frezza1,2, Dr. Marzia Nuccetelli3, Dr. Ilaria Petitta1, Dr. Mariangela Goglia1, Dr. Giulia Greco1, Dr. Francesco Gruosso1,2, Dr. Giovanni Vietri1,2, Dr. Laura Boffa4, Dr. Diego Centonze4,2, Dr. Roberto Massa1,2
1
Unit of Neuromuscular Diseases, Department of Systems Medicine, Tor Vergata University of Rome, Rome, Italy.
2
IRCCS Neuromed, Pozzilli, Italy.
3
Department of Laboratory Medicine, Fondazione Policlinico Tor Vergata, Rome, Italy.
4
Neurology Unit, Fondazione Policlinico Tor Vergata, Rome, Italy
Background: Myotonic dystrophy type 2 (DM2) is a multisystem disorder in which central nervous system involvement is increasingly recognized. However, the cognitive–behavioral profile of DM2 and its relationship with circulating biomarkers of neurodegeneration remain poorly defined. Notably, plasma phosphorylated tau (pTau-181) has not previously been investigated in DM2.
Methods: Patients with genetically confirmed DM2 were consecutively recruited. All participants underwent a comprehensive clinical assessment including motor performance (6-Minute Walk Test [6MWT], quantitative muscle testing [QMT] sum score, Quick Motor Function Test [QMFT]), fatigue and sleep questionnaires, and cognitive and behavioral evaluation using the Edinburgh Cognitive and Behavioral ALS Screen (ECAS). Plasma levels of neurofilament light chain (NfL), pTau-181 and GFAP were measured. Descriptive statistics, group comparisons, and correlation analyses corrected for multiple comparisons (false discovery rate, FDR) were performed.
Results: Seventeen DM2 patients were included (58.8% females), with a mean age of 47.5 ± 19.3 years. Cognitive impairment was observed in 46.7% of patients, predominantly affecting language, memory, verbal fluency, and executive functions, while visuospatial abilities were relatively preserved. Behavioral changes were mild, with apathy being the most frequent feature (44%). No significant sex-related differences were detected across clinical, cognitive, or biomarker measures. Patients with pathological NfL levels (40%) showed significantly worse global and domain-specific cognitive performance, along with higher plasma pTau-181 and GFAP concentrations. FDR-corrected correlation analyses revealed strong associations between older age, reduced muscle strength, cognitive dysfunction, behavioral impairment, and increased NfL and pTau-181 levels.
Conclusion: DM2 is characterized by a clinically relevant cognitive–behavioral involvement closely linked to age, muscle weakness, and biomarkers of neurodegeneration. Elevated NfL and pTau-181 levels were associated with cognitive impairment, supporting an early neurodegenerative component in DM2 and identifying plasma pTau-181 as a potential marker of central nervous system involvement in this disease.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Myotonic Dystrophies
PP01.145
Efficacy and Safety of Metformin in Myotonic Dystrophy Type 1: The MetMyd Study
Dr. Erica Frezza1,2, Dr. Salvatore Rossi3, Dr. Alessia Perna4, Dr. Elisabetta Bucci5, Dr. Giulia Greco1, Dr. Mariangela Goglia1, Dr. Laura Tufano5, Dr. Vittorio Riso3, Dr. Virginia Veronica Visconti6, Prof. Annalisa Botta6, Dr. Marzia Nuccetelli7, Prof. Giuseppe Novelli6, Prof. Giovanni Antonini5, Dr. Antonio Petrucci4, Prof. Gabriella Silvestri3, Prof. Roberto Massa1,2
1
Unit of Neuromuscular Diseases, Department of Systems Medicine, Tor Vergata University of Rome, Rome, Italy.
2
IRCCS Neuromed, Pozzilli, Italy.
3
Institute of Neurology, Department of Neuroscience, Catholic University of the Sacred Heart, Rome, Italy.
4
Center for Neuromuscular and Neurological Rare Diseases, San Camillo-Forlanini Hospital, Rome, Italy.
5
Unit of Neuromuscular Diseases, Sant'Andrea Hospital, Sapienza University of Rome, Rome, Italy.
6
Unit of Medical Genetics, Department of Biomedicine and Prevention, Tor Vergata University of Rome, Rome, Italy.
7
Department of Laboratory Medicine, Tor Vergata University Hospital, Rome, Italy
Background: Metformin is a potential systemic treatment for Myotonic Dystrophy type 1 (DM1) since it can interfere with several pathomechanisms of the disease. We evaluated the effects of a 24-month metformin treatment on mobility and safety in adult DM1 patients.
Methods: METMYD is a multi-center, randomized, triple-blind, two-arm (1:1), placebo-controlled, phase III non-profit clinical trial. The primary endpoint is a better performance at 6MWT at the end of study as compared to baseline. Secondary endpoints included quantitative muscle test, dexterity, fatigue and InQOL. Moreover, circulating alternative splicing products and markers of oxidative stress were measured.
Results: One-hundred and forty-six patients were randomized and 95 performed the final visit. The two arms at baseline did not differ for sex, age, BMI, HOMA index and 6MWT performance. Fifty-one patients (28 metformin, 23 placebo) dropped out for various reasons, mainly diarrhea and non-compliance. No treatment-related SAEs were reported. In the ITT population (n=95), only metformin patients showed a statistically significant improvement of BMI and HOMA index. In the ITT analysis, the mean 6MWT in the metformin arm increased at 24 months by +22.7 m from baseline (p < 0.05), corresponding to +6.3% improvement, compared with +0.47% in the placebo arm (+0.6 m, p = 0.96). In the per-protocol analysis, improvement in the treated group was even greater (+12.4%) without change in the placebo (+0.83%), with a median between-group difference of +36 m favoring metformin, which was statistically significant (p = 0.04).
Conclusion: Metformin induced a partial modification of misplicing in IR and FAS genes and a significantly improvement of Total Antioxidant Capacity. In DM1 patients, metformin is safe and ameliorates mobility. This benefit may depend on different mechanisms such as splicing correction, reduction of oxidative stress, improvement of glucose metabolism. Metformin may be useful in mitigating disease progression in DM1.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Myotonic Dystrophies
PP01.146
Neurofilament Light Chain as a Surrogate Marker for Motor Functions in Myotonic Dystrophy Type 1
Assoc. Prof. Jin-Sung Park1, Assoc. Prof. Jin-Mo Park2
1
Kyungpook National University Chilgok Hospital, Daegu, Korea, Republic of.
2
Dongguk University Hospital, Gyeung-Ju, Korea, Republic of
Background: Myotonic dystrophy type 1 (DM1) is a genetic disease caused by an abnormal expansion of CTG trinucleotide repeats in the dystrophia myotonica-protein kinase (DMPK) gene. Neurofilament light chain (NfL), a biomarker of neuroaxonal damage, has emerged as a promising indicator in various neurological conditions. This study aimed to evaluate the potential of NfL as a biomarker of disease severity by examining its correlation with motor function scales in ambulatory patients with DM1.
Methods: A cohort of 33 patients with DM1 was enrolled and compared to a control group of 31 healthy individuals using plasma NfL levels. Clinical data collected included age, age at disease onset, sex, and disease duration. Laboratory assessments were evaluated along with the 6-minute walk test (6MWT) and the Muscular Impairment Rating Scale (MIRS). In addition, pulmonary function tests and nerve conduction studies (NCS) were also included in the analysis.
Results: We found a significant positive correlation between NfL levels and the MIRS score after adjusting for age (τ = 0.387, p = 0.002). Additionally, a significant negative correlation was observed between age-adjusted NfL levels and 6MWT (τ = −0.344, p = 0.014). Disease duration also demonstrated a statistically significant positive association with NfL levels after age adjustment (τ = 0.334, p = 0.007). A negative correlation was found between NfL levels and peroneal compound muscle action potential (CMAP) amplitudes (τ = −0.310, p = 0.024); however, this did not remain statistically significant after age adjustment (τ = −0.236, p = 0.090).
Conclusion: In conclusion, NfL demonstrated statistically significant correlations with both disease duration and motor function scales in DM1, highlighting its potential as a surrogate biomarker of current disease burden.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Myotonic Dystrophies
PP01.147
Gynecological Disorders in Women With Myotonic Dystrophy Type 1: A Retrospective Study
Dr. Patricia Garay-Albízuri1,2,3, Dr. Roberto Fernández-Torrón1,2,3, Dr. Pablo Iruzubieta1,2,3,4, Dr. Juan José Poza1,2,3, Dr. María José Iñarra Velasco5, Dr. María Lure-Berregi5, Dr. Loreto Martorell6,7
1
Department of Neurology, Donostialdea Integrated Health Organisation, Osakidetza, San Sebastián, Spain.
2
Neuromuscular Diseases Group, Neurosciences Area, Biogipuzkoa Health Research Institute, San Sebastián, Spain.
3
CIBERNED, CIBER, Spanish Ministry of Science & Innovation, Carlos III Health Institute, Madrid, Spain.
4
Department of Neurology and Neurosurgery, Montreal Neurological Hospital and Institute, McGill University, Montreal, Canada.
5
Department of Gynecology, Donostialdea Integrated Health Organization, Osakidetza, San Sebastián, Spain.
6
Department of Genetic and Molecular Medicine-IPER, Hospital Sant Joan de Déu and Institut de Recerca Sant Joan de Déu, Barcelona, Spain.
7
Center for Biomedical Research Network on Rare Diseases (CIBERER), ISCIII, Madrid, Spain
Background: Gynecological disorders are highly prevalent among women with myotonic dystrophy type 1 and have a significant impact on women’s health and quality of life. However, the prevalence of individual gynecological comorbidities in this population has not been systematically assessed.
Methods: We conducted a single-center retrospective study including women with myotonic dystrophy type 1 followed at a tertiary care hospital between January 2011 and November 2025. Data on major gynecological disorders were collected, including the presence of uterine myomas, polyps, gynecological neoplasms and/or history of gynecological surgery, together with other clinical and demographic variables.
Results: A total of 176 women were included, with a median follow-up time of 14.95 years (IQR 14,75–14.95). Ninety-four women (53.9%) presented gynecological disorders, the most frequent being uterine fibroids (47%), followed by endometriosis and endometrial polyps (13.6% each). The presence of gynecological disorders was significantly associated with age using the U Mann-Whitney test. Fifteen patients developed gynecological malignancies, with ovarian cancer being the most common (n = 7). During follow-up, 27,8% of women (N=49) underwent hysterectomy, with multiple uterus fibroids as the leading indication (45% of all hysterectomies); all women with this condition underwent hysterectomy. Twenty-two women of this group (44,9%) had additionally a double anexectomy.
Conclusion: Gynecological disorders are common in women with myotonic dystrophy and represent a relevant source of morbidity in a substantial proportion of patients. These findings highlight the need for a multidisciplinary management approach in this population.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Myotonic Dystrophies
PP01.148
Oculopharyngeal Muscular Dystrophy in a Korean Family with a Pathogenic PABPN1 Gene Duplication
Prof. Hyunjin Ju1, Prof. Sookin Moon2
1
Department of Neurology, Soonchunhyang University Seoul Hospital, Soonchunhyang University College of Medicine, Seoul, Korea, Republic of.
2
Department of Laboratory Medicine, Nowon Eulji Medical Center, Eulji University, Daejeon, Korea, Daejeon, Korea, Republic of
Background: Oculopharyngeal muscular dystrophy (OPMD) is a late-onset neuromuscular disorder caused by short polyalanine expansions in the PABPN1 gene. Although OPMD has been well characterized in certain ethnic populations, genetically confirmed cases in Koreans remain extremely rare, and familial cases have scarcely been reported.
Methods: A 68-year-old woman presented with about five years of progressive proximal weakness, mainly difficulty climbing stairs. On neurological examination, bilateral ptosis, dysphagia, and dysarthria with nasal speech were noted. Her accompanying older brother in his 70s exhibited similar symptoms. Detailed family history taking revealed multiple affected individuals across successive generations involving both sexes, suggesting an autosomal dominant inheritance pattern [Figure 1].
Results: Laboratory studies showed normal thyroid function and serum creatine kinase levels (132 U/L). Anti-acetylcholine receptor antibodies and repetitive nerve stimulation were negative. Needle electromyography did not show abnormal denervation potentials or pathological neurogenic/myogenic motor unit potentials. Thigh MRI revealed diffuse fatty replacement predominantly affecting the posterior compartment muscles bilaterally. Targeted next-generation sequencing identified a heterozygous pathogenic duplication in the PABPN1 gene (NM_004643.3: c.15_23dup, p.Ala9_Ala11dup) in the proband, confirming the diagnosis of OPMD. Additional genetic analysis was performed on the asymptomatic 41-year-old daughter of the affected brother; the absence of the mutation in this individual was confirmed.
Conclusion: This case highlights the critical role of careful neurological examination and deep family history assessment in identifying inherited neuromuscular disorders that may otherwise be misattributed to normal aging. This report expands the clinical and genetic spectrum of OPMD in the Korean population.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Oculopharyngeal Muscular Dystrophy
Images or Table (Optional)
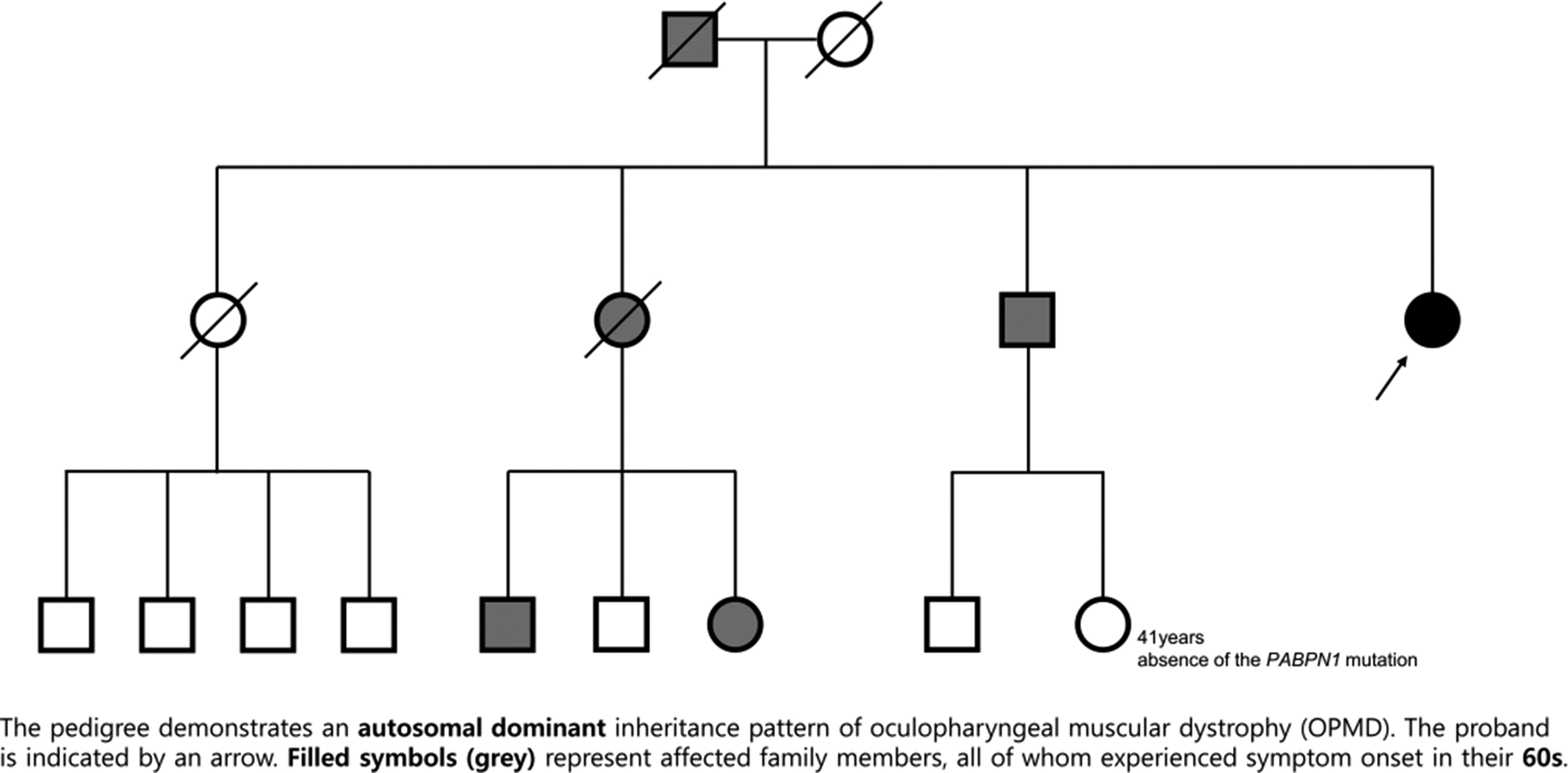
PP01.149
Mitochondrial Dysfunction in Phosphaturic Mesenchymal Tumor-Induced Myopathy and Various Clinical Presentations of Phosphaturic Mesenchymal Tumors
Dr. Ahwon Kim1, Dr. Do-Yeon Lee2, Dr. Sue young Ha2, Prof. Jung-Joon Sung2
1
Myongji Hospital, Goyang, Korea, Republic of.
2
Seoul National University Hopsital, Seoul, Korea, Republic of
Background: Phosphaturic mesenchymal tumors (PMTs) are very rare soft tissue tumors that present with pain, bone fracture, and weakness attributed to hypophosphatemia. To our knowledge, biopsy-confirmed cases of myopathy in PMT have not been previously documented, and the pathophysiology of myopathy in PMT is unknown.
Methods: We reviewed the clinical and pathological characteristics of PMT and investigated the pathophysiology of PMT-induced myopathy. From retrospective chart reviews, 15 patients with biopsy-confirmed PMT were identified from 2004 to 2020 in two tertiary hospitals. In order to investigate the mitochondrial dysfunction in PMT-induced myopathy, the muscle tissues from two patients with or without myopathy, who had limb weakness, were compared with normal muscle tissues on the basis of mitochondrial protein expression.
Results: Most of the patients had osteomalacia or hypophosphatemia. Thirteen of the 15 PMT cases occurred on the left. Nine patients were overweight or obese. One patient had PMT-induced myopathy while the other had intracranial hemorrhage. Immunohistochemistry revealed that the expression of mitofusin 2, which is related to mitochondrial fusion, and ATP5B, mitochondrial ATP synthase subunit, were significantly decreased while the expression of mitochondrial fission factor and dynamin-related protein 1, which is related to mitochondrial fission, were significantly increased in the PMT-induced myopathy compared to the patient who had no myopathy but limb weakness, and the normal controls. PMT-induced hypophosphatemia leads to mitochondrial dysfunction resulting in decreased ATP synthesis.
Conclusion: Clinicians should be aware of various clinical manifestations of PMT, risk of weight gain, and reversibility of symptoms by tumor removal.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Other Myopathies
PP01.150
Clinical and Genetic Profiling of Multisystemic Proteinopathies: Insights From Indian Cohort
Dr. Dipti Baskar1, Prof. Atchayaram Nalini1, Dr. Seena Vengalil1, Dr. Saraswati Nashi1, Dr. Kiran Polavarapu2, Dr. Muddasu Suhasini Keerthipriya1, Dr. Ananthapadmanabha Kotambail1, Dr. Gautham Arunachal1
1
National Institute of Mental Health and Neuro Sciences (NIMHANS), Bengaluru, India.
2
Children's Hospital of Eastern Ontario Research Institute, Ottawa, Canada
Background: Multisystemic proteinopathies (MSPs) are a heterogeneous group of genetically determined disorders characterized by overlapping involvement of motor neurons, skeletal muscle, and other organ systems with shared pathogenic mechanisms of disruptions in protein homeostasis that promote aberrant cytoplasmic protein aggregation in various tissues. Although MSPs are increasingly recognized, data from the Indian population remain sparse, limiting understanding of population-specific genetic diversity and phenotypic expression. This study aims to comprehensively characterize the clinical and genetic spectrum of MSPs in an Indian cohort and identify novel genotype–phenotype correlations.
Methods: Patients with MSPs were included from a quaternary neuromuscular referral center. Detailed clinical phenotyping along with neuroimaging, laboratory and electrophysiological data were analysed. Genetic testing by whole exome sequencing identified pathogenic and likely pathogenic variants across MSP-associated genes, followed by correlation with clinical presentations were done.
Results: A total of 27 patients harboring variants in nine MSP genes were included (Figure-1). The most frequently involved gene was OPTN (n=7), followed by SQSTM1 (n=6) and VCP (n=5). The median age at onset was 42 years (range: 4–65), with a median disease duration of 1.5 years (range: 0.5–22). There was a male predominance (M:F = 19:8). Motor neuron disease (MND) was the most common phenotype, observed in 20 patients (74.1%). Limb-onset MND predominated; however, four patients demonstrated prominent lower limb spasticity consistent with a primary lateral sclerosis phenotype, associated with variants in ANXA11, OPTN, TFG, and VCP.Severe spasticity was particularly notable in OPTN (c.882+2_882+3del, 5′ splice site) and VCP (c.476G>A, p.Arg159His). Additional neurological features associated with MND included hand dystonia in OPTN (c.882+2_ 882+3del, 5’splice site, along with ataxia and behavioural symptoms) and VCP (c.464G>A, p.Arg155His) and parkinsonism in VCP (c.290G>A, p.Gly97Glu). Myopathy was identified in seven patients (25.9%), associated with SQSTM1, VCP, HSPB8 and HNRNPA1. Patterns included proximo-distal weakness (n=4), limb-girdle involvement (n=2), and exertional myalgia with cramps (n=1). Patients with SQSTM1variants c.1184_1187delTTGA (p.Ile395Serfs) and c.1178G>A (p.Arg393Gln) exhibited distinctive finger extensor weakness and macroglossia with tongue infolding. Novel cutaneous manifestations, including hyperpigmentation and blackish plaque-like lesions, were observed in OPTN (c.882+2_ 882+3del) and VCP (c.290G>A, p.Gly97Glu), respectively. None of the patients had cognitive or bone involvement. Novel gene variants were noted in a significant number of patients (n=9, 33.3%). One patient demonstrated a unique compound heterozygous variation in OPTN (c.580G>T, p.Glu194Ter) and C9orf12 (c.166del, p.Ala56LeufsTer6), presenting with rapidly progressive bulbar-onset MND and severe respiratory involvement.
Conclusion: This study represents one of the most comprehensive clinico–genetic analyses of MSPs from the Indian subcontinent. The identification of novel variants, previously unreported neurological and cutaneous features, and a unique oligogenic presentation substantially expands the known phenotypic and genetic spectrum of MSPs. These findings highlight population-specific diversity, beyond classical phenotypes, and provide novel insights with implications for diagnosis, genetic counseling, and future mechanistic and therapeutic studies.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Other Myopathies
Images or Table (Optional)
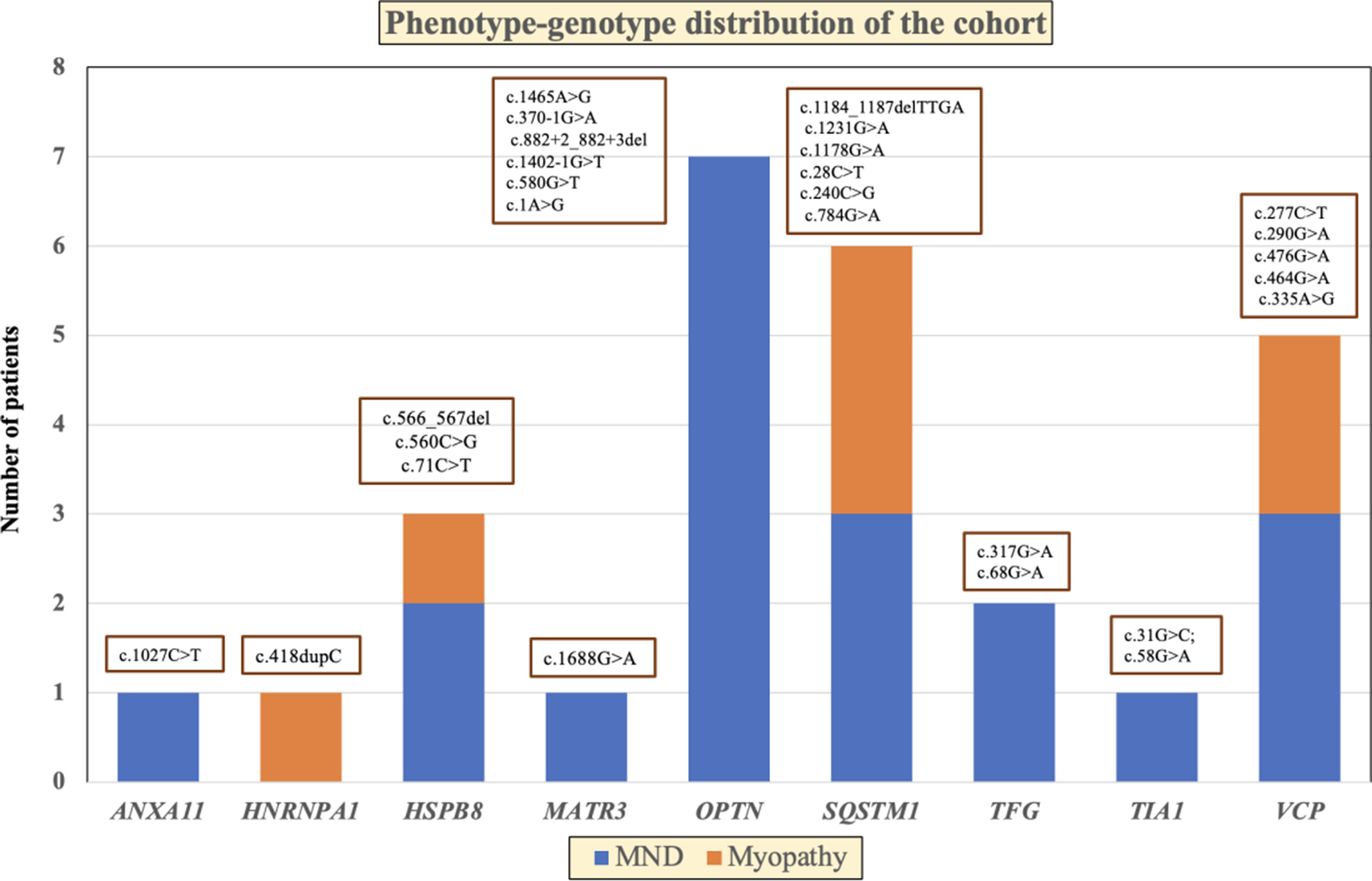
PP01.151
Occult and Overt Myopathy in Adult Cystinosis: Clinical Characterization and Longitudinal Assessment
Dr. Edouard Berling1, Dr. Aude Servais2,3, Prof. Hélène Prigent4, Dr. Clément Guémy1, Dr. Alice Rouyer1, Dr. Claire Lefeuvre1, Prof. Guillaume Nicolas1, Prof. Pascal Laforêt1
1
Service de Neurologie, AP-HP, Hôpital Raymond Poincaré, Centre de référence Nord-Est-Ile-de-France, Garches, France.
2
ERKNet Department of Kidney and Metabolic Diseases, Transplantation and Clinical Immunology, Necker Hospital, AP-HP, Centre of Reference for the French Nationwide MARHEANetwork (CNR-MARHEA), Paris, France.
3
Inserm U1163, Imagine Institute, Paris, France.
4
Physiology Department, Hopital Raymond Poincaré GHU APHP Université Paris Saclay, Garches, France
Background: Cystinosis is a rare lysosomal storage disorder characterized by progressive multiorgan cystine accumulation. With improved survival into adulthood owing to cysteamine therapy and kidney transplantation, late-onset extrarenal complications have become increasingly apparent. Myopathy is a recognized but underdiagnosed manifestation, often evolving insidiously and potentially leading to severe respiratory and swallowing complications. Sensitive tools for early detection and longitudinal monitoring of cystinosis-associated myopathy remain poorly defined.
Methods: We conducted a prospective, longitudinal observational study in adults with genetically confirmed cystinosis recruited from French national reference centers between 2020 and 2025. Participants underwent comprehensive neuromuscular, respiratory, and swallowing assessments at baseline and after 12 months. Muscle involvement was evaluated using clinical examination, Medical Research Council sum score (MRCss), functional motor tests, and upper-limb strength and dexterity measures. Patients were classified as having clinically overt muscle weakness (MRCss <58/60) or no overt weakness. Among those without overt weakness, occult muscle involvement was defined by reduced vital capacity (<80% predicted) and/or swallowing impairment. Longitudinal changes in motor and functional outcomes were analyzed.
Results: Twenty-four adults (median age 36 years) were included. Clinically overt muscle weakness was present in 10 patients (41.7%). Respiratory insufficiency, dysphagia, and sleep apnea were frequent, particularly in patients with overt weakness, but were also observed in those without apparent weakness. Functional motor performance was more impaired in patients with overt weakness, notably affecting distal strength, hand dexterity, and global motor function. Among patients without overt weakness, 57.1% exhibited occult muscle involvement, characterized primarily by respiratory or swallowing abnormalities. In this subgroup, dominant-hand grip and key pinch strength were frequently reduced despite preserved gait and global motor function. Over 12 months, most strength and functional measures remained stable, with only minor declines observed in selected timed motor tasks.
Conclusion: Muscle involvement in adult cystinosis is common and frequently under-recognized, including in patients without clinically overt weakness. Subtle distal strength deficits, particularly in the dominant hand, may signal occult myopathy and associated respiratory or swallowing complications. Short-term progression appears limited, highlighting a window for early detection and preventive intervention. Simple upper-limb strength assessments may represent practical screening tools to guide referral and longitudinal monitoring in this rare population.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Other Myopathies
PP01.152
Fbxo30/MUSA1 is a Novel Critical Regulatory Element for Z-Line Homeostasis and Skeletal Muscle Function
Miss Martina Esposito1,2, Dr. Andrea Armani3, Dr. Davide Steffan2, Dr. Leonardo Nogara1,2, Prof. Bert Blaauw1,2, Dr. Paolo Grumati4, Dr. Sofia De Felice5, Prof. Laura Cendron5, Prof. Marco Sandri1,2
1
Università degli Studi di Padova, Department of Biomedical Sciences, Padova, Italy.
2
Veneto Institute of Molecular Medicine, Padova, Italy.
3
Institute of Neuropathology, UZH, Zurich, Switzerland.
4
Telethon Institute of Genetic and Medicine (TIGEM), Napoli, Italy.
5
Università degli Studi di Padova, Department of Biology, Padova, Italy
Background: Skeletal muscle health and function are guaranteed by a finely tuned balance between synthesis of new proteins and degradation of damaged structures and organelles. However, under catabolic conditions degradative processes exceed de novo protein synthesis and organelle biogenesis leading to muscle atrophy, which is regulated by a specific genetically-encoded program. In the search for new players involved in muscle mass regulation, in the past years we identified Fbxo30/MUSA1, a previously uncharacterized E3 ubiquitin ligase that sits at the crossroads of two fundamental pathways controlling muscle mass; indeed, MUSA1 expression is driven by FoxO3 during catabolic conditions, while it is suppressed by the BMP signaling.
Methods: To better characterize MUSA1 role in skeletal muscle physiology, we generated muscle-specific knock-out mice.
Results: MUSA1 deletion leads to progressive sarcomere disorganization in knock-out muscles. In particular, histological and electron microscope analyses revealed the presence of wide areas within aged knock-out muscle fibers characterized by the presence of indigested proteinaceous material completely disrupting sarcomeres architecture. Moreover, these aggregates appear as enlarged Z-line, resembling nemaline rods, a feature typical of nemaline myopathies. As a consequence, these ultrastructural alterations lead to muscle weakness in aged knock-out animals. In line with these evidences, aged knock-out muscles proteomes show progressive accumulation of proteins important for sarcomeres and cytoskeleton assembly and stabilization, together with Z-line components. For this reason, we speculate that MUSA1 could be involved in some of these proteins turnover. Indeed, we demonstrated by in vitro ubiquitination assay that MUSA1 is responsible for actinin 3 (ACTN3) poly-ubiquitination when coupled with the E2 conjugating enzyme UbcH2.
Conclusion: Concluding, our data support MUSA1 as a novel critical player in controlling Z-line homeostasis together with muscle function. However, further investigations are needed to better characterize MUSA1 substrates and to precisely dissect the molecular pathogenic mechanisms involved in the perspective of therapeutic intervention.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Other Myopathies
PP01.153
Amyloid Myopathy: An Underrecognized but Potentially Treatable Condition
Dr. Carolina Azcona, Dr. Marcelo Rugiero, Dr. Erika Brulc, Dr. Maria Adela Aguirre, Dr. Rocio Blanco, Dr. Diego Perez de Arenaza, Dr. Silvia Christiansen, Dr. Elsa Nucifora
Hospital Italiano, Ciudad Autonoma de Buenos Aires, Argentina
Background: Amyloid myopathy is a rare and likely underdiagnosed manifestation of systemic light-chain (AL) amyloidosis, in which renal and cardiac involvement are more commonly recognized. When skeletal muscle involvement predominates early in the disease course, the condition may be misclassified as an acquired or, less frequently, a genetic myopathy, leading to delayed recognition of the underlying plasma cell dyscrasia. Although early diagnosis and timely initiation of therapy are associated with improved outcomes in AL amyloidosis by limiting irreversible organ damage, data regarding functional skeletal muscle recovery following effective systemic treatment remain scarce.
Methods: We report a 63-year-old woman with systemic AL amyloidosis and progressive muscle weakness. Relevant clinical data were retrospectively reviewed
Results: The patient presented with a 2-year history of progressive, symmetric proximal weakness involving all four limbs and axial muscles, later progressing to distal, bulbar, and respiratory involvement. Due to the severity of weakness, she became wheelchair-dependent and required assistance with activities of daily living. Serum muscle enzymes were elevated (creatine kinase 990 U/L; aldolase 15 U/L). Muscle magnetic resonance imaging revealed muscle edema involving the sartorius, hamstrings, adductor magnus, and tibialis anterior muscles, along with moderate fatty replacement of the gluteal muscles, sartorius, hamstrings, and adductor magnus, with symmetric involvement of both anterior and posterior compartments of the lower limbs.
An inflammatory myopathy was initially suspected by rheumatology; however, there was no response to immunosuppressive therapy, myositis-specific antibodies were negative, and the initial muscle biopsy was non-diagnostic. Whole-exome sequencing revealed no clinically relevant variants, making a genetic myopathy less likely as well. The patient subsequently developed symptoms of heart failure. Cardiac evaluation showed a pseudo-infarction pattern on electrocardiogram, restrictive physiology, and cardiac magnetic resonance imaging findings consistent with amyloid cardiomyopathy. Bone scintigraphy with technetium-labeled pyrophosphate excluded transthyretin amyloidosis. Laboratory studies identified a monoclonal gammopathy with markedly elevated free kappa light chains (3532.09 mg/L) and suppressed lambda light chains (6.62 mg/dL). Bone marrow biopsy confirmed plasma cell dyscrasia, with flow cytometry demonstrating a clonal kappa-restricted plasma cell population. Reevaluation of the muscle biopsy demonstrated Congo red–positive deposits with apple-green birefringence under polarized light within epimysial and perimysial vessels, consistent with amyloid myopathy. Amyloid deposition was also identified in salivary glands. A diagnosis of systemic AL amyloidosis with muscle and cardiac involvement, Mayo stage IV, was established.
Treatment with cyclophosphamide, bortezomib, dexamethasone, and daratumumab resulted in a marked hematological response accompanied by significant improvement in muscle strength and functional capacity.
Conclusion: This case highlights amyloid myopathy as a rare but clinically relevant manifestation of systemic AL amyloidosis that may represent the dominant initial presentation of the disease, leading to diagnostic delay and misclassification as other myopathies. Importantly, it demonstrates that effective treatment of the underlying plasma cell disorder can result not only in hematological response but also in substantial recovery of muscle strength and functional capacity. Maintaining a high index of suspicion and routinely considering amyloid myopathy, particularly in late-onset myopathies, may enable earlier diagnosis and improve outcomes in this potentially lethal yet treatable condition.
Abstract Topic Groups (Submission Categories)
Topic Group 1 - Muscle Diseases. Genetic and Acquired Myopathies: Clinical Features, Pathophysiology, Therapy: Toxic / Endocrine / Other Acquired Myopathies
PP01.154
Peripheral Nerve Involvement in Biallelic SCA27B: A Multicenter Study
Dr. Sara Nagy1,2, Dr. David Pellerin3,4,2, Dr. Pablo Iruzubieta4,5,6, Dr. Giulia Coarelli7, Dr. Claire Ewenczyk7, Dr. Anna Heinzmann7, Dr. Felix Heindl8, Dr. Gianmarco Dalla Zanna9,10, Dr. Astrid Nümann11, Dr. Joana Damasio12, Dr. Andreas Trauschütz13,14, Dr. Teije van Prooije15, Dr. Erik-Jan Kamsteeg16, Prof. Bart van de Warrenburg15, Prof. Alexandra Durr7, Prof. Filippo Santorelli17, Prof. Michael Strupp8, Prof. Matthis Synofzik14,13, Prof. Bernard Brais4, Dr. Mathilde Renaud18,19,20
1
Department of Neurology, University Hospital Basel, University of Basel,, Basel, Switzerland.
2
Department of Neuromuscular Diseases, UCL Queen Square Institute of Neurology, The National Hospital for Neurology and Neurosurgery, University College London, London, United Kingdom.
3
Department of Human Genetics and John P. Hussman Institute for Human Genomics, Dr. John T. Macdonald Foundation, University of Miami Miller School of Medicine,, Miami, United States.
4
Department of Neurology and Neurosurgery, Montreal Neurological Hospital and Institute, McGill University, Montreal, Canada.
5
Department of Neurology and Neurosciences, Donostia University Hospital, Biogipuzkoa Health Research Institute, , Donostia-San Sebastián, Spain.
6
CIBERNED Centro de Investigación Biomédica en Red en Enfermedades Neurodegenerativas-Instituto de Salud Carlos III (CIBER-CIBERNED-ISCIII), Madrid, Spain.
7
Sorbonne Université, Paris Brain Institute (ICM Institut du Cerveau), APHP, Paris, France.
8
Department of Neurology and German Center for Vertigo and Balance Disorders, University Hospital, Ludwig-Maximilians University, Munich, Germany.
9
Department of Neurosciences, Università Cattolica del Sacro Cuore, Rome, Italy.
10
UOC Neurologia Dipartimento Neuroscienze, Fondazione Policlinico Universitario A Gemelli IRCCS, Organi Di Senso E Torace, Rome, Italy.
11
Department of Neurology, Charity University Medicine Berlin, Berlin, Germany.
12
Neurology Department, Centro Hospitalar Universitário de Santo António, Porto, Portugal.
13
German Center for Neurodegenerative Diseases, Tübingen, Germany.
14
Division Translational Genomics of Neurodegenerative Diseases, Hertie-Institute for Clinical Brain Research and Center of Neurology, University of Tübingen, Tübingen, Germany.
15
Department of Neurology, Donders Institute for Brain, Cognition, and Behavior, Radboud University Medical Center, Nijmegen, Netherlands.
16
Department of Human Genetics, Radboud University Medical Center, Nijmegen, Netherlands.
17
IRCCS Fondazione Stella Maris, Via Dei Giacinti, Pisa, Italy.
18
Department of Neurology, CHRU Nancy, Nancy, France.
19
University of Lorraine, Inserm U1256, NGERE, Nancy, France.
20
Department of Medical Genetics, CHRU Nancy, Nancy, Germany
Background: Despite the partial clinical overlap between SCA27B and CANVAS (cerebellar ataxia, neuropathy, vestibular areflexia syndrome), peripheral nerve involvement has not been described as a characteristic feature in patients carrying a monoallelic GAA repeat expansion in FGF14. We investigated whether the prevalence and pattern of neuropathy differ in biallelic SCA27B.
Methods: We reviewed 78 patients with ataxia from 33 centers in 13 countries, stratified by repeat length: group A (both alleles >250 repeats, n=29), group B (one allele >250 repeats with a second allele of 200–250 repeats, n=26), and group C (one allele >200 repeats with a second allele of 150–200 repeats, n=23). Demographic, clinical, and electrophysiological data were analyzed to assess associations between neuropathy, repeat length, and disease duration.
Results: Forty-three patients (55%) underwent nerve conduction studies, confirming neuropathy in 8 (prevalence 19%). Among these cases, one patient belonged to group A, four to group B and three patients to group C. Sixteen additional patients had clinical features suggestive of neuropathy but lacked electrodiagnostic confirmation. Most neuropathies were classified as axonal sensory or sensorimotor, predominantly characterized by reduced vibration sense in the lower limbs and reduced or absent reflexes. When pooled, patients with confirmed neuropathy did not differ significantly in sex, age at onset, current age, disease duration, or repeat length of the larger allele from those without neuropathy; however, the smaller allele showed shorter repeat lengths in those with neuropathy. This pattern persisted when clinically suspected neuropathy cases were also included.
Conclusion: The prevalence of confirmed neuropathy in individuals with biallelic expansions is comparable to that observed in those with monoallelic expansion and in the general population and does not increase with repeat size. Notably, shorter repeats on the smaller allele were more frequently associated with neuropathy. Future studies incorporating updated nerve conduction assessments and longitudinal follow-up are warranted to confirm these findings.
Abstract Topic Groups (Submission Categories)
Topic Group 10 - Hereditary Ataxias/Spastic Paraplegias: N/A
PP01.155
Zilucoplan as Rescue Therapy in Refractory Myasthenia Gravis Exacerbations
Dr. Claudia Vinciguerra1, Dr. Carmen Erra2, Dr. Liliana Bevilacqua1, Dr. Dario Ricciardi2, Dr. Francesco Tuccillo2, Dr. Francesco Habetswallner2, Prof. Paolo Barone1
1
Neurology Unit, University Hospital “San Giovanni di Dio e Ruggi d’Aragona”, Department of Medicine, Surgery and Dentistry “Scuola Medica Salernitana”, University of Salerno, 84131 Salerno, Italy., Salerno, Italy.
2
Clinical Neurophysiology Unit, Azienda Ospedaliera di Rilievo Nazionale-Cardarelli, Naples, Italy., Naples, Italy
Background: Myasthenic crisis (MC) is a life-threatening complication of myasthenia gravis (MG), defined by acute respiratory failure requiring ventilatory support. Impending myasthenic crisis (IMC) describes a rapidly progressive clinical deterioration associated with a high risk of respiratory impairment if prompt therapeutic intervention is not started. In patients with acetylcholine receptor (AChR) antibody–positive MG, complement activation and subsequent membrane attack complex–mediated damage at the neuromuscular junction play a pivotal pathogenic role, rendering complement inhibition a rational therapeutic approach. However, evidence supporting the use of the C5 inhibitor zilucoplan in acute or peri-critical settings remains limited.
Methods: We report two cases of Italian patients with AChR-positive MG who developed severe, treatment-refractory disease exacerbations. One patient experienced a full-blown MC requiring non-invasive ventilatory support, while the second presented with an IMC characterized by rapidly progressive bulbar dysfunction. In both cases, zilucoplan was initiated at a dose of 32.4 mg administered subcutaneously once daily following an poor response to standard rescue therapies, including high-dose corticosteroids and intravenous immunoglobulin (IVIg); one patient had additionally failed multiple prior biologic treatments.
Clinical efficacy was assessed using the Myasthenia Gravis Activities of Daily Living (MG-ADL) and Quantitative Myasthenia Gravis (QMG) scores, together with functional and respiratory evaluations performed before and after treatment initiation. Patients were closely monitored during hospitalization or outpatient management and throughout early follow-up to evaluate clinical outcomes and treatment safety.
Results: The first case involved a 38-year-old man with thymoma-associated MG, refractory to IVIg, plasma exchange, and several targeted therapies. Following initiation of zilucoplan, he exhibited rapid and marked improvement in bulbar and respiratory function within seven days, allowing discontinuation of non-invasive ventilation and safe resumption of oral feeding. The second patient, an 81-year-old man with severe bulbar symptoms and imminent respiratory deterioration, achieved complete resolution of dysphagia and dysphonia within ten days of treatment, thereby avoiding hospital admission and ventilatory support.
Conclusion: Both patients maintained sustained clinical stability during continued zilucoplan therapy, with no treatment-related adverse events observed. These cases suggest that zilucoplan may induce a rapid, robust, and sustained clinical response in patients with refractory AChR-positive MG during both established myasthenic crisis and impending crisis. Its continuous complement inhibition and easy subcutaneous administration may represent significant advantages in both acute rescue and longer-term disease management. Early initiation of zilucoplan may help prevent progression from IMC to MC. Prospective studies are warranted to better define its optimal timing, efficacy, and positioning in the management of MG-related crises.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Advances in the treatment of Neuromuscular Junction diseases
PP01.156
Phase 1b Study of Safety and Efficacy of Adimanebart (ARGX-119) in DOK7 Congenital Myasthenic Syndromes
Prof. Hanns Lochmülller1,2,3, Dr. Lorenzo Maggi4, Dr. Nancy L. Kuntz5, Prof. Steven J. Burden6, Dr. Benjamin Van Hoorick7, Dr. Willem Talloen7, Dr. Kate Lyden8, Dr. Ieuan Clay8, Dr. Yaya Zhai8, Dr. Robert T. Marcotte8, Dr. Roeland Vanhauwaert7, Prof. Jacqueline Palace9
1
Children’s Hospital of Eastern Ontario Research Institute, Ottawa, Canada.
2
The Ottawa Hospital, Ottawa, Canada.
3
Brain and Mind Research Institute, University of Ottawa, Ottawa, Canada.
4
Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy.
5
Lurie Children’s Hospital of Chicago, Chicago, United States.
6
Massachusetts General Hospital, Harvard University, Boston, United States.
7
argenx, Ghent, Belgium.
8
VivoSense, Inc, Newport Coast, United States.
9
John Radcliffe Hospital, Oxford, United Kingdom
Background: Congenital myasthenic syndromes (CMS) are a rare, heterogeneous group of inherited disorders caused by mutations impairing neuromuscular transmission. There are currently no approved treatments. Mutations in the DOK7 gene represent one of the common causes of CMS. Adimanebart (ARGX-119), a humanized, agonistic, monoclonal antibody, specifically targets and activates muscle-specific kinase (MuSK). This may stabilize and improve neuromuscular junction function in DOK7-CMS, reducing muscle weakness/fatigability and improving quality of life (QoL). Here we present results from the ongoing, phase 1b, multicenter, double-blinded, placebo-controlled study (NCT06436742), which evaluated safety, tolerability, pharmacokinetics, immunogenicity, and efficacy of adimanebart in adults with DOK7-CMS.
Methods: Participants underwent intrapatient dose escalation and were randomized 4:1 to intravenous adimanebart or placebo for 6 doses over the 12-week treatment period followed by a ∼7-month follow-up period. The primary endpoint was safety assessment. Efficacy endpoints were standard clinical endpoints of components of the Quantitative Myasthenia Gravis (QMG) score, Myasthenia Gravis Activities of Daily Living (MG-ADL) score, and PROMIS Global Health score, measures of physical function and mobility (eg, Six-Minute Walk Test [6MWT] total distance walked and cadence assessed using digital sensors), and QoL. To evaluate activity at home, participants wore a wrist actigraphy device throughout the study (up to 33 weeks) for up to 24 hours a day (except during prespecified device-charging timepoints). Data from the device were used to assess measures of physical behavior and mobility.
Results: Sixteen participants were randomized to adimanebart (n=13) or placebo (n=3). Adimanebart was well tolerated, with no serious adverse events (AEs), grade ≥3 AEs, or discontinuations due to AEs. A clinically meaningful increase (≥50 m) in median 6MWT distance was reported in participants receiving adimanebart, with consistent improvements in median cadence measured using digital sensors. Improvements occurred in QMG key components (“both legs outstretched,” “both arms outstretched,” and “head lifted”) in the adimanebart arm. Consistent improvements also occurred in MG-ADL over time compared with study baseline in the adimanebart arm. Ambulatory (no wheelchair use at baseline) participants receiving adimanebart demonstrated coherence in response across most endpoints measuring leg function; this coherence in response did not occur in participants in the placebo arm. Actigraphy data suggested that adimanebart-treated participants may show improved activity levels at home compared with those treated with placebo. Additionally, actigraphy data showed clustering of real-world activity measures. Wheelchair users appeared to improve in shorter length activities, whereas ambulatory participants improved in longer timed activities. There was a positive association between changes in 6MWT cadence and real-world preferred cadence at end of treatment, with associations being stronger among ambulatory participants. Additional data from the 7-month follow-up period will be presented.
Conclusion: Results from this phase 1b study demonstrate proof-of-biology for adimanebart in participants with DOK7-CMS, which may correlate to real-world activity.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Advances in the treatment of Neuromuscular Junction diseases
PP01.157
Association Between Measured Variables and Family History of Neuromuscular Disease in Adults Using Macedonian INQoL
Prof. Ivan Barbov1,2, Dr. Goce Kalcev2
1
University Clinic of Neurology, Skopje, North Macedonia, Skopje, North Macedonia.
2
National Alliance for Neuromuscular diseases and Neuroscience, GANGLION Skopje, 1000 Skopje, Republic of North Macedonia, Skopje, North Macedonia
Background: The Individualized Neuromuscular Quality of Life Questionnaire (INQoL) is a disease-specific tool used to assess quality of life in individuals with neuromuscular disorders, evaluating symptom severity and functional limitations.
Methods: The sample included thirty adult patients with various neuromuscular disorders. A t-test was performed to examine differences in measured variables between participants with and without a family history of neuromuscular disease.
Results: The measured variables in the section of the symptoms were: scores of the muscle weakness, pain, fatigue, locking of the muscles, droopy eyelids, double vision, and swallowing difficulty separately. The measured variables of the section life domains were: score of activities, independence, social relationships, emotions, and body image. A score for the quality of life, another measured variable, is also derived from the individual scores in this section using a particular formula. The measured variables in the section of the treatment effects were perceived treatment effects and expected treatment effects. The t-test results indicate that most measured variables did not differ significantly between individuals with and without a family history of neuromuscular disease. Some variables (pain, locking of the muscles, independence and expected treatment effects) approach significance, indicating potential trends that may warrant further investigation.
Conclusion: Findings from this preliminary assessment indicate that the presence of a family history of neuromuscular disease does not significantly affect the measured variables within the studied sample.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Advances in the treatment of Neuromuscular Junction diseases
PP01.158
Adimanebart (ARGX-119): A First-in-Class, Humanized, Agonistic Monoclonal Antibody With Therapeutic Potential in Neuromuscular Junction Disorders
Dr. Jamie Lim1, Dr. Roeland Vanhauwaert1, Dr. Karolien Van Huynegem1, Dr. Ann Swijsen1, Prof. Jan Verschuuren2, Prof. Maartje Huijbers2, Prof. Steven Burden3
1
argenx, Ghent, Belgium.
2
Leiden University Medical Center, Leiden, Netherlands.
3
Massachusetts General Hospital, Boston, United States
Background: The agrin-LRP4-MuSK signaling pathway is essential for neuromuscular junction (NMJ) establishment, maintenance, and function. Diseases that disturb the structure or function of the NMJ cause debilitating and potentially life-threatening neuromuscular conditions such as congenital myasthenic syndromes (CMS), amyotrophic lateral sclerosis (ALS), and spinal muscular atrophy (SMA). CMS can be caused by genetic mutations in the agrin-LRP4-MuSK-DOK7 pathway, which impact development and function of the NMJ. An early pathological feature of ALS is NMJ disassembly, which can cause motor dysfunction and paralysis even in the absence of motor neuron loss. SMA is caused by mutations in SMN1 which disrupt the NMJ and lead ultimately to motor neuron loss.
Current treatments for CMS can lose effectiveness and be associated with central nervous and cardiovascular system-associated side effects following long-term use. Current therapies for ALS offer only modest benefits for survival or motor function, and while targeted therapies for SMA are effective, functional rescue is incomplete. There remains an unmet need for treatment options that improve NMJ stability and muscle function, and slow disease progression in patients with CMS, ALS, and SMA. MuSK agonist antibodies have been shown to preserve neuromuscular synapses in animal models of CMS, ALS and SMA and may offer potential benefit for these and other neuromuscular diseases.
Methods: Adimanebart (ARGX-119) is a humanized agonistic monoclonal antibody that specifically binds and activates MuSK. Adimanebart is in clinical development as a potential targeted therapy to treat CMS, ALS, and SMA.
Results: Agrin, released from motor nerve terminals, binds LRP4 to stimulate LRP4-MuSK association leading to the phosphorylation and activation of MuSK. Recruitment of DOK7 to phosphorylated MuSK further stimulates MuSK dimerization and phosphorylation, resulting in AChR clustering and functional synaptic transmission. Adimanebart binds MuSK, independent of agrin, to promote MuSK dimerization, phosphorylation, and activation, and to stimulate AChR clustering. This leads to a stabilized NMJ and improved synaptic transmission, which could potentially improve or even restore muscle function in patients with neuromuscular disorders such as CMS, ALS, and SMA.
This proposed mechanism of action underpins the rationale for the evaluation of adimanebart during ongoing clinical studies in CMS, ALS, and SMA. A phase 1b, double-blinded, randomized, placebo-controlled study (NCT06436742) is investigating the safety, tolerability, pharmacokinetics, immunogenicity, and efficacy of adimanebart in adult participants with DOK7-CMS (N=16). reALiSe (NCT06441682) is a phase 2a, double-blinded, randomized, placebo-controlled clinical trial investigating the safety and tolerability, preliminary efficacy, pharmacokinetics, and immunogenicity of adimanebart in ∼60 adult patients with ALS. SPARKLE (NCT07287982) is a phase 2, double-blinded, randomized, placebo-controlled, active-treatment extension study designed to assess the safety, tolerability, efficacy, pharmacokinetics, and immunogenicity of adimanebart in pediatric participants (5 to <18 years of age) with SMA.
Conclusion: By stabilizing and preserving NMJ and motor neuron function, facilitating synaptic transmission and promoting motor endplate integrity, adimanebart has therapeutic potential to improve muscle function in patients with CMS, ALS, and SMA.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Advances in the treatment of Neuromuscular Junction diseases
PP01.159
Safety and Efficacy of Claseprubart, an Active C1s Inhibitor, in Patients with Generalized Myasthenia Gravis
Dr. Shahar Shelly1, Assoc. Prof. Stojan Peric2, Dr. Pushpa Narayanaswami3, Dr. Marek Smilowski4, Dr. Agnieszka Slowik5, Dr. Sankalp Gokhale6, Dr. Caitlin Briggs6, Dr. Uzma Siddiqui6, Dr. Simrat Randhawa6, Mr. Matt Truman6, Mr. Luke Hickey6, Dr. Tuan Vu7, Prof. John Vissing8
1
Rambam Medical Center, Haifa, Israel.
2
University of Belgrade, Belgrade, Serbia.
3
Beth Israel Deaconess Medical Center & Harvard Medical School, Boston, United States.
4
Neurologia Śląska Centrum Medyczne, Katowice, Poland.
5
Jagiellonian University, Kraków, Poland.
6
Dianthus Therapeutics, New York, United States.
7
University of South Florida, Tampa, United States.
8
University of Copenhagen, Copenhagen, Denmark
Background: The classical complement pathway plays a significant role in the pathogenesis of generalized myasthenia gravis (gMG). Claseprubart is a monoclonal antibody that targets the classical pathway by inhibiting active C1s (aC1s). The MaGic trial assessed the safety and efficacy of claseprubart in adults with acetylcholine receptor antibody positive (AChR+) gMG.
Methods: MaGic (NCT06282159) is a randomized, double-blind, placebo-controlled Phase 2 study. Sixty-five participants with AChR+ gMG were randomized 1:1:1 to receive: claseprubart 300mg (Q2W), claseprubart 600 mg (Q2W), or placebo for 13 weeks, followed by a 52-week open-label extension and 40-week safety follow-up. Endpoints at 13 weeks included safety, tolerability, efficacy (MG activities of daily living (MG-ADL), Quantitative MG score (QMG), Minimal Symptom Expression (MSE) and MG Composite scale (MGC).
Results: Claseprubart was well tolerated with no serious adverse events, no serious bacterial infections, and no autoimmune activation. Injection site reactions were infrequent and mild to moderate. At Week 13, claseprubart improved MG-ADL by 4.6 (p=0.0113 vs. placebo) for 300 mg, 5.4 (p=0.0006 vs. placebo) for 600 mg, and 2.8 for placebo (no significant difference between active treatment arms). Significant improvement in MG-ADL was observed as early as Week 1. At Week 13, claseprubart 300mg improved QMG by 4.4 (p=0.0144 vs. placebo), claseprubart 600 mg by 4.5 (p=0.0111 vs. placebo), and 2.0 for placebo. MSE was achieved by 37% (300 mg) and 27% (600 mg) versus 14% for placebo. MGC for 300 mg was 8.7 (p=0.0008) and 8.6 (p=0.0008) for 600 mg versus 3.1 for placebo.
Conclusion: With a favorable safety profile, claseprubart demonstrated rapid, statistically significant, and clinically meaningful improvements in MG-ADL, QMG, MGC.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.160
Potential Mechanistic Advantages of aC1s Targeting in Myasthenia Gravis: Upstream Versus Downstream Complement Blockade
Dr. Shahar Shelly1, Dr. Marianna Lalla2, Dr. Yang Zhao2, Dr. Linda Rehaume2, Dr. Jennifer Cross2, Dr. Tuan Vu3
1
Rambam Medical Center, Haifa, Israel.
2
Dianthus Therapeutics, New York, United States.
3
University of South Florida, Tampa, United States
Background: In generalized myasthenia gravis (gMG), pathogenic antibodies activate the classical complement pathway, culminating in membrane attack complex (MAC)-mediated injury at the neuromuscular junction (NMJ). Levels of complement components (C3) have been associated with disease severity in animal models. C5 inhibitors block MAC formation and improve clinical outcomes, without reducing the upstream inflammatory fragments (C3a, C3b), which potentially drive NMJ injury and amplify immune responses in addition to MAC formation. We determined whether active C1s (aC1s) inhibition by claseprubart (DNTH103) suppresses generation of C3a, C3b and MAC in comparison with C5 inhibitor ravulizumab.
Methods: MAC formation was quantified using the Wieslab® Complement Classical Pathway assay in 1% normal human serum (NHS). C3a in supernatants was measured by ELISA and C3b deposition on sensitized human red blood cells in 5% NHS was assessed by flow cytometry. For each assay, we tested dilution series of claseprubart, ravulizumab, and an isotype control.
Results: In head-to-head assays, claseprubart and ravulizumab produced comparable inhibition of MAC formation, confirming similar downstream blockade. Only upstream aC1s inhibition with claseprubart resulted in near-complete reduction of C3a and C3b levels, while ravulizumab did not. Findings were reproduced across three independent experiments, supporting a mechanistic upstream (aC1s)-downstream (C5) inhibition distinction.
Conclusion: Upstream aC1s inhibition with claseprubart prevented MAC formation and reduced C3a and C3b levels, demonstrating broader inflammatory control beyond C5 blockade. Thus upstream inhibition may provide both pathological and immunological advantages.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Advances in the treatment of Neuromuscular Junction diseases
PP01.161
Value of Plasma Neurofilament Light Chain for Monitoring Efficacy in Presymptomatic Spinal Muscular Atrophy Children
Prof. Shanshan MAO
Children‘s hospital, Zhejiang university school of medicine, HANGZHOU, China
Background: While neurofilament light chain is a promising biomarker in spinal muscular atrophy (SMA), its dynamics in presymptomatic patients remain unestablished. This study aimed to analyze the plasma neurofilament light chain (pNfL) as a treatment response biomarker in patients with presymptomatic spinal muscular atrophy (SMA) undergoing nusinersen treatment.
Methods: Eight 5q-SMA patients with three SMN2 copies (four presymptomatic patients from newborn screening and four symptomatic patients) were prospectively enrolled from August 2022 to June 2023. All patients received nusinersen treatment and were followed up for 660 days. pNfL levels were measured at baseline and throughout the treatment, analyzing their temporal changes and correlation with motor function outcomes.
Results: At baseline, presymptomatic patients exhibited higher pNfL levels than symptomatic patients (388.74 ng/L vs. 113.60 ng/L). During the loading phase, pNfL levels decreased markedly in both groups, with greater reductions in presymptomatic patients (94.64% vs 79.50%). All presymptomatic patients achieved age-appropriate motor milestones. Decreased pNfL levels strongly correlated with motor function improvements, as measured by CHOP INTEND (r = −0.548, p < 0.01) and HINE-2 scores (r = −0.635, p < 0.01).
Conclusion: pNfL is a promising biomarker for monitoring treatment response in patients with presymptomatic SMA, highlighting the importance of early diagnosis and treatment through newborn screening.
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: Other Biomarkers
Images or Table (Optional)
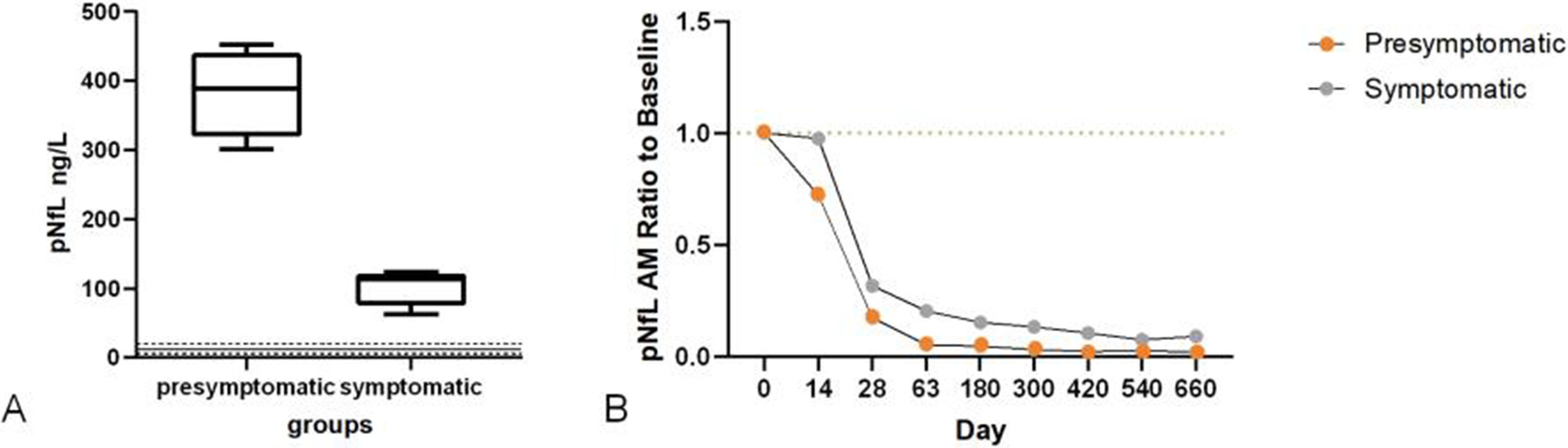
PP01.162
Telitacicept Use in Children With Ocular Myasthenia Gravis: A Case Series Report
Prof. Shanshan MAO
Children‘s hospital, Zhejiang university school of medicine, HANGZHOU, China
Background: Children with ocular myasthenia gravis (OMG) often suffer from relapse under traditional treatment with corticosteroid-related side effects. The biologic agent telitacicept, recently approved for adult patients with generalized MG, has shown good efficacy. This study aims to evaluate the efficacy and safety of telitacicept in children with OMG.
Methods: This is a case series of four children with OMG who were treated with telitacicept in Children's Hospital, Zhejiang University School of Medicine from April 2024 to December 2025. Clinical data were retrospectively collected before and after treatment, including patient demographics, clinical characteristics, scores of quantitative MG scale and MG-activity of daily living scale, as well as laboratory tests. Adverse events were also evaluated.
Results: Among the four patients, two were males and two were females. The median age at onset was 3.2 years (range 1.7 to 10.5 years), and the median course of disease before telitacicept treatment was 8.2 years (range 2.7 to 12.2 years). These children were previously received pyridostigmine bromide, corticosteroids, and nonsteroidal immunosuppressants such as azathioprine and mycophenolate mofetil. However, their symptoms of ptosis, diplopia, and ocular duction limitation did not improve, and adverse effects related to long-term corticosteroid use were observed. Following treatment with telitacicept, the ocular symptoms of these children showed improvement within one month. Subsequently, corticosteroids and other immunosuppressants treatment were successfully discontinued in all children after 5 to 16 months. At the last follow-up, both scores of quantitative MG scale and MG-activity of daily living scale decreased from baseline, indicating a reduction in the severity of the disease and an improvement in quality of life. No adverse drug reactions were reported during treatment period.
Conclusion: Telitacicept demonstrates significant efficacy in improving clinical symptoms of children with OMG and exhibits a relatively good safety profile, which is beneficial in reducing the reliance on corticosteroids and immunosuppressants.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Advances in the treatment of Neuromuscular Junction diseases
PP01.163
Janus Faces of Plectin: A Unified Pleiotropic Neuromuscular Disorder
Dr. Bruno Graziosi, Dr. Enzo Pellacani, Dr. Thainá Louise Rodrigues, Dr. Ana Marina Silva, Dr. Alessandra Tolentino, Dr. Roseli Corazzini, Dr. David Feder, Dr. Alzira Carvalho
Centro Universitário FMABC, Santo André, Brazil
Background: Congenital myasthenic syndromes (CMS) comprise a heterogeneous group of inherited disorders caused by pathogenic variants affecting presynaptic, synaptic, or postsynaptic components of neuromuscular transmission. Among postsynaptic CMS, myasthenic plectinopathy has emerged as an underrecognized entity associated with variants in PLEC, which encodes plectin, a large cytolinker anchoring intermediate filaments to sarcolemmal and synaptic membrane complexes.
Plectin displays marked pleiotropy, and pathogenic PLEC variants have been associated with two OMIM-defined disorders: epidermolysis bullosa simplex with muscular dystrophy (OMIM #226670) and limb-girdle muscular dystrophy type 2Q (OMIM #613723). Although traditionally regarded as distinct nosological entities, increasing evidence suggests that PLEC-related disease represents a pleiotropic neuromuscular spectrum rather than rigidly separated categories.
Classic limb-girdle muscular dystrophy is characterized by progressive scapulopelvic weakness, hyperCKemia, and overt dystrophic changes on muscle biopsy. In contrast, patients with PLEC-related CMS may present with an identical limb-girdle–predominant pattern of weakness but normal creatine kinase levels and only subtle or nonspecific biopsy findings. In such cases, a congenital myasthenic syndrome phenotypically mimics muscular dystrophy. The present report illustrates this overlap and supports a unified pleiotropic model of PLEC-related disease.
Methods: An 11-year-old boy had been followed since two years of age for recurrent lumbar-predominant myalgia, intermittent erythematous skin lesions, constipation, and mild dysphagia. Cutaneous symptoms partially improved with antihistamines and topical corticosteroids. At five years of age, he developed extensive bullous eruptions requiring hospitalization without identifiable triggers.
Over time, he developed symmetrical proximal muscle weakness affecting the shoulder and pelvic girdles (MRC grade 4−), with relative preservation of distal and axial muscles. Examination revealed positive Gowers’ sign, waddling gait, lumbar hyperlordosis, toe walking, frequent falls, and difficulty running. Mild unilateral ptosis was present, without ophthalmoplegia.
Repetitive nerve stimulation showed no decremental response. A comprehensive myopathy gene panel was unremarkable. Whole-genome sequencing identified two heterozygous PLEC variants of uncertain significance in compound heterozygosity. Muscle biopsy revealed minimal, nonspecific dystrophic changes, insufficient to support a primary muscular dystrophy diagnosis, supporting congenital myasthenic plectinopathy.
Results: This case expands the phenotypic spectrum of PLEC-related CMS by highlighting a limb-girdle–predominant presentation with minimal electrophysiological abnormalities and subtle myasthenic features. To date, only two cases have been reported describing this specific phenotypic overlap with available muscle biopsy data, reinforcing the pleiotropic nature of PLEC-related disease rather than strictly segregated nosological entities.
Conclusion: Although the identified variants were classified as variants of uncertain significance, the strong genotype–phenotype correlation, exclusion of alternative etiologies, and consistency with previously reported PLEC-related CMS presentations support their probable pathogenic relevance. Recognition of such atypical phenotypes is clinically important, as PLEC-related disease may benefit from targeted therapy and requires multidisciplinary management. Taken together, these observations support a conceptual shift in which PLEC-related disorders should be approached as a unified pleiotropic disease spectrum encompassing structural, dystrophic, and neuromuscular transmission phenotypes, rather than rigidly separated diagnostic entities defined solely by traditional OMIM classifications.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Congenital Myasthenic syndrome
PP01.164
Challenging Occam’s Razor in Neuromuscular Genetics: Coincident Molecular Diagnoses in Two Patients
Dr. Bruno Graziosi1, Dr. Enzo Pellacani1, Dr. Thaina Louise Rodrigues1, Dr. Pedro Fontana2, Dr. Carolina Correia2, Dr. Ana Marina Silva1, Dr. Alessandra Tolentino1, Dr. Roseli Corazzini1, Dr. David Feder1, Dr. Alzira Carvalho1
1
Centro Universitário FMABC, Santo André, Brazil.
2
Hospital Universitário Oswaldo Cruz, Recife, Brazil
Background: The increasing availability of genomic sequencing has revealed that more than one monogenic disorder may coexist in a single individual, a phenomenon increasingly recognized as a relevant diagnostic challenge in clinical genetics. Large cohorts demonstrate that approximately 4–7% of molecularly diagnosed patients harbor multiple independent genetic conditions, directly challenging the classical application of Occam’s razor in the neuromuscular discipline. We report two unrelated patients evaluated in a tertiary neuromuscular center in whom comprehensive genomic testing disclosed coincident molecular diagnoses explaining complex phenotypes.
Methods: Case 1. A 39-year-old woman developed progressive proximal weakness, dysarthria, dysphonia, fasciculations, hyperreflexia, and muscle atrophy over six months, consistent with combined upper and lower motor neuron involvement. Electroneuromyography demonstrated a preganglionic pattern. Genetic testing identified a variant of uncertain significance in FUS, located in a functionally relevant domain previously associated with sporadic and familial amyotrophic lateral sclerosis, and a pathogenic splice-site variant in NF1. The patient fulfilled diagnostic criteria for amyotrophic lateral sclerosis and also presented multiple café-au-lait macules and cutaneous neurofibromas. Brain and spinal MRI demonstrated multiple nerve sheath tumors consistent with schwannomas. These findings supported two independent molecular diagnoses: FUS-associated amyotrophic lateral sclerosis and neurofibromatosis type 1. Case 2 A one-year-old male infant presented with congenital hypotonia, generalized weakness, dysphagia, and early respiratory failure requiring prolonged invasive ventilation, followed by partial recovery with nocturnal ventilatory dependence. Examination revealed elongated facies, facial paresis, ophthalmoparesis, proximal limb weakness, cleft palate, pectus carinatum, scoliosis, cryptorchidism, short neck, low posterior hairline, and limited cervical mobility. Serum creatine kinase was normal. Electromyography demonstrated a myopathic pattern. Muscle biopsy revealed centrally located nuclei and necklace fibers. Whole-exome sequencing identified a pathogenic variant associated with Klippel–Feil syndrome, while chromosomal microarray demonstrated a pathogenic Xq28 deletion involving MTM1, confirming X-linked myotubular myopathy. Both molecular diagnoses were necessary to explain the combined neuromuscular and skeletal phenotype.
Results: These cases underscore the growing relevance of coincident molecular diagnoses in contemporary neuromuscular practice and highlight the limitations of strict diagnostic parsimony when applied to patients with complex or multisystem phenotypes. Traditionally, Occam’s razor has guided clinical reasoning toward a single unifying diagnosis; however, the expanding use of comprehensive genomic testing has revealed that multiple independent monogenic disorders may coexist and jointly shape the clinical presentation.
Conclusion: These observations reinforce the importance of phenotype-driven, hierarchical interpretation of genomic data, in which discordant or “excess” clinical features prompt reconsideration of diagnostic assumptions rather than being dismissed as atypical manifestations. Recognizing coincident molecular diagnoses is not merely an academic exercise; it has direct implications for prognosis, surveillance strategies, genetic counseling, and family planning. As genomic technologies become increasingly accessible, clinicians must remain vigilant to cognitive biases and embrace a diagnostic framework that accommodates molecular multiplicity in neuromuscular disease.
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: Genetics in Neuromuscular Diseases including Biochemical/Molecular Techniques and next generation sequencing.
PP01.165
Late-Onset Limb-Girdle Phenotype Caused by a Homozygous COLQ Mutation: A Case Report
Dr. Guilherme Barbosa, Dr. Enzo Pellacani, Dr. Roseli Corazzini, Dr. Alzira Carvalho, Dr. David Feder, Dr. Bruno Graziosi
Centro Universitário FMABC, Santo André, Brazil
Background: Congenital Myasthenic Syndrome (CMS) usually present in infancy; however, specific COLQ mutations can lead to late-onset phenotypes that mimic Limb-Girdle Muscular Dystrophy (LGMD). Distinguishing these entities is challenging due to overlapping clinical and neurophysiological features but is quite important for therapeutic success. We report a case of a patient misdiagnosed for decades. We report a case of a patient misdiagnosed for decades, whose correct diagnosis resulted in life-changing improvements.
Methods: A 41-year-old male presented with progressive muscle weakness since childhood, with no family history of neuromuscular disorders. Symptoms began at age 10 with exercise-induced exhaustion and cramps, progressing by age 14–15 to difficulty running and climbing stairs. He developed progressive proximal muscle weakness, starting in the lower limbs and later involving the upper limbs, resulting in wheelchair use since the age of 39. He denied dysphagia, dysphonia, or dyspnea. The patient, a prayer and guitar player, noted he could not keep up with the church choir or playing guitar due to fatigue. Neurological examination revealed significant scoliosis, asymmetric pupillary light reflexes (sluggish on the right), and prominent proximal weakness, from upper and lower limbs, deep tendon reflexes and sensation were normal. EMG and conduction studies showed a myopathic pattern and muscle biopsy presented nonspecific findings. Lower limb MRI (T1) demonstrated significant fatty replacement, and spirometry a restrictive pattern. CPK level was normal. The genetic testing identified a homozygous missense mutation in the COLQ (variant c.1289A>C). Treatment was initiated with Fluoxetine 30 mg and Ephedrine 60 mg, the latest administered in increasing doses. Following treatment, the patient showed significant clinical improvement, regaining the ability to ambulate without assistance.
Results: Misdiagnosing COLQ-CMS as Limb-Girdle Muscular Dystrophy (LGMD) is a frequent pitfall due to the ‘dystrophic-like’ remodeling of the neuromuscular junction. In this case, the pivotal clue lay in the patient's functional history: specific fatigability during sustained repetitive tasks, specifically choir singing and guitar playing, revealed an activity-dependent component masking as fixed weakness.
This suspicion of a synaptic defect guided the search for further signs beyond the standard muscle workup. According to previous studies, the combination of severe scoliosis and pupillary dysfunction provided the specific clinical signature of AChE deficiency.
This case also refines the genotype-phenotype correlation for COLQ. The homozygous c.1289A>C variant spares the patient from classic infantile respiratory crises but leads to insidious limb-girdle weakness. The distinction is therapeutically binary. The favorable response to ephedrine confirms that, in the absence of functional synaptic acetylcholinesterase, upregulating the adrenergic system is the most effective strategy to restore the safety factor of neuromuscular transmission.
Conclusion: This report validates the phenotype-genotype correlation of missense variants with late-onset weakness and highlights the specific contraindication of standard myasthenic treatments in esterase deficiency. The favorable response to sympathomimetics and open-channel blockers demonstrates the pivotal role of Next-Generation Sequencing in transitioning patients from supportive palliative care to targeted disease-modifying therapies.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Congenital Myasthenic syndrome
PP01.166
In Vivo Effects of a MuSK Agonist Antibody Against Mutation-Specific Congenital Myasthenic Syndromes
Dr. Roeland Vanhauwaert1, Ms. Kelly Ho2,3, Dr. Richard Webster4, Dr. Sally Spendiff2, Dr. Julien Oury5, Ms. Lieselot De Clercq1, Ms. Rani Coppejans1, Mr. Bernhardt Vankerckhoven1, Dr. Yin Dong4, Prof. Hanns Lochmülller2,6,7, Prof. Steven J. Burden8, Prof. Lore Mariën9
1
argenx, Ghent, Belgium.
2
Children’s Hospital of Eastern Ontario Research Institute, Ottawa, Canada.
3
University of Ottawa, Ottawa, Canada.
4
Nuffield Department of Clinical Neurosciences, University of Oxford, Oxford, United Kingdom.
5
Tevard Biosciences, Cambridge, United States.
6
The Ottawa Hospital, Ottawa, Canada.
7
Brain and Mind Research Institute, University of Ottawa, Ottawa, Canada.
8
Massachusetts General Hospital, Harvard University, Boston, United States.
9
Agenx, Ghent, Belgium
Background: Congenital myasthenic syndromes (CMS) are caused by impaired neuromuscular junction (NMJ) function, with subtypes arising from mutations in genes including DOK7, Agrn, ColQ and CHRNE. Adimanebart (ARGX-119) is a humanized agonistic monoclonal antibody that specifically binds and activates muscle-specific kinase (MuSK) to stimulate acetylcholine receptor (AChR) clustering and synaptic differentiation. Adimanebart demonstrated improved NMJ function and decreased muscle weakness and fatigability in a DOK7 CMS mouse model. Here, we investigated an adimanebart derivative, 3B2, in CMS mouse models deficient in agrin (Agrnnmf380), the collagen tail of acetylcholinesterase (ColQ
–/–
), or AChR (Chrne–/–; CHRNG+).
Methods: Agrn–, ColQ–, and AChR–CMS were modelled using Agrnnmf380, ColQ knockout (ColQ
–/–
), and Chrne–/–CHRNG+ mice, respectively. Mice received 3B2 (20 mg/kg for first dose, 10 mg/kg thereafter) or isotype control antibody by intraperitoneal injection on post-natal days 5, 15, and 35 (Agrnnmf380), days 22, 29, 36, 43, 50, and 57 (ColQ
–/–
), or weekly from week 2–12 (Chrne–/–CHRNG+). Mice were euthanized and tissues harvested on day 50 (Agrnnmf380), day 66 (ColQ
–/–
), or week 13 (Chrne–/–CHRNG+). Efficacy was assessed by survival duration, weight-gain, muscle strength, and muscle fiber size.
Results: All 3B2-treated Agrnnmf380 mice (n=6) survived and demonstrated increases in body weight over time consistent with wild-type mice (n=6). Of the isotype control-treated Agrnnmf380 mice (n=6), only one survived to study end, increases in body weight plateaued after 15 days. In ColQ
–/–
(3B2, n=13; isotype, n=12; wild-type, n=12) and Chrne–/–CHRNG+ mice (3B2, n=9; isotype, n=13), no difference in body weight was observed between 3B2 or isotype control mice.
Muscle strength significantly improved with 3B2 in Agrnnmf380 versus isotype control-treated mice, as assessed by time to fall in hindlimb suspension on days 7–8 and forelimb grip strength at days 23 and 40. No difference in muscle strength was observed between 3B2 and isotype control-treated ColQ
–/–
and Chrne–/–CHRNG+ mice, as assessed by fore- and hindlimb grip strength at days 32, 45, and 65 (ColQ
–/–
), inverted hanging wire time at days 26, 40, and 54 (ColQ
–/–
), or screen hang time throughout the study (Chrne–/–CHRNG+).
Quadriceps and gastrocnemius muscle fiber area were significantly greater in Agrnnmf380 mice treated with 3B2 versus isotype control, and similar to wild-type mice, while no differences in soleus muscle fiber area were seen between groups. There was no difference in soleus muscle fiber size between treatment groups in ColQ
–/–
mice.
Combining 3B2 with pyridostigmine and salbutamol did not improve phenotypic measures compared with pyridostigmine and salbutamol alone in Chrne–/–CHRNG+ mice.
Conclusion: Administration of 3B2 prevented mortality, restored body weight and muscle fiber size to wild-type levels, and improved muscle strength in an Agrn-CMS mouse model, but did not rescue the CMS phenotype in mouse models of ColQ and AChR deficiency. These data suggest differential effects of adimanebart, dependent upon pathophysiology of CMS subtypes and the expected adimanebart mechanism of action.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Congenital Myasthenic syndrome
PP01.167
The First Korean Case of Congenital Myasthenic Syndrome 10 Responsive to Ephedrine Therapy
Mr. Sangwon Joe1, Miss Yunjung Choi1, Prof. Young-Chul Choi1,2, Prof. Woo-Kyung Kim3, Dr. Jeong Hee Cho4, Prof. Bum Chun Suh5, Prof. Sang Beom Kim6, Prof. Hyung Jun Park1
1
Department of Neurology, Gangnam Severance Hospital, Yonsei University College of Medicine, Seoul, Korea, Republic of.
2
Department of Neurology, SCL Hanaro Leaders Clinic, Seoul, Korea, Republic of.
3
Department of Neurology, Kangdong Sacred Heart Hospital, Hallym University College of Medicine, Seoul, Korea, Republic of.
4
Department of Neurology, National Health Insurance Service Ilsan Hospital, Goyang, Korea, Republic of.
5
Department of Neurology, Kangbuk Samsung Hospital, Sungkyunkwan University School of Medicine, Seoul, Korea, Republic of.
6
Department of Neurology, Kyung Hee University Hospital at Gangdong, Kyung Hee University School of Medicine, Seoul, Korea, Republic of
Background: Congenital myasthenic syndromes (CMS) comprise a heterogeneous group of inherited neuromuscular disorders caused by pathogenic variants in proteins that are essential for the formation, function, and maintenance of the neuromuscular junction. Among these disorders, congenital myasthenic syndrome type 10 (CMS10) is a major subtype resulting from defects in the cytoplasmic adaptor protein DOK7, which is critical for MuSK activation and acetylcholine receptor clustering at the postsynaptic membrane. Although CMS10 has been well documented in Europe, North America, and Japan, it has not previously been reported in the Korean population. In this report, we present the first genetically confirmed case of CMS10 in a Korean patient.
Methods: We retrospectively reviewed the medical records of a patient diagnosed with DOK7 congenital myasthenic syndrome who visited Gangnam Severance Hospital between 2008 and 2025. Clinical, genetic, and electrophysiological characteristics were systematically evaluated.
Results: A 7-year-old girl presented with gait disturbance, frequent falls, and difficulty climbing stairs starting at age 4. Physical examination revealed mild, symmetric proximal weakness; however, ocular, facial, and bulbar functions were preserved. Initial serum creatine kinase (CK) levels were normal (66 IU/L), but needle electromyography suggested generalized myopathic changes, leading to a preliminary diagnosis of congenital myopathy. At age 24, she underwent re-evaluation. Her proximal weakness remained stable and a pulmonary function test revealed a vital capacity 30% below the predicted value. Serum CK level was 35 IU/L but repetitive nerve stimulation test showed a significant decremental response in the trapezius muscle, shifting the diagnostic focus toward an NMJ defect. Whole-exome sequencing identified compound heterozygous pathogenic variants in DOK7 (NM_173660.4: c.[539G>C];[1124_1127dup]). These variants were previously reported as pathogenic variants.
Following the genetic confirmation, treatment with oral ephedrine 50 mg/day was initiated. After one month, she reported significant improvement in motor endurance, particularly in stair climbing and prolonged walking, leading to enhanced independence in daily activities. The adult myopathy assessment tool score increased from 21 to 26, reflecting measurable functional improvement. However, repetitive nerve stimulation showed persistent decremental responses on trapezius muscle. Pulmonary function tests showed no measurable improvement in vital capacity.
Conclusion: This case represents the first reported case of CMS10 in a Korean patient. It demonstrates that some patients initially diagnosed with congenital myopathy may have treatable forms of congenital myasthenic syndrome, and that repetitive nerve stimulation and genetic testing are valuable tools for achieving an accurate diagnosis.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Congenital Myasthenic syndrome
PP01.168
The Epidemiology of Congenital Myasthenic Syndromes: A Comparison of Diagnosed and Genetic Prevalence
Mr. Syed Raza1, Mr. Xin Zhao2, Mr. Nicholas Silvestri3, Mr. Benjamin Van Hoorick1, Mr. François Pajot3, Mr. Connor Buffel2
1
argenx, Ghent, Belgium.
2
ISMS, Zoersel, Belgium.
3
argenx, Boston, United States
Background: Congenital myasthenic syndromes (CMS) are a group of heterogeneous neuromuscular disorders caused by genetic defects at the neuromuscular junction. The few reported diagnosed prevalence estimates vary widely from 2 to 22 per million, reflecting the clinical heterogeneity, evolving disease awareness, and delays in the timely diagnosis of this disease. Prevalence estimates stratified by genetic subtype are particularly sparse, leaving the true burden of disease essentially unknown.
Aims: This study aimed to (1) review the diagnosed prevalence of CMS, with a specific focus on several autosomal recessive genetic subtypes (DOK7, MuSK, AGRN, and LRP4) and (2) estimate the true burden of disease via an epidemiological model driven by genetic prevalence.
Methods: Using PubMed/MEDLINE, recently published abstracts, rare disease registries, and backward and forward snowballing, we conducted a targeted literature review of the diagnosed prevalence, mutation frequency, and age at onset of CMS. We applied the mutation frequency to the diagnosed prevalence of total CMS to estimate subtype-specific diagnosed prevalence. For each country, we identified the most reliable data for both parameters. Where country-level data were lacking, evidence from neighboring or regionally similar settings was applied. We used the Genetic Prevalence Estimator (GeniE) to generate the genetic prevalence of pathogenic and likely pathogenic (PLPV) mutations in the genes of interest. To estimate true prevalence, a country-specific model was developed using genetic prevalence as a proxy for birth prevalence and incorporating assumptions regarding symptom onset and survival. Adjustments were made based on variations in genetic prevalence across ancestries estimated by GeniE. Where applicable, 95% confidence intervals were estimated using the Wilson score method. To account for uncertainty, the lower limits of the 95% confidence intervals around the genetic prevalence were applied.
Results: One hundred twenty-one studies met the inclusion criteria. Only two studies directly reported the diagnosed prevalence for genetic subtypes of interest. Integrating the findings, the diagnosed prevalence was estimated to range from <0.1 to 2.5 per million in Japan and France, respectively. Two of three countries reporting age-specific prevalence showed significantly higher prevalence per million in pediatrics compared to adults (p<0.05). The genetic prevalence was estimated in the total population as 3.58, 0.40, 0.08, and 0.05 per million for DOK7, AGRN, LRP4, and MuSK, respectively. Substantial differences across genetic ancestries were noted for several PLPVs, particularly for AGRN in the East Asian ancestry. The number of diagnosed cases in subtypes of interest was estimated at around 1,100 in Europe and North America. The estimated number of cases based on the genetic prevalence was 3 to 4 times higher in those regions.
Conclusion: Estimating the genetic prevalence of autosomal recessive disease subtypes can offer valuable insights into their true prevalence, especially when genetic subtype-level evidence is unavailable. In CMS, the discrepancy between the diagnosed prevalence and genetic prevalence estimates indicates a substantial under- or misdiagnosis in this heterogeneous and complex disease. Additional epidemiological studies may help to validate these estimates and assess the impact of increasing disease awareness over time.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Congenital Myasthenic syndrome
PP01.169
Sustained Clinical Efficacy and Long-Term Safety of Intravenous Efgartigimod for gMG: ADAPT NXT Part B
Dr. Kristl G. Claeys1,2, Dr. Arjun Seth3, Dr. Ali A. Habib4, Dr. Yessar Hussain5, Dr. Gregory Sahagian6, Dr. Elena Cortés-Vicente7,8, Dr. Jeffrey Guptill9, Dr. Li Liu9, Dr. Rosa H. Jimenez9, Dr. Sirine Bougamra9, Dr. Delphine Masschaele9, Dr. Renato Mantegazza10, Dr. Andreas Meisel11, Dr. Fang Sun12,13, Dr. Shahram Attarian14
1
University Hospitals Leuven, Leuven, Belgium.
2
KU Leuven, Leuven, Belgium.
3
Northwestern University, Chicago, United States.
4
University of California, Irvine, United States.
5
Austin Neuromuscular Center, Austin, United States.
6
The Neurology Center of Southern California, Carlsbad, United States.
7
Hospital de la Santa Creu i Sant Pau, Barcelona, Spain.
8
Biomedical Research Institute Sant Pau, Barcelona, Spain.
9
argenx, Ghent, Belgium.
10
Fondazione IRCCS Istituto Neurologico Carlo Besta (Emeritus), Milan, Italy.
11
Charité – Universitätsmedizin Berlin, Berlin, Germany.
12
University of Pittsburgh School of Medicine, Pittsburgh, United States.
13
Northwestern University Feinberg School of Medicine, Chicago, United States.
14
Timone Hospital University, Marseille, France
Background: Efgartigimod, a human immunoglobulin G1 Fc fragment that blocks the neonatal Fc receptor, was well tolerated and efficacious when administered in fixed-cycles or every-other-week (Q2W) dosing to participants with generalized myasthenia gravis (gMG) during Part A of the Phase 3b ADAPT NXT study (NCT04980495). Part B of ADAPT NXT investigated the long-term efficacy, safety, and tolerability of efgartigimod in participants with gMG receiving Q2W or every-third-week (Q3W) dosing.
Methods: Participants were randomized 3:1 to Q2W or fixed-cycles dosing of 10 mg/kg efgartigimod for 21 weeks in Part A. Fixed-cycles dosing involved 4 once-weekly infusions followed by a 4-week intertreatment period. In Part B, all participants received Q2W dosing during a 105-week extension. Participants could switch to Q3W dosing, depending on clinical assessment.
Results: Sixty-five participants (of 69 in Part A [94.2%]) continued treatment in Part B; 57.8% (37/64) transitioned to Q3W dosing, and 59.5% (22/37) remained on Q3W dosing. Average Q3W treatment duration was 382 days. Mean Myasthenia Gravis Activities of Daily Living (MG-ADL) total score improvements were sustained through Week 126. Overall, 81.2% (56/69) of participants experienced an MG-ADL improvement of ≥5 points, and 56.5% (39/69) achieved minimal symptom expression (MSE; MG-ADL score, 0-1) at any time during the study. In Part B, 51.6% (32/62) achieved MSE. Efgartigimod was well tolerated across dosing regimens; no new safety signals were observed. Data from the final analysis of ADAPT NXT will be presented at the conference.
Conclusion: Efgartigimod demonstrated sustained clinical benefits in participants with gMG during long-term treatment while remaining well tolerated across different dosing schedules.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.170
Impact of Time Since Diagnosis on Inebilizumab Efficacy: Post-Hoc Phase 3 MINT Trial Data Analysis
Dr. Ali Habib1, Dr. James F. Howard Jr.2, Dr. Michael Benatar3, Dr. Emma Ciafaloni4, Dr. M. Isabel Leite5, Dr. Kimiaki Utsugisawa6, Dr. John Vissing7, Dr. Sarah Bray8, Dr. Michaela Schlader-Ratzinger8, Dr. Catherine Najem8, Dr. Sue Cheng8, Dr. Richard J. Nowak9
1
University of California Irvine, Irvine, United States.
2
University of North Carolina, Chapel Hill, United States.
3
University of Miami, Miller School of Medicine, Miami, United States.
4
University of Rochester, Rochester, United States.
5
University of Oxford, Oxford, United States.
6
Hanamaki General Hospital, Hanamaki, Japan.
7
University of Copenhagen, Copenhagen, Denmark.
8
Amgen Inc., Thousand Oaks, United States.
9
Yale University, New Haven, United States
Background: Generalized myasthenia gravis (gMG) is characterized by autoreactive B cells producing anti–acetylcholine receptor antibodies (AChR+), anti–muscle-specific kinase antibodies (MuSK+), or other autoantibodies. The MINT primary endpoint (change in Myasthenia Gravis Activities of Daily Living [MG-ADL] score) was achieved, supporting the efficacy of inebilizumab in gMG. Here, we determine if response to inebilizumab, a monoclonal antibody targeting CD19+B cells, differs by time since diagnosis of generalized myasthenia gravis in the MINT phase 3 trial.
Methods: Participants with AChR+ or MuSK+ seropositive gMG were enrolled in MINT (NCT04524273). Participants underwent a protocol-specified glucocorticoid taper to ≤5 mg/day and were randomized 1:1 to 300mg inebilizumab or placebo for 26 (MuSK+) or 52 (AChR+) weeks. Changes from baseline (CFBs) in MG-ADL and Quantitative Myasthenia Gravis (QMG) scores by time since diagnosis (date of first dose in the randomized, controlled period minus gMG diagnosis date) were examined in this post hoc analysis.
Results: Overall, 238 participants (AChR+, 190; MuSK+, 48) were enrolled and randomized to inebilizumab or placebo. The least squares mean (LSM) CFB in MG-ADL score was overall numerically greater for inebilizumab vs placebo at week (W) 26 for participants with time since diagnosis of <1 year (–5.4 vs –3.0), ≥1y–<4y (–4.3 vs –1.8), and ≥4y (–3.7 vs –2.5); the LSM CFB in QMG score was also numerically greater for inebilizumab vs placebo at W26 (<1y, –5.2 vs –4.0; ≥1y–<4y, –6.4 vs –2.2; ≥4y, –4.0 vs –1.9). Significant improvements from baseline with inebilizumab vs placebo were observed at W52 for MG-ADL (AChR+: <1y, –5.7 vs –2.3; ≥1y–<4y, –5.0 vs –2.7; ≥4y, –4.0 vs –1.5) and QMG (AChR+: <1y, –7.4 vs –1.8; ≥1y–<4y, –8.3 vs –2.9, ≥4y, –3.9 vs –0.5) scores.
Conclusion: The efficacy of inebilizumab appears independent of time since diagnosis, although the small size of subgroups limits firm conclusions.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.171
Safety and Efficacy of rese-cel, an Autologous CD19-CAR T Cell Therapy, in Generalized Myasthenia Gravis
Assoc. Prof. Ali Habib1, Assoc. Prof. Christina Ulane2, Assoc. Prof. Min Kang3, Prof. David Richman4, Prof. Stephan Ciurea1, Prof. Ran Reshef2, Asst. Prof. Madhav Seshadri3, Prof. Mehrdad Abedi4, Dr. Jonathan Hogan5, Dr. Yvonne White5, Dr. Jenell Volkov5, Dr. Daniel Nunez5, Mr. Thomas Furmanak5, Dr. Raj Tummala5, Dr. David Chang5
1
UCI, Irvine, United States.
2
Columbia University, New York, United States.
3
UCSF, San Fransisco, United States.
4
UC Davis, Davis, United States.
5
Cabaletta Bio, Philadelphia, United States
Background: Generalized Myasthenia Gravis (gMG) is a B-cell-mediated disease; most patients have detectable autoantibodies targeting the neuromuscular junction. Available therapies require chronic administration, increasing the risk of side effects, and many patients remain refractory despite treatment with several agents. Rese-cel (formerly CABA-201) is an investigational, fully human, autologous 4-1BB CD19-CAR T cell therapy, designed to deeply and transiently deplete CD19+B-cells following a single, weight-based infusion, potentially enabling an “immune reset” with durable responses. RESET-MG™ (NCT06359041) is a Phase 1/2 trial evaluating the safety and efficacy of rese-cel in 2 independent cohorts of anti-AChR-antibody-positive and anti-AChR-antibody-negative gMG.
Methods: Eligible patients are 18-70 years old with MGFA Classification II-IV gMG and MG-ADL ≥6 despite ≥2 prior/current treatments.
A single rese-cel infusion of 1x106 cells/kg is administered following standard preconditioning with fludarabine and cyclophosphamide. All non-glucocorticoid immunomodulatory agents are discontinued by preconditioning; glucocorticoids and acetylcholinesterase inhibitors are tapered post-infusion. Adverse events, gMG medications and gMG activity were assessed. CAR T cells and B cells were profiled in peripheral blood pre- and post-infusion by digital PCR and flow cytometry, respectively.
Results: As of 11 September 2025, four patients (2 per cohort) have received rese-cel and completed at least 1 month follow-up (Table 1) in the RESET-MG trial.
Rese-cel was well-tolerated with no dose-limiting toxicity or serious infection. One patient had a Grade 2 cytokine release syndrome (CRS), and no immune effector cell-associated neurotoxicity syndrome (ICANS) was observed
The 2 evaluable patients achieved improvements in MG disease activity off of all gMG medications: AChR-neg1 experienced an improvement in MG-ADL 17 to 0 (achieving minimal symptom expression) and QMG from 22 to 5 at 20 weeks post-infusion; AChR-neg2 experienced an improvement in MG-ADL from 14 to 7 and QMG from 21 to 11 at 8-weeks post-infusion. The 3rd patient was not evaluable due to use of a prohibited cytotoxic medication that may have inhibited CAR T activity; the 4th patient has insufficient follow up.
PK and PD profiles were available for 3 adult patients. Rese-cel expanded in all patients and peaked 7 days (median; IQR: 5-10) following infusion at 8 cells/µL (median; IQR: 4-97). Peripheral B cell counts reached a minimum at 7 days (median; IQR: 6.8-7.4) post-infusion with an average depletion duration of 48 days (median; range: 14-81), calculated from the 2 patients who exhibited B cell repopulation at the time of the data cut. Repopulating B cells were mostly transitional naïve, supporting B cell compartment reset.
Conclusion: These initial data suggest that rese-cel is well-tolerated and can lead to immune reset in gMG, allowing patients to achieve meaningful clinical responses off of gMG therapies. Both cohorts are fully enrolled, and additional data will be presented at ICNMD.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
Images or Table (Optional)
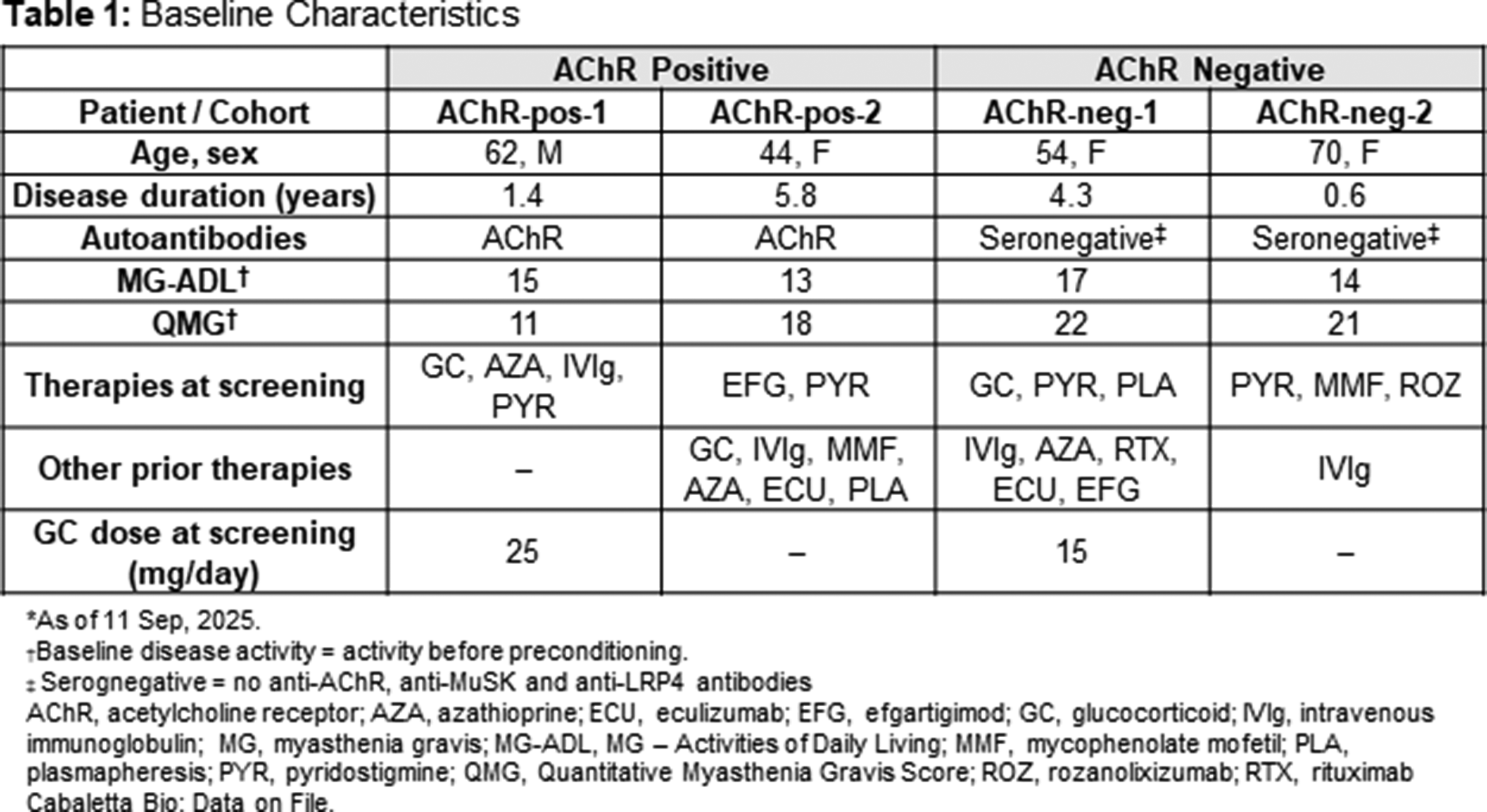
PP01.172
Gender Differences in Myasthenia Gravis in a Neuromuscular Reference Center
Dr. Alicia Alonso-Jiménez1,2, Dr. Willem De Ridder1,2, Prof. Paul Van Schil1, Prof. Jonathan Baets1,2, Prof. Rudy Mercelis1
1
University Hospital of Antwerp, Edegem, Belgium.
2
Faculty of Medicine and Health Sciences, University of Antwerp, Antwerp, Belgium
Background: It is well known that scientific literature has a significant gender gap, as gender differences were not considered until very recently. Women were often excluded from studies, and even when included, the results are rarely analyzed separately by gender. This study aims to investigate gender-specific differences in patients with Myasthenia Gravis (MG) by segregating data from our cohort of patients in Antwerp (Belgium).
Methods: We analyzed the data of our previously published cohort of 163 patients with MG visited in the Antwerp University Hospital between 2019 and 2021, segregating the information by gender to observe any significant differences.
Results: The analysis revealed several notable gender-specific differences. Women experienced a delay in diagnosis of over one year more frequently than men. They also had dysarthria as presenting symptom more often than men. The MGFA scores at maximum severity were higher in women, who also reported more limitations due to the disease and required more treatments to control it. While some differences could be attributed to the younger onset of the disease in women, certain differences were independently influenced by gender
Conclusion: Women experience more limitations due to MG than men and may face a more severe disease course. These differences should be taken into account when determining follow-up and treatment strategies. Additionally, these findings highlight the importance of segregating data by gender in scientific studies to better understand gender-specific differences in disease presentation and management.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.173
Exploring Alternative Operational Definitions for Refractory Myasthenia Gravis Using Real-World Data From MGBase Registry
Prof. Anneke Van der Walt1,2, Prof. Jeannine Heckmann3, Dr. Stefan Blum4, Prof. Elisabeth Chroni5, Prof. Stephen Reddel6,7, Dr. Katherine Buzzard8, Dr. Masoud Etemadifar9, Dr. Matteo Foschi10, Dr. Raed Alroughani11, Dr. Belinda Cruse12, Prof. John Tzartos13, Prof. Pamela McCombe14, Prof. Enrique Gomez-Figueroa15, Dr. Antony Winkel16, Dr. Yi Chao Foong17, Prof. Cavit Boz18, Prof. Allan Kermode19,20, Dr. Soaham Desai21, Dr. Ha Young Shin22, Miss Kelly Kueh23, Dr. Virginia DeLasHeras24, Dr. Aine McConnon25, Dr. Farid Khan26, Prof. Tim Spelman27, Prof. MGBase Study Group28
1
Alfred Health, Melbourne, Australia.
2
School of Translational Medicine, Monash University, Melbourne, Australia.
3
University of Cape Town, Cape Town, South Africa.
4
Princess Alexandra Hospital, Brisbane, Australia.
5
University of Patras, Patras, Greece.
6
University of Sydney, Sydney, Australia.
7
Concord Repatriation & General Hospital, Sydney, Australia.
8
Eastern Health Clinical School, Monash University, Melbourne, Australia.
9
Isfahan University of Medical Sciences, Isfahan, Iran.
10
S.Maria delle Croci Hospital, AUSL Romagna, Ravenna, Italy.
11
Amiri Hospital, Sharq, Kuwait.
12
Royal Melbourne Hospital, Melbourne, Australia.
13
Attikon University Hospital, National & Kapodistrian University of Athens, Athens, Greece.
14
Royal Brisbane and Women's Hospital, Brisbane, Australia.
15
Hospital Civil de Guadalajara, Guadalajara, Mexico.
16
Sunshine Coast University Hospital, Queensland, Australia.
17
Royal Hobart Hospital, Tasmanian Health Service, Hobart, Australia.
18
Karadeniz Technical University, Trabzon, Turkey.
19
Perron Institute for Neurological and Translational Science & Sir Charles Gairdner Hospital, QEIIMC, Nedlands, Australia.
20
University of Western Australia, Perth, Australia.
21
Shree Krishna Hospital, Pramukhswami Medical College, Bhaikaka University, Anand, India.
22
Yonsei University College of Medicine, Seoul, Korea, Republic of.
23
Novartis Corporation (Malaysia) Sdn. Bhd., Petaling Jaya, Malaysia.
24
Novartis Pharma AG, Basel, Switzerland.
25
Novartis Ireland Ltd., Dublin, Ireland.
26
Novartis Pharmaceuticals Corporation, East Hanover, United States.
27
Karolinska Institute, Stockholm, Sweden.
28
MGBase Foundation, Melbourne, Australia
Background: Myasthenia gravis (MG) is a chronic autoimmune neuromuscular disorder characterized by fluctuating muscle weakness. Identifying patients with refractory MG — with inadequate response to standard therapies — is critical for optimizing management and improving outcomes. However, applying standard definitions in real-world databases is challenging due to incomplete or inconsistent data. This study aims to explore alternative operational definitions for refractory MG to better identify patients with higher disease burden in real-world settings.
Methods: We conducted a retrospective analysis using data available in MGBase, a global multicenter registry. Patients were classified into subgroups using operational definitions based on Myasthenia Gravis Activities of Daily Living (MG-ADL), Myasthenia Gravis Foundation of America (MGFA) class, Myasthenia Gravis Composite (MGC), history of MG crisis, and use of rescue therapies (PLEX or IVIG), anchored to time since treatment exposure (≥6 months post-initiation of any MG treatment and ≥6 months post-initiation of biologics). Three operational definitions for refractory MG were explored. Group 1 (Moderate-to-severe gMG): ≥2 episodes of MG-ADL ≥6 and ≥1 episode of MGFA ≥III; Group 2 (High symptom burden MG): ≥2 episodes of MG-ADL ≥6 or ≥2 episodes of MGC ≥10; Group 3 (High risk MG): ≥1 MG crisis (defined as an exacerbation leading to hospitalization and admission to intensive care unit) or ≥4 PLEX/IVIG. Demographics, clinical characteristics, treatment exposures and patient- or clinician-reported outcomes were compared across these subgroups.
Results: Of 867 MG patients, 3.7% met criteria for Group 1, 15.0% for Group 2, and 10.6% for Group 3, despite ≥6 months of treatment. The patients in Group 1 were younger (mean age 43.8 years, 75.0% female) with the highest baseline scores (MG-ADL: 10.9, MGC: 15.3, modified Rankin Scale [MRS]: 2.6) but remained severely symptomatic (MG-ADL: 9.2, MGC: 12.4, MRS: 2.2) at last follow-up despite intensive treatment (75.0% on ≥2 therapies, 71.9% exposed to rituximab, and 75.0% had IVIG). The patients in Group 2 were older (mean age 54.9 years, 64.6% female) and maintained moderate symptoms scores from diagnosis (MG-ADL: 6.6, MGC: 12.9, MRS: 2.1) to last visit (MG-ADL: 6.2, MGC: 8.4, MRS: 1.7), with lower treatment intensity. The patients in Group 3 (50.0 years, 63.0% female) had the lowest symptom scores overall but showed greatest improvement in MGC (11.9 vs 5.8), while having the highest rituximab exposure (78.3%), thymoma presence (25.0%) and AChR antibody positivity (85.9%). Notably, patients with ≥6 months of biologic exposure had the highest MG-ADL and MGC scores at last follow-up (Table), suggesting that biologic therapy is reserved for the most refractory cases, where it may not fully resolve disease burden.
Conclusion: Alternative operational definitions captured 4–15% of MG patients with high disease burden in this cohort, aligning with known refractory MG prevalence (10–15%). Particularly, Group 1 captured patients severely affected at diagnosis with minimal improvement at last follow-up. These flexible operational definitions provide valuable insights into real-world disease burden and treatment patterns, emphasizing the importance of adapting definitions to the strengths and limitations of available data to better identify patients at risk for poor outcomes and guide individualized management strategies.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
Images or Table (Optional)
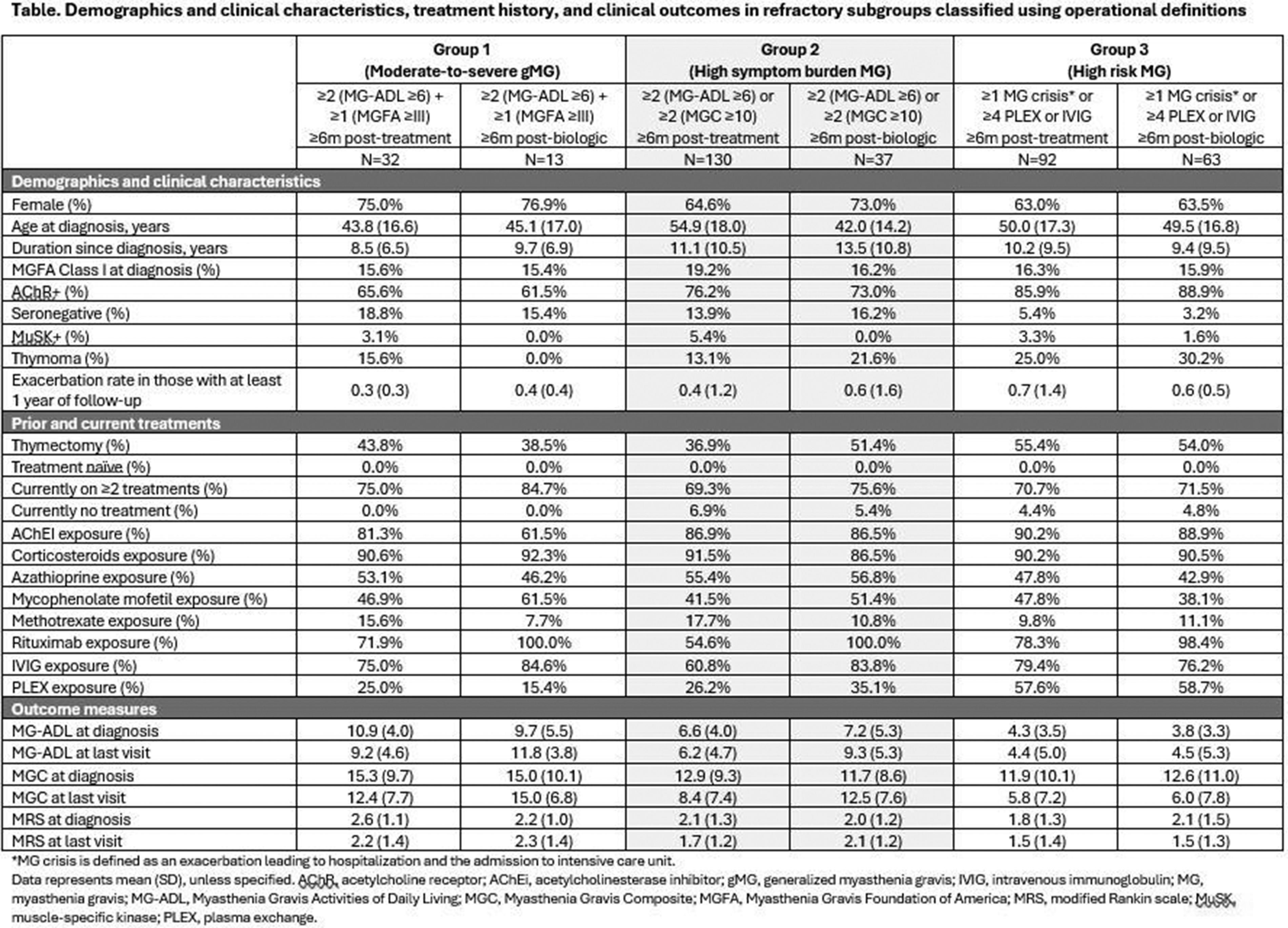
PP01.174
Prevalence and Risk Factors of Perioperative Myasthenia Gravis Exacerbation: A Single-Center Study in Thailand
Dr. Apinya Kroeksattayaporn, Miss Ratiporn Klabsook, Asst. Prof. Theerawat Kumutpongpanich, Assoc. Prof. Kanokwan Boonyapisit, Dr. Pannathat Soontrapa
Division of Neurology, Department of Medicine, Faculty of Medicine Siriraj Hospital, Bangkok, Thailand
Background: Myasthenia gravis (MG) is an autoimmune disorder of the neuromuscular junction characterized by fluctuating skeletal muscle weakness. While surgery is a well-recognized precipitant of perioperative exacerbation and myasthenic crisis, data regarding the prevalence and risk factors of these events in our local settings remain limited. This study aims to determine the prevalence of perioperative MG exacerbation and identify associated risk factors and complications in patients with MG undergoing surgery.
Methods: We conducted a retrospective chart review of adult patients with MG who underwent major surgery at Siriraj Hospital between May 2011 and December 2023. We analyzed clinical characteristics, perioperative factors, and postoperative outcomes to determine the prevalence of MG exacerbation. Univariate and multivariate logistic regression analyses were performed to identify independent predictors of perioperative exacerbation.
Results: A total of 372 patients were included, 73% (271/372) of whom were female. Perioperative MG exacerbation occurred in 31 patients (8.3%), including 17 patients (4.6%) who developed myasthenic crisis. Comorbidities were comparable between groups (p = 0.25). Thymectomy was more frequent in the exacerbation group (83.9% vs 66.6 %, p = 0.048), particularly via the open surgical approach (48.4 vs 19.9%, p = 0.004). In multivariate analysis, independent predictors of perioperative exacerbation were thymectomy (OR 5.7; 95% CI 1.6 -19.5, p = 0.01), intraoperative blood loss >1,000 mL (OR 10.0; 95% CI 1.2-84.5, p = 0.04), perioperative intravenous magnesium sulfate administration (OR 4.74; 95% CI 1.5-15.2, p = 0.01) and perioperative pneumonia (OR 10.5; 95% CI 2.1-51.5, p = 0.004). Patients with exacerbation experienced significantly longer hospital stays (6.0 vs 4.0 days, p <0.001).
Conclusion: Perioperative MG exacerbation remains a critical clinical challenge. Recognizing and mitigating modifiable risk factors are essential for optimizing surgical planning and enhancing perioperative safety in patients with MG.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.175
Long-Term Safety and Efficacy of Nipocalimab: Approximately 2 Years Follow-Up Results From Vivacity-MG3 Open-Label Extension
Dr. Carlo Antozzi1, Dr. Tuan Vu2, Dr. Sindhu Ramchandren3, Dr. Richard J. Nowak4, Dr. Constantine Farmakidis5, Dr. Vera Bril6, Dr. Jan De Bleecker7, Dr. Huan Yang8, Dr. Eduard Minks9, Dr. Jin-Sung Park10, Dr. Mariusz Grudniak11, Dr. Marek Smilowski12, Dr. Teresa Sevilla13, Dr. Sarah Hoffmann14, Dr. Kumaraswamy Sivakumar15, Dr. Panna Sanga3, Dr. Ibrahim Turkoz3, Dr. Yaowei Zhu3, Dr. Marie Fitzgibbon16, Dr. Michel Burcklen17
1
Immunotherapy and Apheresis Unit, Neuroimmunology and Muscle Pathology Unit, Fondazione IRCCS Istituto Neurologico C. Besta, Milan, Italy.
2
Neurology, University of South Florida, Tampa, United States.
3
Johnson & Johnson, NJ, Titusville, United States.
4
Department of Neurology, Yale University School of Medicine, New Haven, United States.
5
Neurology, University of Kansas Medical Center, Kansas City, United States.
6
Neurology, University of Toronto, University Health Network, Toronto, Canada.
7
Neurology, Ghent University Hospital, Ghent, Belgium.
8
Neurology, Xiangya Hospital, Central South University, Changsha, China.
9
First Department of Neurology, Faculty of Medicine, Masaryk University and St. Anne’s Hospital, Brno, Czechia.
10
Department of Neurology, School of Medicine, Kyungpook National University, Kyungpook National University Chilgok Hospital, Daegu, Korea, Republic of.
11
Neurology, Centrum Medyczne, Neuro-Protect, Warszawa, Poland.
12
Neurology, Silesian Neurology Medical Center, Katowice, Poland.
13
Neurology, Hospital Universitari i Politècnic and IIS La Fe/University of Valencia, Valencia, Spain.
14
Charité-Universitätsmedizin, corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Department of Neurology, Neuroscience Clinical Research Center (NCRC) and Integrated Myasthenia Gravis Center, Berlin, Germany.
15
The Neuromuscular Research Center and Neuromuscular Clinic of Arizona, AZ, Phoenix, United States.
16
Johnson & Johnson, NJ, Raritan, United States.
17
Johnson & Johnson, Basel, Switzerland
Background: In patients with generalized myasthenia gravis (gMG), long-term sustained disease control remains an important treatment goal. In a broad population of autoantibody-positive patients with gMG, nipocalimab (a neonatal Fc-receptor blocker), demonstrated substantial improvement from baseline in MG-Activities of Daily Living (MG-ADL) scores over 24 weeks (W) in the double-blind (DB) phase and over 60W of the open-label extension (OLE) phase of the Vivacity-MG3 study. Here we assessed the long-term safety and efficacy of nipocalimab in patients with gMG from the ongoing Vivacity-MG3 (NCT04951622) study OLE phase. Results with additional follow-up are presented up to W96 of the OLE with nipocalimab treatment.
Methods: In Vivacity-MG3, adult patients with gMG (Myasthenia Gravis Foundation of America Class II-IV), inadequately controlled (MG-ADL≥6) on standard-of-care (SOC) therapy, were randomized 1:1 to nipocalimab+SOC or placebo+SOC in 24W DB phase, followed by an option to enter the OLE phase. Efficacy and safety were evaluated in seropositive (anti- acetylcholine receptor [AChR] positive, anti-muscle-specific kinase [MuSK] positive, and/or anti-low density lipoprotein receptor [LRP]4 positive) patients who received ≥1 dose of study treatment.
Results: In the seropositive efficacy analysis set (N=137) during OLE, total duration of treatment (median [range]) for placebo/nipocalimab (n=66) and nipocalimab/nipocalimab (n=71) groups was 69.1W (8–128) and 62.1W (8–128) respectively. The mean (standard deviation [SD]) change-from-baseline (CFB) in MG-ADL scores from DB baseline for those who reached 96W of treatment with placebo/nipocalimab and nipocalimab/nipocalimab groups was –6.69 (3.39) and –6.47 (4.62), and CFB in Quantitative Myasthenia Gravis (QMG) scores was –5.81 (4.11) and –5.97 (4.97) respectively. The mean (SD) percent CFB in IgG for placebo/nipocalimab group was –63.24 (14.31) and for nipocalimab/nipocalimab group was –64.06 (12.91). No new safety concerns were reported in OLE phase.
Conclusion: In autoantibody-positive patients with gMG, long-term treatment with nipocalimab demonstrated sustained disease control through the 24-week phase 3 Vivacity-MG3 study and 96 weeks of the OLE (120 weeks total).
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.176
Long-Term Safety and Efficacy of Subcutaneous Efgartigimod PH20 for Generalized Myasthenia Gravis: ADAPT-SC+ Final Results
Dr. Carlo Antozzi1, Dr. Ratna Bhavaraju-Sanka2, Dr. Jan L. De Bleecker3, Dr. Andreas Meisel4, Dr. Kimiaki Utsugisawa5, Dr. Wan-Yi Huang6, Dr. Rosa Hermina Jimenez6, Dr. Fien M. Verhamme6, Dr. Li Liu6, Dr. Denis Korobko7, Dr. Anna Kostera-Pruszczyk8, Prof. Jan J. G. M. Verschuuren9, Dr. Tuan Vu10, Dr. Heinz Wiendl11, Dr. James F. Howard Jr12
1
Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy.
2
University of Texas Health Science Center at San Antonio, San Antonio, United States.
3
Ghent University Hospital, Ghent, Belgium.
4
Charité – Universitätsmedizin Berlin, Berlin, Germany.
5
Hanamaki General Hospital, Hanamaki, Japan.
6
argenx, Ghent, Belgium.
7
State Budgetary Healthcare Institution of Novosibirsk Region “The State Novosibirsk Regional Clinical Hospital”, Novosibirsk, Russia.
8
Medical University of Warsaw, Warsaw, Poland.
9
Leiden University Medical Center, Leiden, Netherlands.
10
University of South Florida Morsani College of Medicine, Tampa, United States.
11
Institute of Translational Neurology, University Hospital Münster, Münster, Germany.
12
The University of North Carolina, Chapel Hill, United States
Background: Efgartigimod is a human immunoglobulin G1 (IgG1) Fc fragment that blocks the neonatal Fc receptor. During ADAPT-SC, subcutaneous (SC) efgartigimod PH20 (coformulated with recombinant human hyaluronidase PH20) demonstrated noninferior total IgG reduction to intravenous efgartigimod in participants with generalized myasthenia gravis (gMG). ADAPT-SC+ was an open-label extension study evaluating long-term safety and efficacy of efgartigimod PH20 SC in participants with gMG.
Methods: Efgartigimod PH20 SC 1000 mg was administered in cycles of 4 once-weekly injections. Subsequent cycles were initiated based on clinical evaluation (Year 1, ≥4 weeks between cycles; Year 2 and onward, ≥1 week between cycles following protocol amendment). Safety was assessed using incidence and severity of adverse events. Clinical efficacy was assessed using Myasthenia Gravis Activities of Daily Living (MG-ADL). Quality of life (QoL) impact was assessed during Year 1 using Myasthenia Gravis Quality of Life 15-Item Questionnaire, Revised (MG-QoL15r).
Results: During ADAPT-SC+, 180 participants received ≥1 efgartigimod PH20 SC injection (mean [SD] study duration, 2.6 [0.9] years; 459.4 participant years of follow-up). Adverse events were predominantly mild/moderate. Injection site reactions were mild/moderate, did not lead to discontinuation, and decreased in incidence with subsequent cycles. The median (Q1, Q3) average time between treatment cycles decreased from 4.8 (4.3, 7.9) weeks under the original protocol to 4.2 (3.0, 6.5) weeks after the protocol amendment. Efgartigimod remained well tolerated in participants whose cycle intervals shortened under the amended protocol after Year 1. MG-ADL score improvements in anti-acetylcholine receptor antibody–positive (AChR-Ab+) participants were observed by Week 1 and sustained through Week 163; a similar improvement pattern was observed in anti-acetylcholine receptor antibody–negative (AChR-Ab−) participants. Minimal symptom expression (MSE; MG-ADL score, 0-1) was observed at least once in 59.2% (n=84) of AChR-Ab+ participants and in 34.2% (n=13) of AChR-Ab− participants. Of participants who achieved MSE, a majority had periods of sustained MSE, with 88.1% (n=74) of AChR-Ab+ and 69.2% (n=9) of AChR-Ab− participants experiencing MSE at consecutive assessments during ≥4 weeks. Sustained improvements in MG-QoL15r were observed in the period during which it was assessed (Year 1).
Conclusion: Long-term treatment with efgartigimod PH20 SC was well tolerated and led to sustained efficacy and QoL improvements during ADAPT-SC+, including achievement of MSE and sustained MSE in subsets of AChR-Ab+ and AChR-Ab− participants.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.177
Assessing Relationship Between IgG-Levels and Infection Rate in Nipocalimab-Treated Patients with Generalized Myasthenia Gravis (Vivacity-MG3)
Dr. Ghazala Hayat
1
, Dr. Ricardo Rojo Cella
2
, Dr. Robert Edwards
2
, Dr. Marie Fitzgibbon
3
, Mr. Robert Gordon
2
, Dr. Wim Noel
4
, Dr. Zia Choudhry
5
, Miss Kavita Gandhi
3
, Dr. Sindhu Ramchandren
6
, Dr. Kevin L. Winthrop
7
, Dr. Carlo Antozzi
8
1
Department of Neurology, Saint Louis University School of Medicine St Louis, MO, United States.
2
Johnson & Johnson, Spring House, PA, United States.
3
Johnson & Johnson, Raritan, NJ, United States.
4
Johnson & Johnson,, Beerse, Belgium.
5
Johnson & Johnson, Horsham, PA, United States.
6
Johnson & Johnson, Titusville, NJ, United States.
7
Division of Infectious Diseases, School of Medicine, School of Public Health, Oregon Health and Science University, Portland, OR,, United States.
8
Immunotherapy and Apheresis Unit, Neuroimmunology and Muscle Pathology Unit, Fondazione IRCCS Istituto Neurologico C. Besta, Milan, Italy
Background: Nipocalimab, a neonatal Fc receptor inhibitor, demonstrates rapid and substantial immunoglobulin G (IgG) reduction. In the phase 3 Vivacity-MG3 study, nipocalimab (30 mg/kg Day 0 loading-dose, then 15 mg/kg every 2 weeks as maintenance dose) + standard-of-care (SOC) resulted in a median IgG reduction of –69% at week 24, which was associated with rapid and sustained disease control over 24 weeks in a broad population of antibody-positive patients with generalized myasthenia gravis (gMG), and was well-tolerated with an acceptable safety profile. Overall infection incidence was similar between nipocalimab and placebo groups (43% each). The objective of the current analysis was to evaluate whether there is a relationship between reduced IgG levels and infection rates in patients with gMG and treated with nipocalimab + SOC across the double-blind (DB) phase of Vivacity-MG3.
Methods: Receiver Operating Characteristic (ROC) analysis was conducted to assess whether IgG levels (evaluated as a continuous variable) were associated with infection rates in patients from Vivacity-MG3 study. Area-Under-the-Curve (AUC) values were calculated using logistic regression ROC analysis with treatment group, maximum change from baseline in IgG, and baseline IgG as covariates. AUC values range from 0–1, with 0.5–≤0.7 indicating no/poor discrimination (equivalent to chance/poor discrimination) and higher values reflecting predictive association (≥0.8–≤0.9, excellent).
Results: ROC analysis showed AUC (standard error) for patients with lowest post-baseline level of IgG ≤5 g/L (N=94, 105.5 patient-years of exposure): 0.51 (0.062); ≤4 g/L (N=87, 97.9 patient-years-of-exposure): 0.52 (0.064); ≤3 g/L (N=74, 72.4 patient-years-of-exposure): 0.53 (0.070); ≤2 g/L (N=44, 30.0 patient-years-of-exposure): 0.69 (0.082). These values indicate no real predictive association between reduced IgG levels (as low as ≤2 g/L) and infection rates.
Conclusion: These results show no association of occurrence of infections with lowering IgG levels with nipocalimab in patients with gMG in the Vivacity-MG3 study.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.178
Efficacy and Safety of Subcutaneous Efgartigimod PH20 in Adults With oMG: ADAPT OCULUS Interim Results
Dr. Carolina Barnett-Tapia1, Dr. Andreas Meisel2, Dr. James F. Howard Jr3, Dr. Vern C. Juel4, Dr. Chongbo Zhao5, Dr. Jianying Xi5, Dr. Akiyuki Uzawa6, Dr. Stephen W. Reddel7, Dr. Kristl G. Claeys8,9, Dr. Carlo Antozzi10, Dr. Łukasz Rzepiński11,12, Dr. Rodrigo Álvarez-Velasco13, Dr. Oleg Lukash14, Dr. Rosa Hermina Jimenez14, Dr. Fien Gistelinck14, Dr. Fien M. Verhamme14, Dr. Sui H. Wong15
1
University of Toronto, Toronto, Canada.
2
Charité – Universitätsmedizin Berlin, Berlin, Germany.
3
The University of North Carolina, Chapel Hill, United States.
4
Duke University School of Medicine, Durham, United States.
5
Huashan Hospital, Fudan University, Shanghai, China.
6
Chiba University, Chiba, Japan.
7
Concord Hospital, Sydney, Australia.
8
University Hospitals Leuven, Leuven, Belgium.
9
KU Leuven, Leuven, Belgium.
10
Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy.
11
10th Military Research Hospital and Polyclinic, Bydgoszcz, Poland.
12
University of Science and Technology, Bydgoszcz, Poland.
13
Hospital Universitario Ramón y Cajal, Madrid, Spain.
14
argenx, Ghent, Belgium.
15
Moorfields Eye Hospital, London, United Kingdom
Background: Ocular myasthenia gravis (oMG) is an immunoglobulin G (IgG)-mediated autoimmune disease characterized by fluctuating fatigable weakness of the ocular muscles. There is an unmet need for approved, effective treatments for patients with oMG. Efgartigimod PH20 is a human IgG1 antibody Fc fragment coformulated with recombinant human hyaluronidase PH20 that selectively reduces IgG levels by blocking neonatal Fc receptor–mediated IgG recycling. Retrospective analyses of data from patients with generalized MG treated with efgartigimod have indicated an improvement in ocular symptoms. The Phase 3 ADAPT OCULUS trial (NCT06558279) is evaluating the efficacy and safety of subcutaneous (SC) efgartigimod PH20 in adults with oMG.
Methods: In Part A, adults with oMG (defined as Myasthenia Gravis Foundation of America Class I) and a Myasthenia Gravis Impairment Index (MGII) patient-reported outcome (PRO) subcomponent ocular score of ≥6 are randomized 1:1 to receive 4 once-weekly efgartigimod PH20 SC 1000 mg or placebo injections administered via prefilled syringe, followed by 4 weeks of follow-up. Participants may continue the study in Part B (≤2-year open-label extension) to receive cycles of 4 once-weekly efgartigimod PH20 SC injections followed by treatment-free periods. The first 2 cycles of Part B comprise a treatment period followed by a 4-week treatment-free period. Subsequent cycles are initiated based on clinical evaluation, with ≥7 days between treatment periods.
Results: The primary endpoint is change in MGII PRO ocular score from baseline to Week 4. Key secondary endpoints include changes in MGII ocular score (PRO plus physical examination), Myasthenia Gravis Activities of Daily Living ocular domain score, and MGII total score from baseline to Week 4. Safety assessments include incidence and severity of adverse events. Topline results from the primary analysis of Part A will be presented at the conference.
Conclusion: ADAPT OCULUS is the first and largest global Phase 3 clinical trial to address the unmet need for treatment in oMG by evaluating the safety and efficacy of efgartigimod PH20 SC in adults with oMG.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.179
Individual Item-Level Analyses of Myasthenia Gravis-Specific Outcomes During Zilucoplan Treatment in RAISE and RAISE-XT
Dr. Channa Hewamadduma1,2, Dr. Miriam Freimer3, Dr. M. Isabel Leite4, Dr. Angelina Maniaol5, Dr. Kimiaki Utsugisawa6, Dr. Michael D. Weiss7, Dr. Jos Bloemers8, Dr. Babak Boroojerdi9, Dr. Paul Mahoney10, Dr. Natasa Savic11, Dr. James Howard12
1
Academic Neuromuscular Unit, Sheffield Teaching Hospitals NHS Foundation Trust, Sheffield, United Kingdom.
2
Sheffield Institute for Translational Neuroscience (SITraN), University of Sheffield, Sheffield, United Kingdom.
3
Department of Neurology, The Ohio State University Wexner Medical Center, Columbus, Canada.
4
Nuffield Department of Clinical Neurosciences, University of Oxford, Oxford, United Kingdom.
5
Department of Neurology, Oslo University Hospital, Oslo, Norway.
6
Department of Neurology, Hanamaki General Hospital, Hanamaki, Japan.
7
Department of Neurology, University of Washington Medical Center, Seattle, United States.
8
UCB, Brussels, Belgium.
9
UCB, Monheim, Germany.
10
UCB, Slough, United Kingdom.
11
UCB, Bulle, Switzerland.
12
Department of Neurology, The University of North Carolina at Chapel Hill, Chapel Hill, United States
Background: Patients with generalised myasthenia gravis (gMG) experience fluctuating muscle weakness, leading to a diverse range of symptoms that can impact quality of life. In RAISE (Phase 3, NCT04115293), zilucoplan, a potent macrocyclic complement component 5 inhibitor, showed statistically significant and clinically meaningful improvements in myasthenia gravis (MG)-specific outcomes in patients with anti-acetylcholine receptor antibody-positive (anti-AChR Ab+) gMG. These improvements were sustained over 120 weeks in the ongoing, Phase 3, open-label extension, RAISE-XT (NCT04225871). This post hoc analysis assessed change in item scores for MG Activities of Daily Living (MG-ADL), Quantitative MG (QMG) and MG Quality of Life 15-item revised (MG-QoL15r).
Methods: In RAISE, adults with anti-AChR Ab+ gMG were randomised to self-administer once-daily placebo or zilucoplan 0.3 mg/kg. Adults who completed RAISE received zilucoplan 0.3 mg/kg in RAISE-XT. Analyses of MG-ADL, QMG and MG-QoL15r (interim data cut-off: 11 November 2023) included the proportion of patients showing no symptoms, improvement and no change from baseline to Week 12 (RAISE), and from double-blind baseline to Week 120 (RAISE-XT). Change from double-blind baseline (CFB) to Week 120, and safety, were assessed in RAISE-XT.
Results: In RAISE, a greater proportion of patients improved from baseline to Week 12 or had no symptoms at Week 12 across MG-ADL items with zilucoplan than placebo (Table 1). Improvements in the zilucoplan group further increased and were sustained in RAISE-XT to Week 120 (Table 1). At Week 120 (n=73), mean (standard error) CFB for MG-ADL items was: chewing, −0.85 (0.09); swallowing, −0.75 (0.10); talking, −0.86 (0.10); breathing, −0.71 (0.09); double vision, −1.04 (0.14); eyelid droop, −1.44 (0.13); brush hair/teeth, −0.96 (0.11) and rise from chair, −0.79 (0.11). Similar results were observed for QMG and MG-QoL15r. Treatment-emergent adverse events occurred in 97.0% (n=194/200, RAISE-XT) of patients.
Conclusion: Treatment with zilucoplan showed improvements in a greater proportion of patients across individual items in MG-specific outcomes at Week 12 versus placebo. During the extension study, the proportion of patients demonstrating these improvements further increased and were sustained through to Week 120. These data show that treatment with zilucoplan may provide benefit across a broad range of signs and symptoms of MG. Funding: UCB.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
Images or Table (Optional)
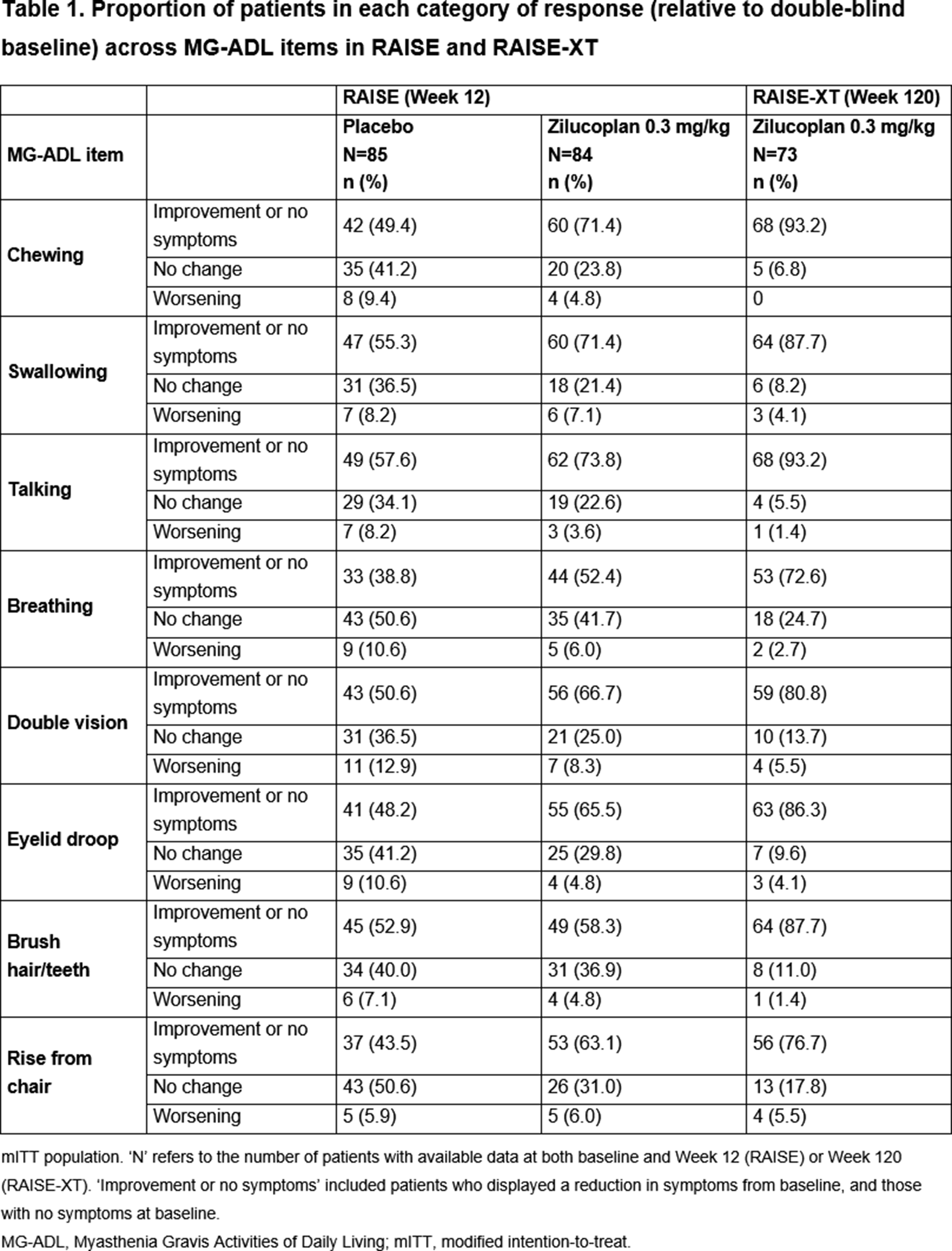
PP01.180
Morphological and Quantitative Analysis of Neuromuscular Junction Pathology in AChR Antibody-Positive Myasthenia Gravis
Dr. Corinna Preusse1,2,3, Miss Katharina Brokamp1, Prof. Andreas Meisel1, Assoc. Prof. Andreas Roos4,5,6, Dr. Paolo Doksani1, Dr. Andreas Hentschel7, Dr. Carsten Dittmayer8, Prof. Markus Schuelke2, Prof. Jens-Carsten Rueckert9, Prof. Matthias Pumberger10, Prof. Friederik Schömig10, Prof. Werner Stenzel3, Prof. Sarah Hoffmann1
1
Charité – Universitätsmedizin Berlin, corporate member of Freie Universität Berlin, Humboldt-Universität zu Berlin, and Berlin Institute of Health (BIH), Department of Neurology and Neuroscience Clinical Research Center, Charitéplatz 1, 10117 Berlin, Germany, Berlin, Germany.
2
Charité - Universitätsmedizin, corporate member of Freie Universität Berlin, Humboldt-Universität zu Berlin and Berlin Institute of Health (BIH), Department of Neuropediatrics and NeuroCure Cluster of Excellence, Augustenburger Platz 1, 13353 Berlin, Germany, Berlin, Germany.
3
Charité - Universitätsmedizin, corporate member of Freie Universität Berlin, Humboldt-Universität zu Berlin, and Berlin Institute of Health (BIH), Department of Neuropathology, Charitéplatz 1, 10117 Berlin, Germany, Berlin, Germany.
4
Centre for Neuromuscular Disorders, Centre for Translational Neuro- and Behavioral Sciences, Department of Pediatric Neurology, University Duisburg-Essen, 45147 Essen, Germany, Essen, Germany.
5
Brain and Mind Research Institute, Children's Hospital of Eastern Ontario Research Institute, Ottawa, ON K1H 8L1, Canada., Ottawa, Canada.
6
Department of Neurology with Heimer Institute for Muscle Research, University Hospital Bergmannsheil, Bochum, Germany, Bochum, Germany.
7
Leibniz-Institut Für Analytische Wissenschaften - ISAS - E.V., 44139, Dortmund, Germany, Dortmund, Germany.
8
Charité – Universitätsmedizin Berlin, corporate member of Freie Universität Berlin, Humboldt-Universität zu Berlin, and Berlin Institute of Health (BIH), Department of Pathology, Charitéplatz 1, 10117 Berlin, Germany, Berlin, Germany.
9
Charité - Universitätsmedizin, corporate member of Freie Universität Berlin, Humboldt-Universität zu Berlin, and Berlin Institute of Health (BIH), Department of Thoracic Surgery, Charitéplatz 1, 10117 Berlin, Germany, Berlin, Germany.
10
Charité - Universitätsmedizin, corporate member of Freie Universität Berlin, Humboldt-Universität zu Berlin, and Berlin Institute of Health (BIH), Center for Musculoskeletal Surgery, Charitéplatz 1, 10117 Berlin, Germany, Berlin, Germany
Background: For over four decades, complement deposition at the neuromuscular junction (NMJ) in acetylcholine receptor antibody-positive myasthenia gravis (AChR-ab⁺-MG) has been recognized as a major contributor to motor endplate destruction. Nevertheless, comprehensive studies addressing the extent, variability, and molecular correlates of NMJ pathology in this condition remain scarce. Given the rapid clinical improvement observed with recent immunotherapies, suggesting that irreversible destruction of NMJ is unlikely, we examined the morphology and molecular profiles of NMJs in AChR-ab⁺-MG using multiple complementary approaches.
Methods: Intercostal muscle (ICM) biopsy specimen from 58 patients with AChR-ab⁺-MG were analyzed and compared to non-disease controls (NDC, biopsy specimen obtained during scoliosis surgery; n=7) using histology, electron microscopy (EM) and transcriptomic and proteomic profiling.
Results: The mean age at biopsy was 48.1 years (SD 16.5), and 64.9% were female. Mean disease duration at the time of biopsy was 4.4 years (SD 3.6). The mean MG-ADL score was 6.9 (SD 3.6), the mean QMG score 11.4 (SD 5.3), and the mean MG-QoL score was 19.2 (SD 9.7), 14% of patients had experienced myasthenic crisis. Regarding ongoing treatments, 91.2% of patients received acetylcholinesterase inhibitors, 50.9% were treated with glucocorticoids, and 52.6% with conventional immunosuppressants. Three patients (5.3%) had not received any MG specific therapy at time of biopsy. Thymic pathology included thymoma (10.5%), thymic hyperplasia (31.6%) and was normal in the remaining patients.
Histological evaluation revealed prominent C5b-9 deposition on endplates in all samples, affecting on average 75% of detectable NMJs (range 33–100%), whereas no C5b-9-positive staining was detectable on the endplates of control intercostal muscle biopsies. Additionally, immune cell infiltration close to the NMJ was detectable, and immunohistochemistry demonstrated occasional CD8⁺ T cells and a more prominent CD68⁺ macrophage infiltration, while B cells were consistently absent.
Transcriptomic analysis by qPCR revealed upregulation of immune-related genes such as IL1B and STAT3, indicating a local pro-inflammatory milieu. Interestingly, this includes increased expression of the B cell chemoattractant BAFF, despite the absence of detectable B cells. Furthermore, proteomic profiling revealed two upregulated and nine downregulated proteins between MG compared to NDC, with many of the reduced proteins linked to extracellular matrix integrity or vesicular trafficking, suggesting impaired structural compensation and synaptic stress in the context of chronic immune-mediated NMJ damage.
Ultrastructural analysis was performed in 44 NMJs and the pre- and post-synaptic morphology including individual fold length, branching and fold areas were assessed. AChR-ab⁺-MG patients muscles harbored NMJ with reduced numbers and short folds as well as a postsynaptic apparatus with small plump folds.
Conclusion: Together, these findings reveal that complement-mediated affection of NMJ in skeletal muscle of AChR-ab⁺-MG is heterogeneous and that even in individual biopsies, not all NMJs demonstrate complement deposition and undergo consecutive structural disruption, and if so, that the destructive process may not be terminal. Hence, our combined histologic, ultrastructural, and molecular data highlight significant intra- and interindividual variability. These observations underscore the importance of examining the precise extent and nature of NMJ involvement in MG and may inform future therapeutic strategies targeting local immune mechanisms.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.181
Thiol–disulfide Homeostasis in Patients with Myasthenia Gravis: Association With Disease Severity and Clinical Parameters
Dr. Mehmet Bal1, Assoc. Prof. Dilek Agircan1, Assoc. Prof. Seyhan Taskin2
1
Harran University Faculty of Medicine, Department of Neurology, Sanliurfa, Turkey.
2
Harran University Faculty of Medicine, Department of Physiology, Sanliurfa, Turkey
Background: Myasthenia Gravis (MG) is an autoimmune neuromuscular disorder characterized by chronic, fluctuating weakness of ocular and skeletal muscles. Oxidative stress has been implicated in MG pathogenesis through damage to acetylcholine receptors (AChRs), which are essential for neuromuscular transmission. Thiol–disulfide homeostasis (TDH) is a dynamic biochemical marker of oxidative stress and has been shown to shift toward oxidation in various neurological disorders, sometimes correlating with disease activity. This study aimed to compare TDH parameters between MG patients and healthy controls and to investigate their association with disease severity and fatigue.
Methods: Patients diagnosed with MG and age- and sex-matched healthy controls were enrolled. Serum native thiol (NT) and total thiol (TT) levels (µmol/L) were measured, and disulfide (SS) levels were calculated using the formula SS = (TT − NT) / 2. The ratios SS/NT, SS/TT, and NT/TT were derived. Clinical assessment of MG patients included the MG Activities of Daily Living (MG-ADL) score, MGFA clinical classification, Fatigue Severity Scale (FSS), and Myasthenia Gravis Quality of Life-15 (MG-QoL15). MG phenotype (ocular or generalized), serological status (AChR or MuSK antibodies), disease duration, and current treatments were recorded. Associations between TDH parameters, disease severity, and fatigue were analyzed.
Results: A total of 30 MG patients and 27 healthy controls were included. Compared with controls, MG patients exhibited significantly lower NT and TT levels, while SS levels were similar between groups. TDH parameters did not differ significantly according to MG phenotype, serological status, treatment modality, or predefined thresholds for fatigue (FSS ≥36) and quality of life (MG-QoL15 ≥20). However, patients with MG-ADL ≥6 showed significantly lower SS, SS/NT, and SS/TT ratios, along with a significantly higher NT/TT ratio. Correlation analysis demonstrated negative correlations between MG-ADL scores and SS, SS/NT, and SS/TT ratios, and a positive correlation between MG-ADL scores and the NT/TT ratio. No significant correlations were found between TDH parameters and FSS or MG-QoL15 scores, whereas FSS and MG-QoL15 were strongly and positively correlated.
Conclusion: MG patients exhibit an altered thiol–disulfide balance characterized by reduced thiol reserves without increased disulfide accumulation, suggesting that oxidative imbalance in MG is primarily driven by thiol depletion. The association between TDH parameters and MG-ADL scores indicates a potential link between oxidative stress and clinical disease severity, independent of MG subtype, serological status, or treatment. In contrast, oxidative stress markers were not associated with fatigue or quality of life. These findings support the role of oxidative stress in MG pathogenesis and suggest that thiol–disulfide homeostasis may serve as a potential biomarker for disease severity, warranting further investigation in larger, longitudinal studies.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
Images or Table (Optional)
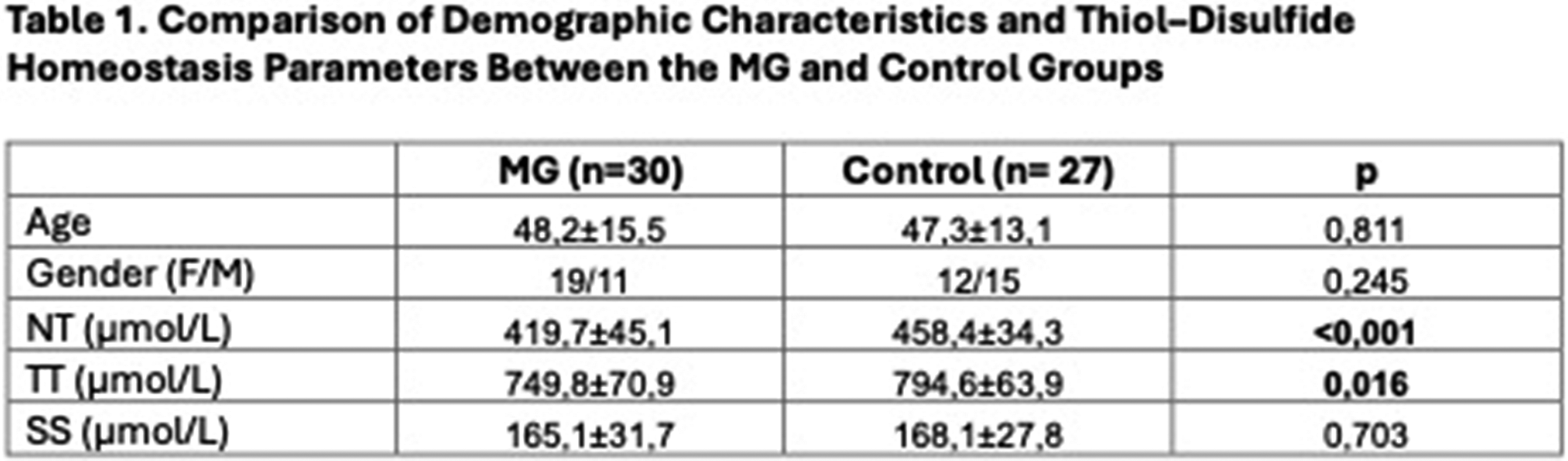
PP01.182
Treatment of Newly Diagnosed AChR - Myasthenia Gravis with Rituximab
Mrs Dimitra Tzavella1,2, Dr. Aigli G Vakrakou1,2, Mrs Eleni Strataki1,2, Mrs Maria Belimezi3, Assoc. Prof. Vasiliki Zouvelou1,2
1
1st Neurology Department, Eginition Hospital, National and Kapodistrian University of Athens, Athens, Greece.
2
ERN EURO-MND, Athens, Greece.
3
Diagnostic Department Hellenic Pasteur Institute, Athens, Greece
Background: Management of newly diagnosed acetylcholine receptor (AChR) – positive generalized myasthenia gravis (gMG) predominantly relies on corticosteroids; however, long-term administration is limited by significant cumulative toxicity. Consequently, there is an unmet need for non-steroidal immunotherapies characterized by rapid onset and favorable safety profiles. Rituximab (RTX), a CD20-targeting monoclonal antibody, represents an adjunct therapy to achieve durable clinical remission while facilitating corticosteroid tapering.
Methods: This single-center, observational study evaluated 32 newly diagnosed AChR-positive gMG who received RTX within 24 months from diagnosis (mean 8 months), as their sole non-steroidal immunosuppressant therapy. Clinical efficacy and safety were longitudinally assessed via Myasthenia Gravis Activities of Daily Living (MG-ADL), MGFA Post-Intervention Status (MGFA-PIS), daily corticosteroid doses, and AChR antibody titers.Follow-up of 25 of 32 patients evaluated for at least one therapeutic cycle and a minimum of 3-month post-RTX. For the rest, 1-month data are available.
Results: The cohort of 32 patients (age range 22–85 years, mean age at RTX initiation: 63 years) included 15 patients (45%) with very-late-onset gMG (≥65 years) and 5 (16%) with thymoma-associated gMG. Patients were categorized into three distinct groups according to the clinical indication for RTX treatment: steroid-sparing effect in well-controlled patients (Group 1, n=22) and clinical improvement in partial respondersor intolerant to steroids (Group 2, n=5), and in refractory cases (Group 3, n=5). Significant clinical improvement was observed, with mean MG-ADL decreasing from 1.9 at baseline to 0.68 at 3 months (64% reduction), 0.3 at 6 months (82%), and achieving 0 at 12 months. Concurrently, the mean daily corticosteroid dose was reduced from 18 mg to 11 mg at 6 months (65% reduction) and 7 mg at 12 months (76% reduction). At 6-months, 56% (n=18) were in pharmacological remission, with 21% (n=7) exhibiting minimal manifestations status. Notably, noclinical deterioration was observed and no rescuetherapy was needed during monitoring. Furthermore, no safety issue was established. AChR antibody titers demonstrated a mean decline of 30% at 6 months and 52% at 12 months.
Conclusion: Our findings underscore the clinical utility of early integration of RTX into the therapeutic algorithm for AChR - gMG, as a steroid-sparing agent and as an early immunosuppressive therapy, with favorable safety profile, for steroid partial responders, including thymoma associated and very-late-onset gMG patients. Early introduction of RTX may accelerate clinical stabilization and substantially mitigate the burden of long-term corticosteroid dependence.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.183
Management of Severe Paediatric Guillain-Barré Syndrome Due to Post-Traumatic Stress Disorder: Case Presentation
Dr. Eleftheria Kokkinou1, Dr. Argiro Kaltsa2, Dr. Doxa-Eleni Sareidaki1, Dr. Anna Moustafellou1, Dr. Iasonas-George Stamatakis1, Mr. Kosmas Lymperatos1, Mr. Dimitris Lignos1, Mr. Nikolaos Mouzis1, Ms. Miranta Pavlou1, Ms. Iliana Psichari1, Ms. Iliana Papadaki1, Prof. Aikaterini Papanikolaou2, Dr. Marina Katsalouli1
1
Department of Neurology, Children's Hospital “Agia Sofia”, Athens, Greece.
2
Department of Child Psychiatry, National and Kapodistrian University of Athens Medical School, Children's Hospital “Agia Sofia”, Athens, Greece
Background: Guillain-Barré Syndrome (GBS) is an acute immune-mediated polyneuropathy that can rapidly progress to life-threatening neuromuscular paralysis in children. While most paediatric patients recover neurologically with timely treatment, severe cases often involve intensive care and prolonged hospitalization—conditions that can lead to significant psychological trauma. Post-traumatic stress disorder (PTSD) is increasingly recognized as a complication in paediatric GBS with the potential to influence both short- and long-term recovery outcomes.
Methods: Case presentation
Results: We report a case of a previously healthy 9-year-old male who initially presenting with ascending paralysis that rapidly progressed to quadriparesis. Despite prompt initiation of standard treatment with intravenous immunoglobulin (IVIG), plasma exchange (PLEX), pain management and intensive rehabilitation care, the patient experienced an extended hospitalization. Neurological function gradually improved; however, the patient remained hospitalized due to persistent refusal to eat, sleep disturbances, nightmares, severe anxiety, avoidance behaviors, irritability, angry outbursts and emotional withdrawal. A multidisciplinary approach was initiated, involving paediatric psychiatry and nutritional support. PTSD was diagnosed clinically and targeted interventions, including psychotherapy, child life support and gradual reintroduction of oral feeding were employed. Following two months of hospitalization, the patient was discharged home with significant neurological and psychological improvement. Ongoing outpatient care was strongly recommended, including continued physical therapy to support neuromuscular recovery and structured psychological support to address residual symptoms of PTSD and prevent relapse.
Conclusion: This case underscores the need for early recognition and integrated management of psychological sequelae, including PTSD, in children recovering from critical illness such as GBS.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Immune-mediated Neuropathies
PP01.184
Management of Paediatric Guillain-Barré Syndrome: Case Presentations
Dr. Eleftheria Kokkinou, Dr. Doxa-Eleni Sareidaki, Dr. Iasonas-George Stamatakis, Dr. Anna Moustafellou, Mr. Dimitris Lignos, Mr. Nikolaos Mouzis, Mr. Kosmas Lymperatos, Ms. Miranta Pavlou, Ms. Ilianna Psichari, Ms. Ilianna Papadaki, Dr. Marina Katsalouli
Department of Neurology, Children's Hospital “Agia Sofia”, Athens, Greece
Background: Guillain-Barré Syndrome (GBS) is an acute immune-mediated neuropathy often triggered by infections. The most paediatric patients respond well to standard treatments. We present three cases of previously healthy children diagnosed with GBS.
Methods: Case presentations
Results: Case 1: A 7-year-old boy presented with sudden refusal to walk and severe bilateral leg pain beginning earlier that day. There was no history of trauma or systemic symptoms. On examination he demonstrated absent deep tendon reflexes in both legs. He was started on IVIG. Serologic testing revealed positive Parvovirus B19 IgM. The patient’s neurological function improved with physical therapy. After a 1 month hospitalization, he was discharged home with near-complete motor recovery and outpatient follow-up arranged for continued rehabilitation.
Case 2: A 9-year-old male developed severe GBS (AMAN variant) and received timely standard treatment and intensive rehabilitation. Serologic test revealed positive Mycoplasma pneumoniae IgM. Despite receiving the above standard treatment and gradually neurological improvement, the patient had a prolonged hospital stay due to exhibition of signs of PTSD.
Case 3: A 3-year-old female presented with rapid-onset quadriparesis and autonomic instability. Her neurological examination showed flaccid paralysis with areflexia. Stool cultures were positive for Campylobacter jejuni and both serum and cerebrospinal fluid (CSF) tested positive for anti-GT1a antibodies. The patient was treated with IVIG, PLEX and received intensive supportive care, including autonomic monitoring, nutritional support, and early physical rehabilitation. Gradual neurological recovery was observed and mechanical ventilation was weaned after clinical stabilization.
Conclusion: These cases highlight the diverse clinical spectrum and variable outcomes of severe paediatric GBS, as well as they underscore the importance of early recognition, prompt immunotherapy and a multidisciplinary approach to optimize recovery.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Immune-mediated Neuropathies
PP01.185
Favorable Outcome in a 2-Year-Old Girl With Anti-MuSK Myasthenia Gravis. a Rare Paediatric Case Presentation
Dr. Eleftheria Kokkinou1, Assoc. Prof. Vasiliki Zouvelou2, Dr. Marina Katsalouli1, Dr. Elissavet Georgiadou3, Dr. Evangelia Lykopoulou3, Dr. Doxa-Eleni Sareidaki1, Dr. Iasonas-George Stamatakis1, Dr. Anna Moustafellou1, Dr. Sotiris Giouroukos3, Prof. Roser Pons3
1
Department of Neurology, Children's Hospital “Agia Sofia”, Athens, Greece.
2
Medical School, National and Kapodistrain University of Athens, Eginition Hopsital, Athens, Greece.
3
First Department of Paediatrics, Medical School, National and Kapodistrian University of Athens, Children's Hospital “Agia Sofia”, Athens, Greece
Background: Myasthenia Gravis (MG) associated with anti-muscle-specific kinase (MuSK) antibodies is a rare subtype of autoimmune MG, particularly uncommon in the paediatric population under 5 years of age. It is often characterized by predominant bulbar, respiratory, and facial muscle weakness and can be refractory to standard treatments. Rituximab, a B-cell depleting monoclonal antibody, has shown efficacy in adult MuSK-MG but its use in very young children is limited. This case highlights the successful use of rituximab in a toddler with severe anti-MuSK MG.
Methods: Case Presentation
Results: A previously healthy 2-year-old girl presented with ptosis, facial weakness, nasal speech, and feeding difficulties over two weeks. Examination revealed fatigable weakness of facial and bulbar muscles, generalized hypotonia, and minimal limb involvement. No respiratory distress was noted. Antibody testing confirmed the presence of anti-MuSK antibodies; acetylcholine receptor antibodies were negative. Initial management with pyridostigmine and corticosteroids yielded minimal improvement. Given the refractory nature and the persistence and severity of symptoms, rituximab was initiated. The patient showed significant clinical improvement within 3 weeks of starting rituximab, with resolution of ptosis and improved swallowing. Steroids were tapered over the following 3 months, and pyridostigmine was gradually discontinued. At 5-year follow-up, she remained in sustained clinical remission, was off all medications, and had regained appropriate weight and developmental milestones.
Conclusion: This case illustrates that early recognition and targeted immunotherapy with rituximab can lead to excellent outcomes in paediatric anti-MuSK MG, even in very young children. Rituximab should be considered in refractory or severe MuSK-positive MG cases in children. Long-term follow-up is essential to monitor for relapse and manage potential immunosuppressive risks.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.186
Effect of Rozanolixizumab on Ocular Symptoms in Generalised Myasthenia Gravis: Analysis of Phase 3 Studies
Dr. Elena Cortés Vicente1, Dr. Ali Habib2, Dr. Zabeen Mahuwala3, Prof. Robert Pascuzzi4, Prof. John Vissing5, Dr. Tuan Vu6, Dr. Jos Bloemers7, Dr. Paul Mahoney8, Dr. Thaïs Tarancón9, Dr. Vera Bril10
1
Neuromuscular Diseases Unit, Hospital de la Santa Creu i Sant Pau, Barcelona, Spain.
2
MDA ALS & Neuromuscular Center, Department of Neurology, University of California, Irvine, Orange, CA, United States.
3
Department of Neuromuscular Medicine, Epilepsy and Clinical Neurophysiology, University of Kentucky, Lexington, KY, United States.
4
Neurology Department, Indiana University School of Medicine, Indiana University Health, Indianapolis, IN, United States.
5
Copenhagen Neuromuscular Center, Department of Neurology, Rigshospitalet, University of Copenhagen, Copenhagen, Denmark.
6
Department of Neurology, University of South Florida Morsani College of Medicine, Tampa, FL, United States.
7
UCB, Brussels, Belgium.
8
UCB, Slough, United Kingdom.
9
UCB, Madrid, Spain.
10
Ellen and Martin Prosserman Centre for Neuromuscular Diseases, Toronto General Hospital, University of Toronto, Toronto, Ontario, Canada
Background: Patients with generalised myasthenia gravis (gMG) may experience disabling ocular symptoms such as diplopia (double vision) and ptosis (eyelid drooping), due to ocular muscle weakness and fatigability. In the Phase 3 MycarinG study (NCT03971422), one 6-week cycle of rozanolixizumab demonstrated greater improvements in ocular items across myasthenia gravis (MG)-specific outcomes than placebo in patients with gMG. Here, we investigate the effect of repeated rozanolixizumab treatment cycles on ocular symptoms, using final data from MycarinG and the completed open-label extension studies MG0004 (NCT04124965) and MG0007 (NCT04650854).
Methods: In MG0004, patients received once-weekly rozanolixizumab 7 mg/kg or 10 mg/kg for ≤52 weeks. In MG0007, patients received an initial 6-week cycle of rozanolixizumab 7 mg/kg or 10 mg/kg, with further cycles administered upon symptom worsening. Final efficacy data were pooled across MycarinG, MG0004 (first 6 weeks) and MG0007 for patients with ≥2 symptom-driven rozanolixizumab cycles. This post hoc analysis evaluated weighted-mean scores at baseline and Day 43 across Cycles 1–13 in ocular items within the MG Activities of Daily Living (MG-ADL), Quantitative MG (QMG), MG Symptoms Patient-Reported Outcome (PRO) Ocular Muscle Weakness and MG Quality of Life Revised 15-items (MG-QoL15r) scales. Means were weighted by sample size at each cycle.
Results: Overall, 129 patients received ≥2 symptom-driven cycles. Weighted-mean ocular item-level scores across 13 cycles at baseline and Day 43 were: MG-ADL double vision, 1.4 and 0.9 (N=939 patient-cycles), eyelid droop, 1.5 and 0.8 (N=939 patient-cycles); QMG double vision, 1.6 and 1.1 (N=924 patient-cycles), ptosis, 1.4 and 0.8 (N=922 patient-cycles); MG Symptoms PRO double vision, 1.3 and 0.9 (N=926 patient-cycles), eyelid drooping, 1.3 and 0.8 (N=927 patient-cycles); MG-QoL15r “trouble using my eyes”, 1.2 and 0.9 (N=890 patient-cycles), respectively.
Conclusion: Long-term rozanolixizumab treatment cycles led to improvements across ocular items in MG-specific outcome measures, suggesting a benefit for patients with gMG experiencing ocular signs and symptoms. Funding: UCB.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.187
Neisseria Mucosa Sepsis in a MG Patient Treated With Ravulizumab: Case Report and Literature Review
Dr. Elena Rossini1, Dr. Laura Tufano1, Dr. Valentina Vera1, Dr. Antonio Lauletta2, Dr. Stefania Morino3, Dr. Luca Leonardi3, Assoc. Prof. Matteo Garibaldi3, Assoc. Prof. Giovanni Antonini2, Dr. Laura Fionda3
1
Department of Neuroscience, Mental Health and Sensory Organs (NESMOS), SAPIENZA University of Rome, Rome, Italy, Rome, Italy.
2
NESMOS Department, Sapienza University of Rome and UniCamillus-St. Camillus International University of Health Science, Rome, Italy., Rome, Italy.
3
Neuromuscular Disease Center, Sant'andrea Hospital, Rome, Italy., Rome, Italy
Background: Complement inhibitors increase susceptibility to infections by capsular-bearing bacteria, particularly Neisseria meningitidis, requiring mandatory vaccination. Non-capsulated Neisseria species are typically commensals of the upper respiratory and urogenital tracts and lack certain virulence factors, which limits the occurrence of invasive disease in humans. Nevertheless, a range of infections attributed to these organisms—including endocarditis, meningitis, pneumonia, peritonitis, and septic arthritis—have been reported in both immunocompromised and otherwise healthy
Methods: We describe the clinical presentation, management, and outcome of a patient with acetylcholine receptor positive (AchR+) thymoma-associated generalized myasthenia gravis (gMG), who developed Neisseria mucosa sepsis while receiving ravulizumab therapy. Additionally, a systematic literature review was conducted to identify published cases of invasive infections caused by uncommon Neisseria species in patients treated with complement inhibitors. Extracted data included underlying disease, immunocompromised status, infecting Neisseria species, clinical severity, treatment, outcomes, and reported vaccination or prophylactic strategies when available.
Results: The 25-year-old patient was receiving ravulizumab infusions every 8 weeks, azathioprine 150 mg daily, and prednisone 15 mg daily. She was fully vaccinated for Neisseria meningitidis serogroup B and ACYW135, with booster doses administered over time.
She was hospitalized for an acute febrile illness with neutropenia, rapidly progressing to septic shock requiring vasoactive support. Blood cultures were positive for Neisseria mucosa. Her last ravulizumab infusion had been administered 7 weeks earlier. Azathioprine therapy was discontinued. She was successfully treated with intravenous ceftriaxone, with complete resolution of symptoms after 10 days of therapy.
A systematic literature review identified 10 additional cases of invasive infections caused by uncommon Neisseria species, including N. sicca (3), N. mucosa/subflava (2), N. cinerea (2), N. mucosa (2), and N. subflava (1), in patients receiving complement inhibitors. Underlying conditions included MG (2), atypical hemolytic uremic syndrome (aHUS; 1), hemolytic uremic syndrome (HUS; 3), classic paroxysmal nocturnal hemoglobinuria (PNH; 2), PNH with aplastic anemia (1), and catastrophic antiphospholipid antibody syndrome (CAPS; 1). Six patients were variably immunocompromised. Five cases progressed to sepsis and one to septic shock. All patients recovered with prompt antibiotic therapy and no reported sequelae however, comprehensive data on vaccination schedule and eventual prophylactic therapy were unavailable for most of the patients
Conclusion: These cases highlight that patients treated with complement inhibitors may remain at risk for invasive infections caused by unusual and non-capsulated Neisseria species, despite anti-meningococcal vaccination. Continuous monitoring of the epidemiology of Neisseria infections, including the collection of international registry data, is needed to better understand their prevalence and the factors that may predispose to their occurrence.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.188
Timing of Thymic and Extrathymic Malignancies Relative to Myasthenia Gravis Onset: Oxford Myasthenia Service Cohort
Miss Francesca Ferguson1, Miss Isobel Cabraal1, Dr. Ezgi Bakircioglu Duman1, Dr. Mohammad Ashraghi1,2, Assoc. Prof. M. Isabel Leite1,3
1
University of Oxford, Oxford, United Kingdom.
2
Chelsea and Westminster Hospital NHS Foundation Trust, London, United Kingdom.
3
Oxford University Hospitals NHS Foundation Trust, Oxford, United Kingdom
Background: While myasthenia gravis (MG) is known to be associated with thymic malignancies, data regarding its association with extrathymic malignancies are sparse. Limited reports describe associations between MG and extrathymic malignancies, including secondary malignancies following thymic malignancy, with prolonged traditional immunosuppressive treatment proposed as a contributing factor. Here, we investigate the type and timing of thymic and extrathymic malignancies in patients with MG receiving traditional immunosuppression using data from the UK Myasthenia Gravis Database (UKMyDb).
Methods: This retrospective observational study included all adults (≥18 years) with MG treated at Oxford University Hospitals and enrolled in UKMyDB, a combined retrospective-prospective registry containing demographic and clinical data. Patients receiving targeted therapies (zilucoplan, rituximab, efgartigimod) or regular intravenous immunoglobulin or plasma exchange were excluded. Timing of malignancy diagnosis relative to MG onset was described across demographic groups. Statistical analysis was performed using R (version 4.5.2).
Results: 416 patients were included (211 male [50.7%], 205 female [49.3%]), with a median age of 65.0 years (IQR 25.3). Thymic malignancy was identified in 25/416 patients (6.0%), and 36/416 patients (8.7%) had an extrathymic malignancy. Among extrathymic malignancies, skin (n = 8) and prostate (n = 7) were most frequent; breast, colorectal, genitourinary (excluding prostate), and haematological malignancies each occurred in n = 3, followed by gynaecological (n = 2), other malignancies (n = 2), and central nervous system malignancies (n = 1). Four patients had more than one malignancy type, including colorectal and skin (n = 1), genitourinary (excluding prostate) with other (n = 1) or skin (n = 1), and prostate and skin (n = 1).
The majority of thymic malignancies (17, 68.0%) were diagnosed within one year before or after the onset of MG. Of extrathymic malignancies, only 4 (10.5%) were diagnosed within this ±1-year interval, with no clear association between malignancy type and proximity to MG onset. For several extrathymic malignancy types, including skin, prostate, breast, and haematological malignancies, the majority of diagnoses occurred after MG onset (60.0%, 57.1%, 60.0%, and 66.7%, respectively); however, numbers were small and findings should be interpreted cautiously. Two patients with thymic malignancy also had a reported extrathymic malignancy, both were women with breast malignancy diagnosed more than 10 years after MG onset. The lower frequency of extrathymic malignancies in the thymic malignancy cohort may reflect younger age at MG onset (median 58.6 years [IQR 17.9] vs 77.1 years [IQR 11.3]).
Conclusion: In this cohort, no clear temporal association was observed between MG onset and the occurrence of extrathymic malignancies, providing no strong evidence to support a paraneoplastic relationship outside of thymic malignancy, although interpretation of these findings is limited by small case numbers. The identification of two cases of breast cancer following thymic malignancy highlights the potential risks associated with radiotherapy and radiation exposure during long-term surveillance. This analysis includes patients exposed to conventional immunosuppressive therapies; further work will assess whether treatment dose or duration is associated with extrathymic malignancy risk, and will explore the low incidence of extrathymic malignancy within the thymic malignancy cohort.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
Images or Table (Optional)
PP01.189
Effect of Nipocalimab on Sustained Myasthenia Gravis Control During Infections: Post-hoc Analysis of Vivacity-MG3 Study
Dr. Carlo Antozzi1, Dr. Gonçalo M.C. Rodrigues2, Dr. Marie Fitzgibbon3, Dr. Robert Edwards4, Dr. Ibrahim Turkoz5, Dr. Michael Kutch5, Dr. Sindhu Ramchandren3, Dr. Ghazala S. Hayat6
1
Immunotherapy and Apheresis Departmental Unit, Neuroimmunology and Muscle Pathology Unit, Fondazione IRCCS Istituto Neurologico C. Besta, Milan, Italy.
2
Johnson & Johnson, Madrid, Spain.
3
Medical Affairs, Johnson & Johnson, Raritan, NJ, United States.
4
Medical Affairs, Johnson & Johnson, Spring House, PA, United States.
5
Medical Affairs, Johnson & Johnson, Titusville, NJ, United States.
6
Department of Neurology, Saint Louis University School of Medicine, Saint Louis, MO, United States
Background: Nipocalimab plus standard-of-care (SOC) treatment demonstrated sustained disease control versus placebo plus SOC in the 24-week double-blind phase of the Vivacity-MG3 (NCT04951622) study in patients with generalized myasthenia gravis (gMG). Although immunoglobulin-G levels were reduced in the nipocalimab group, the occurrence of infectious adverse events were comparable with placebo. Infections can contribute to symptom exacerbations in gMG; therefore, monitoring MG-Activities of Daily Living (MG-ADL) and Quantitative MG (QMG) scores helps to assess the symptom control and therapeutic adequacy during infection episodes. This post-hoc analysis evaluated the efficacy of nipocalimab treatment during/shortly after infection episodes in the double-blind phase of Vivacity-MG3 by measuring changes in MG-ADL and QMG scores.
Methods: Patients with infections/infestations/serious infections at any timepoint in the double-blind phase were analyzed. Pre-infection and post-infection MG-ADL and QMG scores were compared. Pre-infection MG-ADL/QMG score was the most recent observation recorded within 16-days immediately preceding infection start-date. Post-infection MG-ADL/QMG score was the worst observation recorded from the infection start-date through 16-days after the infection end-date.
Results: Infection/infestations were reported in 42.9% in nipocalimab group (infection events=71) and 41.8% in placebo (infection events=59), (Table). The median (IQR) pre-infection to post-infection change in MG-ADL scores was 0.0 (−1.0; 1.0) in nipocalimab versus 1.0 (0.0; 2.0) in placebo, and 0.0 (−1.0; 2.0) in nipocalimab versus 1.0 (−1.0; 1.0) in placebo in QMG scores (Table).
Conclusion: Incidence of infections in nipocalimab arm was comparable to placebo arm despite immunoglobulin-G reduction with nipocalimab. MG-ADL/QMG scores were not altered during infections, demonstrating maintenance of gMG symptom improvement and sustained disease control with nipocalimab.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
Images or Table (Optional)
PP01.190
Follow Up and Management of Patients With Myastheni̇a Gravis
Dr. Melik GURSOY, Dr. Turgay DOLEK, Dr. Tugce AKCADAG CAMAN, Dr. Mahmut Bilal CAMAN, Prof. Gulnihal KUTLU
Mugla SK University, School of Medicine, Department of Neurology, Mugla, Turkey
Background: Myasthenia gravis (MG) is a heterogeneous autoimmune neuromuscular disorder requiring long-term follow-up and individualized treatment strategies. This study aimed to evaluate the demographic, clinical, electrophysiological, and serological characteristics of MG patients followed in a tertiary neurology center.
Methods: Fifty-eight patients with MG who were regularly followed between 2018-2024 were retrospectively analyzed. Data regarding age at diagnosis, sex, disease subtype, antibody status, electrophysiological findings, thymectomy history, and treatments administered during follow-up were recorded.
Results: The majority of patients were male, particularly among those diagnosed after the age of 40, while female predominance was observed in younger-onset cases. Generalized MG was the most common disease subtype. Acetylcholine receptor antibody positivity was high, whereas anti-MuSK positivity was rare. Most patients required combined immunomodulatory treatments, including corticosteroids, immunosuppressive agents, and intravenous immunoglobulin during follow-up.
Conclusion: MG patients followed at our center predominantly presented with generalized disease and high seropositivity rates. The frequent need for combination immunotherapy reflects the clinical burden of MG in a tertiary-care setting.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.191
Real-world Oral Glucocorticoid Dosage and Healthcare Resource Use in gMG Patients Starting Efgartigimod in Japan
Dr. Hirofumi Teranishi1, Dr. Koichi Tsuda1, Dr. Ryohei Aoyagi1, Dr. Yunlong Zheng2, Dr. Jiangyuan Luo2, Dr. Sho Inagaki2, Prof. Masanori P. Takahashi3, Dr. Cécile Blein4
1
argenx, Tokyo, Japan.
2
ZS Associates, Osaka, Japan.
3
Osaka University, Osaka, Japan.
4
argenx BV, Ghent, Belgium
Background: Generalized myasthenia gravis (gMG) is a rare, chronic, autoimmune disorder associated with substantial disease burden. Oral glucocorticoids (GC) remain a mainstay of treatment globally; however, there are growing concerns around the risks of adverse effects associated with long-term use, including serious medical complications. This retrospective cohort study evaluated the real-world impact of intravenous/subcutaneous efgartigimod (EFG) on oral GC dosing, treatment patterns, and healthcare resource utilization (HRU) in adult patients with generalized myasthenia gravis (gMG) in Japan.
Methods: This study used hospital-based claims data from Medical Data Vision. Patients aged ≥15 years who initiated EFG between May 1, 2022, and April 30, 2025, and had ≥1 gMG diagnosis and ≥1 healthcare encounter during the 12 months pre-EFG initiation (baseline) and ≥3 months post-EFG initiation (follow-up) were included. The index date was the first EFG administration. Treatment patterns, including gMG-related oral therapies, fast-acting therapies (FT) (intravenous immunoglobulin, plasma exchange, and intravenous methylprednisolone), biologics, and their combinations; HRU, including hospitalizations and ICU admission; oral GC average daily dose (ADD) were assessed during the baseline and the follow-up periods. Oral GC ADD was calculated as total exposure in prednisolone equivalent dose divided by the number of days per interval. Patients with ≥12 months of follow-up were included for oral GC dose analyses to assess the impact of EFG over time.
Results: Of 539 patients identified with ≥3 months of follow-up, 62% were female; mean age 56.3 years; mean Charlson comorbidity index 3.5; 94% had baseline oral GC exposure; 93% had no baseline biologics use; median duration from the first recorded gMG diagnosis to index was 3.9 years; median follow-up was 443 days; median EFG treatment duration was 246 days. The proportion of patients receiving ≥1 FT administration decreased post-EFG initiation (intravenous immunoglobulin: 52% to 26%; plasma exchange: 11% to 8%; intravenous methylprednisolone: 40% to 31%). At the end of the treatment, 70% of patients either remained on EFG treatment or had not initiated FT or other biologic treatments after their last EFG administration. Hospitalizations per patient per year (PPPY) decreased 37% (1.34 to 0.85); average ICU admissions decreased 53% (5.1% to 2.4%). Among 313 patients (58% of the overall study population) with ≥12 months of follow-up, mean oral GC ADD significantly decreased following EFG initiation (17.0 mg/day at baseline, 8.5 mg/day at follow-up; p<0.001), with considerable decrease in dosing observed as early as the 3rd month (Day 61-90, 9.8 mg/day) in the follow-up. Among patients continuously on EFG treatment at the end of the 12-month follow-up (204 patients), 81% (166 patients) achieved oral GC ADD level ≤10 mg/day, and 44% (90 patients) achieved ≤5 mg/day by the 12th month (Day 330-365) post-EFG initiation (figure 1).
Conclusion: EFG initiation among adult gMG patients in Japan was associated with substantial reductions in oral GC dose, FT use, hospitalizations, and ICU admissions, regardless of prior biologic exposure. These findings support EFG as a valuable option for reducing treatment and healthcare burden in real-world practice.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
Images or Table (Optional)
PP01.192
Exploring Associations Between Patient Factors and Diagnostic Delay in Myasthenia Gravis at a Tertiary Centre
Miss Isobel Cabraal1, Miss Francesca Ferguson1, Dr. Ezgi Bakircioglu Duman1, Dr. Mohammad Ashraghi1,2, Assoc. Prof. M Isabel Leite1,3
1
University of Oxford, Oxford, United Kingdom.
2
Chelsea and Westminster Hospital NHS Foundation Trust, London, United Kingdom.
3
Oxford University Hospitals NHS Foundation Trust, Oxford, United Kingdom
Background: Early diagnosis of myasthenia gravis (MG) is essential for effective disease management and improved outcomes. Despite this, diagnostic delay remains common, with reports suggesting over 25% of patients experiencing delays greater than one year, and 13-14% waiting up to five years. In this retrospective study we assess demographic and clinical laboratory factors associated with prolonged time to diagnosis in a cohort of patients seen at Oxford specialist myasthenia centre.
Methods: Data were extracted from the UK Myasthenia Database (UKMyDb), a secure REDCap-based registry populated from electronic health records. Eligible participants were adults (≥18 years) with recorded dates of symptom onset and MG diagnosis, who had consented for inclusion in UKMyDb at the John Radcliffe Hospital, a tertiary centre in Oxford, UK. Demographic variables analysed included age, sex, ethnicity, serology, and myasthenia subtype. Due to skewed distributions of diagnostic delay, quantile regression was used alongside non-parametric tests to assess associations across the distribution of diagnostic delays and adjust for multiple covariates.
Results: 323 patients met inclusion criteria (163 male, 160 female; with a median age of 63.8 years, IQR 27.2 years). Subtypes included 89 AChR-positive ocular MG, 206 AChR-positive generalised MG, 9 MuSK-positive MG, 11 seronegative ocular MG, and 8 seronegative generalised MG. Median diagnostic delay was 1.94 months (IQR 0.00-6.74), with a maximum delay of 14.24 years. Delays exceeding 6 months occurred in 87 patients (26.9%; 95% CI 22.2-32.2%), and delays over 1 year in 53 patients (16.4%; 95% CI 12.6-21.0%).
Diagnostic delay did not differ significantly between males and females (Wilcoxon rank-sum test: W = 13,544; p=0.54; rank-biserial correlation r_rb = 0.039). No significant differences were observed between serological subgroups (AChR-positive, MuSK-positive, and seronegative; Kruskal-Wallis test: p = 0.13) or across ethnic groups (Kruskal-Wallis χ² = 2.28, df = 4, p=0.68), although MuSK-positive and seronegative subgroups, and several ethnic categories were represented by small numbers. There was a weak negative correlation between diagnostic delay and age at symptom onset (Spearman’s ρ = -0.118, p=0.0346). However, delay did not differ significantly when compared across age groups (18-29, 30-49, 50-59, 60-69, 70-79, ≥80 years; Kruskal–Wallis χ² = 8.66, df = 5, p=0.12). In adjusted quantile regression, ocular MG was associated with shorter diagnostic delay at the median and upper quantiles (τ = 0.50-0.75, β = -0.88 to -3.53 months, p<0.05). Younger age at onset was associated with longer delay only at higher quantiles (τ ≥ 0.75; β = −0.20 to −0.43 months/year, p<0.05). No significant associations were observed for sex, serology, or ethnicity.
Conclusion: Diagnostic delay in myasthenia gravis remains common in this specialist centre cohort, with limited evidence of association with demographic factors. Further analyses examining additional patient- and system-level contributors to diagnostic delay are planned, including organisational factors (e.g. timing of antibody testing) and comorbidities (e.g. fatigue and depression) captured within UKMyDb.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
Images or Table (Optional)
PP01.193
Efgartigimod in Acetylcholine Receptor Antibody Seronegative Generalized Myasthenia Gravis: Initial Results of ADAPT SERON
Dr. James F. Howard Jr1, Dr. Tuan Vu2, Dr. Chongbo Zhao3, Dr. Sushan Luo3, Dr. Sarah Hoffmann4, Dr. Kristl G. Claeys5,6, Dr. Rosa H. Jimenez7, Ms. Ineke Seghers7, Dr. Delphine Masschaele7, Dr. Jeffrey T. Guptill7, Dr. Wan-Yi Huang7, Dr. Łukasz Rzepiński8,9, Dr. Elisabeth Chroni10, Dr. Ali Alshehri11, Dr. Ari Breiner12
1
The University of North Carolina, Chapel Hill, United States.
2
University of South Florida Morsani College of Medicine, Tampa, United States.
3
Huashan Hospital, Fudan University, Shanghai, China.
4
Charité – Universitätsmedizin Berlin, Berlin, Germany.
5
University Hospitals Leuven, Leuven, Belgium.
6
KU Leuven, Leuven, Belgium.
7
argenx, Ghent, Belgium.
8
10th Military Research Hospital and Polyclinic, Bydgoszcz, Poland.
9
University of Science and Technology, Bydgoszcz, Poland.
10
University Hospital of Patras, Rion, Greece.
11
King Faisal Specialist Hospital and Research Centre, Riyadh, Saudi Arabia.
12
The Ottawa Hospital/The University of Ottawa, Ottawa, Canada
Background: Approximately 15%-20% of patients with generalized myasthenia gravis (gMG) are classified as acetylcholine receptor antibody (AChR-Ab) seronegative, which includes those who are muscle-specific tyrosine kinase receptor antibody seropositive (MuSK-Ab+), low-density lipoprotein receptor-related protein 4 receptor antibody seropositive (LRP4-Ab+), and those who lack identifiable antibodies against AChR, MuSK, and LRP4 (triple seronegative). There is an unmet need for approved treatments in this population. Efgartigimod is a human immunoglobulin G1 (IgG1) antibody Fc fragment that reduces IgG levels (including pathogenic autoantibodies) via neonatal Fc receptor blockade. ADAPT SERON is a Phase 3 trial investigating the efficacy and safety of intravenous (IV) efgartigimod in adults with AChR-Ab seronegative gMG.
Methods: Diagnosis of gMG was confirmed by an MG diagnostic adjudication committee. In the double-blinded, placebo-controlled Part A, adult participants were randomized 1:1 to receive 4 once-weekly infusions of 10 mg/kg efgartigimod IV or placebo followed by a 5-week follow-up period. Part B includes an ongoing open-label extension (≤2 years).
Results: Results included 119 participants (n=40, MuSK-Ab+; n=6, LRP4-Ab+; n=73, triple seronegative); 58 received efgartigimod IV, and 61 received placebo. The change in Myasthenia Gravis Activities of Daily Living (MG-ADL) total score from baseline to Week 4 (primary endpoint) was significantly (P=0.007) different between efgartigimod IV and placebo groups (overall population), with least squares mean change from baseline (90% CI) of −3.35 (−3.98 to −2.72) and −1.90 (−2.51 to −1.28) in each group, respectively. Further improvements in MG-ADL total scores were observed during Part B over subsequent treatment cycles across all three subgroups. Efgartigimod IV was well tolerated, with no new safety signals observed. Results from additional analyses will be presented at ICNMD 2026.
Conclusion: Efgartigimod IV demonstrated statistically significant improvement in MG-ADL total score compared with placebo (primary endpoint), with a clinically meaningful change from baseline. Efgartigimod IV was well tolerated in participants with MuSK-Ab seropositive, LRP4-Ab seropositive, and triple seronegative gMG.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.194
Real-World Cardiovascular Burden Among Myasthenia Gravis Patients Across the United States, France, and Japan
Dr. Cécile Blein1, Dr. Jana Podhorna1, Dr. Hirofumi Teranishi2, Prof. Andoni Echaniz-Laguna3, Prof. Jean-Philippe Camdessanché4, Prof. Shahram Attarian5, Dr. Guilhem Solé6, Dr. Akiyuki Uzawa7
1
argenx BV, Ghent, Belgium.
2
argenx Japan, Tokyo, Japan.
3
Department of Neurology, APHP, CHU de Bicêtre, INSERM U1195, Paris-Saclay University, Le Kremlin-Bicêtre, France.
4
Department of Neurology, Neuromuscular Disease Reference Center, Hôpital Nord, University Hospital of Saint-Étienne, Euro-NMD, Saint-Étienne, France.
5
Reference Center for Neuromuscular Disease and ALS, Timone University Hospital, Aix-Marseille University, CHU Timone, Filnemus, Euro-NMD, Marseille, France.
6
Neuromuscular Reference Center AOC, Neurology and Neuromuscular Diseases Department, Pellegrin Hospital, Bordeaux University Hospitals, Bordeaux, France.
7
Chiba University, Chiba, Japan
Background: Myasthenia gravis (MG) is a chronic autoimmune disease affecting the neuromuscular junction. Comorbidities, including cardiovascular (CV) diseases and CV risk factors, are common in MG. These may be further exacerbated by certain MG therapies, including treatments that can affect blood pressure, plasma glucose, or plasma lipid levels. To better understand CV burden among MG patients globally, we compared the prevalence of CV comorbidities and CV risk factors among patients with MG in Japan, France, and the United States (US).
Methods: Patients aged ≥18 years with a confirmed diagnosis of MG were identified using claims data from Medical Data Vision (Japan, 2021–2022), the French national health insurance database (Système National des Données de Santé; France, 2019–2020), and Optum Market Clarity (US, 2022–2023). CV comorbidities and CV risk factors were evaluated using country-relevant diagnostic codes within the specified 2-year periods.
Results: Totals of 10,307, 5210, and 4758 patients with MG were included from Japan, France, and the US, respectively. Mean age was 64.0 years in Japan, 59.6 years in France, and 63.4 years in the US; 56.6%, 55.3%, and 53.3% of patients were female, respectively. Mean Charlson Comorbidity Index was 1.86 in Japan, 2.46 in France, and 2.90 in the US. Most patients with MG in each country had ≥1 CV comorbidity or CV risk factor, including 65.7% in Japan, 56.6% in France, and 93.5% in the US. The proportion with ≥1 CV comorbidity or CV risk factor was highest among patients aged >65 years (73.6% in Japan, 72.0% in France, and 98.1% in the US), but was also substantial among those aged >40–65 years (60.3%, 52.0%, and 92.9%, respectively) and 18-40 years (34.8%, 27.6%, and 72.9%, respectively), particularly in the US (Figure). Across countries and age groups, most patients (80.3–99.5%) with CV comorbidities also had ≥1 CV risk factor. Prevalence of individual CV comorbidities and CV risk factors varied across countries. Overall, the most common CV comorbidities were ischemic heart disease (2.5% Japan, 8.6% France, 26.6% US), heart failure (with hospitalization; 16.5% Japan, 5.8% France, 0.7% US), atherosclerosis/peripheral arterial disease (3.6% Japan, 2.6% France, 13.1% US), cerebrovascular accident (9.0% Japan, 1.7% France, 7.9% US), and angina pectoris (10.7% Japan, 1.7% France, 4.0% US). The most common CV risk factors were hypertension (44.0% Japan, 31.7% France, 70.4% US), hyperlipidemia (40.1% Japan, 9.5% France, 69.7% US), obesity (0.8% Japan, 13.9% France, 48.9% US), and type 2 diabetes mellitus (1.1%, 15.3%, and 34.2%, respectively).
Conclusion: Patients with MG face a significant CV burden across Japan, France, and the US. While overall CV burden was greatest in the US, the majority of patients with MG in all 3 countries had a CV comorbidity or risk factor. These findings support the need for proactive CV risk management in MG patients irrespective of country, and cautious use of MG treatments that could further exacerbate CV risk factors and comorbidities.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
Images or Table (Optional)
PP01.195
Quality of Life in Myasthenia Gravis: Post Hoc Pooled Analysis of Phase 3 Rozanolixizumab Studies
Dr. Jana Zschüntzsch1, Dr. Ali A. Habib2, Dr. Zabeen Mahuwala3, Prof. Renato Mantegazza4, Prof. Robert M. Pascuzzi5, Prof. Sabrina Sacconi6, Prof. John Vissing7, Dr. Jos Bloemers8, Dr. Paul Mahoney9, Dr. Thaïs Tarancón10, Dr. Vera Bril11
1
Department of Neurology, University Medical Center Göttingen, Göttingen, Germany.
2
MDA ALS & Neuromuscular Center, Department of Neurology, University of California, Irvine, Orange, United States.
3
Department of Neuromuscular Medicine, Epilepsy and Clinical Neurophysiology, University of Kentucky, Lexington, United States.
4
Emeritus and Past Director, Department of Neuroimmunology and Neuromuscular Diseases, Fondazione IRCCS, Istituto Nazionale Neurologico Carlo Besta, Milan, Italy.
5
Neurology Department, Indiana University School of Medicine, Indiana University Health, Indianapolis, United States.
6
Université Côte d'Azur, Peripheral Nervous System & Muscle Department, Pasteur 2 Hospital, Centre Hospitalier Universitaire de Nice, Nice, France.
7
Copenhagen Neuromuscular Center, Department of Neurology, Rigshospitalet, University of Copenhagen, Copenhagen, Denmark.
8
UCB, Brussels, Belgium.
9
UCB, Slough, United Kingdom.
10
UCB, Madrid, Spain.
11
Ellen and Martin Prosserman Centre for Neuromuscular Diseases, Toronto General Hospital, University of Toronto, Toronto, Canada
Background: Generalised myasthenia gravis (MG) is a chronic autoimmune disease that can significantly affect patients’ quality of life (QoL). In the randomised, double-blind, Phase 3 MycarinG study (NCT03971422), one 6-week cycle of rozanolixizumab (7 mg/kg or 10 mg/kg) improved MG-QoL 15-item revised (MG-QoL15r) total scores compared with placebo in patients with generalised MG. Here, we evaluate the effect of long-term, cyclic rozanolixizumab treatment on QoL using MG-QoL15r item-level scores.
Methods: Following MycarinG, patients could enrol in open-label extensions MG0004 (NCT04124965) and MG0007 (NCT04650854). In MG0004, patients received once-weekly rozanolixizumab for ≤52 weeks. MG0007 comprised an initial 6-week rozanolixizumab cycle, with further cycles administered upon symptom worsening. Final efficacy data were pooled across MycarinG, MG0004 (first 6 weeks) and MG0007 for patients with ≥2 symptom-driven rozanolixizumab cycles. Change from baseline (CFB) to Day 43 in MG-QoL15r item-level scores across Cycles 1–13 was assessed post hoc using means weighted by sample size.
Results: Overall, 129 patients received ≥2 symptom-driven cycles. Across 13 cycles (N=890 patient-cycles), weighted-mean scores improved between baseline and the end of each 6-week rozanolixizumab cycle for all MG-QoL15r items. Weighted-mean (standard deviation) CFB in item-level scores was: −0.4 (0.7) for “trouble using my eyes”, “trouble eating”, “bothered by limitations in performing work” and “difficulty speaking” (N=889 patient-cycles); −0.3 (0.7) for “limited social activity” and “limited ability to enjoy hobbies/fun activities”; −0.3 (0.6) for “frustrated by my MG”, “trouble meeting the needs of family”, “have to make plans around my MG”, “lost some personal independence”, “trouble walking”, “trouble getting around public places” and “overwhelmed by my MG”; −0.2 (0.5) for “depressed about my MG” and “trouble performing personal grooming”.
Conclusion: Long-term rozanolixizumab treatment cycles led to improvements in all MG-QoL15r item-level scores, demonstrating consistent efficacy across the range of impacts on health-related QoL that MG symptoms have. Funding: UCB.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.196
Clinical Factors Associated With Quality of Life in Patients With Myasthenia Gravis
Prof. Seung Woo Kim, Dr. Jeongju Park
Severance hospital, Seoul, Korea, Republic of
Background: The Myasthenia Gravis Quality of Life 15 revised (MG-QOL15r) is a disease-specific instrument to assess quality of life (QOL) in patients with MG. However, clinical factors associated with MG-QOL15r score remain limited. This study aimed to identify clinical factors associated with QOL in patients with MG, with particular emphasis on the Patient Acceptable Symptom State (PASS).
Methods: This cross-sectional study retrospectively investigated the medical records of patients with MG who visited Severance Hospital between Jul 2025 and Dec 2025. Adult patients with MG who completed the MG-QOL15r questionnaire were included. PASS questionnaire was also completed to assess whether the current disease state would be acceptable if maintained over the next several months. Clinical variables, including disease duration, MG subtype, MGFA classification, MG-ADL score, and current treatments, were collected. Univariate and multivariate linear regression analyses were performed to identify factors associated with MG-QOL15r scores.
Results: A total of 124 patients with MG who conducted MG-QOL15r were included; 65.3% were female, median disease duration was 10.0 (Q1-Q3, 4.0-17.0) years, 77.4% were generalized MG, 43.5% underwent thymectomy, and 27.4% had thymoma. Median MG-QOL15r score was 8.0 (Q1-Q3, 2.0-14.0). On the PASS questionnaire, 47 patients (38.0%) reported an acceptable symptom state, whereas 77 patients (62.0%) did not. The median MG-QOL15r score was 3.0 (Q1–Q3, 1.0–10.0) in patients who reported an acceptable symptom state, compared with 12.0 (5.0–19.0) in those who did not (p < 0.001). In univariate analysis, current MG-ADL score (β = 1.488; p < 0.001), disease duration (β = −0.197, p = 0.029), MGFA classification at nadir (β = 0.203, p = 0.024), current prednisolone dose (β = 0.298, p < 0.001), recent worsening of disease (β = 0.302, p < 0.001), and current IVIG therapy (β = 0.364, p < 0.001) was significantly associated with MG-QOL15r score. In multivariate analysis, higher MG-ADL score (β = 0.615, p < 0.001), higher current prednisolone dose (β = 0.130, p = 0.036) and current IVIG therapy (β = 0.163, p = 0.01) were independently associated with worse MG-QOL15r score.
Conclusion: QOL in MG was closely associated with PAS and current disease burden. Less than 40% of patients considered their symptom state acceptable, highlighting a substantial unmet need despite ongoing treatment. Functional impairment and treatment-related factors were more strongly associated with QOL than historical disease severity. These findings suggest that improving QOL in MG requires not only reducing disease activity and functional disability but also minimizing treatment burden, particularly long-term corticosteroid exposure.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.197
Thymic Hyperplasia in Myasthenia Gravis: Clinical and Lifestyle Determinants and Long-Term Outcomes in Nordic Countries
Dr. Jing Wu1,2,3, Dr. Malin Petterson2,3,4, Miss Wanqing Wu2,3, Dr. Ann Eriksson Dufva2,3,4, Prof. Fang Fang1, Prof. Fredrik Piehl2,3,4, Assoc. Prof. Susanna Brauner2,3,4
1
Institute of Environmental Medicine, Karolinska Institutet, Solna, Sweden.
2
Neuroimmunology Unit, Center for Molecular Medicine, Karolinska Institutet, Solna, Sweden.
3
Department of Clinical Neuroscience, Karolinska Institutet, Solna, Sweden.
4
Department of Neurology, Karolinska University Hospital, Solna, Sweden
Background: Thymic hyperplasia is common in early-onset myasthenia gravis (EOMG; onset <50 years) but much less common in late-onset MG (LOMG; onset ≥50 years). The Nordic MG guidelines recommend thymectomy for patients aged 50-65 years. However, reliable imaging and biomarkers for thymic hyperplasia are not available. Thus, we aimed to identify clinical factors that predict hyperplasia in non-thymomatous patients and to determine whether patients with hyperplasia derive greater benefit from thymectomy in terms of long-term clinical outcomes.
Methods: We identified 416 thymectomized non-thymomatous MG patients in the Swedish MG Register (MGreg), which collected information on clinical characteristics, biological measurements, treatment, and disease activities as part of clinical routine practice. Clinical factors comprised sex, age of disease onset, anti-acetylcholine receptor (AChR) antibody, and disease duration. Information on environmental and lifestyle factors (including education, BMI, physical activity, alcohol consumption, and smoking status) of 208 patients was collected by linking Genes and Environment in the Myasthenia Gravis Study. Rescue treatment was assessed between hyperslasia and normal pathology at 2 and 5 years after diagnosis. Logistic regression was used to estimate the odds ratio (OR) of hyperplasia in relation to clinical and lifestyle factors with 95% confidence intervals (CI).
Results: The majority of the participants were female (n=292; 70.2%), and the mean age at disease onset and thymectomy were 31.7 (SD: 15.5) and 35.6 (SD: 15.4) years, respectively. Patients with hyperplasia were predominantly female, younger, had lower BMI, and were more likely to be anti-AchR positive compared to those lacking thymic hyperplasia.
In the clinical factor model, age at onset and disease duration were associated with a lower odds of hyperplasia (OR 0.91, 95%CI 0.89, 0.93 and OR 0.92, 95%CI 0.83, 1.01, respectively). While female sex (OR 2.24, 95%CI 0.97, 5.17) and anti-AchR positivity (OR 2.71, 95%CI 1.18, 6.20) were associated with higher odds of hyperplasia.
Intriguingly, higher BMI (OR 0.78, 95%CI 0.67, 0.91), regular physical activity (OR 0.22, 95%CI 0.05, 0.90), and low alcohol consumption (OR 0.23, 95%CI 0.06, 0.88) were associated with lower risk of hyperplasia. Former (OR 2.48, 95%CI 0.43, 14.19) and current smokers (OR 2.15, 95%CI 0.46, 9.97) showed a tendency towards a higher risk of hyperplasia.
In terms of clinical outcomes, only 7.7% and 14.2% patients with hyperplasia ever received rescue treatment within 2- and 5-year follow-up compared to patients with normal histology (19.8% and 24%), respectively. Further, patients with hyperplasia tended to receive rescue treatment later than patients with normal pathology (12.5±14.4 vs 21.8±16.9 months).
Conclusion: This study identified potential predictive clinical and lifestyle factors for thymic hyperplasia. Patients aged >50 years more rarely undergo thymectomy in Sweden. Thus, these results are working in progress to be validated in a Finnish cohort where thymectomy frequencies in LOMG are higher. A better outcome of MG after thymectomy is linked to thymic hyperplasia, and therefore, these clinical predictors of hyperplasia should be actively used when selecting patients for thymectomy.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.198
Single Center Real-World Experience With Ravulizumab for Patients With Generalized Myasthenia Gravis in Japan
Dr. Kei Ishizuchi, Dr. Kensuke Okada, Prof. Jin Nakahara
Department of Neurology, Keio University School of Medicine, Tokyo, Japan
Background: In 2022, the Japanese regulatory authority approved the terminal complement C5 inhibitor ravulizumab (RAV) for the treatment of anti-acetylcholine receptor (AChR) antibody-positive generalized myasthenia gravis (gMG). Its indication was however limited to patients whose symptoms were difficult to control with intravenous immunoglobulin therapy or plasmapheresis. In April 2025, the label was expanded and RAV became available as an add-on to the standard-of-care treatments including oral corticosteroids (e.g. prednisolone [PSL]) and/or non-steroidal immunosuppressant therapies (NSISTs). This study was conducted to evaluate the real-world efficacy of RAV in a high-volume center in Tokyo, Japan.
Methods: A retrospective medical chart review was conducted. The data cut-off (DCO) date was January 10th, 2026. Patients who no longer visited the hospital within 8 weeks of the DCO date were excluded. Patients previously enrolled in RAV or eculizumab clinical trials, patients with actively-treated thymoma, and patients harboring C5 polymorphism (c.2654G>A) were excluded from the study.
Results: A total of 25 RAV-treated gMG cases (13 males and 12 females) were identified. 10 cases had previous curative treatment history of thymoma. The average onset age was 47.7±16.1 years old and the average disease duration was 13.0±12.6 years. All patients were currently treated with prednisolone (the average daily dose was 9.5±3.5mg), with 20 patients utilizing concomitant NSISTs (17 tacrolimus and 3 cyclosporine). The average MG-ADL score at baseline was 9.3±4.3, whereas it was 5.7±3.8 under RAV at the last follow-up (the average follow-up duration was 22.8 weeks [range 2-138 weeks]). No meningococcal infection was reported.
Conclusion: Improvements of MG-ADL scores by the add-on RAV in the present cohort were consistent with the previously reported clinical trial results (i.e. the mean MG-ADL score improved by 3.1 on week 26; Vu et al, NEJM Evid (2022)). The above data will be updated, and further analysis will be discussed during the presentation.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.199
Preclinical Characterization of MGAC-007, a First-in-Class Antigen Drug Conjugate for AChR⁺ Myasthenia Gravis
Dr. Kfir Oved1, Dr. Galit Denkberg1, Dr. Sharon Reef1, Dr. Inbar Arman1, Dr. Lena Pinzur1, Dr. Roei D. Mazor1, Dr. Ofer Harel1, Ms. Shir Erez1, Ms. Yael Atiya1, Dr. Reem Dowery1, Prof. Michael Benatar2, Prof. Marc De Baets3, Prof. Gil Wolfe4, Prof. Henry Kaminski5
1
Canopy Immuno-Therapeutics, Yokneam Ilit, Israel.
2
Department of Neurology and ALS Center, University of Miami Miller School of Medicine, Miami, United States.
3
Department of Psychiatry and Neuropsychology, Mental Health and Neuroscience Research Institute, Faculty of Health, Medicine and Life Sciences, Maastricht University, Maastricht, Netherlands.
4
Department of Neurology, Jacobs School of Medicine and Biomedical Sciences, University at Buffalo/SUNY, Buffalo, United States.
5
Department of Neurology and Rehabilitation Medicine, George Washington University, Washington, United States
Background: AChR⁺ MG is an antibody-mediated autoimmune disorder in which a small fraction of B cells produces anti-AChR antibodies that impair neuromuscular transmission and cause fluctuating weakness. Current treatments rely on chronic immunosuppression, increasing infection and malignancy risk while failing to address the disease’s root cause. Canopy Immuno-Therapeutics developed Antigen Drug Conjugates (AgDCs): biologics which employ two complementary mechanisms of action (MoA) that specifically sequester pathogenic autoantibodies (MoA1) and selectively eliminate their autoreactive B-cell sources (MoA2), while preserving protective immunity. Herein, we report the preclinical development of MGAC-007, Canopy’s lead AgDC, supporting its advancement to a Phase I clinical trial in patients with AChR⁺ MG in 2026.
Methods: Ex vivo, in vitro, and in vivo studies were conducted to characterize MGAC-007’s functional activity across its dual mechanisms of action, including assays for autoantibody depletion, B-cell receptor binding and cytotoxicity, and potency evaluation in MG animal models.
Results: In ex vivo assays using sera from AChR⁺ MG patients, MGAC-007 selectively depleted a median of 73% of pathogenic anti-AChR antibodies while preserving protective titers. In vitro, MGAC-007 selectively lysed AChR-reactive hybridoma cells in a BCR-dependent manner, while sparing unrelated BCR clones. In a passive-transfer myasthenic crisis rat model, MGAC-007 reduced circulating pathogenic antibodies by up to 90% within two hours, restored motor function, reduced mortality by 85–100% and normalized muscle histology versus controls. Furthermore, pretreatment with MGAC-007 reduced the AChR-specific titer of mice immunized with AChR by up to 73%, confirming targeted depletion of AChR-specific B cells with autoreactive potential.
Conclusion: Preclinical data strongly support the continued development of MGAC-007, which demonstrates proof-of-concept safety & efficacy as a non-immunosuppressive first-in-class therapy for AChR⁺ MG. Its potential as a targeted immunotherapy will be further evaluated in an upcoming multinational Phase I clinical trial.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.200
Clinical Utility of Therapeutic Drug Monitoring of Mycophenolic Acid in Patients with Myasthenia Gravis
Prof. Kyong Jin Shin1, Prof. Geun Yeol Jo2
1
Department of Neurology, Haeundae-Paik Hospital, Inje University, Busan, Korea, Republic of.
2
Department of Rehabilitation Medicine, Haeundae-Paik Hospital, Inje University, Busan, Korea, Republic of
Background: Although Mycophenolate mofetil (MMF) is a standard steroid-sparing agent for Myasthenia Gravis (MG), its pharmacokinetics vary significantly among individuals. This study aimed to evaluate the relationship between plasma Mycophenolic Acid (MPA) levels and clinical outcomes in MG patients.
Methods: Data from 60 patients with MG treated with MMF were retrospectively analyzed. Plasma concentrations of MPA and its metabolite, MPA-G, were measured. We analyzed the correlation between these levels and clinical parameters, including MGFA classification, MG-activity of daily living (ADL) scores, Anti-acetylcholine receptor (AChR) antibody titers, and the incidence of adverse events (infections).
Results: The mean age of the cohort was 61.2±14.8 years old, with most patients maintaining stable clinical status (MGFA Class 1 or 2). MPA Patients within the therapeutic MPA range (1.0–3.5 ug/ml) tended to show better symptom control, reflected in lower MG-ADL scores. Plasma MPA concentrations showed no significant correlation with age, MMF dosage, BMI, MGFA classification, MG-ADL scores, total leukocyte count, or absolute neutrophil count (ANC). However, MPA levels were significantly higher in female patients (n=38) and exhibited a significant inverse correlation with lymphocyte count. Regarding safety outcomes, no infectious complications were observed in the low MPA group (n=18). In contrast, one patient in the therapeutic MPA group (n=24) and eight patients in the high MPA group (n=18) experienced infections, demonstrating a clear association between elevated MPA levels and increased infection risk. Regarding the clinical manifestations of the infections (n=9), they included enteritis in four patients, and one case each of pneumonia, herpes zoster, pulmonary tuberculosis, aseptic meningitis, and acute pancreatitis. All patients achieved a full recovery following appropriate medical intervention.
Conclusion: The significant inverse correlation between MPA levels and lymphocyte counts provides a mechanistic explanation for the observed clinical outcomes. As MMF selectively inhibits lymphocyte proliferation, the markedly higher incidence of infection in the high MPA group likely stems from excessive lymphopenia. These findings underscore the clinical importance of MPA monitoring, suggesting that maintaining MPA within the therapeutic range is crucial not only for efficacy but also for preventing opportunistic infections by avoiding over-suppression of the immune system. Therapeutic drug monitoring (TDM) of MPA is a valuable approach for optimizing MMF therapy in MG. Monitoring MPA levels allows for personalized dose adjustment, balancing clinical efficacy with the risk of adverse effects.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.201
Effect of Rozanolixizumab on MG-ADL and QMG Items: A Post-Hoc Phase 3 Final Pooled Analysis
Dr. Laura Fionda1,2, Dr. Elena Cortés Vicente3, Dr. Artur Drużdż4, Dr. Ali A. Habib5, Dr. Zabeen K. Mahuwala6, Prof. Renato Mantegazza7, Dr. Robert M. Pascuzzi8, Prof. Sabrina Sacconi9, Dr. Kimiaki Utsugisawa10, Prof. John Vissing11, Dr. Tuan Vu12, Dr. Paul Mahoney13, Dr. Jos Bloemers14, Dr. Thaïs Tarancón15, Dr. Vera Bril16
1
Department of Neurology, Mental Health and Sensory Organs (NESMOS), Faculty of Medicine and Psychology, Sapienza University of Rome, Rome, Italy.
2
Neuromuscular and Rare Disease Centre, Sant'Andrea Hospital, Rome, Italy.
3
Neuromuscular Diseases Unit, Hospital de la Santa Creu i Sant Pau, Barcelona, Spain.
4
Department of Neurology, Municipal Hospital, Poznań, Poland.
5
MDA ALS & Neuromuscular Center, Department of Neurology, University of California, Irvine, Orange, CA, United States.
6
Department of Neuromuscular Medicine, Epilepsy and Clinical Neurophysiology, University of Kentucky, Lexington, KY, United States.
7
Emeritus and Past Director, Department of Neuroimmunology and Neuromuscular Diseases, Fondazione IRCCS, Istituto Nazionale Neurologico Carlo Besta, Milan, Italy.
8
Neurology Department, Indiana University School of Medicine, Indiana University Health, Indianapolis, IN, United States.
9
Université Côte d'Azur, Peripheral Nervous System & Muscle Department, Pasteur 2 Hospital, Centre Hospitalier Universitaire de Nice, Nice, France.
10
Department of Neurology, Hanamaki General Hospital, Hanamaki, Japan.
11
Copenhagen Neuromuscular Center, Department of Neurology, Rigshospitalet, University of Copenhagen, Copenhagen, Denmark.
12
Department of Neurology, University of South Florida Morsani College of Medicine, Tampa, FL, United States.
13
UCB, Slough, United Kingdom.
14
UCB, Brussels, Belgium.
15
UCB, Madrid, Spain.
16
Ellen and Martin Prosserman Centre for Neuromuscular Diseases, Toronto General Hospital, University of Toronto, Toronto, Ontario, Canada
Background: Generalised myasthenia gravis (gMG) is characterised by fluctuating muscle weakness, with treatment responses varying across different muscle groups. In the randomised, double-blind, Phase 3 MycarinG study (NCT03971422), one 6-week cycle of rozanolixizumab significantly improved Myasthenia Gravis Activities of Daily Living (MG-ADL) and Quantitative Myasthenia Gravis (QMG) total scores versus placebo in patients with gMG. Patients could subsequently enrol in open-label extensions MG0004 (NCT04124965), then MG0007 (NCT04650854), or MG0007 directly. Here, we investigate the effect of repeated rozanolixizumab treatment cycles on individual item scores within the MG-ADL and QMG total scores, using final pooled data from the extension studies.
Methods: In MG0004, patients received chronic once-weekly rozanolixizumab infusions for ≤52 weeks. In MG0007, after one initial 6-week cycle (rozanolixizumab 7 mg/kg or 10 mg/kg), subsequent cycles were administered upon symptom worsening (investigator’s discretion). Final efficacy data were pooled across MycarinG, MG0004 (first 6 weeks) and MG0007 for patients with ≥2 symptom-driven cycles (efficacy pool). In this post hoc analysis, MG-ADL and QMG item-level scores were assessed at baseline and Day 43 across 13 cycles, in addition to change from baseline (CFB) to Day 43, using means weighted by sample size.
Results: 129 patients had ≥2 symptom-driven cycles. A reduction in weighted-mean score from baseline was observed at Day 43 for MG-ADL and QMG items (Table 1). For MG-ADL items, weighted-mean (SD) CFB to Day 43 scores (Table 1) were: −0.5 (0.8) for double vision, −0.6 (0.9) for eyelid droop, −0.5 (0.7) for talking and chewing and −0.4 (0.6) for swallowing. For QMG items, weighted-mean (SD) CFB scores (Table 1) were −0.5 (1.0) for double vision, −0.7 (1.1) for ptosis, −0.4 (0.7) for facial muscles, right arm outstretched and left arm outstretched, −0.3 (0.6) for right leg outstretched, −0.4 (0.6) for left leg outstretched and −0.5 (0.9) for speech after counting aloud from 1 to 50.
Conclusion: Long-term rozanolixizumab treatment cycles led to improvements from baseline across ocular, bulbar, respiratory and limb weakness/gross motor items in MG-specific outcomes, demonstrating consistent benefit across a broad range of signs and symptoms. Funding: UCB.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
Images or Table (Optional)
PP01.202
Real-World Evidence on Utilization and Clinical Outcomes of Efgartigimod in Patients With Myasthenia Gravis
Miss Lauren Miguet1, Mr. Martijn De Merlier1, Prof. Kristl G. Claeys1,2
1
Laboratory for Muscle Diseases and Neuropathies, Department of Neurosciences, KU Leuven, and Leuven Brain Institute (LBI), Leuven, Belgium.
2
Department of Neurology, University Hospitals Leuven, Leuven, Belgium
Background: Acetylcholine receptor antibody-positive (AChR-Ab+) generalized Myasthenia Gravis (gMG) often remains inadequately controlled by conventional therapies, negatively impacting patient-reported Myasthenia Gravis Activities of Daily Living (MG-ADL) scores. While advanced therapies like neonatal Fc receptor (FcRn) and terminal complement (C5) inhibitors show efficacy in clinical trials, these highly selected populations may not reflect the heterogeneity of routine clinical practice. A critical knowledge gap remains regarding the real-world effectiveness and treatment patterns of these novel agents in gMG. The study aims to assess the real-world utilization and clinical outcomes of efgartigimod in AChR-Ab+ gMG patients. We aim to characterize treatment patterns, clinical response, the potential for steroid-sparing effects and prolonged drug-free inter-cycle intervals in a cohort of adult patients with AChR-Ab+ gMG in whom conventional treatments have failed.
Methods: We conducted a retrospective, single-center analysis of adult patients with AChR-Ab+ gMG treated with efgartigimod at our tertiary neuromuscular reference center in Leuven, Belgium. Effectiveness was primarily quantified by analyzing longitudinal MG-ADL scores, changes in daily corticosteroid doses, and the optimization of conventional treatments. To evaluate treatment utilization and long-term outcomes, we examined efgartigimod-free inter-cycle intervals, transitions to other advanced therapies, and the need for rescue therapies or hospitalization. In addition, medication-related adverse effects, comorbid conditions, and thymic status were documented.
Results: The study cohort comprised 27 AChR-Ab+ gMG patients (14 male, 13 female) treated with efgartigimod. Median age was 56.0 years [IQR 34.0–66.5] and median disease duration was 7.0 years [IQR 3.5–14.0]. Thymectomy had been performed in 37% of patients due to hyperplasia (18.5%) or thymoma (18.5%). Patients had a median of 2.0 [IQR 1.0–2.5] lifetime myasthenic crises requiring rescue therapy. At baseline, patients presented with a median MG-ADL score of 8.0 [IQR 6.5-10.0] and Myasthenia Gravis Foundation of America (MGFA) severity was distributed across Grade II (26%), Grade III (59%), and Grade IV (15%), with 45% exhibiting predominantly bulbar symptoms (IIb, IIIb, IVb). The study followed patients for up to 7 treatment cycles. A total of 27 patients completed cycle 1, with 9 patients reaching cycle 7 at the time of analysis. Notably, the mean drug-free interval increased progressively from 3.63 weeks after cycle 1 to 4.57 weeks by cycle 7. A sustained reduction in disease burden was observed, evidenced by a consistent decrease in median MG-ADL scores, from 8.0 to 3.0 by cycle 7. In the total cohort, 77.8% of patients achieved a sustained clinical response, with the responder rate increasing to 88.9% among those who completed seven treatment cycles. Four patients (14.8%) switched to a C5-inhibitor zilucoplan, due to lack of response. Corticosteroid sparing was achieved in 48% of patients, while only 7.1% required a dose increase during efgartigimod treatment.
Conclusion: In a real-world setting characterized by high disease burden and bulbar involvement, efgartigimod demonstrates significant clinical effectiveness and a steroid-sparing effect. These findings validate the use of FcRn inhibitors outside of clinical trial parameters and support individualized, cycle-based treatment strategies for refractory AChR-Ab+ gMG.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.203
Novel Therapies in Generalized Myasthenia Gravis: Real-World Experience of Zilucoplan in Spain
Miss Lídia Giramé-Rizzo1, Mr. Daniel Sánchez-Tejerina1, Mr. Jose Alemañ1,2, Dr. Arnau Llauradó1, Mr. Juan Luis Restrepo1, Dr. Maria Salvadó1, Dr. Raúl Juntas1
1
Vall Hebron University Hospital, Barcelona, Spain.
2
Mollet Hospital, Mollet del Vallès, Spain
Background: Myasthenia gravis (MG) is an autoimmune disease affecting the neuromuscular junction of skeletal muscle. The complement system plays a critical role in the pathogenesis of anti–acetylcholine receptor autoantibody–positive MG (MG AChR-Ab+), contributing to postsynaptic membrane damage and impaired neuromuscular transmission. Zilucoplan, a peptide C5 inhibitor, is a novel targeted therapy approved for patients with generalized MG AChR-Ab+ that blocks activation of the terminal complement cascade. The aim of the present study is to describe the real-world experience with Zilucoplan in these patients at a Neuromuscular Disorders Unit in Spain.
Methods: Description of two patients with generalized MG AChR-Ab+ who received Zilucoplan in a real-world setting for 14 months starting in October 2024, after participating in RAISE and RAISE-XT studies. Efficacy was assessed using clinical scores (MG-ADL, QMG, MGC) and by evaluating the need for reduction of concomitant medications at three time periods: T0, defined as the end of the clinical trials; T1, defined as 6 months of follow-up in a real-world setting; and T2, defined as 14 months of follow-up. Safety was also assessed by reporting adverse events at each time point.
Results: Patient 1: A 40-year-old man with thymoma-associated myasthenia gravis (MG), AChR antibody–positive, classified as MGFA class IIIB. Baseline concomitant treatment, defined as medication used prior to trial initiation, included Tacrolimus 6 mg/d, Prednisone 30 mg/d, Pyridostigmine 360 mg/d, and Periodic intravenous immunoglobulins (IVIg) at 1.5 g/kg every 4 weeks. At T0, there was a reduction in Prednisone to 15 mg/d and discontinuation of Periodic IVIg. Patient 2: A 55-year-old woman with early-onset MG, AchR-Ab+, MGFA class IIIB. Baseline concomitant treatment consisted of Tacrolimus 3 mg/d, Mycophenolate mofetil 500 mg/12h, Prednisone 15 mg/48h, Pyridostigmine, and Periodic IVIg at 0.7 g/kg every 6 weeks. At T0, there was a reduction in Tacrolimus to 2 mg/d and discontinuation of Periodic IVIg. Patient 1 and Patient 2 received Zilucoplan at doses of 32.4 mg/d and 23 mg/d, respectively. In terms of efficacy, similar values in MG-ADL, QMG and MGC scales were observed between T0 and T2 in both patients (Figure 1). At T2, both patients had a MGFA Post-intervention Status (MGFA-PIS) of minimal manifestations (MM). Patient 1 could reduce 33.3% of Prednisone dose from T0 to T1 and 25% from T1 to T2. Patient 2 reduced Prednisone dose by 50% at T1, with no changes observed at T2. No IVIg or other rescue treatments were reintroduced in either patient. In terms of safety, at most only mild treatment-related adverse events were observed, and both patients were vaccinated to prevent meningococcal infection (MenACWY and 4CMenB) before starting Zilucoplan.
Conclusion: According to our results, in a real-world setting Zilucoplan is a good novel therapeutic agent for generalized MG AChR-Ab+ in terms of efficacy and safety. Nevertheless, further prospective observational studies are necessary to better define its long-term clinical benefits and safety in MG.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
Images or Table (Optional)
PP01.204
Exploring the Role of Motor Unit Number Index (MUNIX) in Myasthenia Gravis
Dr. Liliana Bevilacqua1, Dr. Ciro Maria Noioso2, Dr. Gabriella Maria Acerra2, Dr. Aniello Iovino1, Dr. Giuseppe Piscosquito1, Dr. Antonella Toriello1, Prof. Paolo Barone2, Dr. Claudia Vinciguerra1
1
U.O.C. Neurologia, A.O.U. “San Giovanni di Dio e Ruggi d’Aragona”, Salerno, Italy.
2
Università degli studi di Salerno, Salerno, Italy
Background: Myasthenia gravis (MG) is a chronic autoimmune disorder characterized by antibody-mediated attack towards different components of the post-synaptic membrane of the neuromuscular junction (NMJ), resulting in skeletal muscles weakness and fatigability. Currently, no objective biomarker of disease progression exists: routine electrophysiological tests such as Repetitive Nerve Stimulation (RNS) and Single Fiber Electromyography (SFEMG) are essential for the diagnostic confirmation of the disease, but play a limited role in monitoring its progression. In fact, to date, only clinical scales can quantitatively monitor the patient's clinical conditions over time. This pilot study explores the role of the motor unit number index (MUNIX) as biomarker of motor unit function and disease monitoring in Myasthenia Gravis (MG), by comparing MUNIX values between patients and healthy controls (HCs), alongside correlations with clinical and electrophysiological measures.
Methods: A cohort of MG patients and HCs were retrospectively enrolled at the Myasthenia gravis center of the University Hospital of Salerno, Italy, from December 2023 to October 2024. Exclusion criteria included history of diabetes, infections, neoplasms, pregnancy, facial palsy and carpal tunnel syndrome. Compound motor action potential (CMAP), MUNIX, and motor unit size index (MUSIX) were assessed in the abductor pollicis brevis (APB) and orbicularis oculi (OO) muscles in the symptomatic side. Clinical severity was evaluated using MGCS, MG-ADL, and MGFA classification. The study was approved by the Ethics Committee of University Hospital of Salerno. Written, informed consent was obtained from all participants. Statistical analyses included ANOVA, Spearman correlations, and multivariate analysis (p<0.05).
Results: 42 MG patients (63.1±12.7 years) and 21 age-matched HCs (59.7±8.7 years) were enrolled. MUNIX values from OO were significantly lower in MG patients while MUSIX was higher. No significant differences were found in MUNIX variables from ABP. Univariate analysis revealed a significant negative correlation between the MUNIX value for the OO muscle and disease duration. When testing this association with logistic regression analysis, MUNIX value for the OO muscle was a significant predictor of disease duration.
Conclusion: MG patients showed reduced MUNIX values, indicating motor unit loss and impaired neuromuscular transmission, while higher MUSIX suggests compensatory remodeling. Correlations with disease duration and clinical severity underscore MUNIX's potential in disease monitoring. Further studies with larger cohorts and longitudinal designs are warranted to validate these findings and explore the predictive value of MUNIX in MG progression.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.205
Environmental Risk Factors for Thymoma-Associated Myasthenia Gravis
Dr. Malin Petersson, Dr. Jing Wu, Prof. Fredrik Piehl, Prof. Lars Alfredsson, Assoc. Prof. Susanna Brauner
Karolinska Institutet, Stockholm, Sweden
Background: Thymoma-associated myasthenia gravis (TAMG), which accounts for approximately 10% of all MG cases, represents a distinct clinical subgroup characterized by more severe disease. Conversely, among patients diagnosed with thymoma, around a third will develop MG. We and others have reported increased MG risk associated with obesity (mainly aged men) and smoking (early-onset MG; EOMG), as well as an inverse association with alcohol consumption. In thymoma, both smoking and alcohol use have also been linked to increased risk. However, to date, no environmental risk factors have been identified specifically for TAMG.
Methods: The Genes and Environment in Myasthenia Gravis study (GEMG) is a cross-sectional questionnaire study, conducted nationwide in Sweden 2018 to 2019. TAMG patients included in the GEMG study were identified by linkage to the nationwide MG registry and selection of cases based on reports of thymectomy pathology outcomes. Population-based controls without MG were matched to cases (15:1), based on year of birth and sex. Possible risk factors were evaluated by conditional logistic regression.
Results: Ninety-one TAMG patients were matched to 1,364 controls. The mean age at onset was 49 (SD 16) years, with 54% females. In a multivariable regression analysis, we found that ever smoking compared to never-smokers was associated with an increased risk of TAMG (OR 1.90, 95% CI 1.14-3.18, p=0.015). Patients who were current smokers at disease had almost twice the risk of developing MG compared to never smokers (OR 2.03, 95% CI 1.08-3.84, p=0.029). Similarly, those who quit smoking compared to never-smokers displayed an OR of 1.82, 95% CI 0.99-3.36, p=0.054. Alcohol consumption was negatively associated with TAMG (drinker compared to non-drinkers OR 0.32, 95% CI 0.18-0.55, p<0.001). Furthermore, fish consumption at least once weekly was also inversely associated with TAMG (OR 0.57, 95% CI 0.33-0.98, p=0.043). Neither body mass index (BMI) at age 20, physical activity nor oral tobacco use was associated with TAMG.
Conclusion: We examined risk factors for TAMG and observed that smoking, particularly smoking at onset, was associated with an increased risk of TAMG. In contrast, alcohol consumption was associated with a reduced risk, opposite to what has been reported for thymoma, suggesting that alcohol may modulate pathways critical for the development of autoimmunity.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.206
Minimal Symptom Expression With Claseprubart, an Active C1S Inhibitor, in Patients With Generalized Myasthenia Gravis
Dr. Marek Smilowski1, Prof. Nils Erik Gilhus2, Dr. Maria Ait-Tihyaty3, Dr. Caitlin Briggs3, Dr. Uzma Siddiqui3, Mr. Luke Hickey3, Dr. Marianna Lalla3, Dr. Said Beydoun4
1
Neurologia Śląska Centrum Medyczne, Katowice, Poland.
2
Haukeland University Hospital, Bergen, Norway.
3
Dianthus Therapeutics, New York, United States.
4
University of Southern California, Los Angeles, United States
Background: The classical complement pathway plays a significant role in generalized myasthenia gravis (gMG) pathology. Claseprubart is a potent monoclonal antibody that selectively targets the classical pathway by inhibiting active C1s (aC1s). Remission is a key goal in gMG and achieving minimal symptom expression (MSE) with claseprubart is a key treatment objective.
Methods: MaGic (NCT06282159), is a global Phase 2, randomized, double-blind, placebo-controlled trial. Patients treated with claseprubart (300mg, Q2W) were evaluated for MSE (MG-ADL ≤1 or QMG ≤3) compared with placebo. p-values were one-sided and nominal significance was assessed at an alpha= 0.1. Outcomes include proportion of patients achieving MSE at Week 13 and anytime during the study, earliest timepoint of MSE achievement, and durability defined as sustained MSE for at least 6 weeks.
Results: At Week 13, 37% of claseprubart treated patients achieved MG-ADL-MSE versus 14% with placebo (OR 3.81; p=0.0550). 43% achieved MG-ADL-MSE at least once during the study versus 14% of placebo (OR: 4.72; p=0.0231). This effect was observed as early as Week 1 (median time to response was Week 3). Sustained remission-like states (for at least 6 weeks) were seen in all patients who achieved MSE. Similar results were observed using QMG minimum symptom expression threshold.
Conclusion: Claseprubart patients were more than four times more likely to achieve MSE than placebo patients and MSE was observed as early as Week 1. Patients who achieved MSE maintained responses for at least 6 weeks. These results highlight the therapeutic potential of aC1s inhibition in AChR+ gMG.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.207
Successful Off-Label Treatment of Aggressive Juvenile Seronegative Generalized Myasthenia Gravis With Efgartigimod Alfa
Dr. Maria Gontika, Dr. Chrysanthi Tsimakidi, Dr. Sotiria Stavropoulou- De Lorenzo, Dr. Aggeliki Psarra, Dr. Eirini-Anastasia Karydi, Dr. Dionysia Gkougka
Penteli's Children Hospital, Athens, Greece
Background: Juvenile seronegative generalized myasthenia gravis (MG) is rare and therapeutically challenging, particularly in patients with aggressive disease and poor response to conventional rescue therapies. Plasma exchange (PLEX) is effective but invasive and associated with possibly significant side-effects. Efgartigimod alfa, an FcRn antagonist that reduces circulating IgG levels and has a mechanism of action analogous to PLEX, represents a less invasive alternative; however, it is currently approved only for adult acetylcholine receptor antibody–positive MG.
Methods: We report an adolescent female (born in 2009) followed since January 2025 for juvenile seronegative generalized MG. Initial symptoms consisted of fluctuating diplopia and left esotropia, first noted in 2021 and initially attributed to strabismus, for which surgical correction was performed in 2024. Following relapse of diplopia, particularly with fatigue, the patient was referred for neurological re-evaluation. Examination revealed fluctuating left ptosis, generalized limb weakness (predominantly left-sided), easy fatigability, and recurrent respiratory tract infections. Baseline scores were 11 on the Quantitative Myasthenia Gravis (QMG) scale and 7 on the Myasthenia Gravis Activities of Daily Living (MG-ADL) scale.
Results: Diagnostic evaluation demonstrated negative MG-related antibodies, normal repetitive nerve stimulation and Desmedt testing, but a positive single-fiber EMG with increased jitter in the left orbicularis oculi muscle, along with a positive pyridostigmine test. Whole-genome sequencing was unremarkable, and thoracic MRI showed mild residual thymic tissue without thymoma. A diagnosis of juvenile seronegative generalized MG was established.
Initial treatment included intravenous immunoglobulin (IVIg), pyridostigmine (up to 240 mg/day), and high-dose corticosteroids (prednisolone up to 1 mg/kg/day). Despite this, the patient developed a highly aggressive disease course with recurrent episodes consistent with impending myasthenic crisis, requiring frequent hospitalizations. IVIg provided only transient benefit, whereas PLEX induced marked and sustained improvement (QMG reduction from 12 to 3), identifying the patient as preferentially PLEX-responsive. However, repeated PLEX necessitated prolonged ICU admissions, and corticosteroid exposure resulted in early Cushingoid features and steroid-induced myopathy, highlighting the need for a steroid-sparing, less invasive therapy. Off-label treatment with efgartigimod alfa was initiated, and the patient received one full treatment cycle. At 3-month follow-up, she remains clinically stable, with a QMG score of 6 and an MG-ADL score of 4. These residual scores are largely attributable to constant diplopia, which cannot be fully or reliably assessed due to prior strabismus surgery. Prednisolone was successfully tapered to 25 mg/day without clinical deterioration, and no rescue therapy was required. Treatment was well tolerated, with no adverse events except for a mild upper respiratory tract infection.
Conclusion: To our knowledge, this is the first reported case of juvenile seronegative generalized MG treated with efgartigimod alfa, representing a double off-label use. The excellent clinical response, meaningful steroid sparing, and favorable safety profile support efgartigimod alfa as a promising therapeutic option in aggressive juvenile seronegative MG, particularly in patients who are PLEX-responsive.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.208
Assessing Relationship Between Immunoglobulin-G Level and Nipocalimab Efficacy using Myasthenia Gravis-Activities of Daily Living Scale
Dr. Zabeen Mahuwala1, Dr. Ruben Faelens2, Dr. Belén Valenzuela Jiménez3, Dr. Martine Neyens2, Dr. Yaowei Zhu4, Dr. Jocelyn H. Leu4, Dr. Marie Fitzgibbon5, Dr. Sindhu Ramchandren6, Dr. Juan-Jose Perez Ruixo2
1
Department of Neurology, University of Kentucky, Lexington, United States.
2
Johnson & Johnson, Beerse, Belgium.
3
Johnson & Johnson, Madrid, Spain.
4
Johnson & Johnson, Spring House, PA, United States.
5
Johnson & Johnson, Raritan, NJ, United States.
6
Johnson & Johnson, Titusville, NJ, United States
Background: In generalized myasthenia gravis (gMG), immunoglobulin G (IgG)-based autoantibodies attack the neuromuscular junction, causing muscle weakness. Nipocalimab, a fully human neonatal fragment crystallizable receptor (FcRn) blocker, prevents IgG recycling and thus lowers levels of circulating IgG antibodies, including pathogenic autoantibodies, thereby improving symptoms of gMG. We explored the longitudinal relationship between IgG level and Myasthenia Gravis-Activities of Daily Living (MG-ADL) score using semi-mechanistic pharmacometrics modeling with data from clinical studies of nipocalimab in gMG and healthy volunteers.
Methods: Data from five phase 1, one phase 2, and one phase 3 (Vivacity-MG3) clinical studies were analyzed to characterize pharmacokinetics ([PK]; 3,429 serum nipocalimab concentrations [n=277]), pharmacodynamics ([PD]; 4,441 serum IgG concentrations [n=421] and 1,247 FcRn receptor occupancy data [n=78]). To establish the longitudinal relationship between IgG and MG-ADL, 2,317 absolute change from baseline (CFB) MG-ADL scores were analyzed in seropositive (anti-acetylcholine receptor, anti-muscle-specific receptor tyrosine kinase, anti-lipoprotein receptor-related protein-4) participants with gMG (n=220). A previously developed nonlinear mixed-effects model was used. The effects of baseline demographics and clinical characteristics on PK, PD, and MG-ADL parameters were investigated.
Results: The previous model captured serum nipocalimab and total serum IgG concentrations of the phase 3 data well. MG-ADL CFB drug effect was linearly related to IgG %CFB at 0.28 points per 10% IgG %CFB. In the patient population, this resulted in approximately 2-points MG-ADL reduction for the nipocalimab-induced 70% IgG reduction when adjusted for placebo. The slope of the regression line was dependent on baseline MG-ADL score. Age, body weight, race, ethnicity, sex, autoantibody status, gMG background therapy and gMG duration did not have a clinically relevant impact.
Conclusion: The model confirmed that the IgG %CFB drives nipocalimab effect on MG-ADL. These results support that total serum IgG reduction is a good predictor for efficacy in gMG, allowing for comparison of clinical effect across FcRn inhibitors.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.209
Efficacy and Safety of Nipocalimab: Switch From 15-mg/kg Every-2-Week to 30-mg/kg Every-4-Week (VIVACITY-MG3 Open-Label Extension)
Dr. Tuan Vu1, Dr. Marie Fitzgibbon2, Dr. Kavita Gandhi2, Dr. Nolan Campbell3, Dr. Yaowei Zhu4, Dr. Jocelyn H. Leu4, Dr. Ibrahim Turkoz3, Dr. Sindhu Ramchandren5
1
Neurology, University of South Florida, Morsani College of Medicine, Tampa, FL, United States.
2
Johnson & Johnson, Raritan, NJ, United States.
3
Johnson & Johnson, Horsham, PA, United States.
4
Johnson & Johnson, Spring House, PA, United States.
5
Johnson & Johnson, Titusville, NJ, United States
Background: The US-Food and Drug Administration (FDA)-approved dosing of nipocalimab for generalized myasthenia gravis (gMG) is 30-mg/kg loading dose, followed by 15-mg/kg every-2-weeks (q2w) for maintenance. In the randomized, double-blind (DB) phase of the VIVACITY-MG3 study, this regimen demonstrated sustained disease control using the Myasthenia Gravis-Activities of Daily Living (MG-ADL) and Quantitative Myasthenia Gravis (QMG) scales. During the open-label extension (OLE), participants from the DB phase were transitioned to 15-mg/kg q2w dosing and had the option to switch to every-4-week (q4w) dosing; this optionality was available for a finite period and was subsequently removed following a protocol amendment. The study reports the efficacy and safety of nipocalimab in participants who switched from 15-mg/kg q2w to 30-mg/kg q4w in OLE of the VIVACITY-MG3 study.
Methods: In OLE, participants on nipocalimab continued treatment; those on placebo switched to nipocalimab. MG-ADL and QMG scores were analyzed pre-switch and post-switch for OLE participants on 15-mg/kg q2w who switched to 30-mg/kg q4w.
Results: Overall, 153 participants entered the OLE; symptom improvements were maintained through a 60-week extension. During the OLE, 25 participants switched to 30-mg/kg q4w dosing, with a median (range) duration of 12.1 (0–48) weeks. In seropositive participants (n=21) (median [range] age: 48.0 [26.0–83.0] years; 61.9% females), the mean (SD) MG-ADL and QMG scores at OLE baseline were 8.3 (3.50) and 14.8 (6.08), respectively. Prior to switch to q4w dosing, the mean (SD) change from OLE baseline (change from baseline [CFB]) was MG-ADL (n=16): −2.4 (3.15) and QMG (n=15): −2.6 (4.21); after switch to q4w dosing, the mean CFB was MG-ADL (n=16): −2.0 (3.03) and QMG (n=15): −2.3 (3.79). Safety data for q4w were comparable with q2w. Infections and infestations were the most-reported adverse events (AEs; 48.0% vs 44.0%). No AEs leading to death were reported.
Conclusion: Temporary 30-mg/kg q4w dosing of nipocalimab showed sustained clinical efficacy and a safety profile consistent with 15-mg/kg q2w dosing. Interpretation is limited by small number of participants and a short duration of follow-up of participants with 30-mg/kg q4w dosing.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.210
Corticosteroid Usage in Patients with Generalized Myasthenia Gravis Receiving Nipocalimab: Phase 3 Vivacity-MG3 Open-Label Extension
Dr. Yuebing Li1, Miss Kavita Gandhi2, Dr. Wisam Karmous3, Dr. Marie Fitzgibbon2, Dr. Ibrahim Turkoz4, Dr. I-Ching Tsai5, Dr. Zia Choudhry6, Dr. Wim Noel7, Dr. Louis Jackson8, Dr. Sindhu Ramchandren6, Dr. Hiroyuki Murai9
1
Neuromuscular Center, Neurological Institute, Cleveland Clinic, Cleveland, OH,, United States.
2
Johnson & Johnson, Raritan, NJ, United States.
3
Johnson & Johnson, Issy-les-Moulineaux, France.
4
Johnson & Johnson, Titusville,, NJ, United States.
5
Johnson & Johnson, Singapore, Singapore.
6
Johnson & Johnson, Titusville, NJ, United States.
7
Johnson & Johnson, Beerse, Belgium.
8
Johnson & Johnson, Horsham, PA, United States.
9
Department of Neurology, International University of Health and Welfare, Narita, Japan
Background: Nipocalimab demonstrated sustained disease control, with improvements in MG-Activities of Daily Living (MG-ADL) and Quantitative MG (QMG) total scores during the 24-week double-blind phase (versus placebo) and through Week-60 of the open-label (OLE) phase of Vivacity-MG3. Corticosteroid dose adjustment was prohibited during the double-blind phase but permitted in the OLE, where all patients received nipocalimab. The objective of the present analysis was to characterize corticosteroid usage, patterns of corticosteroid dose reduction/ discontinuation, and symptom improvements among nipocalimab-treated patients with generalized myasthenia gravis (gMG) during the OLE phase of Vivacity-MG3 (NCT04951622) study.
Methods: Corticosteroid dose reductions to ≤20, ≤15, ≤7.5, ≤10, and ≤5 mg/day prednisone-equivalent were evaluated through OLE data cutoff. Among patients on a low corticosteroid dose (≤10 or ≤5 mg/day), the proportions achieving meaningful clinical improvement (MCI; ≥2-point MG-ADL), minimum symptoms expression (MSE; MG-ADL score=0 or 1), and sustaining these for ≥8-weeks were evaluated.
Results: Of 89 patients receiving corticosteroids at OLE baseline (placebo/nipocalimab:44 [49%], nipocalimab/nipocalimab:45 [51%]), 45% reduced or discontinued corticosteroids by data cutoff; the mean steroid dose reduced from 23 mg/day to 10 mg/day prednisone-equivalent. By OLE data cut-off, 85.4%, 71.9%, 60.7%, 41.6%, and 33.7%, of patients were on ≤20, ≤15, ≤10, ≤7.5, ≤5 mg/day prednisone-equivalents, respectively. Among those on ≤10 mg/day (n=54), 98.1% (n=53) achieved MCI and 44.4% (n=24) achieved MSE; sustained MCI and MSE for ≥8-weeks was achieved by 94.4% (n=51) and 24.1% (n=13), respectively. For those on ≤5 mg/day (n=30), 100.0% (n=30) achieved MCI and 60.0% (n=18) achieved MSE, sustained MCI and MSE for ≥8-weeks was achieved by 93.3% (n=28) and 33.3% (n=10), respectively.
Conclusion: By OLE data cutoff, nearly half of patients receiving corticosteroids at baseline, reduced or discontinued corticosteroids. A substantial proportion of patients who were on low dose corticosteroid (≤10 or ≤5 mg/day) during the OLE sustained MCI and/or MSE for ≥8-weeks. Results support the corticosteroid-sparing effect of nipocalimab and concurrent meaningful clinical improvement and sustained disease control in nipocalimab-treated patients with gMG.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.211
Multiple Responder Analyses With Claseprubart, an Active C1s Inhibitor, in Patients With Generalized Myasthenia Gravis
Dr. Mazen Dimachkie1, Asst. Prof. Francesco Sacca2, Dr. Maria Ait-Tihyaty3, Dr. Caitlin Briggs3, Dr. Uzma Siddiqui3, Mr. Luke Hickey3, Dr. Marianna Lalla3, Dr. Marek Smilowski4
1
University of Kansas Medical Center, Kansas City, United States.
2
Azienda Ospedaliera Universitaria (AOU) Federico II, Naples, Italy.
3
Dianthus Therapeutics, New York, United States.
4
Neurologia Śląska Centrum Medyczne, Katowice, Poland
Background: The classical complement pathway plays a significant role in acetylcholine receptor-positive (AChR+) generalized myasthenia gravis (gMG) pathology. Claseprubart is an investigational, clinical-stage, potent monoclonal antibody engineered to selectively target the classical complement pathway, by selectively inhibiting activated C1s protein. The clinical benefit of claseprubart in gMG may be best supported by improvement across multiple validated scales to capture multi-dimensional improvement beyond single-scale analyses.
Methods: AChR+ gMG patients treated with 300 mg claseprubart subcutaneously once every 2 weeks in the MaGic study (NCT06282159) – a global Phase 2, randomized, double-blind, placebo-controlled trial – were analyzed for responses based on multiple assessments compared to placebo. p-values are one-sided, with nominal significance assessed at an alpha of 0.1. A responder was defined as achieving an improvement in MG-ADL ≥3 and QMG ≥4. Endpoints include proportion of dual MG-ADL and QMG responders at Week 13, time to response, and proportion of responders sustaining response for at least 6 weeks.
Results: 63% of claseprubart patients had dual response improvement in MG-ADL ≥3 and QMG ≥4, versus 14% for placebo (OR 29.00; p=0.0006). Dual responders were seen as early as Week 1. The median time to response was Week 3. All claseprubart patients with dual response at Week 13 had maintained response for at least 6 weeks.
Conclusion: Claseprubart-treated patients were more likely to achieve multi-dimensional response than placebo. Responses emerged early and were sustained, confirming rapid, robust, and durable benefit from claseprubart across functional and quantitative measures from both patient and clinician perspectives.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.212
Clinical and Immunological Features of Thymoma Patients With and Without Myasthenia Gravis: A Pilot Study
Dr. Michela Maria Corbo1, Dr. Francesca Beretta1,2, Dr. Rebecca Taliani1, Dr. Stefano Bongiolatti3, Prof. Luca Voltolini3, Prof. Luca Massacesi1,2, Prof. Valentina Damato1,2, Dr. Gregorio Spagni1,2
1
Department of Neurosciences, Psychology, Drug Research and Child Health (NEUROFARBA), University of Florence, Florence, Italy.
2
Emergency Neurology Unit, Careggi University Hospital, Florence, Italy.
3
Thoracic Surgery Unit, Careggi University Hospital, Florence, Florence, Italy
Background: Thymoma-associated myasthenia gravis (TAMG) is nearly invariably associated with acetylcholine receptor antibodies (AChRAb). However, approximately 15% of thymoma patients have AChRAb in the absence of MG and rarely may develop post-thymectomy MG (PTMG). PTMG represents a rare, understudied, but clinically significant entity, which can be a diagnostic and therapeutic challenge in clinical practice, with yet unclear immunopathogenic mechanisms. This study evaluates the clinical and immunological features of thymoma patients with and without MG to identify predictive biomarkers of PTMG.
Methods: Patients with histologically confirmed thymoma treated in our Centre between 2011 and 2025 were included in the study. Demographic and clinical data were collected, and available serum samples were evaluated for in vitro complement activation using a flow cytometry live cell-based assay.
Results: Fifty thymoma patients (19 women and 31 men; median age at thymectomy: 65 years) were included in the study: 17/50 (34%) TAMG patients, 27/50 (54%) thymoma patients with no MG and AChRAb negative (AChRAb-MG-), 6/50 (12%) AChRAb positive thymoma patients without MG (AChRAb+MG-), of whom 2/6 (33%) developed PTMG. Types AB/B thymomas predominated across groups (n=40/50,80%) without statistically significant difference between MG and non-MG patients (with/without AChRAb). Most had a Masaoka stage I–II (n=43/50, 86%). The majority of patients had maximum MGFA>II (13/19, 68%). Sixteen serum samples were available for further analyses (10 TAMG, 2 PTMG, 4 AChRAb+MG-). Complement activation was significantly lower in AChRAb+MG- thymoma patients compared to TAMG and PTMG patients (p=0.0332, Mann-Whitney U test). In one PTMG patient with available serial serum samples, level of complement activation increased significantly from thymoma diagnosis to MG onset.
Conclusion: In vitro assessment of complement activation could represent a novel biomarker of PTMG. Future studies are warranted to investigate in-depth antibody as well as thymic and B-cells properties in PTMG patients.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.213
A Case of Bulbar-Onset Motor Neuron Disease Emerging in AChR-Positive Myasthenia Gravis
Dr. Muhammed Ameen Noushad, Dr. Ashwin Pinto, Dr. David Allen
Wessex neuroscience centre, Southampton, United Kingdom
Background: Myasthenia gravis (MG) and amyotrophic lateral sclerosis/motor neuron disease (ALS/MND) are pathophysiologically distinct disorders that can both present with bulbar dysfunction and may coexist more often than expected by chance. In patients with established MG, progressive bulbar symptoms are easily misattributed to MG relapse, leading to repeated escalation of immunosuppression and delayed recognition of superimposed ALS/MND
Methods: Clinical records, serial neurophysiology and respiratory function tests were reviewed in a 73-year-old man with AChR-antibody positive generalized MG who later developed progressive bulbar and limb symptoms. Particular attention was paid to temporal evolution of phenotype, treatment responses, electrophysiological patterns and key features differentiating MG relapse from emerging ALS/MND, supported by a comparative table of bulbar-onset ALS versus bulbar MG.
Results: The patient achieved near-complete MG remission (MG-ADL 1) on prednisolone and rituximab but developed steroid toxicity and a non-tuberculous mycobacterial skin infection that constrained further immunosuppression. Two years after MG onset he developed weight loss, fixed dysarthria and dysphagia, exertional dyspnoea, painful cramps, distal weakness and widespread fasciculations, initially interpreted as refractory bulbar MG and treated with additional rituximab, multiple plasma exchanges and IVIG without sustained benefit while vital capacity declined from 2.61 L to 1.42 L (35% predicted). On re-examination he had mixed spastic–flaccid dysarthria, distal hand weakness, brisk reflexes and prominent fasciculations but no ocular MG features. CD19 counts remained depleted following rituximab, and the persistent absence of fatigable ptosis or ophthalmoplegia supported stable, well-controlled MG at the time of reassessment. Repeat neurophysiology demonstrated preserved sensory responses, low-amplitude motor potentials without demyelination, normal repetitive nerve stimulation of limb and trapezius muscles, and widespread active and chronic denervation including tongue and paraspinals, findings incompatible with active MG and diagnostic of bulbar-onset ALS/MND. Immunotherapy was de-escalated; riluzole was initiated, pyridostigmine withdrawn and prednisolone tapered to a low maintenance dose, alongside nocturnal non-invasive ventilation and radiologically inserted gastrostomy, yet his MND continued to progress despite stable MG.
Conclusion: In MG patients, evolution to a non-fluctuating bulbar syndrome with cramps, fasciculations, upper motor neuron signs, limb wasting, respiratory decline and poor response to cholinesterase inhibitors or rescue immunotherapies should strongly prompt evaluation for superimposed ALS/MND. Early use of needle EMG and awareness of distinguishing clinical and neurophysiological features are essential to avoid futile immunosuppression and to initiate timely multidisciplinary MND care, respiratory support and advance care planning.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
Images or Table (Optional)
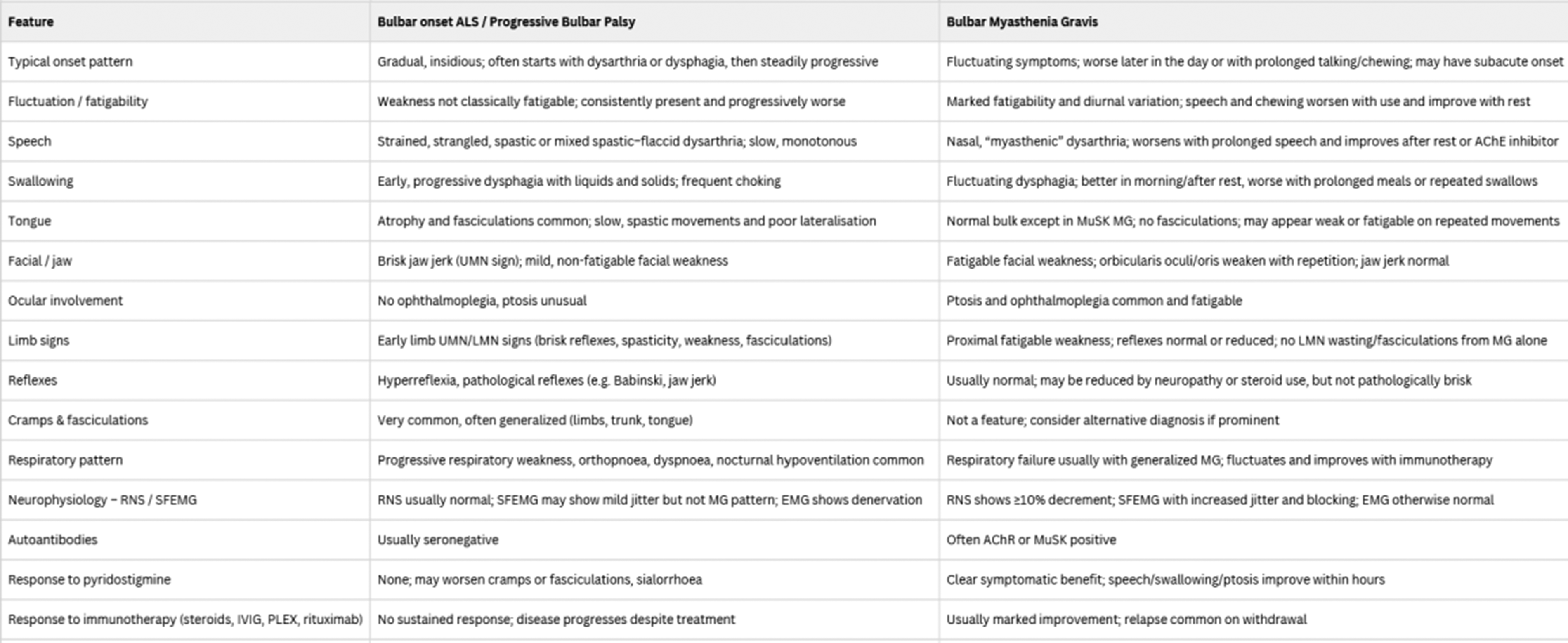
PP01.214
Characterisation of Patients With Generalised Myasthenia Gravis on Zilucoplan Treatment in European Managed Access Programmes
Dr. Natasa Savic1, Dr. Paolo Emilio Alboini2, Dr. Pascal Cintas3, Dr. Sophie Demeret4, Dr. Vincenzo Di Stefano5, Dr. Carmen Erra6, Dr. Laura Fionda7, Dr. Raffaele Iorio8, Dr. Raul Juntas-Morales9, Dr. Emmeline Lagrange10, Dr. Michelangelo Maestri11, Prof. Sabrina Sacconi12, Dr. Fiammetta Vanoli13,14, Dr. Claudia Vinciguerra15, Dr. Karin Annoni16, Dr. Kerina Bonar17, Dr. Grégory Chollet18, Dr. Cristina Lopez19, Dr. Patrik Ohagen20, Dr. Cassandra Slader21
1
UCB, Bulle, Switzerland.
2
Neurology Unit, Fondazione IRCCS Casa Sollievo della Sofferenza, San Giovanni Rotondo, Italy.
3
Department of Neurology, Toulouse Purpan University Hospital, Toulouse, France.
4
Neurological Intensive Care Unit, Pitié Salpêtrière Hospital, AP-HP, Sorbonne University, Paris, France.
5
Department of Biomedicine, Neuroscience and Advanced Diagnostics (BiND), University of Palermo, Palermo, Italy.
6
UOC Neurophysiopathology, AORN Cardarelli, Naples, Italy.
7
Neuromuscular and Rare Disease Centre, UOC Neurologia, Sant’Andrea Hospital, Rome, Italy.
8
Institute of Neurology, Fondazione Policlinico Universitario “A. Gemelli”, IRCCS, Università Cattolica del Sacro Cuore, Rome, Italy.
9
Department of Neurology, Vall d’Hebron University Hospital, Barcelona, Spain.
10
Consultations Maladies Rares Neuromusculaires du CHU Michallon, Grenoble, France.
11
Neurology Unit, Department of Neurosciences, Azienda Ospedaliero Universitaria Pisana, Pisa, Italy.
12
Université Côte d'Azur, Peripheral Nervous System & Muscle Department, Pasteur 2 Hospital, Centre Hospitalier Universitaire de Nice, Nice, France.
13
Department of Neuroimmunology and Neuromuscular Diseases, Fondazione IRCCS, Istituto Nazionale Neurologico Carlo Besta, Milan, Italy.
14
Department of Human Neurosciences, Sapienza University of Rome, Rome, Italy.
15
Neurology Unit, Department of Medicine, Surgery and Dentistry “Scuola Medica Salernitana”, University of Salerno, Salerno, Italy.
16
UCB, Milan, Italy.
17
UCB, Slough, United Kingdom.
18
UCB, Colombes, France.
19
UCB, Barcelona, Spain.
20
UCB, Uppsala, Sweden.
21
UCB, Brussels, Belgium
Background: Myasthenia gravis (MG) is a rare, neuromuscular, autoimmune disease characterised by fluctuating muscle weakness. In the Phase 3 RAISE study (NCT04115293), treatment with zilucoplan, a potent complement component 5 inhibitor, demonstrated statistically significant and clinically meaningful improvements in MG-specific outcomes compared with placebo in patients with generalised MG (gMG); these improvements were sustained up to Week 120 in the open-label extension RAISE-XT study (NCT04225871). Managed Access Programmes (MAPs) enable the use of treatments prior to authorisation for patients with a severe/debilitating/life-threatening condition, where available treatments are deemed inadequate by patients and physicians, and there is no available clinical trial. Here, we describe zilucoplan utilisation patterns and patient characteristics in MAPs initiated under protocolised compassionate use and/or early access authorisation in France, and under compassionate use in Italy and Spain.
Methods: This was an observational, retrospective, longitudinal cohort study of adults with gMG who initiated zilucoplan treatment in MAPs in France, Italy and Spain between December 2021 and November 2024. The primary objective was to describe patient demographics and disease characteristics at treatment initiation. Change from baseline in MG Activities of Daily Living (MG-ADL), Garches and MG Quality of Life 15-Item Revised scores were also assessed when available. All analyses were descriptive.
Results: Overall, 57 patients from France, 32 from Italy and <5 from Spain were included. Mean (standard deviation [SD]) age was 56.5 (19.8), 57.8 (14.9) and 48.8 (13.6) years, respectively. Most patients were female (50.9%, 65.6% and 75.0%, respectively). Additional data were available for France and Italy. Mean (SD) number of years since initial MG diagnosis was 8.7 (9.2) and 11.6 (10.2); 42.1% and 28.1% of patients, respectively, had an MG diagnosis <5 years. Baseline (mean [SD]) MG-ADL scores were 7.3 (4.7) and 9.2 (3.6), respectively. In the 2 years prior to enrolment, MG crises occurred in 57.9% and 9.4% of patients and MG-related hospitalisations occurred in 80.7% and 43.8%, respectively. Prior to enrolment, pyridostigmine use was reported in 73.7% and 96.9% of patients, and corticosteroid use was reported in 75.4% and 100%, respectively. Treatment outcomes data will be presented in the poster.
Conclusion: The patients with gMG who received zilucoplan as part of the MAP had a long disease duration and high rates of prior hospitalisation and crisis, reflecting an unmet clinical need in this population. Funding: UCB.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.215
Steroids Tapering Strategies in Patients With Generalized Myasthenia Gravis Initiating Efgartigimod: STRIVE-gMG Interim Results
Dr. Gordon Smith1, Dr. Nicholas Silvestri2, Ms. Cynthia Qi2, Dr. Hongbo Yang3, Dr. Andrea Corse4, Dr. Ali Habib5, Dr. Tuan Vu6, Dr. Gil Wolfe7, Dr. Lingyun Li2, Dr. Sun Lee3, Dr. Yujie Wu3, Dr. Dongni Ye3, Dr. Dustin Nowacek2, Dr. Jamie Aldridge2, Dr. Arash Mahajerin2, Dr. Glenn Philips2, Dr. James Howard4
1
Virginia Commonwealth University School of Medicine, Richmond, VA, United States.
2
argenx US Inc., Boston, MA, United States.
3
Analysis Group, Inc, Boston, MA, United States.
4
University of North Carolina at Chapel Hill School of Medicine, Chapel Hill, NC, United States.
5
University of California, Irvine, Irvine, CA, United States.
6
University of South Florida Morsani College of Medicine, Tampa, FL, United States.
7
University at Buffalo Jacobs School of Medicine and Biomedical Sciences, Buffalo, NY, United States
Background: Long-term steroids in generalized myasthenia gravis (gMG) is associated with considerable complications. Efgartigimod, the first FcRn antagonist approved in the United States (US) for adults with AChR antibody-positive gMG, offers opportunities for steroid-sparing treatment. Although the MGFA International Consensus Guidance and clinical trial evidence support the potential for reducing steroids with efgartigimod, real-world physician tapering behaviors – specifically when, how, and to what extent neurologists taper steroids – remain poorly characterized. This study examined physician-reported tapering strategies and corresponding patient-level steroids tapering outcomes among individuals with gMG initiating efgartigimod in routine US clinical practice.
Methods: STRIVE-gMG is a retrospective chart review of patients with gMG who were receiving steroids at the time of efgartigimod initiation and completed at least two efgartigimod treatment cycles. Data are collected from neurologists in a specialty panel and from participating medical centers. In addition to patient-level data, participating neurologists reported their typical steroids tapering strategies at practice level, including timing, tapering speed, and target dose. Data collection is ongoing, and this interim analysis reflected initial data obtained from the neurologist panel.
Results: As of November 10, 2025, 15 neurologists contributed data. Participating physicians had an average of 17.3 years of experience treating gMG and managed an average of 23 gMG patients receiving efgartigimod since its approval in 2021. Regarding typical tapering practice, most neurologists (73.3%) reported initiating steroids tapering once a stable clinical response to efgartigimod was achieved, suggesting broad alignment on timing of tapering. However, tapering speed and tapering goals varied, reflecting highly individualized decision-making. Faster tapering (>5 mg/month) was adopted by 46.7% of physicians, whereas 33.3% preferred a slower approach (≤5 mg/month) and 13.3% reported adjusting tapering based on clinical response. Target steroids doses also differed: while the majority targeted a complete discontinuation (33.3%) or low-dose maintenance (0-5 mg/day; 40.0%), some targeted 5-10 mg/day (6.7%) and others indicated variable goals tailored to patient specific factors (13.3%). Forty-one patient charts were abstracted by these neurologists. All patients were AChR positive. At efgartigimod initiation, patients had a mean age of 48.9 years and a mean disease duration of 3.7 years. Most (87.8%) had MGFA class II–III disease, and they had been on their current steroids regimen for an average of 1.1 years (range of 0-6.8 years). All patients tapered steroids following efgartigimod initiation, with mean prednisone-equivalent dose declining from 30.4 mg/day to 4.0 mg/day at 18 months. By the end of follow-up, 68.3% had discontinued steroids entirely and an additional 19.5% had tapered to ≤5 mg/day while remaining on efgartigimod.
Conclusion: Most neurologists identified achievement of a stable clinical response to efgartigimod as the point at which steroids tapering should begin; however, meaningful variation was reported in typical tapering speed and final target dose. These patterns indicate the absence of a standardized tapering protocol and the use of individualized approaches influenced by both patient factors and physician judgment. Across these diverse tapering strategies, substantial reductions in steroids dose were achieved following efgartigimod initiation, supporting its role as an effective steroid-sparing treatment option.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
Images or Table (Optional)
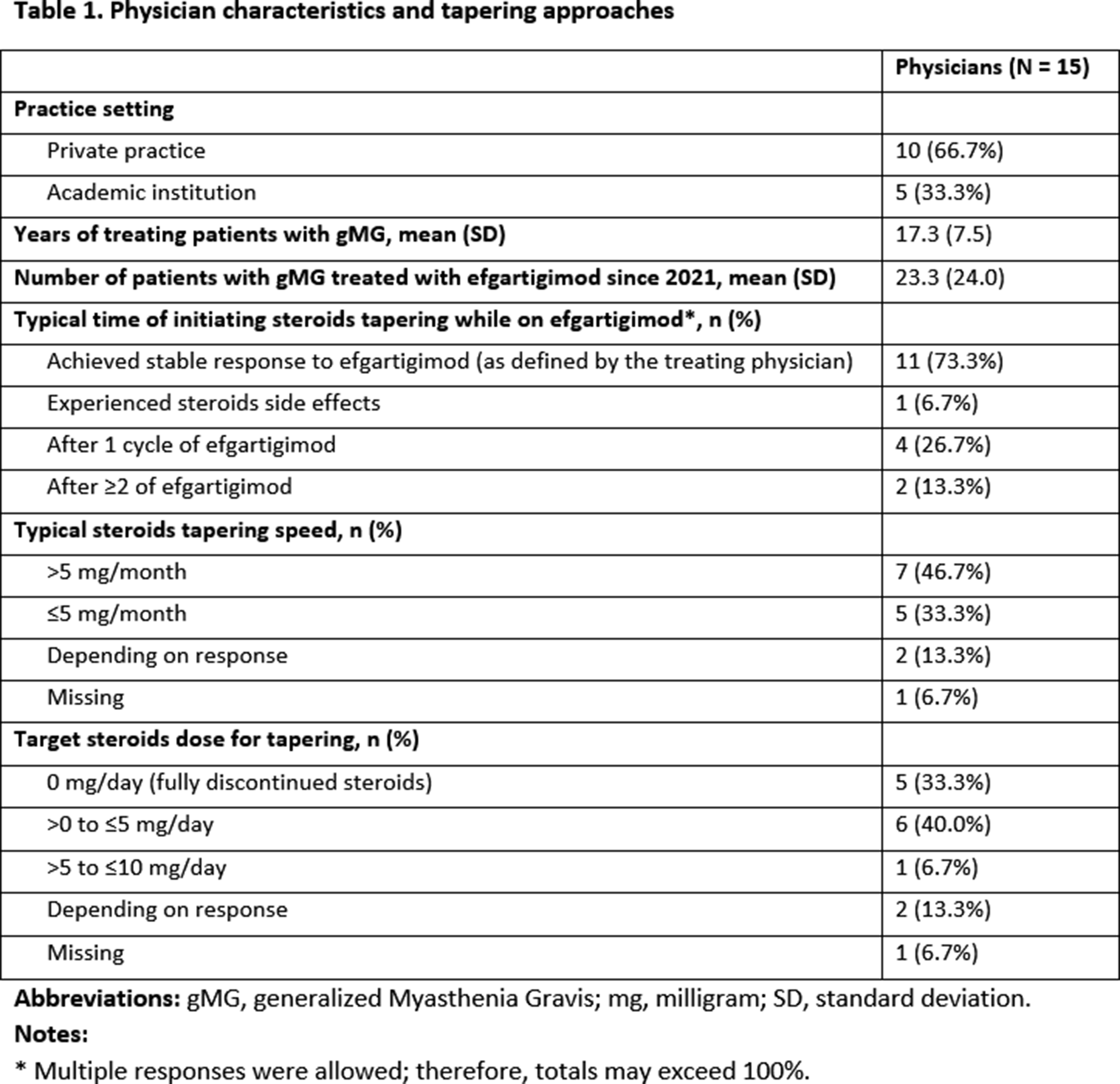
PP01.216
Early FcRn Blockade with Efgartigimod in Generalized Myasthenia Gravis: A German Multicenter Real-World Study
Dr. Niklas Huntemann1, Dr. Axel Haarmann2, Prof. Heidrun H. Krämer3, Prof. Tim Hagenacker4, Dr. Tobias Hegelmaier5, Dr. Norma Krümmer6, Prof. Christiane Schneider-Gold7
1
Medical Faculty, Heinrich Heine University Düsseldorf, Düsseldorf, Germany.
2
University Hospital Würzburg, Würzburg, Germany.
3
Justus Liebig University of Giessen, Giessen, Germany.
4
Center for Translational Neuro- and Behavioral Sciences (C-TNBS), University Medicine Essen, Essen, Germany.
5
Hannover Medical School (MHH), Hannover, Germany.
6
Klinikum Altenburger Land, Altenburg, Germany.
7
St. Josef Hospital, Ruhr-University of Bochum, Bochum, Germany
Background: Generalized myasthenia gravis (gMG) is an autoimmune disorder driven by pathogenic immunoglobulin G (IgG) autoantibodies that disrupt neuromuscular transmission, leading to fluctuating muscle weakness. Efgartigimod, a human Fc IgG1 fragment administered in individualized cyclic treatment courses, blocks the neonatal Fc receptor (FcRn) and accelerates IgG degradation, thereby lowering circulating pathogenic antibody levels. Given that prolonged exposure to pathogenic autoantibodies might amplify neuromuscular junction (NMJ) damage in gMG, prompt FcRn blockade provides a theoretically compelling approach to preserve NMJ integrity and translate into sustained clinical improvement. Real-world evidence on the efficacy and safety of initiating efgartigimod shortly after diagnosis remains limited, prompting the present investigation. This study assessed the clinical efficacy and safety of early treatment intensification with efgartigimod in gMG, defined as therapy initiation within 24 months of diagnosis.
Methods: We conducted a retrospective, multicenter cohort study across seven German specialized MG centers and identified patients who received their first efgartigimod dose within the predefined 24-month period following diagnosis. Outcome measures comprised established MG scores as well as safety parameters. Data were collected retrospectively at baseline (BL) and at Months 1, 3, 6, and 12. Patients with at least two follow-up visits were included. Because individualized dosing cycles can cause fluctuations in the mentioned outcome parameters, the best response observed for each patient during follow-up was used for analysis.
Results: Our cohort comprised 23 gMG patients with a mean latency from diagnosis to first efgartigimod administration of 10.6 months (SD ± 7.7 months). Early initiation of FcRn blockade in the disease course led to rapid and sustained therapeutic benefits; MG-Activities of Daily Living (MG-ADL) scores improved from 8.3 ± 4.3 to 3.4 ± 2.3 after 1 year, with a mean maximal individual reduction of −5.7 ± 3.3 points (P< .001). By Month 12, mean Quantitative Myasthenia Gravis (QMG) score declined from 11.3 ± 6.0 to 3.2 ± 2.8 (P= .0001). At the best individual follow-up visit, 87% of patients achieved a Patient Acceptable Symptom State according to QMG criteria (≤3 points; BL: 35%; P= .0007) and 45% with respect to the MG-ADL (≤2 points; BL: 5%; P= .0039). Clinically meaningful improvement occurred in 87% (QMG) and 73% (MG-ADL) of patients, while minimal symptom expression was achieved in nine (41%) patients. Consistent with these findings, quality of life increased substantially with MG Quality of Life-15 scores, declining from 32.4 ± 10.8 to 19.6 ± 14.0 (P= .0002). Evidence of steroid sparing was demonstrated by a reduction in mean daily prednisolone doses from 25.5 ± 18.7 mg to 5.9 ± 3.8 mg (P= .0005). Efgartigimod treatment was well tolerated, with no withdrawals and no emergence of new safety signals. An updated analysis will be presented at the congress.
Conclusion: Early efgartigimod initiation in gMG was associated with rapid and durable clinical improvement and high responder rates, while substantially reducing corticosteroid dosage. This retrospective multicenter real-world analysis indicates that targeting pathogenic IgG via FcRn blockade early in the disease course can yield meaningful benefits, potentially preventing disease progression and structural NMJ damage.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.217
Myasthenia Gravis in Armenia: Real-World Diagnostic Pathways, Treatment Patterns, and System-Level Gaps
Dr. Kristine Arustamyan1, Dr. Edgar Gevorgyan2, Dr. Mariam Grigoryan1, Dr. Armine Kteyan3,2, Assoc. Prof. Anahit Mehrabyan4, Assoc. Prof. Nune Yeghiazaryan1,2
1
Neurology Department, Erebouni Medical Center, Yerevan, Armenia.
2
Neurology and Neurosurgery Department, Armenian National Institute of Health, Yerevan, Armenia.
3
Center of Medical Genetics and Primary Health Care, Yerevan, Armenia.
4
UNC Hospitals Neurology Clinic, Chapel Hill, NC, United States
Background: Myasthenia gravis (MG) is a chronic autoimmune disorder of the neuromuscular junction that requires timely diagnosis, specialized investigations, and long-term access to immunomodulatory therapy. In Armenia, systematic data on MG remain limited. This study aimed to characterize demographic and clinical features, diagnostic approaches, treatment patterns, and outcomes of patients with MG managed at two tertiary multidisciplinary centers, and to identify key system-level challenges relevant to future improvement of MG care.
Methods: We conducted a longitudinal observational study of patients with MG followed between 2020 and 2025 at Erebouni Medical Centre and the Centre of Medical Genetics in Yerevan, Armenia. Patients were monitored through scheduled outpatient visits and telephone follow-up. Collected variables included age, sex, MG subtype (generalized [GMG] or ocular [OMG]), antibody status, electrophysiological diagnostic methods, chest computed tomography (CT) findings, thymectomy status, prescribed treatments, hospitalizations, and mortality. Diagnostic evaluation included repetitive nerve stimulation (RNS), single-fiber electromyography (SFEMG), serological antibody testing, and radiological assessment of the thymus when available. Descriptive statistics were used to summarize demographic and clinical characteristics. Group comparisons of age were performed using Welch’s two-sample t-test, and associations between categorical variables were assessed using the chi-square test of independence. All analyses were two-sided, with statistical significance defined as p < 0.05.
Results: A total of 62 patients were included, with an age range of 18–80 years. Generalized MG was diagnosed in 50 patients (80.6%), while 12 patients (19.4%) had ocular MG. Anti-AChR antibodies were detected in 44% of GMG and 58% of OMG patients. Antibody testing was not performed in 27 patients. Electrophysiological abnormalities were detected in most cases. Thymic pathology was common: thymoma was identified in 25 patients and thymic hyperplasia in 7. Thymectomy was performed in 23 patients with thymoma and in only two patients with thymic hyperplasia. Four deaths occurred during the study period, none directly related to MG progression or complications. No significant differences in mean age were observed by gender (p = 0.153) or by disease subtype (p = 0.379). Disease subtype was significantly associated with antibody status (p = 0.003), diagnostic modality (p < 0.001), and thymectomy status (p = 0.042). No significant associations were identified between gender and disease subtype or other clinical variables. Chest CT findings were not significantly associated with either disease subtype or gender. Most patients received pyridostigmine and corticosteroids; 19 additionally received azathioprine, and only four were treated with rituximab. Five patients were not receiving pharmacological therapy at the time of analysis. Disease exacerbations requiring hospitalization were common, with 11 patients hospitalized more than once during the previous year and treated with plasmapheresis. Use of intravenous immunoglobulin and biologic therapies was limited due to cost.
Conclusion: MG care in Armenia is characterized by incomplete diagnostic evaluation and restricted access to advanced therapies, largely driven by economic and system-level barriers. Establishing standardized diagnostic pathways, ensuring continuous availability of essential medications, and developing a national MG registry may improve diagnostic accuracy, treatment equity, and long-term outcomes. These findings highlight critical priorities for strengthening MG care in resource-limited settings.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
Images or Table (Optional)
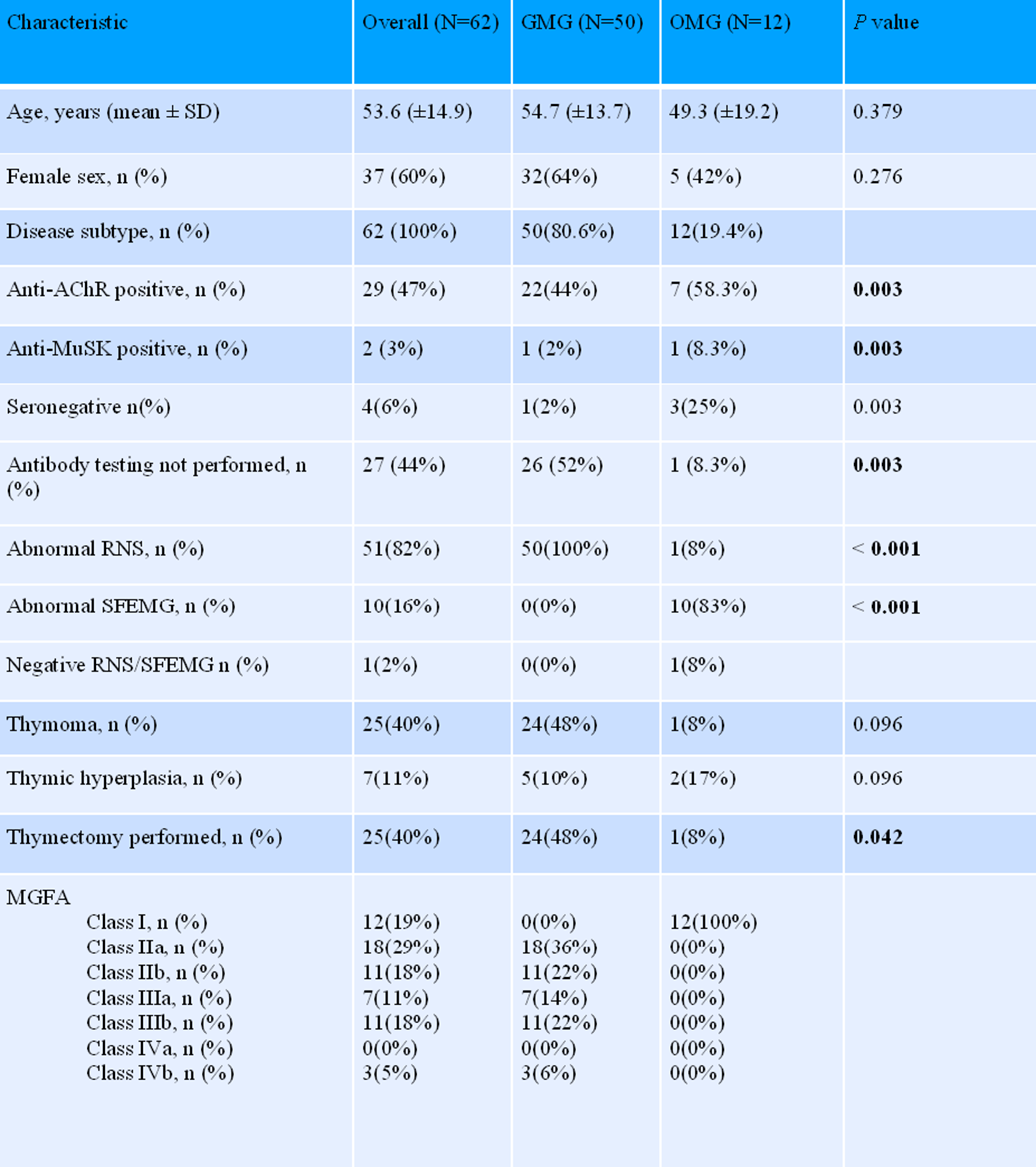
PP01.218
Efgartigimod Provides Rapid Clinical Improvements and Reduces Corticosteroid Use in gMG: Initial Results From PREMIER
Dr. Raffaele Iorio1,2, Mrs Cynthia Qi3, Dr. Min Yang4, Dr. Ashley Anderson5, Dr. Vera Bril6, Dr. Christyn Edmundson7, Dr. Deborah Gelinas8, Dr. Tom Hughes3, Dr. Glenn Phillips3, Dr. Lingyun Li3, Dr. Bruno Martins4, Mrs Cheryl Xiang4, Dr. Kristin Heerlein3, Mrs Cathleen Bergin3, Dr. Boya Lin4, Mrs Jingyi Liu4, Dr. Gil Wolfe9
1
Department of Neuroscience, Università Cattolica del Sacro Cuore, Rome, Italy.
2
Neurology Unit, Fondazione Policlinico Universitario Agostino Gemelli IRCCS, Rome, Italy.
3
argenx US Inc., Boston, United States.
4
Analysis Group, Inc., Boston, United States.
5
Department of Neurology, Houston Methodist, Houston, TX, United States.
6
Ellen and Martin Prosserman Centre for Neuromuscular Diseases, University Health Network, Toronto General Hospital, University of Toronto, Toronto, ON, Canada.
7
Swedish Neuroscience Institute, Seattle, WA, United States.
8
Department of Neurology, UNC School of Medicine, Chapel Hill, NC, United States.
9
Department of Neurology, Jacobs School of Medicine and Biomedical Sciences, University at Buffalo, Buffalo, NY, United States
Background: Generalized myasthenia gravis (gMG) is an autoimmune neuromuscular disorder characterized by weakness in voluntary muscles that significantly impairs patients’ daily functioning. The management of the condition can include long-term corticosteroids, which are associated with significant side effects. Efgartigimod is a first-in-class neonatal Fc receptor blocker indicated for the treatment of gMG in anti-acetylcholine receptor antibody positive adult patients. The Prospective Real-world study of EfgartigiMod for patIents with gEneralized myasthenia gRavis (PREMIER) is an ongoing study characterizing the experiences of patients with gMG treated with efgartigimod in the US.
Methods: Patients were recruited from the enrollees of the efgartigimod patient support program. Adult patients prescribed intravenous (IV) or subcutaneous (SC) efgartigimod for gMG in routine clinical practice in the US were invited to participate. Eligible participants were required to have not received or have no more than two days of exposure to the first dose of efgartigimod, and were ineligible if they were currently enrolled in a clinical trial. Patients were asked to complete a survey at baseline, before efgartigimod initiation, and at Weeks 4, 12, 20, 32, 44 and 56 post initiation. Symptoms and functional status were assessed using MG Activities of Daily Living (MG‐ADL). Patients were also asked to report corticosteroid use and dosage during the 4 weeks preceding each survey. Current analyses report results up to Week 32 due to limited sample size in subsequent weeks.
Results: To date, 143 patients enrolled in PREMIER and initiated efgartigimod, and 52 completed the Week 32 survey at the time of this analysis, with 75.0% remaining on efgartigimod. Patient’s mean (Standard deviation [SD]) age at baseline was 64.8 (14.1), and 48.9% were female. The majority of patients had a history of corticosteroid use (83.9%), and 14.0% had prior use of biologics inhibiting C5 cleavage or neonatal Fc receptors, rituximab or zilucoplan. Baseline mean (SD) MG-ADL was 8.6 (3.4). By Week 4, MG-ADL improved by 4.1 points (p<0.001). Improvements were sustained through Week 32 (Figure 1-a), with larger improvements (4.7 points; p<0.001) observed among those remaining on efgartigimod. The proportion of patients without corticosteroid use increased from 38.5% at baseline to 54.0% by Week 4 (p<0.05 compared to baseline), further increasing to 59.6% by Week 32 (p<0.05) (Figure 1-b). Among the 56.6% (81 of 143) of participants that were using corticosteroids at baseline, 35.7% (10 of 28) became corticosteroid-free by Week 32, and the proportion of patients within this group on high-dose corticosteroids (>10 mg/day) decreased from 69.1% (56 of 81) to 28.6% (8 of 28) at Week 32 (p<0.05).
Conclusion: Initial results from PREMIER demonstrate that patients with gMG treated with efgartigimod improve their symptoms and functional status and reduce corticosteroid use as quickly as Week 4. These benefits were sustained through Week 32. Future data will characterize the long-term experiences of these patients in the real world.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
Images or Table (Optional)
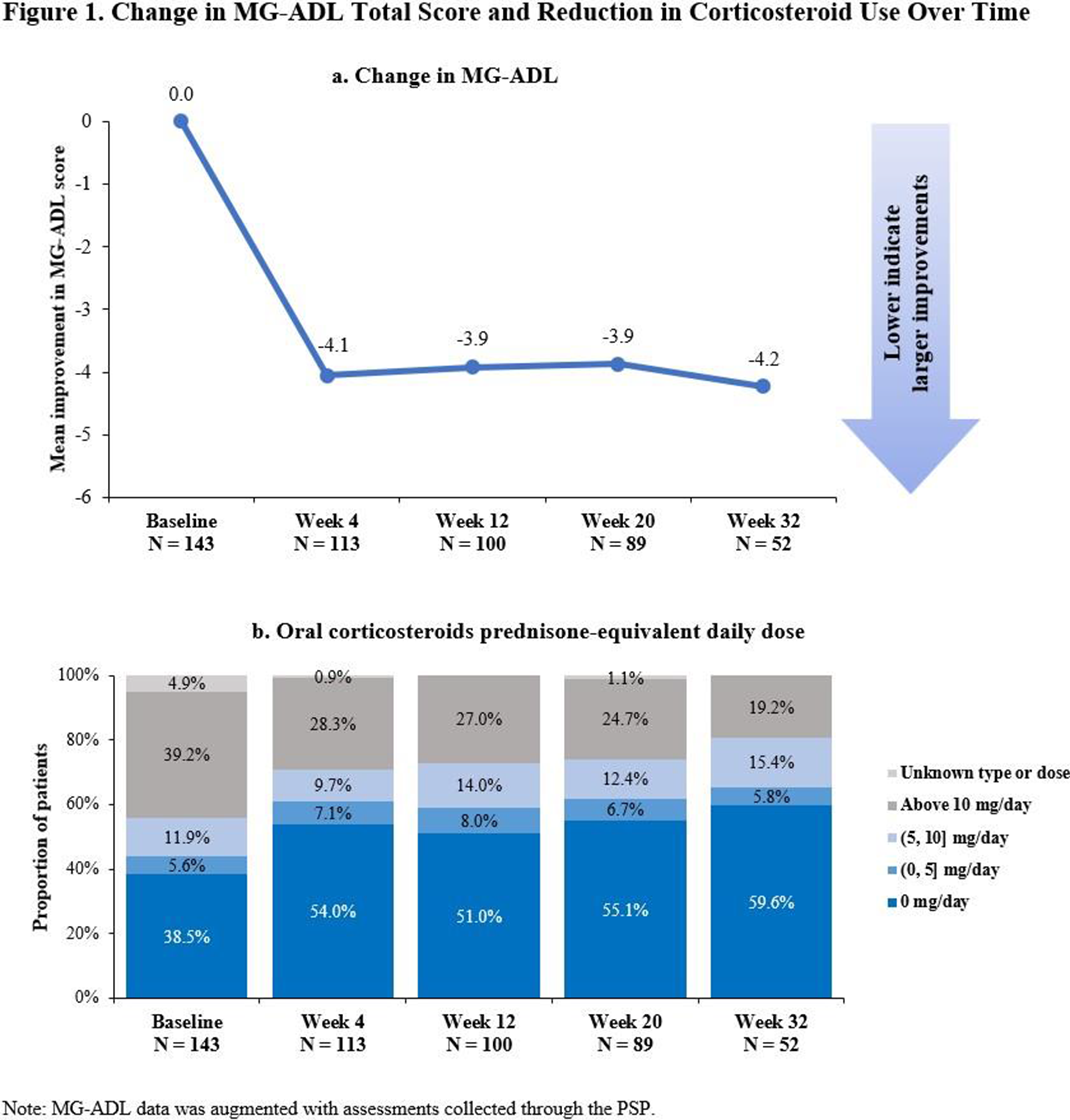
PP01.219
Efgartigimod Improves Ability to Work and Patient Satisfaction in gMG: Initial Results From PREMIER
Dr. Raffaele Iorio1,2, Mrs Cynthia Qi3, Dr. Min Yang4, Dr. Ashley Anderson5, Dr. Vera Bril6, Dr. Christyn Edmundson7, Dr. Deborah Gelinas8, Dr. Tom Hughes3, Dr. Glenn Phillips3, Dr. Lingyun Li3, Dr. Bruno Martins4, Mrs Cheryl Xiang4, Dr. Kristin Heerlein3, Mrs Cathleen Bergin3, Dr. Boya Lin4, Mrs Jingyi Liu4, Dr. Gil Wolfe9
1
Department of Neuroscience, Università Cattolica del Sacro Cuore, Rome, Italy.
2
Neurology Unit, Fondazione Policlinico Universitario Agostino Gemelli IRCCS, Rome, Italy.
3
argenx US Inc., Boston, MA, United States.
4
Analysis Group, Inc, Boston, MA, United States.
5
Department of Neurology, Houston Methodist, Houston, TX, United States.
6
Ellen and Martin Prosserman Centre for Neuromuscular Diseases, University Health Network, Toronto General Hospital, University of Toronto, Toronto, ON, Canada.
7
Swedish Neuroscience Institute, Seattle, WA, United States.
8
Department of Neurology, UNC School of Medicine, Chapel Hill, NC, United States.
9
Department of Neurology, Jacobs School of Medicine and Biomedical Sciences, University at Buffalo, Buffalo, NY, United States
Background: Generalized Myasthenia gravis (gMG) is an autoimmune neuromuscular condition characterized by weakness across various muscle groups, impairing work ability and social engagement. Efgartigimod is a neonatal Fc receptor blocker indicated for the treatment of gMG in adult patients. The ongoing Prospective Real-world study of EfgartigiMod for patIents with gEneralized myasthenia gRavis (PREMIER) characterizes patients’ experiences with efgartigimod in the US.
Methods: PREMIER participants were recruited from the enrollees of the efgartigimod patient support program. Adult patients with gMG prescribed efgartigimod were required to be efgartigimod-naïve or to have initiated treatment within two days of enrollment, and not in a clinical trial. Patients were asked to complete a survey at baseline prior to efgartigimod initiation and at Weeks 4, 12, 20, 32, 44, and 56 post initiation. Satisfaction with disease state was assessed through Patient‐Acceptable Symptom State (PASS). Impact of gMG on ability to work (“affected”/“not affected”) was assessed at baseline, and changes since baseline (“improvement”, “worsening”, “no change”) were evaluated at Week 20. Impact of gMG on personal relationships was measured using a 5-point Likert scale at baseline (“not at all” to “a great deal”), and change since baseline was assessed using a 7-point Likert scale (“very much improved” to “very much worse”). Patient satisfaction with current gMG treatments was assessed via a 7-point Likert scale (“extremely satisfied” to “extremely dissatisfied”), along with ease of efgartigimod dosing schedule (“very difficult” to “very easy”) among patients remaining on treatment. Current analyses report results up to Week 32 due to a limited sample size in subsequent weeks.
Results: Among 143 PREMIER participants initiating efgartigimod to date, mean (standard deviation [SD]) age at baseline was 64.8 (14.1), 48.9% were female, 87.4% were White/Caucasian, and 44.1% had at least a college degree. PASS increased from 25.2% at baseline to 47.8% (p<0.001) at Week 4, and 51.9% (p<0.001) at Week 32 (Figure 1-a). Impaired ability to work due to gMG was reported by 89.6% of patients at baseline. By Week 20, improvements were reported by 42.9% of those answering the question, primarily due to the ability to return to work and increased productivity (both 14.3%). At baseline, 49.0% reported somewhat to greatly impacted relationships with family and friends due to gMG, with 49.6% reporting improvements by Week 4, largely sustained at 40.4% at Week 32. Satisfaction with current gMG treatments (“somewhat” to “extremely satisfied”) increased from 38.5% at baseline to 63.7% (p<0.001) at Week 4, rising to 71.2% (p<0.001) at Week 32 (Figure 1-b). Across all time points, over 80% reported the dosing schedule of efgartigimod easy or very easy to follow.
Conclusion: Initial results from PREMIER show rapid and sustained satisfaction with disease state and gMG treatments after efgartigimod initiation, as early as Week 4, as well as improvements in the ability to work and relationships with family and friends.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
Images or Table (Optional)
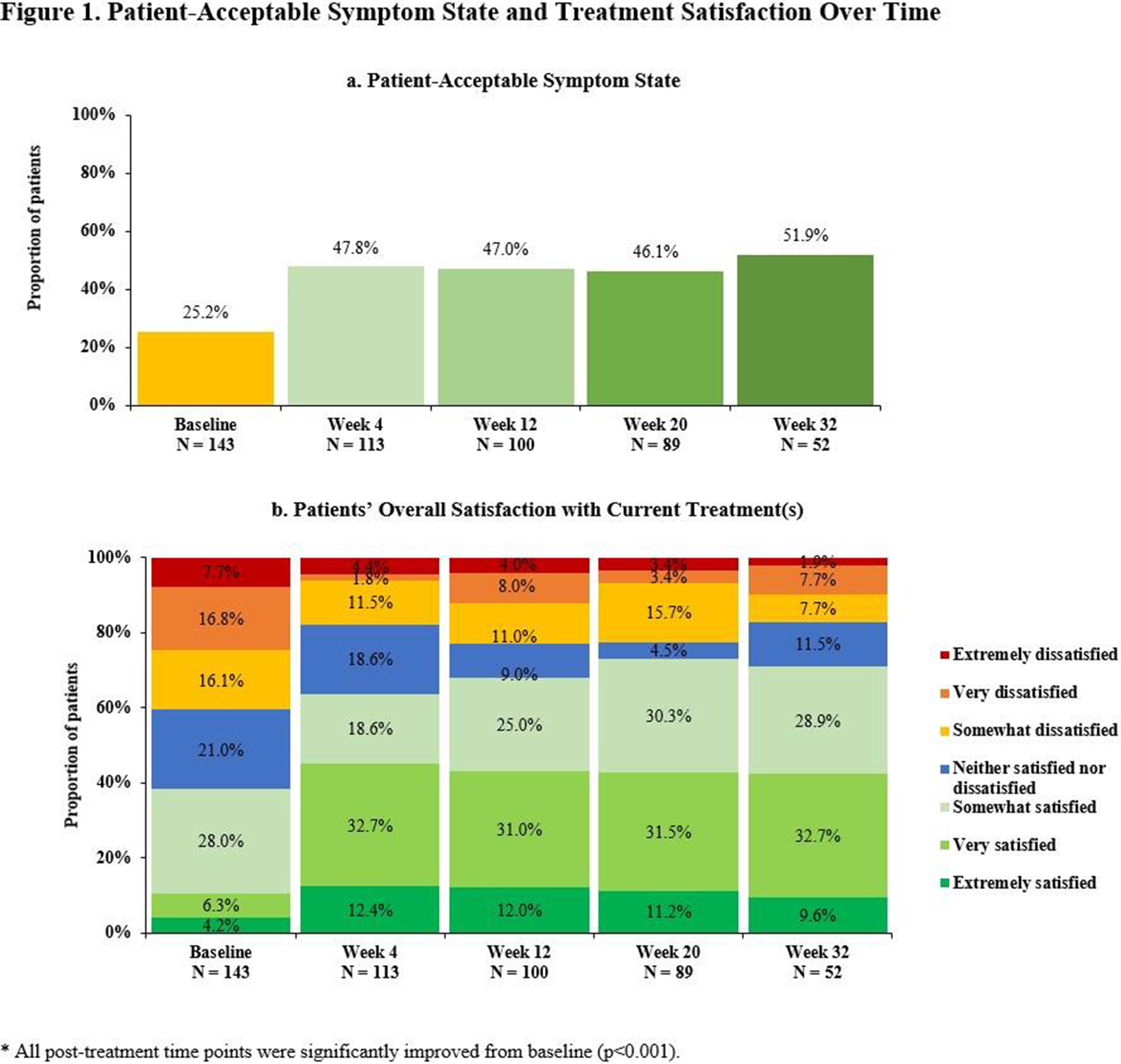
PP01.220
Predictor of Conversion of Myasthenic Crisis from Impending Myasthenic Crisis- a Prospective Study
Dr. Shiny Joy, Dr. Rajesh kumar Singh, Dr. Jasmine Parihar, Dr. Animesh Das, Dr. Arunmozhimaran Elavarasi, Dr. Anu Gupta, Dr. Ayush Aggarwal, Dr. Awadh Kishor Pandit, Dr. Vishnu VY, Dr. Deepti Vibha, Dr. Ajay Garg, Dr. Rohit Bhatia, Dr. Achal kumar Srivastava, Dr. Manjari Tripathi
AIIMS, DELHI, India
Background: Myasthenia Gravis (MG) is an autoimmune disorder that impairs neuromuscular transmission among those affected, 15-20% of patients experience a myasthenic crisis (MC) at least once in their lives.
Methods: This ongoing prospective observational study included patients with MG in crisis or impending crisis (IMC) and assessed their outcome after 3 months.
Results: 55 consecutive patients of myasthenic crisis (MC) and impending myasthenic crisis were recruited. Mean age was 44.84 ± 15.04 years, with male 60%. Out of 55 patients, 39 (70.09%) initially had generalized MG, 7 (12.7)% had ocular MG, 3(5.5%) had bulbar MG, and 6 (10.9%) had oculo-bulbar MG. Acetylcholine receptor (AchR) antibodies were positive in 42( 76.4%), while anti-MuSK antibodies were positive in 9 (16.4%). Thymic abnormalities were present in 27 (49.1%). 46 patients (83.6%) presented with IMC and 9 with direct MC (16.4%). However, out of 46 patients of IMC, 16 further developed MC during admission. So, total no of crisis patients were 25(45.5%) and IMC were 30 (54.5%). The mean duration from conversion to IMC to MC was 2.40 ± 2.79 days. Infection was leading cause of IMC and MC seen in 25 patients (45.5%). Thymectomy was done in 21 patients (38.2%). Infection as a cause was independent predictor of conversion from impending MC to MC (OR 3.0 [1.02- 9.03], p=0.04). The three-month mortality was in 5( 9.1%), with infection as a leading cause.
Conclusion: This study showed that infection was an independent predictor in progression from impending MC to MC.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.221
Complement Inhibitor Use in an Adolescent With Refractory Juvenile-Onset, Thymomatous, Antibody-Positive Myasthenia Gravis
Dr. Rebecca Leung1,2,3, Dr. Laurie McLaughlin1, Dr. Kristen Lefever1, Dr. Lori Mackay1, Assoc. Prof. Stefan Blum1
1
Princess Alexandra Hospital, WOOLLOONGABBA, Australia.
2
Queensland Children's Hospital, South Brisbane, Australia.
3
Sunshine Coast University Hospital, Birtinya, Australia
Background: Juvenile myasthenia gravis (JMG) is a rare antibody-mediated disorder of the neuromuscular junction, with clinical manifestations ranging from isolated ocular involvement to severe generalized weakness that may be life-threatening and require ventilatory support. While many patients respond to conventional immunosuppressive therapies, a subset experience persistent or refractory disease. Emerging targeted therapies, including complement component C5 inhibition and neonatal Fc receptor (FcRn) antagonism, have demonstrated efficacy in adults with refractory generalized myasthenia gravis (gMG); however, data in adolescents with severe JMG remain limited.
Methods: NA
Results: Case: We describe an adolescent female with severe, treatment-refractory JMG associated with malignant thymoma, who demonstrated a marked clinical response to complement inhibition. The patient was initially diagnosed with a complex thymic cyst at age 12, managed with incomplete surgical resection. At age 17, she re-presented with a four-month history of progressive generalized weakness, dysarthria, recurrent falls, and ptosis. Investigations confirmed anti–acetylcholine receptor antibody–positive (AChR-Ab+) gMG. Chest imaging demonstrated residual thymic tissue, and repeat resection identified a WHO type B3 thymoma with invasion into the lung and pericardium. Surgical margins were positive, and the patient subsequently received 30 fractions of adjuvant radiotherapy. Over the following months, she experienced severe generalized disease, with Myasthenia Gravis Composite (MGC) scores ranging from 11 to 21, and three myasthenic crises requiring intensive care admission. This occurred despite aggressive immunotherapy, including high-dose oral prednisolone (up to 50 mg daily), intravenous immunoglobulin, plasma exchange, and rituximab. Ongoing disease activity remained highly disabling, with MGC scores 6 months after therapy initiation of 8–10 at outpatient review, resulting in school absenteeism and significant functional limitation. Treatment-related complications included herpes zoster infection, right subsegmental pulmonary emboli requiring anticoagulation, iron-deficiency anaemia, and corticosteroid-related adverse effects, including Cushingoid features (20 kg weight gain, and hyperglycaemia). Zilucoplan, a subcutaneous, self-administered macrocyclic peptide inhibitor of complement component C5, was initiated approximately nine months after diagnosis. Within three months, the patient’s MGC score improved to 0, allowing plasma exchange to be tapered and discontinued. Oral prednisolone was successfully reduced to 8 mg daily. Disease remission was sustained, with persistent MGC scores of 0, enabling full return to school and regular physical activity, including weight training. Zilucoplan was well tolerated, with no reported adverse effects over a nine-month treatment period.
Conclusion: This case demonstrates the efficacy and tolerability of complement C5 inhibition with zilucoplan in an adolescent patient with severe, thymoma-associated, AChR-Ab+ refractory gMG. Complement inhibition may represent a valuable therapeutic option in selected cases of refractory JMG, facilitating disease control and substantial reduction in exposure to conventional immunosuppressive therapies.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.222
Myasthenia Gravis During Pregnancy: A Descriptive Real-World Study Based on US Claims Data
Dr. Sabrina Guye1, Dr. Bright Dube2, Dr. Jenny Lindroos3, Ms. Elizabeth Packnett4, Dr. Meghan Moynihan5, Mr. Ryan Ross6, Dr. Andreea Lavrov7, Dr. Laura Shaughnessy8, Dr. Lesley Butler9, Prof. Nils Erik Gilhus3,10
1
UCB, Zurich, Switzerland.
2
UCB, Slough, United Kingdom.
3
University of Bergen, Bergen, Norway.
4
Merative, Washington DC, United States.
5
Merative, Ann Harbor, MI, United States.
6
Merative, Los Angeles, CA, United States.
7
UCB, Monheim, Germany.
8
UCB, Durham, NC, United States.
9
UCB, Bulle, Switzerland.
10
Haukeland University Hospital, Bergen, Norway
Background: Myasthenia gravis (MG) is a rare disease with an incidence peak in women of childbearing age. Understanding MG characteristics during pregnancy is essential for effective disease management and family planning. Here, we describe the course of MG before, during, and after pregnancy based on characteristics including exacerbations, myasthenic crisis, disease severity, MG-related healthcare resource utilization (HCRU), and MG-medication.
Methods: A retrospective cohort study utilizing data from two large US administrative claims databases (MarketScan® Medicaid and Commercial Databases) was conducted. Pregnancies in women with an MG diagnosis from 1 January 2013 to 31 December 2023 were identified using validated algorithms. Pregnancy-related periods were defined as pre-conception (180 days before conception), during pregnancy, and post-partum (up to 90 days after pregnancy outcome). Exacerbations were identified based on immunoglobulin use (IG), plasma exchange (PLEX), MG-related hospitalization or emergency department (ED) visit. Myasthenic crisis was defined as a hospital admission where respiratory failure or ventilation, MG diagnosis, and intravenous IG or PLEX was recorded. Disease severity was defined based on exacerbations, myasthenic crises and MG-treatment. MG-related HCRU was assessed using MG-related hospital stays, ED visits, office visits and pharmacy claims. Characteristics are described overall and within the three pregnancy-related periods, without adjusting for variable lengths of the periods.
Results: The results are presented for Medicaid and Commercial Databases, respectively. In total, 168 and 429 pregnancies were included. Over 70% of the women were aged 18-34 years [mean=27.9, standard deviation (SD)=5.8 and mean=32.4, SD=5.3]. Exacerbations were common during all pregnancy-related periods and occurred in 23.2% and 13.8% of pregnancies during pre-conception, in 32.7% and 17.9% during pregnancy and 23.8% and 10.7% post-partum. The average occurrence was 2.85 (SD=2.75) and 3.09 (SD=2.94) exacerbations during the pregnancy period. Overall, 1.8% and 0.2% of pregnancies experienced a myasthenic crisis across all pregnancy periods. In the majority of pregnancies, MG severity was low during pre-conception (76.8% and 85.3%), pregnancy (67.3% and 81.6%), and post-partum (76.2% and 88.3%). There was little variation in MG severity between the three pregnancy-related periods. (See Figure 1). MG-related hospital stays were rare during pre-conception (4.8% and 2.8%), pregnancy (1.8% and 1.2%), and post-partum (0.0% and 0.2%). Whereas MG-related ED visits and office visits occurred more frequently across all periods [MG-related ED visits during pre-conception (13.7% and 4.7%), pregnancy (28.0% and 8.6%), and post-partum (1.2% and 0.0%) and office visits during pre-conception (39.3% and 45.7%), pregnancy (63.1% and 61.5%) and post-partum (1.8% and 1.9%)]. During pregnancy 57.7% and 48.3% used ≥1 MG-medication. Acetylcholinesterase inhibitor use was seen in 38.7% and 30.1%, corticosteroids in 34.5% and 22.6%, and biologics (mainly gamma globulins) in 17.3% and 14.2%.
Conclusion: In one of the largest pregnancy studies among women with MG, most had low disease severity. Despite this, exacerbations were common during all pregnancy-related periods, although myasthenic crisis was rare. In general, about half used MG-medications during pregnancy and overall. MG-related hospital stays were uncommon, while MG-related ED and office visits were more common. Preventing exacerbations and identifying the drivers of HCRU warrants further investigation in pregnant women with MG.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
Images or Table (Optional)
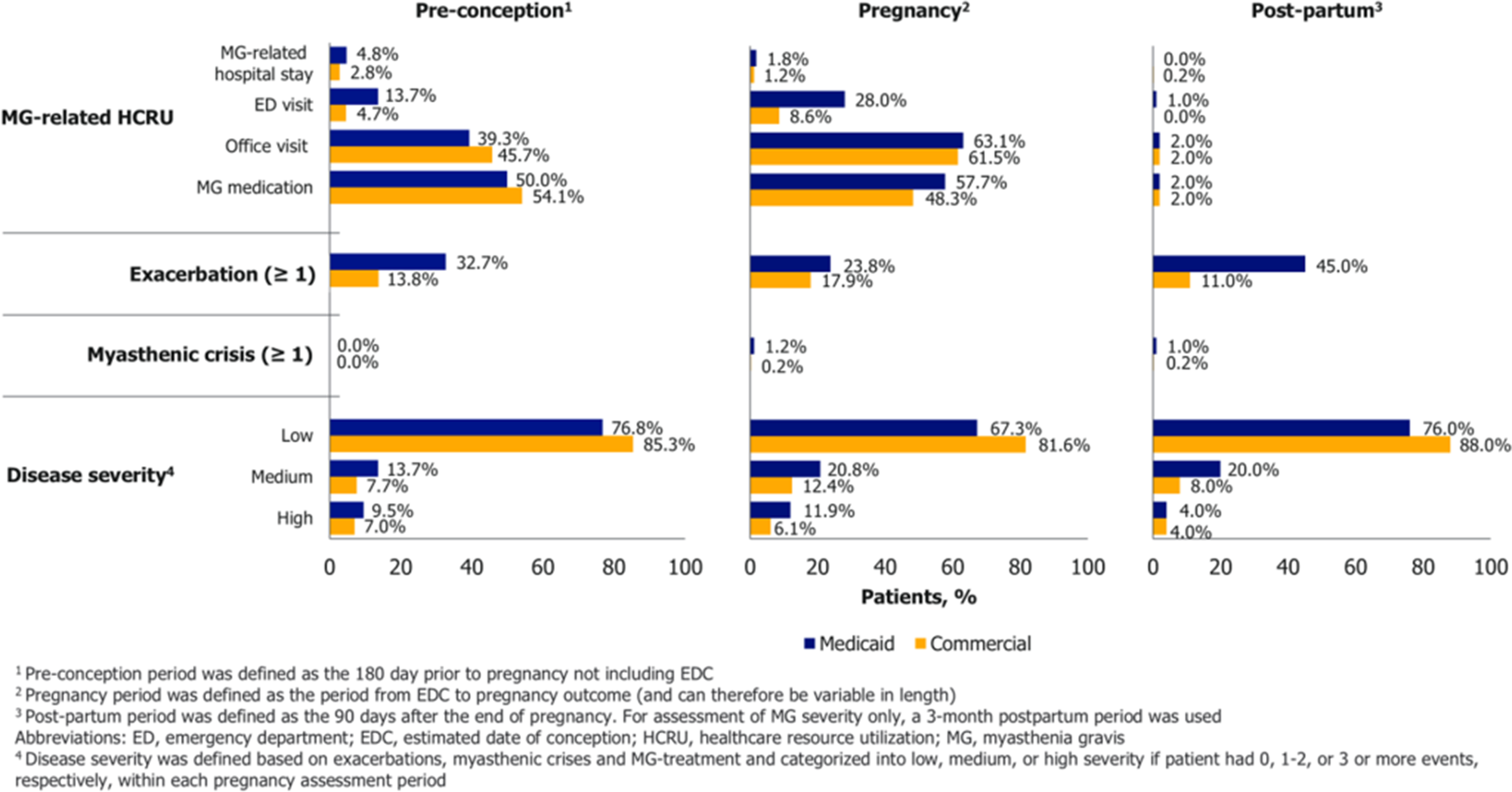
PP01.223
Design of Phase 2a Study Evaluating Empasiprubart Add-On Therapy to Efgartigimod in Adults With gMG
Dr. Sarah Hoffman1, Dr. Jeff Guptill2, Dr. Raphael Bilgraer2, Dr. Anne Gaëlle-Dosne2, Dr. Sophie Steeland2, Dr. Kristin Heerlein2, Dr. Tony Vangeneugden2, Dr. Maria Ramos Masa2, Dr. James F. Howard Jr3
1
Charité – Universitätsmedizin Berlin, Berlin, Germany.
2
argenx, Ghent, Belgium.
3
The University of North Carolina, Chapel Hill, United States
Background: Generalized myasthenia gravis (gMG) is driven by immunoglobulin G (IgG) autoantibodies that target components of the neuromuscular junction and cause pathology through multiple mechanisms. Heterogeneity exists in the relative contributions of these mechanisms among patients, causing gMG to manifest differently in each patient. Despite new therapies, unmet therapeutic needs still exist, with some patients continuing to experience a high disease burden in spite of clinically meaningful improvements during treatment. Efgartigimod, a human IgG1 Fc fragment that blocks neonatal Fc receptor (FcRn)-mediated IgG recycling, alleviates gMG symptoms in many patients. Empasiprubart, an IgG1 antibody, selectively blocks complement component 2 (C2). For patients with a partial response to efgartigimod, empasiprubart add-on therapy may further improve outcomes. This presentation describes the design of ADAPT Forward 1 (NCT07284420), an exploratory, Phase 2a, proof-of-concept study evaluating safety, tolerability, and efficacy of empasiprubart add-on therapy in adult participants with gMG who partially respond to efgartigimod.
Methods: The study consists of 3 parts (total duration, ∼54 weeks). Part A: ∼50 adult participants with acetylcholine receptor antibody–positive (AChR-Ab+) gMG receive 1 efgartigimod (10 mg/kg) treatment cycle (4 once-weekly intravenous [IV] infusions followed by a 4-week treatment-free period). Part B: participants with a partial response to efgartigimod (Myasthenia Gravis Activities of Daily Living [MG-ADL] improvements of ≥2 points, with total remaining ≥5 points) receive 2 cycles of efgartigimod with empasiprubart IV coadministered twice during each cycle. Part C (safety follow-up): participants receive 4 additional cycles of efgartigimod monotherapy. Dosing of empasiprubart was selected based on physiologically-based pharmacokinetic/pharmacodynamic modelling to target sustained free C2 reduction while accounting for the effect of efgartigimod on empasiprubart concentration, since efgartigimod reduces FcRn-mediated recycling of empasiprubart. The dose is supported by data from a Phase 1 drug-drug interaction study examining the effect of a single dose of efgartigimod IV on empasiprubart exposure when coadministered in healthy participants.
Results: Primary endpoints will evaluate safety and tolerability during Parts A and B. Secondary endpoints will evaluate efficacy, including the proportion of participants achieving minimal symptom expression (MG-ADL total score, 0 or 1).
Conclusion: Results of this study will provide proof-of-concept data on empasiprubart as an add-on therapy to efgartigimod for adults with AChR-Ab+ gMG.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.224
Hereditary Spastic Paraplegia Associated With SPTAN1 Mutation
Prof. Sa-Yoon Kang
Department of Neurology, Jeju National University College of Medicine, Jeju, Korea, Republic of
Background: Pathogenic variants in SPTAN1 have been linked to broad phenotypical spectrum. Clinical presentations include cerebellar ataxia, epileptic syndromes, intellectual disability and spastic paraplegia.
Methods: We describe a hereditary spastic paraplegia patient associated with SPTAN1 gene mutation.
Results: A 29-year-old male presented with gait disturbance and weakness of the lower limbs. His gait problem had first appeared when he was 14-year old. Neurological examination revealed spasticity, hyperreflexia, and pathological reflex in both legs, but no abnormality in the upper limbs. Eye movement, cranial nerve, cerebellar and fundoscopic examination showed normal findings. Electrophysiological tests demonstrated no abnormalities. Spine and brain MRI showed unremarkable findings. Genetic study revealed pathogenic variants in SPTAN1 gene. He had no past history of seizure and his Korean Mini-Mental Status Examination score was 30/30. His father showed similar clinical features, but father did not accomplish genetic study.
Conclusion: We report SPTAN1 mutations as a cause of pure hereditary spastic paraplegia. The exact genotype–phenotype correlations remain enigmatic in SPTAN1-related diseases. Although it remains challenging to draw genotype-phenotype correlations for SPTAN1 variants, we suggest SPTAN1 mutations are candidate gene for hereditary spastic paraplegia.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: non 5q SMA/distal SMA/ Hereditary Motor Neuropathies
PP01.225
Myasthenia Gravis Mimicking Miller Fisher Syndrome
Prof. Sa-Yoon Kang
Department of Neurology, Jeju National University College of Medicine, Jeju, Korea, Republic of
Background: Miller Fisher syndrome (MFS) is a rare immune-mediated neuropathy that commonly presents with progressive bilateral external ophthalmoplegia. Myasthenia gravis (MG) is also immune-mediated neuromuscular junction disorder characterized by fluctuating ocular symptoms.
Methods: A 47-year-old woman presented with progressive binocular diplopia and blepharoptosis. She had headache and dizziness after a week later after the diplopia had started. Neurological examination revealed complete bilateral external ophthalmoplegia, ptosis and ataxia, but no areflexia. Cerebrospinal fluid analysis demonstrated no evidence of CNS infection.
Results: A provisional diagnosis of MFS was made, and intravenous immunoglobulin therapy was initiated. After immunoglobulin treatment, ptosis was mildly improved, but diplopia was still remained. Repetitive nerve stimulation test demonstrated post-synaptic neuromuscular junction defects and nerve conduction studies showed mild slowing of sensory and motor nerves in bilateral upper extremities. Serological tests showed normal anti-GQ1b levels, but anti-acetylcholine receptor binding antibody was positive (titer: 6.91 nmol/L). So, we started oral steroids and acetylcholinesterase inhibitor therapy. After that, she showed much improvement of diplopia and ptosis.
Conclusion: This case illustrates that MG can be seen similar to MFS, according to patient’s initial symptoms. Although the predominant ophthalmic features of MFS are complete bilateral external ophthalmoplegia, it should be recognized that myasthenia gravis has variable initial presentation. Such confounding neuro-ophthalmic features require a thorough history, neurological examination, neuroimaging, and serological testing.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.226
Bruton’s Tyrosine Kinase Expression in Peripheral Blood and Thymus of Patients with Myasthenia Gravis
Assoc. Prof. Seung Woo Kim, Mr. Ye Joon Jung, Prof. Ha Young Shin
Yonsei University College of Medicine, Seoul, Korea, Republic of
Background: Myasthenia gravis (MG) is an antibody-mediated autoimmune neuromuscular disorder in which B-cell–driven immune responses play a central pathogenic role. Bruton’s tyrosine kinase (BTK) is a key intracellular signaling molecule downstream of the B-cell receptor and is essential for B-cell activation, differentiation, and survival. Although BTK inhibition has emerged as a promising therapeutic strategy in antibody-mediated autoimmune diseases, the expression and activation status of BTK in human MG, particularly in relation to clinical disease activity and thymic pathology, remain unknown. This study aimed to investigate BTK expression and activation in peripheral blood and thymic tissue from patients with acetylcholine receptor (AChR) antibody–positive MG and to examine their associations with clinical disease activity.
Methods: Peripheral blood mononuclear cells (PBMCs) were obtained from 40 patients with AChR antibody–positive MG and 11 healthy controls. Thymic tissue samples from 22 patients who underwent thymectomy were analyzed. Total BTK and phosphorylated BTK (p-BTK) expression in peripheral B-cell subsets were assessed by intracellular flow cytometry, and BTK expression in thymic tissue was evaluated using immunohistochemistry. Clinical data, including MG subtype, Myasthenia Gravis Activities of Daily Living (MG-ADL) score, and recent clinical course, were collected at the time of sampling. Patients were classified as being in remission, having stable symptomatic disease, or experiencing recent clinical worsening. Associations between BTK or p-BTK expression and clinical variables were evaluated using univariate and multivariate linear regression analyses.
Results: Total BTK and p-BTK expression in PBMCs did not differ significantly between MG patients and healthy controls. However, when patients with MG were stratified by disease status, total BTK expression was higher in patients having generalized symptoms than those in remission (median 19953.5 [Q1–Q3, 15415.8–24403.5] vs 14876.5 [11064.5–15693.3]; p = 0.039). Phosphorylated BTK demonstrated a stepwise increase from remission (111.2 [55.8–264.5]) to ocular MG (352.5 [204.3–681.5]) and generalized MG (612.0 [354.7–782.8]), with significantly higher levels in patients having generalized symptoms compared with healthy controls (294.0 [130.0–567.0]; p = 0.049). Patients with recent clinical worsening showed markedly increased p-BTK expression compared with those in remission (673.0 [322.0–1,044.0] vs 133.0 [62.7–249.0]; p = 0.008). In multivariate analysis adjusting for current prednisolone use and thymectomy status, recent clinical worsening was independently associated with increased p-BTK expression (β = 0.43, p = 0.012), whereas total BTK expression was not. Although p-BTK expression was modestly associated with MG-ADL scores in univariate analysis (p = 0.046), this association was not retained after adjustment for recent disease activity. Immunohistochemical analysis showed BTK expression in 9 of 22 thymic specimens (40.9%), predominantly localized around lymphoid follicular structures.
Conclusion: Activation of BTK signaling, reflected by p-BTK expression, is associated with recent clinical worsening of MG, whereas total BTK expression shows limited clinical relevance. The localization of BTK expression to disease-relevant thymic immune microenvironments further supports a role for BTK signaling in MG immunopathogenesis. These findings provide human tissue–based evidence that BTK activation reflects active immune processes in MG and support BTK signaling as a biologically relevant therapeutic target.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.227
A Pilot Machine Learning Study on Muscle-Specific Diagnostic Contribution of Repetitive Nerve Stimulation in MG
Dr. Sohyun Ahn, Prof. Yangki Minn
Kangnam Sacred Heart Hospital, Seoul, Korea, Republic of
Background: Repetitive nerve stimulation (RNS) is a standard diagnostic tool for myasthenia gravis (MG), and previous studies have suggested higher sensitivity in facial muscles. However, the relative diagnostic contribution of individual muscles has not been quantitatively evaluated.
Methods: RNS decrement data from eight patients with MG and one healthy control were analyzed. Mean decrement values at 2, 3, and 5 Hz were obtained from the abductor digiti minimi, ocularis oculi, nasalis, and trapezius muscles. A Random Forest classifier with leave-one-out cross-validation was used to assess diagnostic performance and feature importance.
Results: Facial muscles demonstrated the greatest diagnostic contribution. Ocularis oculi and nasalis showed the highest feature importance and achieved perfect discrimination in single-feature receiver operating characteristic analysis (AUC=1.0). Trapezius also showed strong diagnostic performance, whereas abductor digiti minimi contributed minimally (AUC=0.75). Patients with generalized MG showed larger and more widespread decrement compared with those with ocular MG.
Conclusion: This pilot study quantitatively confirms the dominant diagnostic role of facial muscle RNS decrement in MG, supporting their prioritization in clinical evaluation. Future studies incorporating waveform analysis and larger cohorts may further improve the diagnostic efficiency of RNST.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.228
RELIEVE: A Phase 3 Study Evaluating Efficacy and Safety of Remibrutinib in Generalized Myasthenia Gravis
Prof. Stephen Reddel1, Prof. Vera Bril2, Prof. James Howard3, Prof. Srikanth Muppidi4, Prof. Kimiaki Utsugisawa5, Prof. John Vissing6, Dr. Weihua Cao7, Dr. Svetlana Jevtic8, Dr. Wendy Su7, Dr. Roman Willi8, Prof. Heinz Wiendl9
1
University of Sydney, Departments of Neurology and Molecular Medicine, Sydney, Australia.
2
Toronto General Hospital, Toronto, Canada.
3
Department of Neurology, University of North Carolina School of Medicine, Chapel Hill, United States.
4
Department of Neurology and Neurological Sciences, Stanford University School of Medicine, Stanford, United States.
5
Department of Neurology, Hanamaki General Hospital, Hanamaki, Japan.
6
Copenhagen Neuromuscular Center, Department of Neurology, Rigshospitalet, University of Copenhagen, Copenhagen, Denmark.
7
Novartis Pharmaceuticals Corporation, East Hanover, United States.
8
Novartis Pharma AG, Basel, Switzerland.
9
Department of Neurology and Neurophysiology, University of Freiburg, Freiburg, Germany
Background: Generalized myasthenia gravis (gMG) is a rare, chronic autoimmune disorder caused by autoantibodies against neuromuscular junction (NMJ) components, leading to debilitating muscle weakness and other clinical manifestations. Inhibition of Bruton’s tyrosine kinase (BTK), resulting in reduced activation of B cells and innate immune cells, offers a potential mechanism to modulate the immune response and reduce the autoimmune attack on the NMJ. Remibrutinib is a novel, highly selective and potent, covalent, oral BTK inhibitor with a promising pharmacological and safety profile. Here, we present the design of the Phase 3 RELIEVE study that aims to evaluate the efficacy and safety of remibrutinib in people living with gMG who are on stable, standard-of-care (SOC) treatment.
Methods: RELIEVE is a randomized, double-blind, placebo-controlled, multicenter Phase 3 study. The study will enroll patients aged 18 to 75 years diagnosed with gMG (Myasthenia Gravis Foundation of America disease class II–IV) who are either acetylcholine receptor positive (AChR+), muscle-specific tyrosine kinase positive (MuSK+), or double-seronegative (AChR− and MuSK−), with a Myasthenia Gravis Activities of Daily Living (MG-ADL) score ≥6 (>50% non-ocular), and who are on stable SOC treatment. Eligible participants will be randomized 1:1 to receive either remibrutinib or placebo during the 6-month double-blind core treatment period, followed by an open-label extension with remibrutinib treatment for up to 60 months. The primary endpoint is the change from baseline to Month 6 in MG-ADL total score. Key secondary endpoints include assessments of Quantitative MG (QMG), Minimal Symptom Expression (MSE), and MG Composite (MGC), among others.
Results: The ongoing study will enroll approximately 180 eligible participants. Further details of the study design will be presented at the congress.
Conclusion: The pivotal Phase 3 RELIEVE study will investigate the efficacy and safety of remibrutinib versus placebo in adult patients with gMG across serotypes.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.229
Usability of Zilucoplan Auto-Injector in Generalised Myasthenia Gravis: A Human Factors Validation Study
Dr. Sukru Karali1, Mrs Barbara Domanska1, Dr. Natasa Savic2, Dr. Miriam Freimer3, Mr. Gilles Vinçonneau2
1
UCB, Slough, United Kingdom.
2
UCB, Bulle, Switzerland.
3
Department of Neurology, The Ohio State University Wexner Medical Center, Columbus, OH, United States
Background: Zilucoplan is a complement component 5 inhibitor approved for treatment in patients with generalised myasthenia gravis (gMG). It is currently self-administered with a pre-filled syringe. Auto-injectors (AIs) are self-administration devices designed to automatically inject a defined dose of medication, which may be a preferred option for some patients. The zilucoplan AI is button-free and contains a pre-filled syringe enclosed in the device so that the needle is not visible. The objective of this human factors validation study was to assess use-related safety and effectiveness of the zilucoplan AI.
Methods: Participants were divided into 5 categories: patients with gMG or those exhibiting ocular symptoms (injection-experienced and injection-naïve); caregivers (injection-experienced and injection-naïve); and healthcare professionals (HCPs). Participants were required to simulate use of the AI by injecting into a manikin (HCPs and caregivers) or skin pad attached to their body (patients) in a simulated use environment, without training. Instructions for Use (IFU) were available, but participants were not directed to use them.
Results: In total, 100 participants were recruited in the United States of America (injection-experienced patients, n=21; injection-naïve patients, n=22; injection-experienced caregivers, n=25; injection-naïve caregivers, n=17; and HCPs, n=15). Overall, 90% (n/N=90/100) of participants performed a successful injection with the AI on their first attempt, without training. Observed use errors did not introduce new risks beyond those previously assessed during risk management. The use errors were attributed to prior experience with other injection devices, IFU layout and use environment influences; minor changes were made to the IFU to address these use errors.
Conclusion: The data from this human factors validation study demonstrate that the zilucoplan AI is safe and effective for use in patients with gMG. No new hazards or use-related risks were identified. Funding: UCB.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.230
Hospitalisation, Crisis and ICU Trends in Myasthenia Gravis: Nationwide Study, Spain 2016–2022
Dr. Tania Garrido-Hernández1, Dr. María del Mar Martínez-Salmerón1, Dr. Marta Rodríguez-Camacho1, Mr. Javier Del-Águila-Mejía2,3, Dr. Beatriz Vélez-Gómez1
1
Torrecárdenas University Hospital, Almería, Spain.
2
2National Center for Epidemiology, Carlos III Health Institute, Madrid, Spain.
3
Epidemiology and Public Health Biomedical Network Research Consortium (CIBERESP), Madrid, Spain
Background: Myasthenia gravis (MG) is a chronic autoimmune disorder increasingly affecting older adults, with rising prevalence and comorbidity burden across Europe. However, contemporary nationwide data describing MG-related hospital utilisation in European healthcare systems remain limited. This study examined temporal trends and severity-related hospital outcomes in Spain between 2016 and 2022.
Methods: We conducted a retrospective, population-based analysis using the Spanish Minimum Basic Data Set, including all adult hospital episodes with a primary diagnosis of MG (ICD-10-ES G70.00, G70.01). Hospital admissions, day-hospital visits and emergency department (ED) encounters were evaluated. Variables included demographics, markers of disease severity (intensive care unit [ICU] admission and myasthenic crisis), in-hospital mortality and comorbidities. Temporal trends were analysed using Poisson regression.
Results: A total of 20,251 MG-related episodes corresponding to 6,308 unique patients were identified. Hospital admissions declined by 2.3% annually, whereas day-hospital and ED utilisation increased markedly from 2018 onwards, particularly among older adults. Despite this shift toward ambulatory management, in-hospital mortality remained low and stable (mean 2.6%). ICU admissions increased from 8.2% to 13.7% of hospitalised patients. Myasthenic crisis accounted for 4.5% of admissions and was strongly associated with ICU utilisation, representing 39% of all ICU stays.
Conclusion: MG care in Spain is undergoing a sustained transition toward ambulatory management without evidence of worsening short-term hospital outcomes. Despite an ageing patient population and rising ICU utilisation, in-hospital mortality remained stable, supporting the safety of outpatient-based MG care models. These findings highlight the need for integrated clinical data to inform future healthcare planning in MG.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.231
Characterisation of Patients With Generalised Myasthenia Gravis on Rozanolixizumab Treatment in European Managed Access Programmes
Dr. Thaïs Tarancón1, Dr. Paolo Emilio Alboini2, Dr. Silvia Falso3, Dr. Rita Frangiamore4, Dr. Matteo Gastaldi5, Dr. Melania Guida6, Dr. Solange Kapetanovic Garcia7,8, Dr. Nicasio Rini9, Dr. Carmelo Rodolico10, Dr. Elena Rossini11, Prof. Sabrina Sacconi12, Dr. Guilhem Solé13, Dr. Karin Annoni14, Dr. Kerina Bonar15, Dr. Grégory Chollet16, Dr. Cristina Lopez17, Dr. Patrik Ohagen18, Dr. Cassandra Slader19
1
UCB, Madrid, Spain.
2
Neurology Unit, Fondazione IRCCS Casa Sollievo della Sofferenza, San Giovanni Rotondo, Italy.
3
Department of Neuroscience, Catholic University of the Sacred Heart, Rome, Italy.
4
Neuroimmunology and Muscle Pathology Unit, IRCCS Carlo Besta Neurological Institute, Milan, Italy.
5
IRCCS Mondino Foundation, Neuroimmunology Research Unit, Pavia, Italy.
6
Neurology Unit, Department of Neurosciences, Azienda Ospedaliero Universitaria Pisana, Pisa, Italy.
7
ALS and Neuromuscular Unit, Hospital Universitario Basurto, Bilbao, Spain.
8
NAT-RD Research Group, IIS Biobizkaia, Barakaldo, Spain.
9
Department of Biomedicine, Neuroscience and Advanced Diagnostics (BiND), University of Palermo, Palermo, Italy.
10
Department of Clinical and Experimental Medicine (DIMED), University of Messina, Sicily, Italy.
11
Department of Neurology, Mental Health and Sensory Organs (NESMOS), Faculty of Medicine and Psychology, Sapienza University of Rome, Rome, Italy.
12
Université Côte d’Azur, Peripheral Nervous System & Muscle Department, Pasteur 2 Hospital, Centre Hospitalier Universitaire de Nice, Nice, France.
13
Centre de Référence des Maladies Neuromusculaires AOC CHU de Bordeaux, Bordeaux, France.
14
UCB, Milan, Italy.
15
UCB, Slough, United Kingdom.
16
UCB, Colombes, France.
17
UCB, Barcelona, Spain.
18
UCB, Uppsala, Sweden.
19
UCB, Brussels, Belgium
Background: Generalised myasthenia gravis (gMG) is a rare, chronic, autoimmune disease, characterised by fluctuating muscle weakness. MycarinG (Phase 3, NCT03971422) and its open-label extension studies (MG0004 [NCT04124965] and MG0007 [NCT04650854]), showed that the efficacy of rozanolixizumab, a neonatal Fc receptor blocker, was maintained over repeated treatment cycles across multiple myasthenia gravis (MG)-specific outcomes in patients with gMG. Managed Access Programmes (MAPs) enable the use of treatments prior to authorisation for patients with a severe/debilitating/life-threatening condition, where available treatments are deemed inadequate by patients and physicians, and there is no available clinical trial. Here, we describe rozanolixizumab utilisation patterns and patient characteristics in MAPs, initiated under protocolised compassionate use in France, Italy and Spain.
Methods: This was an observational, retrospective, longitudinal cohort study of adults with gMG initiating rozanolixizumab treatment in MAPs in France, Italy and Spain between November 2022 and May 2025. The primary objective was to describe baseline demographics and disease characteristics of patients at treatment initiation. Change from baseline in MG Activities of Daily Living (MG-ADL), Garches and MG Quality of Life 15-Item Revised scores were also assessed when available. All analyses were descriptive.
Results: Overall, 12 patients from France, 16 from Italy and <5 from Spain were included. Mean (standard deviation [SD]) age was 73.7 (14.6), 53.8 (14.0) and 61.5 (16.5) years, respectively. Most patients were female (58.3%, 87.5% and 50.0%, respectively). Additional data were available for Italy: mean (SD) number of years since initial MG diagnosis was 10.8 (10.6); 43.8% of patients had an MG diagnosis <5 years and baseline (mean [SD]) MG-ADL score was 7.4 (2.6). In the 2 years prior to enrolment, MG crises occurred in 6.3% of patients and MG-related hospitalisations occurred in 43.8%. Prior to enrolment, 56.3% of patients had received pyridostigmine and all patients had received corticosteroids. Treatment outcomes data will be presented in the poster.
Conclusion: The baseline characteristics of the patients who entered the rozanolixizumab MAPs were indicative of a population with moderate-to-severe gMG. Funding: UCB.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.232
Do We Really Need Non-Steroidal Immunosuppressants in Generalized Myasthenia Gravis Treated With New Targeted Therapies?
Dr. Valentina Vera1, Dr. Elena Rossini1, Dr. Stefania Morino2, Dr. Luca Leonardi2, Prof. Giovanni Antonini1, Dr. Antonio Lauletta1, Dr. Francesca Forcina1, Dr. Laura Tufano1, Dr. Demetrio Marando1, Dr. Matteo Garibaldi1,2, Dr. Fionda Laura2
1
Department of Neuroscience, Mental Health and Sensory Organs (NESMOS), SAPIENZA University of Rome, Rome, Italy.
2
Neuromuscular and Rare Diseases Centre, Neurology Unit, Sant’Andrea Hospital, Rome, Italy
Background: Myasthenia gravis (MG) is a chronic autoimmune neuromuscular disorder characterized by fluctuating muscle weakness and fatigability. In recent years, the therapeutic landscape of generalized MG (gMG) has been transformed by the introduction of targeted biological therapies, including complement inhibitors (CI) and anti–neonatal Fc receptor (FcRn) agents which have demonstrated rapid and sustained efficacy in patients with refractory or poorly controlled disease. Evidence from randomized clinical trials and real-world studies has largely been derived from patient populations receiving background non-steroidal immunosuppressive therapies (NSISTs), frequently in combination with corticosteroids. However, the clinical relevance of continuing concomitant NSIST therapy in patients treated with targeted agents remains uncertain. This study aimed to evaluate whether background NSIST use influences clinical scale outcomes, corticosteroid reduction, and disease stability in patients with gMG receiving targeted therapies.
Methods: We conducted a single-center, retrospective, observational study at the Neurology Unit of Sant’Andrea Hospital, Rome. Forty-one patients with gMG who initiated treatment with CI or FcRn inhibitors were included. Collected data included demographic and clinical characteristics, thymic pathology, comorbidities and concomitant therapies. Clinical assessments were conducted at baseline and during follow-up using the MG Activities of Daily Living (MG-ADL) scale and the Quantitative MG (QMG) score. Achievement of Minimal Symptom Expression (MSE), MG Foundation of America Post-Intervention Status (MGFA-PIS), and changes in corticosteroid dosage from baseline to last available follow-up were also evaluated. Treatment discontinuations and treatment-related adverse events were additionally recorded.
Results: Patients were stratified according to concomitant use of NSIST: 20 patients were receiving background NSIST (Group 1) whereas 21 patients were not (Group 2). The distribution of complement inhibitors and anti-FcRn therapies was balanced between the two groups. Baseline demographic characteristics werewere overall comparable. Disease duration at initiation of biologic therapy and mean follow-up duration were similar between groups (17.47±20.59 vs 15.9±9.12 months). At baseline, mean MG-ADL scores were comparable between Group 1 and Group 2 (9.10±4.36 vs 8.24±3.38) as were baseline corticosteroid dosages (22.17±13.81 mg vs 22.06±16.43). Mean baseline QMG scores was slightly higher in Group 1 compared to Group 2 (14.00±5.14 vs 10.86±4.82). MG-ADL and QMG scores showed a statistically significant improvement for both groups at every time point compared to baseline. No significant differences in clinical scale improvement during follow-up were observed comparing the two groups. The achievement of MSE, improvement in MGFA-PIS and reduction in mean corticosteroid dose from baseline to the last available follow-up did not differ significantly between the two groups. Treatment discontinuation occurred in 5 of 20 patients in Group 1 and 7 of 21 patients in Group 2.
Conclusion: Our pilot analysis indicates that concomitant use of NSIST does not appear to be associated with greater improvement in clinical outcomes, enhance corticosteroid-sparing effects or increased disease stability. These findings suggest that, in the context of highly effective targeted therapies, background NSIST may not confer additional benefit for all patients. Larger, prospective studies are needed to better define the role of concomitant chronic immunosuppression and to optimize treatment strategies in gMG.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.233
Prevalence and Predictors of Sleep Disturbances in anItalian Cohort of Patients With Myasthenia Gravis
Dr. Veronica Iovino, Dr. Michelangelo Maestri Tassoni, Dr. Alessandro Colitta, Dr. Alba Cepele, Dr. Aurora Lo Iacono, Dr. Melania Guida
Department of Clinical and Experimental Medicine, Neurology Unit, University of Pisa, Pisa, Italy
Background: Myasthenia Gravis (MG) is an autoimmune neuromuscular disorder resulting from impaired neuromuscular transmission1. Previous literature reported a variable prevalence of sleep disturbances in MG patients(24%-64%). Moreover, MG outcome measures have been inconsistently associated with sleep parameters2-3. Within this context, this cross-sectional study aims to: investigating the prevalence of self-reported sleep disturbances in an Italian cohort of MG patients; exploring possible associations between such disturbances and MG outcome measures.
Methods: We administered a battery of self-report questionnaires to a cohort of patients with AChRAb or MuSKAb-positive MG, who are being followed at the MG Clinic of Pisa University Hospital: Pittsburgh Sleep Quality Index(PSQI), Reduced Morningness-Eveningness Questionnaire(rMEQ), Epworth Sleepiness Scale(ESS), Berlin Questionnaire(BQ), Insomnia Severity Index(ISI), single-item screens for Restless Legs Syndrome(RLS) and REM Sleep Behavior Disorder(RBD1Q), the Hospital Anxiety and Depression Scale(HADS), and the Fatigue Severity Scale(FSS). Demographic and clinical data—including MG subtype and MGFA classification—were extracted from the medical records. MG-related functional status and quality of life were assessed using the MG Activities of Daily Living scale(MG-ADL) and the 15-item Revised MG Quality of Life scale(MG-QoL-15r). Clinical severity was measured using Myasthenia Gravis Composite score(MGC). Descriptive statistics was computed. Multivariate, linear or binomial modelling was employed to explore possible associations between sleep and circadian parameters (the dependent variables) and MG outcome measures. To avoid multicollinearity, only one MG outcome measure was included in each model as an independent variable, along with possible confounders, i.e., severity of anxiety and depression, daily steroid dosage, age, and BMI.
Results: Sixty-three MG patients from 13 Italian regions were enrolled (mean age 47 years; mean BMI 25.8; 43 females). Widespread sleep disturbances emerged, including poor perceived sleep quality (94% of recruited patients), subthreshold to severe insomnia (48%), excessive daytime sleepiness (16%), high pre-test OSAS risk (16%), and presence of RLS and RBD symptoms (49% and 16%, respectively). Morning type was the most represented circadian typology (46%). In regression models, ESS was predicted by MGC(p=0.010) and MG-ADL(p=0.021), while high pre-test OSAS risk was predicted by MG-ADL(p=0.005) and MG-QoL-15r(p=0.014). Furthermore, MG-ADL predicted the presence of RLS(p=0.045) and RBD symptoms(p=0.037). Finally, all MG outcome measures significantly predicted fatigue severity.
Conclusion: Self-reported sleep disturbances are widespread among MG patients. Moreover, MG outcome measures are associated with partially diverging sleep disruption patterns in MG, irrespective of age, BMI, daily steroid dosage, anxiety and depression symptoms. Finally, chronotype is not associated with MG outcome measures.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.234
Impact of Published Single-Fiber EMG Reference Values on Diagnostic Classification in Myasthenia Gravis
Dr. Wayne Zhong1, Mr. Artor Pogosean2, Dr. Anna Rostedt Punga2, Dr. Bhaskar Roy1
1
Yale New Haven Hospital, New Haven, United States.
2
Uppsala University and Clinical Neurophysiology, Uppsala, Sweden
Background: Single-fiber electromyography (SFEMG) is a highly sensitive diagnostic tool for myasthenia gravis (MG), but its interpretation relies on jitter reference values that vary substantially across publications. We quantified the effects of published mean and paired jitter cutoffs on diagnostic sensitivity, specificity, and misclassification in patients evaluated for suspected MG.
Methods: In a retrospective two-center cohort of adults (≥18 years) undergoing SFEMG for suspected neuromuscular junction disorder, we analyzed frontalis and orbicularis oculi studies using published mean and individual-pair jitter values. Diagnostic performance was assessed across published reference values using sensitivity, specificity, and Youden J statistics. Misclassification patterns were examined across thresholds.
Results: We included 153 adults (mean age 55.5 ± 17.7 years; range 18-95), 88/153 were female, and 62/153 were clinically diagnosed with MG. Diagnostic performance varied substantially across published standards. For frontalis, the mean jitter cutoffs of 34.5 µs (Kokubun et al.) and 53.1 µs for the individual pair upper limit of normal (ULN) (Benatar et al.) maximized sensitivity-specificity (Table 2). For orbicularis oculi, the mean jitter cutoff of 31.0 µs (Sanders AANEM) and 49.7 µs individual pair ULN (Farrugia et al.) maximized sensitivity-specificity (Table 3).
Conclusion: SFEMG diagnostic classification in MG is sensitive to reference values, and the interpretation of a study can vary across different published standards. A diagnosis of MG often leads to prolonged immunosuppressive therapies and biologics, which may have unwanted adverse effects. A large-scale, population-based study with multi-center data is critical for developing consensus-based values to limit diagnostic uncertainty in SFEMG.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
Images or Table (Optional)
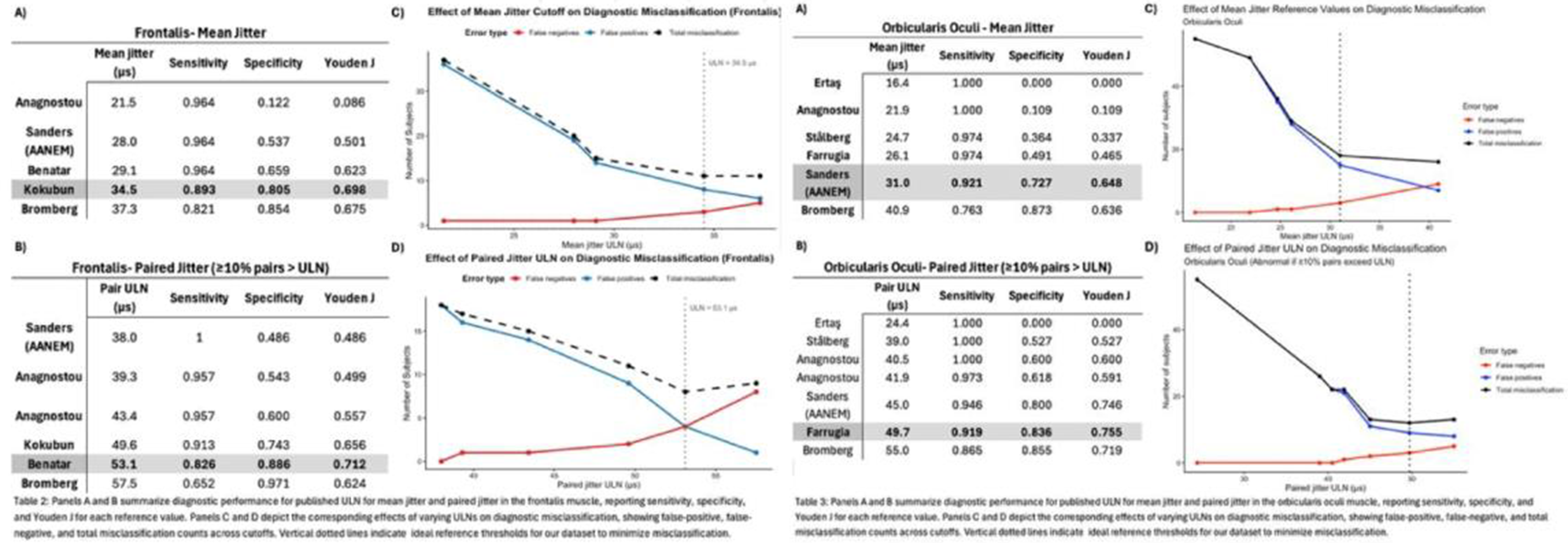
PP01.235
Efficacy of Nipocalimab in Patients with Myasthenia Gravis and Lower Baseline Activity of Daily Living
Dr. Carlo Antozzi1, Dr. Wim Noel2, Dr. Wisam Karmous3, Dr. Marie Fitzgibbon4, Dr. Kavita Gandhi5, Dr. Ibrahim Turkoz5, Dr. Michael Kutch6, Dr. Sindhu Ramchandren7
1
Immunotherapy and Apheresis Unit, Neuroimmunology and Muscle Pathology Unit, Fondazione IRCCS Istituto Neurologico C. Besta, Milan, Italy.
2
Johnson & Johnson, Diegem, Belgium.
3
Johnson & Johnson, Issy-les-Moulineaux, France.
4
Johnson & Johnson, Raritan, United States.
5
Johnson & Johnson, Horsham, United States.
6
Cytel Inc., Cambridge, United States.
7
Johnson & Johnson, Titusville, United States
Background: Myasthenia gravis (MG) is a chronic autoimmune disease characterised by fluctuating muscle weakness and fatigability, which may worsen without effective treatment. In VIVACITY-MG3 (NCT04496507) study, nipocalimab added to standard-of-care (SOC), demonstrated greater improvement in Myasthenia Gravis–Activities of Daily Living (MG-ADL), and Quantitative Myasthenia Gravis (QMG) scores versus placebo (+SOC). This post hoc analysis evaluated whether efficacy could be achieved in patients with lower baseline symptom burden (MG-ADL scores: 6–9).
Methods: Analysis included seropositive patients treated with nipocalimab or placebo with baseline MG-ADL score below cohort median score of 9. Efficacy was evaluated by changes in MG-ADL (meaningful clinical improvement [MCI]: ≥2-point improvement; substantial clinical improvement [SCI]: ≥3-point improvement), and QMG (MCI: ≥3-point improvement; SCI: ≥4-point improvement) scores at week (W)24. Two-sample t-tests compared changes from baseline between groups.
Results: In this population, baseline demographics were generally balanced between nipocalimab and placebo-treated patients (Table). At W24, MG-ADL reductions (mean [SD]) from baseline were greater with nipocalimab (−4.5 [2.64]) versus placebo (−2.3 [2.37]); difference: −2.23 (standard error [SE]: 0.588); 95%CI (−3.41; −1.06); p<0.001. QMG reductions (mean [SD]) were also greater with nipocalimab (−5.2 [4.45]) versus placebo (−1.9 [3.69]); difference: −3.38 (SE:0.986); 95%CI (−5.34; −1.41); p=0.001. A greater proportion of patients receiving nipocalimab met MCI and SCI criteria for MG-ADL and QMG at W24 versus placebo (Table).
Conclusion: In patients with baseline MG‑ADL scores 6–9, nipocalimab demonstrated greater efficacy versus placebo, achieving MCI and SCI in MG‑ADL and QMG at W24. These results support the benefit of nipocalimab for sustained disease control in patients with gMG with lower baseline MG-ADL scores.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
Images or Table (Optional)
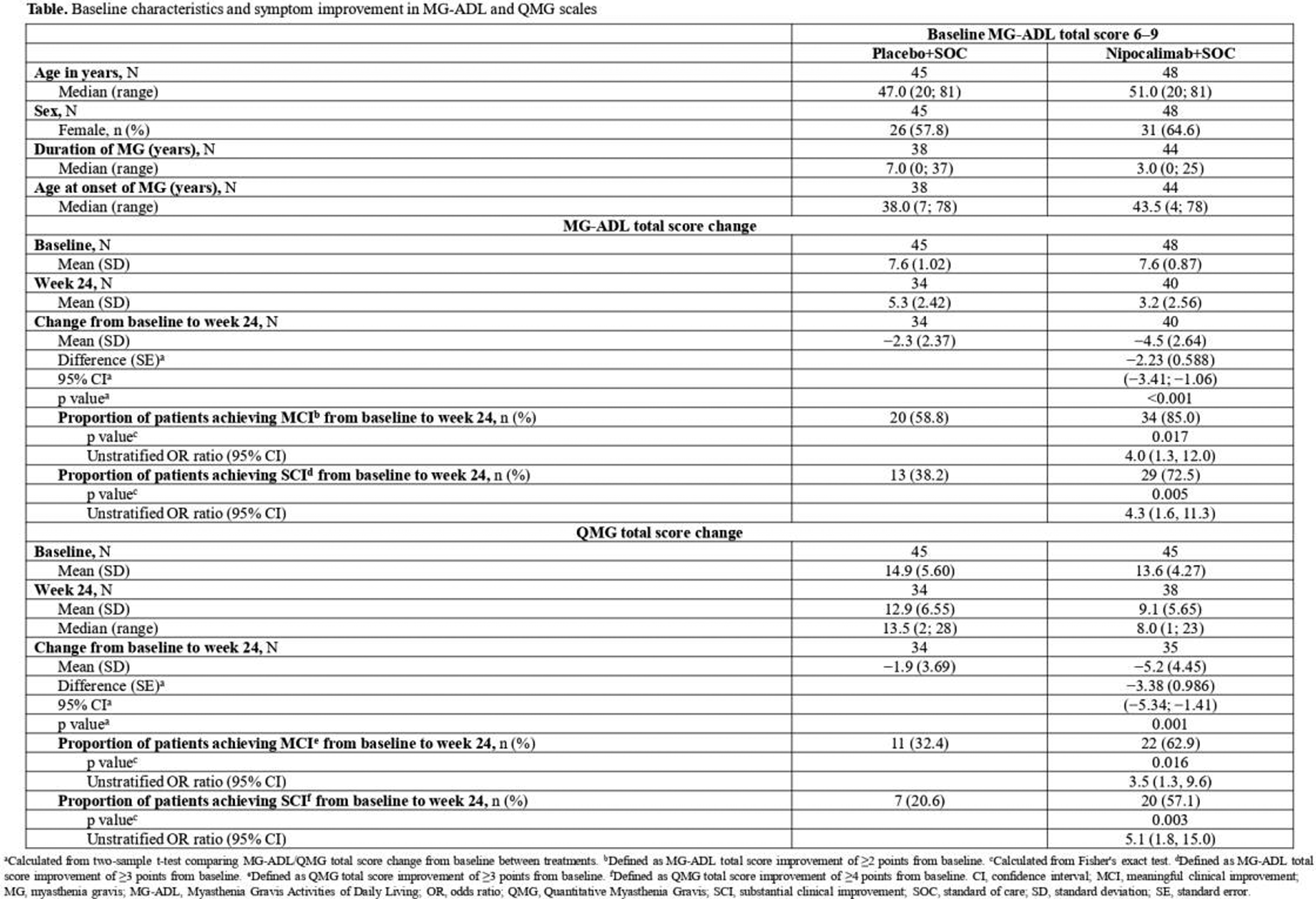
PP01.236
Efficacy of Nipocalimab in Patients Early in Their Disease Course of Generalized Myasthenia Gravis
Dr. Carlo Antozzi1, Dr. Wim Noel2, Dr. Marie Fitzgibbon3, Dr. Wisam Karmous4, Dr. Kavita Gandhi5, Dr. Ibrahim Turkoz5, Dr. Michael Kutch6, Dr. Sindhu Ramchandren7, Dr. Elena Cortés-Vicente8
1
Immunotherapy and Apheresis Unit, Neuroimmunology and Muscle Pathology Unit, Fondazione IRCCS Istituto Neurologico C. Besta, Milan, Italy.
2
Johnson & Johnson, Beerse, Belgium.
3
Johnson & Johnson, Raritan, United States.
4
Johnson & Johnson, Issy-les-Moulineaux, France.
5
Johnson & Johnson, Horsham, United States.
6
Cytel Inc., Cambridge, United States.
7
Johnson & Johnson, Titusville, United States.
8
Unitat Patologia Neuromuscular, Servei Neurologia, Hospital Santa Creu i Sant Pau, Barcelona, Spain
Background: Myasthenia gravis (MG) is a chronic autoimmune condition characterized by fluctuating muscle weakness and fatigue. Nipocalimab added to standard-of-care (SOC) showed greater improvement than placebo (+SOC) in Myasthenia Gravis–Activities of Daily Living (MG-ADL), and Quantitative Myasthenia Gravis (QMG) scores in VIVACITY-MG3 study (NCT04496507). This post hoc analysis evaluated whether efficacy was achievable in patients with generalized MG (gMG) diagnosed earlier (≤5 years) before study initiation.
Methods: Analysis included seropositive patients treated with nipocalimab or placebo who had been diagnosed with gMG within previous 5 years (below the cohort median of 5 years). Efficacy at week (W)24 was assessed using MG-ADL (meaningful clinical improvement [MCI]: ≥2-point improvement; substantial clinical improvement [SCI]: ≥3-point improvement) and QMG (MCI: ≥3-point improvement; SCI: ≥4-point improvement) scores. Two-sample t-tests compared baseline-to-W24 changes between groups.
Results: Baseline demographics were generally balanced between nipocalimab and placebo-treated patients (Table). At W24, MG-ADL reductions (mean [SD]) from baseline were greater with nipocalimab (−4.9 [2.88]) versus placebo (−2.7 [2.46]); difference: −2.22 (standard error [SE]: 0.76); 95%CI (−3.74; −0.70); p=0.005. Similarly, QMG reductions (mean [SD]) from baseline were greater with nipocalimab (−5.1 [4.14]) versus placebo (−2.3 [3.20]); difference: −2.85 (SE:1.08); 95%CI (−5.03; −0.66); p=0.012. A greater proportion of patients receiving nipocalimab met MCI and SCI criteria for MG-ADL and QMG at W24 versus placebo (Table).
Conclusion: In patients diagnosed with gMG within 5 years before study initiation, nipocalimab demonstrated greater efficacy versus placebo, achieving MCI and SCI in MG-ADL and QMG at W24. These findings highlight the benefit of nipocalimab in recently diagnosed patients with gMG.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
Images or Table (Optional)
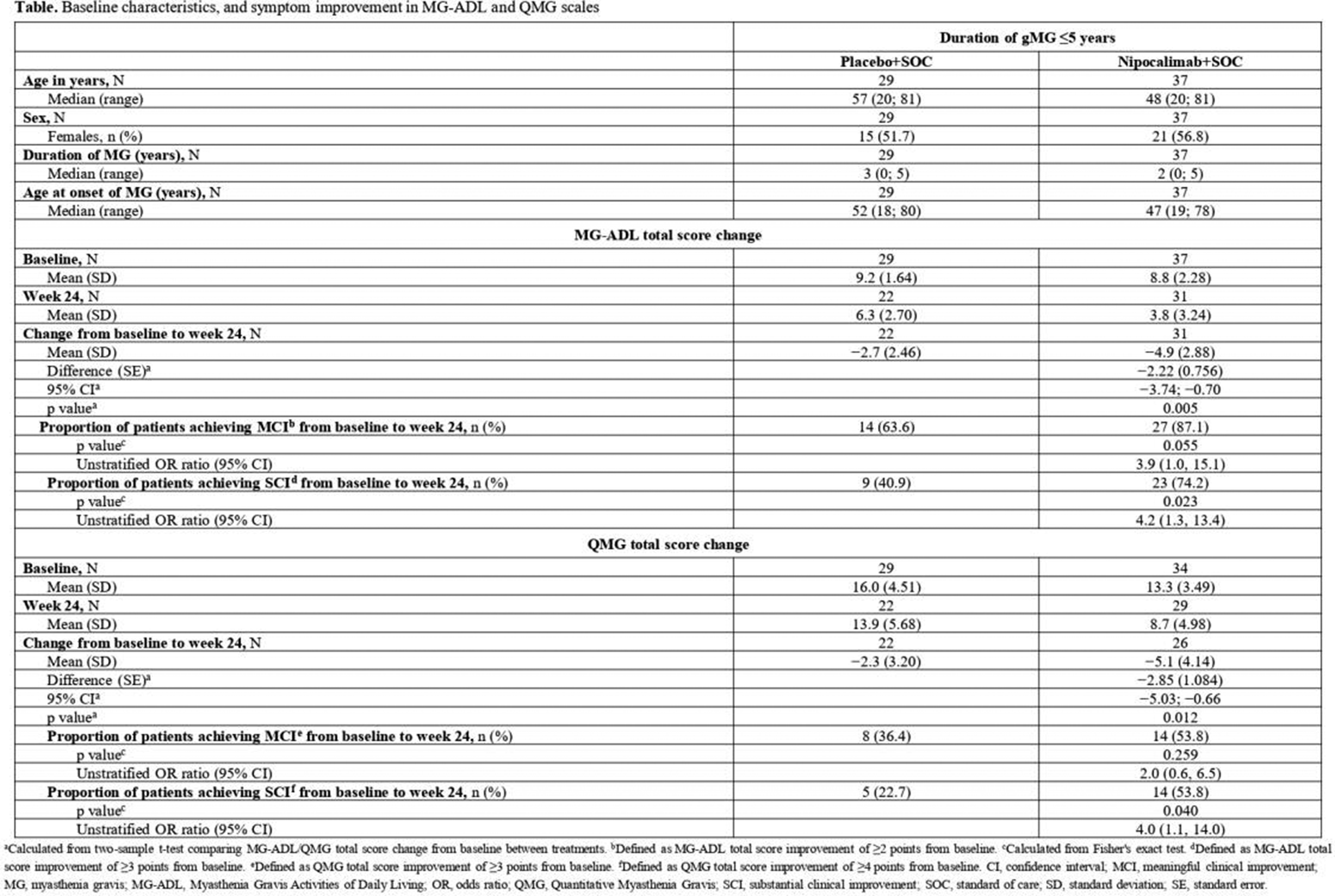
PP01.237
Treatment and Outcomes in a Real-World Australian Myasthenia Gravis Cohort: Baseline Findings from VALUE-MG
Dr. Yifat Biran1,2, Dr. Xin Zhang1, Dr. Gozde Aydin1, Dr. Adam Irving1, Dr. Laurie McLaughlin3, Assoc. Prof. Stefan Blum3, Dr. Belinda Cruse4, Assoc. Prof. Katherine Buzzard5, Assoc. Prof. Stephen Reddel6, Prof. Dennis Petrie1, Assoc. Prof. Anneke Van der Walt1,2
1
Monash University, Melbourne, Australia.
2
Bayside Health - Alfred, Melbourne, Australia.
3
Metro South Health - Princess Alexandra Hospital, Brisbane, Australia.
4
Royal Melbourne Hospital, Melbourne, Australia.
5
Eastern Health, Melbourne, Australia.
6
University of Sydney - Brain and Mind Centre, Sydney, Australia
Background: High-quality longitudinal data on treatments and outcomes for people with Myasthenia Gravis (MG) is limited, but is required for informed healthcare funding decisions. VALUE-MG is an ongoing, prospective study designed to integrate these data in order to develop health economic models that will determine the cost-effectiveness of MG therapies in Australia.
Methods: Participants were recruited from five hospitals across Australia over an 18 month period. VALUE-MG links de-identified data from various resources, such as clinical data from the MGBase registry, primary healthcare use and costs from national datasets, as well as quality-of-life (QoL) and productivity data collected via electronic patient-reported outcome measures (ePROMs). ePROMs used are: Myasthenia Gravis Activity of Daily Living (MG-ADL), Myasthenia Gravis Quality of Life-15 revised (MG-QoL15r), EuroQol-5 Dimensions-5 Levels (EQ-5D-5L), and Work Productivity and Activity Impairment-General Health (WPAI-GH). ePROMs were administered quarterly from date of enrolment (i.e., baseline) via secure, web-based Research Electronic Data Capture (REDCap). Baseline analyses were conducted on a sample of 207 participants, using descriptive statistics, correlations, and scatter plots.
Results: Baseline cohort demographics showed an equal distribution between males and females, with mostly middle-aged (40–69 years; 46.5%) or older (≥70 years; 40.8%) individuals. At time of diagnosis, antibody sub-types were predominantly acetylcholine receptor (AChR) positive (68.3%) and seronegative (24.6%), with few Muscle-Specific Kinase (MuSK) positive (5.6%) participants, and their disease severity spanned all MGFA classes (I: 48.2%, II: 31.2%, III: 13.4%, IV: 4.5%, V: 1.8%). Clinician-reported baseline disease severity and functional status were generally mild, with mean (±SD) scores of 2.83±3.48 for Myasthenia Gravis Composite (MGC) and 2.45±2.86 for MG-ADL. Patients reported moderate disease burden at baseline, as reflected by QoL, work and productivity measures with mean (±SD) scores of: 4.32±4.05 for MG-ADL, 8.57±7.36 for MG-QoL15r, 0.86±0.19 for EQ-5D-5L utility (Australia value set), and 0.28±0.27 for WPAI-GH work impairment (among employed participants). Although patient-reported MG-ADL scores seemed significantly higher than clinician-reported ones (4.32 and 2.45, respectively), the two measures were moderately correlated (r=0.64, p<0.001). Scatter-plot visualisation demonstrated an overall positive association with considerable dispersion around the fitted line. Together, results indicate alignment but non-interchangeability between patient and clinician assessments of MG-related functional impairment.
Conclusion: The VALUE-MG observational study captures a diverse, real-world MG population that is still being followed-up. Baseline data demonstrate meaningful associations between disease severity, quality of life, and work productivity, while highlighting important differences between patient- and clinician-reported outcomes. These findings support the selected outcome measures for the study and will inform the development of decision-analytic economic models to evaluate the cost-effectiveness of MG therapy strategies, ultimately providing a robust evidence base for clinicians, policymakers, and people living with MG.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.238
Rituximab in Myasthenia Gravis: A Real-World Study Using Inverse Probability of Treatment Weighting
Dr. Yong Lin Wang1, Dr. Chao Zhu1, Dr. Mahima Kapoor1, Prof. Gary Cutter2, Assoc. Prof. Carolina Barnett-Tapia3, Prof. Helmut Butzkueven1, Dr. WenWen Zhang1, Dr. Gabor Lovas4, Dr. Csilla Rozsa4, Prof. Jeannine Heckmann5, Assoc. Prof. Stefan Blum6, Dr. Laurie McLaughlin6, Dr. Katherine Buzzard7, Dr. Yi Chao Foong8, Prof. Elisabeth Chroni9, Asst. Prof. Dimitra Veltsista9, Dr. Belinda Cruse10, Dr. Mina Botrous1, Dr. Stephen Reddel11, Assoc. Prof. Mastura Monif1, Assoc. Prof. Anneke van der Walt1
1
School of Translational Medicine, Monash University, Melbourne, Australia.
2
Department of Biostatistics, University of Alabama in Birmingham, Birmingham, United States.
3
Division of Neurology, Department of Medicine, University Health Network and University of Toronto, Toronto, Canada.
4
Department of Neurology, Jahn Ferenc Hospital, Budapest, Hungary.
5
Neurology Research Group, Neuroscience Institute, University of Cape Town, Cape Town, South Africa.
6
Neurology, Princess Alexandra Hospital, Wooloongabba, Australia.
7
Department of Neurosciences, Box Hill Hospital, Box Hill, Australia.
8
Neurology, Royal Hobart Hospital, Tasmanian Health Service, Hobart, Australia.
9
Neuromuscular Unit, Neurology Department, University of Patras, Patras, Greece.
10
Neurology, Royal Melbourne Hospital, Melbourne, Australia.
11
Brain and Mind Centre, Camperdown, Australia
Background: The role of rituximab in the treatment of myasthenia gravis (MG), particularly among patients with acetylcholine receptor (AChR) antibody positivity, remains uncertain due to limited randomized controlled evidence and heterogeneous observational data. While rituximab is often used in refractory MG, its comparative effectiveness against other non-steroidal immunosuppressive therapies (NSISTs) requires further clarification.
Methods: We conducted a retrospective cohort study of AChR-antibody-positive MG patients treated with rituximab or a second NSIST. The primary outcome was time to achieving a composite clinical endpoint representing a patient acceptable symptom state (PASS): Myasthenia Gravis Composite (MGC) score ≤ 3, daily corticosteroid dose ≤ 5 mg prednisolone equivalent, and no rescue therapy use in the preceding month. To reduce confounding by indication and improve causal comparability between treatment groups, inverse probability of treatment weighting (IPTW) based on propensity scores was used to balance baseline covariates. Cox regression was applied to estimate the effect of treatment on time to achieving composite endpoint.
Results: 169 patients entered into the time-to-event analysis, and after IPTW adjustment, baseline characteristics between treatment groups were well balanced. There was no statistical difference in the hazard ratio between rituximab compared to a second NSIST in achieving the composite clinical outcome (HR = 1.27, 95% CI 0.66-2.45, p = 0.48). Sensitivity analyses yielded consistent findings.
Conclusion: In this IPTW-adjusted analysis, rituximab did not improve attainment of clinical improvement compared to a second NSIST in AChR-antibody-positive MG.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenia Gravis
PP01.239
A New Clinical Entity: Retrospective Case Study of Immune Checkpoint Inhibitor Induced Myasthenia Gravis
Dr. Ge Xiong1, Dr. Tianhong Li1, Dr. Han Lee1, Dr. Vihar Patel1, Dr. Lee-Way Jin1, Dr. Ning Wu2, Dr. Sophie Teng1, Dr. Ricardo Maselli1, Dr. David Richman1
1
University of California Davis, Sacramento, United States.
2
Kaiser Permanente Roseville Medical Center, Roseville, United States
Background: Immune checkpoint inhibitors (ICI) represent a breakthrough in cancer treatment, but immune-related adverse events (irAEs), including neurological complications, have been increasing with the increased ICI usage. Neuromuscular irAEs include myositis, myasthenia gravis (MG), peripheral neuropathy. Although neuromuscular irAEs are uncommon, mortality is significant (could be >20%). ICI induced MG (iMG) appears to have a unique clinical presentation, different from spontaneous MG. In this case series study, we aim to characterize the clinical features of iMG to help achieve timely management of this disorder.
Methods: We have retrospectively analyzed twelve cases with the clinical diagnosis of iMG presenting from Jan 2020 to Dec 2025 in the Sacramento metropolitan area
Results: History and physical examination showed that all cases of iMG had fatigable weakness. The common symptoms were similar to spontaneous MG: eyelid ptosis, diplopia, dysarthria, dysphagia, extremity weakness and difficulty breathing. Only one case had ocular myasthenia gravis, the remaining eleven had generalized MG (Myasthenia Gravis Foundation of America Class III to IV). The patients’ mean age was 72.6 years (range 38-87), nine of twelve were men. The most common cancer was renal cell carcinoma (5/12), followed by bladder cancer (2/12), melanoma (2/12), squamous cell carcinoma (2/12, one was cutaneous in thoracic area, one was tonsil cancer) and colon cancer (1/12). The most used ICI was Pembrolizumab (5/12, 41.67%), followed by Nivolumab (4/12). Four cases underwent dual ICIs or ICI combined with chemotherapy. The time from initiation of ICI treatment to symptom onset ranged from 3 days to 6 weeks. Two had positive acetylcholine receptor antibodies, all were negative for MuSK and LRP4 antibodies. Other autoantibodies were detected in some seronegative MG cases: one had striated muscle antibody (titer 1:160), one had ANA (titer 1:160) as well as positive SSA, and one had GAD antibody (>250 IU/ml). Seven cases underwent EMG/NCS study: only two exhibited decremental CMAP amplitudes to low frequency repetitive nerve stimulation. Ten iMG cases presented with overlap syndromes: five had myositis and myocarditis, four had myositis, one had myocarditis and hepatitis. Two received muscle biopsy, each of which demonstrated multifocal muscle fiber necrosis in clusters. High-dose steroids, intravenous immunoglobulin, and pyridostigmine were the most commonly used treatments. Two cases with triple syndrome (MG+myositis+myocarditis) received Abatacept and Roxolitinib with mild to moderate improvements. Despite some clinical improvement to the above treatments, the mortality for iMG was 50%.
Conclusion: This case series study indicates that: 1) iMG usually presents with more severe symptoms than spontaneous MG; 2) the rates of positive MG antibodies and of abnormal repetitive nerve stimulation responses are much lower than in spontaneous MG; 3) overlap syndromes are common in iMG, 4) along with increased mortality. Therefore, we propose that iMG is a new clinical entity with unique features that may have a different pathophysiology. We are engaged in further analysis to determine the underlying mechanism, with the aim of achieving early diagnosis and decreased mortality.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenic Syndromes
PP01.240
Clinical Features in Patients With Neuroimmunological Disorders Associated With Antibodies to Voltage-Gated Calcium Channel
Asst. Prof. Makoto Samukawa, Prof. Makito Hirano, Dr. Genki Hoshino, Dr. Hiroto Nakamura, Asst. Prof. Toru Michiura, Asst. Prof. Hitoshi Namura, Asst. Prof. Hanami Sakata, Asst. Prof. Yuta Fukumoto, Asst. Prof. Keisuke Yoshikawa, Dr. Yuko Yamagishi, Asst. Prof. Motoi Kuwahara, Prof. Kazumasa Saigoh, Prof. Yoshiyuki Mitsui, Prof. Susumu Kusunoki, Prof. Yoshitaka Nagai
Kindai University Hospital, Sakai, Japan
Background: Lambert-Eaton myasthenic syndrome (LEMS) is an immune-mediated neurological disorder characterized by proximal limb muscle weakness and autonomic dysfunction. Antibodies to voltage-gated calcium channel (VGCC) are positive in 85 to 95 % in patients with LEMS. Approximately 10% of cases of LEMS are associated with cerebellar ataxia (CA), termed paraneoplastic cerebellar degeneration-associated LEMS (PCD-LEMS). Notably, 3,4-diaminopyridine (3,4-DAP) has been approved for coverage under public insurance as the first medicine for LEMS in Japan in September 2024. The clinical criteria for early diagnosis remain insufficiently shared and understood, although it becomes even more important. Therefore, we here report the clinical features of patients with antibodies to VGCC including LEMS in our institution.
Methods: We collected clinical and therapeutic information of patients with neuroimmunological disorders carrying antibodies to VGCC who admitted in our hospital between April 2007 and December 2025, and evaluated clinical information, treatment methods and outcome. Diagnosis of LEMS was based on the diagnostic criteria from the MG/LEMS Clinical Practice Guidelines 2022 (supervised by Japanese Society of Neurology).
Results: During the above period, nine patients (six men) with neurological disorders had antibodies to VGCC. The mean age at onset was 66.5 years (range 42–78). We diagnosed eight patients with LEMS. The remaining patient was diagnosed with immune-mediated CA with antibodies to VGCC because she did not meet the LEMS diagnostic criteria, without myasthenic symptoms and electrophysiological abnormalities on a repetitive nerve stimulation test. Bulbar symptoms were present in eight patients, and muscle weakness was observed in five. Autonomic dysfunction was present in seven, with bladder and bowel dysfunction and orthostatic hypotension being highly frequent. CA was observed in six. Tumors included six cases of small cell lung cancer and one case of small cell esophageal cancer. Of the six patients presenting with CA, one patient had progression of only CA symptoms for eight years after onset, and no tumor was detected for 15 years. Treatment details were as follows: chemotherapy only in three patients (two effective), intravenous immunoglobulin (IVIg) only in a patients (ineffective), 3,4-DAP after intravenous methylprednisolone (IVMP) in a patient (effective), 3,4-DAP with 40mg/day of oral prednisolone after IVIg and IVMP in a patient (effective), chemotherapy after IVIg in a patient (effective), 40mg/day of oral prednisolone after IVIg in a patient (effective), and 3,4-DAP with 25mg/day of oral prednisolone after IVMP in a patient without LEMS (effective in bulbar symptoms and CA).
Conclusion: In this study, CA was observed more frequently than previous reports estimated to be around 10%. All patients treated with 3,4-DAP showed improvement. It was noteworthy that therapeutic efficacy with 3,4-DAP was observed even in CA where this treatment was usually ineffective in previous reports. This study may add to the understanding of neuroimmunological disorder associated with VGCC antibodies.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenic Syndromes
PP01.241
Genetic Landscape and Innovative Therapeutic Approaches Concerning a Large Algerian Series of Congenital Myasthenic Syndromes
Prof. Mohamed Islam Kediha1,2, Dr. Damien Sternberg3, Prof. Bruno Eymard4, Prof. Lamia Ali Pacha1,2
1
Mustapha university hospital, Algiers, Algeria.
2
Benyoucef Benkhedda medical school, Algiers, Algeria.
3
Pitié Salpetrière hospital, Paris, France.
4
Rothschild foundation, Paris, France
Background: Congenital myasthenic syndromes (CMS) are disorders that exhibit a wide range of phenotypic and genotypic diversity. To date, at least 35 genes have been identified, with the majority being autosomal recessive, which relates to the level of consanguinity and endogamy, still prevalent in Algeria. This research involves analysing a group of Algerian families from both phenotypic and genetic perspectives. This series represents the first cohort study of CMS in Algeria and in north Africa.
Methods: We analyzed the clinical and paraclinical phenotypes of our families based on data from neurological examinations, general somatic examinations (particularly pleuropulmonary), EMG tests to detect decrement and double response of motor potential, and genetic studies of these families. These patients were scored using the Garches myasthenic score and the QMG (Quantitative myasthenai gravis) score before and after treatment.
Results: We have gathered data from 33 families (71.7%) and detected various mutations (including 5 novel ones) across different identified genes (COLQ, COL13A1, CHRNE, CHRNA1, CHRND, GMPPB, and RYR1). Two families have been diagnosed with Escobar syndrome, while 11 families are currently under investigation. The most common mutations occur in the CHRNE gene, with the '1293insG' mutation, primarily found in North Africa, being the predominant one identified. The phenotype is classified as moderate and generally shows a good response to pyridostigmine treatment. The initial step should involve identifying the founder mutation in all cases of congenital myasthenic syndromes (CMS) in North Africa.The pharmacological approach is well-established, tailored to each specific gene and the underlying pathophysiological mechanisms of the post-synaptic genes, including receptor deficiencies or kinetic anomalies.Several novel treatments have also been explored such as Salbutamol, Fluoxetine, and corticosteroids. Additionally, palliative care delivered within a multidisciplinary framework is a crucial aspect of the treatment plan.
Conclusion: Our series is the largest ever studied in Africa on CMS. We have identified several genes and new mutations, providing a genetic map of these conditions in Algeria and North Africa. We have also tested certain repurposed and innovative therapies, which have led to considerable functional improvement in some patients.
Abstract Topic Groups (Submission Categories)
Topic Group 2 - Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy: Myasthenic Syndromes
PP01.242
Application of Gait Training Robots According to the Functional Level of Patients With Guillain-Barré Syndrome
Dr. Hara Jeon
National Health Insurance Service Ilsan Hospital, Goyang, Korea, Republic of
Background: Gait training robots are categorized into exoskeleton-type, end-effector-type, and wearable-type devices. It is well established that robot-assisted gait training (RAGT) is beneficial for patients with Guillain-Barré syndrome (GBS) who experience gait disorders. RAGT requires training by selecting an appropriate type of gait training robot, considering the patient's functional level; however, there is no established protocol for this. In this study, we would suggest which gait training robot is appropriate for patients with GBS depending on the patient's gait function.
Methods: The gait training robots used for patients were as follows. We applied an exoskeleton-type Walkbot, an end-effector type Morning Walk, and a wearable-type Angel Legs according to the patients’ functional level. The patient's functional level was classified using the Functional Ambulation Categories (FAC) and Berg Balance Scale (BBS). For 24 patients with GBS who received RAGT, the types of gait training robots applicable to the patients were summarized according to their functional level.
Results: There were 10 patients who applied Walkbot, 12 patients who applied Morning Walk, and 2 patients who applied Angel Legs. The FAC of patients who applied the Walkbot was 0∼1, and their average BBS was 4.5 points. The FAC of patients who applied the Morning Walk was 1∼3, and their average BBS was 7.9 points. The FAC of patients who applied the Angel Legs was 2 and 3, and their average BBS was 22.5 points. The application range of the Walkbot was suitable for FAC 0 to 1. If the patient can control the trunk somewhat, the Walkbot could provide gait experience even in cases where lower limb strength was zero. Morning Walk and Angel Legs were suitable for patients with FAC 1 to 3, and if body weight support was consistently measured less than 10% during RAGT using Morning Walk, the gait training robot was changed to Angel Legs. However, Angel Legs, which is a mobile gait training robot, requires higher balance ability compared to fixed gait training robots such as Walkbot or Morning Walk.
Conclusion: By investigating the scope of application of exoskeleton, end-effector, and wearable-type gait training robots according to GBS patients’ functional level, the continuity of RAGT could be suggested. In the future, there will be a need to develop more precise training protocols for GBS patients according to their functional level and the type of gait training robot.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Advances in treatment of Peripheral Neuropathies
PP01.243
Beyond the Nerve: Uncovering Skeletal Muscle Involvement in SORD Neuropathy
Assoc. Prof. Andreas Roos1, Dr. Heike Kölbel1, Dr. Andreas Hentschel2, Dr. May Tiet3, Dr. Maike Dohrn4, Dr. David Muhmann1, Dr. Jelle van den Ameele33, Ms. Lei Chen1, Prof. Ulrike Schara1, Prof. Joachim Weis5, Prof. Rita Horvath3
1
University Medicine Essen, Department of Pediatric Neurology, Essen, Germany.
2
Leibniz-Institute for Analytical Science -ISAS- e.V., Dortmund, Germany.
3
Department of Clinical Neurosciences, School of Clinical Medicine, John Van Geest Centre for Brain Repair, University of Cambridge, Cambridge, United Kingdom.
4
RWTH-Aachen University Hospital, Institute of Neurology, Aachen, Germany.
5
RWTH-Aachen University Hospital, Institute of Neuropathology, Aachen, Germany
Background: Biallelic pathogenic variants in SORD, encoding sorbitol dehydrogenase, have recently been identified as a frequent cause of autosomal recessive axonal Charcot-Marie-Tooth disease (CMT2). Although the disease is primarily considered a peripheral neuropathy, emerging evidence suggests a direct involvement of skeletal muscle. The extent and molecular basis of this muscle pathology remain insufficiently understood.
Methods: We investigated skeletal muscle biopsies from four genetically confirmed CMT-SORD patients, including one individual with marked post-exercise CK elevation (13,000 U/L). An integrative approach was applied, combining histopathology, electron microscopy, quantitative proteomics, immunofluorescence, and serum biomarker analyses. Findings were compared with control muscle samples and with data from other rare recessive CMT subtypes.
Results: Histological analysis revealed features of chronic denervation, including grouped fiber atrophy, fiber-type grouping, and central nuclei. Ultrastructural examination demonstrated mitochondrial abnormalities and expansion of the sarcoplasmic reticulum. Proteomic profiling identified 220 significantly dysregulated proteins in CMT-SORD muscle compared with controls, affecting mitochondrial complex I components, redox enzymes, and metabolic regulators. This proteomic signature was distinct from that observed in CMT associated with pathogenic GAN or GDAP1 variants. Immunofluorescence confirmed increased expression of CYTC, PGC-1α, and PERM1, indicating mitochondrial stress and compensatory metabolic activation. Serum analyses showed that GDF-15 and FGF-21 were not reliable biomarkers for CMT-SORD.
Conclusion: SORD deficiency induces molecular and structural alterations in skeletal muscle that extend beyond secondary denervation. Impaired sorbitol metabolism, oxidative stress, and mitochondrial dysfunction appear to contribute to disease pathology. The coexistence of neurogenic and intrinsic myopathic features in SORD-related CMT2 highlights the need for therapeutic strategies targeting both neuronal and muscular compartments.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Hereditary Peripheral Neuropathies
PP01.244
Novel SIGMAR1 Missense Variant: A Case Report and Literature Review
Dr. Bianca Rugginini1, Dr. Sabrina Ravaglia2, Dr. Michele Giovanni Croce1, Dr. Simone Gana3, Prof. Enza Maria Valente3,4, Dr. Luca Diamanti2, Dr. Paolo Prunetti5, Prof. Giuseppe Cosentino1,5, Dr. Elisa Vegezzi2
1
University of Pavia, Department of Brain and Behavioural Sciences, Pavia, Italy.
2
IRCCS Mondino Foundation, Pavia, Italy.
3
Neurogenetics Research Center, IRCCS Mondino Foundation, Pavia, Italy.
4
University of Pavia, Department of Molecular Medicine, Pavia, Italy.
5
Translational Neurophysiology Research Unit, IRCCS Mondino Foundation, Pavia, Italy
Background: Biallelic variants in SIGMAR1 (OMIM #605726) are associated with distal hereditary motor neuropathy (dHMN), also known as Jerash type. This condition is characterized by early-onset distal weakness, pyramidal signs, and foot deformities.
Methods: We performed a comprehensive literature review of SIGMAR1-related dHMN and report a case carrying a novel biallelic missense variant. A PubMed search using selected keywords was conducted, and relevant original articles and reviews were included.
Results: Fifteen publications describing 56 patients with detailed phenotypic data were identified. The cohort included 24 females and 32 males of Middle Eastern, Asian, and European origin, harboring homozygous or compound-heterozygous missense, truncating, or splice-site variants. Patients typically presented with childhood-onset, progressive, symmetric distal weakness and pyramidal signs, with disease severity ranging from mild impairment to wheelchair dependence. We describe a 49-year-old Italian woman, born to consanguineous parents, who developed gait difficulties and clumsiness at age 3, followed by progressive distal weakness and atrophy of the upper limbs in adulthood. Neurological examination revealed symmetric, predominantly distal lower-limb weakness (Medical Research Council grade 2/5), pes cavus and varus deformities with hammer toes, absent deep tendon reflexes except for brisk patellar reflexes, and preserved sensation. Nerve conduction studies showed a length-dependent axonal motor neuropathy, with evidence of chronic and active denervation predominantly affecting distal lower limbs on needle electromyography. Brain and spinal MRI were unremarkable. Clinical exome sequencing identified a novel homozygous missense variant in exon 1 of SIGMAR1 (c.139C>T; p.Arg47Trp). Although currently classified as a variant of uncertain significance, its pathogenic role is supported by absence from population databases (gnomAD), high evolutionary conservation (phyloP = 5.67), and multiple in silico prediction tools (CADD = 32).
Conclusion: The clinical and electrophysiological features of our patient closely mirror those reported in SIGMAR1-related dHMN. Together with supportive genetic evidence, this finding expands the mutational spectrum of SIGMAR1, although functional studies are required to confirm pathogenicity.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Hereditary Peripheral Neuropathies
PP01.245
Acute Brachial Plexopathy as a Presenting Feature for Hereditary Neuropathy With Liability to Pressure Palsies
Dr. Ravi Ambati, Dr. Dev Nathani
Joondalup Health Campus, Perth, Australia
Background: Acute painless proximal upper limb weakness presents a broad diagnostic challenge. We report the case of a 29-year-old woman, LB, who noted painless right shoulder weakness and sensory changes upon waking. There were no preceding infective or traumatic symptoms. Persistence of symptoms over the next three weeks led to presentation to hospital. She had no significant past medical history, or relevant family history, and was not taking any regular medications.
Examination: Significant findings included grade 4 weakness of right shoulder abduction, external rotation, and flexion. Pinprick perception was reduced over the right lateral shoulder and lateral proximal arm.
Initial Investigations: MRI cervical spine and right brachial plexus was normal. MRI right shoulder demonstrated subtle T2 hyperintensity within the supraspinatus muscle belly. Chest radiography was unremarkable. LB was initially felt to have an idiopathic brachial neuritis (Parsonage-Turner), noting that painless forms of this have been reported in the literature. She was discharged for outpatient rehabilitation and nerve conduction studies (NCS) and her symptoms gradually improved over eight weeks. At follow-up 7 months later, there was slight weakness of shoulder external rotation (elbow flexed). Previously noted sensory examination findings had mostly resolved but there was new finding of impaired light touch perception in the right hand in an ulnar nerve distribution. At this time, NCS (see image) of the upper and lower limbs demonstrated a generalised sensorimotor neuropathy with mixed demyelinating and axonal features. Conduction velocity slowing was most prominent at common entrapment sites. Peripheral nerve ultrasound showed enlargement of bilateral ulnar and median nerves at the cubital tunnels and proximal carpal tunnels respectively.
Methods: N/A
Results: Consequently, additional history was obtained. LB reported recurrent pressure-induced paraesthesia and longstanding positional numbness raising strong suspicion for a diagnosis of hereditary neuropathy with liability to pressure palsies. Targeted genetic testing was requested which confirmed that LB was heterozygous for a pathogenic variant in the PMP22 gene (c.281dup).
Conclusion: This case underscores the importance of a systematic approach to acute proximal upper-limb weakness, recognizing that acute presentations localising to the brachial plexus may, particularly if painless, reflect underlying hereditary neuropathies. It also highlights that, although uncommon, hereditary neuropathy with liability to pressure palsies can present as an acute brachial plexopathy.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Hereditary Peripheral Neuropathies
Images or Table (Optional)
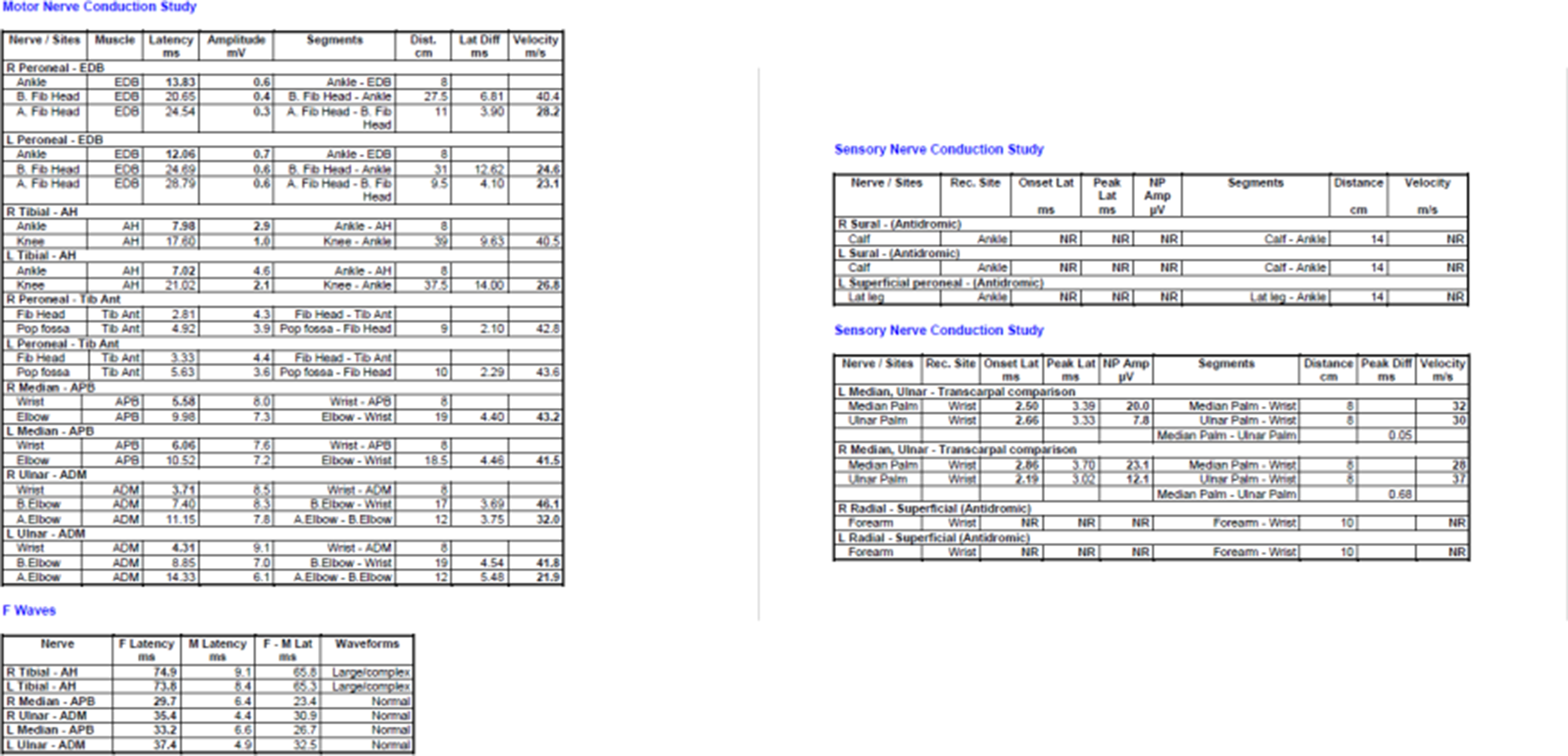
PP01.246
Infra-Clinical Small-Fiber Impairment in Hereditary Transthyretin Amyloidosis with Polyneuropathy: A Latin American Phenotypic Study
Prof. Edicson Ruiz-Ospina1,2, Prof. Sandra Milena Castellar-Leones1,2,3, Prof. Jorge Arturo Diaz-Ruiz1,2, Dr. Cristian Correa-Arrieta2, Dr. Diana Ramirez-Montaño4, Dr. Eduardo Echeverry4, Dr. Juan David Lopez5, Dr. Ana Laura Castro6, Dr. Juan Pablo Muñoz7, Dr. Diana Luzuriaga-Carpio8, Dr. Dario Zambrano-Vera9, Dr. Edison Vasquez10, Dr. Daniel Cesar Chávez10, Prof. Fernando Ortiz-Corredor1,2,3
1
Universidad Nacional de Colombia, Bogota, Colombia.
2
CIFEL, Bogota, Colombia.
3
Hospital Universitario Nacional de Colombia, Bogota, Colombia.
4
Clinica Imbanaco, Cali, Colombia.
5
Fundacion Valle de Lili, Cali, Colombia.
6
Hospital Universal Cartago, Cartago, Costa Rica.
7
Caja Costarricense de Seguro Social, Alajuela, Costa Rica.
8
Hospital General Manuel Ygnacio Monteros-IESS, Loja, Ecuador.
9
Hospital de Especialidades Carlos Andrade Marín-IESS, Quito, Ecuador.
10
Hospital Teodoro Maldonado Carbo-IESS, Guayaquil, Ecuador
Background: Hereditary transthyretin amyloidosis with polyneuropathy (hATTR-PN) is characterized by marked clinical heterogeneity, which frequently delays diagnosis and complicates individualized management. In Latin America, detailed phenotypic characterization remains limited, partly due to unequal access to specialized functional assessments and regional genetic variability. This study aimed to identify clinical sub-phenotypes of hATTR-PN in a multi-country Latin American cohort, with particular emphasis on small-fiber dysfunction (cold detection threshold [CDT] and heat pain threshold [HPT]) as an infra-clinical marker of disease progression.
Methods: We evaluated 105 patients from Colombia, Ecuador, and Costa Rica. Hierarchical cluster analysis using Ward’s D2 method was applied to a multidimensional dataset comprising 30 clinical, functional, sensory, and imaging variables. Clinical severity and symptoms were assessed using the Neuropathy Impairment Score (NIS), Norfolk Quality of Life–Diabetic Neuropathy (QOL-DN), COMPASS-31, and the Neuropathic Pain Scale (NPS). Functional performance was measured with the Nine-Hole Peg Test (NHP), monopodal static balance (MSB), Timed Up and Go (TUG), and the 5-item Sit-to-Stand test (SST5). Small-fiber function was assessed through CDT and HPT. High-resolution ultrasound was used to quantify the cross-sectional area (CSA) of the vagus nerve, brachial plexus, and peripheral nerves (median, ulnar, tibial, peroneal, and sural). Cluster stability was evaluated using bootstrap resampling (B = 100).
Results: Four distinct phenotypic clusters were identified. Cluster 2 (n = 34) showed the highest stability (Bootmean = 0.91) and represented a sub-clinical profile, with a median NIS of 0.00 and preserved MSB and NHP performance. Cluster 4 (n = 29; Bootmean = 0.71) corresponded to a “small-fiber predominant” phenotype, characterized by a high prevalence of infra-clinical sensory abnormalities (abnormal foot CDT in 72.4% and abnormal foot HPT in 58.6%), despite a median NIS of 0.00. Cluster 1 (n = 24; Bootmean = 0.51) included the most severe cases, with the greatest impairment in functional tests (NHP median: 28.7 s; MSB median: 4.1 s), higher autonomic involvement (COMPASS-31 median: 17.0), and vagus nerve CSA enlargement. Cluster 3 (n = 18; Bootmean = 0.63) demonstrated a distinct, focal infra-clinical state characterized by isolated sensory dissociation, with predominant HPT involvement in the hands (88.9% abnormal) while cold detection and distal lower-limb thresholds remained largely preserved. Significant differences across clusters were observed for NIS, NPS, NHP, MSB, and most ultrasound measures (p < 0.001), except for brachial plexus diameter (p = 0.373).
Conclusion: This analysis identifies clinically meaningful hATTR-PN phenotypes within a Latin American cohort. Comparison of Clusters 3 and 4 offers important insight into early disease stages: Cluster 3 reflects a localized infra-clinical state with predominant HPT involvement in the hands, whereas Cluster 4 represents a more advanced infra-clinical stage with widespread distal small-fiber dysfunction affecting both CDT and HPT. These findings support small-fiber impairment as the earliest marker preceding motor involvement, functional decline, and overt nerve enlargement, highlighting the importance of small-fiber assessment for early diagnosis in Colombian, Ecuadorian, and Costa Rican populations.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Hereditary Peripheral Neuropathies
Images or Table (Optional)
PP01.247
Toward Standardized Multidisciplinary Assessment of Charcot-Marie-Tooth Disease: Integrating MUNIX, Clinical Scales and Plasma Biomarkers
Dr. Francesco Gruosso1,2,3, Dr. Mariangela Goglia1,2, Dr. Erica Frezza1,2,3, Dr. Giulia Greco1,2, Dr. Giovanni Vietri1,2,3, Dr. Laura Boffa1,2, Prof. Diego Centonze1,3, Dr. Camilla Rocchi1,2, Dr. Antonio Petrucci4, Prof. Roberto Massa1,2,3
1
Department of Systems Medicine, University of Rome Tor Vergata, Rome, Italy.
2
Neuromuscular Diseases Unit, Tor Vergata University Hospital, Rome, Italy.
3
Unit of Neurology, IRCCS Neuromed, Pozzilli, Italy.
4
Center for Neuromuscular and Neurological Rare Diseases, San Camillo Forlanini Hospital, Rome, Italy
Background: Charcot-Marie-Tooth disease (CMT) is a spectrum disorder with variable progression and severity, but reliable biomarkers for predicting severity are lacking. This study aimed to widely evaluate whether combining neurophysiological measures, plasma biomarkers, and standardized clinical scales may identify independent predictors of severity across CMT subtypes and distinguish phenotype-specific patterns of disease progression.
Methods: Forty-six genetically confirmed CMT patients (PMP22, NEFL, GDAP1, MFN2, GJB1, HSBP1, LRSAM1, MPZ, KIF1A) performed standardized assessment at two Italian referral centres: clinical scales (CMTNSv2, NIS, ONLS, MRC) and quality-of-life measures (SF-36, MAM-16); neurophysiological evaluation, including conventional tests(CMAP, SNAP, MNCV) and advanced quantitative techniques: Motor Unit Number Index (MUNIX) in the abductor digiti minimi muscle, reflecting motor unit loss quantitatively; Motor Unit Size Index (MUSIX), assessing denervation-reinnervation dynamics. Plasma biomarkers (neurofilament light chain [NfL], phosphorylated-tau181 [p-tau181]) were measured in 22 patients via advanced immunoassays. Stratification by electrophysiological phenotype enabled phenotype-specific analysis (axonal 56.5%, demyelinating 43.5%), with multivariable regression models identifying independent severity predictors within and across phenotypes.
Results: MUNIX was considerably reduced in the whole cohort compared to normal reference values (57.7±34.2 vs ∼158±40 units, p<0.001), demonstrating a marked motor unit denervation characteristic of CMT pathophysiology. Notably, axonal phenotype showed significantly higher MUNIX (69.7±33.2) than demyelinating forms (40.2±28.4, p=0.005). Furthermore, MUNIX negatively correlated with clinical disability parameters, for instance, CMTNSv2 total severity (r=-0.467, p=0.002), suggesting how MUNIX may be an objective, quantitative marker of motor reserve. MUSIX (mean 103.6±48.4 μV) was elevated 52.4% above normal values, reflecting compensatory motor unit remodelling through collateral sprouting. NfL was elevated in 17.4% of patients and p-tau181 in 22.7%, but neither correlated significantly with clinical severity measures (NfL-CMTNSv2: ρ=-0.156, p=0.481; p-tau181-CMTNSv2: ρ=-0.102, p=0.713). This finding suggests these biomarkers may reflect acute denervation events rather than cumulative chronic disease burden, as in other disorders, such as amyotrophic lateral sclerosis (ALS). Multivariable regression showed MUNIX and disease duration as independent severity predictors (R²=0.650, p=0.011). Phenotype-stratified models found strong prediction in demyelinating forms (R²=0.862, p=0.037), in contrast to axonal ones (R²=0.530, p=0.450).
Conclusion: MUNIX emerges as the most robust, objective neurophysiological predictor of clinical severity in CMT disease, greater than plasma biomarkers and electrophysiological patterns alone when evaluated in univariate analysis. Phenotype-specific assessment is essential for precision monitoring and stratification: demyelinating forms benefit from MUNIX-based severity prediction with remarkably robust models (R²=0.862) more than axonal phenotypes, which require complementary biomarker strategies, as traditional neurophysiological measures and cross-sectional plasma markers incompletely capture severity drivers in these heterogeneous forms. Plasma biomarkers (NfL, p-tau181), although elevated in a minority of cases, may reflect acute denervation cascades rather than chronic disease burden, and their longitudinal utility warrants prospective validation. This integrated phenotype-stratified multimodal assessment, combining quantitative motor unit analysis, genetic and electrophysiological stratification, plasma biomarkers, and clinical standardized scales, provides a comprehensive framework for individualized outcome assessment and monitoring in genetically diverse CMT. Future longitudinal studies and clinical trials should employ phenotype-stratified models to enhance patient selection, outcome prediction, and treatment response monitoring in CMT.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Hereditary Peripheral Neuropathies
PP01.248
Nerve Ultrasound, Clinical and Electrophysiological Characterization of Pediatric Neuralgic Amyotrophy, HNPP and Mononeuropathies
Dr. Hanna Küpper1, Dr. Veronka Horber1, Dr. Tobias Haack2, Prof. Alexander Grimm3, Prof. Hendrik Rosewich1,4
1
Neuropediatric Department, University Hospital Tübingen, Tübingen, Germany.
2
Institute of Genetics, University Hospital Tübingen, Tübingen, Germany.
3
Neurology Department, University Hospital Tübingen, Tübingen, Germany.
4
Neuropediatric Department, University Hospital Göttingen, Göttingen, Germany
Background: Neuralgic amyotrophy and hereditary neuropathy with liability to pressure palsies (HNPP) are rare in childhood and youth. Consequently, only few studies have been published and little is known about their clinical, electrophysiological and imaging course at the pediatric age. A good diagnostic characterization as well as sensitive outcome parameters, however, are very important as a timely diagnosis and early, adapted treatment initiation are crucial concerning the clinical outcome. High-resolution nerve ultrasound (HRNUS) has emerged as a promising diagnostic method for evaluating neuropathies. Due to its adaptive nature, it is ideally suited for children. The here presented monocentric, longitudinal cohort study provides a thorough description of pediatric patients with neuralgic amyotrophy, HNPP and mononeuropathies, with a particular focus on ultrasound and treatment outcome.
Methods: High-resolution nerve and muscle ultrasound (Canon aplio a ultrasound system (Canon Medical Systems GmbH, Neuss, Germany)) was applied with a high-frequency linear array probe (18Mhz) with standardized imaging settings, in adjunction to detailed clinical and electrophysiological examinations at presentation and follow-up under treatment. The study included 7 patients with hereditary neuropathy with liability to pressure palsies (HNPP), 5 patients with neuralgic amyotrophy, 2 patients with neonatal plexus palsy and 12 patients with mononeuropathies of different etiologies (age range 5 months-17years 11 months). Follow-up ranged from 1 month to 3 years.
Results: Patients with HNPP presented mostly with a mononeuropathy while ultrasound depicted nerve enlargement at multiple entrapment sites. This frequently prompted a further diagnostic work-up and finally resulted in the genetic confirmation. Most patients improved with physiotherapy and orthotics, however neurosurgical decompression was sometimes necessary. Patients with neuralgic amyotrophy tended to have a more widespread clinical complaint, mostly in the upper extremity. Nerve ultrasound depicted enlargement of peripheral arm nerves as well as the brachial plexus. Most patients had a good effect of steroid treatment, however refractory courses were also noted. Mononeuropathy of unknown etiologies comprised mostly pressure palsies of the peroneal nerve at the fibula which showed good improvement after physiotherapy. Focal nerve enlargement were noted in more severe cases and improved in the course.
Conclusion: This cohort study depicted mostly a good clinical recovery of (mono-)neuropathic presentations in pediatric patients with HNPP, neuralgic amyotrophy and mononeuropathies of different etiologies. However, refractory courses were also noted. Nerve and muscle ultrasound served as a helpful tool for establishing the diagnosis and guiding the treatment evaluation, in adjunction to electrophysiological studies. Especially in children, it has a great potential to improve the diagnostic sensitivity in these rare disorders and, consequently, establish timely and adequate treatment.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Hereditary Peripheral Neuropathies
Images or Table (Optional)
PP01.249
Hereditary Neuropathy With Liability To Pressure Palsy (HNPP) Presenting As Bilateral Long Thoracic Nerve Injuries
Dr. Katrina Bernardo, Dr. Kenneth Chao
NYU Langone, East Meadow NY, United States
Background: Scapular winging has been recognized as a cause of shoulder dysfunction that leads to painful difficulties with elevation of the arm and lifting objects. The most common causes reported include long thoracic nerve injury resulting in medial winging and spinal accessory nerve injury causing lateral winging. These injuries typically result from mechanical compression or stretch injuries. Genetic causes are rare. HNPP also known as tomaculous neuropathy, is an autosomal dominant disease characterized by recurrent but typically painless entrapment palsies. Genetically it is associated with a 1.5 Mb deletion of locus 17p11.2 which contains the gene for peripheral myelin protein-22 (PMP22). HNPP usually presents with mononeuropathies affecting the median, ulnar, perineal nerve and least commonly the brachial plexus.
Methods: NA
Results: EMG: There is evidence of chronic denervation changes in the bilateral serratus anterior muscles, worse on the right. There is no abnormalities of tested nerves of the brachial plexus and muscles of the upper extremities including trapezius and rhomboid muscles.
MRI Brachial Plexus: Within normal limits
Athena Diagnostics: Positive PMP22, Exon Deleted 1.5
is evidence of chronic denervation changes in the bilateral serratus anterior muscles, worse on the right. There is no abnormalities of tested nerves of the brachial plexus and muscles of the upper extremities including trapezius and rhomboid muscles.
MRI Brachial Plexus: Within normal limits
Athena Diagnostics: Positive PMP22, Exon Deleted 1.5
Conclusion: This case report details a rarely described clinical presentation of HNPP as bilateral long thoracic mononeuropathies in a 20 year old male without severe pain. There have been a few reported cases of related brachial plexus involvement as the only expression of HNPP. A more commonly considered genetic cause affecting the brachial plexus include hereditary neuralgic amyotrophy (HNP), with attacks of plexopathy preceded by severe pain in the affected arm distinguishing HNA from HNPP. This case demonstrates the importance of an expanded differential in cases of younger patients presenting with scapular winging and/or brachial plexopathies.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Hereditary Peripheral Neuropathies
PP01.250
Plasma Phosphorylated Tau181 in Amyotrophic Lateral Sclerosis: Relationships With Clinical Measures and Disease Progression
Dr. Mariangela Goglia1, Dr. Erica Frezza1,2, Dr. Giulia Greco1, Dr. Francesco Gruosso1,2, Dr. Giovanni Vietri1,2, Dr. Laura Boffa1, Dr. Marzia Nuccetelli3, Prof. Diego Centonze4,2, Prof. Roberto Massa1,2
1
Neuromuscular Diseases Unit, Department of Systems Medicine, Tor Vergata University, Rome, Italy.
2
Unit of Neurology, IRCCS Neuromed, Pozzilli, Italy.
3
Division of Clinical Biochemistry and Clinical Molecular Biology, University of Rome Tor Vergata, Rome, Italy.
4
Department of Systems Medicine, Tor Vergata University, Rome, Italy
Background: Amyotrophic lateral sclerosis (ALS) is a neuromuscular disorder characterized by the progressive degeneration of upper and lower motor neurons. Recent studies have reported increased levels of blood phosphorylated tau181 (p-tau181) in patients with ALS compared to controls. According to the literature lower motor neuron (LMN) damage may represent a potential peripheral source of p-tau181 elevation. This hypothesis was supported by analyses of clinical and electromyographic measures of LMN impairment, as well as by neuropathological studies. These findings have raised concerns regarding the specificity of blood p-tau181 as a biomarker. More recently, skeletal muscle tissue has been proposed as an alternative peripheral source of p-tau. Despite these recent findings, our understanding of p-tau181 in ALS remains limited, and it is still unclear whether it could serve as a potential biomarker of lower motor neuron damage or offer insights into disease progression and prognosis.
Methods: The aim of this study was to evaluate blood p-tau181 levels in patients with ALS in relation to clinical parameters and disease progression rate. In this study we recruited patients diagnosed with ALS and a healthy control group. Patients underwent the ALS functional rating scale-revised (ALSFRS-R), MRC sum score and Penn upper motor neuron score (PUMNS). Disease progression rate was calculated as (48 – ALSFRS-R)/disease duration (months) and patients were stratified into fast and slow progressors. Plasma p-tau181 was detected both in patients and control group by CLEIA assay (Lumipulse).
Results: Eighteen patients (11 males and 7 females, 14 with spinal and 4 with bulbar onset) and 18 healthy controls were recruited. Median age was 65.8 years (IQR 13.8). Median disease duration was 12 months (IQR 11.5). Median ALS-FRS-R score was 40 (IQR 6.0). Eleven out of eighteen patients were slow progressors. Median p-tau181 level was 3.99 pg/mL (IQR 3.75) in ALS patients and 1.08 pg/mL in the control group (IQR 0.7). P-tau181 levels were significantly increased in ALS patients compared to the control group (p < 0.001). We found no correlations between ALS clinical parameters and p-tau181 levels. A negative correlation between p-tau181 and disease progression rate was observed (rho -0.5; p = 0.03). Moreover, slow progressors had higher p-tau181 values than fast progressors (p = 0.01).
Conclusion: According to recent studies, plasma p-tau181 levels were significantly higher in ALS patients compared with healthy controls. No associations were observed between p-tau181 levels and clinical measures. In contrast, p-tau181 showed a significant inverse correlation with disease progression rate, with higher levels observed in slow progressors compared to fast progressors. These results suggest that p-tau181 might be linked to slower functional decline, potentially reflecting predominant lower motor neuron involvement, a pattern frequently associated with slower disease progression. At the same time, its behaviour could reflect both the extent of lower motor neuron damage and the degree of denervation, suggesting that plasma p-tau181 may follow different trajectories depending on the stage of disease. Larger cohorts and longitudinal studies are required to clarify whether plasma p-tau181 could reflect or influence the rate of functional decline in ALS.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: Biomarkers in MND
PP01.251
A Longitudinal Evaluation of Cardiovascular Autonomic Reflex Testing in Hereditary Transthyretin Amyloidosis
Dr. Mariangela Goglia1, Dr. Erica Frezza1,2, Dr. Giulia Greco1, Dr. Francesco Gruosso1,2, Dr. Giovanni Vietri1,2, Dr. Laura Boffa1, Prof. Diego Centonze3,2, Prof. Roberto Massa1,2, Dr. Camilla Rocchi4
1
Neuromuscular Diseases Unit, Department of Systems Medicine, Tor Vergata University, Rome, Italy.
2
Unit of Neurology, IRCCS Neuromed, Pozzilli, Italy.
3
Department of Systems Medicine, University of Rome Tor Vergata, Rome, Italy.
4
Neurology Unit, Policlinico Tor Vergata, Rome, Italy
Background: Hereditary transthyretin amyloidosis (hATTR) is a systemic disease predominantly affecting peripheral nerves and heart. Autonomic dysfunction is an undervalued feature of hATTR caused by small nerve fiber involvement. Autonomic involvement is often subtle and undetected if not specifically investigated. We conducted an observational, retrospective, longitudinal study in order to assess cardiovascular autonomic function in patients carrying ATTR pathogenic mutations starting at presymptomatic stage.
Methods: Patients underwent to cardiovascular reflex test (CRTs) including head-up tilt test (HUTT), Valsalva manoeuvre (VM), deep breathing, cold face and hand-grip test. CRTs were conducted in Policlinico Tor Vergata Autonomic Unit under continuous blood pressure (BP) and heart rate (HR) monitoring. On the same day patients performed nerve conduction studies (NCS) to assess large fiber involvement. A 60 months (5 years) median follow up time was considered.
Results: 10 patients were enrolled in the study (6 female and 4 male). All patients carried Val30Met TTR mutation. Median age was 50 years at first evaluation. At first assessment (T0) none of the patients had polyneuropathy at NCS; carpal tunnel syndrome was detected in 4 out of 10 subjects. CRTs revealed lower respiratory sinus arrythmia at deep breathing test (Δ I-E) in 2 out of 10 patients. Other CRTs were within the normal ranges.
At 5 years (T5) follow up, 2 out of 10 patients developed an axonal length-dependent motor-sensory polyneuropathy. Deep breathing (Δ I-E), was out of normal ranges in 4 subjects with significantly lower values in T5 compared to T0 evaluation (p = 0.05). Five and three patients had pathological responses to cold face and hand grip test, respectively.
During HUTT, HR changes at the third minute significantly decreased between T0 and T5 (p = 0.006). During VM, a decrease of Valsalva ratio was observed in T5 assessment (p = 0.07). Cold face test revealed significant lower responses in both diastolic blood pressure (DBP) and HR at T5 (p = 0.03 and 0.06, respectively). SBP and HR were also significantly decreased during hand grip test at T5 (p = 0.01, 0.008).
Conclusion: Longitudinal assessment of CRTs in hATTR patients revealed an involvement of autonomic system also in asymptomatic subjects without polyneuropathy. Involvement of cardiovagal response could be considered as an early marker of autonomic dysfunction. Sympathetic adrenergic dysfunction was also observed at follow up evaluation. A complete battery of standardized CRTs could be useful to detect initial peripheral nervous system involvement in hATTR.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Hereditary Peripheral Neuropathies
PP01.252
Investigating Sympathetic Small Fibers Using the Skin Wrinkle Test in Patients with Amyotrophic Lateral Sclerosis
Dr. Otto Hernandez Fustes1,2, Miss Thalita Aparecida dos Santos1, Mr. Webert Alex Santos Benetti1, Mr. Edson Hideki Kawamura Junior1, Dr. Paulo Jose Lorenzoni1,2, Dr. Cláudia Suemi Kamoi Kay2, Dr. Renata Dal-Pra Ducci Cirino1,2, Dr. Paula Raquel do Vale Pascoal Rodrigues2, Dr. Rosana Herminia Scola1
1
Federal University of Paraná,, Curitiba, Brazil.
2
Clinics Complex Hospital at Federal University of Paran, Curitiba, Brazil
Background: The skin wrinkling test (SWT) is a noninvasive clinical method used to assess sympathetic small fibers associated with vasomodulation, pain, and temperature regulation. Amyotrophic lateral sclerosis (ALS), a condition recently described as not only motor―but also sensory―in nature, has been evaluated using SWT. Studies have revealed that the SWT index reflects the density of intraepithelial fibers in skin biopsy.
Methods: This observational, cross-sectional study was conducted at a tertiary care hospital and included 27 patients diagnosed with ALS and 109 volunteers. All participants underwent the SWT, involving immersion of the right hand in water (temperature, 40°C) for 30 min. Skin wrinkling was assessed by 3 observers using 2 distinct scales. All were undergoing drug treatment with riluzole. The time of disease evolution and ALSFRS-R functional scale were recorded.
Results: In relation to the study samples, regarding age, the ALS group was statistically different from the control group (p<0.001) but with no statistical difference in relation to the paired group (p=0.403), indicating that the ALS group was homogeneous in relation to the paired. Interobserver agreement was excellent (p<0.001) across all 3 groups; significant agreement was also found between the scales. Quantitative analysis revealed that the ALS group exhibited significantly lower mean scores on Scale 1 (8.23 versus [vs.] 12.78; p=0.001) than controls, and on Scale 2 (0.52 vs. 0.86, and 0.84; p<0.002) than both controls and matched controls. Qualitatively, patients diagnosed with ALS exhibited a higher frequency of negative results (p=0.001 and p=0.013 for Scales 1 and 2, respectively).
Disease severity was not associated with a greater probability of the test having a positive or negative result on scale 1 (p=0.329) or 2 (p=0.914). Similarly, the test results were not associated with the length of ALS progression (p=0.283 and p=0.779, respectively)
Conclusion: A relevant result of our study was the pilot investigation on the level of agreement between different SWT evaluators for the same individual. Our evaluators, comprising 2 initial evaluators and 1 tiebreaker evaluator, qualitatively and quantitatively graded the results produced by the SWT in each study participant. Using the ICC for interobserver reliability, we found excellent agreement between the 2 initial evaluators, with p<0.001 for the control, paired, and ALS groups.
SWT demonstrated excellent inter-rater agreement. Significant differences were observed between patients with ALS and controls, indicating the involvement of sympathetic fine fibers in patients with ALS. These findings highlight the utility of the SWT as a tool for assessing the sensory components of ALS.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: ALS: Epidemiology and Clinical Features,
PP01.253
Nocturnal Oximetry in Neuromuscular Diseases
Dr. João Guilherme de Toledo Justo1, Dr. Eduardo Hummelgen1, Miss Lara Godoy2, Prof. Sara Giampá3, Prof. Otto Hernandez Fustes2,4
1
Hospital do Rocio, Campo Largo, Brazil.
2
Universidade Federal do Paraná, Curitiba, Brazil.
3
Biologix Institute, São Paulo, Brazil.
4
Hospital de Reabilitação, Curitiba, Brazil
Background
Introduction: Neuromuscular diseases (NMDs) comprise a group of disorders that topographically affect the spinal cord, nerve roots, plexuses, peripheral nerves, the neuromuscular junction, and muscles. Respiratory system involvement in these diseases may lead to acute and/or chronic respiratory failure related to weakness of the respiratory musculature. Chronic restrictive respiratory failure is the most common ventilatory disorder in chronic neuromuscular diseases, characterized by pulmonary hypoventilation, with nocturnal hypoventilation being the earliest clinical manifestation. Nocturnal oximetry is a simple method for screening and diagnosing nocturnal hypoventilation and has proven to be more comfortable, inexpensive, and accessible, as it objectively measures intermittent hypoxia.
Objectives: To analyze and compare features detectable by nocturnal oximetry in patients with NMDs.
Methods: Ambulatory nocturnal oximetry was performed using a wireless oximeter (Oxistart, Biologix Sistemas Ltd., Brazil) with an embedded accelerometer, placed on the second digit. The Oxistart firmware acquires 100 samples per second, generating beat-to-beat raw SpO₂ data with a resolution of 0.1%. A moving time average of four cardiac beats was applied. Data obtained by Oxistart are transferred via a smartphone application to the cloud, where they are automatically analyzed using a proprietary algorithm. The following variables were analyzed: age, oxygen desaturation index (ODI), number of desaturations, time spent at specific oxygen saturation levels, and sleep apnea. The study was approved by the Ethics Committee of the Workers’ Hospital Complex (approval number: 7,501,240). Patients received detailed explanations regarding the study and the examination protocol and subsequently signed an informed consent form.
Results: Twenty-two consecutive patients with confirmed diagnoses were evaluated and divided into two groups: one with polyneuropathies and another with other NMDs, with 11 patients in each group, predominantly male (54.55%). The mean ODI was higher in the polyneuropathy group (25.1) compared with the other NMD group (18.9), as was the number of desaturations (144.5 vs. 122.8). Regarding sleep apnea classification, the proportion of patients classified as severe was identical in both groups (18.18%). In the polyneuropathy group, a higher number of patients were classified as normal (4 patients; 36.36%).
Conclusion: Nocturnal oximetry showed abnormalities in 72.7% of the patients studied. There were no statistically significant differences in the analyzed variables between the two groups. These preliminary results support the continuation of this study to determine the value of nocturnal oximetry in NMDs as a tool for the early detection of hypoxemia.
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: Cardiac and respiratory issues in neuromuscular diseases.
PP01.254
Historical Notes on Spinocerebellar Ataxia Type 2. Contributions from Cuban Physicians
Dr. Otto Hernandez Fustes1,2, Dr. Carlos Arteaga Rodriguez3,4, Dr. Hélio Ghizoni Teive1
1
Federal University of Paraná, Curitiba, Brazil.
2
Clinics Complex Hospital at Federal University of Paraná, Curitiba, Brazil.
3
Positivo University, Curitiba, Brazil.
4
Pontifical Catholic University of Paraná, Curitiba, Brazil
Background: The term “ataxia” refers to a lack of coordination in movements and was recognized in nineteenth-century medicine, initially as a manifestation of other neurological diseases. In 1863, the German physician Nicolas Friedreich made the first clinical description of a familial hereditary ataxia, characterized by childhood onset, autosomal recessive inheritance, and progressive neurological symptoms. Then in 1893, the French neurologist Pierre Marie identified another type of hereditary ataxia with a late-onset autosomal dominant pattern, distinct from that described by Friedreich. These contributions established the conceptual foundations for hereditary ataxias today.
In Cuba, during the second half of the nineteenth century (colonial period), local physicians began documenting cases of “locomotor ataxia,” a term coined by Duchenne de Boulogne in 1864.
Cuban Heredoataxia, the original description of spinocerebellar ataxia type 2 (SCA2), is an autosomal dominant inherited neurodegenerative disease that affects the central nervous system, particularly the cerebellum, brainstem, and spinal cord.
Methods: This qualitative, historical-descriptive study analyzes the historical aspects of Cuban Heredoataxia.
Methodological Procedures
a) Literature and Documentary Review
A systematic review of the scientific and documentary literature was conducted, including articles indexed in PubMed, SciELO, LILACS, and Google Scholar. The keywords used were: Cuban Heredoataxia, spinocerebellar ataxia type 2, history of medicine in Cuba, Cuban population genetics, and neurodegenerative diseases.
b) Interviews with Experts
Results: Spinocerebellar ataxia type 2 (SCA2) has a worldwide distribution, with its first clinical description in 1971, in India, by Wadia & Swami. However, SCA2 is most frequently found in Cuba (in the province of Holguín) and is recognized worldwide as Cuban heredoataxia. In 1989, Orozco et al. published an outstanding paper on SCA2 describing its clinical, neuropathological, and biochemical features. This article proposes a historical analysis of Cuban Heredoataxia, based on a bibliographical and documentary review, with an understanding of the scientific path of research since its identification in the 1970s related to contemporary research strategies. The role of the four main Cuban researchers—Estrada, Valles, Orozco, and Pérez—is highlighted in the recognition of this rare disease.
Conclusion: The first Cuban contributions to understanding Cuban Heredoataxia, currently known as spinocerebellar ataxia type 2, range from the historical identification of cases and families in Holguín to detailed clinical descriptions and large-scale population studies. The contributions of Cuban neurologists, supported by international collaborations, allowed delineation of the clinical profile, genetic pattern, and regional concentration of the disease. Cuban Heredoataxia offers a unique case in the history of Latin American medicine, combining high local incidence, a structured medical response, and a socially engaged approach. The historical study of the disease allows reflection on the links between genetics, territory, and public policies, highlighting the role of science in contexts of limited resources.
Abstract Topic Groups (Submission Categories)
Topic Group 9 – History: N/A
PP01.255
Late-Onset Hereditary Transthyretin Amyloidosis (Attr) with Polyneuropathy in an Elderly Person
Dr. Otto Hernandez Fustes1,2, Dr. Ádria Rodrigues da Silva2, Dr. Cláudia Suemi Kamoi Kay2, Dr. Paulo Jose Lorenzoni1,2, Dr. Renata Dal-Pra Ducci Cirino1,2, Dr. Paula Raquel do Vale Pascoal Rodrigues2, Dr. Erika Christina Silva2, Dr. Rosana Hermínia Scola1
1
Federal University of Paraná, Curitiba, Brazil.
2
Clinical Complex Hospital at Federal University of Paraná, Curitiba, Brazil
Background: Hereditary transthyretin amyloidosis (ATTRv) is an autosomal dominant genetic disorder caused by mutations in the transthyretin (TTR) gene. Mutations in the TTR gene lead to structural instability of the protein, resulting in tetramer dissociation, release of mutant (or even wild-type) monomers, aggregation into amyloid fibrils, and extracellular deposition. These deposits compromise multiple organs, most notably the peripheral nervous system and the heart. Over 130 TTR variants have been identified, most of which are pathogenic. The most common worldwide is Val30Met—endemic in regions such as Portugal, Brazil. The clinical features of ATTRv-PN depend on the types of nerve fibers affected, which in turn are influenced by whether the disease presents with early-onset (<50 years) or late-onset (≥50 years), as well as by the disease stage.
Methods: A case presentation.
Results: An 82-year-old man with a history of hypertension, undergoing treatment for heart failure, presented with chronic diarrhea associated with hypotension, dehydration, and decline in overall condition. He reported a two-year history of progressive muscle weakness initially affecting the distal region of the right lower limb, later involving the contralateral limb, accompanied by paresthesias. Approximately one year prior, he developed difficulty grasping objects, dysphagia, dysphonia, and recurrent diarrheal episodes. Physical examination revealed tetraparesis, predominantly distal and slightly asymmetric, inability to ambulate, and axial instability while seated; absent patellar and Achilles reflexes; globally reduced tactile and pain sensation, more pronounced up to the knees and elbows, with loss of joint position sense and reduced vibration sense to the patellar level. Electroneuromyography revealed an axonal sensorimotor polyneuropathy. Minor salivary gland biopsy revealed material compatible with amyloid, and genetic testing for hereditary transthyretin (TTR) amyloidosis identified the V30M variant.
Conclusion: In the present study, we draw attention to the late onset of clinical manifestations in ATTR with polyneuropathy (PN), presenting an 82-year-old man with a length-dependent, predominantly distal, asymmetrical axonal sensorimotor polyneuropathy associated with marked autonomic symptoms and cardiac involvement.
In the context of hereditary transthyretin amyloidosis, recognizing late-onset cases is crucial, as these patients often present with manifestations that pose particular diagnostic challenges in primary healthcare settings. Such cases are frequently misdiagnosed as idiopathic axonal neuropathies or other more common conditions in the elderly population. Therefore, a high index of suspicion should prompt healthcare professionals to refer patients promptly to specialized centers.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Hereditary Peripheral Neuropathies
PP01.256
CMT-1C c.334G>A LITAF Mutation: A Case Report of Clinical Features and Electrodiagnostic Findings
Prof. Pariwat Thaisetthawatkul, Assoc. Prof. Mara Seier
Department of Neurological Sciences, University of Nebraska Medical Center, Omaha, NE, United States
Background: Charcot-Marie-Tooth type 1C (CMT 1C) is a rare, dominantly inherited neuropathy caused by mutation in the lipopolysaccharide-induced tumor necrosis factor (LITAF) gene. The frequency has been estimated to be about 0.5% of all CMT1 cases. Here we report a genetically confirmed case of CMC-1C with mild sensory symptoms and signs without motor weakness or gait disturbance.
Methods: A case report
Results: A 69-year-old male, with underlying Parkinson disease responding to carbidopa-levodopa treatment and diabetes mellitus, started having numbness, coldness and neuropathic pain in both feet for 5 years prior to evaluation. The sensory symptoms later progressed to both hands. There were no cranial nerve symptoms, limb weakness, falls or loss of balance. Family history revealed CMT of unknown type in his sister and mother. Neurological exam revealed clinical signs of Parkinsonism including tremor, bradykinesia and rigidity but normal cranial nerve functions, no motor weakness, normal deep tendon reflexes, absent vibration sense from toes to knees but normal pin and proprioceptive senses, normal cerebellar functions and ability to walk by himself with mild stooping posture. Electrodiagnostic test revealed findings of chronic demyelinating neuropathy with uniform, marked slowing with conduction blocks and dispersion between distal and proximal limb recording in motor conduction studies. Laboratory investigation revealed normal CSF analysis and unremarkable blood tests. Genetic testing showed pathogenic heterozygous c. 334G>A mutation of LITAF gene diagnostic of CMT1C.
Conclusion: We report a case of CMT1C, a rare form of CMT1, that presents without motor weakness, and mild clinical signs with predominantly sensory symptoms without or with minimal gait disturbance consistent with previous reports. The symptoms and signs in CMT1C were reported to be generally milder than those of CMT1A. Electrodiagnostic findings in CMT1C shows uniform, marked slowing in demyelination ranges and may include conduction blocks and dispersion in motor conduction studies.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Hereditary Peripheral Neuropathies
PP01.257
Single-Centre Experience on Genotypic and Phenotypic Features of Southern Brazilian Patients With Hereditary Neuropathies
Prof. Paulo José Lorenzoni, Mr. Lucas Cesar Werneck, Mr. Igor Baroni Cardoso, Prof. Otto Jesus Hernandez Fustes, Dr. Paula Raquel do Vale Pascoal Rodrigues, Prof. Renata Dal-Pra Ducci, Dr. Claudia Suemi Kamoi Kay, Prof. Rosana Herminia Scola
Universidade Federal do Paraná, Curitiba, Brazil
Background: Hereditary neuropathies comprise a group of genetically determined disorders that primarily affect the peripheral nerves and exhibit marked clinical heterogeneity. Their genetic aetiology is highly diverse, with more than 100 genes described, and the prevalence of specific subtypes varies according to inheritance pattern and geographical origin.
Methods: This study analysed a cohort of patients with hereditary neuropathies diagnosed at a single referral centre for neuromuscular diseases in Southern Brazil. The aim was to contribute to disease characterisation and to determine the distribution of neuropathy subtypes in this region, thereby defining the genetic profile of affected patients.
Results: A total of 86 patients with suspected hereditary neuropathy were included. Next-generation sequencing panel analysis identified pathogenic or likely pathogenic variants in 48 patients; however, four individuals harbouring variants in genes associated with other disorders (SCN10A, ATP7A, MYH14, and SIGMAR1) were excluded. Among the remaining 44 patients, variants were identified in the following genes: PMP22 (n = 19), TTR (n = 9), GJB1 (n = 6), MORC2 (n = 3), SPTLC1 (n = 1), LRSAM1 (n = 1), ATP1A1 (n = 1), SH3TC2 (n = 1), MFN2 (n = 1), GDAP1 (n = 1), and POLG (n = 1). Distinct variants affecting different exons were observed across most genes. Recurrent findings included duplication of exons 1–5 in PMP22, recurrent TTR variants (p.Val50Met, p.Val142Ile, and p.Val127Ile), and the GJB1 frameshift variant p.Trp132Valfs*14.
Conclusion: This study provides a comprehensive phenotypic and genotypic characterisation of hereditary neuropathies in a Southern Brazilian cohort. The distribution of genetic subtypes was compared with national and international data, contributing to a better understanding of the regional genetic landscape of hereditary neuropathies.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Hereditary Peripheral Neuropathies
PP01.258
Multidimensional Analysis of hATTR Polyneuropathy Burden: A Latin American Cohort Study
Prof. Sandra Milena Castellar-Leones1,2,3, Dr. Cristian Correa-Arrieta2, Prof. Edicson Ruiz-Ospina1,2, Prof. Jorge Arturo Diaz-Ruiz1,2, Dr. Diana Luzuriaga-Carpio4, Dr. Dario Zambrano-Vera5, Dr. Daniel César-Chávez6, Dr. Edison Vasquez6, Dr. Juan Pablo Muñoz7, Dr. Ana Laura Castro8, Dr. Eduardo Echeverry9, Dr. Juan David Lopez10, Dr. Diana Ramirez-Montaño9, Prof. Fernando Ortiz-Corredor1,2,3
1
Universidad Nacional de Colombia, Bogota, Colombia.
2
CIFEL, Bogota, Colombia.
3
Hospital Universitario Nacional de Colombia, Bogota, Colombia.
4
Hospital General Manuel Ygnacio Monteros-IESS, Loja, Ecuador.
5
Hospital de Especialidades Carlos Andrade Marín-IESS, Quito, Ecuador.
6
Hospital Teodoro Maldonado Carbo-IESS, Guayaquil, Ecuador.
7
Caja Costarricense de Seguro Social, Alajuela, Costa Rica.
8
Hospital Universal Cartago, Cartago, Costa Rica.
9
Clinica Imbanaco, Cali, Colombia.
10
Fundación Valle de Lili, Cali, Colombia
Background: Hereditary transthyretin amyloidosis with polyneuropathy (hATTR-PN) is a progressive, multisystemic, and life-threatening condition. In Latin America, the disease exhibits high genetic diversity and heterogeneous clinical manifestations, which are traditionally assessed through linear neurological scales. However, these conventional metrics may not fully capture the multidimensional spectrum of patient burden. This study aims to map the clinical landscape of hATTR-PN in a cohort from Colombia, Costa Rica, and Ecuador, identifying specific burden domains to guide personalized medical and rehabilitative interventions.
Methods: We analyzed a cohort of 105 patients, integrating demographic data with ten multidimensional functional and clinical variables. To characterize neurological impairment, we utilized the Neuropathy Impairment Score (NIS) and the Norfolk Quality of Life-Diabetic Neuropathy (QOL_DN) scale. Autonomic dysfunction was assessed using the Composite Autonomic Symptom Score 31 (Compass31), while neuropathic pain was quantified via the Numerical Pain Scale (NPS). Physical performance and functional mobility were evaluated through the Timed Up and Go (TUG) and the Five-Times Sit-to-Stand (SST5) tests. Postural stability was specifically measured using the Monopodal Static Balance (MSB) test, recorded as the maximum duration of a one-legged stance in seconds. To identify the underlying structures of disease impact, we performed a Principal Component Analysis (PCA), mapping a “Burden Space” to evaluate the influence of mutation type and age as vectors within the distribution of physical, functional, and autonomic disability. Statistical significance for age and sex was assessed via Spearman correlation and Wilcoxon tests, while genotype influence was validated using Monte Carlo permutation tests to account for low-frequency mutations.
Results: The cohort (mean age 49.47 years; 55.2% female) presented a diverse genetic profile led by Ser43Asn (33.3%), Val50Met (24.8%), and Val142Ile (22.9%). PCA identified two axes explaining 65.9% of the variance: PC1 (55.2%) represents the “Physical-Functional Core” and PC2 (10.7%) the “Sensory-Autonomic Axis.” Age emerged as the primary driver of physical burden (p < 0.001), showing a direct correlation with progression toward the functional core. While sex (p = 0.093) and country of origin (p = 0.077) showed marginal trends, the physical burden remained relatively consistent across the region. Genetic mapping confirmed that while genotype does not dictate absolute physical severity (p = 0.55), specific mutations like Ser43Asn exhibit a broader phenotypic cloud. Notably, Monopodal Static Balance (MSB) identified a unique stability-deficit cluster, likely driven by individual clinical particularities such as orthopedic comorbidities, sensory paresthesias, or edema, marking a critical area for early intervention independent of gait capacity.
Conclusion: The global burden of hATTR-PN in Latin America is multidimensional and primarily driven by age-related functional decline. We identified three actionable clinical phenotypes: 1) Age-dependent functional decline requiring progressive physical rehabilitation, 2) Genotype-specific variability (notably in Ser43Asn) requiring multidisciplinary monitoring, and 3) Stability-specific burden identified through MSB, requiring early fall-prevention strategies. This mapping allows for personalized interventions tailored to the patient’s specific clinical coordinates and comorbidities within the Latin American burden landscape.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Hereditary Peripheral Neuropathies
Images or Table (Optional)
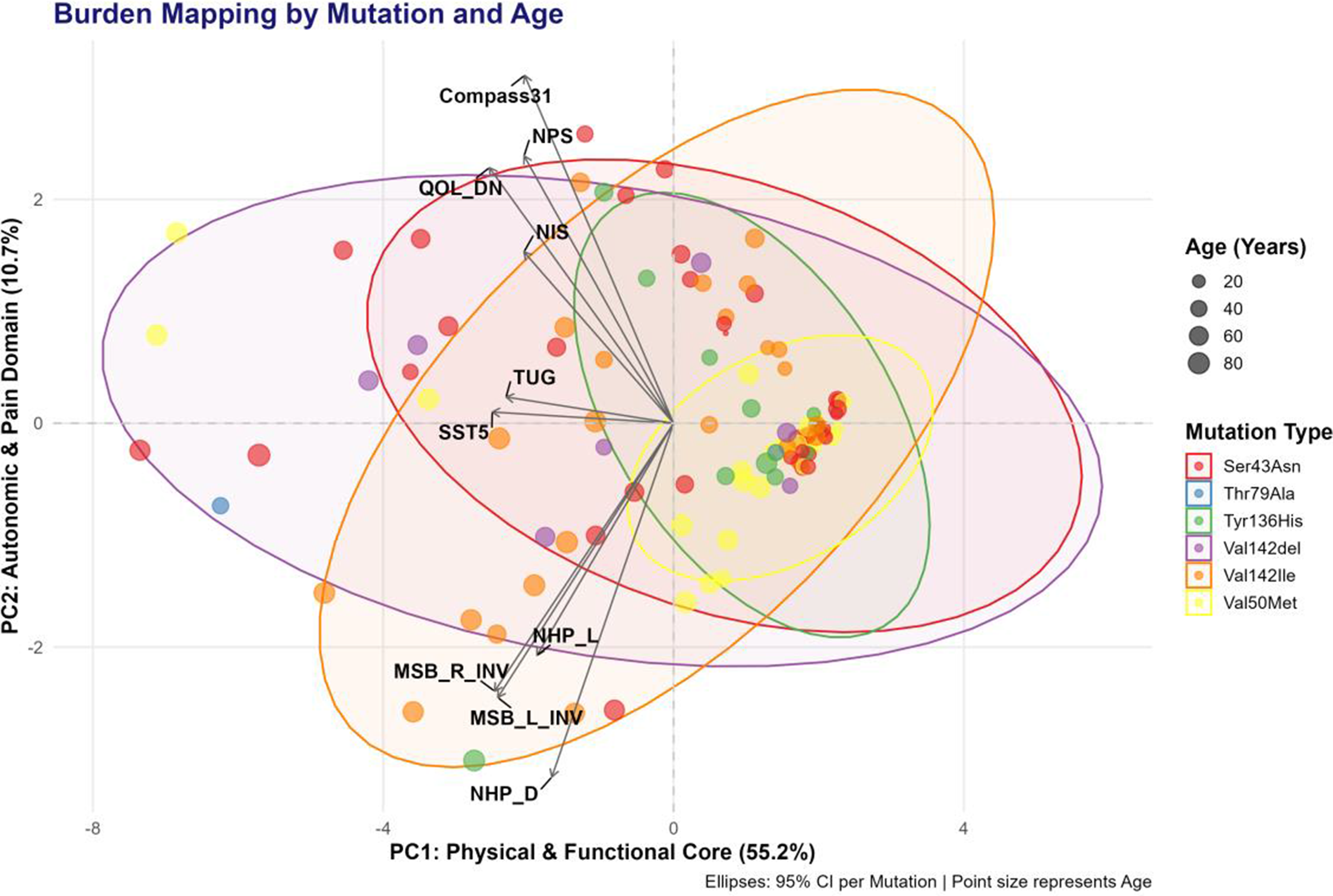
PP01.259
The Genetic Profile of Hereditary Neuropathies in Southern Africa
Dr. Tarin Europa1, Dr. Stanley Zimba2, Dr. Niki Floudiotis1, Dr. Somwe Wa Somwe2, Dr. David Bearden3, Dr. Michelle Kvalsund3, Prof. Jeannine Heckmann1, Dr. Melissa Nel1
1
University of Cape Town, Cape Town, South Africa.
2
University Teaching Hospital, Lusaka, Zambia.
3
University of Rochester Medical Center, New York, United States
Background: Genetic data in Africans with hereditary neuropathies, collectively called Charcot-Marie-Tooth disease (CMT), is sparse. Genetic testing for CMT in South Africa (SA) is mostly limited to PMP22 screening for CMT1, but no testing is available in Zambia (ZM). To date, CMT1, the most common hereditary neuropathy in the Global North, has not been described in individuals with black African ancestry. Aim: We report the genetic profile of patients with suspected CMT, enrolled in the International Centre for Genomic Medicine in Neuromuscular Diseases (ICGNMD) Consortium at two centres in Cape Town, SA and Lusaka, ZM.
Methods: Between 2019 and 2025, consecutive patients with suspected CMT were enrolled. Electrophysiological tests categorised patients into demyelinating CMT1 (median nerve conduction velocity <25 m/s,), axonal CMT2 (>45 m/s,) or intermediate CMTi (25-45 m/s). Additional phenotypes included those with either sensory or motor only electrophysiological phenotypes, or “complex” for those with >2 additional neurological/multisystemic features. PMP22 assays were performed for probands with CMT1/CMTi, RFC1 assays for those with possible sensory ataxic neuropathy, and the remainder underwent next generation sequencing (NGS).
Results: Overall, we report on 71 probands. In 52 SA probands, with median age at symptom onset of 17 years (IQR 10;40), genetic diagnoses were determined in 50%. In six demyelinating neuropathies, five with European ancestry and one admixed ancestry, five showed a PMP22 duplication and one PMP22 deletion. One family with late-onset demyelinating neuropathy had an MPZ variant of uncertain significance.Only one European ancestry family was diagnosed with cerebellar ataxia, neuropathy and vestibular areflexia syndrome (CANVAS). Of the axonal CMTs, the most frequent causes were heterozygous MFN2 pathogenic variants in three probands (admixed ancestry), and compound heterozygous variants in MPV17 (p.Gln36Ter and p.Arg125Trp) in four probands with African ancestry. The remainder comprised single cases of axonal neuropathy (GJB1, ATP1A1, MORC2, HSPB1, GAN and ADPRS), and “complex” neuropathy syndromes (PEX11B, SERAC1, RFT1, TDP1 and ATM). In 19 ZM probands, with median age at symptom onset of 5 years (IQR 2; 11) and axonal CMT, genetic diagnoses were determined in 47% by NGS. No cases with demyelinating neuropathy were found. The most frequent axonal CMT was due to MFN2 variants, diagnosed in four probands. The remaining solved cases comprised single probands with axonal CMT and variants in DYNC1H1, VWA1, NDRG1, MME and TDP1. A homozygous TDP1 variant (p.His493Arg) was shared between a ZM proband and a proband from Malawi (but living in SA), both with sensory ataxic neuropathy.
Conclusion: The genetic profile of CMT in Africa differs from the global North. The most outstanding feature is the absence of demyelinating neuropathies or pathogenic PMP22 variants in those with African ancestry. Overall, MFN2-CMT was the most common (10%) but the MPV17-associated axonopathy was more frequent in cases from SA. The mutational profile of CMT in this heterogenous cohort of participants living in Southern Africa, suggested that NGS provides the best diagnostic yield.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Hereditary Peripheral Neuropathies
PP01.260
Focal CIDP Variants with Brachial Plexus Involvement Compared to Neuralgic Amyotrophy – Similarities and Differences
Assoc. Prof. Alexander Grimm1, Dr. Katharina Kneer2, Dr. Helene Hurth3, Assoc. Prof. Martin Schuhmann3, Dr. Nathalie Winter2, Dr. Jan-Hendrik Stahl2
1
University hospital of Neurology, Tübingen, Germany.
2
University hospital of Neurology, Tuebingen, Germany.
3
University hospital of Neurosurgery, Tuebingen, Germany
Background: CIDP variants and neuralgic amyotrophy (NA) may present with similar symptoms that make differentiation difficult, i.e. focal palsies, sensory disturbances and pain. We therefore compared imaging tools, electrophysiology, disease course, and treatment response in treatment-naive and newly diagnosed patients suffering from either of these disorders.
Methods: CIDP and NA with primary brachial plexus and arm nerve involvement were included in our study. All patients underwent CSF and blood analysis, MR neurography, high-resolution ultrasound (HRUS), and electrophysiological studies (EDx). HRUS was performed on the median, the ulnar, the radial nerve along its course, as well as from nerve roots C5 and C6. All patients were initially treated with steroids.
Results: We included 22 patients (11 CIDP and 11 NA patients). MRI neurography of the brachial plexus was considered abnormal in 9 NA and 7 CIDP patients, whereas HRUS abnormalities could be differentiated in generalized and diffuse enlargements, seen in 11 NA patients, and focal enlargement, seen in 2 CIDP patients. Peripheral nerve enlargement affected proximal nerve segments in both patient groups equally, nerve enlargement in distal nerve segments was more often seen in CIDP patients, but did not statistical significance.
Lumbal puncture was unremarkable in 11 patients (7 NA, 4 CIDP), proteinaemia was seen in 5 patients (2 NA, 3 CIDP) and pleocytosis in 2 patients, both with CIDP. The differences were not statistically significant.
Surprisingly, in 8/10 focal variants of CIDP, EDx was not demyelinating but rather axonal. In NA, EDx was always predominantly axonal (p>0.05).
9/10 CIDP patients initially improved to repeated steroid pulse, however ongoing treatment switched to IVIG or others was needed in 8/10 patients.
7 out of 11 patients with relapsing-remitting NA did not sufficiently improve to repeated steroids in all affected nerve segments (p<0.001 compared to CIDP), constrictions and fascicular entwinements were found in all NA patients and were limited to this patient group.
Conclusion: Differentiation between NA and focal CIDP variants remains clinically challenging. MRI and HRUS often show similar abnormalities, whereby a diffuse and generalized enlagement is more often seen in NA patient. Further, differentiation between both disease entities may be possible by identifying distal nerve enlargement or nerve constrictions in a relapsing treatement course.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Immune-mediated Neuropathies
PP01.261
Riliprubart in CIDP: Time and Magnitude of Response Analysis from a Phase 2 Trial
Dr. Annie Dionne1, Dr. Richard A. Lewis2, Dr. Jie Lin3, Dr. Yi Lu4, Dr. Miguel Alonso-Alonso5, Dr. Asif Paker5, Dr. Luis Querol6,7
1
CHU de Quebec Universite Laval, Quebec, Canada.
2
Department of Neurology, Cedars Sinai Medical Center, Los Angeles, United States.
3
Department of Neurology and Rare Disease Center, Huashan Hospital, Fudan University, Shanghai, China.
4
Sanofi R&D, Evidence Generation and Decision Science, Cambridge, MA, United States.
5
Sanofi, Cambridge, MA, United States.
6
Neuromuscular Diseases Unit, Department of Neurology, Hospital de la Santa Creu i Sant Pau, Barcelona, Spain.
7
Centro para la Investigación Biomédica en Red en Enfermedades Raras (CIBERER), Madrid, Spain
Background: Riliprubart, a first-in-class humanized IgG4-monoclonal antibody, selectively inhibits activated-C1s in classical complement pathway and can be self-administered subcutaneously via auto-injector. In Phase-2 trial (NCT04658472), riliprubart demonstrated favorable safety and encouraging efficacy in chronic inflammatory demyelinating polyradiculoneuropathy (CIDP). Here, we present time and magnitude of response analyses to characterize clinical benefits.
Methods: The open-label Phase-2 trial evaluated riliprubart across three groups: Standard-of-care (SoC)-Treated, SoC-Refractory, and SoC-Naïve. Participants received 24-week treatment (Part-A), followed by an optional 52-week treatment-extension (Part-B). Primary endpoint (Part-A) evaluated percentage of participants relapsing in SoC-Treated (≥1-point increase in adjusted Inflammatory Neuropathy Cause and Treatment [INCAT] disability score) and percentage of participants responding in SoC-Refractory/Naïve groups (≥1-point decrease in adjusted INCAT score). This post-hoc analysis focused on INCAT responders (≥1-point decrease) in Part-A, and evaluated time-to-first-response, and proportion of participants with deep response (≥2-point decrease), overall, and by baseline INCAT score (3-4 versus ≥5).
Results: Among 43 INCAT responders in Part-A, first INCAT response was observed as early as Week-2 with ≥44% of first responses achieved by Week-4, and ≥89% by Week-12 in all three groups. At the last INCAT assessment through Week-24, 44% (19/43) responders achieved deep response with higher rates in participants with severe baseline impairment (INCAT ≥5: 69% [11/16]) versus moderate impairment (INCAT 3-4: 38% [8/21]).
Conclusion: Riliprubart provided rapid and substantial improvement in INCAT score with majority of responders achieving first response by Week-12 and nearly half of responders achieving deep response (≥2-point decrease); supporting Phase-3 development of riliprubart as a potential treatment for people with CIDP.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Immune-mediated Neuropathies
PP01.262
Efgartigimod in Adults With Active Idiopathic Inflammatory Myopathy: The ALKIVIA+ Phase 3 Open-Label Extension Trial
Dr. Rohit Aggarwal1, Dr. Anthony A. Amato2, Dr. Ingrid E. Lundberg3, Dr. Andrew L. Mammen4, Dr. Jiri Vencovský5, Mr. Sebastian Rodriguez6, Dr. Paul Duncombe6, Dr. Bas van der Woning6, Dr. Hector Chinoy7,8
1
University of Pittsburgh Medical Center, Pittsburgh, United States.
2
Brigham and Women’s Hospital Harvard Medical School, Boston, United States.
3
Karolinska Institutet and Karolinska University Hospital, Stockholm, Sweden.
4
Muscle Disease Section, National Institute of Arthritis and Musculoskeletal and Skin Disease, National Institutes of Health, Bethesda, United States.
5
Institute of Rheumatology, Research Department, and Department of Rheumatology, 1st Medical Faculty, Charles University, Prague, Czechia.
6
argenx, Ghent, Belgium.
7
Salford Royal Hospital, Northern Care Alliance NHS Foundation Trust, Manchester Academic Health Science Centre, Salford, United Kingdom.
8
Division of Musculoskeletal and Dermatological Sciences, Faculty of Biology, Medicine and Health, The University of Manchester, Manchester, United Kingdom
Background: Idiopathic inflammatory myopathy (IIM), a rare, debilitating, systemic rheumatic disease, has limited treatment options. Efgartigimod is a human immunoglobulin G1 (IgG1) antibody Fc fragment, with increased affinity for the neonatal Fc receptor (FcRn), that selectively reduces IgG by blocking FcRn-mediated IgG recycling without impacting antibody production or other parts of the immune system. Efgartigimod does not lower albumin levels and does not increase low-density lipoprotein cholesterol levels. The ALKIVIA trial assessed the safety and efficacy of subcutaneous (SC) efgartigimod versus placebo in individuals with active IIM receiving background therapy. Interim data from the open-label extension ALKIVIA+ trial are reported.
Methods: In ALKIVIA (NCT05523167; a seamless phase 2/3, randomized [1:1], double-blinded, placebo-controlled, parallel-group, multicenter trial), adults received weekly efgartigimod (1000 mg) co-formulated with recombinant human hyaluronidase (rHuPH20) SC (EFG) or placebo rHuPH20 SC (PBO). After the 24-week placebo-controlled period, eligible participants either enrolled in ALKIVIA+ (NCT05979441) and received EFG or entered a 56-day safety follow-up. The ALKIVIA+ primary endpoint is long-term safety and tolerability (treatment-emergent adverse events [TEAEs], serious adverse events [SAEs], and adverse events of special interest [infections]). Secondary endpoints include changes over time in the 2016 American College of Rheumatology/European Alliance of Associations for Rheumatology (ACR/EULAR) Myositis Response Criteria’s Total Improvement Score (TIS); the proportion of participants with minimal (TIS ≥20), moderate (TIS ≥40), and major (TIS ≥60) clinical improvement over time; and changes from baseline in Physician Global Assessment of Disease Activity (MDGA), Patient Global Assessment of Disease Activity (PGA), Patient Global Impression of Severity (PGI-S), and manual muscle testing-8 (MMT8).
Results: In ALKIVIA, 89 participants received EFG (n=47) or PBO (n=42), with 69 entering ALKIVIA+ at Week 24; 35 continued EFG (EFG-EFG; mean age: 57.7 years, mean time since diagnosis: 4.60 years), and 34 switched to EFG (PBO-EFG; 54.9 and 4.81 years, respectively).
At Weeks 24 to 52, for PBO-EFG, TIS increased from 38.96 to 49.62, and for EFG-EFG, it remained stable (50.67 to 52.19) (Image 1A). At Week 52, minimal, moderate, and major TIS improvements were achieved by 93.8% versus 93.9%, 75.0% versus 66.7%, and 37.5% versus 33.3% of EFG-EFG versus PBO-EFG participants, respectively. Mean changes from baseline in MDGA, PGA, and MMT8 scores at Week 52 were similar for participants receiving EFG-EFG versus PBO-EFG (–3.56 vs –3.10, –2.56 vs –1.89, and 16.3 vs 17.1, respectively). At Week 28, PGI-S symptoms improved in 9.4% versus 26.0% of those receiving EFG-EFG versus PBO-EFG and worsened in 15.6% versus 8.8% of participants in these groups, respectively.
The incidence of adverse events was comparable between EFG-EFG (TEAE: 33/35 [94.3%], SAE: 7/35 [20.0%]) and PBO-EFG (TEAE: 31/34 [91.2%], SAE: 6/34 [17.6%]) (Image 1B). In general, the incidence of TEAEs was consistent for both groups. Only 1 severe infection was noted in the EFG-EFG group. Generally, the TEAE incidence remained consistent throughout ALKIVIA and ALKIVIA+.
Conclusion: Interim ALKIVIA+ data show a favorable safety profile over 52 weeks for EFG, with notable improvements in efficacy for participants who switched to EFG. These findings support the ongoing phase 3 evaluation of EFG in ALKIVIA+.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Immune-mediated Neuropathies
Images or Table (Optional)
PP01.263
Lateral Dorsal Cutaneous Sural Entrapment - an Uncommon Neuralgic Syndrome of the Foot
Assoc. Prof. Antony Winkel
Sunshine Coast Health, Sunshine Coast, Australia. Griffith University, Sunshine Coast, Australia
Background: Entrapment neuropathy is a common cause of neuropathic pain, although outside of the tarsal tunnel syndrome, foot entrapment syndromes are uncommon.
Methods: Here, we present the case of a 50-year-old lady with no risk factors for nerve entrapment or neuropathy, who presented with entrapment of the lateral dorsal sural nerve, causing refractory neuropathic foot pain until surgical intervention provided symptomatic relief.
Results: A 50-year-old lady, who was otherwise well with no significant medical or family history, presented with a 3-year history of right foot pain, radiating into the lateral dorsal foot. There was no inciting trauma or injury. Assessment at another institution failed to identify an aetiology, and she was treated with escalating doses of gabapentinoids after normal nerve conduction studies. Despite this, her symptoms were ongoing and debilitating and she sought further review.
On review at our institution, her symptoms were identified to be concordant with the lateral dorsal cutaneous sural nerve territory. Additional nerve conduction studies using a modified published technique demonstrated a mild relatively prolonged sensory peak latency on the symptomatic side. Focal neuromuscular ultrasound was not available at the time, but MRI of the area showed T2 hyperintensity of the lateral dorsal cutaneous sural nerve branch. Surgical exploration revealed a fibrous band and thickened nerve at the entrapment site, and this was released and surgical neurectomy performed.
At last follow up 2 years since surgery, the patient is on low-dose gabapentin (300mg/day) and apart from residual non-debilitating sensory disturbance is pain-free.
Conclusion: This case reinforces the importance of tailoring neurophysiological assessment to the symptomatic issue. Non neuromuscular physicians should not be reassured by 'normal' routine nerve conduction studies unless the neurophysiologist has explicitly considered the possible symptomatic structures.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Small fibers and painful neuropathies
PP01.264
High Dose Intravenous Immunoglobulin for Multifocal Motor Neuropathy Resulting in Hyperviscosity and Digital Ischaemia
Assoc. Prof. Antony Winkel1,2, Dr. Andrew Clarke1,2
1
Sunshine Coast Health, Sunshine Coast, Australia.
2
Griffith University, Sunshine Coast, Australia
Background: Multifocal motor neuropathy (MMN) is an immune-mediated neuropathy that typically requires high doses of intravenous immunoglobulin (IVIG). Pending trials underway currently for novel therapeutic agents, other treatment options are very limited, with traditional immunotherapies used in CIDP often ineffective in this condition. High-dose IVIG carries a known thrombotic risk, though hyperviscosity and associated ischaemia is rarely reported.
Methods: Here we report a case of a 46 year old lady who, following the diagnosis of MMN and institution of IVIG therapy, required additional management for secondary digital ischaemia likely due to an increase of plasma viscosity due to IVIG in conjunction with an IgM-kappa paraprotein. The management is discussed.
Results: The patient, a 46-year-old otherwise well lady presented in November 2021 with subtle left finger drop without sensory symptoms. Over several months this progressed to a radial motor deficit in the left hand, without sensory findings on examination, and then left median motor dysfunction and left fibular motor dysfunction followed. Nerve conduction studies and EMG showed focal motor changes with patchy conduction block at non-entrapment sites in these nerves and MRI with contrast showed T2 hyperintensity and contrast enhancement of the brachial plexus extending into the left radial nerve through the arm. A diagnosis of multifocal motor neuropathy with conduction block (MMN) was made and IVIG induction at 2g/kg produced significant improvement, with maintenance titrated to 1g/kg 2-weekly to maintain optimal motor function.
An IgM-kappa paraprotein was detected as part of the initial work up, and Haematology review diagnosed monoclonal gammopathy of uncertain significance (MGUS) after further assessment, including bone marrow sampling. It was considered likely, however, that the immune dysregulation associated with the paraprotein may have contributed to the MMN.
In July of 2023 the patient presented with new, subacute digital ischaemia in the hands with distal ulceration and dusky fingertips. Arterial supply to the upper limbs was normal on CT angiography and doppler ultrasound, and screening for other causes of thrombophilia or hyperviscosity were normal. Total IgG was elevated at 18-20 g/L. The known IgM-kappa paraprotein was stable at 3-4g/L, with no manifestations of haematological malignancy or plasma cell dyscrasia after repeat Haematology assessment. A secondary Raynaud's type phenomenon due to increased plasma viscosity from IVIG and the paraproteinaemia was considered.
Intravenous phosphodiesterase infusion improved the digital ischaemia and allowed transition to oral therapy. Digital botulinum toxin was added as a second--line vasodilator, with further improvement and resolution of the ischaemia. A trial of rituximab was provided over 24 months given the M-protein, in the hope that this may allow IVIG dose-reduction. Unfortunately, no benefit was seen. Currently, the patient remains on her high-dose IVIG with 3-monthly botulinum and oral sildenafil, with good MMN symptom-control and no active digital ischaemia.
Conclusion: This case illustrates the challenges of MMN in the setting of limited therapeutic options and significant other-organ dysfunction contributed to by high-dose IVIG and comorbidity. Trials of novel therapeutic agents, especially focusing on complement pathways, are welcomed as a potential alternative option in future for similar cases.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Immune-mediated Neuropathies
PP01.265
Phase 2 Trial of Riliprubart in CIDP: Patient-reported Quality-of-life, Pain and Fatigue Outcomes at Week-76
Dr. Asif Paker1, Dr. Shahram Attarian2, Dr. Hans-Peter Hartung3,4,5,6, Dr. Yi Lu7, Dr. Miguel Alonso-Alonso1
1
Sanofi R&D, Neurology Development, Cambridge, MA, United States.
2
Neuromuscular Disease and ALS Reference Center, Timone University Hospital, Aix-Marseille University, CHU Timone, Marseille Cedex 05, France.
3
Department of Neurology, Faculty of Medicine, Heinrich-Heine-University, Düsseldorf, Germany.
4
Brain and Mind Center, University of Sydney, Sydney, NSW, Australia.
5
Department of Neurology, Medical University of Vienna, Vienna, Austria.
6
Department of Neurology, Palacky University Olomouc, Olomouc, Czechia.
7
Sanofi R&D, Evidence Generation and Decision Science, Cambridge, MA, United States
Background: People living with chronic inflammatory demyelinating polyradiculoneuropathy(CIDP) experience fatigue, weakness and sensory abnormalities, affecting quality-of-life(QoL). Riliprubart, a first-in-class, humanized, IgG4-monoclonal antibody, selectively inhibits activated-C1s within the classical complement pathway and is self-administered subcutaneously via auto-injector. A Phase-2 trial(NCT04658472) evaluating riliprubart in CIDP demonstrated decreased fatigue-severity and improved QoL at week-48. Here we assess effect of riliprubart on fatigue, pain and QoL up to week-76.
Methods: Open-label trial evaluating riliprubart across three groups: Standard-of-Care(SoC)-Treated(N=48), SoC-Refractory(N=18), and SoC-Naïve(N=12). Participants underwent 24-week treatment(Part-A), followed by optional treatment-extension(Part-B:52-weeks). At week-76, changes from baseline in health-related QoL were descriptively analyzed using EuroQol-Visual Analogue Scale(EQ-VAS; range 0-100, higher score indicate better outcomes) and health utility index value(range <0[as bad or worse than dead] to 1[full-health]), pain through EQ-5D-5L Dimension(range 1[no pain/discomfort] to 5[extreme pain/discomfort]), and fatigue using Modified Rasch-built Fatigue Severity Scale(R-FSS; range: 0-21, higher score indicate greater fatigue-severity).
Results: At week-76, QoL, pain and fatigue were assessed in 37 SoC-Treated, 11 SoC-Refractory and 6 SoC-Naïve participants. Mean[SD] change from baseline in EQ-VAS(SoC-Treated:12.1[18.4], SoC-Refractory:19.1[21.8], SoC-Naïve:8.5[16.2]) and health utility index value(SoC-Treated:0.18[0.28], SoC-Refractory:0.21[0.18], SoC-Naïve:0.05[0.22]) suggested QoL improvement across all groups. Higher proportion reported no pain versus baseline(SoC-Treated:27.0%[10/37] vs 12.8%[6/47], SoC-Refractory:45.5%[5/11] vs 16.7%[3/18], SoC-Naïve:33.3%[2/6] vs 16.7%[2/12]). Mean[SD] R-FSS changes indicated reduced fatigue across SoC-Treated(-3.7[7.6]), SoC-Refractory(-2.2[9.5]), and SoC-Naïve(-3.3[4.2]) groups.
Conclusion: Riliprubart indicated sustained decrease in fatigue and pain, and improved QoL in people with CIDP, along with clinical improvement. Two randomized, placebo-controlled Phase-3 studies(MOBILIZE, VITALIZE) are ongoing to confirm these findings.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Immune-mediated Neuropathies
PP01.266
Clinical and Metagenomic Characterization of Gut Microbiome in Guillain-Barré Syndrome
Asst. Prof. Byeol-A Yoon, Assoc. Prof. Jung-Hwa Seo
Dong-A University, Busan, Korea, Republic of
Background: Guillain-Barré Syndrome (GBS) is an acute post-infectious polyneuropathy often triggered by Campylobacter jejuni via molecular mimicry. While gut dysbiosis is known to influence immune homeostasis, its specific role in GBS remains poorly defined. This study aimed to elucidate the interplay between the gut microbiome, C. jejuni infection, and clinical phenotypes to identify microbial markers for potential therapeutic interventions.
Methods: We analyzed fecal and blood samples from 111 GBS patients and 108 healthy controls (HC) using metagenomic shotgun sequencing. Clinical severity, GBS subtypes, and serological markers including anti-ganglioside and anti-C. jejuni antibodies were correlated with microbial profiles and functional gene compositions.
Results: A total of 111 GBS patients (median age 62.5 years) were enrolled. The clinical distribution consisted of 25% AIDP, 33% axonal subtypes (AMAN and AMSAN), 22% Miller Fisher syndrome, and 20% other variants including acute bulbar palsy and MFS overlapping syndrome. Seropositivity was 58.6% for anti-ganglioside antibodies and 55% for anti-C. jejuni antibodies. Patients with C. jejuni infection demonstrated significantly lower MRC sum scores at nadir, indicating higher initial clinical severity. Metagenomic analysis revealed a distinct microbial landscape in GBS patients characterized by the enrichment of Escherichia coli and significantly higher alpha diversity of antibiotic resistance genes, such as pmr and tetM. Functional profiling showed enhanced pathways for oxidative stress response (glutathione metabolism) and biofilm formation, suggesting microbial adaptation to the host’s inflammatory environment.
Conclusion: Our findings demonstrate a clear association between gut microbiome dysbiosis, C. jejuni seropositivity, and GBS pathogenesis. The correlation between specific microbial shifts, such as increased resistance genes, and clinical severity—particularly in axonal subtypes—suggests that the gut environment may influence disease progression. These results provide a robust scientific basis for targeting the gut-nerve axis through microbiome-based adjunct therapies to improve clinical outcomes in GBS patients.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Immune-mediated Neuropathies
PP01.267
Riliprubart Phase 2 Trial Results: Efficacy Analysis Using Immune Neuropathy-No Evidence of Disease Activity Framework
Dr. Jeffrey A Allen1, Dr. Claudia Sommer2, Dr. Manuel Nunez3, Dr. Karen M. Lynch3, Dr. Yi Lu4, Dr. Li Xiong3, Dr. Miguel Alonso Alonso3, Dr. Asif Paker3, Dr. Luis Querol5,6
1
Department of Neurology, Division of Neuromuscular Medicine, University of Minnesota, Minneapolis, Minnesota, United States.
2
Neurologische Klinik und Poliklinik, Universitätsklinikum Würzburg, Würzburg, Germany.
3
Sanofi, Cambridge, MA, United States.
4
Sanofi R&D, Evidence Generation and Decision Science, Cambridge, MA, United States.
5
Neuromuscular Diseases Unit, Department of Neurology, Hospital de la Santa Creu i Sant Pau (IR SANT PAU), Barcelona, Spain.
6
Centro para la Investigación Biomédica en Red en Enfermedades Raras (CIBERER), Madrid, Spain
Background: Riliprubart, a first-in-class, humanized IgG4-monoclonal antibody, selectively inhibits activated-C1s in classical complement pathway and is self-administered subcutaneously. Phase 2 trial demonstrated encouraging efficacy and safety in chronic inflammatory demyelinating polyradiculoneuropathy (CIDP). Adapted from the established multiple sclerosis model, GBS|CIDP Foundation has proposed Immune Neuropathy-No Evidence of Disease Activity (IN-NEDA) as a composite endpoint tailored for CIDP with IN-NEDA-1 defined as no patient-perceived change and either no decline in disability or no decline in impairment while IN-NEDA-2 requires no patient-perceived change and no decline in both disability and impairment for a duration of 6 months or more. This framework offers structured outcome assessment, to guide treatment decisions and enhance patient care through disability/impairment evaluation. The goal of this study was to conduct an exploratory analysis of Phase 2 results of riliprubart applying the proposed IN-NEDA framework.
Methods: NCT04658472, an open-label, Phase 2 trial evaluated riliprubart across three groups: Standard-of-care (SoC)-Treated, SoC-Refractory and SoC-Naïve. Participants underwent 24-week treatment (Part-A) with optional 52-week treatment-extension (Part-B). This post-hoc analysis evaluated durability of non-deterioration in (trial) participants using a composite approach incorporating disability and impairment scales aligned with the IN-NEDA framework.
Results: Findings exploring different approaches will be presented at the congress.
Conclusion: These results will help illustrate the applicability of the IN-NEDA framework in CIDP, supporting its role as a comprehensive approach for assessing sustained disease control.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Immune-mediated Neuropathies
PP01.268
CAPTIVATE Trial Design: A Pivotal Phase 3 Study of Claseprubart in Chronic Inflammatory Demyelinating Polyneuropathy
Dr. Jeffrey Allen1, Dr. Luis Querol2, Dr. Filip Eftimov3, Assoc. Prof. Stojan Peric4, Assoc. Prof. Tina Dysgaard5, Prof. Yusuf A. Rajabally6, Assoc. Prof. Thomas Harbo7, Prof. Eduardo Nobile-Orazio8, Assoc. Prof. Hans Katzberg9, Dr. David Cornblath10, Dr. Jon Katz11, Dr. James Sheffield12, Mr. Luke Hickey12, Dr. Bethany Beazley12, Dr. Nuria Carrillo12, Dr. Richard Lewis13
1
University of Minnesota, Minneapolis, United States.
2
Hospital de la Santa Creu, Barcelona, Spain.
3
Amsterdam UMC, Amsterdam, Netherlands.
4
University of Belgrade, Belgrade, Serbia.
5
University of Copenhagen, Copenhagen, Denmark.
6
University of Aston, Birmingham, United Kingdom.
7
Aarhus University Hospital, Aarhus, Denmark.
8
Università degli Studi di Milano, Milan, Italy.
9
Institute of Medical Science, Toronto, Canada.
10
Johns Hopkins University School of Medicine, Baltimore, United States.
11
California Pacific Medical Center, San Francisco, United States.
12
Dianthus Therapeutics, New York, United States.
13
Cedars Sinai Medical Center, Los Angeles, United States
Background: Chronic inflammatory demyelinating polyneuropathy (CIDP) is a heterogeneous disorder in which complement dysregulation contributes to pathogenesis. Claseprubart (DNTH103) is a potent monoclonal antibody that selectively binds to active C1s, inhibiting the classical complement pathway. CAPTIVATE Phase 3 study (NCT06858579) evaluates claseprubart in a broad population of adults aged 18–75 with a diagnosis of CIDP.
Methods: CAPTIVATE is a global, multicenter, randomized, double-blind, placebo-controlled Phase 3 study assessing the efficacy and safety of claseprubart in adults aged 18–75 with a diagnosis of CIDP per 2021 EAN/PNS guidelines confirmed by an independent adjudication panel. Eligible participants are responders to standard-of-care (SOC) therapy (immunoglobulins [Ig] or oral corticosteroids [OCS]), refractory to SOC, or treatment-naïve. In SOC responders, Ig will be discontinued 1 week before dosing and OCS tapered and continued.
Part A is open-label with an initial intravenous loading dose followed by biweekly subcutaneous claseprubart up to 13 weeks. Responders in Part A (≥1-point improvement in adjusted Inflammatory Neuropathy Cause and Treatment [INCAT]) are randomized (N=64/arm) into a placebo-controlled, double-blind period for 52 weeks (Part B). The primary endpoint is time to relapse in Part B with key secondary endpoints evaluating Inflammatory Rasch-built Overall Disability Scale (I-RODS) and grip strength changes. Those who complete or relapse (≥1-point worsening in adjusted INCAT) in Part B may enter an optional open-label extension (up to 104 weeks). Upon discontinuation, participants enter a 40-week safety follow-up.
Results: CAPTIVATE was launched in 2025 and is actively enrolling participants in North and South America, Europe, and Asia.
Conclusion: CAPTIVATE is an innovative, rigorous Phase 3 study evaluating claseprubart, an active C1s inhibitor administered as a convenient biweekly injection, in CIDP. The design prioritizes patient safety, avoids complete washout of SOC, minimizes exposure to ineffective therapy, and provides continued access as part of an optional open-label extension.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Immune-mediated Neuropathies
PP01.269
Impact of Efgartigimod on Inflammatory Neuropathy Cause and Treatment Scores: ADHERE/ADHERE+ Post Hoc Analysis
Dr. Christian Eggers1, Dr. Ratna Bhavaraju-Sanka2, Dr. Patrick Kwon3, Prof. Eduardo Nobile-Orazio4, Prof. Thomas Skripuletz5, Dr. Arne De Roeck6, Dr. Arie Gafson6, Dr. Anneleen Remmerie6, Prof. Pieter van Doorn7
1
Kepler University Hospital, Linz, Austria.
2
UT Health San Antonio, San Antonio, United States.
3
NYU Grossman School of Medicine, New York, United States.
4
University of Milan, Milan, Italy.
5
Hannover Medical School, Hanover, Germany.
6
argenx, Ghent, Belgium.
7
Erasmus University Medical Center, Rotterdam, Netherlands
Background: The pivotal ADHERE study (NCT04281472) and open-label extension ADHERE+ (NCT04280718) assessed efficacy and safety of efgartigimod PH20 subcutaneous (SC) in chronic inflammatory demyelinating polyradiculoneuropathy (CIDP).
Methods: Enrolled participants with active CIDP (standard treatments withdrawn during run-in) received open-label, weekly efgartigimod PH20 SC 1000 mg (stage-A). Responders were randomized (1:1) to weekly efgartigimod PH20 SC or placebo (stage-B). Participants with clinical deterioration in stage-B, ongoing in stage-B at 88th relapse, or who completed ADHERE could enter open-label ADHERE+. The Inflammatory Neuropathy Cause and Treatment (INCAT) score is the standard measure of arm and leg disability in CIDP (maximum score=10; minimal clinically important difference: decrease of ≥1 point). Minimal symptom burden in CIDP currently remains undefined. This post hoc analysis aims to contribute to the characterization of minimal symptom burden by reporting the percentage of stage-A responders reaching an INCAT ≤1 (no or minimal disability) by ADHERE stage-B last assessment and during ADHERE+.
Results: At ADHERE stage-A baseline, stage-A responders had an INCAT score of 4.6 (standard deviation, 1.6; n=220); no participants had an INCAT ≤1 at ADHERE run-in baseline (n=191) or stage-A baseline (n=220). At stage-A last assessment, 28/221 (12.7%) stage-A responders had an INCAT ≤1 (13 were subsequently randomized to placebo). At stage-B last assessment, 20/111 (18.0%) efgartigimod-treated and 7/109 (6.4%) placebo-treated stage-A responders had an INCAT ≤1. The percentage of participants at ADHERE+ Week 12, 24, and 36 with an INCAT ≤1 increased to 20.3% (38/187), 26.0% (46/177), and 24.4% (42/172), respectively. 68/196 (34.7%) stage-A responders reached INCAT ≤1 at any point in ADHERE+.
Conclusion: Over one-third of stage-A responders achieved an INCAT ≤1 at any time in ADHERE+. Nearly 1 in 5 efgartigimod-treated stage-A responders reached INCAT ≤1 at ADHERE stage-B last assessment, showing minimal symptom burden (i.e., no disability [INCAT=0], walking without support [INCAT=1]).
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Immune-mediated Neuropathies
PP01.270
Multifocal Intraneural Perineurioma Mimicking Acquired Demyelinating Neuropathy
Dr. Elke Schipani, Dr. B. Kalei Chang, Ms. JaNean Engelstad, Dr. William Litchy, Dr. Michelle Mauermann, Dr. B. Matthew Howe, Dr. Robert Spinner, Dr. P. James Dyck
Mayo Clinic, Rochester, United States
Background: Intraneural perineurioma is a rare, benign peripheral nerve tumor of youth that typically presents as a focal, motor-predominant neuropathy with axonal features. Demyelinating-appearing electrophysiologic findings and multifocal involvement are uncommon and may lead to a misdiagnosis of chronic inflammatory demyelinating polyneuropathy (CIDP).
Methods: A 15-year-old male presented with slowly progressive, asymmetric weakness involving the right upper and left lower limbs. Symptoms began around age 9 with right finger extensor weakness and exercise intolerance, with the eventual development of left foot drop. Despite prolonged treatment with IVIg and later subcutaneous immunoglobulin, disease progression was felt to have slowed but did not remit.
Examination demonstrated high arched feet, a steppage gait, and distal predominant weakness in the right upper and left lower limbs, though mild, patchy weakness was noted in the other limbs. Reflexes were diminished in the right upper limb compared to the left, but lower limb reflexes were symmetric but diminished at the ankles. Sensation was essentially normal.
Results: Serial nerve conduction studies over five years leading up to and inclusive of our evaluation demonstrated multifocal abnormalities, including low-amplitude or absent CMAPs and SNAPs, temporal dispersion, partial motor conduction block, slowed conduction velocities, prolonged distal latencies, and absent F-waves. Nerve conduction studies met the 2021 EAN/PNS electrodiagnostic criteria for motor conduction block, with >30% CMAP amplitude drop between the proximal and distal sites in two non-tibial nerves (ulnar and fibular) with preserved distal CMAP amplitudes (Figure 1A). Formal criteria for CIDP were not fulfilled due to the absence of qualifying temporal dispersion, prolongation of distal latencies, and widespread sensory demyelinating features. Needle EMG revealed chronic denervation with large polyphasic motor units and reduced recruitment affecting multiple elements of the right brachial plexus and, to a lesser degree, the left L4–5 distribution.
MRI of the brachial and lumbosacral plexus demonstrated multifocal, fusiform nerve enlargement with marked T2 hyperintensity and avid, homogeneous enhancement involving the brachial plexus (Figure 1C) and bilateral sciatic nerves (Figure 1D), accompanied by denervation-related muscle edema and fatty atrophy. Extensive laboratory, CSF, genetic, and metabolic evaluations were unrevealing.
Targeted fascicular biopsy of the right middle trunk revealed diagnostic features of intraneural perineurioma, including concentric perineurial cell proliferation with preserved nerve architecture and characteristic immunohistochemical staining (EMA-positive, S100-negative perineurial cells; Figure 1E-G).
Conclusion: This case highlights multifocal intraneural perineurioma as an important mimic of demyelination, capable of producing conduction block–like electrophysiologic features and MRI findings suggestive of an acquired process. Recognition of the characteristic imaging pattern, treatment refractoriness, and pathologic confirmation is essential to avoid prolonged immunotherapy and to guide appropriate management.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Immune-mediated Neuropathies
Images or Table (Optional)

PP01.271
Immune-mediated Sensory Predominant Polyradiculoneuropathy Initially Mimicking Myelopathy
Dr. Eunbyol Hwang1, Assoc. Prof. Jung Hwang Lee2
1
Department of Neurology, Incheon St. Mary’s Hospital, College of Medicine, The Catholic University of Korea, Seoul, Korea, Republic of.
2
2Department of Neurology, Seoul St. Mary’s Hospital, College of Medicine, The Catholic University of Korea, Seoul, Korea, Republic of
Background: Immune-mediated polyradiculoneuropathies can present with subacute, ascending sensory disturbances that clinically mimic spinal cord disorders. Early recognition is critical to avoid misdiagnosis and delay in appropriate immunomodulatory treatment. We report a case of immune-mediated sensory-predominant polyradiculoneuropathy that initially presented with features suggestive of myelopathy, including ascending sensory loss and spinal root enhancement on magnetic resonance imaging (MRI).
Methods: A 36-year-old woman presented with a 2-month history of gradually progressive sensory loss in the right leg, more prominent on the lateral aspect, which had been treated as lumbar disc disease at a local clinic with epidural injection, without improvement. Half a day later, similar sensory loss developed in the left leg. The next day, sensory disturbance ascended to the level of the umbilicus, and shortly thereafter, she developed numbness and tingling in both hands.
On admission, she reported hypoesthesia below the upper abdomen and all four limbs, with sensory loss, neuropathic pain, and gait instability. Neurological examination revealed reduced light touch sensation below C6∼C8 dermatomes, with preserved muscle strength and deep tendon reflexes. Spinal MRI demonstrated diffuse enhancement of the rootlets and nerve roots below the T7 level, without evidence of cord compression or intrinsic cord lesion. Cerebrospinal fluid (CSF) analysis revealed normal cell counts (WBC 0, RBC 0) and mildly elevated protein (80.3 mg/dL). Nerve conduction studies (NCS) demonstrated sensory polyneuropathy with relatively preserved motor conduction. The patient was treated with a 5-day course of high-dose intravenous methylprednisolone, followed by outpatient follow-up with oral steroid taper (prednisolone 5 mg) and repeat NCS.
Results: After high-dose steroid therapy, her sensory symptoms and gait instability improved significantly, and she was discharged. During outpatient follow-up, clinical symptoms continued to improve, with gradual recovery of sensation in the limbs and trunk. Repeat NCS demonstrated improvement in sensory nerve action potentials compared to the initial study, supporting a favorable response to immunomodulatory treatment. The pattern of ascending sensory loss, spinal root enhancement, and CSF protein elevation without pleocytosis, along with clinical and electrophysiological improvement after steroids, was consistent with an immune-mediated sensory-predominant polyradiculoneuropathy.
Conclusion: This case illustrates an immune-mediated, sensory-predominant polyradiculoneuropathy that initially mimicked myelopathy due to ascending sensory loss and spinal root enhancement on MRI. The clinical response to high-dose steroids and improvement on follow-up NCS support an immune-mediated mechanism. In patients presenting with subacute, ascending sensory disturbances, immune-mediated polyradiculoneuropathy should be considered in the differential diagnosis, even when the initial presentation suggests a spinal cord disorder. Early neuroimaging, CSF analysis, and NCS are essential to differentiate between myelopathy and immune-mediated polyradiculoneuropathy and to guide timely immunotherapy.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Immune-mediated Neuropathies
PP01.272
Chronic Inflammatory Demyelinating Polyradiculoneuropathy in France (REVEAL-CIDP): Healthcare Resource Use and Disease Management
Dr. Mariana Ciumas1, Dr. Emilien Delmont2, Prof. Gwendal Le Masson3, Dr. Cécile Blein1, Dr. Clémence Arvin-Berod1, Ms. Clémentine Lacueille4, Prof. Andoni Echaniz-Laguna5
1
argenx BV, Ghent, Belgium.
2
Referral Centre for Neuromuscular Diseases and ALS, Hospital La Timone, Aix-Marseille University, Marseille, France.
3
Department of Neurology (Nerve-Muscle Unit), AOC National Reference Center for Neuromuscular Disorders, ALS Center, University Hospital of Bordeaux (CHU Bordeaux), Bordeaux, France.
4
Horiana, Bordeaux, France.
5
Department of Neurology, APHP, CHU de Bicêtre, INSERM U1195, Paris-Saclay University, Le Kremlin-Bicêtre, France
Background: Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) is a rare, disabling neuropathy characterized by progressive sensory and motor deficits. Data regarding disease management and healthcare resource use among patients with CIDP in Europe and the United States is scarce and heterogeneous and limited to regional or subset populations. Using the French national health insurance claims database (SNDS), this study provides the first French nationwide analysis of CIDP management, resource utilization, and socio-economic impact.
Methods: A retrospective, longitudinal cohort study was conducted using SNDS data (2011–2023). Adults with incident CIDP were identified based on the following criteria: (i) ≥1 hospitalization and/or long-term disease status with an ICD-10 code G61.8 during 2016–2023; (ii) an electrodiagnostic exam (EDX) within 90 days before or after the first diagnosis; (iii) a second medical event (hospitalization or long-term disease status) with ICD-10 code G61.8 in the year after EDX; (iv) without ≥2 differential diagnoses after the second diagnosis of CIDP; and (v) without a CIDP diagnosis in the 2011–2015 period. The index date was the date of the first CIDP-related claim during the inclusion period (2016–2023). Patients were followed from the index date to the end of the study period or death.
Results: Among 3775 incident CIDP patients (median follow-up 3.4 years), mean age was 61.7 years, 67% were men, mean age-adjusted Charlson Comorbidity Index (CCI) score was 2.6, and 16% had an age-adjusted CCI score ≥5. Despite 83% of patients being treated with immunoglobulin (Ig) therapy during follow-up, a substantial proportion still experienced significant healthcare needs and functional limitations. Specifically, 97% had at least one CIDP-related hospitalization, 93% required nurse visits, 50% physiotherapy, and 69% neurology consultations. Almost two-thirds of patients (65%) were treated for depression and/or pain. Over half (57%) of patients needed walking aids (orthotics, walking sticks, walking frame, or wheelchair), with 37% of patients having prescriptions for walking aids during the first year after diagnosis (Figure). Among working-age patients, 32% (600/1859) took sick leave during the first year after diagnosis, with a median duration of 195.5 days, decreasing to 13% (81/629), with a median duration of 16.0 days, five years after diagnosis.
Conclusion: Despite the widespread use of Ig, a substantial proportion of French CIDP patients experience significant functional limitations and socio-economic burden. High rates of hospitalizations, nurse and specialist visits, use of walking aids, and treatments for depression and pain persist. More than half of working-age patients require the use of sick leave, and more than half need walking aids. These findings highlight an unmet need for more effective and alternative therapies.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Immune-mediated Neuropathies
Images or Table (Optional)
PP01.273
Cytokine Profiles Associated With IgG4 Class Switching in NF-155 Antibody–Positive Autoimmune Nodopathy
Dr. Hyun Gi Kim1, Ms. MinGi Kim2, Prof. Ha Young Shin2
1
Department of Radiology and Research Institute of Radiology, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea, Republic of.
2
Department of Neurology, Yonsei University College of Medicine, Seoul, Korea, Republic of
Background: Autoimmune nodopathy (AN), associated with antibodies against paranodal and nodal proteins, represents a distinct subgroup of immune-mediated neuropathies characterized by unique clinical features and treatment responses. Among these, anti–neurofascin-155 (NF-155) antibody–positive AN is well recognized to predominantly involve the IgG4 isotype. IgG4 class switching is driven by specific immunoregulatory pathways, particularly involving T follicular helper (Tfh) cells, regulatory cytokines, and B-cell survival factors. However, the cytokine milieu associated with IgG4 class switching in NF-155 antibody–positive AN remains poorly defined. Clarifying cytokine profiles related to IgG4 differentiation may provide insights into disease mechanisms and identify potential therapeutic targets distinguishing NF-155 antibody–positive AN from seronegative chronic inflammatory demyelinating polyneuropathy (CIDP).
Methods: We conducted a cross-sectional serum cytokine analysis including patients with NF-155 antibody–positive AN (n = 10), patients with seronegative CIDP (n = 21), and healthy controls (n = 7). Serum samples were collected and stored at −80 °C. Cytokines implicated in IgG4 class switching were quantified.
Analytes included IL-4, IL-5, IL-10, IL-13, IL-21, IFN-γ, APRIL, BAFF, GITR, GITRL, and TGF-β1, TGF-β2, and TGF-β3. IL-4, IL-5, IL-10, IL-13, IL-21, IFN-γ, APRIL, BAFF, and GITR were analyzed using the Human Premixed Multi-Analyte Kit (R&D Systems). GITRL was measured using the Human Immuno-Oncology Checkpoint Protein Panel 1 (Merck Millipore), and TGF-β isoforms were quantified using the TGF-β 1,2,3 Magnetic Bead Kit (Merck Millipore). All assays were performed on the MAGPIX (Luminex) platform according to the manufacturers’ instructions. Cytokine concentrations were compared among groups to identify differential immunological profiles associated with NF-155 antibody–positive AN.
Results: Serum GITRL levels differed significantly among healthy controls, seronegative CIDP patients, and NF-155 antibody–positive AN patients (Kruskal–Wallis H = 9.13, p = 0.010). Median GITRL concentrations were highest in the NF-155 antibody–positive group [28.75 pg/mL (IQR 21.88–48.0)], compared with seronegative CIDP patients [16.5 pg/mL (IQR 13.0–20.5)] and healthy controls [14.5 pg/mL (IQR 13.5–16.25)]. Post-hoc pairwise comparisons with Bonferroni correction demonstrated significantly higher GITRL levels in NF-155 antibody–positive patients compared with healthy controls (adjusted p = 0.006). Differences between seronegative CIDP patients and healthy controls were not statistically significant. Although GITRL levels tended to be higher in NF-155 antibody–positive patients than in seronegative CIDP patients, this difference did not reach statistical significance after correction (adjusted p = 0.064). No significant group differences were observed for other analyzed cytokines, including IL-4, IL-5, IL-10, IL-13, IL-21, IFN-γ, APRIL, BAFF, GITR, or TGF-β isoforms.
Conclusion: In patients with NF-155 antibody–positive autoimmune nodopathy, serum GITRL levels were selectively elevated. These findings suggest a potential role for the GITR–GITRL pathway in the immunopathogenesis of IgG4-dominant NF-155–associated disease. Larger studies are warranted to validate these observations and to elucidate the mechanistic relevance of GITRL signaling in NF-155 antibody–positive autoimmune nodopathy.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Immune-mediated Neuropathies
PP01.274
CASE REPORT: Chronic Inflammatory Demyelinating Polyneuropathy Initially Misdiagnosed as Subacute Paraneoplastic Polyneuropathy
Dr. Ieva Glāzere1,2,3, Dr. Gundega Ķauķe1,3, Dr. Marija Roddate1, Dr. Staņislavs Mironovs1,3, Assoc. Prof. Viktorija Ķēniņa1,2
1
Pauls Stradins Clinical University Hospital, Department of Neurology, Centre of Rare Neurological Disorders, Riga, Latvia.
2
Riga Stradins University, Department of Biology and Microbiology, Riga, Latvia.
3
Pauls Stradins Clinical University Hospital, Department of Neurology, Laboratory of Neurophysiology, Riga, Latvia
Background: Chronic inflammatory demyelinating polyneuropathy (CIDP) is a rare autoimmune neuropathy, often challenging to diagnose due to its ability to mimic other peripheral nerve disorders, both – immune and hereditary. Typical CIDP can present acutely in up to 10-15% of patients with rapid progresssion within 4 weeks and initially could be diagnosed with Guillain Barre syndrome (GBS). Additionally, various recently diagnosed disorders in the same patient may perplex the diagnosis even more.
Methods: A retrospective analysis of one clinical case was conducted at Pauls Stradiņš Clinical University Hospital, by reviewing medical records, performed between 2020 and 2026. A written informed consent was obtained by authors from the patient prior to submission of the case report.
Results: We present a clinical case with a previously healthy 37-year-old female who initially manifested with acute onset sensory disturbances and motor weakness in both lower legs. Her symptoms progressed over the time of two weeks until she was unable to walk without aid. Notably, she did not report vision disturbances, facial weakness, shortness of breath or dysarthria, but did have a tingling sensation in her tongue. Two weeks prior to the hospitalisation her son has had a varicella zoster infection. Neurological examination revealed symmetrically reduced muscle strength in her limbs, with a Medical Research Council (MRC) score of 4/5 in both upper limbs proximally, and 3/5 in both lower limbs proximally, 2/5 distally. Additionally, she exhibited absent tendon reflexes, impaired superficial and deep perception in all limbs. Therefore, a preliminary diagnose of GBS was made. Lumbar puncture was performed, cerebrospinal fluid (CSF) workup showed extremely elevated protein level (4.6 g/L), with normal cell count. Spinal MRI results revealed massive contrast enhancement in cauda equina. Screening of infectious diseases, as well as other autoimmune/systemic disorders was negative. The patient received 5 plasma exchange procedures, which resulted in good clinical response – legs distally 3/5, proximally – 4/5. Four weeks later she returned to the hospital with reoccurring weakness, particularly in legs. She received intravenous immunoglobulin treatment (IVIG, 2g/kg) as a treatment related relapse. Explicit re-examination of the patient was performed, and an early stage (T1N0M0) breast cancer (Grade 2 ductal carcinoma) was diagnosed. She underwent surgical removal and was started on Tamoxifen 20 mg per day. The patient recovered neurologically with mild sensory disturbances in feet and no motor deficit. She was asymptomatic for four years, since 2025 she started to experience frequent leg muscle cramps, 6 months later the weakness of the lower legs re-appeared (MRC 2/5) and re-evaluation was completed. Nerve ultrasound revealed enlarged cervical roots and multiple nerves in arms and legs. Nerve conduction studies supported the diagnosis of CIDP. The IVIG treatment was started, which stabilised the patient and improvement her clinical symptoms.
Conclusion: CIDP can clinically manifest with subacute onset motor weakness and sensory disturbances, and in the presence of newly diagnosed cancer, it can mimic paraneoplastic polyneuropathy. It is essential to perform a wide work-up, including nerve ultrasound, and re-evaluated patients history data to provide timely precise diagnosis and treatment.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Immune-mediated Neuropathies
PP01.275
The Spectrum of Polyneuropathies and Polyradiculoneuropathies Requiring Recurrent Inpatient Admissions Over a 9-Year Period
Dr. Inga Suna1,2, Dr. Elisa Andzeva3
1
Riga East Clinical University Hospital, Riga, Latvia.
2
Faculty of Medicine and Life Sciences, University of Latvia, Riga, Latvia.
3
Pauls Stradins Clinical University Hospital, Riga, Latvia
Background: The prevalence of peripheral neuropathy in the general population ranges from 1% to 7%, increasing with age but also affecting younger individuals. Management primarily focuses on treating the underlying aetiology (Castelli et al., 2020). Autoimmune neuropathies represent a heterogeneous group of peripheral nervous system disorders, complicating accurate diagnosis in clinical practice (Caballero-Ávila et al., 2025). The diagnostic and treatment course of the patients is complex, often involving recurrent exacerbations, treatment adjustments, and the need for improved patient adherence to achieve optimal long-term disease control. The aim on the study was to determine and characterize aetiology and causes of repeated hospitalization of patients with polyneuropathies, polyradiculoneuropathies over a 9-year period at Riga East Clinical University Hospital.
Methods: A retrospective study involved medical records of patients diagnosed with polyneuropathy, polyradiculoneuropathies and autoimmune neuropathies treated at the Neurology and Neurosurgery Clinic of Riga East Clinical University Hospital between 2014 and 2023. Data analysis was performed with Microsoft Excel and IBM SPSS Statistics 22 software.
Results: Medical records of 511 patients with polyneuropathies and polyradiculoneuropathies were included in the study. Of all the patients, 50.9% (n=260) were female and 49.1% (n=251) were male. Mean age was 54.8 years (18-90). The average hospital stay was 11 days (SD± 8,51). Based on aetiology, the majority of patients 40.1% (n=205/511) had immune-mediated neuropathy (Guillain-Barré syndrome (GBS), chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) or multifocal motor neuropathy (MMN)) and 38,6% (n=197/511) had idiopathic polyneuropathy. Rest of the patients (21.3%, n=109/511) had polyneuropathy associated with endocrine or systemic disease, infectious disease, toxic factors, vitamin deficiency and hereditary cause. Additional analysis focused on the patients requiring recurrent hospital admissions. During the period from 2014 to 2023, 7.4% (n=38/511) of the patients with polyneuropathy, polyradiculoneuropathies were hospitalized more than once; 60.5% (n = 23/38) were hospitalized twice, 23.7% (n = 9/38) three times, and 10.5% (n = 4/38) four times. The highest number of recurrent inpatient admissions for a single patient was 16, followed by 11 times for another patient. The indications for recurrent hospitalizations were as follows: clinical exacerbation of CIDP in 57.9% (n=22/38), progression after GBS in 15.8% (n=6/38), deterioration of MMN in 10.5% (n=4/38), deterioration of idiopathic polyneuropathy in 10.5% (n=4/38); deterioration of paraproteinemic polyneuropathy in 5.3% (n=2/38). The most frequently hospitalized patient with CIDP was ultimately diagnosed with colon cancer and the outcome was fatal.Three patients with MMN needed repeated training for the use of subcutaneous immunoglobulin therapy, but 2 patients received treatment with Rituximab. All the patients with immune-mediated neuropathies received combined therapy, including intravenous immunoglobulins, plasma exchange and glucocorticosteroids.
Conclusion: Most patients with polyneuropathies and polyradiculoneuropathy are successfully managed on an outpatient basis (92.6%). Recurrent inpatient admissions are rarely required (7.4%) for management of therapy. Neoplastic CIDP is even rarer and can be fatal, thus the possibility of this form of CIDP should be considered, when treating immune-mediated neuropathies, especially in case of many exacerbations and ineffective therapy.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Immune-mediated Neuropathies
PP01.276
Beyond Nonsystemic Vasculitic Neuropathy: Evidence for a PNS–CNS Vasculitis Spectrum
Prof. Seungkeun Lee, Prof. Jee-Eun Kim
Soonchunhyang University Bucheon Hospital, Seoul, Korea, Republic of
Background: Nonsystemic vasculitic neuropathy (NSVN) is regarded as a single-organ vasculitis limited to the peripheral nervous system (PNS), yet emerging evidence suggests that neurological involvement may represent a spectrum rather than discrete entities.
Methods: We present a case in which initially isolated vasculitic neuropathy evolved to involve the skin and central nervous system(CNS), supporting the concept of a PNS-CNS vasculitis spectrum as a distinct neurological phenotype of vasculitis.
Results: A 63-year-old woman presented with subacute left posterior calf–sole pain and left foot drop three months prior to admission. One month later, similar pain developed in the right posterior calf and foot, followed by right hand tingling and weakness. Neurological examination revealed left ankle dorsiflexion and eversion weakness and right finger abduction weakness, sensory loss in the right ulnar nerve territory, and diffusely hypoactive deep tendon reflexes. Nerve conduction studies demonstrated asymmetric sensorimotor polyneuropathy consistent with mononeuritis multiplex. Extensive laboratory evaluation for vasculitis, paraneoplastic, and autoimmune etiologies, spine MRI, abdomen & chest CT and cerebrospinal fluid analysis, were unremarkable. She was diagnosed with nonsystemic vasculitic neuropathy and treated with oral prednisolone, resulting in gradual improvement. However, during tapering, she developed severe ischemic toe pain with color change and ulceration, along with acute dysarthria. Brain MRI revealed an acute right corona radiata infarction, while CT angiography showed no steno-occlusive disease. Relapsing vasculitic neuropathy with skin and central nervous system involvement was suspected, and treatment with high-dose corticosteroids and azathioprine led to sustained improvement without relapse.
Conclusion: Rather than representing discrete entities such as NSVN or primary CNS vasculitis, this presentation suggests a continuous PNS-CNS vasculitis spectrum, representing a neurological phenotype of vasculitis characterized by preferential small-vessel involvement of neural tissues, with or without cutaneous manifestations. Single-organ vasculitis may reflect an early or incomplete expression within a broader vasculitic continuum rather than a static disease entity.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Immune-mediated Neuropathies
PP01.277
A Case of Acute Bickerstaff Brainstem Encephalitis: Diagnostic Challenges and Therapeutic Response to Early Immunotherapy
Prof. Joong-Yang Cho
Department of Neurology, Inje University College of Medicine, Ilsan Paik Hospital, Go-yang, Korea, Republic of
Background: Bickerstaff brainstem encephalitis (BBE) is a rare autoimmune neurological disorder characterized by the clinical triad of ophthalmoplegia, ataxia, and impaired consciousness. While it shares overlapping features with Miller Fisher syndrome (MFS) and Guillain-Barré syndrome (GBS), BBE is distinguished by central nervous system (CNS) involvement. We report a case of BBE following acute gastroenteritis, emphasizing the importance of clinical suspicion even in the presence of unremarkable initial laboratory and imaging findings.
Methods: Case : A 25-year-old previously healthy male presented with acute onset of dizziness and gait instability following a severe episode of gastroenteritis (AGE) seven days prior. Initial neurologic examination showed ataxia and dysarthria without significant limb weakness. Brain MRI and CSF analysis (WBC 0/μL, Protein 24 mg/dL) were unremarkable. Nerve conduction studies (NCS) showed only delayed H-reflex and F-wave latencies, while the blink reflex revealed absent bilateral R2 responses.
Results: Initially, the patient was managed under the diagnosis of Guillain-Barré syndrome with intravenous immunoglobulin (IVIG). Despite initiating IVIG, the patient’s condition rapidly deteriorated within 24 hours. He developed a drowsy mental status, severe dysarthria, bilateral ptosis, and total ophthalmoplegia (CN III, IV, VI). Notably, the patient exhibited hyperreflexia, suggesting CNS involvement. Based on the rapidly progressive brainstem symptoms and impaired consciousness, BBE was strongly suspected. High-dose methylprednisolone was added to the IVIG regimen. Following combined immunotherapy, the patient’s consciousness cleared, and his neurological deficits, including ataxia and bulbar weakness, showed significant improvement. He was subsequently transferred to a rehabilitation facility for continued recovery.
Conclusion: This case highlights that BBE can present with rapidly progressive consciousness disturbances and brainstem signs, mimicking GBS but differentiated by CNS involvement such as drowsy mental status and hyperreflexia. As initial CSF, MRI, and even anti-ganglioside antibody tests (which were negative in this case) may not be definitive, early clinical identification and prompt treatment are crucial for improving clinical outcomes.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Immune-mediated Neuropathies
PP01.278
Intravenous Immunoglobulin to Subcutaneous Efgartigimod PH20 Transition in CIDP: A Phase 4 Study in Progress
Dr. Arjun Seth1, Dr. Andrew Lerman2, Dr. Anneleen Remmerie3, Dr. Arne De Roeck3, Dr. Katerina Anokhina3, Dr. Arie Gafson3, Dr. Yessar M. Hussain4
1
Northwestern Medicine, Chicago, United States.
2
Grove Neurology, Miami, United States.
3
argenx, Ghent, Belgium.
4
Austin Neuromuscular Center, Austin, United States
Background: Efgartigimod is a human immunoglobulin G1 antibody Fc fragment that blocks FcRn, reducing pathogenic IgG autoantibody levels. Results from the ADHERE trial (NCT04281472) demonstrated a significant, clinically meaningful benefit of efgartigimod PH20 subcutaneous (SC) in participants with chronic inflammatory demyelinating polyradiculoneuropathy (CIDP), regardless of prior CIDP therapy. Participants in ADHERE underwent a washout period during which they were required to show disease worsening before initiation of efgartigimod. Since disease worsening is not required for efgartigimod initiation outside of the clinical trial setting, research on the transition from intravenous immunoglobulin (IVIg) to efgartigimod without disease worsening will inform clinical practice. This phase 4, open-label, multicenter trial (NCT06637072) in the US investigated whether transitioning to efgartigimod PH20 SC within 1 week after last IVIg dose is a safe and effective approach in CIDP.
Methods: Twenty-three participants ≥18 years of age diagnosed with CIDP and treated with stable IVIg doses (0.5–2 g/kg once every 3–6 weeks for ≥3 doses) were enrolled. After a ≤3-week screening period, participants received efgartigimod PH20 SC 1000 mg once weekly for a 12-week treatment period within 1 week of stopping IVIg. The primary endpoint was the percentage of participants who continued receiving efgartigimod PH20 SC during the treatment period. Secondary endpoints included changes from baseline in quality-of-life assessments, perception of disease improvement/severity, treatment satisfaction, and safety/tolerability. Following study completion, participants may have received treatment at the investigator’s discretion per their routine clinical practice and completed a safety follow-up visit 4 weeks after the final study dose of efgartigimod PH20 SC.
Results: The study commenced December 10, 2024. Last patient last visit is expected by end of February 2026. Topline results will be reported.
Conclusion: This study evaluated an approach to transitioning from IVIg to efgartigimod PH20 SC within 1 week after last IVIg infusion in patients with CIDP.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Immune-mediated Neuropathies
PP01.279
Understanding the Patient Journey in Multifocal Motor Neuropathy (MMN): Descriptive Insights From the iMMersioN Study
Dr. Kristl G. Claeys1,2, Dr. Bhaskar Roy3, Dr. Channa Hewamadduma4,5, Dr. Stojan Peric6, Dr. Luis Querol7,8, Dr. Emma Persson9, Ms. Stéphanie Cadour10, Dr. Olivier Van de Steen9, Dr. Clémence Arvin-Bérod9, Dr. Jeffrey A. Allen11
1
University Hospitals Leuven, Leuven, Belgium.
2
Laboratory for Muscle Diseases and Neuropathies, KU Leuven, and Leuven Brain Institute (LBI), Leuven, Belgium.
3
Yale University School of Medicine, New Haven, United States.
4
Academic Neuromuscular Unit, Sheffield Teaching Hospitals Foundation NHS Trust, Sheffield, United Kingdom.
5
Sheffield Institute for Translational Neuroscience (SITRAN), School of Population Medicine and Population Health, University of Sheffield, Sheffield, United Kingdom.
6
University of Belgrade, Faculty of Medicine, Neurology Clinic, University Clinical Center of Serbia, Belgrade, Serbia.
7
Neuromuscular Diseases Unit, Hospital de La Santa Creu I Sant Pau, Universitat Autònoma de Barcelona, Barcelona, Spain.
8
Centro Para La Investigación Biomédica en Red en Enfermedades Raras (CIBERER), Madrid, Spain.
9
argenx, Ghent, Belgium.
10
argenx (via PPD, part of Thermo Fisher Scientific), Ghent, Belgium.
11
University of Minnesota, Minneapolis, United States
Background: Multifocal motor neuropathy (MMN) is a rare, chronic, peripheral neuropathy characterized by progressive and disabling asymmetric limb weakness without sensory loss, primarily caused by immune-mediated complement activation, motor nerve conduction block, and axonal degeneration. Patients with MMN often experience challenges in obtaining a timely diagnosis, accessing appropriate care, and managing the ongoing functional and psychosocial burden of disease. Existing literature is largely restricted to small cohorts and retrospective analyses, suggesting an unmet need for data on patient experience, burden, and health care utilization. This analysis aims to characterize the MMN patient journey using baseline data from adult participants enrolled in the iMMersioN study.
Methods: iMMersioN (NCT05988073) is a global, prospective, longitudinal study enrolling participants from ≥105 sites across 19 countries. Participants with MMN will be followed for at least 24 months and site visits will coincide with regular MMN treatment visits (approximately every 3 months). No investigational medicinal product will be administered. Planned analyses include time from symptom onset to diagnosis, hospitalization patterns, and health care provider visit patterns between definite, probable, and possible MMN (based on European Federation of Neurological Societies/Peripheral Nerve Society [EFNS/PNS] categories) and across geographical distribution.
Results: Baseline demographic and clinical characteristics of the non-North American cohort (N=314), comprising patients from Europe (n=267), East Asia (n=33), and the rest of the world (n=14), will be described, including age, sex distribution, disease duration, EFNS/PNS diagnostic classification, and patterns of limb involvement (eg, upper vs lower limb involvement and the number of limbs affected). Hospitalization patterns (eg, mean number of hospitalizations and mean duration of stays) and analyses of time from symptom onset to diagnosis will be presented, stratified by EFNS/PNS classification and by geographic region. Patterns of health care provider visits, such as the number and type of providers seen, will likewise be examined by EFNS/PNS classification and geographic region. These analyses will improve our understanding of the patient journey, highlighting regional and diagnostic subgroup variations in disease recognition, management, and health care resource utilization.
Conclusion: The presented data from the iMMersioN study are expected to deliver a detailed, multinational perspective on the clinical journey experienced by patients with MMN. Characterizing baseline features, time to diagnosis, hospitalization patterns, and health care resource utilization across geographic and diagnostic subgroups may inform the development of optimized diagnostic strategies and care approaches, ultimately improving outcomes for patients with MMN worldwide.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Immune-mediated Neuropathies
PP01.280
Combined Cyclophosphamide and Rituximab as Rescue Therapy in Atypical CIDP Refractory to First-Line Immunotherapy
Dr. Laura Kennelly, Dr. Orna O'Toole
Cork University Hospital, Cork, Ireland
Background: Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) most commonly affects older adults and typically responds to intravenous immunoglobulin (IVIG). Fulminant presentations mimicking Guillain–Barré syndrome (GBS) are rare. We report a young patient with severe, antibody-negative, treatment-refractory CIDP who achieved sustained recovery following combined cyclophosphamide and rituximab therapy.
Methods: We report the clinical presentation, investigations, treatment course, and outcome of a 36-year-old man presenting with rapidly progressive distal paraesthesia and limb weakness following a mild gastroenteritis. Serial neurological examinations, cerebrospinal fluid analysis, nerve conduction studies, spinal MRI, antibody testing, nerve biopsy, and genetic testing were performed. Therapeutic responses to IVIG, plasma exchange, corticosteroids, rituximab, and cyclophosphamide were documented.
Results: The patient initially presented with mild distal weakness, areflexia, and preserved sensation and coordination but deteriorated despite IVIG to quadriplegia with bulbar and respiratory involvement, requiring intensive care admission, plasma exchange, and tracheostomy. Cerebrospinal fluid demonstrated marked albuminocytologic dissociation (protein 1800 mg/L). Nerve conduction studies showed a severe demyelinating sensorimotor polyneuropathy with absent lower-limb motor responses. MRI of the spine was normal. Ganglioside and nodo-paranodal antibodies were repeatedly negative, and nerve biopsy confirmed demyelination without vasculitis or amyloid. Following minimal response to pulsed corticosteroids and relapse despite rituximab, pulsed cyclophosphamide was introduced, resulting in sustained clinical improvement. The patient progressed from quadriplegia to independent ambulation and return to work within four months, with electrophysiological evidence of remyelination.
Conclusion: This case illustrates a rare, severe, antibody-negative CIDP refractory to standard immunotherapies. Combined cyclophosphamide and rituximab achieved disease stabilisation and marked functional recovery, supporting its consideration as rescue therapy in selected cases of fulminant CIDP.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Immune-mediated Neuropathies
PP01.281
A Case of Acute Flacid Paralysis in a Child With Kaposiform lymphangiomatosis/Kasabach–Merritt Phenomenon
Dr. Maria Kovalchuk, Dr. Lili Khachatryan, Dr. Elizaveta Tuzova
Dmitry Rogachev National Medical Research Center Of Pediatric Hematology, Oncology and Immunology., Moscow, Russia
Background: Acute flacid paralysis (AFP) in children requires a prompt exclusion of infectious reasons and should be differentiated with Guillain-Barre syndrome (GBS), transverse myelitis and acute demyelinating encephalomyelitis.
Methods: A 2.9 y.o. child with kaposiform lymphangiomatosis with Kasabach–Merritt phenomenon was treated with rapamycin and a liposomal form of doxorubicin and prednisone during his hospitalization, when suddenly he developed spasticity in his upper limbs and labs showed decreased serum calcium. In the next 36 hours he was found to have flacid paralysis. CSF analysis showed lymphocytosis up to 190 cell/mcl and protein elevation up to 0.73. Because of incomplete polio immunisation of the baby, spinal cord MRI with contrast enhancement was performed, but did not show any abnormality. Brain MRI was also normal. All infectious causes were excluded, including normal stool specimenand absent non polio enteroviruses. Nerve conduction studies showed primary demyelination of motor and sensory nerves. The baby was regarded as acute inflammatory demyelinating polyneuropathy (AIDP) and treated with human IVIgin standard dose.
Results: The boy recovered completely within few weeks. No reason was detected for persisting CSF lymphocytosis, being elevated for more than a month. Few weeks later the baby developed severe meningitis.
Conclusion: AFP in children requires first of all exclusion of polio and non polio enterovirus reasons. Other rare infectious should also be considered. GBS is one of the most benign causes for AFP in children arising even with rare conditions such as vascular tumor associated with a thrombocytopenic condition. Such children may demonstrate other unusual clinical and laboratory symptoms and require special attention.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Immune-mediated Neuropathies
PP01.282
Long-Term Safety and Efficacy Data of Empasiprubart in Multifocal Motor Neuropathy: Phase 2 ARDA+ Study
Dr. Luis Querol1,2, Assoc. Prof. Thomas Harbo3, Dr. Stojan Peric4, Asst. Prof. Yessar M. Hussain5, Assoc. Prof. Pietro E. Doneddu6, Ms. Stéphanie Cadour7, Dr. Emma K. Persson8, Dr. Miodrag Vujcic8, Prof. Jeffrey A. Allen9, Prof. Eduardo Nobile-Orazio6, Prof. Shahram Attarian10, Prof. Chafic Karam11, Prof. Hans Katzberg12, Dr. Mark Stettner13, Prof. Simon Rinaldi14, Prof. W. Ludo van der Pol15
1
Universitat Autònoma de Barcelona, Barcelona, Spain.
2
Centro De Investigación Biomédica en Red en Enfermedades Raras (CIBERER), Madrid, Spain.
3
Aarhus University, Aarhus, Denmark.
4
University of Belgrade, Belgrade, Serbia.
5
Austin Neuromuscular Center, Austin, TX, United States.
6
IRCCS Humanitas Research Hospital, University of Milan, Milan, Italy.
7
argenx (via PPD, part of Thermo Fisher Scientific), Ghent, Belgium.
8
argenx, Ghent, Belgium.
9
University of Minnesota, Minneapolis, MN, United States.
10
Hôpital La Timone, Marseille, France.
11
University of Pennsylvania, Philadelphia, PA, United States.
12
Toronto General Hospital, University Health Network, University of Toronto, Toronto, ON, Canada.
13
Essen University Hospital, University Duisburg-Essen, Essen, Germany.
14
University of Oxford, Oxford, United Kingdom.
15
University Medical Center Utrecht, Utrecht, Netherlands
Background: Multifocal motor neuropathy (MMN) is a rare, chronic, predominantly complement-driven neuropathy leading to axonal degeneration and progressive, disabling, asymmetric limb weakness. Empasiprubart (EMPA) binds C2, blocking the classical and lectin complement pathways while leaving the alternative pathway intact. The ongoing, phase 2 ARDA+ study (NCT05405361) is assessing the long-term safety and efficacy of intravenous EMPA in adults with MMN.
Methods: Participants completing the 16-week, double-blind ARDA trial (NCT05225675) entered the long-term open label-extension (OLE; ARDA+) and received EMPA at the same dose as in their original ARDA cohort (EMPA or placebo [PBO]). Those discontinuing ARDA or ARDA+ entered a 15-month safety follow-up. Intravenous immunoglobulin (IVIg) is not permitted throughout ARDA+. The primary endpoint was safety. Efficacy endpoints at Week 64 included absolute changes from ARDA baseline in grip strength (GS, most-affected hand), MMN Rasch-Built Overall Disability Scale (MMN-RODS), and modified Medical Research Council 14-sum score (mMRC-14).
Results: Interim results are reported (cutoff: June 4, 2025 [1 year after the last participant rolled over]; median treatment duration: 74 weeks) for this analysis endpoint. Cohort 1 included 17 participants on EMPA/EMPA (received EMPA in ARDA and continued EMPA in ARDA+), and 7 on PBO/EMPA (received PBO in ARDA and switched to EMPA in ARDA+); cohort 2 included 18 on EMPA/EMPA and 9 on PBO/EMPA. At the time of data cutoff, 44 of 51 participants had at least 1 year of treatment with EMPA and remained on treatment. Cohort 2 participants were older with a longer disease duration vs cohort 1; aside from these differences, baseline characteristics were generally well balanced. EMPA demonstrated a consistent safety profile, with no deaths and no new safety concerns. At Week 64, median change (Q1–Q3) from ARDA baseline in GS was 16.7 kPa (2.7–30.7) in EMPA/EMPA and 8.3 kPa (4.0–27.0) in PBO/EMPA in cohort 1, and 44.7 kPa (18.0–53.3) and 10.2 kPa (2.7–19.3) in cohort 2, respectively. Median MMN-RODS (Q1–Q3) increased by 9.0 (0.0–23.0) in EMPA/EMPA and 18.0 (-2.0–7.0) in PBO/EMPA in cohort 1, and 13.5 (6.0–32.5) and 0.5 (-2.0–4.0) in cohort 2. Median mMRC-14 (Q1–Q3) improved by 5.0 (0.0–9.0) in EMPA/EMPA and 8.0 (2.0–16.0) in PBO/EMPA in cohort 1, and 7.5 (3.0–11.0) and 2.0 (-1.0–4.0) in cohort 2.
Conclusion: In ARDA+, participants receiving EMPA maintained improvements across measured efficacy and safety endpoints, while those who switched from PBO experienced new improvements after EMPA initiation, supporting complement system involvement in MMN pathophysiology.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Immune-mediated Neuropathies
PP01.283
Telemedicine-Based INCAT Assessment in CIDP: Reliability and Agreement Between Nurse and Neuromuscular Expert Virtual Evaluations
Dr. Pietro Emiliano Doneddu1,2, Dr. Mazen M. Dimachkie3, Dr. Karissa Gable4, Dr. Arjun Seth5, Dr. Yuyao Sun6, Mr. Brien Hawley7, Dr. Federico Bonofiglio8, Dr. Karen Lynch9
1
Humanitas University, Via Rita Levi Montalcini, Pieve Emanuele (MI), Italy.
2
Neuromuscular and Neuroimmunology Unit, IRCCS Humanitas Research Hospital, Via Manzoni 56, 20089 Rozzano, Milan, Italy.
3
The University of Kansas Hospital, Kansas City, KS, United States.
4
Division of Neuromuscular Medicine, Duke University, Durham, NC, United States.
5
Northwestern Medicine, Chicago, IL, United States.
6
University of Kentucky, Lexington, KY, United States.
7
Clinical Innovation Strategy Implementation, Sanofi, Morristown, NJ, United States.
8
DCVM and EGDS, Sanofi, Cambridge, MA, United States.
9
Sanofi, Cambridge, MA, United States
Background: The Inflammatory Neuropathy Cause and Treatment (INCAT) scale is a widely implemented disability measure in chronic inflammatory demyelinating polyradiculoneuropathy (CIDP). Recent advances in telemedicine, combined with patient-centric care approaches addressing geographic, mobility, and logistical challenges, have driven demand for virtual INCAT assessment to facilitate remote monitoring across clinical trials, registry studies, and routine clinical practice. We aim to evaluate inter-rater reliability of virtual INCAT assessments among multiple assessors and examine systematic agreement between registered nurse (RN)-administered assessments and neuromuscular experts’ evaluations to establish if RN-assessments can reliably replicate expert findings.
Methods: As part of the ORBIT-CIDP study (Observational, Real-world, Digital Biomarker, and Integrated Treatment Outcomes in CIDP; NCT06968975), we conducted a virtual INCAT validation sub-study. ORBIT-CIDP planned to enroll 200 participants with ≥3 months of therapy and residual disability for virtual INCAT assessments by RNs using a telehealth platform. 20 participants were randomly selected at the time of evaluation. These virtual sessions were video recorded with consent and de-identified videos underwent independent INCAT scoring by 7 neuromuscular specialists. Outcomes included 1) general agreement among all experts and scoring reliability, 2) targeted agreement and reliability between RNs and any other expert. Interobserver agreement was measured by Kendall W coefficient, Fleiss kappa, and mean absolute deviation. Interobserver reliability was measured by intraclass correlation coefficients. Pairwise agreement/reliability (RN vs expert) was measured similarly.
Results: To be presented during the congress.
Conclusion: The ORBIT-CIDP study will provide important evidence on the reliability of virtual INCAT assessments by establishing inter-rater concordance between clinical experts and RNs. If validated, telehealth-based INCAT assessment could transform CIDP patient evaluation by enabling remote monitoring in clinical trials, expanding access in resource-limited settings, and reducing patient burden. This study will lay the groundwork for future studies comparing virtual and in-person assessments to establish virtual INCAT as a reliable and accessible standard of care.
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: Outcome measures and clinimetry
PP01.284
Empasiprubart Versus Placebo (EMNERGIZE) or Immunoglobulin (EMVIGORATE) in Chronic Inflammatory Demyelinating Polyradiculoneuropathy: Study Designs
Prof. Simon Rinaldi1, Prof. Thomas H. Brannagan2, Dr. Pietro Emiliano Doneddu3,4, Dr. Karissa L. Gable5, Dr. Luis Querol6, Prof. Mark Stettner7, Dr. Kevin Budding8, Dr. Susan Ellor8, Dr. Hafedh Haddad8, Dr. Martin Markov8, Dr. Reena Patel8, Dr. Olivier Van de Steen8, Dr. Inge Van de Walle8, Prof. Jeffrey A. Allen9
1
University of Oxford, Oxford, United Kingdom.
2
Vagelos College of Physicians & Surgeons, Columbia University Irving Medical Center, Columbia University, New York, United States.
3
Neuromuscular and Neuroimmunology Unit, IRCCS Humanitas Research Hospital, 20089 Rozzano, Milan, Italy.
4
Humanitas University, 20072 Pieve Emanuele, Milan, Italy.
5
Duke University Medical Center, Duke University, Durham, United States.
6
Neuromuscular Diseases Unit, Hospital de La Santa Creu I Sant Pau, Universitat Autònoma de Barcelona, Barcelona, Spain.
7
University Medicine Essen, Essen, Germany.
8
argenx, Ghent, Belgium.
9
University of Minnesota, Minneapolis, United States
Background: Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) is a rare immune-mediated neuropathy involving complement-driven macrophage-mediated demyelination. Empasiprubart binds complement C2, blocking activation of the classical and lectin complement pathways. Two phase 3, randomized, double-blinded studies in participants with CIDP will compare intravenous (IV) empasiprubart versus placebo (EMNERGIZE, NCT07091630) and IV empasiprubart versus IV immunoglobulin (IVIg) (EMVIGORATE, NCT06920004).
Methods: EMNERGIZE (placebo-controlled) will randomize ∼160 adults with CIDP 2:1 to receive either empasiprubart or placebo on Days 1 and 8, then once every 4 weeks in a 24-week double-blind treatment period (Part A) (Figure 1). EMVIGORATE (double-dummy) will randomize ∼218 adults with CIDP 1:1 on stable maintenance IVIg to receive either empasiprubart or continue the stable IVIg dose in a 24-week double-blind treatment period (Part A) (Figure 2). Both studies are followed by a 4-week double-blind rollover, 23-month open-label period (Part B) and 15-month safety follow-up.
Results: The primary endpoint for both studies is a reduction of >=1 point versus baseline in adjusted Inflammatory Neuropathy Cause and Treatment (aINCAT) score at Week 24. Key secondary endpoints (Week 24) include changes from baseline in Inflammatory Rasch-built Overall Disability Scale score, Medical Research Council sum score, dominant hand grip strength, Timed Up and Go, and time to reduction of >=1 point versus baseline in aINCAT score. The EMVIGORATE study commenced on August 22, 2025, while EMNERGIZE commenced on September 16, 2025. Enrollment site information from both studies will be presented.
Conclusion: These two global phase 3 studies will evaluate the efficacy and safety of empasiprubart in participants with CIDP. Similar designs and endpoints with complementary study populations support the potential role of empasiprubart as a reference treatment across patients with CIDP, regardless of treatment status.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Immune-mediated Neuropathies
Images or Table (Optional)
PP01.285
ORBIT-CIDP: A Real-World CIDP Study Leveraging Digital and AI-Powered Patient-Centric Innovation
Dr. Karen Lynch1, Dr. Molly Scannell Bryan2, Dr. Karissa Gable3, Dr. Arjun Seth4, Dr. Pietro Emiliano Doneddu5,6, Dr. Yuyao Sun7, Dr. Jan C. Schuller8, Mrs Melissa Dupont1, Dr. Ashley Cogell2, Dr. Alex Seluzhytsky1
1
Sanofi, Cambridge, MA, United States.
2
PicnicHealth, San Francisco, CA, United States.
3
Division of Neuromuscular Medicine, Duke University, Durham, NC, United States.
4
Northwestern Medicine, Chicago, IL, United States.
5
Humanitas University, Via Rita Levi Montalcini, Pieve Emanuele (MI), Italy.
6
Neuromuscular and Neuroimmunology Unit, IRCCS Humanitas Research Hospital, Via Manzoni 56, 20089 Rozzano, Milan, Italy.
7
University of Kentucky, Lexington, KY, United States.
8
Enovalife, Cœur Défense A – 110 Esplanade du Général de Gaulle – 92931 Paris La Défense Cedex, France
Background: Despite standard-of-care treatments, people with chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) often experience residual disability, mobility limitations, and neurological deficits. These challenges increase the burden of regular, in-person clinical assessments required for clinical research. ORBIT-CIDP (Observational, Real-world, Digital Biomarker, and Integrated Treatment Outcomes; NCT06968975), a first-of-its-kind study, addresses this gap by implementing an innovative telehealth-based approach that integrates artificial intelligence (AI), digital biomarkers, and patient-centric data collection methods. Here we aim to describe the ORBIT study.
Methods: This study will characterize the clinical evolution, quality-of-life, and outcomes in people with CIDP using a direct-to–patient approach. The study will enroll 200 adults (≥18 years) from the United States with CIDP who have received therapy for ≥3 months and have residual disability, impairment, or neurologic deficits. Exclusion criteria include participation in interventional clinical trials. The primary objective is to characterize changes in disability over up to 2 years using the patient-reported Inflammatory Rasch-built Overall Disability Scale (IRODS) score. The secondary objective is to characterize change in disability by the adjusted Inflammatory Neuropathy Cause and Treatment (INCAT) score, including assessing the reliability of virtually administered INCAT. The study will also assess novel digital biomarkers including video-AI movement assessments and insole-derived gait analysis.
Results: To date, 124 participants have been enrolled in ORBIT-CIDP study. Of which 62.1% are female, 82.3% are white, and 89.5% are not Hispanic or Latino. Additional data will be presented at the congress.
Conclusion: ORBIT-CIDP pioneers a direct-to–patient approach in CIDP research, enabling real world, disability assessment and feasibility of virtual INCAT administration. By integrating novel digital biomarkers- including AI-enabled video movement and insole-based gait analysis- this study reduces the burden of clinical visits while potentially validating remote monitoring methods, ultimately accelerating patient-centered research in CIDP.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Immune-mediated Neuropathies
PP01.286
Long-Term Purity Profile of IQYMUNE®: A Direct Result of Quality by Design
Ms. Aurélie Hazotte1, Ms. Linda Baptista1, Mr. Stéphane Boyer1, Dr. Rabye Ouaja2
1
LFB Biotechnologies, Les Ulis, France.
2
LFB Biomédicaments, Les Ulis, France
Background: Quality by Design (QbD) is a systematic approach for incorporating quality into pharmaceutical products. This analysis examines the link between the application of QbD and the long-term purity profile of IQYMUNE®, a plasma-derived intravenous immunoglobulin (IVIg).
Methods: The development of IQYMUNE® was reviewed in accordance with a defined QbD process. This included defining a target product quality profile, identifying critical quality attributes such as purity and key impurities, and conducting risk assessments. Subsequent manufacturing data relating to the purity profile of IQYMUNE® were evaluated over the last decade (2016-2025).
Results: Purity was a primary driver of the QTPP. The QbD process designed and optimized multiple manufacturing steps (e.g., caprylic acid fractionation, chromatography) specifically to achieve high purity by removing impurities. The control strategy, derived from QbD outputs, combined process controls at critical steps with end-product testing. Over a decade after IQYMUNE® launch, analysis of 387 batches shows a mean purity of 99.01% (SD=0.42), with a 95% confidence interval between 98.70% and 99.33%, demonstrating exceptional process stability and output consistency (Figure 1).
The high level of purity of IQYMUNE®, sustained over the years after obtaining its initial marketing authorization, is a direct consequence of its QbD foundation. The systematic, science-based approach ensured deep process understanding, established a robust control strategy, and maintained product quality over its lifecycle. This case underscores QbD's value in achieving predictable, long-term quality for complex therapeutics.
Conclusion: The sustained, exceptional purity of IQYMUNE® over time is a direct result of its foundational Quality by Design development. This demonstrates that a systematic QbD approach successfully builds and maintains high, consistent product quality throughout a product's lifecycle.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Immune-mediated Neuropathies
Images or Table (Optional)
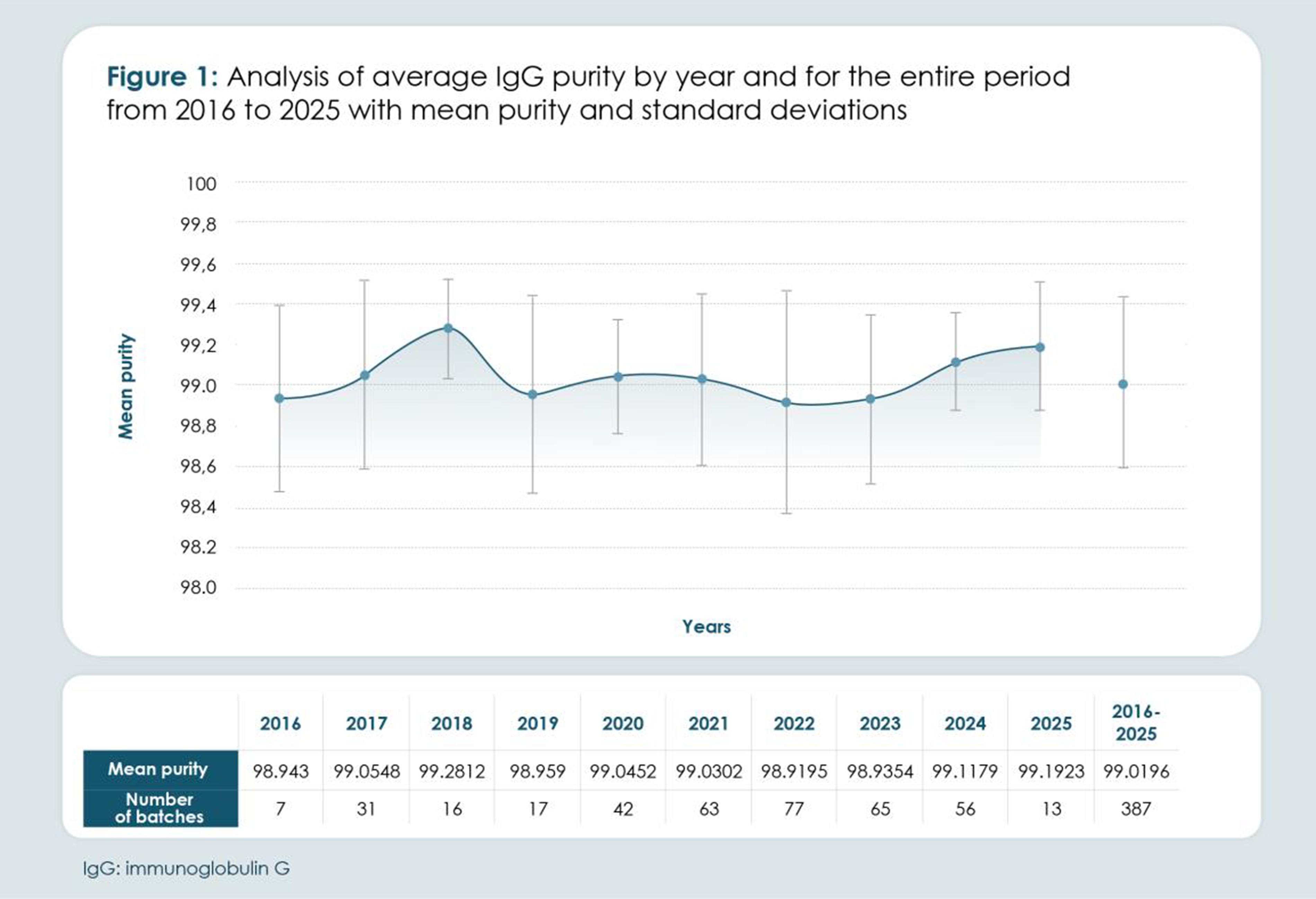
PP01.287
Double-Positive, Double Trouble: Anti–NF186 and Anti-FGFR3 Seropositivity in an Atypical Immune Neuropathy
Mrs Shivi Gunawardane, Dr. David Saxon, Dr. Anastassia Kerasidis, Dr. Oluwaseyi Olulana, Dr. Hassan Khuram, Dr. Nicholas Streicher, Dr. Yasir Al-Khalili
Georgetown University, Washington DC, United States
Background: Autoimmune nodopathies associated with Neurofascin antibodies can resemble CIDP but lack demyelination and respond poorly to IVIG. In contrast, patients with anti-FGFR3 antibodies present with painful sensory neuropathy, frequently non-length dependent with dorsal root ganglion or small fiber predominance. We present an unusual case of seropositivity for anti-NF186 and anti-FGFR3.
Methods: A 71-year-old woman with Hashimoto’s thyroiditis and lumbar radiculopathy developed progressive, predominantly left-sided lower-extremity spasticity with painful paresthesias and thoracolumbar numbness radiating to the legs (“hot knife”) with intermittent cold sensations. Symptoms worsened with weight bearing, stairs, and cold weather. She remained independently ambulatory but was limited by pain, gait instability, and intermittent urinary urgency with incontinence.
She reported intermittent, self-limited episodes of abrupt generalized weakness with transient blurry vision/diplopia and falls; one episode occurred during use of an external neuromodulator and resolved after deactivation. Visual hallucinations were also reported.
Exam showed intact cranial nerves; strength 5/5 except ankle dorsiflexion 4/5; intact pain/temperature; diffuse areflexia, 1+ brachioradialis; and normal Romberg. CSF protein was markedly elevated (352 mg/dL). EMG/NCS showed an axonal sensorimotor neuropathy without demyelination, with mildly reduced sensory amplitudes, mildly slowed median/ulnar velocities, absent right fibular F-waves, limited needle testing due to hypertonicity.
Workup was unrevealing (no M-spike; negative LGI1, CASPR2, NMDA-R, GAD65; mildly elevated B6). INCAT remained 0 despite 10 IVIG infusions and pulse steroids, though sensory symptoms partially improved. WashU testing identified anti-NF186 and anti-FGFR3 IgG, prompting IVIG discontinuation and planned rituximab.
Results: Discussion: This case highlights dual seropositivity for anti–neurofascin-186 (NF186) and anti-FGFR3 antibodies—an association that, to our knowledge, has not been previously reported. These antibody-mediated neuropathies share overlapping features including limb weakness, painful paresthesias, sensory ataxia, and gait instability, and the patient’s phenotype likely reflects contributions from both immune targets. Markedly elevated CSF protein, episodic diplopia suggesting cranial nerve involvement, and diffuse areflexia align with an anti-NF186–associated nodopathy, while autonomic symptoms (notably bladder dysfunction) and fatigue have been described in anti-FGFR3–associated polyneuropathy. Electrodiagnostic testing demonstrated a sensorimotor axonal neuropathy without clear demyelinating features, a pattern reported in both entities. IVIG is often less effective in autoimmune nodopathies, though anti-NF186 cases show variable responses. In anti-FGFR3 polyneuropathy, symptomatic benefit has been reported, but consistent functional improvement remains unestablished. In contrast, B-cell–depleting therapy, most notably rituximab, has shown favorable outcomes in both conditions. Given this patient’s lack of meaningful improvement with corticosteroids and IVIG, escalation to rituximab is next therapeutic step.
Conclusion: This case shows how antibody testing can reframe an “atypical CIDP.” The patient’s painful paresthesias, gait instability, diffuse areflexia, autonomic/bladder symptoms, episodic cranial/visual complaints, markedly elevated CSF protein, and axonal sensorimotor neuropathy without demyelination—plus dual anti-NF186 and anti-FGFR3 seropositivity and poor response to IVIG/steroids—support an antibody-associated immune neuropathy/nodopathy rather than classic CIDP. These antibodies are clinical tools, not standalone diagnoses: interpret them in context, and if discordant, confirm with repeat/alternative testing, reassess EDX/CSF, exclude mimics, and follow longitudinally. Staying current with evolving literature guides timely, targeted therapy.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Immune-mediated Neuropathies
PP01.288
Easily Missed: A Case Series of Lewis-Summer Syndrome
Dr. Tanvir Khosla1, Assoc. Prof. Ge Xiong1, Dr. Ning Wu2, Asst. Prof. Sophie Teng1
1
University of California, Sacramento, United States.
2
Kaiser Permanente, Roseville, United States
Background: Lewis-Sumner Syndrome (LSS), also known as multifocal acquired demyelinating sensory and motor (MADSAM) neuropathy, is a rare asymmetric demyelinating polyneuropathy that falls within the spectrum of chronic inflammatory demyelinating polyneuropathy (CIDP). LSS is characterized by segmental demyelination and remyelination due to immune attacks on the myelin sheath by autoantibodies and inflammatory cells. The pathology is similar to classic CIDP but more localized, leading to multifocal conduction blocks. Unlike classic CIDP, LSS primarily affects the upper limbs and follows multifocal distribution, mimicking mononeuropathy multiplex. It may be misdiagnosed as compression neuropathy, multifocal motor neuropathy, vasculitic neuropathy, even hereditary neuropathies.
Methods: We have compiled a list of 6 patients that have been diagnosed with LSS from 2018 to 2025 at Sacramento Metropolitan area. We summarized the clinical features, neurological examination, lab tests, EMG/NCS study findings as well as imaging abnormalities. All cases have varying clinical presentations with different time courses to achieve the right diagnosis and various treatment responses.
Results: All the patients in this case series presented with asymmetric sensorimotor deficits. Weakness and paresthesia occur in patchy nerve distributions, most commonly affecting the median and ulnar nerves. Reflexes are diminished or absent in all extremities for 4 cases. Out of 6 patients, 3 had lower extremity onset and 3 with upper extremity onset of symptoms. One presented with all distal extremities at the onset. Six cases were slow onset but one had acute onset. The time from onset to confirmed diagnosis ranged from 5 months to 7 years. One case was misdiagnosed as motor neuron disease, one case was first considered to have cervical radiculopathy, one case was initially diagnosed with multiple compression neuropathies. The acute onset case was initially diagnosed as Guillain Barre syndrome (GBS). This case continued progressing over 8 weeks with 2 relapses in 4 months. EMG and nerve conduction studies are crucial for diagnosis. Key findings include multifocal conduction block, prolonged distal motor latency, and temporal dispersion, suggesting primary demyelination neuropathy. Sensory and motor responses are affected asymmetrically. Neuromuscular ultrasound could offer clues of focal fascicles abnormalities at non compression sites that may help improve diagnosis yield. All cases had been treated with intravenous immunoglobulin (IVIg) treatment. Four cases have steady improvements after IVIg treatment. One patient responded to IVIg for 2 years then deteriorated without clear triggering factors. He did not respond to combined steroids and IVIg then started Rituximab. One patient had severe allergic reactions to Ig with Dressler syndrome, and refused steroids/plasmapheresis. He was treated with azathioprine for more than 5 years and slowly progressed. Now this case has been on efgartigimod. The acute onset case responded to steroids and plasmapheresis during exacerbations and has been improving with IVIg.
Conclusion: Lewis-Sumner Syndrome is a rare but treatable immune-mediated neuropathy with distinct clinical and electrophysiological features. Early recognition and targeted immunotherapy, particularly IVIg, can significantly improve outcomes. Further strategies have been considered in this case series with the addition of high-resolution neuromuscular ultrasound to assist in diagnosis.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Immune-mediated Neuropathies
PP01.289
Evaluation of the Effect of Empasiprubart on Nerve Morphology in Multifocal Motor Neuropathy: Study Design
Dr. Stephan Goedee1, Ms. Els de Paepe2, Dr. Emma K Persson2, Dr. Olivier Van de Steen2, Dr. Miodrag Vujcic2, Dr. Sergio Barrera-Sierra2, Dr. Ludo van der Pol3
1
Brain Center Rudolph Magnus, University Medical Center Utrecht, Utrecht, Netherlands.
2
argenx, Ghent, Belgium.
3
University Medical Center, Utrecht, Netherlands
Background: Multifocal motor neuropathy (MMN) is rare, chronic, autoimmune neuromuscular disease that is predominantly complement driven. MMN is associated with slow, progressive, and disabling asymmetric limb weakness in the absence of sensory loss. Over the past decade, ultrasound has gained increasing popularity as a diagnostic tool in inflammatory neuropathies, owing to its wide availability and its capacity to provide high-resolution, accurate visualization of peripheral nerves and nerve roots. A characteristic ultrasonographic and magnetic resonance finding in MMN is nerve enlargement; however, data describing changes in nerve thickness following therapy are limited. Empasiprubart is a first-in-class, humanized, monoclonal antibody that binds complement factor C2, blocking downstream activation of both the classical and lectin complement pathways. Empasiprubart is being developed as a treatment for adult patients with MMN. It demonstrated a favorable safety profile and clinical proof of concept in patients with MMN during the phase 2 ARDA trial. A phase 3 study evaluating the efficacy of intravenous (IV) empasiprubart versus intravenous immunoglobulin (IVIg) in adult participants with MMN is ongoing.
Methods: This phase 1b, open-label, single treatment group study will enroll up to 30 adults aged ≥18 years. The main cohort will include participants with a definite or probable MMN diagnosis per European Federation of Neurological Societies (EFNS)/Peripheral Nerve Society (PNS) 2010 criteria who are receiving maintenance IVIg. The study will also include an ancillary cohort, enrolling participants with possible MMN per EFNS/PNS criteria and/or participants not receiving maintenance IVIg. Key inclusion criteria include confirmed nerve enlargement per ultrasound at screening and a Rasch-built Overall Disability Scale for Multifocal Motor Neuropathy (MMN-RODS) centile score of ≤90. Key exclusion criteria are any coexisting condition that could interfere with the outcome assessments, clinical signs/symptoms suggestive of neuropathies other than MMN, and previous empasiprubart treatment. The study will consist of a screening period (up to 8 weeks), a 48-week treatment period and a 60-week safety follow-up period. Participants will be administered IV empasiprubart on Days 1 and 8 (loading dose) then every 4 weeks from Day 29 during the treatment period. Nerve ultrasound will be performed on Day 1 and screening and Weeks 12, 24, and 48.
Results: This study will evaluate the effect of empasiprubart IV on nerve morphology by ultrasound in adults with MMN. The primary endpoint will be the change from baseline in nerve size at Week 24. Secondary endpoints will include evaluation of empasiprubart on muscle strength/function (modified Medical Research Council-14 sum score, grip strength), manual dexterity (9-hole peg test) and patient-reported outcomes (MMN-RODS) in addition to safety. Exploratory endpoints will include analysis of translational biomarkers (complement activity, neuroinflammation, and autoantibodies).
Conclusion: This phase 1b, open-label, single treatment group study will evaluate the effect of empasiprubart IV on nerve morphology by ultrasound and changes in clinical outcome measures in adults with MMN.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Immune-mediated Neuropathies
PP01.290
Giant Nerve Root Hypertrophy and Pseudofroin’s Syndrome in Chronic Inflammatory Demyelinating Polyneuropathy
Dr. Tanya Banerjee, Dr. Karthik Vinay Mahesh, Dr. Ritu Shree, Dr. Abeer Goel, Prof. Manish Modi
Post Graduate Institute of Medical Education and Research, Chandigarh, India
Background: Chronic inflammatory demyelinating polyneuropathy (CIDP) is a heterogeneous immune-mediated neuropathy with variable clinical, electrophysiological, and radiological manifestations. Nerve root hypertrophy has been described in CIDP but is typically mild to moderate. Marked or “giant” nerve root enlargement resembling plexiform neurofibromas is uncommon and may lead to diagnostic confusion. Severely raised cerebrospinal fluid (CSF) protein levels with pseudofroin’s syndrome and papilledema and secondary complications such as hydrocephalus are rarely reported in this context.
Methods: We describe a case series of three patients presenting with relapsing or progressive sensorimotor lower motor neuron quadriparesis who fulfilled electrophysiological criteria for CIDP. All patients underwent detailed clinical evaluation, nerve conduction studies, CSF analysis, spinal magnetic resonance imaging (MRI), myeloma and inflammatory work-up to rule out secondary causes of CIDP, antibody testing for nodal and paranodal antibodies where available, and genetic testing to exclude hereditary neuropathies. Treatment responses and clinical outcomes were assessed following immunotherapy.
Results: The case series comprised of two males and one female aged 49–55 years. None of them had diabetes mellitus. All three patients demonstrated marked enlargement of spinal nerve roots on MRI, involving cervical and/or lumbosacral regions, with appearances initially reported as plexiform neurofibromas in two of the cases.
One patient additionally showed extensive involvement of intercostal and cranial nerves, including bilateral trigeminal and lower cranial nerves. CSF analysis revealed strikingly elevated protein levels in all patients (range 487–1116 mg/dL), with albuminocytological dissociation and features of pseudofroin’s syndrome, including papilledema. One patient developed communicating hydrocephalus with seizures and altered sensorium, which resolved solely after ventriculoperitoneal shunting combined with immunotherapy. Neurofascin-140 antibodies were positive in one patient, while antibody testing was negative or not performed in the remaining cases, highlighting immunological heterogeneity among the cases. Genetic testing, including whole exome sequencing where performed, did not support a hereditary neuropathy despite radiological mimicry.
All patients showed clinically meaningful responses to immunotherapy, including corticosteroids, intravenous immunoglobulin, mycophenolate mofetil, rituximab, and cyclophosphamide, with improvement from severe disability to assisted or independent ambulation.
Conclusion: This case series expands the recognized clinical and radiological spectrum of CIDP by highlighting extreme nerve root hypertrophy mimicking neurofibromatosis, severe CSF hyperproteinorrachia with pseudofroin’s syndrome, and extremely rare secondary complications such as hydrocephalus. Awareness of this presentation is critical to avoid misdiagnosis and delays in treatment. These findings underscore the importance of considering CIDP in patients with giant nerve root enlargement and reinforce the role of timely immunotherapy in preventing irreversible neurological morbidity.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Immune-mediated Neuropathies
Images or Table (Optional)
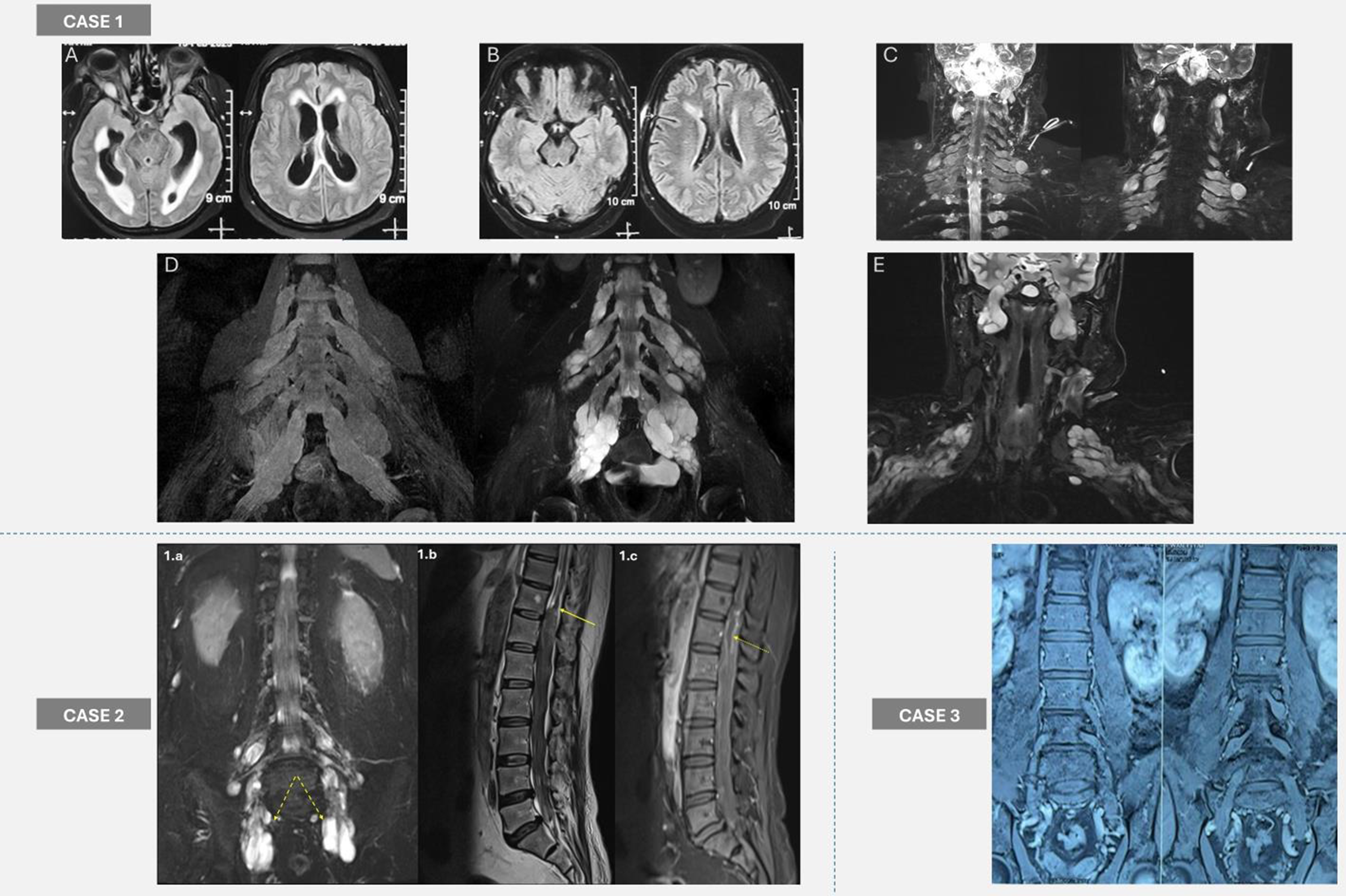
PP01.291
Electrophysiological Evaluation of Oropharyngeal Dysphagia in Chronic Inflammatory Demyelinating Polyneuropathy
Asst. Prof. Zeynep Tanriverdi1, Dr. Aysen Suzen Ekinci1, Dr. Ebru Boluk2, Assoc. Prof. Sehnaz Arici1, Dr. Sevgin Gundogan1, Prof. Yaprak Secil1
1
İzmir Katip Celebi University Ataturk Education and Research Hospital Neurology Department, IZMIR, Turkey.
2
Izmir Sehir Hospital Neurology, IZMIR, Turkey
Background: Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) is the most common chronic immune-mediated demyelinating neuropathy. The clinical examination shows progressive symmetric proximal and distal muscle weakness, sensory loss, and decreased or absent deep tendon reflexes. Cranial nerve and autonomic involvements are less commonly observed in CIDP. In the literature, dysphagia has been described in separate cases diagnosed with CIDP or concurrent myasthenia gravis and CIDP. However, there are no controlled studies about dysphagia. Our research is the first to focus specifically on evaluating dysphagia in patients with CIDP. The aim of this study is to perform an electrophysiological evaluation of oropharyngeal dysphagia in patients with CIDP and to identify changes in cardiac and respiratory responses during swallowing.
Methods: The study included 23 healthy individuals and 26 patients being followed up in our neuromuscular disease outpatient clinic or newly diagnosed and hospitalized in the neurology clinic. Patients with other diseases causing dysphagia were excluded. Only “typical CIDP” patients were included in the study. The diagnosis of CIDP was established based on 2021 EFNS/PNS guideline. All patients and normal controls were examined neurologically and physically. A questionnaire was administered to assess clinical dysphagia condition. Swallowing difficulties and demographic data were recorded. Dysphagia limit (DL) and sequential water swallowing (SWS) tests, were used. Heart rate and respiration frequency during SWS were calculated
Results: The mean age of the patient group was 54.85 ± 16.78 years (range 20-79). In the control group, the mean age was 48 ± 8.57 years (range 40-68). Among the 26 patients, 19 (73.1%) were previously diagnosed with CIDP. Cranial nerve examination revealed facial diplegia in 2 patients (7.7%) and nasal speech in 1 patient (3.8%). The dysphagia limit was >20 mL of water in all normal subjects. Four patients with CIDP demonstrated a decreased dysphagia limit (DL) on electrophysiological tests. Two of the patients with facial diplegia and nasal speech had low DL. In the pre-test questionnaire, three patients reported dysphagia; of these, two also demonstrated a reduced DL during electrophysiological tests. Aspiration was observed in only one patient (3.8%), who also had facial diplegia. While there was no significant difference in the total duration of swallowing between the groups, the swallowing apnea duration (p:0,003) and the number of swallows (p:0,024) were significantly prolonged in all patients with CIDP compared to the normal subjects. The swallowing apnea duration (p:0,003) and the number of swallows (p:0,024) were significantly prolonged in all patients with CIDP compared to the normal subjects.
Conclusion: This study showed us CIDP can cause dysphagia. Sometimes clinically healthy patients have subclinical dysphagia. Routine electrophysiological evaluations generally focus on peripheral nerves, standardized, objective assessment of bulbar involvement is rarely performed in CIDP. Our data demonstrate that dysphagia, though infrequent, does occur in CIDP and is reliably detectable with electrophysiological assessment.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Immune-mediated Neuropathies
PP01.292
Acute Ophthalmoplegia, Facial Weakness and Paraesthesia After Sea-Whelk Consumption – a Case of Paralytic Shellfish Poisoning
Dr. Daryl Yin Keong Lo1, Dr. Elizabeth Ming Jing Tan2, Dr. Yee Sean Teng3, Dr. Kelvi Kaibin Kuan4, Dr. Zhibin Tan1
1
Department of Neurology, National Neuroscience Institute (Singapore General Hospital Campus), Singapore, Singapore.
2
Department of Emergency Medicine, Singapore General Hospital, Singapore, Singapore.
3
Department of Internal Medicine, Singapore General Hospital, Singapore, Singapore.
4
Department of Emergency Medicine, Changi General Hospital, Singapore, Singapore
Background: A 44-year-old lady presented to the emergency department with acute onset binocular diplopia associated with facial and limb paraesthesias 30 minutes after consumption of cooked sea whelk. Physical examination revealed alternating exotropia worse on right gaze, as well as difficulty sustaining upward gaze. There was isolated mild weakness of the bilateral orbicularis oculi. There was generalised hyporeflexia. Pinprick sensation was diminished over all fingertips. The rest of the neurological examination was normal. Bedside pulmonary function tests were normal.
Magnetic resonance imaging of the brain, third cranial nerves and intracranial vessels was unremarkable. Acetylcholine receptor, muscle-specific kinase and ganglioside antibodies were negative. Thyroid function tests were normal. Nerve conduction studies and electromyography, performed on day four of symptoms as an early baseline, were normal. Toxicology testing for paralytic shellfish toxins was not immediately available.
Methods: The patient was managed supportively in the high dependency unit. She showed a rapid improvement of symptoms with initial resolution of her visual and sensory symptoms by day three, followed by resolution of orbicularis oculi weakness on day four, allowing for discharge from hospital. Interval ophthalmology examination at six weeks confirmed improvement of her exotropia.
Results: Paralytic shellfish poisoning is the most common form of biotoxin poisoning which occurs after the consumption of contaminated shellfish. It occurs due to the accumulation within shellfish of a neurotoxin known as saxitoxin, which cannot be destroyed by gastric acid or through food preparation. Saxitoxin blocks voltage-gated sodium channels and modifies channel gating of potassium channels, leading to a rapid onset of neurological symptoms, typically within 30 minutes to 3 hours of shellfish consumption. Sensory disturbances such as tingling and numbness typically occur first, usually starting over the tongue and lips then spreading to the face and distal extremities. Other sensory deficits involving distal touch, vibration and position sense are also possible. In severe cases, there can be ataxia, dysphagia, flaccid paralysis and respiratory failure. Nerve conduction study (NCS) findings may be normal or show abnormalities resulting from the transient ion channel blockade, with resolution of abnormalities within two to three weeks.
Laboratory testing for saxitoxin requires specialised expertise and is not readily available. The diagnosis is hence clinical and other diagnoses with similar presentations should be excluded. Food-borne botulism is distinguished by absence of sensory involvement, presence of cholinergic symptoms and a slower temporal onset. Acute inflammatory demyelinating polyneuropathy has neurophysiological abnormalities which persist for weeks, with positive anti-ganglioside antibodies and elevated CSF protein. Amnestic shellfish poisoning is distinguishable by prominent encephalopathy with severe memory loss, whilst neurotoxic shellfish poisoning causes neuroexcitatory effects rather than paralysis. Ciguatera has characteristic cold allodynia, whereas tetrodotoxin poisoning may feature fixed mydriatic pupils.
Treatment is supportive. Recovery occurs between two days to two weeks, although prolonged symptoms lasting up to three months may occur. Most patients recover fully, however mortality is up to 10% and typically occurs within 12 hours of symptom onset.
Conclusion: This case illustrates the importance of bedside clinical assessment in diagnosing saxitoxin poisoning – a rare syndrome with potentially serious neurological manifestations.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Metabolic, Toxic and Iatrogenic Neuropathies
PP01.293
Metabolic Variability and Inflammation-Derived Biomarkers for Diabetic Polyneuropathy Risk Prediction: A Systematic Review
Prof. Jong Seok Bae
Kangdong Sacred Heart Hospital, Seoul, Korea, Republic of
Background: Diabetic polyneuropathy (DPN) affects over 50% of diabetic individuals worldwide and represents a major microvascular complication. Early detection and risk stratification are critical, yet conventional risk factors alone do not fully explain DPN development. This systematic review synthesizes evidence on emerging biomarkers that enhance DPN risk prediction and early detection. This study systematically reviews biomarkers associated with DPN risk prediction and early detection, categorized by pathophysiological mechanisms.
Methods: A comprehensive search of MEDLINE and Embase (2009–2024) using terms related to diabetic neuropathy, biomarkers, glycemic variability, insulin resistance, dyslipidemia, and systemic inflammation identified studies examining associations between novel biomarkers and DPN development. Studies were stratified by biomarker category: (1) glycemic variability, (2) insulin resistance indices, (3) lipid variability, and (4) systemic inflammatory markers. Risk of bias was assessed using the Newcastle-Ottawa Scale.
Results: Twenty-eight studies met inclusion criteria. Four major biomarker categories emerged: (1) Glycemic Variability—Coefficient of variation in fasting plasma glucose (CV-FPG) and HbA1c variability (CV-HbA1c) independently predicted DPN (HbA1c cutoff 15.15%, sensitivity 66.7%, specificity 65.7%); hemoglobin glycation index (HGI) showed no association; (2) Insulin Resistance—Estimated glucose disposal rate (eGDR) showed promise, while direct DPN outcome data for the triglyceride-glucose (TyG) index remain limited; (3) Lipid Variability—Visit-to-visit variability in HDL cholesterol, triglycerides, and remnant cholesterol associated with increased DPN risk; (4) Systemic Inflammation—Systemic immune-inflammation index (SII), neutrophil-to-lymphocyte ratio (NLR), and platelet-to-lymphocyte ratio (PLR) demonstrated independent DPN associations.
Conclusion: Multiple pathophysiologically distinct biomarker categories improve DPN risk prediction beyond conventional parameters. Integration of these indices may enhance clinical risk stratification. Future prospective studies with standardized DPN diagnostic criteria across diverse populations are needed for clinical implementation.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Metabolic, Toxic and Iatrogenic Neuropathies
PP01.294
Multimodal Neurophysiological Correlates of Disease Severity in Diabetic Peripheral Neuropathy
Asst. Prof. Juhee Chae, Prof. Sun-Young Oh
Department of Neurology, Jeonbuk National University College of Medicine and Hospital, Jeonju, Korea, Republic of
Background: Diabetic peripheral neuropathy (DPN) is a common disabling complication of diabetes mellitus. There is growing evidence that neuropathic processes from diabetes may affect not only peripheral somatic nerves but also the vestibular and autonomic systems. However, few studies have explored how DPN severity relates to different neurophysiological measures, including vestibular reflex parameters and autonomic sudomotor function. This study aimed to analyze associations between DPN severity and several neurophysiological measures: nerve conduction parameters, vestibular function, and autonomic function. Clarifying these associations may help us understand the complex mechanisms underlying DPN.
Methods: In this cross-sectional study, we enrolled patients with DPN and healthy controls (HCs), and performed nerve conduction study, vestibular function tests (including cervical vestibular evoked myogenic potentials [cVEMP], ocular vestibular evoked myogenic potentials [oVEMP], and video head impulse test [vHIT]), as well as quantitative sudomotor axon reflex testing (QSART). We collected clinical data (age, gender, BMI, lipid profile, HbA1c, etc.). Standardized nerve conduction studies were performed, and composite scores for motor, sensory, and overall severity were calculated based on latency and amplitude abnormalities.
Results: This study included 43 patients with DPN and 37 healthy controls. DPN showed a significantly higher age, diabetes duration, and prevalence of dyslipidemia and hypertension than HCs. DPN patients had significantly higher motor, sensory, and overall nerve conduction severity scores, longer cVEMP and oVEMP latencies, and reduced amplitudes. Within the DPN group, proximal leg QSART latency was associated with motor, sensory, and overall severity (r = 0.35–0.38, p = 0.012–0.024), while distal leg latency was significantly associated with sensory and overall severity (r = 0.31–0.38, p = 0.011–0.040). Increasing nerve conduction severity score was associated with delayed cVEMP latencies, and the overall severity score was correlated with both P1 (r = 0.30, p = 0.051) and N1 latencies (r = 0.26, p = 0.089). Vestibulo-ocular reflex gains were not correlated with DPN severity.
Conclusion: Our findings show that disease severity in DPN is associated with latency-dominant neurophysiological abnormalities across the systems. An increase in the nerve conduction severity score was significantly correlated with QSART latencies, particularly in the lower extremity, a pattern consistent with the length-dependent manifestation of DPN. In addition, delays in cVEMP latencies suggest subclinical involvement of vestibulo-collic pathways in DPN. Together, these results indicate a significant association between DPN and vestibular dysfunction, potentially reflecting shared neuropathic mechanisms that concurrently affect multiple pathways and emphasize the value of integrated neurophysiological assessment in patients with DPN. A multimodal approach may improve characterization of disease severity and help identify individuals at increased risk of balance impairment and falls, thereby informing more comprehensive clinical evaluation and targeted management strategies.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Metabolic, Toxic and Iatrogenic Neuropathies
PP01.295
A Case of Ado-Trastuzumab Emtansine induced Nerve Vasculitis
Dr. Rachel Knopp, Dr. Marcus Pinto
Mayo Clinic, Rochester, United States
Background: Peripheral neuropathy is a recognized adverse effect of Ado-Trastuzumab Emtansine (T-DM1). However, an immune-mediated neuropathy triggered by this agent has not been previously reported.
Methods: A case report of vasculitic neuropathy triggered by T-DM1.
Results: A 64-year-old woman with stage IA, grade II, triple-positive infiltrating ductal carcinoma of the breast underwent bilateral mastectomy with sentinel node evaluation and received two cycles of adjuvant T-DM1, followed by prophylactic letrozole. Shortly after her second infusion of T-DM1, she developed severe burning pain in her hands and feet, followed by subacute numbness, sharp pain, and weakness in the right hand, impairing her grip strength and balance. She presented to the Neurology Clinic five months after symptom onset, reporting fatigue, a 20-pound weight loss, and continuous worsening of her symptoms. Neurologic examination revealed moderate-to-severe weakness in the right radial and ulnar nerve distribution, asymmetric mild distal lower limb weakness, and sensory loss affecting light touch, vibration, and proprioception following the right radial and ulnar distribution superimposed on a length-dependent pattern. Reflexes were reduced in the upper limbs and absent in the lower limbs. EMG/NCS demonstrated a length-dependent sensorimotor axonal neuropathy, and subacute right ulnar and radial mononeuropathies. Laboratory evaluation showed elevated neurofilament light chain (195 pg/mL; normal ≤28.8 pg/mL), but was otherwise unremarkable including a negative HIV, ANCA panel, cryoglobulins, ENA, connective tissue disease panel, hepatitis panel, and autoimmune/paraneoplastic axonal neuropathy panel. A right superficial radial nerve biopsy was diagnostic of nerve microvasculitis. The patient was treated with intravenous methylprednisolone 500 mg daily for 3 days, followed by weekly dosing for 11 weeks. At three-month follow-up, the patient's pain had resolved, her weakness was markedly improved, and neurofilament light chain normalized.
Conclusion: T-DM1 may rarely trigger vasculitic neuropathy. Clinicians should suspect an immune-mediated etiology in chemotherapy-associated neuropathies when presenting in a multifocal or asymmetric distribution, and/or symptoms progress beyond three months after discontinuation of the agent.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Metabolic, Toxic and Iatrogenic Neuropathies
PP01.296
Sural-to-Radial Amplitude Ratio (SRAR) in Patients with Diabetic and Chemotherapy-Induced Polyneuropathy
Dr. Maria Kovalchuk1, Assoc. Prof. Eugenia Druzhinina2, Miss Sofya Vorobeva3, Miss Evgeniya Anatolyevna Tikhomirova3
1
Dmitry Rogachev National Medical Research Center Of Pediatric Hematology, Oncology and Immunology, Moscow, Russia.
2
Pirogov Russian National Research Medical University, Moscow, Russia.
3
Scientific and Practical Psychoneurological Center named after Z. P. Solovyov, Moscow, Russia
Background: Diabetic and chemotherapy-induced polyneuropathies (CIPN) are examples of length-dependent sensory axonal polyneuropathy (PNP). A classic early clinical manifestation of this type of PNP is distal sensory loss in the lower extremities. For instrumental confirmation of this complication, electrodiagnostic testing (nerve conduction studies (NCS)) is performed; the findings typically include a reduction in sensory nerve action potential (SNAP) amplitude. It was previously believed that a decreased sural-to-radial amplitude ratio (SRAR)—i.e., the ratio of sural SNAP amplitude to superficial radial SNAP amplitude—of <0.21 could serve as an early sign of this sensory length-dependent PNP.
Methods: We retrospectively analyzed clinical and electrophysiological data from 29 pediatric patients with diabetes mellitus (DM) (16 females and 13 males; median age 16 years, interquartile range [IQR] 13.5–17), 56 adult patients with DM (42 females and 14 males; mean age 60 ± 13.8 years), and 21 pediatric patients receiving chemotherapy (CT). In each group, sensory nerve conduction parameters of the sural and superficial radial nerves were assessed, the sural-to-radial amplitude ratio (SRAR) was calculated, and groups were compared statistically. Patients with a sural sensory nerve action potential (SNAP) amplitude of 0 μV were excluded from the study.
Results: In the pediatric DM group, the mean (M) sural sensory nerve action potential (SNAP) amplitude was 14.36 μV (SD ± 5.9), and the mean superficial radial SNAP amplitude was 24.44 μV (SD ± 8); the mean SRAR was 0.64 (SD ± 0.29). Nineteen patients had a DM duration of <10 years, and ten patients had a duration of ≥10 years. No statistically significant differences in SRAR were observed between these subgroups according to disease duration.
In the pediatric CT group, the mean sural SNAP amplitude was 12.3 μV (SD ± 4.1), the mean superficial radial SNAP amplitude was 26.6 μV (SD ± 7), and the mean SRAR was 0.59 (SD ± 0.22). The difference in SRAR between pediatric patients with DM and those receiving CT was not statistically significant.
In the adult DM group, the mean sural SNAP amplitude was 7.81 μV (SD ± 4.68), the mean superficial radial SNAP amplitude was 20.04 μV (SD ± 9.26), and the mean SRAR was 0.38 (SD ± 0.21). A statistically significant difference in SRAR was found between children and adults with DM.
Conclusion: The sural-to-radial amplitude ratio (SRAR) did not differ significantly between pediatric patients with diabetes mellitus (DM) according to disease duration, nor between children with DM and those receiving chemotherapy (CT). It can be hypothesized that, when the sural sensory response (SNAP) is preserved, SRAR does not provide an advantage for detecting early signs of axonal polyneuropathy.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Metabolic, Toxic and Iatrogenic Neuropathies
PP01.297
Chronic Ataxic Neuropathy With Disialosyl Antibodies Responsive to Zanubrutinib
Dr. Benedetta Tierro1, Asst. Prof. Andrea Visentin2, Dr. Alessandro Salvalaggio1, Dr. Gabbriella Cacchio'3, Dr. Susanna Ruggero1, Dr. Francesco Logullo4, Prof. Chiara Briani1
1
Neurology Unit, Department of Neurosciences, University of Padova, Padova, Italy.
2
Hematology Unit, Department of Medicine, University of Padova, Padova, Italy.
3
3Neurology Unit AST AP, Mazzoni Hospital, Ascoli Piceno, Italy.
4
Neurology Unit, Pesaro Hospital, Pesaro, Italy
Background: To describe a patients with chronic ataxic neuropathy with disialosyl antibodies (CANDA) with clinical and neurophysiological improvement after zanubrutinib therapy.
Methods: A 62-year-old-man started complaining of sensory symptoms in his hands and feet extending over time to the knees and elbows. Nerve conduction studies were consistent with diffuse sensory axonal ganglionopathy. Blood tests revealed an IgM 𝜆 paraprotein expression of a Waldenström’s macroglobulinemia. Anti-MAG and anti-neuronal antibodies were negative. Anti-ganglioside IgM antibodies (GD1b; GD2, GD3, GT1a, GT1b, GQ1b) were positive.
Neurological examination disclosed pinprick, and tactile hypoesthesia with stocking-glove distribution. The patient was responsive to intravenous immunoglobulin (IVIg) but reported end-dose fluctuations. Rituximab was started but soon discontinued for clinical worsening so IVIg therapy was resumed.
Results: The patient was started on zanubrutinib, a second generation Bruton's tyrosine kinase (BTK) inhibitor, while continuing IVIg with global improvement and absence of end of dose efficacy. After 15 months, besides a clinical and hematological amelioration, a dramatic neurophysiological improvement also occurred. Antibodies to IgM GM1 and GM2 were not detectable, whereas anti-disialosyl antibodies GD1a and GD1b persisted unchanged.
Conclusion: To the best of our knowledge, this is the first patient with Waldenström-associated CANDA with clinical, hematological and neurophysiological benefit after zanubrutinib therapy.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Neuropathies Associated with Monoclonal Gammopathy / Paraneoplastic
PP01.298
The Significance of Carpal Tunnel Syndrome for Diagnosing AL-Amyloidosis Associated With Multiple Myeloma
Mrs Liubov Pikus1, Dr. Natalia Chernova1,2, Mrs Eleonora Makunina1, Ms. Zemfira Bekoeva1
1
Moscow Multidisciplinary Clinical Center «Kommunarka», Moscow, Russia.
2
Russian Medical Academy of Continuing Professional Education, Moscow, Russia
Background: AL-amyloidosis is the most common systemic amyloidosis, 5,000 new cases are diagnosed annually in Europe with a frequency of 10 per 1,000,000 people [Merlini G., Palladini G, 2013]. Usually, a small and slowly proliferating clone of aberrant B cells secretes monoclonal free light chains (FLCs), which are improperly folded and deposited in tissues in the form of extracellular amyloid fibrils [Gerts M.A., Dispenzieri., 2020]. AL-amyloidosis can debut as an independent disease and complicate the course of multiple myeloma (MM) [Thimm A. et al., 2022]. The most common target organs are the heart, kidneys, and peripheral nervous system. The average life expectancy of patients with AL-amyloidosis after diagnosis ranges from 4 months to 3 years, which is associated with late diagnosis due to the variety and non-specificity of clinical manifestations. Heart damage is the most common cause of death in AL-amyloidosis [Merlini G., Palladini G., 2013]. Given the tendency of amyloid to deposit in soft tissues, patients may also develop carpal tunnel syndrome (CTS), which is bilateral in 21% [Marcus V. P. et al.,2020] and may precede other clinical manifestations of AL-amyloidosis [Namiranian D., Geisler., 2022]. Alertness towards amyloidosis and the detection of early signs of the disease can prevent irreversible damage to target organs [Merlini G., 2017].
Methods: 18 patients (16 women and 2 men) with multiple myeloma and amyloidosis, confirmed by the results of histological and immunohistochemical studies of subcutaneous fat biopsy, were treated at Moscow Multidisciplinary Clinical Center (MMCC) «Kommunarka» in the period from 2023 to 2025. The median age was 73 (63-88) years, and patients over 70 years of age predominated. Neurological examination was performed on all patients, Nerve conduction studies (NCS) was performed. The CTS was identified based on the results of NCS and ultrasound of the median nerves. Damage to organs and tissues was determined according to laboratory and instrumental studies.
Results: Carpal tunnel syndrome was detected in 14 (78%) of the 18 patients. 7 patients had unilateral CTS, and 7 had bilateral CTS. According to clinical and NCS data, CTS was accompanied by neuropathy in 10 patients. Neuropathy was detected in all patients with bilateral CTS (3 people – axonal sensory, 3 – axonal motor-sensory, 1 – probable small fiber neuropathy (SFN)).In patients with unilateral CTS neuropathy was detected in 3 out of 7 (1– sensory, 1–motor-sensory, and 1 – SFN).
Conclusion: Carpal tunnel syndrome in patients with multiple myeloma and AL-amyloidosis is common and can be established before signs of damage to other organs are detected. Both two- and one-sided involvement is of diagnostic importance. There may be a combination of carpal tunnel syndrome with neuropathy, especially often this combination is observed with bilateral CTS. The diagnosis of CTS in patients at risk of AL-amyloidosis is important for its early verification.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Neuropathies Associated with Monoclonal Gammopathy / Paraneoplastic
Images or Table (Optional)
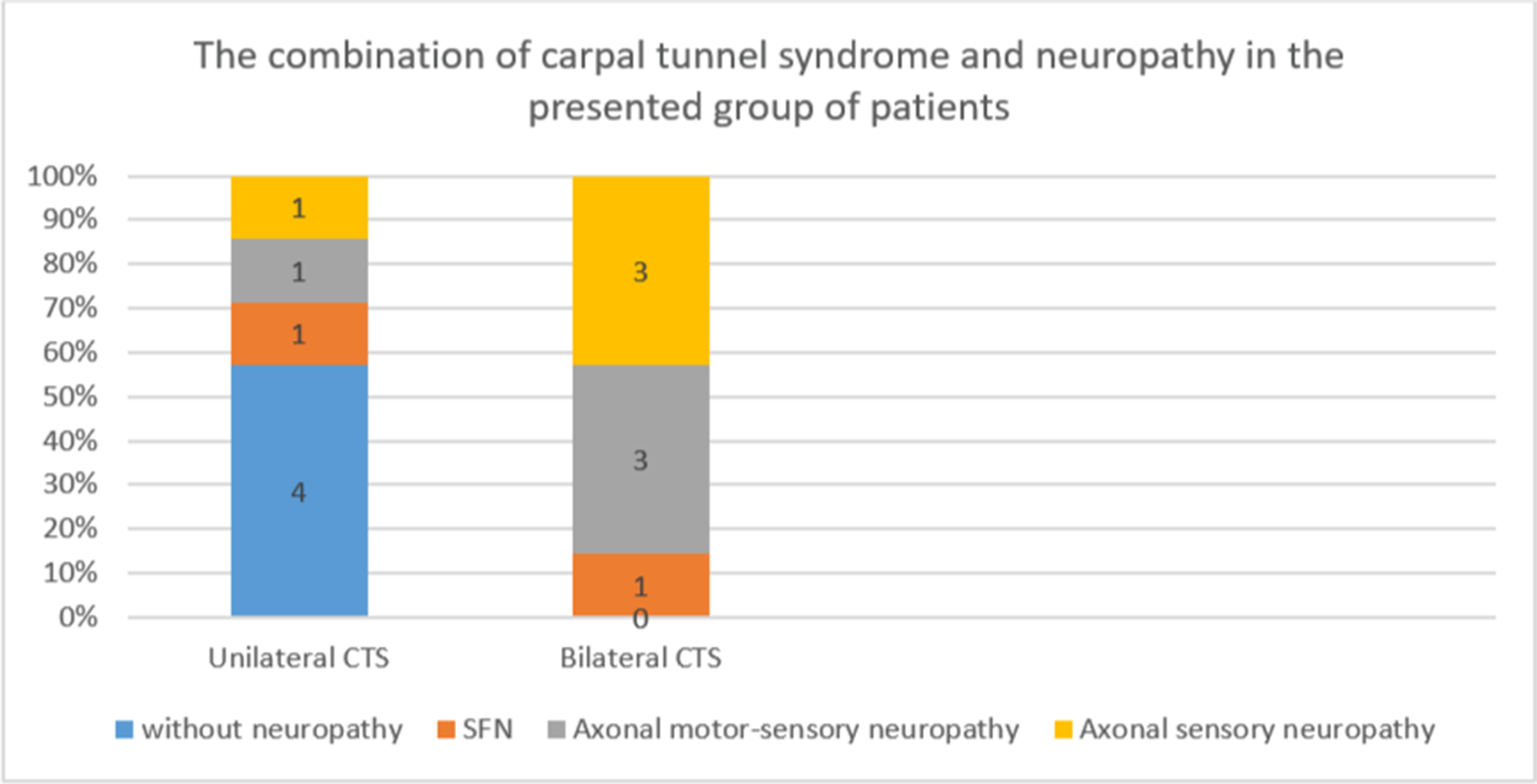
PP01.299
Intravascular Large B-Cell Lymphoma Mimicking Guillain–Barré Syndrome
Prof. Sunyoung Kim
Ulsan University Hospital, Ulsan, Korea, Republic of
Background: Intravascular large B-cell lymphoma (IVLBCL) is a rare aggressive extranodal lymphoma characterized by proliferation of malignant B cells within small vessel lumina. Neurologic manifestations may mimic immune-mediated neuropathies, leading to diagnostic delay.
Methods: A 62-year-old woman with a history of hypertension and seropositive rheumatoid arthritis initially presented with anal and left posterior thigh pain, followed by fever and a diagnosis of influenza A. Two days later, she developed abrupt bilateral leg weakness, sensory impairment below the L1 dermatome, and bowel and bladder dysfunction. Initial neurologic examination showed Medical Research Council (MRC) grade 2 weakness in the lower extremities, decreased anal tone, intact cranial nerve function, and preserved deep tendon reflexes (DTRs). Cerebrospinal fluid (CSF) analysis demonstrated albuminocytologic dissociation but with decreased glucose and markedly elevated LDH. Nerve conduction studies (NCS) revealed reduced bilateral tibial and peroneal CMAP amplitudes, prolonged left median distal latency, and absent F and H reflexes. Spine MRI showed enhancement of the cauda equina from L1 to S3 and inflammatory changes in the epidural space from C7 to S1.
On hospital day (HD) 3, as the patient’s bilateral lower-extremity strength deteriorated to MRC grade 1 and the knee and ankle DTRs progressed to areflexia, a 5-day course of intravenous immunoglobulin (IVIG) was initiated. Despite IVIG, further deterioration occurred on HD 6, including limitation of extraocular movements, bilateral upper-extremity weakness (MRC grade 3), impaired cheek puffing, and a confusional state. Repeat NCS on HD 6 revealed further decreased CMAP amplitudes in the tibial and peroneal nerves, suggestive of acute motor axonal polyradiculoneuropathy.
On HD 7, she developed respiratory difficulty and a drowsy mental state and was transferred to the intensive care unit. Arterial blood gas analysis revealed a pO₂ of 67 mmHg, a lactate level of 11.6 mmol/L, and a SaO₂ of 93.3% despite supplemental oxygen. Chest CT and abdominal–pelvic CT were performed due to worsening pneumonia. Abdominal–pelvic CT revealed infiltrative involvement of the cervix, vagina, adnexa, adrenal glands, and kidneys, with mild splenomegaly—findings strongly suggestive of systemic lymphoma
Results: A cervical biopsy performed on HD 12 showed CD20-positive squeezed cells within vascular lumina, with limited diagnostic interpretability, but raising suspicion for IVLBCL. Despite broad-spectrum antibiotics and corticosteroid treatment, multiorgan failure progressed, and the patient expired on HD 15.
Conclusion: This case demonstrates that IVLBCL may initially present as a Guillain-Barré syndrome. Markedly elevated LDH, reflecting tumor-related tissue destruction, along with atypical CSF findings (elevated protein, low glucose, high LDH) and poor response to IVIG should raise suspicion for IVLBCL. Early systemic imaging and tissue biopsy are essential for timely diagnosis and treatment in patients with rapidly progressive neuropathy and unexplained systemic abnormalities.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Neuropathies Associated with Monoclonal Gammopathy / Paraneoplastic
PP01.300
Immunoglobulin Light-Chain Amyloid Neuropathy Mimicking a Lower Motor Neuron Disorder: A Case Report
Dr. Tanish Modi1, Dr. Brendan Putko2, Dr. Kamal Shouman1, Dr. Marcus Pinto1
1
Mayo Clinic, Rochester, United States.
2
University of Alberta, Edmonton, Canada
Background: Immunoglobulin light-chain (AL) amyloidosis is the most common cause of amyloid neuropathy, with peripheral nerve involvement occurring in up to one‑third of patients. The neuropathy is typically sensory predominant, and autonomic symptoms are frequent. Rarely it may mimic a motor neuron disorder.
Methods: We describe a patient with AL amyloidosis presenting with progressive weakness resembling a lower motor neuron disorder.
Results: A 69-year-old man presented with a 4-year history of slowly progressive weakness without sensory or autonomic symptoms. Initial manifestations included chewing fatigue, progressive dysphonia, and dysphagia. He later developed bilateral but asymmetric (right > left) hand weakness with atrophy. These findings were initially attributed to C8 radiculopathies and carpal tunnel syndrome. He also experienced bilateral rotator cuff injuries resulting in persistent shoulder girdle weakness. Carpal tunnel release and C7–T1 bilateral foraminotomies did not result in clinical improvement. Neurological examination demonstrated asymmetric mild-to-moderate distal-predominant weakness and atrophy in the upper limbs (right > left) and right lower limb with normal reflexes and normal sensory exam. Electromyography showed active and chronic denervation in the bulbar, cervical, thoracic, and lumbosacral segments, consistent with a diffuse anterior horn cell disorder, superimposed by a mild sensory neuropathy. Laboratory evaluation revealed markedly elevated kappa free light chains (131 mg/dL; normal 0.33–1.94) and an abnormal kappa/lambda ratio of 655 (normal 0.26-1.65). Serum mass spectrometry (mass-fix) identified a small IgG kappa monoclonal gammopathy. Subcutaneous fat aspirate was positive for amyloid, and mass spectrometry confirmed kappa AL amyloidosis. Bone marrow biopsy demonstrated 15–20% clonal plasma cells. There was no evidence of renal involvement, lytic bone lesions, or other systemic manifestations. The patient initiated treatment with daratumumab-CyBorD. Neurological function remained stable over six months, but only a partial hematologic response was achieved. Given the atypical presentation, a right vastus lateralis muscle biopsy and sural nerve biopsy were obtained. Muscle biopsy revealed prominent interstitial amyloid deposition with mild myopathic features, and mass spectrometry analysis confirmed kappa AL amyloidosis. The sural nerve biopsy identified small perivascular amyloid deposits and mild decreased density of myelinated fibers. With biopsy-confirmed neuromuscular amyloid infiltration, treatment was escalated to autologous hematopoietic stem cell transplantation. Due to persistent partial hematologic response, the patient subsequently underwent chimeric antigen receptor T-cell therapy, resulting in a complete hematologic response and mild improvement in bulbar and upper limb symptoms after 2 months.
Conclusion: AL amyloidosis should be considered in the differential diagnosis of lower motor neuron syndromes, even in the absence of sensory or autonomic involvement. The mechanism underlying predominant motor involvement in this case remains uncertain. Tissue confirmation of amyloid deposition in both muscle and nerve guided escalation of therapy and contributed to improved clinical outcomes.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Neuropathies Associated with Monoclonal Gammopathy / Paraneoplastic
PP01.301
POEMS Syndrome: A Multisystem Presentation
Dr. Tara Hamilton, Dr. Orna O'Toole
Cork University Hospital, Cork, Ireland
Background: Poems is a rare condition. The prevalence is estimated to be approximately 0.3 cases per 100’000 people. It often can be difficult to diagnose and it is easily missed given the multiple different signs/symptoms that it can present with.
Methods: We had a case of a 49 year old female who presented with abdominal distension and discomfort. She then represented with hypertension. She had a medical background of depression, Castleman's syndrome and right parotid gland resection in 2021 for pleomorphic adenoma. She has no family history of malignancy or hereditary neuropathies.
She had a CTTAP as part of her investigations which revealed low level para aortic adenopathy and mild splenomegaly. Her serum protein electrophoresis showed an IgA lambda monoclonal band in the beta region. She had a bone marrow biopsy done which showed increased plasma cells in the lymphoid cluster. CD56 positivity was appreciated. In situ hybridization for kappa and lambda showed light restriction with dominant lambda positivity. Overall these features were consistent with a plasma cell neoplasm which appears to involve ∼10% of the overall marrow cellularity. VEGF levels then came back raised at 6975.
She was started on daratumumab, lenalidomide and dexamethasone for her plasma cell dyscrasia.
She then developed new sensory symptoms involving reduced sensation in her fingers and then her toes as well as a deeper muscle ache in both calves.
Her neurological exam found reduced sensation in all modalities in upper limbs to the level of her metacarpophalangeal joint and in her lower limbs with slight asymmetry extending higher to the knee on the right versus mid-calf on the left. Her power was preserved, as were her reflexes. Her gait exam was normal. She had visible acrocyanosis in her fingers and toes.
Results: She had nerve conduction studies done which showed evidence of a mixed neuropathy with demyelinating neuropathy. This along with her clinical picture of splenomegaly, Castleman's syndrome, elevated VEGF levels, acrocyanosis and an IgA lambda paraprotein supported a diagnosis of POEMS (polyneuropathy, organomegaly, endocrinopathy, monoclonal plasma cell disorder and skin changes).
Her sensory symptoms were not bothersome enough to warrant medications. She is currently awaiting an autologous stem cell transplant.
Conclusion: This case is a good example of how much work it takes and multiple disciplines are needed to help diagnose this POEMS syndrome. This patient had input from hematology, endocrinopathy, neurology and neurophysiology. The diagnostic criteria include major and minor criteria with mononeuropathy and monoclonal plasma cell proliferative disorder being mandatory criteria. These findings should prompt you to look for the other possible features of POEMS. The pathophysiology of POEMS is not fully understood. Treatment focuses on managing the monoclonal plasma cell proliferative disorder as was the case with our patient.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Neuropathies Associated with Monoclonal Gammopathy / Paraneoplastic
PP01.302
A Diagnostic Odyssey of Paraneoplastic Sensory Neuronopathy
Dr. Sohyun Ahn, Prof. Yangki Minn
Kangnam Sacred Heart Hospital, Seoul, Korea, Republic of
Background: Paraneoplastic sensory neuronopathy is a rare neurological manifestation of malignancy, characterized by the selective destruction of dorsal root ganglion (DRG) neurons. Because it often presents with localized sensory symptoms, it is frequently misdiagnosed, leading to unnecessary surgical interventions.
Methods: A 48-year-old male with a history of lumbar surgery presented with progressive numbness in both lower and upper extremities. Prior to neurological consultation, he had undergone multiple procedures, including vertebroplasty, L4-5 laminoplasty, and an ulnar nerve release at the elbow, none of which alleviated his symptoms. Neurological examination revealed non-length dependent sensory loss, sensory ataxia, and generalized areflexia. Nerve conduction studies showed a profound loss of sensory nerve action potentials with preserved motor conduction, diagnostic of SNN. While extensive paraneoplastic antibody panels and brain MRI were negative, a whole-body PET-CT identified FDG-avid lymphadenopathy in the axilla and groin. Biopsy confirmed Hodgkin lymphoma. Following systemic chemotherapy, the patient achieved oncological remission, which has been maintained for five years.
Results: This case highlights the “diagnostic pitfalls” of paraneoplastic sensory neuronopathy . The presentation of sensory symptoms in the extremities often mimics focal entrapment or spinal pathology, as evidenced by this patient’s three unsuccessful surgeries. The key red flags were the non-length dependent progression and the “pure sensory” involvement on NCS. Notably, unlike some other paraneoplastic syndromes, sensory neuronopathy associated with Hodgkin lymphoma can be antibody-negative and may show poor response to immunotherapy even after successful tumor treatment, reflecting irreversible DRG neuronal loss.
Conclusion: Clinicians should maintain a high index of suspicion for sensory neuronopathy in patients with progressive, multi-focal sensory loss that does not respond to conventional treatments. Early electrophysiological evaluation and malignancy screening are crucial to prevent unnecessary surgeries and to ensure timely diagnosis of underlying hematological malignancies.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Neuropathies Associated with Monoclonal Gammopathy / Paraneoplastic
PP01.303
Prevalence and Phenotypic Correlates of Vestibulopathy in Peripheral Neuropathy and Ganglionopathy
Dr. Fatemeh Rezania1,2, Dr. Lauren Ross1,2, Mr. Murray Worner1, Assoc. Prof. David Szmulewicz3,2, Assoc. Prof. Leslie Roberts1,2
1
St Vincent's Hospital, Melbourne, Australia.
2
The University of Melbourne, Melbourne, Australia.
3
The Royal Victorian Eye and Ear Hospita, Melbourne, Australia
Background: Vestibular neuropathies are often under-recognised in patients with peripheral neuropathy or ganglionopathy, as ataxia is frequently attributed solely to proprioceptive loss despite possible vestibular or cerebellar contributions. We investigated vestibular function across neuropathies and ganglionopathies using video head impulse testing (vHIT). We hypothesised that unrecognised vestibular involvement is common and that specific clinical or neurophysiological features could serve as red flags for vestibulopathy.
Methods: We studied 30 patients with confirmed peripheral neuropathy or sensory ganglionopathy who underwent the Michigan Neuropathy Screening Instrument (MNSI) questionnaire and the Michigan Diabetic Neuropathy Score (MDNS) clinical examination, which included sensory, motor, and reflex testing, along with gait assessment and the Romberg test. Nerve conduction study findings were reviewed to define the neurophysiological phenotype and the number of abnormal nerves (NCS severity). Semicircular canal function was assessed using video head impulse testing (vHIT), which quantifies the vestibulo-ocular reflex (VOR). VOR gain, calculated as the eye/head velocity area ratio, was considered abnormal if <0.8 for horizontal canals or <0.7 for vertical canals.
Results: Twelve of 30 participants (40%) had dysimmune neuropathy, while 7/30 (23%) had hereditary neuropathy, including 4 with RFC1 repeat expansion disorder, 2 with Charcot–Marie–Tooth disease type 1A (CMT1A), and 1 with hereditary neuropathy with liability to pressure palsies (HNPP). Three patients (10%) had metabolic/toxic/nutritional neuropathy, and 8 (26%) were idiopathic.
Nineteen of 30 patients (63%) had abnormal VOR gain in at least one canal, including 4/4 (100%) with RFC1, 6/12 (50%) with dysimmune neuropathy, 2/3 (66%) with metabolic/toxic/nutritional neuropathy, 1/3 (33%) with CMT1A/HNPP, and 6/8 (75%) with idiopathic neuropathy.
In multivariable logistic regression using NCS phenotype axes, none of the individual axes significantly altered the odds of vestibulopathy relative to the sensory, non–length-dependent, axonal baseline. However, this baseline composite phenotype showed a high absolute probability of vestibulopathy (OR 5.37, p=0.038). Consistently, in categorical NCS phenotype analysis, no single NCS pattern was directly associated with vestibulopathy (p>0.13).
In univariate logistic regression, vestibulopathy was strongly associated with ataxia/Romberg positivity (OR 7.1, p=0.042), while neuropathy severity measures and disease duration did not predict vestibular involvement (p>0.3). Conversely, ataxia/Romberg positivity was associated with greater MDNS sensory score (OR 1.47, p=0.015), but not with MDNS motor score or other severity measures.
VOR gain in individual canals or canal planes showed no association with disease duration or neuropathy severity (p>0.3), except for MDNS motor score, which showed a moderate positive correlation with higher (better) VOR gain for both lateral canals (p<0.05), with a similar but weaker pattern for vertical canals.
Conclusion: Vestibulopathy is common in patients with peripheral neuropathy and ganglionopathy and clusters within the sensory ataxic phenotype rather than reflecting overall neuropathy burden. A composite sensory, non–length-dependent, axonal NCS pattern shows high baseline vestibular involvement, although no single NCS feature predicts vestibulopathy in isolation. Vestibular involvement is strongly associated with ataxia, while sensory burden predicts ataxia but not vestibulopathy directly, supporting an indirect, ataxia phenotype-mediated relationship. These findings support targeted vestibular screening and rehabilitation in patients with disproportionate sensory ataxia.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Others
PP01.304
Ischemic Neuropathy Possibly as a Complication of Percutaneous Coronary Intervention: A Case Report
Prof. Geun-Young Park, Dr. Jisun Bae, Prof. Sun Im
Bucheon St. Mary’s Hospital, College of Medicine, The Catholic University of Korea, Bucheon-si, Korea, Republic of
Background: Ischemic neuropathy is an uncommon cause of peripheral nerve injury, resulting from compromised blood flow or oxygen delivery to the vasa nervorum. We report a rare case of ischemic multiple mononeuropathies of the right lower limb that developed after percutaneous coronary intervention (PCI).
Methods: A 57-year-old male presented to the emergency department of our hospital in May 2023 with dizziness. Electrocardiography (ECG) performed in the emergency department showed ST-elevation myocardial infarction (STEMI). Immediate PCI was performed via the right femoral artery, and a temporary pacemaker was inserted through the right femoral vein. Following the procedure, the patient was hemodynamically stable; however, he subsequently reported pain and weakness in the right lower limb during ambulation. Contrast-enhanced computed tomography (CT) of the tibia, contrast-enhanced three-dimensional CT angiography and venography, and lumbar spine magnetic resonance imaging (MRI) were performed, and the patient was referred to us for electrodiagnostic examination. CT revealed complete occlusion of the right popliteal, proximal posterior tibial, anterior tibial, and peroneal arteries. The patient reported generalized pain, swelling, and weakness in the right lower limb. Muscle strength was evaluated using the Medical Research Council (MRC) scale (0–5). Strength in the right lower limb was as follows: hip flexors and knee extensors, grade 5; ankle dorsiflexors, grade 3; first toe extensors, grade 2; ankle plantar flexors, grade 3. Light touch sensation was reduced over the posterior aspect of the right calf. Purplish discoloration and bullae were observed on the distal portions of the second to fourth toes and the dorsum of the right foot.
Results: Initial electromyography (EMG) was performed 4 weeks after symptom onset. EMG was performed. The findings on NCS are consistent with multiple mononeuropathies of the right lower limb, confined to the distal segment below the knee. These lesions are presumed to be associated with ischemia resulting from complete occlusion of the popliteal, anterior tibial, posterior tibial, and peroneal arteries due to a post-PCI embolic event, which is thought to have induced multiple neuropathies involving the distal nerves of the lower limb. Follow-up NCS and EMG were performed in the outpatient clinic in September 2025. Motor strength in the right lower limb showed recovery: grade 5 in the hip flexors and knee extensors, grade 4 in the ankle dorsiflexors, grade 3 in the first toe extensor, and grade 5 in the ankle plantar flexors (MRC scale). On the follow-up study, improvement across multiple nerves was observed, with increased amplitudes in the right peroneal, sural, superficial peroneal, and saphenous nerves compared with the initial examination. On needle EMG, previously observed abnormal spontaneous activities were no longer present. Polyphasic, large-amplitude MUAPs were seen in the right PL, TP, EHL, and GCM, with reduced recruitment and a reduced interference pattern noted in the PL, TP, and EHL.
Conclusion: Ischemic neuropathy caused by acute arterial occlusion following PCI is a rare complication. Early recognition of post-PCI arterial thrombosis is crucial for initiating immediate revascularization therapy. Prompt recognition and timely management are key to preventing motor deficits and optimizing functional recovery.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Others
PP01.305
The Muscle Cramp Impact Index: A Patient-Centered Scale for The Assessment of Muscle Cramps
Prof. Hans Katzberg1, Prof. Vera Bril1, Prof. Lorne Zinman1, Dr. Agessandro Abrahao1, Prof. David Cherney1, Dr. Hemant Shah1, Dr. Meg Mendoza2, Dr. Nouf Alfaidi3, Dr. Sara Alnajjar4, Assoc. Prof. Carolina Barnett-Tapia1
1
University of Toronto, Toronto, Canada.
2
University Health Network, Toronto, Canada.
3
King Faisal Specialist Hospital and Research Centre, Riyadh, Saudi Arabia.
4
King Abdulaziz National Guard Hospital, Riyadh, Saudi Arabia
Background: Muscle cramps are common, variably disabling, and lack a comprehensive validated tool to assess theirmultidimensional impact. Prior cramp studies have relied primarily on cramp frequency and intensity, despitequalitative work demonstrating that sleep disturbance, daytime functioning, and mental health are also importantto patients. Guided by these findings, this study aimed to develop and validate a patient-reported outcomemeasure, the Muscle Cramp Impact Index (MCII), to assess the impact of muscle cramps across diverseetiologies and clinical settings.
Methods: This multi-site, prospective scale-development and validation study conducted in neuromuscular, ALS,hepatology, and kidney clinics from 2018–2022. Item generation was based on a prior qualitative work, literaturereview, and cognitive interviews. Content validity was assessed by 27 international experts. Adults experiencingmuscle cramps within the preceding two weeks were enrolled. Items were evaluated for missing data, floor/ceilingeffects, and exploratory factor analysis (EFA). Test–retest reliability was assessed in clinically stable patients over atwo-week interval using weighted kappas and intraclass correlation coefficients (ICC). Construct validity wasexamined using predefined hypotheses with established clinical measures.
Results: A total of 105 patients (40% female; mean age 60.0 ± 14.2 years) completed field testing. Eighteen draft itemswere refined to 16 after patient and expert review. All items demonstrated low missingness (<6%). EFA identifiedthree domains-cramp frequency/distribution, sleep interference, and interference with daily life - and two itemswere removed for floor effects or low factor loadings, yielding a final 14-item measure (score range 0–42). TheMCII demonstrated moderate correlations with EQ5D, Epworth Sleepiness Scale, Beck Depression Inventory, andSleep Disturbance scores. Test–retest reliability was excellent (n=57; ICC 0.83, 95% CI 0.72–0.89).
Conclusion: The MCII is a patient-centered, psychometrically robust measure capturing the multidimensional impact of musclecramps. Further evaluation of responsiveness and validation in additional cramp populations is warranted tosupport broader implementation.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Others
PP01.306
Ultrasonographic Localization of Ulnar Neuropathy With Normal or Isolated Sensory Nerve Conduction Studies
Prof. Jae-Young An
Department of Neurology, St. Vincent’s hospital, College of Medicine, The Catholic University of Korea, Suwon, Korea, Republic of
Background: Ulnar neuropathy is primarily evaluated using nerve conduction studies (NCS). However, patients may continue to experience characteristic clinical symptoms despite normal or only minimally abnormal electrophysiological findings. In such nonlocalizing cases, accurate lesion localization and etiological assessment remain challenging. High-resolution ultrasonography enables direct visualization of peripheral nerve morphology and may provide diagnostic information beyond conventional electrophysiology.
Methods: We retrospectively analyzed five patients with clinical features consistent with ulnar neuropathy whose routine NCS were either normal or demonstrated only an isolated reduction in distal ulnar sensory nerve action potential (SNAP) amplitude. Short-segment nerve conduction studies were nonlocalizing in some patients. All patients underwent ultrasonographic evaluation of the ulnar nerve, and one patient was followed longitudinally over a 2.4-year period.
Results: Despite persistent ulnar neuropathic symptoms, none of the patients showed consistent lesion localization on NCS. In contrast, ultrasonography identified definite focal structural abnormalities of the ulnar nerve in all cases. Although ultrasonographic abnormalities were consistently detected within the elbow region, the site of maximal cross sectional area (CSA) enlargement varied among patients. Maximal nerve enlargement was observed at different locations along the ulnar nerve within the elbow, including the retrocondylar groove, the medial epicondyle, and the region between the humeral and ulnar heads of the flexor carpi ulnaris, indicating heterogeneity in the location of structural involvement rather than a single predominant site of compression. CSA values ranged from 14 to 36 mm². In one patient with only minimal NCS abnormalities, marked focal nerve enlargement was detected and subsequently confirmed surgically as a schwannoma. In the longitudinally followed patient, a previously observed conduction block resolved spontaneously over time without intervention, whereas ultrasonography demonstrated persistent focal nerve enlargement corresponding to ongoing clinical symptoms.
Conclusion: This case series highlights a clear discordance among clinical symptoms, electrophysiological findings, and ultrasonographic localization in ulnar neuropathy. Although limited by the small number of cases, our findings suggest that the inability of NCS to localize pathology, together with the variable distribution of maximal structural abnormalities within the elbow, underscores the essential role of ultrasonography as a complementary diagnostic tool in patients with ulnar symptoms and nonlocalizing or electrophysiologically equivocal NCS.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Others
PP01.307
Meralgia Paraesthetica Associated with Weight Loss
Dr. Linda Jaffe
Queens Neurosciences Institute, Honolulu, Hawaii, United States
Background: Belts and tight-fitting clothing, obesity, and pregnancy are all common causes of compression of the Lateral Femoral Cutaneous Nerve (LFCN) also known as Meralgia Paraesthetica (MP.) But the LFCN can be vulnerable to other sources of traction as it passes beneath the inguinal ligament. We sought to consider if MP, much to our surprise, could be caused by weight loss.
Methods: A review of the literature and commonly used references such as UpToDate was completed to search for the possibility that mechanical factors involved with loss of weight might contribute to MP. This effort was undertaken when a 63 year old non-Diabetic man presented to our clinic with the classic features of LFCN syndrome without any of the commonly associated risk factors and rather than weight gain, he had lost 30 pounds during the pandemic.
Results: The most commonly identified risk factors for MP are obesity, Diabetes Mellitus, and older age, pregnancy, increased abdominal girth such as in ascites. Mechanical traction on the nerve due to prolonged leaning against a table or bench, carrying heavy objects supported by the groin, various exercises have also been associated. Postoperative MP is also cited as cause of the compressive neuropathy. But weight loss is not described.
Conclusion: Meralgia Paraesthetica is a mononeuropathy of the LFCN and like other neuropathies, injury can be due to various causes with the ultimate result being local ischemia. We report on a case of such injury occurring due to mechanical effects from weight loss rather than weight gain.
(This poster was presented at WCN Rome 2021 during the virtual meeting.)
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Others
PP01.308
Neuromuscular Involvement in Connective Tissue Disease
Dr. Nurul Fadli1,2, Dr. Adrian Ridski Harsono1,2, Asst. Prof. Winnugroho Wiratman1,2,3, Dr. Faisal Parlindungan4, Dr. Dinda Larastika Riyanto2, Asst. Prof. Luh Ari Indrawati2,3, Asst. Prof. Astri Budikayanti2,3, Assoc. Prof. Fitri Octaviana2,3, Asst. Prof. Manfaluthy Hakim2,3, Asst. Prof. Ahmad Yanuar Safri2,3
1
Neurology Department Universitas Indonesia Hospital, Depok, Indonesia.
2
Neurology Department Faculty of Medicine Universitas Indonesia, Depok, Indonesia.
3
Neurology Department Cipto Mangunkusmo General Hospital, Jakarta, Indonesia.
4
Internal Medicine Department Universitas Indonesia Hospital, Depok, Indonesia
Background: Connective tissue disease (CTD) is a group of systemic inflammatory diseases that attack connective tissue in various organs due to autoantibody processes. CTD can involve various organ systems, with one manifestation being the neuromuscular system, which can worsen morbidity and quality of life in patients. However, neuromuscular involvement in CTD is often underrecognized. This study aimed to determine the prevalence, clinical characteristics, and types of neuromuscular involvement in patients with CTD.
Methods: A retrospective, descriptive, cross-sectional study was conducted using secondary data from patients diagnosed with CTD who had neuromuscular symptoms and underwent electrodiagnostic testing at the neurology clinic of the University of Indonesia Hospital. Data were collected from medical records during the period of May 2019 to July 2025. The data collected included demographics, clinical symptoms,neurological examination, nerve conduction study and electromyography, ancillary testing and final diagnosis of the patients.
Results: A total of 44 patients with CTD underwent electrodiagnostic examinations during the study period. The majority were female (97.6%), with a mean age of 41.29 ± 1.908 years. The types of CTD included systemic lupus erythematosus (31.7%), Sjögren’s syndrome (26.8%), rheumatoid arthritis (26.8%), and overlap syndrome (14.6%). Neurological symptoms reported by patients included sensory impairment (61%), pain (61%), weakness (41.5%), tremor and disequilibrium. Neuromuscular involvement confirmed by abnormal electrodiagnostic findings were found in 21 patients (51.2%), including disorders of the peripheral nerves (neuropathies) in 15 patients, neuromuscular junction (myasthenia gravis) in 1 patient, and muscles (myositis) in 5 patients.
Conclusion: Neuromuscular involvement is a common manifestation in patients with CTD. Neurological evaluation should be considered as part of routine assessment in patients with CTD for earlier detection and management.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Others
PP01.309
Unveiling CTS: Ultrasound Detection of Median Neuropathy Despite Normal NCS - A Case Report
Dr. Sang Beom Kim1, Dr. Bum Chun Suh2, Dr. Yang-Ki Minn3, Dr. Hee-Jung Song4, Dr. Jin Yong Lee1
1
Department of Neurology, Kyung Hee University Hospital at Gangdong, Seoul, Korea, Republic of.
2
Department of Neurology, Kangbuk Samsung Hospital, Sungkyunkwan University School of Medicine, Seoul, Korea, Republic of.
3
Department of Neurology, Kangnam Sacred Heart Hospital, Hallym University College of Medicine, Seoul, Korea, Republic of.
4
Department of Neurology, Chungnam National University Sejong Hospital, Sejong, Korea, Republic of
Background: Carpal tunnel syndrome (CTS) is recognized as the most prevalent form of entrapment neuropathy. It occurs due to the compression of the median nerve as it passes beneath the transverse carpal ligament in the wrist. Typical clinical manifestations include numbness, tingling in the fingers, and significant pain that often intensifies at night. In chronic or advanced stages, patients may experience thenar atrophy and a noticeable decline in fine motor skills. While diagnosis is generally based on patient history and electrodiagnostic tests, such as nerve conduction studies, these standard tests do not always capture the full clinical picture. When results from nerve conduction tests appear normal despite persistent symptoms, neurosonography becomes an essential tool for identifying rare anatomical abnormalities or nerve tumors that might be mimicking CTS.
Methods: This case involves a 66-year-old female housewife who sought medical attention for a ten-year history of stabbing pain and numbness in her left palm. Her condition had deteriorated significantly over the final year. Her symptoms were particularly triggered by hand-clapping and were notably worse during the night. These symptoms led to functional impairments, including difficulty holding a book while reading, challenges opening glass bottle caps, and difficulty lifting or maneuvering a shopping cart. Despite these challenges, physical examination showed no observable muscle atrophy in her thumb. Her medical history included hyperlipidemia, for which she took atovastatin, but she had no history of tobacco or alcohol use. Neurological testing revealed a mild decrease in grip strength in her left hand. Both Phalen’s sign and Tinel’s sign were positive in the left hand. Because CTS was the primary suspicion, an upper extremity nerve conduction study was performed; however, the results were entirely normal. To investigate further, clinicians performed neurosonography to evaluate the anatomical integrity of the median and ulnar nerves. The ultrasound revealed that the cross-sectional area of the median nerve was mildly increased, measuring 10.7 mm2 at the left carpal tunnel. The wrist:forearm ratio was also slightly elevated at 1.5. Most importantly, the scan identified a hypoechoic mass measuring 3.8 x 8.4 mm in the symptomatic region of the left palm. A subsequent MRI of the hand provided more detail, showing a heterogeneously contrast-enhancing mass measuring 2.3 x 1.6 x 1.1 cm located between the third metacarpal and the flexor tendons.
Results: The patient was referred for surgical excision, where a white mass measuring 2.2 x 1.5 x 1.3 cm was successfully removed. Histopathological analysis confirmed the mass was positive for S-100 but negative for other markers like SMA and CD 34. This confirmed a diagnosis of schwannoma, a benign nerve sheath tumor, with no evidence of malignancy. Ten months following the surgery, the patient’s recovery was complete. Her motor skills returned to normal, and the persistent numbness, tingling, and pain were entirely resolved.
Conclusion: This case underscores the importance of using neurosonography for differential diagnosis when standard electrodiagnostic results are inconclusive.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Others
PP01.310
Vessel Wall Characteristics in Compressive Trigeminal Neuralgia and Its Correlation With Vascular Risk Factors
Dr. karthik Vinay Mahesh, Dr. Vineet Dixit, Dr. Abeer Goel
Post graduate institute of medical education and research, chandigarh, India
Background: Trigeminal neuralgia (TGN) is commonly attributed to neurovascular compression, but neurovascular contact is also frequently seen in asymptomatic individuals. This suggests that additional vascular factors, such as vessel wall pathology and systemic vascular risk, may influence symptom generation. Vessel wall imaging (VWI) provides a novel approach to evaluate arterial wall characteristics beyond luminal anatomy.
Methods: This was a prospective case–control study including 100 patients with TGN and 17 age- and sex-matched controls with asymptomatic neurovascular conflict of the trigeminal nerve. All participants underwent high-resolution MRI with 3D CISS, TOF angiography, and vessel wall imaging. Neurovascular compression was graded (0–3) based on nerve–vessel relationship. Basilar artery parameters including lumen diameter, wall thickness, lumen-to-wall ratio, and tortuosity index were assessed. Clinical pain severity was evaluated using the Penn Facial Pain Scale. Vascular risk factors and ASCVD scores were recorded. Statistical comparisons were performed between compressive cases and controls.
Results: Among TGN patients, 77% demonstrated neurovascular compression. Cases were significantly older than controls (p = 0.046). Vessel wall thickness, lumen-to-wall ratio, and trigeminal nerve segment length did not differ significantly between compressive cases and controls. Pain severity did not show a significant correlation with compression grade (Spearman rho = 0.11, p = 0.27). Although compressive cases showed higher prevalence of vascular risk factors and higher mean ASCVD scores, these differences were not statistically significant.
Conclusion: Neurovascular compression is common in trigeminal neuralgia; however, macroscopic vessel wall and nerve morphometric parameters alone do not fully explain clinical severity. The findings suggest a multifactorial pathophysiology in which microstructural neural changes and dynamic vascular factors may play a greater role than static imaging features. Vessel wall imaging is feasible in TGN and may provide complementary insights, warranting further large-scale studies
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Others
PP01.311
Machine Learning-Based Skin Nerve Morphometry for Diabetic Neuropathy: Diagnostic and Clinical Implications
Dr. Hsueh-Wen Hsueh1,2,3, Mr. Yao-Yu Wu4, Mr. Tzu-I Chuang4, Ms. Cheng-Chen Lin5, Dr. Ti-Yen Yeh5, Ms. Yi-Hui Kao5, Prof. Herng-Hua Chang6, Prof. Chi-Chao Chao7, Prof. Sung-Tsang Hsieh1,8
1
Department of Neurology, National Taiwan University Hospital, Taipei, Taiwan.
2
Department of Anatomy and Cell Biology, National Taiwan University College of Medicine, Taipei, Taiwan, Taipei, Taiwan.
3
Department of Neurology, National Taiwan University Hospital Hsin-Chu Branch, Hsin-Chu, Taiwan.
4
School of Medicine, National Taiwan University, Taipei, Taiwan.
5
Department of Anatomy and Cell Biology, National Taiwan University College of Medicine, Taipei, Taiwan.
6
Department of Engineering Science and Ocean Engineering, National Taiwan University, Taipei, Taiwan.
7
Department of Neurology, National Taiwan University Hospitalt, Taipei, Taiwan.
8
Department of Anatomy and Cell Biology, National Taiwan University College of Medicine, Taipei, Taiwant, Taipei, Taiwan
Background: Diabetic neuropathy is common and imposes a substantial disease burden on the general population, in particular, affecting small-diameter sensory nerves, small fiber neuropathy (SFN). The degeneration of intraepidermal nerve fiber (IENF) on the skin impairs the protection of painful insult, which leads to a high risk of wound and amputation. Traditionally, the assessment of skin innervation by counting IENFs normalized to the length of the epidermis, IENF density (IENFd), has been the standard. However, the quantification is labor-intensive and time-consuming, and thus hampers the diagnosis of SFN. Advance in machine learning-based image analysis had the potential to facilitate the entire process of quantification without sacrificing the accuracy and reliability. This study aimed to (1) develop and validate new IENF biomarkers with the aid of machine learning algorithms for the diagnosis of SFN in diabetes, and (2) compare its diagnostic performance and clinical significance with the standard of IENFd.
Methods: This study recruited patients with diabetic neuropathy and the control subjects for comparison. Area-based morphometry of IENF (IENFa) parameters were developed using machine learning system for automatic quantification, including three algorithms: Weka-based automatic annotation, edge detection, and custom-convolution methods. The IENFa parameters were further refined according to the area or perimeter of the epidermis as IENFa/A and IENFa/P respectively. The diagnostic performance was assessed according to Receiver Operating Characteristic (ROC) analysis. The clinical implications of the various IENFa parameters were examined by exploring the correlation to metabolic profiles and the electrophysiological studies.
Results: The cohorts, diabetic neuropathy (n=48) and control subjects (n=63), were comparable in the age and gender [age: 59.3 ± 12.5 vs. 56.8 ± 13.0; male gender: 31 (65%) vs. 40 (63%)]. IENFa parameters of different algorithms were inversely correlated with the age and only the IENFd and IENFa/A were sex-dependent in the control group. All IENFa parameters showed equivalent performance according to (1) the correlation with IENFd and (2) the diagnosis of IENFd-based SFN using the area-under-curve (AUC) of ROC analysis, i.e., AUC: 0.91-0.95, P > 0.05. Furthermore, the IENFa biomarkers were significantly correlated to the sural sensory nerve action potential (SNAP) amplitudes, indicating the parallel involvement of small fiber (IENF) and large fiber (SNAP amplitudes) in diabetic neuropathy.
Conclusion: The IENFa using the automatic annotation is time-saving and showed comparable performance to IENFd in the diagnosis of SFN in diabetes. Furthermore, this IENFa also reflect the parallel involvement of large fiber nerves of diabetic neuropathy, showing its efficiency and reliability. Given IENFa-based measure reflects the total area of all IENFs, the results also imply global axonal atrophy in diabetic neuropathy.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Small fibers and painful neuropathies
Images or Table (Optional)
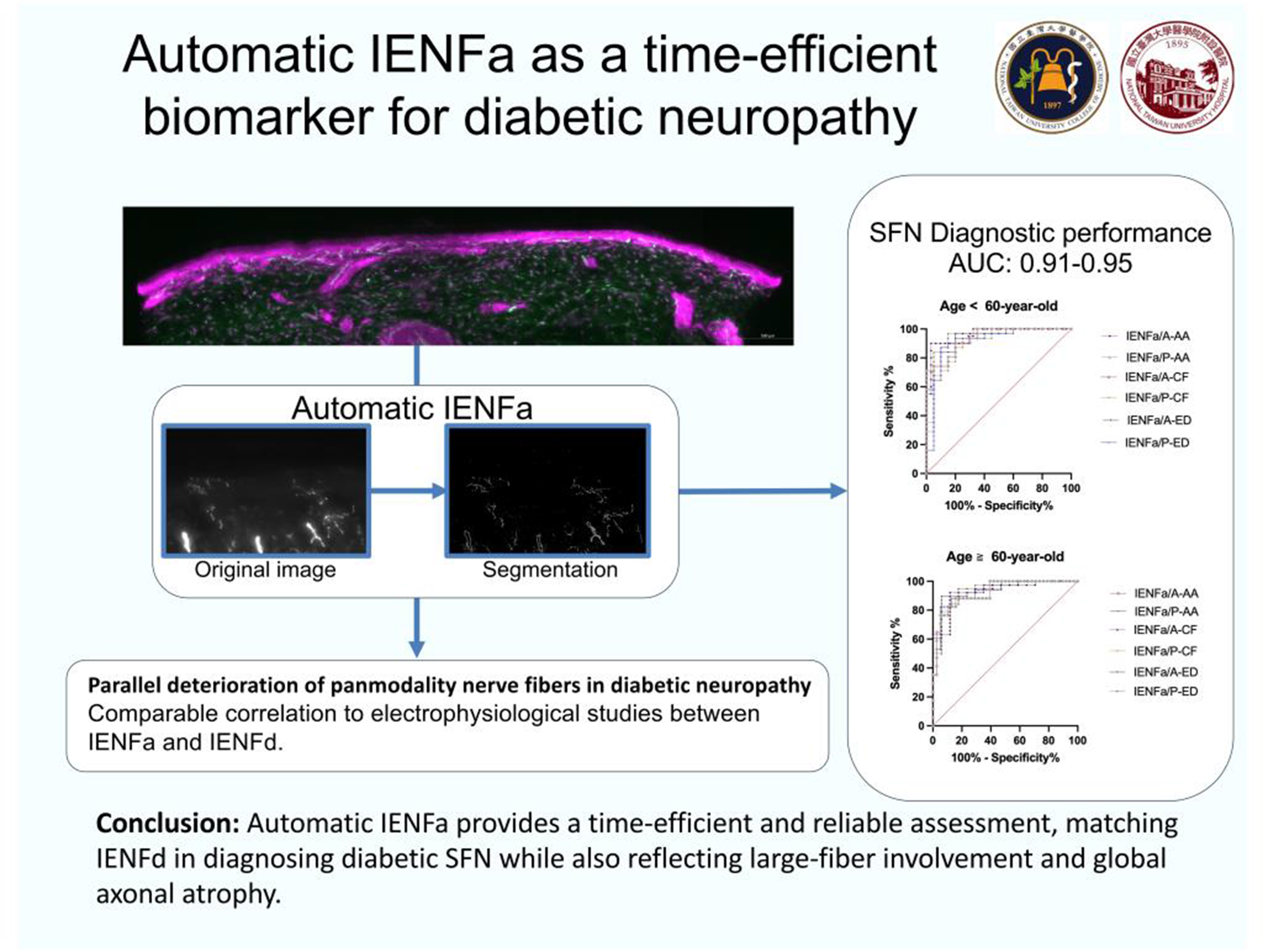
PP01.312
Small Fiber Neuropathy and Autonomic Dysfunction in Sjogren’s Syndrome
Miss Reka Hejas1, Miss Nikolett Orban2, Miss Krisztina Fulop2, Assoc. Prof. David Varga1, Dr. Mirtill Rohonczi1, Dr. Andrea Sipos1, Prof. Endre Pal1
1
University of Pecs, Department of Neurology, Pécs, Hungary.
2
University of Pecs, Depathment of Pathology, Pécs, Hungary
Background: Small-fiber neuropathy (SFN) is a common condition associated with Sjogren’s syndrome (SS). Neuropathic pain is the leading feature of SFN, and the frequency of autonomic dysfunction is unknown.
Methods: Clinical characteristics, histological findings (quantitation of nerve fiber density in the epidermis and around sweat glands in punch biopsies), and findings of autonomic tests (tilt table, 30/15 ratio, Valsalva test, handgrip test, sympathetic skin reflex (SSR), and temperature sensation threshold tests) were investigated in 24 patients with SS and compared with 12 CMT1 (Charcot-Marie-Tooth) patients.
Results: The Valsalva and handgrip tests were the most commonly abnormal (67% and 84% in SS and CMT, respectively). The temperature threshold was abnormal in more than half of the patients. The tilt table test was positive in only one-third of the patients. Low intraepidermal nerve fiber density (IENFD) is associated with abnormal Valsalva results and disturbances in temperature sensation. In patients with neuropathic pain, IENFD was lower than that in those without significant pain. The SSR response was abnormal in at least one extremity in all cases and was related to the diminished innervation of sweat glands in skin biopsy samples. Self-administered questionnaires showed abnormal results in 83% of patients with SS and 100% of patients with CMT. In four patients with SS, autonomic tests were positive despite normal IENFD.
Conclusion: Our study proves that autonomic dysfunction is common among patients with SFN due to either SS or CMT. Questionnaires are sensitive to the recognition of autonomic dysfunction.
Abstract Topic Groups (Submission Categories)
Topic Group 3 - Peripheral Neuropathies, including Cranial Nerves: Clinical Features, Pathophysiology, Therapy: Small fibers and painful neuropathies
PP01.313
Virtual Emergency Care Use in Neuromuscular Disease: Foundations for Multidisciplinary Support
Mrs Emily Farrugia1,2, Dr. Hazel Heng1,2, Dr. Stephen Quick1,2, Prof. Adam Semciw2,1
1
Northern Health, Melbourne, Australia.
2
La Trobe University, Melbourne, Australia
Background: The Victorian Virtual Emergency Department (VVED) is the largest virtual emergency service in Victoria, free for all Victorians requiring urgent but non-critical care as an alternative to in-person care. It remains a novel pathway to managing patients at home who may require more timely medical and support across metropolitan, rural and remote areas across the state. Virtual models of healthcare are rapidly expanding, yet their reach for patients with neuromuscular diseases requiring time-sensitive care, irrespective of funding, age, location or cultural differences, remains unclear. Studies published to date have explored preferences for virtual care and/or remote patient monitoring in comparison to in-person care, as well as barriers and enablers to the mode of these assessments. However, no published studies have examined virtual care use for patients with neuromuscular diseases in the emergency care setting, and evidence within the Australian context is particularly limited. Understanding how patients with neuromuscular diseases use virtual health services is a critical first step to identifying gaps in service delivery, access and scope for advancing multidisciplinary support. This study aimed to explore the uptake, acuity and timing of all presentations to the VVED by patients with neuromuscular diseases and identify relevant referral reasons and usage patterns. A secondary aim was to compare physical emergency department re-presentation rates and outcomes across different VVED referral pathways and identify patient subgroups with higher re-presentation rates.
Methods: A retrospective audit of service data from July 2022 to June 2025, analysing usage patterns and outcomes for patients with neuromuscular diseases including presentations, geographical distribution, admission rates to a physical emergency department and acuity following virtual consultation as well as demographic characteristics using descriptive statistics and pre-defined VVED metrics. Demographic statistics will include age, gender and primary language spoken at home. Re-presentation rates will be calculated as proportions with 95% confidence intervals. Kaplan-Meier survival analysis and Cox proportional hazards regression will examine time to re-presentation.
Results: Between July 2022 and December 2025, n=2,115 presentations with neuromuscular diseases, including Parkinson’s Disease and Multiple Sclerosis, were identified. Analysis is underway to determine the proportion of patients managed virtually without the need for physical emergency department attendance, presenting complaints, and factors associated with the need to transfer to physical emergency department care.
Conclusion: As healthcare systems increasingly adopt virtual-first models, ensuring these innovations meet the needs of patients with neuromuscular diseases is critical. This study will provide foundational evidence to guide the development of tailored virtual pathways for the neuromuscular disease community, with potential for integration into existing multidisciplinary care models.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: Advances in treatment of Motor Neuron Diseases
PP01.314
Risks and Benefits of Gastrostomy in Non-Motor Neurone Disease Progressive Neurological Diseases: A Systematic Review
Ms. Lauren Roberts1,2, Dr. Gina Trakman2, Dr. Kate Furness2, Ms. Emily Farrugia1,2
1
Northern Health, Melbourne, Australia.
2
La Trobe University, Melbourne, Australia
Background: In contrast to Motor Neurone Disease, for which evidence-based guidelines exist, evidence regarding the risks and benefits of gastrostomy insertion and use in progressive neurological diseases, excluding Motor Neurone Disease, is lacking. As a result, gastrostomy decision‐making discussions can arise as a matter of debate amongst clinicians working in the field of neurology. This review aimed to synthesise current evidence on the effects of gastrostomy in Parkinson's disease and parkinsonism, Multiple Sclerosis and Huntington's disease.
Methods: Three databases (Web of Science, Ovid Medline and Embase) were searched for research on the impacts of gastrostomy, including survival, complication rates, and nutrition changes, in adults with progressive neurological diseases, excluding Motor Neurone Disease. Quality was assessed using the Academy of Nutrition and Dietetics Quality Criteria Checklist: Primary Research, and a narrative synthesis was conducted.
Results: Twelve (n = 12) studies were eligible for inclusion, with either retrospective cohort (n = 12) or cross‐sectional (n=1) design, examining the effects of percutaneous endoscopic gastrostomy (PEG) (n = 8), radiologically inserted gastrostomy (RIG) (n = 1), or gastrostomy not further defined (n = 4) in adults with Parkinson’s (n = 6), parkinsonism (n = 3), Multiple Sclerosis (n = 3), or Huntington’s Disease (n = 3). There was variability in outcomes and comparators with inconclusive results. Quality was assessed as positive (n = 3) or neutral (n = 9). Survival post‐gastrostomy insertion ranged from 13.8 to 31.3 months, and in some cases, appeared to be improved by the presence of gastrostomy. Gastrostomy insertion in patients with progressive neurological disease appears to increase health service utilisation and is associated with varying rates of major and minor associated complications. Nutrition‐related outcomes were scarcely reported and did not reflect the potential meaningful impact on the population of interest.
Conclusion: Gastrostomy appears to improve survival in the population cohort, however, the quality of data on this is limited. The review identified an association between gastrostomy and reduced likelihood of discharge home in the population of interest. Gastrostomy appears to be associated with low levels of major complications in the cohort of interest, and varying levels of minor complications. Patients with Huntington’s Disease appear to be at higher risk of dislodgements and tube site infections, potentially due to increased choreiform movements challenging tube-related use and care. The review highlights the need for further, high quality, research to determine the benefits of gastrostomy on well‐defined nutrition‐related outcomes and survival in this patient cohort, as well as the prevalence of relevant gastrostomy‐related complications. In practice, it is hoped this review will provide support to decision‐making practices regarding gastrostomy insertion in this patient cohort.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: Advances in treatment of Motor Neuron Diseases
PP01.315
Immersive Technology and AI Voice Synthesis: Advancing Equity in Motor Neuron Disease
Mr. Lok Man WAN, Ms. Irene Heung Lan CHAN, Mr. Wai Kin CHING
Occupational Therapy Department, North District Hospital, Hong Kong
Background: Motor neuron disease (MND), including amyotrophic lateral sclerosis (ALS), progressively impairs speech and motor function while typically preserving cognition. Demoralization and suicidal ideation rates significantly exceed those in advanced cancer, yet communication barriers severely restrict access to psychosocial interventions. These inequities limit holistic care, leading to loss of autonomy, social isolation, and diminished quality of life. Artificial Intelligence (AI) voice synthesis and immersive technology offer targeted solutions to restore naturalistic expression, meaningful engagement, and equitable support in advanced MND. The objective of this study was to apply AI voice synthesis and immersive technology to advance equity and autonomy in advanced MND. Drawing on occupational therapy's enabling occupation framework, the platform integrated patient-specific voice synthesis with eye-gaze-navigated immersive memory re-creation to enable independent exploration of cherished memories, preserve personal identity, facilitate authentic communication, and support precise expression of needs and emotions, ultimately enhancing quality of life and access to person-centered care.
Methods: A single-case exploratory design followed a 58-year-old participant with advanced ALS (preserved cognition, anarthria, eye-gaze dependent) over 12 weeks in an occupational therapy-led intervention. The co-designed platform featured: (1) AI voice synthesis from the patient's preserved recordings to retain auditory identity and enable natural, emotive narration; (2) immersive, photo-realistic virtual re-creation of a treasured personal memory; and (3) real-time eye-tracking for autonomous navigation. Sessions progressed from calibration to self-directed exploration, reflective storytelling via synthesized voice, and creation of shareable family narratives. Data were collected through semi-structured interviews, usage logs, and the McGill Quality of Life Questionnaire (MQOL), analyzed thematically and descriptively for feasibility, expressive efficacy, autonomy restoration, and quality-of-life outcomes.
Results: The participant independently navigated the immersive memory environment and utilized AI voice synthesis to narrate experiences, emotions, and preferences in a rich, context-embedded manner. This enabled multimodal, naturalistic expression of needs and healthcare priorities. MQOL scores improved notably in existential wellbeing (+32%) and perceived support (+36%). Thematic analysis highlighted restored autonomy through self-guided immersion, reinforced self-identity via personal historical reconnection, and greater confidence in articulating needs. No adverse cognitive or emotional effects occurred. Family members reported enhanced insight and relational closeness through shared synthesized content.
Conclusion: AI voice synthesis combined with immersive technology offers a feasible, low-burden approach to advancing equity and autonomy in motor neuron disease. By preserving identity, enabling independent engagement with meaningful memories, and supporting natural tone and pronunciation in expressing needs and emotions, it reduces frustration and embarrassment from slurred speech, mitigates social isolation, promotes equitable access to tailored psychosocial care, and markedly improves quality of life despite severe physical disability. Guided by occupational therapy principles, this patient-centered innovation shows strong potential as a transformative model for progressive neurological conditions and merits larger-scale evaluation for broader implementation.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: Advances in treatment of Motor Neuron Diseases
PP01.316
Evaluation of the Efficacy of Home-Based tDCS in ALS Patients: A Randomized, Placebo-Controlled Clinical Trial
Dr. Magda Quagliotto1, Dr. Jacopo Della Toffola1, Dr. Edoardo Ricci1, Miss Martina Baldissera1, Dr. Alessio Bratina2, Prof. Paolo Manganotti1,2, Assoc. Prof. Alberto Benussi1,2
1
Neurology Unit, Department of Medical, Surgical and Health Sciences, University of Trieste, Trieste, Italy.
2
Neurology Unit, Hospital Care Department of Medicine, Azienda Sanitaria Universitaria Giuliano Isontina, Trieste, Italy
Background: Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease affecting upper and lower motoneurons, causing gradual muscle weakness and progressively impairing motor and bulbar functions. Disease progression can be monitored using neurofilament light chain (NfL) levels, biomarkers of neuronal damage1. Currently, ALS remains incurable: the only approved medications (riluzole, edaravone) provide only modest survival benefits. Non-invasive brain stimulation (NIBS) techniques represent emerging approaches aimed at modulating cortical hyperexcitability, a relevant pathogenetic mechanism in ALS. Transcranial direct current stimulation (tDCS) exerts long-lasting effects on synaptic plasticity in the motor cortex through modulation of NMDA receptors and GABAergic systems, potentially offering therapeutic benefit. Preliminary studies have demonstrated transient improvements in muscle strength and survival following cortico-spinal tDCS administered over two weeks2,3. A recent systematic review evaluating NIBS techniques in ALS found that prolonged tDCS showed significant improvements in efficacy, tolerability, and adherence4. However, robust evidence remains limited due to study heterogeneity, small sample sizes, and lack of standardized biomarkers.
Methods: This single-center, prospective, double-blind, randomized, sham-controlled superiority trial evaluates the effects of a 16-week home-based tDCS program. Inclusion criteria comprise: age >18 years; probable or definite ALS diagnosis according to current clinical criteria; disease duration ≤24 months; disease progression in the previous 3 months; ALSFRS-R respiratory item score ≥2; stable riluzole or edaravone treatment for ≥1 month; availability of a trained caregiver; informed consent. Exclusion criteria include implanted electrical devices, intracranial metallic foreign bodies, epilepsy, and pregnancy. Forty patients will be randomized 1:1 to receive either real tDCS (bilateral anodal motor cortex stimulation with cathodal cervical spinal stimulation at 4 mA for 20 minutes) or sham stimulation, 5 days/week for 16 weeks. Clinical evaluation will occur at baseline (T0), 16-weeks (T1), 32-weeks (T2) and 48-weeks (T3). Assessments include the ALSFRS-R, muscle strength via handheld dynamometry, quality of life (ALSAQ-40, EQ-5D-5L), caregiver burden (CBI), and plasma NfL levels. The protocol was approved by the local ethics committee (ASUGI, 371/2024H, approved 18.11.24) and registered at ClinicalTrials.gov (NCT07006571).
Results: The primary endpoint is the difference in ALSFRS-R score at T1. Secondary endpoints include ALSFRS-R changes at T2 and T3, muscle strength, ALSAQ-40, EQ-5D-5L, CBI scores, plasma NfL levels, and survival from symptom onset.
Conclusion: Home-based tDCS represents a potentially significant innovation in ALS management, offering a non-invasive, safe, and accessible therapeutic option that may improve patient quality of life and stimulate further research in neurodegenerative diseases.
References:
1 Weydt P et al., Neurofilament levels as biomarkers in asymptomatic and symptomatic familial amyotrophic lateral sclerosis. Ann Neurol. 2016 Jan;79(1):152-8. doi: 10.1002/ana.24552.
2 Benussi A et al., Cortico-spinal tDCS in ALS: A randomized, double-blind, sham-controlled trial. Brain Stimul. 2019 Sep-Oct;12(5):1332-1334. doi: 10.1016/j.brs.2019.06.011.
3 Benussi A et al., Cortico-spinal tDCS in amyotrophic lateral sclerosis: A randomized, double-blind, sham-controlled trial followed by an open-label phase. Brain Stimul. 2023 Nov-Dec;16(6):1666-1676. doi: 10.1016/j.brs.2023.11.008.
4 Della Toffola J et al., Non-Invasive Brain Stimulation for Amyotrophic Lateral Sclerosis: Current Evidence and Future Perspectives. Medicina (Kaunas). 2025 Sep 17;61(9):1685. doi: 10.3390/medicina61091685.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: Advances in treatment of Motor Neuron Diseases
Images or Table (Optional)
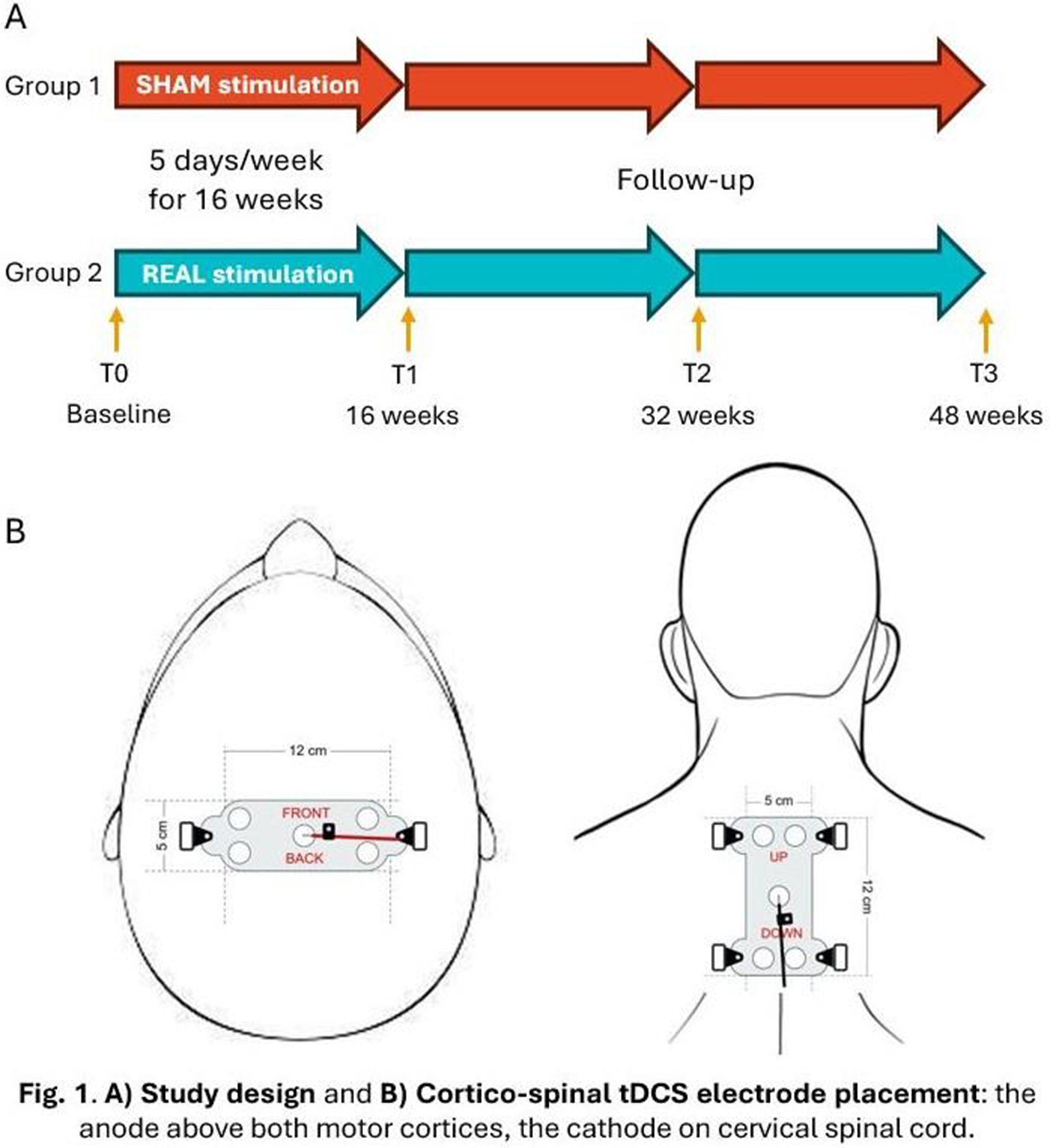
PP01.317
Spontaneous Resolution of Cognitive Symptoms in Morvan Syndrome: A Case Report and Literature Review
Dr. Marco Guglielmi1, Dr. Eleonora Pacucci1, Dr. Valeria Delmonte1, Dr. Gianni Difonzo1, Dr. Eustachio D'Errico2, Dr. Teresa Francavilla2, Dr. Angela Fraddosio2, Dr. Stefania Cataldo2, Dr. Antonio Iaffaldano2, Prof. Damiano Paolicelli1
1
University of Bari “Aldo Moro”, Bari, Italy.
2
Azienda Ospedaliera Universitaria Consorziale Policlinico di Bari, Bari, Italy
Background: A 66-year-old male with a history of arterial hypertension, obstructive sleep apnea syndrome, remote Hodgkin lymphoma and cognitive dysfunction diagnosed as Alzheimer’s disease in 2017 (which fully resolved within 8 months since onset) presented with a subacute onset of motor weakness, spasticity and diffuse and abundant fasciculations beginning in March 2025. This presentation raised suspicion for a peripheral nerve hyperexcitability disorder.
Methods: During hospitalization, the patient underwent neurological examination, blood tests, cerebrospinal fluid (CSF) analysis, electromyography (EMG), motor evoked potentials (MEPs), somatosensory evoked potentials (SEPs), and brain and spine MRI with and without contrast. Therapeutic intervention with intravenous immunoglobulins (IVIG, 0.4 g/kg/day for 5 days) was administered.
Results: Neurological examination revealed paraparesis with spasticity, hyperreflexia and widespread fasciculations. EMG demonstrated diffuse persistent fasciculations and myokymias and sporadic acute denervation activity, suggesting peripheral nerve hyperexcitability in combination with lower motor neuron involvement. MEPs showed abnormal corticospinal tract conduction, while SEPs revealed preserved sensory conduction. MRI imaging and CSF analysis were unremarkable. Blood tests showed positive anti-LGI1 autoantibodies. Following IVIG therapy, a mild clinical improvement was observed, particularly in muscle tone and frequency of fasciculations.
The combination of diffuse fasciculations, EMG findings of peripheral hyperexcitability, absence of structural CNS pathology, partial clinical response to immunotherapy and previous transient cognitive dysfunction supported a diagnosis of Morvan syndrome. The sporadic presence of acute denervation signals raised consideration for concomitant lower motor neuron dysfunction, but no alternative etiology was identified. Furthermore, we performed a review of 21 cases published in English literature (papers found by bibliographic search on PubMed and Google Scholar on January 9, 2026). Out of 21 patients, 6 (29%) had cognitive symptoms at the time of diagnosis and no patient ever experienced a spontaneous resolution of cognitive symptoms. The most common symptoms were insomnia (19 out of 21 patients, 90%), followed by dysautonomia (18 out of 21 patients, 86%), paresthesias, muscle cramps, limb weakness and REM sleep behavioural disorder. 19 out of 21 patients (90%) tested positive for anti-VGKC autoantibodies.
Conclusion: The clinical, neurophysiological and radiological findings were most compatible with Morvan syndrome. Immunoglobulin therapy produced partial symptomatic improvement. The patient was discharged with symptomatic and immunomodulatory treatmentand a follow-up plan including serial laboratory and clinical monitoring. Furthermore, the literature review suggests that a spontaneous resolution of cognitive symptoms is unlikely in Morvan syndrome, therefore more data is needed in order to fully understand the prevalence of this phenomenon.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: Advances in treatment of Motor Neuron Diseases
PP01.318
Dermal Fibroblasts From Amyotrophic Lateral Sclerosis Patients as a Tool to Study Autophagy-Lysosome Pathway Alterations
Dr. Giovanni Vietri1,2,3, Dr. Francesco Gruosso4,2,5, Dr. Mariangela Goglia1,2, Dr. Erica Frezza1,2, Dr. Giulia Greco1,2, Dr. Camilla Rocchi1,2, Prof. Diego Centonze1,3, Dr. Laura Boffa1,2, Dr. Francesca Ciaiola6, Dr. Alida Spalloni6, Dr. Roberta De Mori7, Prof. Roberto Massa1,2,3, Prof. Patrizia Longone6
1
Department of Systems Medicine, University of Rome Tor Vergata, Rome, Italy.
2
Neuromuscular Diseases Unit, Tor Vergata University Hospital, Rome, Italy.
3
Unit of Neurology, IRCCS Neuromed, Pozzilli (IS), Italy.
4
Department of Systems Medicine, University of Rome Tor Vergata, Roma, Italy.
5
Unit of Neurology, IRCCS Neuromed, Rome, Italy.
6
Molecular Neurobiology Unit, Experimental Neurology, Fondazione Santa Lucia, Rome, Italy.
7
Stem Cell Facility, Fondazione Santa Lucia, Rome, Italy
Background: Amyotrophic lateral sclerosis (ALS) is increasingly recognized as a multisystemic disorder in which disturbances of proteostasis and cellular homeostasis are not strictly confined to motor neurons. Increasing evidence points to dysfunction of the autophagy–lysosome pathway (ALP), encompassing macro-autophagy and chaperone-mediated autophagy, as a key pathogenic pathway across sporadic and genetic ALS. In this context, accessible patient-derived cellular models may enable patient-specific profiling and quantitative approaches to patient stratification. We investigated whether primary dermal fibroblasts from ALS patients reflect ALP-related cellular and molecular alterations observed in the human disease.
Methods: We performed a monocentric observational pilot study at a tertiary ALS center. ALS patients (mild-to-moderate disease stages) and matched healthy controls were enrolled after informed consent, with exclusion of major metabolic comorbidities. Skin punch biopsies were obtained and primary fibroblast cultures were established under standardized conditions. Proliferation rate was assessed counting at 0, 24, 48, and 96 hours. ALP-related markers were evaluated using immunoblotting using antibodies against autophagy-related markers, normalized to β-actin and quantified by computerized densitometry. Qualitative protein distribution was assessed through immunofluorescence for autophagy markers, acquiring confocal images at 63× (Zeiss LSM800). Statistical analyses were conducted in Prism; group comparisons used unpaired t-tests with significance at p ≤0.05.
Results: Nine biopsies were collected: five ALS fibroblast lines managed to produce a complete colony, four cultures showed a reduced proliferation rate and clonal production. The ALS cohort included sporadic and genetic forms with heterogenous genotype. Compared with matched healthy controls, ALS fibroblasts showed an early and persistent slower growth rate, with significantly reduced cell numbers at 24 hours (p ≤0.01) and 96 hours (p ≤0.05). Preliminary western blot and immunofluorescence data confirmed an altered autophagy pathway, consistent with altered lysosomal trafficking and impaired vesicle turnover.
Conclusion: Patient-derived dermal fibroblasts in ALS display convergent abnormalities across proliferation, autophagic pathways, and lysosomal organization, supporting an early peripheral ALP defect compatible with a multisystemic model of ALS. Despite the small sample size and reduced culture survival, the agreement across independent assays and the observed inter-patient variability suggest that fibroblast-based multi-marker assays may capture biologically meaningful heterogeneity useful for patient stratification and, in future larger cohorts, could be linked to clinical severity and progression rate and integrated with established and emerging fluid biomarkers.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: ALS: Biology, Pathophysiology, Genetics
PP01.319
Young-Onset Amyotrophic Lateral Sclerosis at a Tertiary Center in Korea
Dr. Jhee LEE
Department of Neurology, Seoul National University Hospital, Seoul, Korea, Republic of
Background: Young-onset amyotrophic lateral sclerosis (yALS), defined by symptom onset before age 45, may differ from typical-onset ALS in genetic architecture, phenotype, and trajectory. We characterized the clinical features, genetic findings, and outcomes of yALS at a large tertiary center in Korea.
Methods: We retrospectively reviewed consecutive patients diagnosed with ALS at the Department of Neurology, Seoul National University Hospital between 01/01/2025 and 12/31/2025. Of 109 total ALS cases, 18 met the age-at-onset criterion for young-onset ALS (yALS; <45 years). Diagnosis adhered to the revised El Escorial and Gold Coast (2019) criteria. Standardized workup included neurological examination, electrophysiology, neuroimaging, and targeted laboratory testing to exclude mimics. An ALS multigene panel was performed in 15 of the 18 yALS patients using next-generation sequencing, and repeat-primed PCR was used to test for C9orf72.
Results: Of 109 patients with ALS, 18 (16.5%) met criteria for young-onset ALS (yALS), with a male-to-female ratio of 3:1. Limb onset was the predominant presentation; bulbar onset was observed in two patients, both of whom were men. Genetic testing identified variants most frequently in SOD1 (n = 3) and FUS (n = 2). Variants of uncertain significance were detected in seven patients. No C9orf72 repeat expansions were identified.
Conclusion: In this single-center cohort, yALS accounted for 16.5% of ALS cases. Pathogenic or potentially relevant variants were enriched in SOD1 and FUS. Clinically, yALS was characterized by predominantly limb-onset disease.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: ALS: Biology, Pathophysiology, Genetics
PP01.320
Dysphagia in Patients With Spinocerebellar Ataxia Type 8-Related Amyotrophic Lateral Sclerosis and in SCA8-Mouse Models
Prof. Makito Hirano1, Dr. Makoto Samukawa1, Ms. Chiharu Isono2, Dr. Tomoyasu Matsubara3, Prof. Yuishin Izumi4, Dr. Yuko Saito3, Dr. Shigeo Murayama3, Prof. Yuji Higashimoto5, Assoc. Prof. Mamoru Nagano6, Prof. Laura P. W. Ranum7, Prof. Yoshitaka Nagai1
1
Department of Neurology, Kindai University, Faculty of Medicine, Sakai, Japan.
2
Division of Rehabilitation Medicine, Kindai University Hospital, Sakai, Japan.
3
Department of Neuropathology (the Brain Bank for Aging Research), Tokyo Metropolitan Institute for Geriatrics and Gerontology, Tokyo, Japan.
4
Department of Neurology, Tokushima University Graduate School of Biomedical Sciences, Tokushima, Japan.
5
Department of Rehabilitation Medicine, Kindai University Faculty of Medicine, Sakai, Japan.
6
Department of Anatomy, Kindai University Faculty of Medicine, Sakai, Japan.
7
Center for Neurogenetics, Department of Molecular Genetics & Microbiology, College of Medicine, McKnight Brain Institute, Genetics Institute and Fixel Institute, University of Florida, Gainesville, United States
Background: We previously identified non-coding CTA/CTG repeat expansions (>50 repeats) in the gene causative for spinocerebellar ataxia type 8 (SCA8) in ∼3% of Japanese patients with sporadic ALS. Seven patients with SCA8-related ALS so far accumulated in our cohort (N = 230). Three patients (43%) had bulbar-onset ALS, and two (29%) had cervical-onset ALS with acute bulbar palsy, while among non-SCA8-related ALS patients 20% presented with bulbar onset and 3% with cervical onset. This study was aimed to clarify swallowing functions in patients with SCA8-associated ALS and in an SCA8-transgenic mouse model with 121 CTA/CTG repeats.
Methods: Videofluorography (VF) was used to evaluate swallowing functions in five of the seven patients. For the evaluation of swallowing functions in mice, a barium contrast agent identical to that used for humans was administered orally to one-year-old-mice, followed immediately by anesthesia and computed tomography analyses. We also conducted pathological analyses in one patient and in heterozygous and homozygous mouse models.
Results: VF examination revealed oral and pharyngeal phase disorders in all patients, with severe aspiration confirmed in 2 of 5 patients. In mice, no aspiration was observed in wild-type, but aspiration was observed in significantly more mice (50%) among heterozygotes or homozygotes with severer tendency in homozygotes (p < 0.05, Kruskal-Wallis test and post-hoc analysis). Pathological analyses in an autopsied patient revealed the degeneration of the neurons in the hypoglossal nucleus with phosphorylated TDP43 accumulation in the remaining neurons. In homozygous mice, the number of motor neurons in the hypoglossal nucleus was reduced.
Conclusion: We found that repeat expansions in the gene for SCA8 affected swallowing functions in humans as well as in mouse models. We propose that swallowing function may serve as a biomarker for SCA8-related ALS for future therapeutic interventions.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: ALS: Biology, Pathophysiology, Genetics
PP01.321
Juvenil Amyotrophic Lateral Sclerosis in Two Algerian Sisters Expends the Phenotypic Spectrum of SYNE1-Related Disorders
Asst. Prof. Ouissem Benchaabi, Prof. Sonia Nouioua, Asst. Prof. Farroudja Ramdane-Cherif
EHS Cherchell, TIPAZA, Algeria
Background: SYNE1-related disorders are a group of genetic conditions,with a broad phenotypic spectrum, most commonly presenting as adult-onset Autosomal Recessive Cerebellar Ataxia (ARCA1) or SYNE1-ataxia with possible motor neuron disease, cognitive deficits (Cerebellar Cognitive Affective Syndrome), muscle weakness, and severe neonatal hypotonia with joint contractures in rare cases(SYNE1-deficient arthrogryposis multiplex congenita). To date, a pure juvenile-onset ALS phenotype has not been clearly established as part of the SYNE1-related disease spectrum.
Methods: We performed detailed clinical, neurophysiological and genetic analysis, by neuromuscular panel next generation sequencing, in two sisters with juvenil amyotrophic lateral sclerosis (JALS) followed by variant interpretation according to current pathogenicity criteria to describe a novel clinical phenotype associated with SYNE1 mutations by reporting a juvenil-onset amyotrophic lateral sclerosis and intellectual disability without cerebellar signs over time
Results: We report a consanguineous family with two sisters, aged 16 and 13 years, presenting with juvenile-onset motor neuron disease fulfilling clinical and electrophysiological criteria for ALS. In both patients, disease onset occurred at approximately four years of age, with progressive distal and proximal muscle weakness evolving over time and associated with combined upper and lower motor neuron signs, leading to severe functional impairment. In addition to motor involvement, both sisters exhibited early-onset intellectual disability. One patient presented with congenital hearing loss. Notably, no cerebellar manifestations were observed during the follow-up. Next-generation sequencing-based genetic analysis, including a neuromuscular panel of genes, identified a pathogenic variant in the SYNE1 gene: a homozygous nonsense variant, c.20227C>T (p.Gln6743Ter).
Juvenile-onset amyotrophic lateral sclerosis (JALS) is a rare motor neuron disorder defined by symptom onset before the age of 25 years. Very early-onset forms, occurring before the age of 10 years, are particularly uncommon and are most often associated with autosomal recessive genetic etiologies. JALS is characterized by the progressive degeneration of upper and lower motor neurons and may be associated with additional neurological or neurodevelopmental manifestations.
$ù=ùPMhe SYNE1 gene (OMIM *608441) k;pà)omencodes nesprin-1, a large nuclear envelope protein involved in nuclear positioning and cytoskeletal organization. Pathogenic variants in SYNE1 are classically associated with autosomal recessive cerebellar ataxia type 1 (ARCA1), but an expanding phenotypic spectrum has been described, including spastic paraplegia, motor neuron disease with overlapping features, intellectual disability, and multisystem neurodegeneration.
To date, a pure juvenile-onset ALS phenotype has not been clearly established as part of the SYNE1-related disease spectrum. In this context, reporting novel clinical presentations is essential to improve genotype–phenotype correlations and refine genetic diagnostic strategies in early-onset motor neuron diseases.
Conclusion: This report expands the clinical spectrum of SYNE1-related disorders by identifying juvenile-onset ALS as a novel associated phenotype. Our findings support the inclusion of SYNE1 in next-generation sequencing panels gene for ALS, particularly in juvenile cases and in the presence of associated neurodevelopmental features. Further studies are needed to better define phenotype genotype correlations in SYNE1-related motor neuron diseases.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: ALS: Biology, Pathophysiology, Genetics
PP01.322
Swallowing Function in Patients With SOD1-Related Amyotrophic Lateral Sclerosis
Ms. CHIHARU ISONO1, Prof. MAKITO HIRANO2, Asst. Prof. MAKOTO SAMUKAWA2, Prof. Kazumasa Saigoh3, Prof. Yuji Higashimoto4, Prof. Yoshitaka Nagai2
1
Division of Rehabilitation Medicine, Kindai University Hospital, OSAKA, Japan.
2
Department of Neurology, Kindai University Faculty of Medicine, OSAKA, Japan.
3
Division of Clinical Genetics, Kindai University Hospital, OSAKA, Japan.
4
Department of Rehabilitation Medicine,Kindai University Faculty of Medicine, Japan, OSAKA, Japan
Background: Mutations in SOD1 are the second most common cause of familial amyotrophic lateral sclerosis (ALS), found in approximately 20-30% of familial cases, as well as 2-3% of sporadic cases. While swallowing function in SOD1-related ALS is thought to be relatively preserved compared to non-SOD1-related ALS, its detailed findings remain largely unknown. The objective of this study was to assess swallowing function in patients with SOD1-ALS using videofluoroscopic examination (VF).
Methods: Genetic testing in our cohort (116 sporadic cases and 10 familial cases) revealed that three patients with sporadic ALS and four with familial ALS had SOD1 mutations. VF were performed in three women with a mean age at onset of 53.7 ± 4.9 years, all of whom presented with lower-limb onset. VF evaluation was based on the scale established by the Japanese Society of Dysphagia Rehabilitation, which can separately evaluate oral and pharyngeal phases in addition to total scores, the international scale Dysphagia Outcome and Severity Scale (DOSS), and Penetration-Aspiration Scale (PAS).
Results: Mild oral phase disorders were observed in the VF. Two patients had silent penetration without aspiration during the pharyngeal phase. All patients continued oral intake, and none developed aspiration pneumonia or required gastrostomy for feeding.
Conclusion: In general, dysphagia is a critical factor affecting prognosis in ALS. Our study suggests that in SOD1-related ALS, swallowing function varies depending on the mutation and may be milder than expected for the duration of disease.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: ALS: Epidemiology and Clinical Features,
PP01.323
Analysis of Vocal Parameters in Als Patients: A Preliminary Study
Dr. Ester Franchi1, Dr. Barbara Risi1,2, Dr. Nesaiba Ait Allali1, Dr. Giulia Gilberti1, Dr. Lucia Ferullo1, Dr. Filomena Caria1, Dr. Loris Poli3, Prof. Alessandro Padovani3,4, Prof. Massimiliano Filosto1,3,4
1
NeMO-Brescia Clinical Center for Neuromuscular Diseases, Brescia, Italy.
2
Department of Molecular and Translational Medicine, University of Brescia, Brescia, Italy.
3
Unit of Neurology, ERN EURO-NMD Center ASST Spedali Civili, Brescia, Italy.
4
Department of Clinical and Experimental Sciences, University of, Brescia, Italy
Background: Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease that frequently affects bulbar motor control, leading to impairments in speech and voice. Acoustic voice analysis may represent a non-invasive approach for monitoring functional changes; however, longitudinal data across different ALS phenotypes remain limited.
The objective of the study is to longitudinally characterize acoustic voice parameters in patients with spinal-onset ALS and to explore their descriptive co-variation with motor-functional status, respiratory measures, quality of life, and clinical phenotype (mixed vs. flail-arm).
Methods: Five adults with spinal-onset ALS were enrolled (three with mixed phenotype and two with flail-arm phenotype) and evaluated at baseline and after 3, 6, 9, and 12 months. Acoustic parameters were extracted using Praat software (version 6.2.22), including jitter, shimmer, maximum phonation time (MPT), fundamental frequency (F0), and degree of voice breaks (DVB). Motor and functional status (ALSFRS-R, Barthel Index), respiratory function (forced vital capacity [FVC], peak expiratory flow [PEF]), and quality of life (ALSAQ-40, QOL-DYS) were assessed. Analyses were limited to descriptive statistics.
Results: At baseline, shimmer values were markedly elevated in both phenotypes (median [Q1–Q3]: 16.3% [12.3–17.3] in mixed; 16.8% [14.5–17.2] in flail-arm), while median jitter ranged from 1.01% (0.29–1.25) in mixed to 1.39% (0.91–1.88) in flail-arm patients. MPT was shorter in flail-arm patients (median 6.4 s [4.7–8.13]) compared with mixed-phenotype patients (9.9 s [5.6–28.6]).
Over follow-up, mixed-phenotype patients showed a reduction in shimmer values (median [Q1–Q3]: 2.9% [2.7–13.7] at 12 months) and variable MPT, whereas flail-arm patients exhibited persistently shorter MPT and higher DVB. Lower ALSFRS-R total scores (median [Q1–Q3]: 20 [16–24] at 12 months in flail-arm patients) tended to co-occur with greater vocal instability. FVC percentage values remained moderately reduced over time without consistent parallel changes in acoustic measures. From baseline (T0) to T4, worsening in QOL-DYS scores was observed, accompanied by a parallel increase in shimmer.
Conclusion: In this preliminary study, acoustic voice parameters displayed phenotype-specific longitudinal patterns and showed parallel descriptive changes with motor-functional impairment and communication-related quality of life. No consistent descriptive association was observed with respiratory decline. Further longitudinal studies in larger cohorts are warranted to clarify phenotype-related voice changes in ALS over time.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: ALS: Epidemiology and Clinical Features
PP01.324
Ten-Year Trajectories of Pre-diagnostic Medical Conditions Preceding Amyotrophic Lateral Sclerosis: A Nationwide Korean Study
Assoc. Prof. eun bin cho1,2, Mr. Bongseong Kim3, Prof. Kyungdo Han3, Dr. Seung Ho Choo4, Asst. Prof. Soonwook Kwon5, Prof. Dong Wook Shin6, Prof. Ju-Hong Min4
1
Department of Neurology, College of Medicine, Gyeongsang National University, Jinju, Korea, Republic of.
2
Gyeongsang National University Changwon Hospital, Changwon, Korea, Republic of.
3
Department of Statistics and Actuarial Science, Soongsil University, Seoul, Korea, Republic of.
4
Department of Neurology, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea, Republic of.
5
Department of Neurology, Inha University Hospital, Incheon, Korea, Republic of.
6
Department of Family Medicine & Supportive Care Center, Samsung Medical Center, Sungkyunkwan University School, Seoul, Korea, Republic of
Background: Prodromal manifestations of amyotrophic lateral sclerosis (ALS) remain incompletely characterized at the population level.
Methods: In a nationwide Korean case–control study using National Health Insurance data (2011–2021), 9,435 individuals with ALS and 94,350 age-, sex-, and index year–matched controls were analyzed. Adjusted rate ratios (aRRs) for 23 pre-diagnostic medical conditions were estimated across 0–1, 1–2, 2–5, and 5–10 years before ALS diagnosis using Poisson regression. Temporal trajectories of log-transformed aRRs over ten consecutive 1-year intervals were evaluated using linear regression with false discovery rate (FDR) correction.
Results: Radiculopathy (5–10 years: aRR 1.11, 95% CI 1.06–1.17; annual percentage change [APC] 16.8%), obstructive sleep apnea (2.67, 2.03–3.52; 15.4%), depression (1.49, 1.38–1.61; 14.9%), insomnia (1.12, 1.03–1.21; 11.4%), anxiety (1.32, 1.24–1.41; 11.3%), constipation (1.12, 1.04–1.22; 9.3%), and diabetes mellitus (1.06, 1.00–1.11; 6.7%) demonstrated significant progressive enrichment up to a decade before diagnosis (FDR p<0.05). Other fractures and benign prostatic hyperplasia demonstrated progressive enrichment beginning 2–5 years before diagnosis (1.32, 1.13–1.54; 8.8% and 1.19, 1.09–1.29; 6.7%, respectively). In contrast, anemia (5–10 years: 1.21, 1.08–1.35) and asthma (5–10 years: 1.18, 1.05–1.32) exhibited early enrichment without consistent temporal escalation. Schizophrenia and bipolar disorder also exhibited early enrichment with heterogeneous, non-monotonic patterns.
Conclusion: A defined cluster of psychiatric, sleep-related, and musculoskeletal conditions, along with constipation and diabetes mellitus, shows long-standing and progressively increasing enrichment up to a decade before ALS diagnosis, supporting their role as prodromal features or early manifestations of ALS. In contrast, anemia and asthma show early and sustained elevation without progressive escalation, suggesting potential underlying biological associations.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: ALS: Epidemiology and Clinical Features,
PP01.325
5q-Associated Spinal Muscular Atrophy in Austria: Epidemiology and Co-Morbidities
Dr. Omar Keritam1,2, Dr. Gudrun Zulehner1,2, Dr. Raphael Wurm1,2, Mr. Felix Gruber3, Dr. Jakob Rath1,2, Dr. Martin Krenn1,2, Dr. Daniel Bormann1,2, Dr. Marcus Erdler4, Dr. Simone Mahal5, Dr. Anna Wiesenhofer5, Dr. Magdalena Gosk-Tomek5, Dr. Mika Rappold6, Dr. Anna Hüpper5, Dr. Katia Vettori5, Dr. Theresa Antonia Griedl7, Dr. Christian Kiss7, Dr. Valeriu Gold7, Prof. Julia Wanschitz8, Dr. Anna Hotter8, Dr. Vera Kleinveld8, Dr. Corinne Horlings8, Dr. Michael Gräßl9, Dr. Anette Schwerin-Nagl9, Dr. Johannes Troger10, Dr. Susanne Grinzinger11, Dr. Eva Stögmann12, Dr. Petra Müller13, Dr. Dieter Langenscheidt14, Prof. Barbara Plecko9, Prof. Fritz Zimprich1,2, Dr. Raffi Topakian13,15, Dr. Christian Eggers16, Prof. Stefan Quasthoff7, Dr. Florian Knipp5, Prof. Günther Bernert17, Dr. Matthias Baumann18, Prof. Wolfgang Löscher8, Prof. Hakan Cetin1,2
1
Department of Neurology, Medical University of Vienna, Vienna, Austria.
2
Comprehensive Center for Clinical Neurosciences and Mental Health, Medical University of Vienna, Vienna, Austria.
3
Austrian Federation of Social Insurances, Vienna, Austria.
4
Department of Neurology, Klinik Donaustadt, Vienna, Austria.
5
Department of Paediatrics, Klinik Favoriten, Vienna, Austria.
6
Primary Care Centre Nepomuk, Vienna, Austria.
7
Department of Neurology, Medical University of Graz, Graz, Austria.
8
Department of Neurology, Medical University of Innsbruck, Innsbruck, Austria.
9
Department of Paediatrics and Adolescent Medicine, Medical University of Graz, Graz, Austria.
10
Department of Neurology, Klinikum Klagenfurt, Klagenfurt, Austria.
11
Department of Neurology, Paracelsus Medical University, Salzburg, Austria.
12
Department of Paediatrics, Landesklinikum Mödling, Mödling, Austria.
13
Department of Neurology, Academic Teaching Hospital Wels-Grieskirchen, Wels, Austria.
14
Department of Neurology, Landeskrankenhaus Rankweil, Rankweil, Austria.
15
Clinical Research Institute for Neurosciences, Johannes Kepler University, Linz, Austria.
16
Department of Neurology, Johannes Kepler University, Linz, Austria.
17
Department of Paediatrics, Medical University of Vienna, Vienna, Austria.
18
Division of Paediatric Neurology, Department of Paediatrics I, Medical University Innsbruck, Innsbruck, Austria
Background: Spinal muscular atrophy (SMA) is a genetic motor neuron disease caused by homozygous deletions or other pathogenic variants in the SMN1 gene, resulting in the degeneration of lower motor neurons associated with progressive weakness and muscle wasting. The development and approval of disease-modifying therapies (DMT) in recent years has fundamentally transformed the management, as well as the clinical and epidemiological landscape of patients with SMA.
Methods: In this study, hospitalisation and prescription databases together with the capture-recapture method are utilised to estimate the prevalence of SMA in Austria and to identify co-morbidities associated with SMA. Patients were matched to healthy controls (1:10) by sex, age and district of residence.
Results: We estimated a total population of 236 patients (95% CI 231-241) with SMA in Austria and a corrected prevalence of 2.5/100,000 persons in the year 2023. Patients with SMA showed significantly higher use of several medication classes, particularly therapies related to respiratory and infectious conditions. Healthy controls, by contrast, exhibited higher prescribing rates of cardiovascular medications, including beta-blockers and ACE inhibitors, as well as lipid-lowering agents and muscle relaxants.
Conclusion: Our findings provide a robust nationwide estimate of SMA prevalence in Austria and highlight a distinct pattern of co-morbidities in the era of disease-modifying therapies, supporting informed healthcare planning and long-term management of patients with SMA.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: Spinal Muscular Atrophies: Epidemiology and Clinical Features,
PP01.326
Increasing Prevalence of Amyotrophic Lateral Sclerosis in Austria Correlates With Reduced Mortality
Dr. Omar Keritam1,2, Dr. Raphael Wurm1,2, Dr. Daniel Bormann1,2, Mr. Felix Gruber3, Dr. Martin Krenn1,2, Dr. Gudrun Zulehner1,2, Dr. Jakob Rath1,2, Prof. Fritz Zimprich1,2, Prof. Hakan Cetin1,2
1
Department of Neurology, Medical University of Vienna, Vienna, Austria.
2
Comprehensive Center for Clinical Neurosciences and Mental Health, Medical University of Vienna, Vienna, Austria.
3
Austrian Federation of Social Insurances, Vienna, Austria
Background: Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder characterised by the progressive loss of motor neurons, leading to muscle weakness, respiratory failure, and ultimately death within a few years of disease onset. In Austria, the average annual incidence and prevalence of ALS between 2009 and 2011 were 3.2/100,000 person-years and 9.1/100,000 persons, respectively. Although recent studies suggest that the global burden of the disease has increased over past decades, the trajectory of epidemiological metrics in Austria after 2011 remain unknown.
Methods: Hospital discharge records and riluzole prescription databases were used to identify ALS cases between 2016 and 2023. The capture–recapture method was then applied to estimate the incidence and prevalence of ALS in Austria. Survival was analysed in dependence of age, gender, and riluzole/physiotherapy. Furthermore, the relationship between disease prevalence and the ratio of deaths to prevalent cases (death-to-case ratio) in a given year was analysed using Pearson’s correlation.
Results: A total of 3,344 patients were identified during the study period, of whom 44.3% were female. While the corrected annual incidence remained stable (5.2/100,000 person-years in 2016 and 4.8/100,000 person-years in 2023), the prevalence increased steadily over the same period, rising from 8.6 to 14.5/100,000 individuals. Median survival was 605 days (95% CI 569-638). Younger age at diagnosis, male sex and physiotherapy were associated with longer survival. The death-to-case ratio decreased during the study period, from 0.32 in 2016 to 0.22 in 2023, which correlated negatively with the corresponding prevalence numbers (r=-0.85, p=0.0019).
Conclusion: In this study, we report on the epidemiology of ALS in Austria between 2016 and 2023. Notably, while the annual incidence remained stable over the study period, our findings indicate an increase of prevalence, most likely due to reduced mortality rates. Our results underscore the need for national healthcare policies to address the challenges posed by the growing disease burden.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: ALS: Epidemiology and Clinical Features,
PP01.327
Incidence and Prevalence of Amyotrophic Lateral Sclerosis in Curitiba, Brazil
Dr. Otto Jesus Hernandez Fustes1, Miss Nathalia Faria1, Miss Bruna Maçaneiro1, Dr. Claudia Suemi Kamoi Kay1, Dr. Renata Dal-Pra Ducci Cirino1, Dr. Paulo Jose Lorenzoni1, Dr. Paula Raquel do V.P. Rodrigues1, Dr. Luiza Fraga1, Dr. Rosana Herminia Scola1, Dr. Paula Silvia Rosignoli2
1
Universidade Federal do Paraná, Curitiba, Brazil.
2
Secretaria da Saúde do Paraná, Curitiba, Brazil
Background
Introduction: Amyotrophic Lateral Sclerosis (ALS) is a chronic, degenerative, and progressive disease that primarily affects upper and lower motor neurons. It has an unknown, multifactorial etiology and an invariably fatal outcome. Epidemiological studies have reported a prevalence ranging from 0.8 to 10.32 cases per 100,000 people. This variability can be explained by methodological differences, changes in diagnostic criteria over time, and the lack of adequate disease notification systems in several countries, such as Brazil. Incidence studies, which show less variability because they can only be conducted under optimal conditions of disease documentation, report rates between 1.5 and 2.5 cases per 100,000 people per year. However, most epidemiological studies have been conducted in North America and Europe. Therefore, it is essential to seek data from other regions of the world to better understand the distribution and potential determinants of the disease. The objective of this study was to determine the incidence and prevalence of individuals with amyotrophic lateral sclerosis in the population of Curitiba from 2019 to 2024.
Methods: This study employed a cross-sectional cohort design. Demographic data, including sex and age, were obtained from the State Health Department for patients with ALS receiving treatment in the municipality of Curitiba. The public health system provides treatment with riluzole, prescribed by neurologists, to all patients diagnosed with ALS.
Results: Curitiba is the capital of the state of Paraná, located in southern Brazil, with an area of 435.036 km². It is situated at 25°25′40″ south latitude and 49°16′23″ west longitude, with a north–south extension of 35 km and an east–west extension of 20 km. According to 2022 data from the Brazilian Institute of Geography and Statistics (IBGE), the city has a population of 1,773,718 inhabitants. During the study period (2019–2024), 159 patients receiving riluzole treatment were identified. The estimated prevalence ranged from 2.46 to 3.80 cases per 100,000 inhabitants, showing an upward trend over time, with a peak observed in 2024 (68 active cases). Annual incidence ranged from 0.06 to 0.22 new cases per 10,000 inhabitants. A male predominance was observed, accounting for 59.7% of the sample (95 patients), with an approximate male-to-female ratio of 1.5:1. The mean age at treatment initiation was 63.4 years, ranging annually from 60.8 to 65.4 years, and was slightly higher in males than in females.
Conclusion: Data from Curitiba corroborate global epidemiological patterns, demonstrating incidence and prevalence rates consistent with the literature. Male predominance and the affected age range reinforce the classic disease profile. The observed increase in prevalence rates in recent years suggests possible improvements in diagnosis, increased patient survival, or greater access to pharmacological treatment provided by the public health system. These findings highlight the importance of continuous registry systems for planning healthcare actions aimed at rare neurodegenerative diseases.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: ALS: Epidemiology and Clinical Features,
PP01.328
Short-Term Outcome of Tofersen Treatment in Patients with SOD1-ALS at a Tertiary Academic Center
Assoc. Prof. Aysha Alshareef, Dr. Manal Alsulami, Assoc. Prof. Ahmad Abuzinadah
King Abdulaziz University Hospital, Jeddah, Saudi Arabia
Background: Tofersen is an antisense oligonucleotide designed to promote RNase-mediated degradation of SOD1 mRNA, leading to reduced synthesis of both wild-type and mutant SOD1 protein. It has shown promising results across different physical function scales. In this study, we evaluated the short-term outcome of tofersen on clinical paramters across physcial function scales including (Revised Upper Limb Module (RULM), Hammersmith Functional Motor Scale-Expanded (HFMSE), and the Revised Amyotrophic Lateral Sclerosis Functional Rating Scale (ALSFRS-R).
Methods: A retrospective, single-center study analyzed 10 patients with genetically confirmed SOD1-ALS who received intrathecal tofersen. All patients carried the SOD1 D77V mutation. The mean age was 47 years (range 28–61), and patients received a mean of 14 intrathecal doses (range 7–24). Pre- and post-treatment scores were compared for RULM, HFMSE, and ALSFRS-R. Paired comparisons were performed using the Wilcoxon signed-rank test for non-normally distributed outcomes and paired t-tests where appropriate.
Results: Nine patients had complete RULM and HFMSE data, and all ten had ALSFRS-R data. RULM showed a trend toward improvement (median change +1; Wilcoxon Z = –1.89, p = 0.059), but did not reach statistical significance. HFMSE changes were small and not significant (median change 0; p = 0.686). ALSFRS-R showed a slight numerical improvement (mean change +0.7 ± 2.8) without statistical significance (p = 0.454). Individual assessments showed improvement in ALSFRS-R scores in four out of ten patients. No patient demonstrated marked functional decline over the observation period. (Table 1)
Conclusion: Tofersen was associated with stabilization of motor function across multiple clinical scales, with improvement observed in some patients. Importantly, all patients carried the SOD1 D77V mutation, a subtype linked to rapid functional decline, typically losing ∼0.8–1.0 ALSFRS-R points per month (∼10–12 points/year) in natural history. In contrast, our cohort showed a mean gain of +0.7 points over 3–6 months, and no patient experienced decline—an outcome highly unexpected for this aggressive genotype. Although statistical significance was not reached, likely due to the small sample size, this pattern of stability and improvement aligns with emerging observational data and suggests a clinically meaningful effect of tofersen even in severe SOD1 variants. Larger, genotype-stratified prospective studies are warranted to confirm these findings.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: ALS: Therapy
Images or Table (Optional)
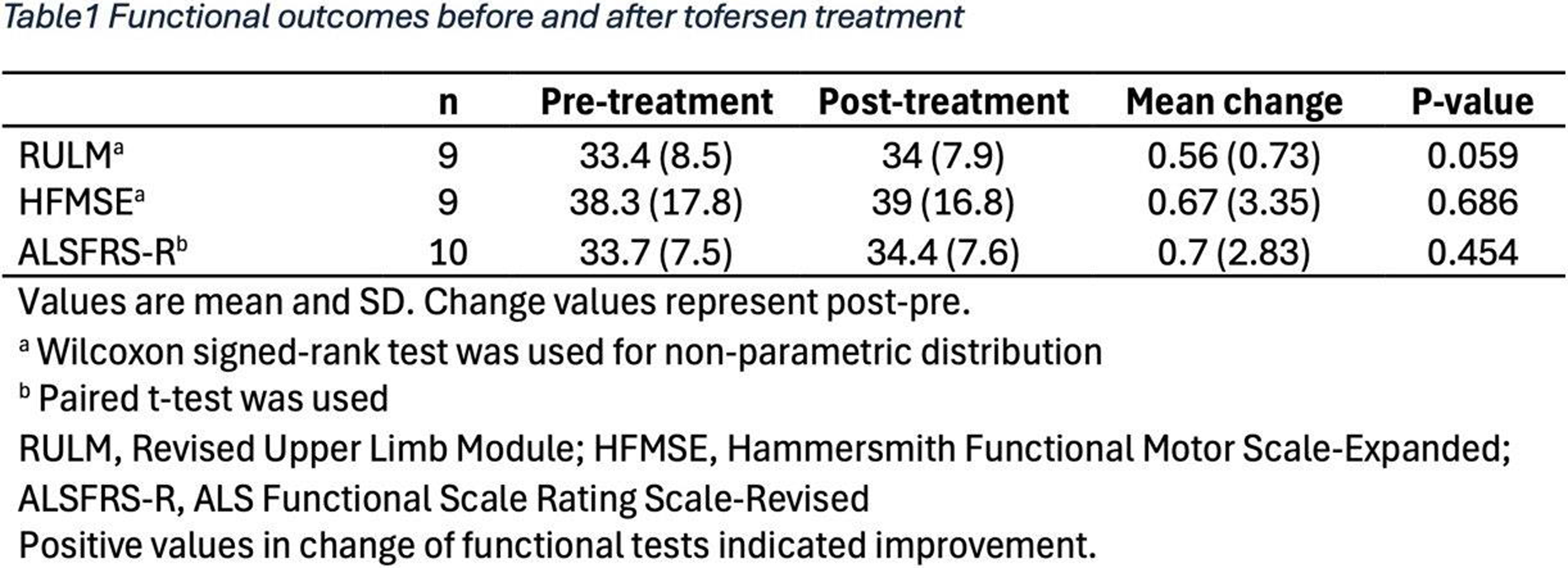
PP01.329
Preclinical Safety and Efficacy of Intrathecal INS1202 AAV9-SOD1-shRNA in Mice and Nonhuman Primates
Dr. Laura Ferraiuolo1, Dr. Erin Hurley1, Dr. Mark Stahl1, Dr. Gretchen Thomsen1, Ms. Hana Julazadeh1, Dr. Jannigje Kok1, Dr. Cleide Dos Santos Souza1, Ms. Ashley Fox2, Mr. Ryan Weiss1, Dr. Vivian Ko1, Ms. Haydee Gutierrez1, Ms. Melissa McAlonis-Downes3, Mr. Robert Cano1, Dr. Binh Chu1, Dr. Angélique Braen4, Dr. Junguo Zhou4, Dr. W. David Arnold5, Dr. Stephen J. Kolb2, Dr. Allan Kaspar1, Dr. Brian Kaspar1
1
Insmed Gene Therapy LLC, San Diego, CA, United States.
2
Ohio State University Wexner Medical Center, Columbus, OH, United States.
3
University of California, San Diego, CA, United States.
4
Insmed Incorporated, Bridgewater, NJ, United States.
5
University of Missouri, Columbia, MO, United States
Background: INS1202 is an engineered adeno-associated virus 9 (AAV9) vector that drives expression of a short hairpin RNA (shRNA) construct targeting human superoxide dismutase 1 (SOD1) mRNA, being evaluated for the potential treatment of amyotrophic lateral sclerosis (ALS).
Methods: SOD1G93A mice treated with single ascending doses of INS1202 or vehicle via intracerebroventricular administration at postnatal day (p)1 were assessed for disease onset and survival, motor skills, and muscle force through p200 (scheduled study end). Plasma neurofilament levels were evaluated at p50, p120, and p200 in all animals alive at the respective timepoints. Tissues were collected for biodistribution, histological examination, and protein analysis. Nonhuman primates (NHPs) were administered single ascending doses of INS1202 or vehicle via lumbar intrathecal injection to assess biodistribution and safety in a 3-month GLP-compliant toxicology study. To assess efficacy in a human disease in vitro model, astrocytes directly reprogrammed from SOD1-ALS (n=1) and sporadic ALS (n=10) human patient fibroblasts were treated with INS1202 or vehicle and co-cultured with healthy control mouse GFP-positive motor neurons (MNs).
Results: SOD1G93A mice treated with INS1202 (n=7-12/sex/group) showed a dose-dependent improvement in median survival. Median age at death in the vehicle control group was p130, while 93% of mice in the highest dose group were still alive at study end (p200). INS1202-treated SOD1G93A mice gained and sustained physiological weight (p<0.0001), retained muscle function per the rotarod test (p<0.0001), and retained muscle force per the muscle physiology Aurora system (p<0.0001). Additionally, compared with vehicle-treated mice, INS1202-treated SOD1G93A mice exhibited significantly lower plasma neurofilament levels at three timepoints: p50, when mice are asymptomatic (p<0.01, n=6/group), p120±10 when vehicle-treated mice showed signs of paralysis (p<0.001, n=12-15/group), and at p200, end of study. INS1202 treatment of SOD1G93A mice preserved lumbar MN numbers (n=6/group, p=0.0006) and preserved staining intensity of IBA1 (n=4/group, p<0.0001) and GFAP (n=6/group, p<0.0001) in-line with levels observed in wild-type mice. At the highest dose, INS1202 treatment reduced the levels of human mutant SOD1 protein (n=6/group, p<0.0001) by 46% in the lumbar spinal cord of SOD1G93A mice.
In the 3-month GLP-compliant NHP toxicology study, biodistribution via droplet digital PCR (ddPCR) demonstrated vector genome delivery to all spinal cord segments, brainstem, and motor cortex, with lower distribution to peripheral organs including liver, compared with what is reported with intravenous administration of AAV9. INS1202 was well tolerated at all doses with single intrathecal administration in NHPs and the No-Observed-Adverse-Effect Level was determined to be the highest dose evaluated. Additionally, INS1202 treatment was neuroprotective in astrocyte-MN co-cultures from patients with SOD1-ALS, as well as in co-cultures from 7 of 10 patients with sporadic ALS negative for SOD1 and other known mutations.
Conclusion: Administration of INS1202 improved survival and motor function and ameliorated hallmarks of neurodegeneration in SOD1G93A mice. INS1202 was well tolerated in NHPs and showed potential for treating patients with SOD1-ALS and sporadic ALS negative for SOD1 and other known mutations. Based on these efficacy and safety data, three ascending doses of INS1202 are being evaluated in a phase 1 first-in-human clinical trial.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: ALS: Therapy
PP01.330
Quantitative Assessment of Motor Band Sign as a Neuroimaging Biomarker in Amyotrophic Lateral Sclerosis
Dr. Eunhee Sohn1, Dr. Sooyoung Kim1, Dr. Eun Kyoung Lee2, Dr. Kyomin Choi3, Dr. Jeeyoung Oh3, Dr. Seong-il Oh4, Dr. Jin Myoung Seok5, Dr. Kyong Jin Shin6, Dr. Eungseok Oh1, Dr. Hee Jin Chang1, Dr. Dayoung Kim3, Dr. Dallah Yoo4, Dr. Tae-Beom Ahn4, Dr. Jinse Park6, Dr. Jinyoung Youn7, Dr. Jin Whan Cho7, Dr. Byoung Joon Kim7
1
Chungnam National University Hospital, Daejeon, Korea, Republic of.
2
Chungnam National University Sejong Hospital, Sejong, Korea, Republic of.
3
Konkuk University Medical Center, Seoul, Korea, Republic of.
4
Kyung Hee University Hospital, Seoul, Korea, Republic of.
5
Seoul Hospital, Ewha Womans University College of Medicine, Seoul, Korea, Republic of.
6
Haeundae Paik Hospital, Inje University College of Medicine, Busan, Korea, Republic of.
7
Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea, Republic of
Background: Motor band sign (MBS), defined as low signal intensity along the motor cortex, has been proposed as an imaging marker of neurodegeneration. Previous studies have reported an association between MBS and upper motor neuron degeneration as well as disease progression rate in amyotrophic lateral sclerosis (ALS). However, these studies primarily relied on visual rating methods, which may introduce variability and compromise reliability. Therefore, we aimed to quantitatively assess the MBS and investigate its relationship with clinical features of ALS.
Methods: Using data from the Korean Research Network for Motor Neuron Disease and Spinocerebellar Ataxia (K-MoSCA), we retrospectively collected clinical data and brain MRI scans. A total of 34 patients with ALS (mean age 61.2 ± 10.8 years, M:F = 2.4:1) and 35 controls (mean age 65.2 ± 8.7 years, M:F = 2.2:1) were enrolled. MRI and clinical evaluations conducted within 3 months were analyzed, and participants without susceptibility-weighted imaging were excluded. Clinical variables included onset age, sex, disease duration, ALS Functional Rating Scale (ALSFRS), delta ALSFRS, upper motor neuron (UMN) symptom score, MRC sum score, and CMAP sum score of nerve conduction studies.
Motor band hypointensity ratio (MBHR) was calculated for medial, intermediate, and lateral regions of the motor cortex bilaterally. MBHR was defined as the ratio between signal intensity in the motor cortex ROI and the adjacent subcortical ROI. Correlations between MBHR and clinical features were analyzed, and each motor cortex region was compared with its corresponding functional domain.
Results: All motor cortex regions showed significant negative correlations with delta ALSFRS (Rt. medial r = −0.368, p = 0.032; Rt. intermediate r = −0.457, p = 0.007; Rt. lateral r = −0.423, p = 0.013; Lt. medial r = −0.540, p < 0.001; Lt. intermediate r = −0.461, p = 0.006; Lt. lateral r = −0.422, p = 0.013). MBHR of the right intermediate region correlated with UMN score (r = −0.347, p = 0.044). Disease duration was positively correlated with MBHR in the intermediate cortex (Rt. intermediate r = 0.389, p = 0.023; Lt. intermediate r = 0.384, p = 0.025). In ROC analysis, MBHR of the bilateral intermediate cortex demonstrated good diagnostic performance. The left intermediate cortex showed the highest diagnostic performance with 100% specificity, 64.7% sensitivity (cutoff value = 89.6, AUC = 0.874). MBHR of the right intermediate cortex also showed high diagnostic performance (specificity = 82.9%, sensitivity = 79.4, cutoff value = 91.5, AUC = 0.869). There were no correlations between each motor cortex region and corresponding functional domain.
Conclusion: Quantitative evaluation of the motor band sign provides a reliable and objective biomarker that reflects upper motor neuron involvement and disease progression rate in ALS. Among all regions, the bilateral intermediate motor cortex showed the higher diagnostic performance, suggesting its potential clinical utility in assessing both upper motor neuron involvement and disease severity. Further prospective validation studies are needed to establish MBHR as a standardized neuroimaging marker in ALS.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: Biomarkers in MND
PP01.331
Identification of Glial Biomarkers in ALS Using Single Nucleus Transcriptomic analysis
Ms. Jin-Ah Kim1,2, Ms. Do-Yeon Lee3, Prof. Jung-Joon Sung1, Prof. Jong-Il Kim1
1
Seoul National University Hospital, Seoul, Korea, Republic of.
2
Genomic medicine Institute, Seoul, Korea, Republic of.
3
Seoul National University, Seoul, Korea, Republic of
Background: Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease characterized by the selective loss of motor neurons. Although accumulating evidence indicates that glial cell dysfunction plays a crucial role in ALS pathogenesis, neurofilament light chain, which reflects neuroaxonal damage, is predominantly used in ALS. Here, we aimed to identify novel glial biomarkers from activated microglia and astrocytes through single-nucleus RNA sequencing (snRNA-seq) and proteomics analysis of patient serum.
Methods: We performed snRNA-seq of the spinal cord from hSOD1 G93A transgenic mice at pre-symptomatic (P60), peri-symptomatic (P90), and progressive (P120) stages. Differential gene expression (DEG) analyses were conducted for microglia and astrocytes, respectively, compared to littermate controls. Protein expression changes were identified using Western blot analysis. Human relevance was supported by analyzing publicly available snRNA-seq datasets from sporadic ALS patients.
Results: In microglia, Apoe was the most significantly upregulated, evident from the pre-symptomatic stage. Western blot analysis demonstrated that APOE protein also rose prior to symptom onset and became markedly elevated according to disease progression. Apoe upregulation is assumed to be associated with enhanced lipid transport during the clearance of myelin debris resulting from oligodendrocyte degeneration. In astrocytes, Fn1 was significantly upregulated, consistent with its key role in reactive astrogliosis and glial scar formation. Importantly, both APOE and FN1 increases were confirmed in spinal cord tissues from sporadic ALS patients.
Conclusion: By integrating snRNA-seq data from an ALS mouse model and human tissues, we identified candidate biomarkers reflecting active glial cells. While ApoE did not show significant changes in serum, its high systemic levels could potentially mask disease-related alterations, suggesting that additional validation in CSF is required.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: Biomarkers in MND
PP01.332
Autonomic Biomarkers Associated With Disease Severity and Duration in Amyotrophic Lateral Sclerosis
Assoc. Prof. Jinwoo Park1,2, Prof. Byung-Jo Kim1
1
Korea University, Seoul, Korea, Republic of.
2
Vanderbilt University, Nashville, United States
Background: Amyotrophic lateral sclerosis (ALS) is increasingly recognized as a multisystem neurodegenerative disorder with involvement of the autonomic nervous system. However, the characteristics of cardiovascular autonomic dysfunction and their clinical relevance across disease stages remain incompletely defined. The Valsalva maneuver provides a physiologically grounded assessment of baroreflex-mediated autonomic control and may offer sensitive biomarkers for ALS-related autonomic involvement.
Methods: In this cross-sectional study, 27 drug-naïve patients with ALS (El Escorial criteria: possible, n = 3; probable, n = 13; definite, n = 11) and 24 age- and sex-matched healthy controls were enrolled. Cardiovascular autonomic function was evaluated using baroreflex sensitivity indices (adrenergic [BRSa] and vagal [BRSv]), pressure recovery time (PRT), heart rate variability during deep breathing (HRVdb), and the Valsalva ratio (VR). Head-up tilt testing (HUTT) was also performed. Clinical severity and disease burden were assessed using the ALS Functional Rating Scale–Revised (ALSFRS-R) and disease duration.
Results: In this cross-sectional study, 27 drug-naïve patients with ALS (El Escorial criteria: possible, n = 3; probable, n = 13; definite, n = 11) and 24 age- and sex-matched healthy controls were enrolled. Cardiovascular autonomic function was evaluated using baroreflex sensitivity indices (adrenergic [BRSa] and vagal [BRSv]), pressure recovery time (PRT), heart rate variability during deep breathing (HRVdb), and the Valsalva ratio (VR). Head-up tilt testing (HUTT) was also performed. Clinical severity and disease burden were assessed using the ALS Functional Rating Scale–Revised (ALSFRS-R) and disease duration.
Conclusion: Patients with ALS demonstrate early-stage sympathetic hyperactivity, reflected by elevated adrenergic baroreflex sensitivity, which appears to attenuate with disease progression. Pressure recovery time may serve as a marker of disease severity and cumulative disease burden. Autonomic parameters, particularly those derived from the Valsalva maneuver, show strong discriminatory potential and may be useful as biomarkers for early detection and staging of ALS.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: Biomarkers in MND
PP01.333
Choroid Plexus Enlargement Is Associated With Disease Progression in Amyotrophic Lateral Sclerosis
Assoc. Prof. Seol-Hee Baek1, Dr. Woo-Suk Tae2, Assoc. Prof. Jin-Woo Park3, Prof. Byung-Jo Kim3,2
1
Department of Neurology, Korea University Ansan Hospital, Korea University College of Medicine, Ansan, Korea, Republic of.
2
Brain Convergence Research Center, Korea University Medical Center, Seoul, Korea, Republic of.
3
Department of Neurology, Korea University Anam Hospital, Korea University College of Medicine, Seoul, Korea, Republic of
Background: Cerebrospinal fluid (CSF) plays a critical role in maintaining brain homeostasis through mechanical protection, metabolic support, and clearance of metabolic waste. The glymphatic system, a recently identified pathway that facilitates CSF–interstitial fluid exchange, is considered a major brain waste clearance mechanism and has been implicated in the pathogenesis of neurodegenerative diseases. In the brain, the choroid plexus (CP) is a key structure responsible for CSF production and homeostasis. Thus, alterations in the CP may be associated with disrupted CSF dynamics and impaired glymphatic function. Accordingly, this study aimed to investigate whether the CP is altered in patients with amyotrophic lateral sclerosis (ALS) compared with normal controls (NCs).
Methods: We analyzed 25 patients with ALS and 43 age- and sex-matched NCs. Collected demographic and clinical data included age, sex, disease duration, ALS functional rating scale-revised (ALSFRS-R) score, and ΔALSFRS-R. The ΔALSFRS-R was calculated using the following equation: (48–ALSFRS-R score)/disease duration (months). Three-dimensional T1-weighted MRI was acquired using a 3-T scanner, and CP segmentation was performed with FreeSurfer. Generalized linear models with covariate was performed to compare CP volume between ALS and NCs. Age-adjusted partial correlation analyses were conducted to assess associations between CP volume fraction and clinical variables. CP volume fraction was calculated as the sum of right and left CP volume divided by total intracranial volume (TIV).
Results: Age and sex did not differ between groups (p=0.340, and p=0.803, respectively). After adjustment for age, right and left CP volumes were significantly larger in the ALS group than in the NC group (p=0.004, and p=0.046, respectively), whereas TIV did not differ between the groups (p=0.919). In addition, CP volume fraction was higher in the ALS group than in the NC group (0.092% vs. 0.070%; p=0.004). CP volume fraction was positively correlated with age (r=0.442; p=0.027). Age-adjusted partial correlation analyses revealed that CP volume fraction was positively correlated with disease duration (r=0.482; p=0.017) and negatively correlated with ΔALSFRS-R (r=-0.444; p=0.034). However, no significant correlation was observed with the ALSFRS-R score (r=-0.053; p=0.812).
Conclusion: This study revealed CP volume enlargement in patients with ALS compared to NCs, and CV volume fraction was negatively correlated with ΔALSFRS-R. These findings may reflect an association between impaired glymphatic function and disease progression in ALS. Further studies with larger cohorts are needed to validate these findings.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: Biomarkers in MND
PP01.334
Electrophysiological Origin of Repeater F-waves and Their Correlation with Clinical Severity in Polio Survivors
Dr. Akiko Hachisuka1, Dr. Tatsuya Abe2, Dr. Tetsuo Komori3, Dr. Satoru Saeki1
1
University of Occupational and Environmental Health, Rehabilitation Medicine, Kitakyushu, Japan.
2
Department of Neurology, National Hospital Organization Hakone Hospital, Odawara, Japan.
3
Tokyo Healthcare University, Tokyo, Japan
Background: Repeater F-waves (RFs) are characteristic findings in polio survivors, and our previous studies have established their occupancy rate of RFs (ORF) as a potential diagnostic parameter. This study aimed to further clarify the electrophysiological origin of RFs by analyzing amplitude and latency, and to determine their relationship with the clinical severity of paralysis.
Methods: Forty-three polio survivors and twenty healthy age- and height-matched controls underwent bilateral F-wave studies of the median and tibial nerves, elicited by 100 stimuli. Parameters including F-wave persistence (FP), ORF, mean amplitude, and minimal latency were estimated. Healthy controls were used only to define normal reference ranges (mean ± 2 SD), and analyses of F-wave amplitude and latency were limited to polio survivors. Based on these reference ranges, subjects were categorized into four stages: Stage A (normal FP and ORF), Stage B (normal FP, abnormal ORF), Stage C (abnormal FP, normal ORF), and Stage D (abnormal FP and ORF). Normal ORF ranges were defined as 0–25.4% (median) and 0–3.8% (tibial) based on our previous study. Clinical severity of polio was classified as mild, moderate, or severe based on the National Rehabilitation Hospital classification.
Results: F-waves were obtained from 82/86 upper limbs (62 mild, 14 moderate, 6 severe) and 76/86 lower limbs (12 mild, 18 moderate, 46 severe). In both nerves, RFs exhibited significantly higher mean amplitudes and prolonged minimum latencies compared to non-RFs (p<0.01). Specifically, in the median nerve, RFs showed higher amplitude (167.3±144.8μV vs. 82.8±81.1μV) and longer latency (27.2±3.0ms vs. 25.6±2.8ms). In the tibial nerve, RFs also showed higher amplitude (219.3±161.2μV vs. 160.8±143.0μV) and longer latency (47.6±4.6ms vs. 44.4±4.7ms).
Regarding the relationship with clinical severity, in the median nerve, 62.9% of the mild group were already classified as Stage B or D. As severity progressed to moderate and severe, the proportion of Stage A (normal) decreased from 33.9% to 0%, while the proportion of Stage D (abnormal FP and ORF) significantly increased. In the tibial nerve, 75.0% of the mild group were rated as Stage B or D. In the severe group, Stage A and B (normal FP) disappeared, with Stage D becoming the predominant classification. Across all severities, Stage C (isolated FP reduction) was rarely observed, indicating that an increase in RFs typically precedes a reduction in FP.
Conclusion: RFs, with their higher amplitude than non-RFs, might be derived from reinnervated motor units. Given their electrophysiological characteristics, RFs may reflect the activity of reinnervated motor units, potentially associated with a predominance of type I fiber, as reported in polio survivors. With the progression of polio severity, RFs increased, followed by a reduction in FP. These findings suggest that RFs could indicate early signs of motor unit pathology in polio survivors.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: Post poliomyelitis Syndrome
PP01.335
Adult SMA REACH: Safety and Efficacy Profiles of Nusinersen and Risdiplam in Adults With SMA
Miss Elena Karkkainen, Miss Jess Page, Mr. Aleks Carver, Miss Laura Simms, Mr. Robert Muni Lofra, Assoc. Prof. Chiara Marini Bettolo
Newcastle University - John Walton Muscular Dystrophy Research Centre, Newcastle Upon Tyne, United Kingdom
Background: Advancements in treatment options aimed at modifying disease progression in SMA in recent years have been made, with the introduction of drug treatments Nusinersen and Risdiplam. There has been a reported improvement in motor and pulmonary function tests in SMA patients both post-Nusinersen and post-Risdiplam treatment based on real-world observational data. Most studies have focused on evaluating each drug individually in clinical trials or real-world settings, but comparative analyses are limited in adults. Adult SMA REACH is a longitudinal observational data collection study that collects RWD during routine clinical visits across 18 different sites in the UK. The study includes patients aged ≥16 years with genetically confirmed 5q SMA.
Methods: Utilising data collected from the 460 patients enrolled in this study, we aim to perform an analysis comparing the efficacy and safety profiles of both interventions within the UK adult SMA population, while also identifying correlations with clinical phenotype, non-SMA medications, and patient-perceived benefits as well as identifying differences in the treated cohorts. The primary objective is to assess the efficacy and safety of Nusinersen and Risdiplam in adults, focusing on the following areas:
Changes in functional assessments will be derived from functional scales such as RHS, ATEND, HFMSE, EK2, RULM, WHO and 6MWT.
Changes in respiratory outcomes including FVC and PCF.
Analysis of safety profiles based on the frequency of adverse events recorded during treatments.
Baseline and follow-up visit scores will be summarised using descriptive statistics for safety and efficacy profiles. Repeated measures ANOVA will be used to evaluate changes in functional assessments and respiratory outcome over time. Subgroup analysis will be performed to compare outcomes by SMA type, SMN2 copy number, and functional status. Chi squared tests will be used to compare frequencies of adverse events between subgroups.
Results: The overall population included 410 SMA patients, with near-equal distribution of sex consisting of 45.5% females and 52.7% male participants. A small number of patients had unspecified sex (1.9%), due to missing data. Patient distribution by SMA type was as follows: type 3 with 206 (54.8%) participants, type 2 with 158 (42.0%) participants and type 1 with 4 (1.1%) participants. There were no patients with type 4 after the applying the exclusion criteria. WHO functional status at baseline distributed the patients as follows: 33.8% non-sitters, 46.8% sitters and 19.4% walkers. Overall results show stabilisation of disease after starting treatment, however further analysis includes detailed changes in functional assessments and respiratory outcomes over time.
Conclusion: The data presented will represent the first direct comparison of these treatments in the adult UK population using real-world evidence, and aims to compare the efficacy and safety profiles of both interventions within the UK adult SMA population providing critical insights that will support improved patient outcomes and inform future treatment decisions.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: Spinal Muscular Atrophies: Treatment
PP01.336
UK SMA Patient Registry: Characterise Disease Burden and Treatment Impact in Spinal Muscular Atrophy
Miss Elena Karkkainen, Mr. Aleks Carver, Miss Jess Page, Miss Laura Simms, Miss Stephanie Tanner, Ms. Dionne Moat, Mr. Robert Muni Lofra, Assoc. Prof. Chiara Marini Bettolo
Newcastle University - John Walton Muscular Dystrophy Research Centre, Newcastle Upon Tyne, United Kingdom
Background: The UK SMA Patient Registry, established in 2008, represents a well-defined cohort of individuals living with Spinal muscular atrophy (SMA) in the United Kingdom and Ireland. The registry has 688 participants: 474 adults (≥16 years); 214 paediatric (<16 years). The registry is a valuable tool for the collection of SMA data through patient-reported outcome measures (PROMs). PROMs capture the SMA patients’ perspectives about their quality of life and the impact of their condition.In April 2022, the registry implemented a range of PROMs in order to contribute to Managed Access Agreement (MAA) data collection supporting the national regulatory review of recently emerged SMA treatments Nusinersen and Risdiplam, aiming to collect PROMs data from 100 patients receiving each respective treatment.
Methods: In April 2022, the registry implemented the following PROMs:
- EQ-5D-5L
- EQ-5D-Y-3L
- Patient Global Impression of Severity (PGI-S)
- Patient Global Impression of Improvement (PGI-I)
- SMA Independence Scale – Upper Limb Module (SMAIS-ULM)
- Free-text box.
The registry simultaneously launched a pilot study in collaboration with the national Adult SMA REACH and SMA REACH UK clinical networks to support the collection of PROMs data through the registry.
Results: Since their implementation, PROMs questionnaires have been completed by 257 adults and by the caregivers of 106 paediatric patients in the registry. In total, the registry has collected 3764 entries to PROMs questionnaires.
In collaboration with the UK’s national SMA REACH clinical networks, PROMs data collected from patients receiving SMA treatment has been aligned with SMA REACH clinical data and submitted to UK regulatory authorities for consideration as part of the treatment reviews.
Participants’ first submission of PROMs data post treatment initiation will be presented in comparison to their most recent submission of PROMs data post treatment initiation.
Conclusion: The PROMs data presented indicates that participants reported stabilised or improved scores when comparing their first PROMs entry post treatment initiation to their most recent PROMs entry post treatment initiation. The registry’s presented data proves that PROMs add value to clinician-reported outcomes and offer a different perspective, demonstrating that it is worth continuing the effort of collecting the patient voice.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: Spinal Muscular Atrophies: Treatment
PP01.337
Adult SMA REACH: A Ready-Made Infrastructure to Support Research and Improvement Initiatives in SMA
Miss Elena Karkkainen, Miss Jess Page, Miss Laura Simms, Mr. Aleks Carver, Ms. Dionne Moat, Dr. Robert Muni Lofra, Asst. Prof. Chiara Marini Bettolo
Newcastle University - John Walton Muscular Dystrophy Research Centre, Newcastle Upon Tyne, United Kingdom
Background: Adult SMA REACH a Research and Clinical Hub that established a collaborative clinical network in 2020 across 18 clinical sites in the UK, patient advocacy groups, regulators, and industry. It also established a longitudinal observational real-world data (RWD) collection study, collecting clinical data and outcome measures from adult SMA patients in the UK. SMA treatments Nusinersen and Risdiplam are available through Managed Access Agreements (MAA); Adult SMA REACH is responsible for capturing and reporting data to UK regulatory authorities to support their review of drug efficacy. The final data cuts for both MAA’s have now passed and Adult SMA REACH is in a position to grow; as a ready-made infrastructure to inform research questions in the UK, it is already currently supporting a number of initiatives. The aims of Adult SMA REACH are to better understand the natural history of adult SMA, evolving phenotypes and the impact of new therapies. Data is collected from clinical sites via a centralised online database. Using innovative data modelling techniques, we created an automated software for data validation, consistency checks, completeness analysis and treatment tracking. This allows us to continuously monitor evolving datasets, maximising the quality of collected data. Anonymised data can be provided to support research following submission of a data request which must receive approval from the steering committee.
Methods: Data is collected from clinical sites via a centralised online database. Using innovative data modelling techniques, we created an automated software for data validation, consistency checks, completeness analysis and treatment tracking .This allows us to continuously monitor evolving datasets, maximising the quality of collected data. Anonymised data can be provided to support research following submission of a data request which must receive approval from the steering committee .Data request forms are available on request from the Adult SMA REACH team .
Results: As of 10 /03 /2025, Adult SMA REACH contains data on more than 460 patients with more than 2200 visits entered .Of the total patients, 418 have provided consent for their data to be used in academic research . Visits entered have an average completeness of 87 %across mandatory data items .
Conclusion: Data collected via Adult SMA REACH has supported research related to access to care; analysis of age at diagnosis, prevalence, and mortality; baseline characterisation of adults; and safety/efficacy analyses of available treatments. In addition to this, it is supporting the SMA Care UK initiative which aims to update and implement the Standards of Care for SMA patients. A pregnancy sub-study has also recently been established to characterise the effects of Nusinersen on pregnant women and the infants born to them. Adult SMA REACH is a valuable infrastructure to support SMA research, ultimately optimising patient care, advancing therapy approvals and broadening the understanding of the disease.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: Spinal Muscular Atrophies: Epidemiology and Clinical Features
PP01.338
Experience of Algeria to Diagnosis and Management Adult Spinal Muscular Atrophy
Asst. Prof. HALLAL SIHAM1, Dr. BENCHAABI OUISSEM2, Prof. NOUIOUA SONIA2, Prof. YARGUI LYECE1
1
CHU MUSTAPHA, ALGIERS, Algeria.
2
EHS CHERCHEL, BLIDA, Algeria
Background: Spinal muscular atrophy (SMA) is a form of motor neuron disease caused by a mutation in the survival motor neuron of gene (SMN1), which results in a wide disease spectrum affecting children and adults. There are four subtypes of SMA depending on age of onset and maximum acquired motor function.
Spinal Muscular Atrophy (SMA) in adults (often Type 4) is a genetic condition causing progressive muscle weakness, typically starting after age 18 with slow progression, usually mild, affecting leg muscles first, but with normal life expectancy and rare impact on breathing/swallowing, managed with physical therapy, supportive care.
Methods: Our work represents a retrospective descriptive study of 10 years of diagnosis spinal muscular atrophy in adults (January 2015- December 2025), performed in the only referral laboratory that diagnoses SMA in Algeria by amplification with ACRS method to search a deletion homozygote of exon 7.
Results: We analyze a total of 1620 patients, which 526 cases were confirmed as SMA. The frequency of the disease in our series was 33%
21 % of them are type 4, with an average age of symptoms apparition at 31 years.
42 % of our patients were born into a consanguineous marriage.
The most frequent reasons for consultation were functional impotence of the lower limbs, and fatigue, and fasciculations.
Conclusion: The adult form of SMA is a frequently suspected etiology in neuromuscular consultations.
Diagnosing adult SMA involves ruling out other neuromuscular conditions with tests like EMG, muscle enzyme assay, and DNA testing, focusing on symptoms like progressive muscle weakness.
In our country treatments were not yet available, management is multidisciplinary, with a key goal of avoid disability, preventing complications like scoliosis, respiratory issues.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: Spinal Muscular Atrophies: Epidemiology and Clinical Features,
PP01.339
Understanding Neurologist Perspectives on the Clinical Meaningfulness of ‘Any-Point Differences’ on the HFMSE in SMA
Dr. Leslie Nelson1, Ms. Natalie Land2, Dr. Melissa Maravic2, Dr. Thomas Brown3, Dr. Christabella Cherubino3, Dr. Mouhamed Gueye3
1
Department of Physical Therapy, University of Texas Southwestern Medical Center, Dallas, United States.
2
Precision AQ, New York, United States.
3
Scholar Rock, Inc., Cambridge, United States
Background: SMA is a neuromuscular disorder marked by the irreversible degeneration of spinal motor neurons, resulting in progressive muscle atrophy, weakness, and loss of motor function. Although motor function scales such as the HFMSE are commonly used to evaluate treatment effectiveness in patients with SMA, it can be challenging to determine whether changes in motor function scores are clinically meaningful. As a result, individual insights from patients or their caregivers are often required to obtain a more accurate understanding of treatment impact.
Methods: To gain deeper insights into how functional changes on the HFMSE are perceived in terms of clinical meaningfulness, we conducted 60-minute, web-based, semi-structured interviews among patients, their caregivers, and providers (neurologists/physical therapists) experienced with treating SMA and utilizing the HFMSE to monitor motor function. Here we report outcomes from interviews with neurologists (n=11), highlighting their interpretation of what constitutes clinically meaningful change for the HFMSE and functional tasks that may be the most meaningful.
Results: For interviewed neurologists, the mean age (SD) was 50.8 (9.9) years, with most specializing in neuromuscular medicine (7/11; 63.6%), followed by pediatric neurology (3/11; 27.3%), clinical neurophysiology (2/11; 18.2%), epilepsy (1/11; 9.1%), and/or vascular neurology (1/11;9.1). The most common clinical settings of practice were universities/academic medical centers (4/11; 36.4%) and multi-specialty group practices (3/11; 27.3%). All 11 interviewed neurologists (100%) reported they spend at least 80% of their work week providing direct care to patients and indicated they were ‘very familiar’ with the HFMSE (100%), with 10 (90.9%) reporting they received specific training on its administration. When defining clinically meaningful change, neurologists frequently framed it from the patient’s perspective, generally characterizing it as a change that clearly enhances a patient’s functioning, independence, and capacity to perform everyday activities. They also noted that any point improvement on functional tasks within the HFMSE reflects a meaningful gain in independence and quality of life, with the most impact being noted for patients who were previously unable to perform those tasks. One neurologist highlighted this when discussing the lifting hands to head task, “They [patients] would have more autonomy…This means that they can do certain tasks better such as feeding themselves…” Another neurologist noted the importance of patients making small improvements in the standing task stating, “Standing alone, you can probably take a shower that way by yourself, dress by yourself. That's big.” A separate neurologist also noted the significance of any point improvement in the sitting to laying task, emphasizing the impact on quality of life, “[Improvement] makes a tremendous change in their quality of life…their motor functioning and their ability to perform the motor work.”
Conclusion: According to neurologists’ responses, clinically meaningful change is best understood through a patient-centered lens, with improvements in daily functioning and independence being assessed in relation to each patient’s goals and lived experience. Neurologists generally agreed that any point improvement, regardless of functional task, can meaningfully affect a patient’s independence and quality of life, highlighting the clinical and patient-focused value of recognizing and tracking small changes.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: Spinal Muscular Atrophies: Epidemiology and Clinical Features,
PP01.340
Design of a Non-interventional Study to Assess Patient Perspectives Inspinal Muscular Atrophy (SMA-Perceptions Study)
Dr. David Gómez-Andrés1, Dr. Paz Castro2, Dr. Jorge Mauriño2, Mrs Angeles Terrancle2, Mrs Fátima Jerez2, Dr. Mónica Povedano3
1
Pediatric Neurology, Vall d´Hebron Institut de Recerca (VHIR), Hospital Universitari Vall d'Hebron, Universitat Autònoma de Barcelona, Barcelona, Spain.
2
Medical Departament, Roche Farma, Madrid, Spain.
3
Department of Neurology, Hospital Universitari de Bellvitge, Institut d´Investigació Biomèdica de Bellvitge-IDIBELL, Barcelona, Spain
Background: The therapeutic landscape of spinal muscular atrophy (SMA) has changed in recent years with the introduction of new therapies. Despite these advances and the availability of validated scales, current assessment tools often fail to capture the full spectrum of symptoms and signs that significantly affect patients’ daily lives and health-related quality of life. In addition, they may lack the sensitivity needed to detect subtle but clinically meaningful improvements from the patient’s perspective. Therefore, there is a clear need to study the quality of life of this population in a broader context, covering all ages and individual characteristics, to obtain a more comprehensive understanding of the disease burden from the patient and the caregiver perspective.
Methods: To evaluate the perception of patients regarding their quality of Life, decision-making, and care, as reported by both them and their caregivers, to capture data that reflects the unique characteristics of the healthcare system and sociocultural environment.
Results: We present the design of a cross-sectional, non-interventional study. Approximately 100 patients are planned to be enrolled across Spain. Patients aged ≥ 2 years with a confirmed diagnosis of SMA will be included. The study will be based on a single data collection point, coinciding with a medical follow-up visit. A battery of validated clinical outcomes assessments (COAs) will be administered to the patient or their caregiver. These assessments are designed to evaluate several key domains specific to SMA patients across different age groups: 2 to 5 years, 6 to 15 years, and 16 years and up. Quality of Life (using the INQoL or PedsQoL); level of participation in shared decision-making (using SDM-Q-9, SURE DCS, DRS, and CPS); quality of chronic care (using PACIC); and psychological and emotional factors, identification of symptoms, and treatment satisfaction, among others.
The primary outcome will be the mean total quality of life score. Correlation factors between the COAs and clinical/demographic variables will be determined. Furthermore, factors associated with quality of life (INQoL/PedsQoL) will be analyzed using correlation and linear regression analyses with other COAs, specifically grouping them into psychological factors, uncertainty/concerns, and care/decision experience.
Conclusion: The collected data is expected to provide a more comprehensive and nuanced view of the SMA patient population in Spain across all ages and their individual characteristics. Ultimately, the findings are anticipated to contribute to identifying areas for improvement, guiding clinical practice towards more patient-centered care, and allowing for the enhancement of meaningful outcomes in the context of emerging treatments.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: Spinal Muscular Atrophies: Epidemiology and Clinical Features,
PP01.341
Assessment and Management of Bulbar Function in Spinal Muscular Atrophy in Spain
Dr. Paz Castro1, Dr. Laura Carrera-García2, Dr. Vanessa Ejarque3, Dr. Ana María León4, Mrs Cristina Puig5, Mrs Carmen Álvarez1, Dr. Jorge Mauriño1, Mrs Sofía García-López1, Dr. Andrés Nascimento2
1
Medical Department, Roche Farma, Madrid, Spain.
2
Neuromuscular Unit, Department of Neurology, Hospital Sant Joan de Déu, Barcelona, Spain.
3
Department of Gastroenterology, Hepatology and Nutrition, Hospital San Joan de Déu, Barcelona, Spain.
4
Phoniatrics and Speech Therapy Unit, Hospital Universitari Vall d´Hebron, Barcelona, Spain.
5
Institut de Recerca Sant Joan de Déu, Barcelona, Spain
Background: Bulbar dysfunction is a frequent and clinically relevant complication in spinal muscular atrophy (SMA), affecting quality of life and health outcomes. Systematic assessment is limited by a lack of standardized tools; addressing these gaps is crucial for optimizing comprehensive patient-centred-care. This study describes the current management for bulbar dysfunction in SMA patients in Spain.
Methods: A non-interventional, cross-sectional pilot study was conducted including healthcare professionals (HCPs) with expertise in SMA. Participants were invited to the study by the national SMA registry CuidAME and completed a structured electronic survey, including information regarding their demographic characteristics, and practice-related variables. A descriptive analysis was conducted.
Results: The study included 37 HCPs (62.2% female), primarily pediatric neurologists (37.8%) and neurologists (21.6%), with a mean of 9.6 years ± 5.8 of experience. Most (86.5%) work in multidisciplinary teams at public, tertiary centers (89.2%). HCPs placed great importance (mean 8.9/10) on bulbar function and the systemic effect of disease modifying treatments (mean 8.7/10). While 56.8% of HCPs gave a high priority to bulbar function, motor evaluation still received more dedication (2.8/6).
The most widely recognized bulbar domains were dysphagia (97.3%), voice and speech (89.2%), and fatigability (89.2%) which directly aligned with the main aspects evaluated: dysphagia (97.3%), voice and speech 89.2% and fatigue (83.8%). Swallowing assessment is a common practice (94.4%), performed by speech-language therapists (82.4%) and gastroenterologists (70.6%), particularly in SMA types 1 and 2.
Evaluation combines instrumental methods (videofluoroscopy: 96.8%) with non-instrumental scales (Functional Oral Intake Scale: 55.9%, Egen Klassifikation 2: 50.0%), with intervention programs available in 85.3% of these centers.
The evaluation is specifically performed in SMA type 1 (94.1%) and SMA type 2 (82.4%) patients. It is noteworthy that only 47.1% perform it in type 3 and 32.4% in presymptomatic patients.
Voice and speech evaluations are conducted in 59.5% of centers, with 77.3% of them offering specific interventions.
HCPs working in centers with a multidisciplinary team more frequently used instrumental assessment of bulbar function (93.5% vs. 50%; p=0.022), and videofluoroscopy (87.5% vs. 40%; p=0.037) compared to their counterparts, and were more likely to recommend bulbar rehabilitation (87.1% vs. 33.3%; p=0.013).
Regarding professional well-being, participants predominantly showed high empathy (45.9%) and openness in evidence-based innovations (27.0%). Bulbar prioritization is associated with a significantly higher total empathy (Jefferson Scale of Empathy) score (p=0.032). Openness to Innovations is significantly associated with a lower procrastination (Pure Procrastination Scale) score (p=0.030).
Conclusion: This study suggests strong awareness and commitment among HCPs managing bulbar function in SMA, characterized by a multidisciplinary approach and established evaluation and rehabilitation. However, motor evaluation still dominates time, and dysphagia evaluation is critically limited in SMA Type 3 and presymptomatic patients. A significant correlation links bulbar prioritization with higher medical empathy. Standardization of assessment protocols is still required to optimize care across the full SMA spectrum.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: Spinal Muscular Atrophies: Epidemiology and Clinical Features,
PP01.342
DYNC1H1-Related Spinal Muscular Atrophy With Arthrogryposis and Congenital Fractures
Dr. Evgeniya Melnik1, Prof. Vladimir Kenis2, Dr. Kseniya Zabudskaya1, Dr. Vadim Tsargush3, Prof. Sergei Nikitin1, Prof. Elena Dadali1
1
Research Centre for Medical Genetics, Moscow, Russia.
2
H. Turner National Medical Research Centre for Children's Orthopedics and Trauma Surgery of the Ministry of Health of the Russian Federation, Saint Petersburg, Russia.
3
Limited Liability Company “My Medical Center Advanced Technologies”, Krasnodar region, Sirius, Russia
Background: Dyneinopathies are a group of DYNC1H1-related disorders caused by dysfunction of lower motor neurons in the anterior horns of the spinal cord and/or motor structures of the brain. The DYNC1H1 gene encodes the heavy chain of cytoplasmic dynein 1, a key motor protein required for retrograde axonal transport along microtubules and normal neurodevelopment. Pathogenic variants in DYNC1H1 lead to a broad phenotypic spectrum ranging from neurodevelopmental disorders with central nervous system involvement to predominantly peripheral motor neuron disease resembling spinal muscular atrophy (SMA), typically with lower limb predominance, and combined senso-motor neuropathies such as Charcot–Marie–Tooth disease type 20. Increasing evidence indicates that DYNC1H1-related disease may also include significant neuro-orthopedic manifestations, including congenital long-bone fractures, hip dysplasia/dislocation, arthrogryposis, and spinal deformities. However, the combination of SMA-like motor neuron disease with congenital multiple arthrogryposis and congenital fractures remains rare and insufficiently characterized, highlighting the need for further clinical and genetic characterization of these presentations.
Methods: We report three unrelated male patients aged 6 months to 13 years presenting with a rare phenotype of DYNC1H1-related disease. All patients underwent comprehensive clinical evaluation, neurophysiological testing, and neuroimaging. Muscle MRI was performed to assess muscle involvement patterns and fatty replacement. Electromyography (EMG) was used to evaluate the type and distribution of neuromuscular impairment. Genetic testing included whole-exome sequencing (WES) in all cases. Identified variants were confirmed by Sanger sequencing, and their de novo origin was established through parental testing.
Results: Two patients carried a previously reported pathogenic variant in the tail domain of DYNC1H1: NM_001376.5:c.791G>A, p.(Arg264Gln). The third patient harbored a novel variant, NM_001376.5:c.1822C>A, p.(Leu608Met), located in the stalk domain. Clinically, all patients demonstrated generalized hypotonia and predominant lower limb weakness, multiple joint contractures from birth, hip dislocations, foot deformities, and congenital fractures of long bones. Muscle MRI revealed marked and early fatty infiltration of the lower limb muscles, detectable already in infancy. EMG showed features consistent with a generalized neurogenic process, supporting motor neuron involvement. The combination of SMA-like weakness, congenital multiple arthrogryposis, and fractures represents a distinctive and rare DYNC1H1-related phenotype.
Conclusion: These three cases expand the phenotypic spectrum of DYNC1H1-related disease and broaden the known variant landscape, including identification of a novel DYNC1H1 variant. Our observations highlight the importance of including DYNC1H1 in NGS diagnostic panels when SMA-like presentations are accompanied by arthrogryposis and congenital fractures. Additionally, early and extensive fatty infiltration of lower limb muscles on MRI should be considered during orthopedic decision-making and long-term management planning in affected patients.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: Spinal Muscular Atrophies: Pathophysiology, Genetics
PP01.343
RAINBOWFISH: 3-Year Efficacy and Safety Data of Risdiplam in Infants With Presymptomatic Spinal Muscular Atrophy
Prof. Enrico Bertini1, Prof. Maria Mazurkiewicz-Bełdzińska2, Prof. Laurent Servais3,4, Prof. Michelle A Farrar5, Dr. Dmitry Vlodavets6, Dr. Edmar Zanoteli7, Prof. Mohammad Al-Muhaizea8, Prof. Alexandra PQC Araújo9, Dr. Leslie Nelson10, Miss Manni Kuthiala11, Dr. Ksenija Gorni12, Dr. Heidemarie Kletzl13, Dr. Francis Warren14, Dr. Eleni Gaki14, Mr. Dave Summers14, Dr. Marianna Manfrini12, Prof. Richard S Finkel15
1
Research Unit of Neuromuscular and Neurodegenerative Disorders, Bambino Gesù Children’s Research Hospital IRCCS, Rome, Italy.
2
Department of Developmental Neurology, Chair of Neurology, Medical University of Gdańsk, Gdańsk, Poland.
3
MDUK Oxford Neuromuscular Centre, Department of Paediatrics, University of Oxford, Oxford, United Kingdom.
4
Division of Child Neurology, Centre de Références des Maladies Neuromusculaires, Department of Pediatrics, University Hospital Liège & University of Liège, Liège, Belgium.
5
Sydney Children’s Hospital Network and UNSW Medicine, UNSW Sydney, Sydney, Australia.
6
Russian Children Neuromuscular Center, Veltischev Clinical Pediatrics and Pediatric Surgery Research Institute of Pirogov Russian National Research Medical University, Moscow, Russia.
7
Department of Neurology, Faculdade de Medicina, Universidade de São Paulo, São Paulo, Brazil.
8
Neuroscience Centre of Excellence, King Faisal Specialist Hospital & Research Center-Riyadh, Riyadh, Saudi Arabia.
9
Pediatrics Department, Faculty of Medicine, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil.
10
Department of Physical Therapy, University of Texas Southwestern Medical Center, Dallas, TX, United States.
11
Pharma Development, Safety, F. Hoffmann-La Roche Ltd, Basel, Switzerland.
12
PDMA Neuroscience, F. Hoffmann-La Roche Ltd, Basel, Switzerland.
13
Roche Pharmaceutical Research and Early Development, Roche Innovation Center Basel, Basel, Switzerland.
14
Roche Products Ltd, Welwyn Garden City, United Kingdom.
15
Center for Experimental Neurotherapeutics, St Jude Children’s Research Hospital, Memphis, TN, United States
Background: Risdiplam is a centrally and peripherally distributed, oral survival of motor neuron 2 (SMN2) pre‑mRNA splicing modifier approved for the treatment of spinal muscular atrophy (SMA). RAINBOWFISH (NCT03779334) is a global, open-label, single-arm, multicentre, Phase 2 study assessing the efficacy, safety, pharmacokinetics and pharmacodynamics of risdiplam in infants with genetically diagnosed and presymptomatic SMA from birth to 6 weeks of age (at first dose), regardless of SMN2 copy number or baseline compound action potential (CMAP).
Methods: RAINBOWFISH enrolled 26 infants: eight infants had two SMN2 copies, 13 infants had three SMN2 copies and five infants had ≥4 SMN2 copies. The primary efficacy (PE) population (n=5) had two SMN2 copies and baseline CMAP amplitudes ≥1.5 mV. Drug dosage was adjusted to achieve a target exposure of approximately 2,000 ng∙hr/mL.
This analysis assesses the efficacy and safety of risdiplam in children with presymptomatic SMA after 3 years of treatment.
Results: The primary endpoint was met after 12 months of risdiplam treatment, with 4/5 (80%) infants in the PE population able to sit without support for ≥5 seconds (Item 22 of the Gross Motor Scale of the Bayley Scales of Infant and Toddler Development, third edition [BSID-III]).
Twenty-three children completed 3 years of treatment with risdiplam (data cut-off: 10 February 2025). The vast majority of children who reached Year 3 (21/23, 91%) were able to sit, stand and walk without support, and once achieved, no children lost any abilities. All children who completed 3 years of risdiplam treatment were able to swallow and 96% of children were fed exclusively orally. Cognitive function and speech development were similar to that of typically developing children without SMA in the same age range.
There were no treatment-related serious adverse events reported up to Year 3. No child required respiratory support outside of an illness and the majority of children (21/26) did not require any hospitalisations up to Year 3. There were no withdrawals from treatment between Years 2 and 3.
Conclusion: The majority of children treated with risdiplam before the onset of SMA symptoms, maintained motor milestones and bulbar function and showed cognitive skills similar to children without SMA after 3 years of treatment.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: Spinal Muscular Atrophies: Treatment
PP01.344
Clinical Outcomes of SMA Type 1 Patients Treated With Zolgensma in Brazil Public Health System
Dr. Cristina Iwabe1, Dr. Felipe Franco da Graça1, Dr. Marcondes Cavalcante França Jr1, Dr. Juliana Gurgel-Giannetti2, Dr. Flávia Nardes dos Santos3, Dr. Vanessa Luiza Romanelli Tavares4, Dr. Tertuliana Medeiros Mota dos Reis5
1
UNICAMP, Campinas, Brazil.
2
UFMG, Belo Horizonte, Brazil.
3
UFRJ, Rio de Janeiro, Brazil.
4
Instituto Jo Clemente, São Paulo, Brazil.
5
UPC - MA, Maranhão, Brazil
Background: Spinal Muscular Atrophy (SMA) is a rare, severe genetic disorder characterized by progressive degeneration of motor neurons, leading to profound muscle weakness and high mortality in its most severe forms. Early and highly specialized therapeutic interventions are essential to modify the natural history of the disease. Recent advances in gene therapy have transformed SMA management, with onasemnogene abeparvovec (Zolgensma®) representing one of the most innovative disease-modifying treatments currently available. Its single-dose administration and mechanism targeting the underlying genetic defect confer major clinical relevance, particularly within public health policy frameworks.
In Brazil, the incorporation of Zolgensma® into the Unified Health System (Sistema Único de Saúde – SUS) represents a landmark achievement in rare disease care, considering its high cost and the strict eligibility criteria for its use.
Methods: Patients with a confirmed diagnosis of SMA type 1, either presymptomatic or oligosymptomatic, aged up to 6 months, were included. Participants may or may not have received prior disease-modifying therapies. All patients were followed at the Neuromuscular Diseases Outpatient Clinic of the Hospital de Clínicas, University of Campinas (UNICAMP), Brazil.
Results: Four patients with SMA type 1 were enrolled, including two females and two males. Two patients were presymptomatic, diagnosed through prenatal screening, and two were oligosymptomatic at the time of treatment. All patients were younger than 6 months of age at the time of gene therapy infusion. The presymptomatic patients had previously received SMN protein replacement therapy.
No adverse events were observed following infusion, and no clinically significant laboratory abnormalities were detected. Escalation of corticosteroid dosage was not required during the treatment period. Three of the four patients have already completed corticosteroid tapering, with a mean duration of steroid use of approximately 60 days.
Motor function was assessed by a physiotherapist specialized in neuromuscular disorders using the CHOP INTEND scale before infusion and one month after treatment. One presymptomatic patient, previously treated with protein replacement therapy, achieved the maximum CHOP INTEND score prior to infusion and maintained this score at follow-up. The remaining patients demonstrated improvements ranging from 2 to 10 points in total CHOP INTEND scores after gene therapy.
Conclusion: Early diagnosis of SMA is a critical determinant of therapeutic efficacy, as rapid and irreversible motor neuron loss occurs early in the disease course. Identification of affected infants—particularly through newborn screening—enables timely initiation of treatment before significant neuromuscular impairment, thereby optimizing motor outcomes.
In this context, the availability of Zolgensma® within the Brazilian Unified Health System represents a major advance in public health policy for rare diseases. Access to a one-time gene replacement therapy through the SUS allows early and equitable intervention, contributing to improved motor function, greater functional independence, and better overall quality of life for patients with SMA.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: Spinal Muscular Atrophies: Treatment
PP01.345
SMA Type 1 Diagnosed by Newborn Screening Post Disease-Modifying Treatments in Hong Kong Chinese Children
Ms. Mei Wun Cheung1, Dr. Sophelia Hoi Shan Chan2,3, Miss Angel Wing Lam Tung1, Dr. Stephen Wing Wai Chan1, Mr. Ming Chung Poon1
1
Allied Health Department (Physiotherapy) of Hong Kong Children's Hospital, Hong Kong SAR, China.
2
Department of Pediatrics and Adolescent Medicine, The University of Hong Kong, Hong Kong SAR, China.
3
Paediatric Neurology Team, Department of Paediatrics and Adolescent Medicine, Hong Kong Children's Hospital, Hong Kong SAR, China
Background: Spinal muscular atrophy (SMA) is a neuro-degenerative disease characterized by progressive muscle weakness, atrophy and paralysis. SMA with two SMN2 copies (SMA type 1) typically have disease onset before 6 months of age and rarely achieve independent sitting. Most died before age 2. Disease-modifying treatments have significantly altered their natural disease course. In Hong Kong, SMA newborn screening (NBS) started in 2022, enabling early diagnosis and treatment initiation for better clinical outcomes. Real-world data on post-treatment outcomes of NBS-diagnosed SMA are needed. This study aims to evaluate the clinical outcomes of patients with NBS-diagnosed SMA carrying two SMN2 copies after being treated with disease-modifying therapies.
Methods: We collected demographic and clinical data – age, sex, symptom-onset age, treatment-initiation age, and clinical status including feeding, breathing, musculoskeletal, and motor function - at pre-treatment and post-treatment follow-ups, according to protocol. Motor functions were evaluated using (1) Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP-INTEND), (2). Bayley-4 Motor Scale (Growth Scale Value, GSV), (3) HINE-2. (4) Motor milestone achievement age. Data collection spanned from 2022 to November 2025. All patients received same standard of care including regular multidisciplinary evaluation and physiotherapy.
Results: Four eligible patients were included. One remained pre-symptomatic; others showed symptom onset on days 1, 4, and 15 of life. All started disease-modifying treatment between two weeks and one month of life, with three being symptomatic at treatment initiation. Follow-up ranged over at least one year. Motor assessments showed noticeable improvements: CHOP-INTEND scores increased by a mean of 34.75±12.63; Bayley fine-motor, gross-motor GSV increased by 37.75 ±14.31 and 24.5 ±8.74 respectively; HINE-2 increased by 19±4.97 points. Three patients (75%) achieved independent sitting before age 1, two (50%) achieved independent standing by 15 and 29 months, and one (25%) achieved independent walking by 33 months. Independent standing/walking occurred earlier than crawling. Three (75%) maintained oral feeding, one (25%) required gastrostomy feeding. Two (50%) started nocturnal non-invasive ventilation (NIV). Two (50%) had mild scoliosis before age 2 with spinal brace. All (100%) had pronated feet in standing.
Conclusion: NBS allowing early initiation of disease-modifying treatments improves the muscle strength and motor functions of SMA with 2 SMN2 copies. In our three patients with treatment started at the symptomatic stage, delay in motor milestone related to transitional maneuvers such as rolling, crawling, and floor to stand, was observed, suggesting truncal and proximal instability. They also developed respiratory, feeding and musculoskeletal complications. Proactive physiotherapy training on transitional maneuvers, respiratory maintenance, and early introduction of hydrotherapy, is recommended. An advanced initiation of treatment within the first week of life may increase the likelihood of treatment prescribed at a pre-symptomatic stage in affected babies with 2 SMN2 copies diagnosed through NBS. A global effort to develop a new standard of care including rehabilitation strategies and early introduction of spinal brace and foot orthosis for the new phenotype of SMA in the era of disease-modifying treatments, is urgently needed.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: Spinal Muscular Atrophies: Treatment
PP01.346
Indirect Treatment Comparison of OAV101 IT vs Risdiplam and Nusinersen in SMA DMT-Experienced Patients
Mr. Nicholas Riley1, Mr. Nicolas Ballarini2, Ms. Almuth Marx3, Mr. Orlando Dohring4, Ms. Grace McCarthy5, Ms. Anja Haltner6, Ms. Sarah Walsh6, Mr. Chris Drudge6, Mr. Maxwell Jones6, Ms. Roya Gavanji6, Ms. Nikethana Srikanth6, Ms. Monica Duong6, Mr. Paul Spin6
1
Novartis, Basel, Switzerland.
2
Novartis, East Hanover, United States.
3
Novartis, Nuremberg, Germany.
4
Novartis, London, United Kingdom.
5
Novarts, Dublin, Ireland.
6
EVERSANA, Burlington, Canada
Background: Spinal muscular atrophy (SMA) is a severe neuromuscular disorder leading to muscle atrophy and loss of motor function. The approval and use of disease modifying treatments (DMTs), such as nusinersen, risdiplam, and intravenous onasemnogene abeparvovec (OAV101 IV) have significantly changed the treatment landscape for SMA over the past decade. Intrathecal onasemnogene abeparvovec (OAV101 IT) is a one-time gene replacement therapy for SMA that recently received Food and Drug Administration (FDA) approval and is under regulatory review in other countries. Following regulatory approval, the next priority is to secure market access and reimbursement by demonstrating comparative effectiveness against currently available chronically administered SMA therapies. As there are a lack of head-to-head trials for SMA DMTs, this will require conducting an indirect treatment comparison (ITC) to assess the comparative efficacy of OAV101 IT versus existing chronically administered SMA therapies.
Objective: To estimate the comparative efficacy of OAV101 IT versus risdiplam or nusinersen in SMA DMT-experienced patients using matching-adjusted indirect comparisons (MAICs) to fulfill evidence requirements in addressing the assessment scope requested as part of the Joint Clinical Assessment (JCA) submission in the European Union and support other market access activities for OAV101 IT.
Methods: Clinical trials of SMA DMT-experienced patients were identified via a systematic literature review. Pivotal trials were selected for an in-depth feasibility assessment based on patient population overlap with OAV101 IT and relevance to an ITC. Pre-specified criteria such as age at screening, symptomatic status, functional status, naivety to OAV101 IT, EMA approval status, baseline characteristics, outcome data, and study design were used to determine whether comparator studies were appropriate for an ITC. Among studies included in the broader assessment, cross-study heterogeneity was evaluated for study characteristics, eligibility criteria, baseline and outcome characteristics, and data availability. Due to limited clinical data for prior DMT patients, the assessment concluded SAPPHIRE was the only appropriate study for a DMT-experienced ITC versus OAV101 IT, using its control arm as a proxy for DMT-experienced patients.
An unanchored MAIC was performed with individual patient data for STRENGTH (NCT05386680; OAV101 IT), which enrolled a DMT-experienced patient population, and published summary level data for the control arm from SAPPHIRE (NCT05156320; prior and continued nusinersen or risdiplam + placebo). Relative efficacy was evaluated using change from baseline (CFB) in Hammersmith Functional Motor Scale–Expanded (HFMSE) and Revised Upper Limb Module (RULM) scores.
Results: For OAV101 IT versus nusinersen or risdiplam + placebo, the MAIC showed numerical CFB improvement favouring OAV101 IT in HFMSE (mean difference [MD]: 2.16; 95% confidence interval [CI]: -0.06, 4.38; p=0.057) and RULM (MD: -0.26; 95% CI: -1.64, 1.12; p=0.713).
Conclusion: In the absence of direct head-to-head trials and limited clinical data for DMT-experienced patients, this analysis demonstrates that treatment with OAV101 IT results in comparable improvement in motor milestones relative to nusinersen and risdiplam in SMA DMT-experienced patients. These findings support OAV101 IT as a viable option for SMA DMT-experienced patients who cannot continue or tolerate their current long-term therapy, or prefer to choose a one-time treatment option.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: Spinal Muscular Atrophies: Treatment
PP01.347
Indirect Treatment Comparisons of OAV101 IT vs Risdiplam and Nusinersen in SMA DMT-Naïve Patients
Mr. Nicholas Riley1, Mr. Nicolas Ballarini2, Ms. Almuth Marx3, Mr. Orlando Dohring4, Ms. Grace McCarthy5, Ms. Anja Haltner6, Ms. Sarah Walsh6, Mr. Chris Drudge6, Mr. Maxwell Jones6, Ms. Roya Gavanji6, Ms. Nikethana Srikanth6, Ms. Monica Duong6, Mr. Paul Spin6
1
Novartis, Basel, Switzerland.
2
Novartis, East Hanover, United States.
3
Novartis, Nuremberg, Germany.
4
Novartis, London, United Kingdom.
5
Novartis, Dublin, Ireland.
6
EVERSANA, Burlington, Canada
Background: Spinal muscular atrophy (SMA) is a neuromuscular disorder leading to muscle atrophy and loss of motor function. The treatment landscape for SMA has evolved significantly with the use of disease-modifying therapies (DMTs). Intrathecal onasemnogene abeparvovec (OAV101 IT) is a one-time gene replacement therapy for SMA that recently received Food and Drug Administration (FDA) approval and is under regulatory review in other countries. Comparative efficacy or safety of OAV101 IT in patients with SMA versus existing chronically administered SMA therapies, such as risdiplam and nusinersen, have not been assessed in any head-to-head clinical trials. Indirect treatment comparisons (ITCs) are therefore critical for estimating relative efficacy and safety.
Objective: To estimate the relative efficacy and safety of OAV101 IT versus risdiplam and nusinersen in SMA DMT-naïve patients using anchored matching-adjusted indirect comparisons (MAICs) to fulfill evidence requirements in addressing the assessment scope requested as part of the Joint Clinical Assessment (JCA) submission in the European Union and support other market access activities for OAV101 IT.
Methods: Randomized controlled trials of SMA DMT-naïve patients were identified via a systematic literature review. Pivotal trials were selected based on an in-depth feasibility assessment identifying overlap with OAV101 IT trial populations, using criteria such as age at screening, symptomatic and ambulatory status, and naivety to DMTs. Indirect comparisons were performed using individual patient data from STEER (NCT05089656; OAV101 IT), which enrolled a DMT-naïve patient population, and published summary level data from SUNFISH part 2 (NCT02908685; risdiplam) and CHERISH (NCT02292537; nusinersen). Relative efficacy and safety were primarily evaluated using change from baseline (CFB) in Hammersmith Functional Motor Scale–Expanded (HFMSE) and Revised Upper Limb Module (RULM) scores, and proportion of patients with treatment-emergent adverse events (TEAEs) and serious TEAEs (STEAEs).
Results: For OAV101 IT versus risdiplam, MAIC results showed a numerical improvement in HFMSE favouring OAV101 IT (mean difference [MD]: 1.93; 95% confidence interval [CI]: -0.25, 4.10); p=0.0825). MAIC results for CFB in RULM favoured risdiplam (MD: -1.46; 95% CI: -3.38, 0.46; p=0.1367). Safety comparisons of OAV101 IT versus risdiplam indicated numerical favorability in proportions of patients with at least one TEAE (risk ratio [RR]: 1.11; 95% CI: 0.93, 1.33; p=0.2610) and at least one STEAE (RR: 0.73; 95% CI: 0.25, 2.12; p=0.5614). For OAV101 IT versus nusinersen, MAIC results showed comparable improvements in HFMSE (MD: -0.13; 95% CI: -2.93, 2.66; p=0.9252) and RULM (MD: -1.15; 95% CI: -3.32, 1.01; p=0.2975), both favoring nusinersen. Safety comparisons of OAV101 IT versus nusinersen indicated numerical favorability in proportions of patients with at least one TEAE (RR: 1.12; 95% CI: 0.96, 1.30; p=0.1518) and at least one STEAE (RR: 1.26; 95% CI: 0.43, 3.74; p=0.6709).
Conclusion: In the absence of direct head-to-head trials for SMA DMTs, these analyses demonstrate that treatment with OAV101 IT results in similar motor milestone improvement and OAV101 IT has a similar safety profile relative to risdiplam and nusinersen in the SMA DMT-naïve patient population. These findings support OAV101 IT as a viable first-line gene-replacement therapy, expanding treatment options for patients with SMA.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: Spinal Muscular Atrophies: Treatment
PP01.348
Neuron-Specific Lrp4 Overexpression Rescues Impaired Neuromuscular Junction Formation via BMP Signaling in Drosophila SMA Model
Dr. Rei Takada1, Dr. Taro Matsuoka1, Dr. Yosuke Kimura1, Dr. Amane Matsuura1, Dr. Yosuke Miyamoto1, Prof. Masafumi Morimoto1, Assoc. Prof. Tomohiro Chiyonobu2, Assoc. Prof. Hideki Yoshida3, Asst. Prof. Takenori Tozawa1
1
Department of Pediatrics, Graduate School of Medical Science, Kyoto Prefectural University of Medicine, Kyoto, Japan.
2
Department of Molecular Diagnostics and Therapeutics, Graduate School of Medical Science, Kyoto Prefectural University of Medicine, Kyoto, Japan.
3
Department of Applied Biology, Kyoto Institute of Technology, Kyoto, Japan
Background: Disease-modifying therapies and newborn screening have led to improved survival outcomes in Spinal Muscular Atrophy (SMA). However, a key challenge remains that Survival Motor Neuron (SMN) replacement therapies alone are insufficient for recovering neuromuscular junction (NMJ) dysfunction, which precedes neurodegeneration. Consequently, SMN-independent therapies combined with SMN replacement therapy are gaining attention. We focus on low-density lipoprotein receptor-related protein 4 (Lrp4) as a therapeutic target molecule for restoring NMJ formation abnormalities. Using a Drosophila SMA model, we will verify the ameliorative effect of enhancing Bone Morphogenetic Proteins (BMP) signaling through forced Lrp4 expression on NMJ formation abnormalities.
Methods: We utilized a neuron-specific Smn knockdown line by using elav-GAL4 and a compound heterozygous line by crossing two heterozygous mutant alleles, Smn73Ao which contains a point mutation in YG box and Smnf01109 which is predicted to introduce a premature stop codon. Furthermore, we achieved neuron-specific overexpression of Lrp4 in this Smn compound heterozygote using the GAL4-UAS system. The motor function was evaluated using a crawling assay for larvae and a climbing assay for adults. The immunostaining analysis is performed to assess the morphological abnormalities in NMJ. To assess BMP signaling activity, we measured the fluorescence intensity of phosphorylated Mad (pMad), a downstream effector of the signaling pathway, at the NMJ, the anterior Corner Cell (aCC) motor neuron nuclei and muscle nuclei.
Results: In similar to Smn knockdown lines, the Smn compound heterozygotes exhibited loss of motor function in both larvae and adults, morphological abnormalities at the NMJ and reduced expression of pMad suggesting decreased BMP activity. Neuron-specific overexpression of Lrp4 in the Smn compound heterozygotes restored NMJ morphology and BMP activity, although it did not improve motor function. Besides, we found that the expression of pMad was reduced at the aCC motor neuron nuclei and muscle nuclei in the Smn compound heterozygotes. Notably, this reduction was also restored by neuronal-specific Lrp4 overexpression.
Conclusion: Our study showed that neuron-specific Lrp4 overexpression can partially rescue the SMA pathology by restoring NMJ morphology through the recovery of impaired BMP signaling in Drosophila SMA model. These results suggest that Lrp4 may be one of the candidate molecules for SMN-independent therapies in SMA and the therapeutic target for other neuromuscular disorders impaired BMP signaling.
Abstract Topic Groups (Submission Categories)
Topic Group 4 - Motor Neuron Diseases: Spinal Muscular Atrophies: Treatment
PP01.349
Histopathology in Mitochondrial Cardiomyopathies
Prof. Anders Oldfors1, Dr. Malin Nilsson1, Assoc. Prof. Carola Hedberg-Oldfors2
1
Department of Clinical Pathology, Sahlgrenska University Hospital, Gothenburg, Sweden.
2
Department of Clinical Genetics and Genomics, Sahlgrenska University Hospital, Gothenburg, Sweden
Background: Mitochondrial disorders are diseases of the respiratory chain that lead to impaired energy production and tissue dysfunction. Virtually any organ may be affected, resulting in a broad clinical spectrum. Cardiomyopathy has been reported as an isolated and severe manifestation of mitochondrial disease but is rarely suspected based on heart failure alone. Moreover, mitochondrial cardiomyopathies are generally not detected using conventional routine staining of endomyocardial biopsies. Diagnosis of mitochondrial disorders typically relies on genetic, biochemical, and histopathological analyses of skeletal muscle biopsy. In contrast, the assessment of mitochondrial protein expression, enzymatic function, and ultrastructural morphology in myocardial tissue has not been extensively studied, despite its potential utility for identifying isolated mitochondrial cardiomyopathy—an entity that may be underdiagnosed among cardiomyopathies.
Methods: Nine patients with a genetically confirmed diagnosis of mitochondrial cardiomyopathy and available cardiac muscle tissue were included. Genetic defects comprised mtDNA point mutations affecting tRNA and protein-coding genes, single large-scale mtDNA deletions, and nuclear gene mutations in AARS2, PARS2, and AGK (Sengers syndrome). Cardiac material included endomyocardial biopsies, heart explants, and post-mortem specimens. Immunohistochemistry was performed on fresh-frozen and formalin-fixed paraffin-embedded sections to assess the expression of selected subunits of respiratory chain complexes I–V. Enzyme histochemistry was applied to fresh-frozen tissue to evaluate the activity of complexes II and IV. Mitochondrial ultrastructure was examined by electron microscopy. All findings were compared with control myocardial tissue.
Results: Enzyme histochemistry was available in seven cases; six demonstrated complex IV deficiency, while none showed complex II deficiency. Immunohistochemical analysis revealed complex I deficiency in seven of nine cases and complex IV deficiency in three cases. Respiratory chain deficiencies typically displayed a mosaic pattern among cardiomyocytes. Electron microscopy showed nonspecific mitochondrial abnormalities without distinctive ultrastructural features.
Conclusion: Our findings support the concept that myocardial tissue can provide histopathological evidence of mitochondrial cardiomyopathy in the majority of cases. Immunohistochemistry and enzyme histochemistry targeting respiratory chain complexes I and IV appear particularly informative. Further studies are warranted to validate these methods and to determine their diagnostic value in the routine evaluation of endomyocardial biopsies.
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: Cardiac and respiratory issues in neuromuscular diseases.
PP01.350
Blink Reflex Recording as a Diagnostic Tool for Facial Involuntary Movements
Prof. Bum Chun Suh1, Prof. Won Tae Yoon1, Prof. Sang-Beom Kim2
1
Department of Neurology, Kangbuk Samsung Hospital, Sungkyunkwan University School of Medicine, Seoul, Korea, Republic of.
2
Department of Neurology, Kyung Hee University Hospital at Gangdong, Kyung Hee University College of Medicine, Seoul, Korea, Republic of
Background: Facial involuntary movements encompass a broad clinical spectrum, from brief twitching to sequelae of Bell’s palsy, hemifacial spasm, and blepharospasm. These phenomena may arise from peripheral nerve irritation, aberrant reinnervation, or central hyperexcitability. The blink reflex assesses the trigemino-facial pathway and may help clarify underlying mechanisms. This study evaluated the diagnostic contribution of extended blink reflex recordings in patients presenting with facial involuntary movements.
Methods: We conducted a retrospective review of patients who underwent blink reflex testing between January 2024 and July 2025. Beyond the conventional zygomatic branch recording, additional electrodes were placed over the buccal and mentalis muscles to detect signs of abnormal reinnervation. Because these muscles do not typically respond to supraorbital stimulation, any elicited response was interpreted as hyperexcitability.
Results: Eighty-eight patients (mean age 56.1 ± 14.8 years; 65.9% female) were included. Seven had a prior history of facial palsy (ipsilateral 3, contralateral 2, bilateral 2). Abnormal reflex activity was observed in the buccal muscle in 34.1% of patients (ipsilateral R1 33%, ipsilateral R2 31%, contralesional-stimulation R2 25%) and in the mentalis muscle in 30% (24/80). Facial nerve compressive lesions were identified on MRI in 7/79 patients (8.8%), all of whom demonstrated hyperexcitability on blink reflex testing.
Conclusion: Extended blink reflex testing that includes buccal and mentalis recordings may serve as a valuable adjunct in assessing facial involuntary movements. Both additional muscle channels showed similar sensitivity in detecting aberrant reinnervation. Neuroimaging is recommended when hyperexcitable responses are present. Contralesional R2 responses, in particular, may indicate broader facial motor neuronal hyperexcitability rather than localized ephaptic transmission.
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: Electrophysiology
PP01.351
Long Exercise Test: A Simple Diagnostic Tool for the Etiological Evaluation of Rhabdomyolysis
Dr. Emilie Retailleau, Dr. Clément Guémy, Prof. Pascal Laforet, Prof. Guillaume Nicolas
Hopital Raymond Poincaré, Garches, France
Background: Early identification of a genetic etiology of rhabdomyolysis is essential for optimized patient management and prevention of complications. However, establishing this diagnosis remains challenging, owing to the lack of readily available bedside diagnostic tools and the current reliance on increasingly large genetic panels with complex interpretative issues. The long exercise test (LET), performed as part of electrophysiological assessment, reflects muscle responsiveness to excitation and reproduces patient symptoms following sustained muscle contraction. Its diagnostic value has been well established in periodic paralysis and congenital myotonia, and more recently in McArdle disease. However, its role in the diagnostic workup of rhabdomyolysis has not yet been studied.
Methods: We retrospectively analyzed the clinical, biological, genetic, and electrophysiological data of 43 patients with a history of rhabdomyolysis followed in three hospitals of the Assistance Publique–Hôpitaux de Paris (AP-HP). Particular attention was paid to LET results according to the etiology of rhabdomyolysis.
Results: In patients with a single episode of rhabdomyolysis and a negative genetic panel, where an acquired origin was suspected, LET results were normal. In contrast, 72.4% of patients with an identified genetic cause showed significant abnormalities. An early decrement was observed in glycogen storage diseases and VLCAD deficiency, whereas a late decremental response was found in LCHAD deficiency and channelopathies (RYR1, RYR3, CACNA1S). Rather, patients carrying variants of uncertain significance in genes associated with channelopathies showed normal LET results. Patients with CPT2 deficiency showed more heterogeneous abnormalities.
Conclusion: These findings suggest that the long exercise test may provide additional diagnostic arguments in the evaluation of rhabdomyolysis, particularly when metabolic myopathies or channelopathies are suspected. It may also contribute to the interpretation of variants of uncertain significance identified through genetic testing.
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: Electrophysiology
PP01.352
Diagnostic Accuracy of Quantitative Electromyography in Idiopathic Inflammatory Myopathies: A Comparative Analysis with Muscle Biopsy
Dr. Piergiorgio Lasorella1, Dr. Dario Zoppi2, Dr. Anna Russo2, Dr. Francesca Vallefuoco2, Dr. Martina De Maria2, Dr. Rosario Russo2, Dr. Virginia Boemia2, Dr. Anita Marciano E Ortolano3, Assoc. Prof. Teresa Somma3, Asst. Prof. Lucia Ruggiero2
1
Presidio Ospedaliero “Lorenzo Bonomo”, Andria, Italy.
2
Department of Neurosciences, Reproductive Sciences, and Odontostomatology, University of Naples “Federico II”, Naples, Italy.
3
Department of neurosciences, reproductive and Odontostomatological sciences, division of neurosurgery, university of Naples “Federico II”, Naples, Italy
Background: Idiopathic inflammatory myopathies (IIM) are a heterogeneous group of autoimmune neuromuscular disorders characterized by muscle inflammation, progressive weakness and variable patterns of muscle fiber damage. Muscle biopsy remains the gold standard for diagnosis and subclassification, yet it is invasive and may be limited by sampling error and interobserver variability. Quantitative electromyography (qEMG) offers an objective and minimally invasive method to assess motor unit characteristics and muscle membrane instability, potentially providing valuable diagnostic information. However, the relationship between qEMG findings and histopathological features in IIM has not been fully established. Defining the diagnostic performance of qEMG and its correlation with biopsy findings could improve the accuracy and reliability of the neurophysiological evaluation in patients with suspected inflammatory myopathy.
Methods: We retrospectively analyzed 40 patients diagnosed with IIM who underwent both qEMG and muscle biopsy for suspected IIM, and 20 patients with histologically confirmed non-inflammatory myopathy were enrolled as a pathological control group. The qEMG protocol involved the analysis of at least 10 Motor Unit Potentials (MUPs) from the tibialis anterior, rectus femoris and biceps brachii. Key parameters included MUP duration, myopathic pattern (on the Interference Pattern, IP) and the amount of fibrillation potentials (quantified using a point-based scoring system). Histological assessment was performed using a scoring system that incorporated various histopathological features. Diagnostic performance was calculated using Sensitivity (Sn), Specificity (Sp), Positive Predictive Value (PPV), and Negative Predictive Value (NPV).
Results: qEMG identified a myogenic pattern characterized by a significant reduction in MUP duration and increased polyphasia in 35% cases for the biceps brachii, 32.5% for the rectus femoris, and 27.5% for the anterior tibialis. Sensitivity for identifying the myogenic pattern ranged from 27.5% to 35.0% across the muscles tested, while the presence of fibrillation potentials (grades 3 - 4) was notably more frequent in the biceps brachii (45%) and rectus femoris (40%) compared to the anterior tibialis (15.0%). Moreover, the prevalence of fibrillation potentials was significantly higher in the IIM group compared to the pathological control group (TA: 59.5% vs 50%; RF: 67.5% vs 40%; BB: 72.5% vs 35%, respectively). Finally, a strong correlation was observed between the reduction in MUP duration and the severity of muscle fiber atrophy/necrosis, particularly between the rectus femoris and biceps brachii (r = 0.95). Histopathological analysis revealed muscle fiber hypotrophy in 92.5% of cases and necrotic fibers in 90%, with a median biopsy score of 8.5.
Conclusion: Multi-MUP qEMG demonstrates high diagnostic accuracy and strong correlation with histopathological markers of inflammation and fiber damage. While it cannot replace biopsy for definitive subtyping, qEMG provides objective, quantifiable data that significantly enhances the reliability of neurophysiological assessment in the diagnostic workup and differential diagnosis of IIM.
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: Electrophysiology
PP01.353
Electrophysiological Characteristics of Subclinical Diabetic Polyneuropathy in Adolescents with Type 1 Diabetes Mellitus
Prof. Hyung-Soo Lee1, Assoc. Prof. Jungmin So2
1
National Medical Center, Seoul, Korea, Republic of.
2
Ansan Hospital, Korea University, Ansan, Korea, Republic of
Background: Early detection of diabetic polyneuropathy (DPN) is essential for the prevention and management of complications in type 1 diabetes mellitus (T1DM), as it is in type 2 diabetes mellitus (T2DM). However, the electrophysiologic characteristics of DPN in T1DM—particularly during the subclinical stage—remain less well characterized than those in T2DM.
Methods: We retrospectively reviewed the medical records of patients aged 10–19 years with type 1 diabetes mellitus (T1DM) who underwent screening for diabetic polyneuropathy (DPN) in our electrodiagnostic laboratory between January 2022 and February 2024. Patients were excluded if they had a history of hereditary or otherwise known polyneuropathy, conditions associated with an increased risk of polyneuropathy (e.g., specific infections, systemic vasculitis, malignancy, or exposure to chemotherapy), or self-reported sensory or motor symptoms identified through a questionnaire. Motor nerve conduction studies (NCSs) were performed in the median, ulnar, peroneal, and posterior tibial nerves, while sensory NCSs were conducted in the median, ulnar, superficial peroneal, and sural nerves. Diabetic polyneuropathy was diagnosed when two or more independent electrophysiologic parameters exceeded the established reference values.
Results: A total of 107 patients were included in the analysis, with a mean age of 17.02 years (range, 10.5–19.3 years); 51 were male, and 56 were female. Of these, 56 patients were diagnosed with diabetic polyneuropathy (DPN), while 51 did not meet the diagnostic criteria. There was no significant difference in mean age between the DPN and non-DPN groups (17.51 vs. 16.12 years, p = 0.648). Although the duration of diabetes tended to be longer in the DPN group, this difference did not reach statistical significance (8.47 vs. 6.98 years, p = 0.176).
All patients in the DPN group demonstrated prolonged terminal latency in the ulnar nerve. The next most frequent abnormalities were reduced motor conduction velocity in the distal segment of the ulnar nerve (38.1%), followed by reduced motor conduction velocity in the posterior tibial nerve (23.9%). Additional common abnormalities included reduced sensory conduction velocity and decreased sensory nerve action potential amplitudes in the superficial peroneal and sural nerves, reduced motor conduction velocity in the posterior tibial and peroneal nerves, and prolonged terminal latency in the median nerve, each observed in 19.0% of patients. No abnormal nerve conduction study findings were observed in the non-DPN group.
Conclusion: These findings suggest that subclinical DPN in T1DM represents a distinct electrophysiological feature, differing from both overt DPN and subclinical DPN observed in type 2 diabetes mellitus. Therefore, further investigation is warranted to better characterize the electrophysiological features of subclinical diabetic polyneuropathy in this population.
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: Electrophysiology
PP01.354
Clinical and Electrophysiological Aspects About Two Cases of Parsonage Turner Syndrom
Prof. LALA BOUNA SECK1,2, Dr. ZEINA JOUBAILY2, Prof. MAMADOU MOUSTAPHA SARR3, Dr. ARAME GAYE2, Dr. HENRIETTE SENGHOR2, Prof. MARIEME SODA DIOP SENE2,4, Prof. ANNA MODJI BASSE FAYE2,4, Prof. NGOR DIAGNE4,2, Dr. NDEYE FATOU NDOYE SALL2, Dr. MAMADOU SY2
1
GASTON BERGER UNIVERSITY, SAINT LOUIS, Senegal.
2
FANN NATIONAL UNIVERSITY HOSPITAL CENTER, DAKAR, Senegal.
3
THIES UNIVERSITY, THIES, Senegal.
4
CHEIKH ANTA DIOP UNIVERSITY, DAKAR, Senegal
Background: Parsonage Turner is a rare condition. Its electrophysiological exploration informs on the extend of the subclinical nervous lesions, although the exploration of some trunks is rather delicate. Our aim is to describe clinical and elctrophysiological aspects of a rare syndrom.
Methods: We conducted a descriptive study, after collection for one year on the database of the clinical neurophysiology laboratory of the neurological service of the Fann National University Hospital Center of Dakar, of data from patients admitted with a suspicion of Parsonage Turner syndrom. We analyzed under privacy their demographical, clinical and electrophysiological characteristics, using a data collection sheet.
Results: We studied two patients records (A and B), 53 years old female and a 61 years old male. They have been each admitted for motor weakness of the right upper limb, set in a painful context. For patient A, it ocurred 2 months earlier and motor and sensitive neurography were normal on the right median, ulnar and radial nerves. On the right axillary nerve, the amplitude was decreased and distal latency preserved. On detection of the corresponding arm, curves were normal on trapezius, brachial triceps, brachioradialis, flexor carpi ulnaris and first dorsal interosseus muscles, while on deltoidus and brachial biceps, we found fibrillations at rest and neurogenic curves during effort. Pain was persistent at more than two months.
For patient B, motor and sensitive neurography were normal on the right median, ulnar and radial nerves. On the right axillary nerve, the amplitude was decreased and distal latency preserved. On detection of the corresponding arm, curves were normal on trapezius, brachial triceps, brachioradialis, and first dorsal interosseus muscles, while on deltoidus, we found fibrillations at rest, not on the brachial biceps and neurogenic curves during effort on both.
Conclusion: During Parsonage Turner syndrom, we can have clinico-electrical discrepancy. Sensory trunks exploration can be limited during electroneuromyography and this shows the interest of somatosensory evoked potentials to evaluate the functionnal prognosis regarding sensory damages.
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: Electrophysiology
PP01.355
Longitudinal Electrophysiological Changes Predict Clinical Outcome in Chronic Inflammatory Demyelinating Polyradiculoneuropathy
Dr. Sadıka Özdemir, Dr. Ebru Özbezen Kızıltan, Dr. Sezin Alpaydın Baslo, Dr. Aysun Soysal
Bakirkoy Prof. Dr. Mazhar Osman Training and Research Hospital for Mental Health and Neurological Diseases, University of Health Sciences, Istanbul, Turkey, istanbul, Turkey
Background: Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) shows a variable clinical course ranging from sustained remission to relapsing disease. Although electrophysiological studies are essential for diagnosis, their longitudinal evolution and prognostic value in long-term follow-up are not fully established.
Methods: We retrospectively analyzed clinical and electrophysiological data of CIDP patients followed between 2000 and 2024. Patients with available baseline and follow-up nerve conduction studies were included. Functional outcomes were assessed using the Medical Research Council (MRC) sum score, modified Rankin Scale (mRS), and INCAT disability score. Remission, stable disease, and relapse were defined using standardized clinical criteria. Longitudinal electrophysiological changes were analyzed using repeated-measures general linear models, and correlations with clinical outcomes were assessed using appropriate statistical tests.
Results: Forty-two CIDP patients were included, with a median follow-up of 57.6 months (range: 3–240). The overall remission rate was 71.1%. Patients achieving remission showed significant longitudinal improvement in CMAP amplitudes, whereas relapsing patients demonstrated persistent electrophysiological abnormalities. Conduction blocks detected at baseline largely resolved or evolved into temporal dispersion on follow-up, consistent with remyelination under treatment. Importantly, baseline axonal involvement—particularly reduced lower-limb CMAP amplitudes—was significantly associated with higher long-term disability scores (higher INCAT and mRS, lower MRC scores). The presence of diabetes mellitus was associated with a reduced likelihood of remission.
Conclusion: Longitudinal electrophysiological changes in CIDP closely reflect clinical course and functional outcome. While demyelinating features may improve with treatment, baseline axonal damage is a strong predictor of long-term disability. Serial nerve conduction studies provide clinically meaningful prognostic information and should be considered an integral part of long-term disease monitoring in CIDP.
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: Electrophysiology
PP01.356
Systemic Botulism Mimicking Generalized Myasthenia Gravis After Cosmetic Botulinum Neurotoxin Injection: A Case Report
Dr. Tatsuya Abe1, Dr. Kei Okuba2, Dr. Hitoshi Kawasaki2, Dr. Syugo Fujita2, Dr. Yasuo Ito2, Dr. Genko Oyama2, Dr. Tomihiro Imai1
1
Department of Neurology, National Hospital Organization Hakone Hospital, Odawara, Japan.
2
Department of Neurology, Saitama Medical University, Moroyama, Japan
Background: In recent years, off-label intramuscular injections of botulinum neurotoxin type A (BoNT/A) for cosmetic purposes have become widely prevalent in East Asia to improve visual appearance. However, the actual number of patients receiving this treatment is not fully understood, and attention must be paid to undesirable side effects. This paper reports complications associated with these BoNT/A injections.
A 24-year-old woman presented to our hospital approximately 30 days after receiving a total of 800 U of botulinum toxin type A (BoNT/A) for cosmetic purposes via intramuscular injection, complaining of fatigue, dysphagia, inability to raise her arms, and dyspnea. Weakness was observed in the neck flexor muscles, deltoid, trapezius, and quadriceps femoris. Grip strength decreased to approximately 10 kg. Jaw clenching and shoulder muscle weakness fluctuated during the day. Serological tests were negative for anti-AchR and MuSK antibodies, whereas respiratory function test noted a significant reduction in vital capacity.
Methods: We performed electromyography (EMG) 30 days after her symptom was revealed, to evaluate and diagnose the pathology, including nerve conduction study, and repetitive nerve stimulation. Given the possibility of an immune-mediated neuromuscular junction disorder could not be ruled out, IVIG and pyridostigmine were administered as symptomatic therapy. Subsequent outpatient follow-up revealed that improvement in fatigue took approximately 90 days. In addition, follow up EMG was undergone 180 days after symptom onset.
Results: EMG demonstrated a marked decrease in compound muscle action potential (CMAP) amplitude. Low-frequency repetitive nerve stimulation (LF-RNS) in the nasalis and trapezius muscles revealed an abnormal decrement of the CMAP. Furthermore, this decrement of CMAP manifested as “progressive decrement” toward the latter part of the evoked response and was accentuated not only by exercise loading but also by high-frequency repetitive nerve stimulation (HF-RNS). A second EMG study performed 180 days after symptom onset showed increased CMAP amplitude in the nasalis muscle and trapezius muscle compared to the previous examination, and abnormal decrement was improved in LF-RNS at rest. However, accentuated “progressive decrement” was still observed after exercise loading and after HF-RNS. In addition, needle EMG showed short duration and polyphasic MUPs with normal recruitment, and single-fiber EMG revealed a marked increase in jitter in the deltoid muscle.
Conclusion: Clinicians should be aware of the risks associated with intramuscular injections of BoNT/A for cosmetic purposes and should consider the possibility of severe botulism, including respiratory dysfunction, occurring as a side effect. This case highlights important electromyographic features of iatrogenic botulism following BoNT/A intramuscular injection and suggests the potential for persistent symptoms even after recovery. Furthermore, “progressive decrement” following exercise loading or HF-RNS may be useful in identifying iatrogenic presynaptic impairment caused by botulinum toxin type A injection into muscles.
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: Electrophysiology
PP01.357
A Spectrum of Rapid-Onset Motor–Bulbar Disability and Tocilizumab Treatment Outcomes in RANBP2-ANE1
Dr. Ljelja Muaremoska Kanzoska1, Prof. Todor Arsov2
1
University clinic for pediatric diseases, Skopje, North Macedonia.
2
Faculty of Medical Sciences, University “Goce Delchev”, Shtip, North Macedonia
Background: RANBP2-associated acute necrotizing encephalopathy type 1 (ANE1) is an infection-triggered neuroinflammatory disorder caused by monoallelic pathogenic RANBP2 variants, typically inherited in an autosomal-dominant pattern with incomplete penetrance and strikingly variable intrafamilial clinical expressivity. While often labeled as “encephalopathy,” survivors commonly experience abrupt, high-burden acquired motor–bulbar dysfunction, with variable respiratory involvement, translating into substantial rehabilitation needs. We aimed to define the spectrum of motor–bulbar disability and rehabilitation burden within a single RANBP2-ANE1 pedigree carrying the shared pathogenic variant c.1754C>T (p.Thr585Met), and to report IL-6 receptor blockade (tocilizumab) as escalation therapy in one family member.
Methods: Retrospective descriptive review of six affected children from one family carrying the familial RANBP2 variant c.1754C>T (p.Thr585Met), presenting with clinical and imaging features consistent with ANE1. Analysed variables included infection trigger, acute neurological phenotype, supportive and immunomodulatory interventions, and functional outcomes emphasizing mobility, bulbar impairment (swallowing/communication), and respiratory dependence.
Results: Clinical expression included a spectrum from fulminant disease with fatal outcomes to survival with severe persistent disability, but also asymptomatic (non-penetrant) cases. Functional trajectories included early death after rapid neurological decline; survival with major cognitive and motor sequelae; severe permanent spastic motor disability requiring long-term rehabilitation; and an extreme respiratory-care trajectory with progression to chronic unresponsive state and prolonged mechanical ventilation following influenza-triggered ANE1. Collectively, these outcomes delineate a neuromuscular spectrum from loss of independent ambulation, contracture risk, dysphagia, and respiratory dependence - despite differing acute presentations, illustrating variable expressivity within a single family.
The most recent (sixth) case is a 3.5-year-old girl from the extended family who developed severe acute encephalopathy with prominent unilateral pyramidal deficit after laboratory-confirmed influenza A infection, with neuroimaging typical for ANE and molecular confirmation of the familial RANBP2 variant. She received antiviral therapy and standard immunomodulation with escalation to single-dose tocilizumab (12 mg/kg). After IL-6 blockade, rapid improvement in consciousness and interaction was observed. She is currently on multidisciplinary rehabilitation program with standard physiotherapy and specialized dysphagia-focused therapy.
Conclusion: RANBP2 associated ANE1 can manifest as a wide spectrum of rapid-onset motor–bulbar symptoms with variable respiratory involvement and major disability burden. Early recognition (family history and prompt imaging) and rapid molecular confirmation allow early treatment (immunomodulatory) escalation. In that context, IL-6 receptor blockade escalation was associated with meaningful early neurological recovery allowing early rehabilitation. Presymptomatic testing of family members enables anticipatory counseling and prompt clinical management with treatment escalation.
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: Genetics in Neuromuscular Diseases including Biochemical/Molecular Techniques and next generation sequencing.
Images or Table (Optional)
PP01.358
Personalized Medicine From Birth: Nationwide Newborn Screening for Spinal Muscular Atrophy in Serbia
Asst. Prof. Milos Brkusanin, Mr. Nemanja Garai, Mr. Nemanja Radovanović, Asst. Prof. Jovan Pešović, Dr. Lana Radenković, Mrs Suzana Matijašević Joković, Miss Vanja Obadović, Miss Sanja Madić, Miss Ksenija Obadović, Prof. Goran Brajušković, Prof. Dušanka Savić Pavićević
University of Belgrade - Faculty of Biology, Centre for Human Molecular Genetics, Belgrade, Serbia
Background: Spinal muscular atrophy (SMA) is among the most severe inherited neuromuscular disorders and, in the absence of timely intervention, a leading genetic cause of infant mortality. The availability of disease-modifying therapies has fundamentally changed the natural history of SMA, with the greatest clinical benefit achieved when treatment is initiated presymptomatically. This has positioned newborn screening (NBS) for SMA as both a medical necessity and a public health priority.
Methods: In 2021, Serbia initiated its first genetic newborn screening program for SMA, centralized at the Faculty of Biology, University of Belgrade, a national reference center for SMA diagnostics and research since 1997. During a 17-month pilot phase, 12,000 newborns from two maternity hospitals were screened using dried blood spots and real-time PCR to detect SMN1 exon 7 absence, with confirmatory MLPA analysis and SMN2 copy number determination.
Results: Following the successful pilot, a nationwide SMA NBS program was launched on September 15, 2023. The program currently covers 52 public and 6 private maternity hospitals across Serbia. By January 16, 2025, a total of 141,101 newborns had been screened, identifying 22 infants with SMA. Of these, 20 received immediate disease-modifying therapy: 8 infants with 2 SMN2 copies, 8 with 3 copies, and 4 with 4 copies. Two infants with 5 SMN2 copies are under active clinical surveillance. To date, treated infants remain predominantly asymptomatic.
Clinical decision-making is coordinated by a national multidisciplinary Expert SMA Commission, ensuring individualized treatment strategies based on genetic findings, clinical assessment, and laboratory parameters. The program has enabled the first reliable estimate of SMA incidence in Serbia (1:6,414 live births) and demonstrates the feasibility and clinical impact of presymptomatic diagnosis within a middle-income healthcare system.
Conclusion: The Serbian experience highlights how structured newborn screening workflows, centralized diagnostics, and personalized therapeutic approaches can transform SMA from a devastating, life-limiting disease into a manageable condition. This program serves as a practical model for integrating precision medicine into national public health systems through close collaboration between academia, patient organizations, industry partners, and governmental institutions.
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: Genetics in Neuromuscular Diseases including Biochemical/Molecular Techniques and next generation sequencing.
PP01.359
Advancing Clinical Trials in Myotonic Dystrophy Type 1: Refining Radiological, Clinical and Patient-Reported Outcome Measures
Miss Louise Iterbeke1, Dr. Lotte Huysmans2,3, Dr. Kobe Bamps4,5, Dr. Ronald Peeters6, Ms. Veerle Goosens6, Prof. Frederik Maes2,3, Prof. Patrick Dupont7, Prof. Kristl Claeys1,8
1
Laboratory for Muscle Diseases and Neuropathies, Department of Neurosciences, KU Leuven, and Leuven Brain Institute (LBI) and Leuven Institute for Rare Diseases (Leuven.IRD), Leuven, Belgium.
2
Department ESAT/PSI, KU Leuven, Leuven, Belgium.
3
Medical Imaging Research Center, University Hospitals Leuven, Leuven, Belgium.
4
Department of Cardiovascular Sciences, KU Leuven, Leuven, Belgium.
5
Department of Cardiology, University Hospitals Leuven, Leuven, Belgium.
6
Department of Radiology, University Hospitals Leuven, Leuven, Belgium.
7
Laboratory for Cognitive Neurology, Department of Neurosciences, KU Leuven, and Leuven Brain Institute (LBI), Leuven, Belgium.
8
Department of Neurology, University Hospitals Leuven, Leuven, Belgium
Background: Adult-onset Myotonic Dystrophy type 1 (DM1) is a progressive multisystemic disorder primarily characterized by distal muscle weakness and myotonia. As disease-modifying therapies advance, clinical trials require reliable, non-invasive and sensitive outcome measures. In this longitudinal study, we delineated the natural history of adult-onset DM1 and identified robust outcome measures for future clinical trials by assessing quantitative muscle MRI, clinical, and patient-reported outcome measures in patients with adult-onset DM1.
Methods: Thirty-three patients with adult-onset DM1 and 33 age- and sex-matched healthy controls were assessed at baseline, 12 months, 18 months and 24 months. Quantitative muscle MRI of the lower limbs included 6-point Dixon imaging to quantify proton density fat fraction (PDFF %) and water T2 (T2H2O) to evaluate disease activity in the muscles. A convolutional neural network enabled automated 3D segmentation of 18 proximal and 10 distal leg muscles. Clinical outcome measures included Motor Function Measure-32 (MFM32), 6-Minute Walk Distance (6MWD), 10-Meter Walk Test (10MWT), 30-Second Sit-to-Stand (30SSS), 9-Hole Peg Test and hand opening time (myotonia). Strength was measured by Medical Research Council Sum Score (MRCSS), handgrip, key pinch and tip pinch strength and peak cough flow. Patient-reported outcomes (PROM) included DM1-ActivC, Brief Pain Inventory (BPI), Individualized Neuromuscular Quality of Life (INQoL), and Fatigue and Daytime Sleepiness Scale (FDSS).
Results: Three patients completed the baseline visit only for medical reasons and were excluded from further analyses. Baseline MRI revealed significant higher PDFF (%) in patients with DM1 compared to their matched controls (Distal leg: 31.8% vs 5.9%, p<0.001; Proximal leg: 13.0% vs 7.6%, p<0.001). In patients, muscle fat replacement followed a distinct pattern. Early and severe fat replacement in the posterior distal leg (soleus, gastrocnemius), sparing of the popliteus and tibialis posterior, and progressive involvement of the anterolateral distal and proximal leg. Longitudinal analysis showed significant increases in PDFF (%) across 9/10 distal leg muscles and 15/18 proximal leg muscles at 12, 18, and 24 months. The distal leg muscles demonstrated the largest 24-month mean change (+3.5%, p<0.001), with the flexor hallucis longus muscle showing the highest effect size (SRM = 1.7). Proximal leg muscles showed a smaller but significant progression (+1.5% at 24 months, p<0.001). Conversely, T2H2O remained stable over time. Patients with adult-onset DM1 scored significantly worse on all clinical outcome measures compared to their controls, and demonstrated progressive, significant deterioration in motor function (MFM32) and muscle strength (MRC sum score), with significant declines detected as early as 6 months. DM1-activC significant deteriorated at 12, 18, and 24 months (-5.6, p<0.01; -6.2, p<0.001; -10.3, p<0.001). The total quality of Life, measured by the INQoL, showed significant decline at 12 and 18 months (5.4, p<0.05; 6.9, p<0.01). Leg PDFF (%) correlated significantly with functional measures (10MWT, 6MWD, MFM32, 30SSS, MRC sum score) but was independent of CTG repeat length.
Conclusion: Muscle MRI fat fraction, MFM32, MRC Sum Score, DM1-ActivC and INQoL emerged as sensitive and robust outcome measures to monitor the disease progression in adult-onset DM1. These measures are well-suited for implementation in future clinical trials to show therapeutic effect.
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: MRI
PP01.360
Responsiveness to Change of Quantitative Whole-Body MRI in FSHD: Results From ReDUX4 OLE
Dr. Per Widholm, Dr. Markus Karlsson, Dr. Mary Folz
AMRA Medical AB, Linköping, Sweden
Background: To facilitate detection of treatment effects in heterogenous and slowly progressing muscle diseases, there is a need for more objective and responsive biomarkers that can accurately describe the small changes in muscle composition anticipated during the timespan of a clinical trial. Changes that often are too small to be captured by functional tests such as time-up-and-go (TUG). Utilizing whole-body MRI, a framework consisting of standardized scan-protocols and image analysis pipeline including new composite biomarkers has previously been developed. By utilizing the innate high reproducibility of MRI together with an enrichment strategy where muscles with a high probability of near-term progression is identified and combined into composite scores, the aim is to provide more responsive biomarkers that can be used as endpoints in multi-site clinical trials in neuromuscular disorders. These biomarkers were included as secondary endpoints in a 48-week phase 2b study of Losmapimod (ReDUX4) in FSHD with an optional open label extension (OLE) study.
Methods: Whole-body quantitative MRI were acquired using fat-water separated imaging (Dixon). Lean Muscle Volume (LMV), Muscle Fat Fraction (MFF), and Muscle Fat Infiltration (MFI) were quantified in 36 muscles covering arms, shoulder girdles, thorax, and legs. The muscles were divided in three different categories (A, B, C) reflecting the probability of near-term progression based on the baseline fat content where B-muscles are anticipated to have the highest progression. Whole-body composite scores for LMV, MFF, MFI were generated for both A- and B-muscles. The mean change from baseline at weeks 48, 96, and 120 were calculated using paired t-tests and reported as percent change for LMV and absolute change in percentage points (pp) for MFF and MFI. The responsiveness of the biomarkers were assessed using standardized response mean (SRM).
Results: The change from baseline in B-muscle whole-body composite LMV/MFF/MFI (mean±SE [SRM]) were -5.37%±0.60% [1.1], p<0.001 / 1.38pp±0.25pp [0.67], p<0.001 / 0.27pp±0.09pp [0.38], p<0.003 at week 48; -8.85%±0.80% [1.34 ], p<0.001 / 3.11pp±0.42pp [0.89], p<0.001 / 0.80pp±0.13pp [0.73], p<0.001 at week 96; and -10.93%±0.94% [1.46], p<0.001 / 4.57pp±0.55pp [1.04], p<0.001 / 1.13pp±0.18pp [0.8], p<0.001 at week 120. The corresponding changes observed in A-muscles were -0.84%±0.49% [0.21], p=0.09 / 0.06pp±0.14pp [0.05], p=0.6 / 0.07pp±0.06pp [0.14], p=0.2; -2.23%±0.55% [0.48], p<0.001 / 0.57pp±0.16pp [0.41], p<0.001 / 0.37pp±0.07pp [0.63], p<0.001; -3.27%±0.62% [0.66], p<0.001 / 0.92pp±0.19pp [0.6], p<0.001 / 0.50pp±0.08pp [0.82], p<0.001.
Conclusion: Describing progression in heterogenous and slowly progressing diseases such as neuromuscular disorders is challenging and increases both cost and complexity in drug development. By utilizing whole-body MRI to identify muscles with an increased likelihood of near-term progression, the responsiveness of the biomarkers can increase significantly, potentially enabling earlier detection of disease progression or treatment response.
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: MRI
PP01.361
Expanding Spectrum of Molecular Landscape of Nemaline Rod Myopathy
Dr. MEHAR SHARMA, Dr. Aishwarya Dhall, Dr. vaishali suri
AIIMS, delhi, India
Background: Congenital myopathies are rare muscle diseases, predominantly present at birth as generalised hypotonia, floppy infants and non-progressive muscle weakness. This is a heterogeneous group, and many gene variations are described.
Methods: During a period of 11 years, 2997 muscle biopsies were received in our tertiary referral centre for various reasons. Various modalities like histochemistry, immunohistochemistry, ultrastructural examination and NGS was performed for diagnosis.
Results: Based on ultrastructural examination 8 cases were diagnosed as nemaline myopathy comprising of less than 1% of all muscle diseases. Age ranged from 5months to 60 years ( mean age 19 years) and onset of symptoms is variable. NGS show variations in ACTA1 gene 1 case, Nebulin 2 cses, KLHL 1 in 1 patient, TTN in 2 cases, KBTBT in 1 case and KLHL 1 patient
Conclusion: Morphological changes in Nemaline rod myopathy are consistently uniform but molecular variations are heterogenous. Predominantly present in early life but can present at any age. This molecular spectrum of nemaline rod myopathy is expanding.
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: Neuromuscular Pathology: Muscle and Nerve Biopsy
PP01.362
Increase of Number and Genic Type of eccDNA in Spinal Cord of ALS Murine Model
Dr. Daniela Gerovska1, Dr. Julie B Noer2, Dr. Yating Qin2, Dr. Quratul Ain3, Dr. Donjetë Januzi3, Prof. Matthias Schwab3, Dr. Otto W Witte3, Dr. Marcos J Araúzo-Bravo1,4, Dr. Alexandra Kretz3
1
Biogipuzkoa Health Research Institute, San Sebastian, Spain.
2
University of Copenhagen, Copenhagen, Denmark.
3
Jena University Hospital, Jena, Germany.
4
Basque Foundation for Science, IKERBASQUE, Bilbao, Spain
Background: ALS is a progressive neurodegenerative disorder characterized by motor neuron loss and pronounced genome instability. Mutations in genes such as SOD1 contribute to defects in DNA damage response and repair, highlighting chromosomal vulnerability as a central feature of ALS pathophysiology. Extrachromosomal circular DNAs (eccDNAs) are circular DNA molecules derived from chromosomal sequences that can arise from genome instability, yet their abundance and genic composition in ALS-affected tissues remain largely unexplored. We investigated the number and genic type of eccDNAs in the spinal cord of a genotoxic stress–related murine ALS model expressing human SOD1G93A.
Methods: Cervical spinal cord tissue from nine symptomatic hSOD1
G93A
mice and ten non-transgenic controls was processed for eccDNA isolation using rolling circle amplification combined with linear DNA digestion. Purified eccDNAs were sequenced using high-throughput short-read technology, and mapped eccDNAs were analyzed with the DifCir computational approach, which quantifies the production of eccDNAs per gene (DPpGCs). EccDNA abundance, length distribution, and genic origin were compared between ALS and control tissues. To assess functional relevance, eccDNA-producing genes were integrated with proteomic profiles from the same spinal cord samples and cross-referenced with ALS risk genes from genome-wide association studies (GWAS).
Results: Analysis revealed a six-fold increase in the number of genomically distinct eccDNAs in ALS spinal cord relative to controls, demonstrating enhanced circular DNA formation under disease-associated genotoxic stress. Beyond this quantitative increase, ALS tissue exhibited a disease-specific shift in eccDNA composition. We identified 225 upregulated DPpGCs (up-DPpGCs), defined as genes generating more eccDNAs from distinct genomic regions in ALS than in controls. Notably, the top six up-DPpGCs showed at least 89% recurrence across samples, indicating high reproducibility of the genic eccDNA signature. Among the 225 upregulated eccDNA-producing genes in ALS spinal cord, 83 (37%) correspond to neural coding genes previously identified as recurrent DNA double-strand break loci, indicating that eccDNAs arise disproportionately from genome-fragile regions in the hSOD1G93A model.
Integration with proteomic data identified 42 corresponding differentially expressed proteins (DEPs), 19 of which were associated with ALS risk in GWAS. Functional annotation revealed that up-DPpGCs and their protein counterparts predominantly contribute to neuron-specific functions. Gene set enrichment analysis indicated significant overrepresentation of the adenylate cyclase–modulating G protein pathway, suggesting that changes in genic eccDNA production intersect with neuronal signaling networks relevant to ALS pathogenesis.
Conclusion: Our findings demonstrate both a substantial increase in eccDNA number and a distinct alteration in genic composition in the spinal cord of an ALS murine model. The identified up-DPpGCs correspond with proteomic changes and known ALS risk genes, providing an integrated view of genome instability and its molecular consequences. This study establishes eccDNAs as a previously underappreciated molecular feature of ALS and highlights their potential as biomarkers of genome stress, offering a new dimension for understanding neurodegenerative mechanisms and exploring therapeutic avenues.
Reference: Gerovska et al.A distinct circular DNA profile intersects with proteome changes in the genotoxic stress-related hSOD1G93A model of ALS. Cell Biosci. 2023 ;13(1):170. doi: 10.1186/s13578-023-01116-1.
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: Other Biomarkers
PP01.363
Exploring MSOT as a Non-Invasive Biomarker for Myotonic Dystrophy Type 2
Ms. Jin Wang1,2, Dr. Stefanie Meyer1, Dr. Janek Gröhl3, Ms. Luca Eisenblätter1, Ms. Paula Schwabe1, Ms. Hannah Bruex1, Ms. Alica Stobbe1, Ms. Meret Dreyer1, Ms. Katharina Kief1, Prof. Mathias Bähr1, Dr. Stefanie Glaubitz1, Dr. Jana Zschüntzsch1
1
University Medical Center Goettingen, Clinic for Neurology, Robert-Koch-Straße 40, 37075 Goettingen, Goettingen, Germany.
2
University Medical Center Goettingen, Heart & Brain Center Goettingen, Robert-Koch-Straße 42, 37075 Goettingen, Goettingen, Germany.
3
ENI-G, a Joint Initiative of the University Medical Center Göttingen and the Max Planck Institute for Multidisciplinary Sciences, Göttingen, Germany, Goettingen, Germany
Background: Myotonic dystrophy type 2 (DM2) is a multisystem neuromuscular disorder characterized by progressive muscle involvement with substantial inter-individual heterogeneity. The emergence of targeted therapies highlights the urgent need for objective, non-invasive biomarkers to support disease monitoring and therapeutic evaluation. Multispectral optoacoustic tomography (MSOT) is a hybrid imaging modality combining laser excitation with ultrasound detection to visualize endogenous chromophores such as collagen, hemoglobin, and lipids, thereby enabling non-invasive quantitative assessment of muscle microstructure without the need for contrast agents.
Methods: In this prospective, cross-sectional study, we enrolled eight genetically confirmed DM2 patients and six age- and sex-matched healthy volunteers (HV). Recruitment of additional healthy controls is ongoing. MSOT imaging of selected skeletal muscles was performed using the Acuity Echo system (iThera Medical GmbH, Munich, Germany). Imaging parameters related to tissue composition were extracted as candidate biomarkers. Clinical evaluation included standardized strength testing (Medical Research Council scale, handgrip dynamometry), functional performance measures (6-minute walk test (6MWT), timed up and go test (TUGT)), and patient-reported outcomes (ACTIVLIM, EQ-5D, SSQ). Analyses covering between-group comparisons and exploratory correlations between MSOT-derived metrics and clinical severity are ongoing.
Results: The interim analyses includes eight DM2 patients and six HV. MSOT imaging was successfully completed and well tolerated in all participants. Participants were predominantly female in both cohorts, DM2=6/8 (75.0%) vs HV=5/6 (83.3%) with a mean age of 59.0±16.5 years in DM2 and 53.0±13.1 years in HV. In the preliminary results, BMI differed between groups, with an average BMI of 29.0±7.6 in DM2 compared to 26.1±4.1kg/m² in HV. DM2 patients showed impaired mobility compared to HV (6MWT: DM2= 438.5±116.7m, HV=548.3±71.3 m; TUGT: DM2=8.6±4.0s, HV=5.6±0.5s). Quantitative analysis of MSOT-derived parameters is ongoing, and detailed results will be presented at the meeting.
Conclusion: This study represents the first application of MSOT imaging in DM2. MSOT offers a feasible, non-invasive method for assessing muscle involvement and may provide complementary insights to established clinical and functional outcome measures.
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: Other Biomarkers
PP01.364
Orthogonal Methods Demonstrate Pharmacodynamic Responses in Duchenne Muscular Dystrophy Patients Treated with BMN 351
Mr. Kevin Larimore1, Ms. Annie Greenslade1, Ms. Ara Yu1, Mr. Schwend Santos1, Mr. Muhammed Hamir1, Mrs Kristen Jahr1, Mrs Lucy Crockett1, Mr. Jeremy Van Vleet1, Mr. Bert Blank1, Mrs Huiyu Zhou1, Mrs Yulan Qi1, Mr. Joshua Henshaw1, Mr. Andrew Melton1, Ms. Kristin Obrochta-Moss2
1
BioMarin Pharmaceutical Inc., Novato, CA, United States.
2
BioMarin Pharmaceutical Inc., Novato, United States
Background: BMN 351 is an antisense oligonucleotide therapeutic designed to exclude DMD exon 51 during mRNA splicing in muscle and induce synthesis of functional, near full-length dystrophin protein in amenable patients with Duchenne muscular dystrophy (DMD). 351-201 (NCT06280209) is an ongoing Phase 1/2 clinical trial of BMN 351 in boys with DMD. Three key pharmacodynamic biomarkers were measured in participant muscle biopsy tissue to evaluate early patient responses to treatment.
Methods: DMD mRNA exon 51 skipping was measured by quantitative droplet digital PCR (ddPCR), while dystrophin protein expression was measured by quantitative immune-affinity ultra-performance liquid chromatography tandem mass-spectrometry (IA-UPLC-MS/MS) and semi-quantitative western blot. DMD mRNA exon 51 skipping provides a proximal measure of target engagement and pharmacodynamic activity. IA-UPLC-MS/MS data provides quantitative measurement of near full-length dystrophin protein expression resulting from DMD mRNA exon 51 skipping, while western blot analysis confirms IA-UPLC-MS/MS measurements and provides qualitative analysis of the size and diversity of the dystrophin protein isoforms present before and after treatment. The IA-UPLC-MS/MS method includes absolute quantitation of both dystrophin and alpha-actinin 2 proteins. The latter was used to normalize dystrophin data based on myofiber content in each biopsy. The absolute dystrophin and alpha-actinin 2 IA-UPLC-MS/MS measurements from each sample were used to calculate % of normal myofiber-normalized dystrophin concentrations relative to the median of a panel of 19 normal human muscle samples measured in triplicate, with data reported as % of normal.
Results: The ddPCR data demonstrated dose-dependent increases in muscle DMD mRNA exon 51 skipping that correlated with dose-dependent increases in near full-length dystrophin protein measured by both IA-UPLC-MS/MS and western blot.
Conclusion: Together, the data from these orthogonal methods provide a robust data set demonstrating early patient responses and support the potential for clinical benefit in patients treated with BMN 351.
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: Other Biomarkers
PP01.365
Muscle Mass and Strength Changes in Patients With Peripheral Neuropathy and Central Nervous System Disorders
Dr. Sung-Ju Hsueh1, Dr. Kai Chieh Chang2
1
National Taiwan University Hospital Bei-Hu Branch, Taipei, Taiwan.
2
National Taiwan University Hospital Yunlin Branch, Yunlin County, Taiwan
Background: Studies had shown that both peripheral neuropathy (PN) and central nervous system (CNS) disorders, such as Parkinson’s disease (PD), could results in the loss of muscle mass and strength in patients. These changes could be measured using bioelectrical impendance analysis (BIA) and hand grip as markers, respectively. However, comparison of muscle mass changes and strength among patients with different diseases were seldom reported. We investigate whether different etiologies may result in different pattern of muscle mass and strength changes in patients with neurological disorders.
Methods: Patients with PN and PD were enrolled from a general neurology clinic, in which we routinely recorded grip strength using a hand dynamometer and performed body composition analysis using an Accuniq BC380 multi-frequency BIA device (Selvas Healthcare, Daejeon, Republic of Korea) at the patients’ first encounter. Clinically, PD was diagnosed based on the Movement Disorder Society diagnosis criteria, while PN was diagnosed based on neurological examinations by neurologists and electrophysiological studies with results compatible with peripheral nervous system dysfunction. Appendicular skeletal muscle mass (ASM) was measured using BIA and was further adjusted with height according to the Asian Working Group for Sarcopenia (AWGS) 2019 consensus (height-adjusted muscle mass, HAMM) to better represent the patients’ state of muscle health. Patients not able to stand without support, with significant deformities, with history of cerebrovascular diseases or traumatic brain injury resulting in significant functional impairment, or declined to be evaluated were excluded. After the clinical examinations, the patients received medical treatment and exercise instructions based on the etiology and the attending neurologist’s clinical judgements. Follow-up evaluations were performed if deemed to be necessary by the neurologist.
Results: 18 patients with PN and 12 patients with PD were included for analysis. No significant difference in age and gender distribution was noted among the two groups. Six of the PN patients had diabetic neuropathy. 6 patients with PN and 2 patients with PD fulfill the criteria for decreased muscle mass based on the AWGS 2019 consensus according to the HAMM measured by BIA. Significant correlation with grasp strength was observed between the patients’ age (p=0.012) and HAMM (p=0.036), while there was no significant correlation between the patients’ age and HAMM. No significant difference of HAMM and grip strength were observed between PN and PD patients, and between patients with diabetic neuropathy and non-diabetic PN. Among the 10 PN and 11 PD patients who were under medical treatment and received follow-up evaluations more than 5 months after the initial evaluation, the average monthly changes of HAMM were not significant different between the two groups of patients.
Conclusion: Patients with either PN or PD could have changes in muscle strength measured by grip strength related to loss of ASM, and the pattern of changes are similar. Since both the loss of muscle mass and decreased strength may result in disability in either PN or CNS disorders, routine evaluation of these parameters in patients with various neurological disorders should be considered to better understand the factors that may contribute to the disability of patients.
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: Other Biomarkers
PP01.366
STEP Study Design: Validating the Syde® Device to Measure Motor Function in Late-Onset Pompe Disease
Dr. Andrew Oldham1, Dr. Tan P. Pham2, Ms. Dina Elsouda2, Ms. Eugénie Kluczka3, Dr. Jader Baima4, Mr. Ian Keyzor5, Dr. Mark Roberts1
1
Salford Royal NHS Foundation Trust, Salford, United Kingdom.
2
Astellas Pharma, Inc., Northbrook, IL, United States.
3
Sysnav, Vernon, France.
4
Astellas Gene Therapies, San Francisco, CA, United States.
5
Astellas Pharma Europe Ltd., Addlestone, United Kingdom
Background: In Pompe disease, acid alpha-glucosidase deficiency leads to lysosomal glycogen accumulation, causing progressive neuromuscular degeneration over time. Despite improvements in standard of care (SOC) with enzyme-replacement therapy (ERT), some individuals experience continued muscular and respiratory decline. The 6-minute walk test (6MWT) is the current gold standard endpoint in trials of ambulatory individuals with neuromuscular disorders; however, this measure is challenging for those with significant motor impairments and may not accurately reflect treatment effect. The stride velocity 95th centile (SV95C) is a clinical outcome assessment (COA) endpoint that quantifies an individual’s fastest stride speed over a defined period. SV95C is captured using a wearable device, such as the Syde® device (Sysnav, Vernon, France), enabling continuous monitoring of daily movement. SV95C is the first wearable digital COA qualified by the European Medicines Agency as a primary endpoint for use in trials of neuromuscular diseases, as an alternative to 6MWT (Servais et al, 2023). STEP, a non-interventional, longitudinal, observational pilot study aims to assess the validity and reliability of SV95C (captured using the Syde® device) in measuring physical and motor function in participants with late-onset Pompe disease (LOPD).
Methods: Approximately 22 adult ambulant participants (≥18 to <65 years) with genetically diagnosed LOPD treated with ERT for ≥2 years will be enrolled from the UK. Eligible participants will continue to receive ERT as usual. Participants will be followed for approximately 12 weeks, with visits scheduled relative to participant’s ERT infusion schedule (Figure). Functional performance tests and clinician-reported outcomes (ClinROs) will be assessed on-site (Visits 1, 4); patient-reported outcomes (PROs) will be assessed on-site (Visits 1, 4) and at-home (Visits 2, 3). Participants will be required to wear the Syde® device daily while awake during weeks 1–4 and 9–12. PROs and ClinROs will be assessed using generic and Pompe disease-specific questionnaires. The relationship between SV95C and 6MWT (and other functional performance tests, PROs, and ClinROs) will be tested using regression analyses, correlation and interclass correlation analyses, and Bland-Altman tests.
Results: The primary outcome is to evaluate the relationship, including equivalence and correlation, between SV95C and 6MWT. Secondary/exploratory outcomes include evaluating the relationship between SV95C and other functional performance tests (100-meter walk timed test, 4-stair climb), PROs and ClinROs, assessing test-retest reliability, validity (including convergent and known-group validity) and responsiveness of SV95C using functional performance tests and PROs and ClinROs, describing participants’ responses for PROs and ClinROs, assessing changes in fatigue and physical function between intervals of ERT by comparing SV95C and PROs by number of days since last ERT infusion, describing participants’ baseline demographic and clinical characteristics, and evaluating adherence to and usability of the Syde® device.
Conclusion: This study will assess whether SV95C, as measured by the Syde® device, is a valid and reliable endpoint for measuring physical and motor function in participants with ambulant LOPD. The findings may help reduce trial participation burden and expand options for evaluating the impact of therapies for individuals with LOPD.
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: Outcome measures and clinimetry
Images or Table (Optional)
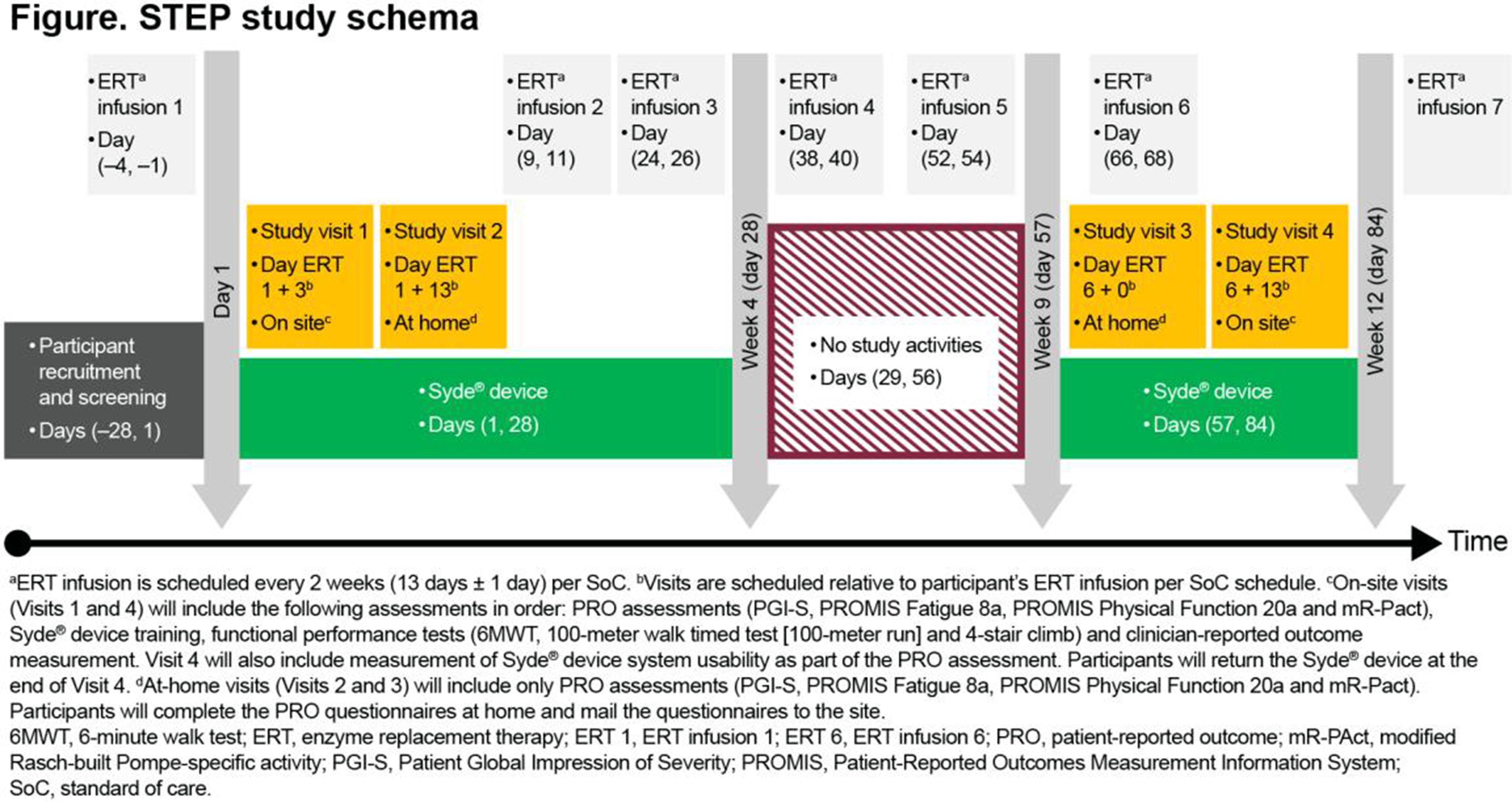
PP01.367
LEOPARD-DMD: Interval Results from a Duchenne Muscular Dystrophy Natural History Study
Dr. Chad Heatwole1, Ms. Preshetha Kanagaiah1, Mrs Jennifer Weinstein1, Ms. Christina Shupe1, Ms. Charlotte Engebrecht1, Ms. Judith Monickaraj1, Ms. Anika Varma1, Mr. Spencer Rosero2, Ms. Charlotte Irwin1, Ms. Karnavaal Al-Rubayie1, Ms. Alicia Brockt1, Mrs Peggy Auinger1, Mrs Debra Guntrum1, Dr. Emma Ciafaloni1
1
University of Rochester, Rochester, United States.
2
University of Utah, Salt Lake City, United States
Background: In preparation for upcoming therapeutic trials with patients with Duchenne muscular dystrophy (DMD), there is a need for sensitive outcome measures that can detect clinically relevant changes in disease burden overtime. To satisfy this need we previously developed and validated the regulatory-grade caregiver-reported DMDCR-HI as a potential outcome to serially quantify patient disease burden. The DMDCR-HI can be used for ambulatory or non-ambulatory patients and consists of individual subscales that measure the symptomatic domains most important to patients, as reported by the caregiver. Additional information is needed regarding the performance metrics of the DMDCR-HI in the context of a longitudinal clinical study.
Methods: We are in the process of conducting a 24-month, remote, longitudinal study with caregivers and individuals with DMD. Caregivers of individuals with DMD are completing the DMDCR-HI, the Proxy-Reported PedsQL (PedsQL), an outcome preference survey, and a global impression of change questionnaire at 6-month intervals. As part of this study, we are evaluating: 1) the ability of the DMDCR-HI to measure disease progression in DMD; 2) the survey preference for the DMDCR-HI compared to the PedsQL; and, 3) the meaningful score difference of the DMDCR-HI and its subscales.
Results: 92 DMD caregivers (mean child age: 12.3 years (range: 2 to 21)) enrolled at baseline. After 12 months, the DMDCR-HI total score demonstrated a 5.15 point change from baseline (p-value:0.0068) in addition to detecting disease progression in the following subscales: shoulder and arm function (10.46 points, p-value:0.0002), gastrointestinal health (9.48 points, p-value:0.0115), heart health (7.33 points, p-value:0.0256), activity participation (7.27 points, p-value:0.0085), mobility (6.94 points, p-value:0.0157), ambulation (6.83 points, p-value:0.0134), finger and hand function (6.29, p-value:0.0131), breathing (6.03 points, p-value:0.0183), and core and truncal strength (5.20 points, p-value:0.05). The PedsQL did not demonstrate any significant change over 12 months. A total of 65% of caregivers preferred the DMDCR-HI over the PedQL (p-value:0.03). Figure 1 shows 12-month data regarding caregiver preferences for the DMDCR-HI vs. the PedsQL. To date, all 12-month data has been collected with participants currently completing their 18 month and final 24-month assessments. Meaningful score difference data based on final data collection is forthcoming.
Conclusion: The DMDCR-HI is a validated outcome measure that is able to quantify point-in-time multifactorial disease burden in patients with DMD. Longitudinally, the DMDCR-HI is able to detect disease progression over a 12-month period and is preferred by users. Data from our study will provide insight into what a meaningful change in DMDCR-HI score is, and will be useful when interpreting changes in DMDCR-HI scores during future therapeutic and regulatory trials.
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: Outcome measures and clinimetry
Images or Table (Optional)
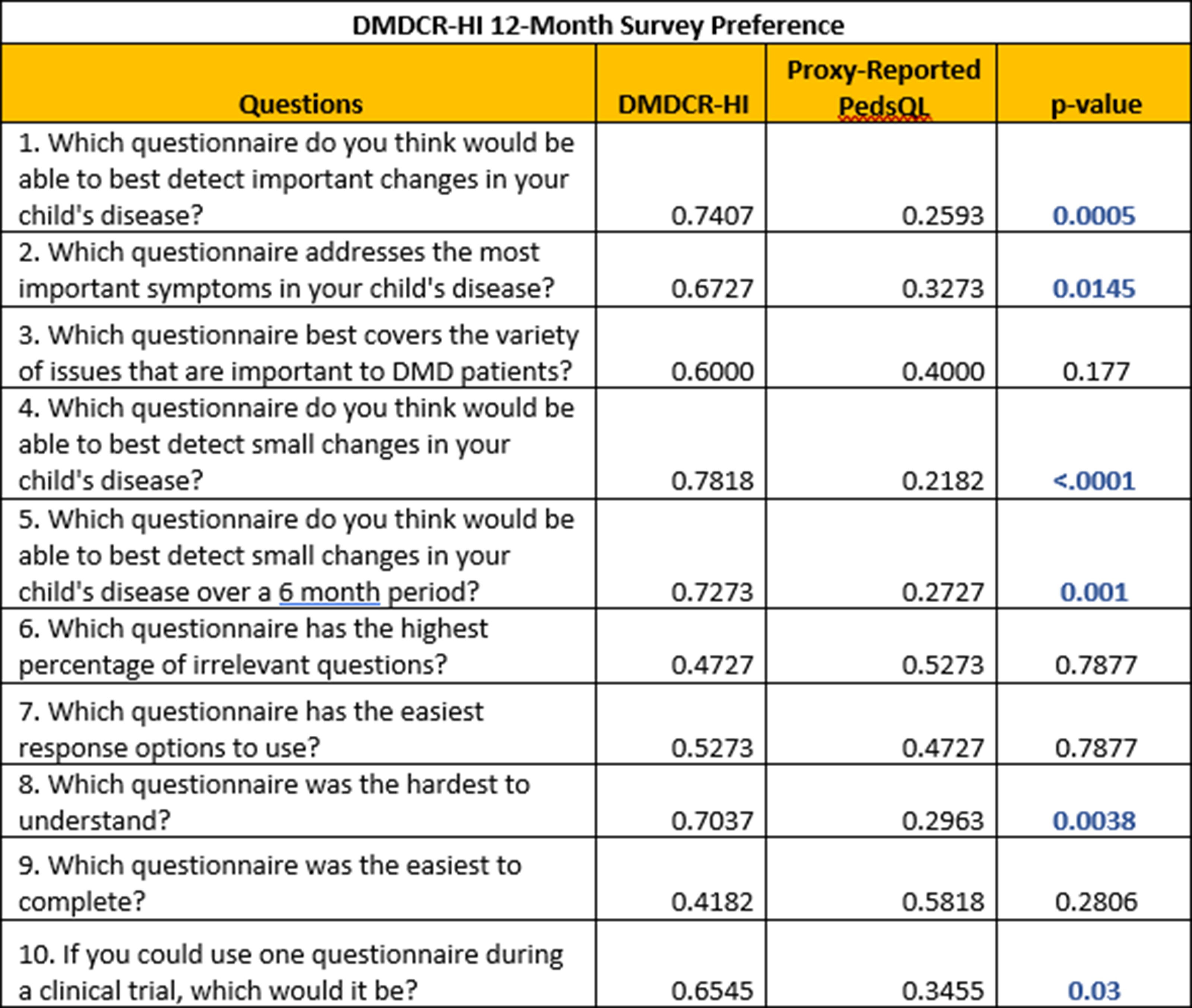
PP01.368
Smartphone-Based Assessment of Shoulder Motion in Patients With Facioscapulohumeral Muscular Dystrophy
Mr. Jonathan Street1,2, Dr. Tecla Bonci3,4, Dr. Alexander Jakubiec3,4, Mr. Adrien Juraver5, Dr. Laura Castillo6, Dr. Óscar Reyes6, Dr. Shibeshish Belachew5, Dr. Channa Hewamadduma7,8, Dr. Claudia Mazzà5
1
Academic Unit of Neuromuscular Disorders, Sheffield Teaching Hospitals, Sheffield, United Kingdom.
2
Sheffield Institute of Translational Neuroscience, University of Sheffield, Sheffield, United Kingdom.
3
School of Mechanical, Aerospace and Civil Engineering, Sheffield, United Kingdom.
4
Insigneo Institute, Sheffield, United Kingdom.
5
Indivi AG, Basel, Switzerland.
6
Indivi AG, Córdoba, Spain.
7
Sheffield Teaching Hospitals Foundation Trust, Sheffield, United Kingdom.
8
Sheffield Institute for Translational Neurosciences (SITRAN), University of Sheffield, Sheffield, United Kingdom
Background: Facioscapulohumeral muscular dystrophy (FSHD) affects the face, shoulders, trunk, and upper and lower limb muscles with symptoms varying in severity and rate of progression. Despite the increasing number of candidate therapeutics for FSHD from pre-clinical studies, heterogeneity of muscle involvement, shortage of robust FSHD biomarkers, and a lack of responsiveness to change seen in traditional outcome measures challenge the implementation of informative FSHD clinical trials.
Wearable sensors are increasingly adopted in neurological and neuromuscular diseases to augment the accuracy of measures recorded during established tests of upper and lower limb motion. This is achieved by transforming sensor data into objective numerical outcomes, called sensor-derived measures (SDMs), which comprehensively quantify the loss of ability and the response to treatment. In this study, we evaluated the feasibility and accuracy of applying such an approach to the data collected with the motion sensors included in a smartphone to assess the shoulders range of motion and muscles fatigability during the execution of an upper limb motor task, designed specifically for capturing relevant aspects of FSHD related functional limitations.
Methods: Twelve participants (Sex: 4 female, and 8 male; Age: Mean 48yr (SD 14.9); FSHD Clinical Score (FCS): Mean 7.4 (SD 2.6)) were asked to perform a test called “Traffic Controller”, during which they had to first separately explore their maximum ranges of shoulder flexion and abduction, and then stably hold their arm in front of them at shoulder level for 30s. Tests were repeated twice per side. While performing the motion, participants were asked to hold a phone (iPhone 16e) in their hand. The phone was used to record the sensor motion data (linear accelerations, angular velocities and orientation, sampling rate 50Hz). Besides recording the movement data, the phone provided detailed audio guidance on when and how to perform the various phases of the test. Instructions also included asking participants to keep their arm straight and the phone orientation constant, to ensure consistency between tests. As a preliminary analysis, the maximum angles during the flexion and lateral abduction phases were extracted as SDMs (Figure 1). Their values were compared against those obtained from a marker-based motion capture system (Vicon). Furthermore, the correlation of the SDMs with the FCS was investigated.
Results: All participants completed the test without significant burdens. Results supported the feasibility of reliably estimating shoulder maximum flexion (ICC(2,1)=0.95) and abduction angles (ICC(2,1)=0.96) from the phone data. Comparison between the phone and the marker-based systems showed a correlation of 0.76 and 0.83 for the flexion and abduction angles, respectively. The data from both movements appeared to be strongly correlated with the FCS (flexion: right: r=0,79; left r=0.79; abduction: right: r=0,68; left r=0.70).
Conclusion: The Traffic Controller proved to be a feasible structured test to estimate shoulder mobility in patients with FSHD using a Smartphone. While the study is still ongoing and further validation is needed to prove their generalisability, these pilot results pave the way for remotely monitoring treatment effects within a clinical trial setting involving patients with FSHD.
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: Outcome measures and clinimetry
Images or Table (Optional)
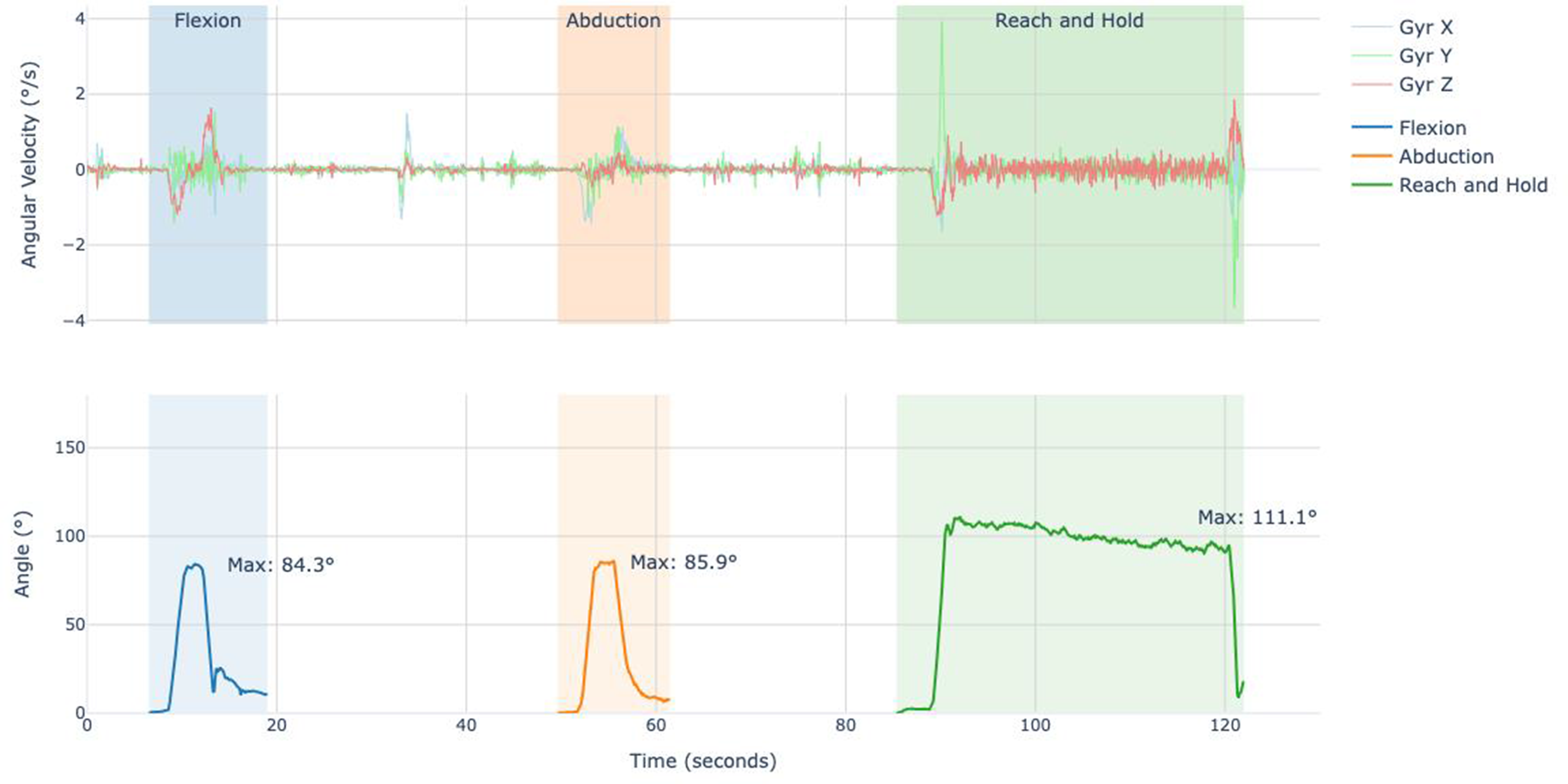
PP01.369
Evaluation of Ambulation With a Wearable Sensor in Patients With CMT
Dr. Margaux Poleur1, Dr. Stéphanie Delstanche1, Dr. Manon Hustinx1, Mr. Guillaume Parinello2, Ms. Noor Benmhammed1, Ms. Thao Nguyen Le Lam2, Ms. Camille Bisson2, Mr. Nicolas Bovy1, Dr. Céline Cluzeau2, Ms. Charline Dubois1, Mr. Damien Eggenspieler2, Prof. Laurent Servais3,4
1
University department of neurology, CHR Citadelle, Liege, Belgium.
2
Sysnav, Vernon, France.
3
Department of Paediatrics, University and University Hospital of Liege, Liege, Belgium.
4
University of Oxford, Oxford, United Kingdom
Background: Charcot-Marie-Tooth disease (CMT) is the most common inherited neuropathy and is characterized by marked genetic and phenotypic heterogeneity, predominantly affecting distal muscles and sensory function. Progression is typically slow and often occurs over several decades. Current clinical outcome measures lack robustness and sensitivity to detect change over the course of a clinical trial. Wearable digital health technology (wDHT) enables passive collection of ambulation data in daily living. The qualification of a wDHT-derived variable, stride velocity 95th centile (SV95C), as a primary endpoint in Duchenne muscular dystrophy by the European Medicines Agency raised interest in applying similar approaches in other neuromuscular disorders. We hypothesized that SV95C and digital variables derived from the same wDHT, Syde, could provide a representative and sensitive assessment of motor function in daily life of patients with CMT.
Methods: Patients with CMT were enrolled in two natural history studies, ActiLiege-Next and ActiLiege-Adult, and underwent clinical evaluation every 6 months. Participants wore one sensor at each ankle daily for the first 3 months and then for 1 month every 6 months. We started by investigating a portfolio of digital outcomes including SV95C, maximal walking distance (WD95C), and number of strides per hour (NbStrides/h) and will expand to other variables that could better evaluate gait impairment due to distal weakness. The reliability of digital measures was assessed by comparing two 2-week periods of each recording month using intraclass correlation coefficient (ICC). Discrimination between patients and controls (enrolled in a separate cohort) was determined using a Mann Whitney U test. Correlations of digital measures with 6-minute walk test (6MWT), 10-meter walk test (10MT), CMT examination score version 2 (CMTESv2), and overall neuropathy limitation score (ONLS) were tested using Spearman’s rank correlation coefficient. Longitudinal changes were assessed using Wilcoxon signed-rank test.
Results: To date, 24 patients were enrolled (median age [range]: 36 years [6-85]) and compared to 45 healthy volunteers (40 years [5-84]). Most subjects were adherent to wearing the ankle sensors: >85% of them recorded at least 50 hours of data at baseline. Analysis of the first 12 enrolled patients showed that SV95C reliability was excellent with intraclass correlation coefficients of 0.99 for patients and 0.90 for controls at baseline, whereas reliability of WD95C and NbStrides/h varied between 0.64 and 0.99. Mean baseline SV95C was significantly higher in controls than in patients (p<0.001), but WD95C and NbStrides/h baseline values were similar between the two groups. SV95C correlated strongly with 6MWT (r = 0.70), 10MT (r = −0.94), ONLS (r = −0.75), and CMTESv2 (r = −0.52). Preliminary longitudinal analysis in the first 3 patients with 6-month follow-up data showed limited changes from baseline for both digital and conventional clinical measures. All available longitudinal results will be presented at the congress.
Conclusion: Further work is needed to confirm the good metric properties observed for SV95C in a larger population, to evaluate sensitivity to detect change versus gold standards and to expand the set of valid wearable-derived variables with measures meaningful for CMT.
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: Outcome measures and clinimetry
PP01.370
Synthetic Data as a Research Enabler in CIDP: A Registry-Based Validation Study
Prof. Alessio Signori1, Dr. Erika Schirinzi2, Prof. Fiore Manganelli3, Prof. Dario Cocito4, Dr. Yuri Falzone5, Prof. Chiara Briani6, Prof. Anna Mazzeo7, Prof. Angelo Schenone8, Dr. Vincenzo Di Stefano9, Dr. Giuseppe Cosentino10, Prof. Girolama Alessandra Marfia11, Dr. Luana Benedetti8, Dr. Marinella Carpo12, Prof. Massimiliano Filosto13, Dr. Luca Leonardi14, Dr. Marco Luigetti15, Dr. Sabrina Matà16, Dr. Giuseppe Piscosquito17, Dr. Tiziana Rosso18, Dr. Marta Lucchetta19, Prof. Gabriele Siciliano2, Prof. Giuseppe Lauria Pinter20, Prof. Maurizio Inghilleri21, Dr. Teresa Cantisani22, Dr. Francesca Notturno23, Dr. Dario Ricciardi24, Prof. Francesco Habetswallner24, Dr. Elisa Vegezzi25, Dr. Alberto De Lorenzo26, Dr. Claudia Lozi26, Dr. Alessandro Salvalaggio27, Dr. Camilla Strano5, Dr. Luca Gentile7, Dr. Giorgia Mataluni11, Prof. Filippo Brighina9, Dr. Barbara Risi13, Dr. Loris Poli28, Dr. Francesca Forcina29, Dr. Federica Moret21, Dr. Fabrizio Canale30, Dr. Emanuele Cassano31, Dr. Eleonora Della Bella20, Prof. Eduardo Nobile-Orazio26, Prof. Pietro Emiliano Doneddu26
1
Department of Health Sciences, University of Genoa, Genoa, Italy.
2
Department of Clinical and Experimental Medicine, Neurological Clinic, University of Pisa, Pisa, Italy.
3
3.Department of Neurosciences, Reproductive and Odontostomatological Sciences, University of Naples Federico II, Naples, Italy.
4
Dipartimento Scienze Cliniche e Biologiche, Università di Torino, Turin, Italy.
5
Neurology Unit, IRCCS San Raffaele Scientific Institute, Milan, Italy.
6
Neurology Clinic, Department of Neurosciences, University of Padova, Padua, Italy.
7
Unit of Neurology and Neuromuscular Diseases, Department of Clinical and Experimental Medicine, University of Messina, Messina, Italy.
8
Department of Neuroscience, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health (DiNOGMI), University of Genoa, Genoa, Italy.
9
Department of Biomedicine, Neuroscience, and Advanced Diagnostic (BIND), University of Palermo, Palermo, Italy.
10
Department of Brain and Behavioral Sciences, University of Pavia, Campus della Salute, Policlinico San Matteo, Pavia, Italy.
11
Dysimmune Neuropathies Unit, Department of Systems Medicine, Tor Vergata University of Rome, Rome, Italy.
12
ASST Bergamo Ovest-Ospedale Treviglio, Treviglio, Italy.
13
Department of Clinical and Experimental Sciences, NeMO-Brescia Clinical Center for Neuromuscular Diseases, University of Brescia, Brescia, Italy.
14
Neuromuscular and Rare Disease Centre, Department of Neuroscience, Mental Health and Sensory Organs (NESMOS), SAPIENZA University of Rome, Sant'Andrea Hospital, Rome, Italy.
15
Department of Neurosciences, Università Cattolica del Sacro Cuore, Rome, Italy.
16
Department of Neurological and Psychiatric Sciences, Azienda Ospedaliero-Universitaria di Careggi, Florence, Italy.
17
Neurology Unit, University Hospital “San Giovanni di Dio e Ruggi d'Aragona”, Salerno, Italy.
18
UOC di Neurologia, Ospedale San Bassano, Vicenza, Italy.
19
UOC Neurologia, Ospedale Santa Maria della Misericordia, Rovigo, Italy.
20
Unit of Neuroalgology, IRCCS Foundation 'Carlo Besta' Neurological Institute, Milan, Italy.
21
Department of Human Neuroscience, Sapienza University of Rome, Rome, Italy.
22
Servizio di Neurofisiopatologia, Azienda Ospedaliera di Perugia, Perugia, Italy.
23
Servizio di neurofisiopatologia, Ospedale SS di Pescara, Perugia, Italy.
24
Clinical Neurophysiology Unit, Cardarelli Hospital, Naples, Italy.
25
Neuroimmunology Laboratory and Neuroimmunology Research Section, IRCCS Mondino Foundation, Pavia, Italy.
26
Neuromuscular and Neuroimmunology Unit, IRCCS Humanitas Research Hospital, Rozzano-Milan, Italy.
27
Neurology Clinic, Department of Neurosciences, University of Padova, Padua, Italy.
28
Unit of Neurology, ASST Spedali Civili, Brescia, Italy.
29
Neuromuscular and Rare Disease Centre, Department of Neuroscience, Mental Health and Sensory Organs (NESMOS), SAPIENZA University of Rome, Sant'Andrea Hospital, Rome, Italy.
30
Department of Neurosciences, Reproductive and Odontostomatological Sciences, University of Naples Federico II, Naples, Italy.
31
Neurology and Stroke Unit, Ospedale del Mare, ASL Napoli 1 Centro, Naples, Italy
Background: Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) is a rare, heterogeneous, and treatable immune-mediated neuropathy. Research in CIDP is constrained by low prevalence, small cohorts, and restricted data sharing due to privacy regulations, limiting reproducibility and secondary analyses. Synthetic data (SD) generated through regression-based and generative artificial intelligence models offer a potential solution by preserving the statistical structure of real-world datasets while eliminating re-identification risk.
Methods: To validate the use of regression-based synthetic data derived from the Italian CIDP Registry, using a previously published multicenter case–control study on lifestyle and dietary factors as a reference framework. We aimed to determine whether SD could accurately reproduce (i) marginal distributions of variables, (ii) univariable associations with CIDP risk, and (iii) the multivariable joint structure of key predictors. We analyzed the matched dataset of 390 individuals (195 CIDP cases, 195 controls) from the original study. Synthetic datasets were generated using the synthpop package in R, stratified by sex to preserve the original 1:1 matching scheme. Binary and multinomial regression models were used to synthesize predictors and outcome variables. Fidelity was assessed through information-theoretic distance measures (Total Variation, Jensen–Shannon, Hellinger), χ² tests with Cramér’s V, and propensity score mean squared error (pMSE). Privacy was evaluated using exact match rates and nearest-neighbor Hamming distances. Univariable and multivariable logistic regression models were replicated in real and synthetic datasets.
Results: The synthetic data preserved case–control balance and sex distribution. Marginal distributions closely matched the real data, with minimal discrepancies confined to a few multi-level dietary variables. Distance metrics and goodness-of-fit tests indicated high fidelity (Total Variation typically 0.01–0.05; Cramér’s V ≤0.09). No identical matches between real and synthetic records were observed; nearest-neighbor analyses showed low disclosure risk (median Hamming distance 5). Univariable associations with CIDP risk were highly concordant between datasets. Protective effects of higher fish and rice intake were preserved in both univariable and multivariable models (e.g., fish intake: real OR 0.52 vs SD OR 0.44; rice intake: real OR 0.45 vs SD OR 0.43).
Conclusion: Regression-based synthetic data generated from the Italian CIDP Registry reliably preserve the statistical structure and clinically meaningful associations of real-world case–control data, while effectively eliminating the risk of patient re-identification. This strategy overcomes key barriers imposed by small sample sizes and data-sharing constraints in rare diseases, enabling secure dissemination of high-fidelity datasets. By facilitating independent validation, secondary analyses, and innovative study designs, synthetic data provide a robust foundation for collaborative, reproducible, and precision-oriented research in CIDP and other rare peripheral neuropathies.
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: Outcome measures and clinimetry
PP01.371
Non-Physician–Administered Neuropathy Symptom and Disability Scores for Screening of Diabetic Distal Symmetric Polyneuropathy
Dr. Jakkrit Amornvit1,2, Mr. Tharadon Deepracha3, Ms. Phuettha Sangrut3, Mr. Peerakan Inkhao3, Ms. Wanicha Boonyuen3, Ms. Benjamat Pensrisirikul3, Ms. Supaporn Krittanupong3
1
Division of Neurology, Department of Medicine, King Chulalongkorn Memorial Hospital, Thai Red Cross Society, Bangkok, Thailand.
2
Faculty of Medicine, Chulalongkorn University, Bangkok, Thailand.
3
Chula Neuroscience Center, King Chulalongkorn Memorial Hospital, Thai Red Cross Society, Bangkok, Thailand
Background: Despite recommendations for annual neuropathy and foot screening, early detection of diabetic distal symmetric polyneuropathy (DSPN) remains inconsistent in routine practice, particularly in high-volume outpatient settings. A key barrier is not the availability of bedside tools, but limited time and a shortage of trained examiners to apply standardized assessments at scale. A workflow-ready screening approach that can be reliably delivered by non-physician healthcare professionals may help close this implementation gap.
Methods: We conducted a cross-sectional diagnostic accuracy study at a Thai tertiary hospital. Adults with type 1 or 2 diabetes (n=78, age 30–65) without previously documented neuropathy were enrolled. The Neuropathy Symptom Score (NSS; 15-item symptom questionnaire) and Neuropathy Disability Score (NDS; brief bedside exam of distal sensation and ankle reflexes) were translated into Thai using forward–backward procedures and administered by trained nurses/health staff. A blinded reference standard was performed independently by a neurologist, including standardized neurological examination and nerve conduction studies to determine DSPN. Feasibility (administration time), internal consistency (Cronbach’s α), test–retest and inter-rater reliability, and criterion validity against the reference standard were assessed. Diagnostic performance (sensitivity, specificity, predictive values, AUC) was evaluated for NSS, NDS, and prespecified combined screening rules.
Results: The Thai NSS/NDS instruments demonstrated excellent content validity and reliability. NSS showed high internal consistency (α=0.93) and strong 4-week test–retest stability (r=0.84). NDS inter-rater reliability was high (ICC=0.83). DSPN was confirmed in 45% of participants (35/78) by the reference standard; none had been previously diagnosed. Approximately 50% of newly identified DSPN cases were asymptomatic by symptom screening (NSS <3) despite objective distal sensory/reflex deficits on examination and/or nerve conduction studies. NSS and NDS scores were significantly higher in participants with DSPN than those without (mean NDS 5.0 vs 1.8, p<0.001). A composite screening rule (either NSS ≥5 or NDS ≥3) identified 88% of DSPN cases (sensitivity) with ∼70% specificity. A stricter dual-positive rule increased specificity to >90% but missed mild cases. Discrimination was good for both tools (AUC 0.80 for NSS, 0.84 for NDS). Nurses completed NSS/NDS assessments in under 10 minutes.
Conclusion: Task-shifted implementation of NSS and NDS by non-physician staff was feasible and showed good diagnostic performance against a blinded neurologist and nerve conduction reference standard for DSPN. The Thai-translated NSS/NDS are reliable and valid, and their use in routine diabetes visits may expand screening coverage, support earlier risk stratification, and facilitate timely preventive care and referral pathways in settings where specialist time is limited.
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: Outcome measures and clinimetry
PP01.372
Revolutionizing Gait Quality Monitoring in People with CIDP: An Instrumented Shoe Insole Solution
Dr. Karen M. Lynch1, Dr. Karissa L. Gable2, Dr. Matthew P. Mavor3, Dr. Mohammad H. Akhavanfar3,4, Dr. Kristen H.E. Beange4, Dr. Ryan B. Graham3, Dr. Jan C. Schuller5, Dr. Alex Seluzhytsky1
1
Sanofi, Cambridge, MA, United States.
2
Neuromuscular Division, Department of Neurology, Duke University Medical Center, Durham, NC 27710, United States.
3
School of Human Kinetics, Faculty of Health Sciences, University of Ottawa, Ottawa K1N 6N5, Ontario, Canada.
4
Celestra Health Systems, Ottawa K2K 0G7, Ontario, Canada.
5
Enovalife, Cœur Défense A – 110 Esplanade du Général de Gaulle – 92931 Paris La Défense Cedex, France
Background: Biomechanical gait assessments offer an objective alternative to clinical gait evaluations. However, they rely on non-transportable technology, requiring patients to travel and providing infrequent snapshots of walking ability. Using wearable technology, such as insoles, continuous monitoring in free-living conditions becomes possible, enabling development of digital biomarkers that comprehensively describe gait quality. This study aims to develop a digital biomarker that passively assesses gait quality in people with Chronic Inflammatory Demyelinating polyradiculoneuropathy (pwCIDP) using instrumented shoe insoles.
Methods: Up to two-hundred mobile pwCIDP will perform 5- to 15-minute walks 3-times per week for 12-months under free-living conditions while wearing insoles (pressure, accelerometer, gyroscope) streaming to a smartphone app; to date, 107 participants have been recruited. First, insole data will undergo human activity recognition; classified ‘walk’ data are segmented into standardized lengths and analyzed for spatiotemporal and sensor waveform features. Participants will undergo remote Inflammatory Neuropathy Cause and Treatment score and Inflammatory Rasch-built Overall Disability scale assessments at six- and three-month intervals, respectively. Meaningful gait features will be derived by correlating with assessment scores and t-tests to identify significant differences from previously collected controls. By training a support vector machine to classify controls vs pwCIDP, a gait composite index score (gCI; 0-100%) will be developed and benchmarked against the most informative features and assessment scores. Locally estimated scatterplot smoothing regressions will be used to identify meaningful trends in gCI scores over time.
Results: Data collection is ongoing. Trend analysis for all participants with ≥6 months of walking will be presented; a preliminary gCI will be developed and benchmarked using all recruited participants.
Conclusion: Establishing a gCI for pwCIDP will enable longitudinal monitoring of disease progression from free-living walking bouts, reshaping how neurologists may understand and evaluate disease progression (i.e., improvement, worsening, maintenance) and treatment response (i.e., exercise, pharmacological, assistive devices).
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: Outcome measures and clinimetry
PP01.373
Transitory Symptom Worsening in Facioscapulohumeral and Becker Muscular Dystrophies: Do They Exist and Matter?
Dr. Nurit Birman Har-Noy1, Dr. Jacopo Luca Casiraghi2, Dr. Andrea Lizio2, Dr. Silvi Cadri3, Dr. Michela Coccia3, Dr. Andrea Barp4, Dr. Riccardo Zuccarino4, Dr. Filomena Caria5, Prof. Massimiliano Filosto6, Prof. Valeria Ada Sansone1
1
Neurorehabilitation Unit University of Milan The NeMO Clinical Center, Milan, Italy.
2
Neuromuscular Omnicentre (NEMO), Fondazione Serena Onlus, Milan, Italy.
3
Neuromuscular Omnicentre (NEMO), Fondazione Serena Onlus, Ancona, Azienda Ospedaliero-Universitaria Delle Marche, Ancona, Italy.
4
Neuromuscular Omnicentre (NEMO), Fondazione Serena Onlus, Trento, Ospedale Riabilitativo Villa Rosa, Pergine Valsugana Italy, Trento, Italy.
5
Neuromuscular Omnicentre (NEMO), Fondazione Serena Onlus, Brescia, Italy.
6
NeMO-Brescia Clinical Center for Neuromuscular Diseases; Department of Clinical and Experimental Sciences, University of Brescia; ERN Euro-NMD Center ASST Spedali Civili, Brescia, Italy, Brescia, Italy
Background: Patient-reported outcome measures (PROs) are essential for assessing disease burden and tracking changes over time. Neuromuscular diseases exhibit heterogeneous phenotypic presentations. A preliminary exploratory study at the NeMO Center in Milan (77 patients with FSHD and BMD) suggested that episodes of transitory symptom worsening may be a clinically relevant phenomenon. This expanded multicenter study investigates the prevalence, characteristics, and impact of self-reported episodes of transitory neuromuscular symptom worsening in patients with facioscapulohumeral muscular dystrophy (FSHD) and Becker muscular dystrophy (BMD), compared to healthy controls.
Methods: We developed a patient-reported questionnaire and distributed it anonymously online in April and May 2025 to adult patients with FSHD and BMD followed at the NeMO Center in Milan, Italy. In July 2025, the study was expanded to additional NeMO centers in Italy (Ancona, Brescia, and Trento). A further group of 18 healthy controls also completed the questionnaire. Symptoms of fatigability, focal muscle weakness, and pain were classified as episodic if they occurred in addition to chronic neuromuscular impairment, lasted 1–14 days, and returned to baseline or near-baseline levels.
Results: A total of 145 patients responded (104 FSHD, 41 BMD). Overall, 58 patients (40%) reported episodes of transitory symptom worsening, significantly higher than healthy controls (3/18, 17%). Episodes were reported by 43% of FSHD and 32% of BMD patients. Within the FSHD group, 87% of patients who reported episodes had baseline pain or fatigue, compared with 68% who had baseline symptoms but no episodes. Only FSHD patients reported episodes lasting longer than 7 days (20% experienced 8-14 day episodes), demonstrating greater heterogeneity in episode duration. The annual frequency was higher in FSHD: 29% experienced more than 6 episodes per year, compared with only 8% of BMD patients. Episodes interfered with daily routine in 62-68% of patients, with 41-57% requiring increased bed rest and 24-43% reducing social activities. Importantly, 50-62% changed their ongoing routine after episodes. Disease-specific triggers emerged: excessive physical activity predominated in BMD (87.5%) and FSHD (60%), while psychological stress was more common in FSHD (50%) and climate changes in BMD (62.5%).
Conclusion: This multicenter study confirms that transitory symptom worsening is a clinically relevant phenomenon in FSHD and BMD, significantly more often than in healthy controls. Episodes substantially impact daily living and lead to behavioral modifications. FSHD patients experience a higher annual burden due to increased frequency and longer duration of episodes. Disease-specific trigger patterns suggest different underlying mechanisms. These findings support the development of PROs that capture symptom fluctuations, not only progressive decline. Clinical trial implications include performing baseline assessments multiple times, using digital measures and wearables for continuous patient monitoring, or incorporating questions about current versus baseline status into trial PROMs. Personalized interventions such as adjusted physical activity and psychological support are suggested. Larger studies are needed to validate these findings and explore prevention strategies.
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: Outcome measures and clinimetry
PP01.374
Efficacy of Anti-Osteoporotic Drugs and Vitamin D/calcium Supplementation in NMD: A Systematic Review
Dr. Sara Liguori1, Dr. Francesca D'Andrea1, Dr. Marco Paoletta2, Assoc. Prof. Antimo Moretti1, Prof. Giovanni Iolascon1
1
Department of Medical and Surgical Specialties and Dentistry, University of Campania “Luigi Vanvitelli”, Naples, Italy.
2
Department of Mental and Physical Health and Preventive Medicine, University of Campania “Luigi Vanvitelli”, Naples, Italy
Background: Neuromuscular diseases (NMDs) represent a heterogeneous group of musculoskeletal system disorders primarily affecting the anterior horn cells of the spinal cord, peripheral nerves, neuromuscular junctions, or skeletal muscles. Although most NMDs are rare, with an estimated prevalence of 1–10 cases per 100,000 individuals, collectively they impact a substantial number of patients. Clinically, NMDs are characterized by progressive reductions in muscle strength, mobility, and bone mineral density (BMD), often associated with fatigue, loss of ambulation, and impaired functional performance. Novel therapies, including gene-based and symptomatic treatments, have mitigated severe manifestations and slowed disease progression. Despite these advances, emerging complications—such as increased fall risk, fragility fractures, scoliosis, and contractures—have become more prominent, prompting interest in optimizing bone health in NMD patients. This study aimed to evaluate the effectiveness of current interventions for bone health management in this population, considering both pharmacological and nutritional strategies.
Methods: A systematic review was conducted by searching PubMed, Embase, and Scopus for studies published until 22 May 2024. The search focused on studies assessing the effects of anti-osteoporotic drugs and/or vitamin D or calcium supplementation on BMD in NMD patients. The methodological quality of the included studies was assessed using the PEDro scale for randomized controlled trials (RCTs) and the JBI‑QES (Joanna Briggs Institute – Qualitative Evidence Synthesis) tool for non‑randomized experimental studies. The protocol was registered on PROSPERO.
Results: Of 363 articles retrieved, 28 met inclusion criteria: 6 interventional studies, 14 observational studies, 5 case reports, and 3 case series. Overall, 687 NMD patients aged 0–70 years were included. Duchenne muscular dystrophy was the most prevalent diagnosis (66.6%), followed by spinal muscular atrophy (SMA). The most reported interventions were vitamin D supplementation (68.9%) and calcium (55.1%). Among anti-osteoporotic agents, zoledronate was most commonly employed (37.9%), followed by pamidronate (20.6%) and alendronate (17.2%). Regarding efficacy in improving BMD, vitamin D and calcium supplementation alone were insufficient to prevent progressive bone loss. Zoledronate and alendronate demonstrated consistent positive effects in most studies, while Pamidronate showed partial efficacy, though methodological limitations affected interpretation. Risedronate and neridronate did not demonstrate consistent benefits. Limited evidence suggested denosumab effectively improved BMD in small patient samples, whereas teriparatide maintained stable BMD values.
Conclusion: This analysis highlights a lack of data on bone health management in NMD, particularly regarding specific age groups and rarer subtypes. These gaps emphasize the necessity of a targeted, evidence-based approach to prevent and treat bone loss. Early therapeutic interventions, guided by the biological rationale of anti-osteoporotic agents, represent a critical strategy to optimize bone health in NMD, in order to preserve functional capacity, skeletal integrity, and quality of life. Future research should focus on longitudinal outcomes, comparative efficacy of agents and individualized strategies to preserve bone health and quality of life in NMD population.
Abstract Topic Groups (Submission Categories)
Topic Group 8 - Patient Related Issues: Palliative Care
PP01.375
Chronic Pain and Postural Instability in Individuals With Neuromuscular Diseases: A Cross-Sectional Study
Dr. Sara Liguori1, Dr. Francesca D'Andrea1, Dr. Marco Paoletta2, Assoc. Prof. Antimo Moretti1, Prof. Giovanni Iolascon1
1
Department of Medical and Surgical Specialties and Dentistry, University of Campania “Luigi Vanvitelli”, Naples, Italy.
2
Department of Mental and Physical Health and Preventive Medicine, University of Campania “Luigi Vanvitelli”, Naples, Italy
Background: Neuromuscular diseases (NMDs) comprise a heterogeneous group of disorders characterized by progressive impairment of muscle strength, motor control, and functional mobility. Pain is a highly prevalent and frequently under-recognized symptom in patients with NMDs. Beyond its direct impact on physical function and psychosocial well-being, pain has been shown to interfere with sensorimotor processing, proprioceptive acuity, and postural strategies, contributing to balance deficits and an increased risk of falls. The aim of this study was to further investigate the association between pain and postural stability in adults with NMD through an objective stabilometric assessment.
Methods: In this cross-sectional study, we included patients aged >18 years with a confirmed diagnosis of NMD. The assessment protocol comprised the collection of anthropometric data, pain evaluation using the Brief Pain Inventory (BPI), and balance assessment performed with a ProKin stabilometric platform. Postural stability tests were conducted under eyes-open (EO) and eyes-closed (EC) conditions to better challenge proprioceptive control mechanisms. From these tests, sway area (mm²) was derived, defined as the area encompassing all center of pressure (CoP) displacement points during postural oscillations. The NMD population was stratified according to the absence of pain (group 0) or presence of pain (group 1). The association between pain status and sway area under EO and EC conditions was analyzed using the chi-square test and odds ratio (OR).
Results: A total of 39 patients with NMD were included, with a mean age of 45.8 ± 16.4 years and a mean BMI of 25.0 ± 4.26 kg/m². Eleven patients reported no pain on BPI assessment, whereas 28 reported the presence of pain. Sway area parameters under EO and EC conditions were stratified according to reference cut-off values reported in the literature. In group 0, 3 patients showed altered sway area under EO conditions and 4 under EC conditions, whereas in group 1, 8 patients showed alterations under EO conditions and 16 under EC conditions. Statistical analysis revealed that patients with pain exhibited a significantly higher prevalence of abnormal sway under EC conditions compared with those without pain (72.2% vs 27.8%, p = 0.015). Furthermore, patients with pain had a 5.2-fold increased risk of pathological sway area under EC conditions compared with group 0 (95% CI: 1.3–20.5).
Conclusion: In this cohort of patients with NMD, the presence of pain was associated with an increased sway area with eyes-closed, indicating impaired balance compared with patients without pain. These findings suggest that pain may play a relevant role in proprioceptive dysfunction and balance impairment in patients with NMD.
Abstract Topic Groups (Submission Categories)
Topic Group 8 - Patient Related Issues: Palliative Care
PP01.376
Musculoskeletal Impairment in Adults With Neurofibromatosis Type 1: An Observational Study
Dr. Sara Liguori1, Dr. Marco Paoletta2, Assoc. Prof. Antimo Moretti1, Prof. Giovanni Iolascon1
1
Department of Medical and Surgical Specialties and Dentistry, University of Campania “Luigi Vanvitelli”, Naples, Italy.
2
Department of Mental and Physical Health and Preventive Medicine, University of Campania : “Luigi Vanvitelli”, Naples, Italy
Background: Neurofibromatosis type 1 (NF1) is an autosomal dominant genetic disorder with an estimated incidence of 1 in 3,000 live births. It is mainly characterized by neurocutaneous manifestations and an increased risk of benign and malignant tumors, but it is a multisystem disorder involving several organs and tissues. NF1 is caused by loss-of-function mutations in the NF1 gene on chromosome 17q11, which encodes neurofibromin, a protein regulating cell proliferation and differentiation through multiple intracellular signaling pathways.¹ Recent evidence indicates that neurofibromin also plays a role in muscle cell growth, bone extracellular matrix formation, and bone metabolism regulation.² Consequently, its dysfunction may contribute to musculoskeletal abnormalities in NF1, including bone dysplasia, hypotonia, and muscle weakness, potentially promoting physical inactivity and reducing quality of life.³ Despite their clinical relevance, musculoskeletal manifestations in NF1 remain poorly investigated. This study aimed to characterize musculoskeletal impairment iin a cohort of adult NF1 patients
Methods: This observational study included patients with NF1. Data collected included age, body mass index (BMI), and history of fragility fractures. Muscle strength was assessed using a portable dynamometer, physical performance with the Short Physical Performance Battery (SPPB), and gait parameters with a BTS G-Walk® inertial sensor. Quality of life was assessed using the SF-36, and physical activity using the IPAQ, expressed as weekly metabolic equivalent of task (MET) minutes. Physical activity was classified as “inactive” (<700 MET/week), “sufficiently active” (700–2519 MET/week), or “active/very active” (>2520 MET/week), corresponding to groups 0–2. Bone metabolism was assessed through laboratory tests and DXA (GE Lunar), measuring bone mineral density (BMD) at the lumbar spine (L1–L4), left femoral neck, and total body less head.
Results: We recruited 83 patients (37 M; 46 F) with an average age of 40.61 ± 15.45 years and an average BMI of 24.34 ± 4.31 kg/ m2. The mean handgrip strength was 27.94 ± 10.17 kg. On analysis of the level of PA, 17 patients (20.5%) were found to be inactive, 38 patients (45.8%) sufficiently active, and 28 patients (33.7%) active or very active. On comparing the patients stratified by PA, statistically significant differences were found between Group 0 and Group 2 for the three sub-items of the SPPB (p < 0.05). Thirty patients completed the densitometric examination (14 M; 16 F); of these, 19 (63.3%) showed normal mean values for sex and age, 7 had values consistent with a diagnosis of osteopenia (23.3%), while 4 had a diagnosis of osteoporosis/reduced values for sex and age. When comparing the densitometric parameters by level of PA, no statistically significant differences were found between the three groups (p >0.05).
Conclusion: Our data provided a musculoskeletal characterization of a cohort of adults with NF1. Although many patients were physically active, muscle strength remained below that of the general population, and bone health was compromised in one-third of the cohort. Promoting regular physical activity is crucial for improving musculoskeletal health in NF1 patients. Future research should focus on long-term effects of physical activity on bone strength in this population.
Abstract Topic Groups (Submission Categories)
Topic Group 8 - Patient Related Issues: Palliative Care
PP01.377
The Role of Magnesium in Muscle Health and Neuromuscular Diseases
Dr. Sara Liguori1, Dr. Marco Paoletta2, Assoc. Prof. Antimo Moretti1, Prof. Giovanni Iolascon1
1
Department of Medical and Surgical Specialties and Dentistry, University of Campania “Luigi Vanvitelli”, Naples, Italy.
2
Department of Mental and Physical Health and Preventive Medicine, University of Campania “Luigi Vanvitelli”, Naples, Italy
Background: Magnesium (Mg) is an alkaline earth metal essential for numerous cellular processes, acting as a cofactor for over 300 enzymatic reactions. In the human body, approximately 60% of magnesium is stored in bones, while 30% is distributed across muscles and soft tissues. Primary nutritional magnesium deficiency is uncommon, typically occurring only when low intake is combined with excessive losses, such as during prolonged diarrhoea. Hypomagnesemia initially manifests with symptoms including weakness, anorexia, fatigue, nausea, and vomiting. In more severe cases, it may lead to muscle cramps, hypertension, and vasospasms due to secondary increases in intracellular calcium. Several muscle disorders, including sarcopenia, inflammatory myopathies, and neuromuscular diseases, are frequently associated with significant hypomagnesemia. Despite widespread magnesium supplementation in patients with neuromuscular conditions, robust clinical evidence supporting this intervention remains limited, and current guidelines do not provide clear recommendations. The aim of this scoping review was to examine the role of magnesium in skeletal muscle tissue, focusing on its biological effects as well as clinical and therapeutic implications.
Methods: This scoping review was conducted in accordance with the PRISMA-ScR (Preferred Reporting Items for Systematic Reviews and Meta-Analyses Extension for Scoping Reviews) guidelines. A systematic search was performed on PubMed using the following keywords: “Magnesium” OR “Magnesium Compounds” AND “Duchenne Muscular Dystrophy” OR “Becker Muscular Dystrophy” OR “Myasthenia Gravis” OR “Charcot-Marie-Tooth Disease” OR “Spinal Muscular Atrophy” OR “Glycogen Storage Disease Type II” OR “neuromuscular diseases” OR “skeletal muscle”. We considered all the articles published until 21th May 2024.
Results: A total of 305 articles were identified through the PubMed search. Based on title and abstract screening, 275 articles were excluded. Full-text review led to the exclusion of an additional 10 articles, leaving 20 studies that met the inclusion criteria and were included in the discussion. Preclinical studies in animal models have demonstrated that magnesium intake can modulate multiple metabolic pathways involved in muscle homeostasis. For example, a study in chickens showed that a magnesium-deficient diet resulted in reduced muscle and serum magnesium levels, mitochondrial dysfunction, and increased oxidative stress. Other preclinical studies reported that magnesium supplementation can prevent corticosteroid-induced muscle atrophy and counteract sarcopenia. Clinical studies in healthy volunteers with normal magnesium levels have shown that oral supplementation does not significantly alter muscle or serum magnesium concentrations. Conversely, in patients with conditions such as cystic fibrosis or alcoholic liver disease, magnesium supplementation has been associated with improved muscle strength. Additionally, in athletes, magnesium supplementation appears to reduce exercise-induced muscle damage, and in sarcopenic patients, it can improve muscle mass. Notably, no studies were identified investigating magnesium supplementation specifically in patients with neuromuscular diseases.
Conclusion: The available evidence suggests that adequate magnesium intake supports musculoskeletal health, particularly by maintaining or improving muscle mass. Magnesium may also help counteract muscle atrophy, highlighting its potential as a therapeutic strategy for sarcopenia and other age-related muscle disorders. However, no studies have investigated magnesium supplementation in patients with neuromuscular diseases, indicating a significant gap in the literature and a promising avenue for future research.
Abstract Topic Groups (Submission Categories)
Topic Group 6 - Basic Sciences in Neuromuscular Diseases: Muscle Structure / Muscle Development / Muscle Growth
PP01.378
Clinical and Functional Characterization of a Pediatric Cohort With Neurofibromatosis Type 1
Dr. Sara Liguori1, Dr. Francesca D'Andrea1, Dr. Marco Paoletta2, Assoc. Prof. Antimo Moretti1, Prof. Giovanni Iolascon1
1
Department of Medical and Surgical Specialties and Dentistry, University of Campania “Luigi Vanvitelli”, Naples, Italy.
2
Department of Mental and Physical Health and Preventive Medicine, University of Campania “Luigi Vanvitelli”, Naples, Italy
Background: Neurofibromatosis type 1 (NF1) is an autosomal dominant genetic disorder with an estimated incidence of approximately 1 in 3,000 individuals.¹ It is caused by mutations in the NF1 gene located on chromosome 17q11, which encodes the neurofibromin protein. Clinically, NF1 is characterised by the development of benign tumours affecting the central and peripheral nervous systems, including the brain and spinal cord, as well as other organs, the skin, and the skeletal system.² From a clinical perspective, NF1 presents with marked phenotypic heterogeneity, with manifestations varying according to the organs and tissues involved. This variability may result in a wide range of functional impairments that can affect motor performance, autonomy, and overall independence in daily activities. The aim of the present study was to characterise a cohort of paediatric patients with NF1 in terms of muscle strength, gait performance, fatigue, level of autonomy, manual dexterity, balance, and health-related quality of life, as assessed from both the patients’ and parents’ perspectives.
Methods: In this observational study, we included patients with a diagnosis of NF1 aged between 3 and 18 years. Anthropometric data, including body weight, height, and body mass index (BMI), were collected. Handgrip strength was evaluated by a portable dynamometer (HGS). Ambulatory capacity was examined using the Functional Ambulation Categories (FAC). Fatigue was assessed with the Fatigue Severity Scale, manual dexterity with the Nine Hole Peg Test, and balance with the Pediatric Balance Scale (PBS). Health-related quality of life was evaluated from both the patient’s and the parent’s perspectives using the Pediatric Quality of Life Inventory (PedsQL) 4.0.
Results: Twenty-seven patients with NF1 (16 males, 11 females) were recruited, with a mean age of 12.85 ± 3.03 years and a mean BMI of 22.15 ± 3.74 kg/m². Handgrip strength assessment showed that 21 of 26 patients (80%) had reduced strength compared to normative values for age and sex. Manual dexterity was impaired in 15 of 25 patients (60%) relative to reference values. Gait assessment indicated that 26 patients were classified in FAC category 5 (indipendent), while 1 patient was in category 3 (dependent on supervision). Fatigue, measured by the Fatigue Severity Scale (FSS), had a mean score of 21.7 ± 12.72 (total score 63). Balance, assessed with the Pediatric Balance Scale (PBS), was generally preserved (mean 54.65 ± 2.31). Health-related quality of life, as reported by parents, had a mean score of 25.78 ± 17.06, while patient-reported quality of life had a mean of 20.56 ± 13.4, substantially lower than the European normative mean of 80.3 ± 4.3.
Conclusion: In this cohort of paediatric patients with NF1, reduced muscle strength, impaired manual dexterity, and increased fatigue were observed, which may contribute to the perception of lower health-related quality of life. Notably, patients reported a poorer quality of life compared to their parents’ assessments, highlighting potential discrepancies between reported outcomes.
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: Outcome measures and clinimetry
PP01.379
Multidimensional Evaluation of Functioning in a Pediatric Population With X‑linked Hypophosphatemic Rickets: Observational Study
Dr. Sara Liguori1, Dr. Francesca D'Andrea1, Dr. Marco Paoletta2, Assoc. Prof. Antimo Moretti1, Prof. Giovanni Iolascon1, Prof. Anna Grandone3
1
Department of Medical and Surgical Specialties and Dentistry, University of Campania “Luigi Vanvitelli”, Naples, Italy.
2
Department of Mental and Physical Health and Preventive Medicine, University of Campania “Luigi Vanvitelli”, Naples, Italy.
3
Department of Woman, Child and of General and Specialized Surgery, University of Campania, Naples, Italy
Background: X-linked hypophosphatemic rickets (XLH; RC0170) is a rare genetic disorder affecting approximately 1 in 47,000 individuals. It results from inactivating mutations in the PHEX gene on the X chromosome, causing elevated blood level of fibroblast growth factor 23 (FGF23), which increases renal phosphate excretion and reduces 25-hydroxyvitamin D₃ activation. Chronic hypophosphatemia impairs bone mineralization, leading to rickets in childhood and osteomalacia in adulthood. Clinically, XLH is characterized by short stature, skeletal deformities, dental dysplasia, chronic musculoskeletal pain, enthesopathies, and reduced functional mobility. In 2019, the Italian Medicines Agency (AIFA) approved burosumab, an anti-FGF23 monoclonal antibody, demonstrating higher efficacy and improved tolerability than conventional therapy.¹ However, data on muscle strength and functional performance in pediatric patients treated with burosumab remain limited. The present study aimed to characterize this patient cohort using a standardized rehabilitative assessment protocol to guide personalized rehabilitation and monitor long-term treatment response.
Methods: Pediatric patients with XLH receiving burosumab were enrolled and underwent a comprehensive clinical and instrumental evaluation. Isometric handgrip strength was measured with a portable Jamar dynamometer, and upper limb manual dexterity assessed with the Nine Hole Peg Test. Spatiotemporal gait parameters were evaluated through the 10-Meter Walking Test, while lower limb power with the Jump test, using the BTS BAIOBIT® wireless inertial sensor. Static balance was examined via the IPODO baropodometric platform and Pediatric Balance Scale (PBS). Body composition was assessed with iDEXA and bioelectrical impedance analysis (BIA), and bone mineral density determined through standard iDEXA densitometry. Fatigue was evaluated with the Pediatric Quality of Life Inventory Multidimensional Fatigue Scale (PedsQL™ MFS), including general, sleep/rest, and cognitive domains.
Results: Twelve pediatric patients affected by XLH were enrolled (3 males, 9 females; mean age 7.58 ± 3.84 years; mean BMI 18.14 ± 2.33 kg/m²). Ten patients were within normal height ranges, though all fell below expected means for age. Handgrip strength, adjusted for sex and height, was within reference ranges in eight of ten patients. Manual dexterity was preserved in seven of eleven patients, while static balance was maintained in eleven of twelve. In the 10-meter walking test, right and left step cycle quality indexes averaged 90.0 ± 9.55% and 89.5 ± 11.51%. Jump test results showed mean maximum force 0.48 ± 0.22 kN and mean total power 619.72 ± 306.31 W. Baropodometric assessment revealed asymmetrical weight distribution in 50% of patients, with predominant forefoot loading in all (mean right forefoot 78.78 ± 18.15%). Height-adjusted DXA Z-scores were positive. iDEXA revealed appendicular muscle mass below age-adjusted means in seven of nine patients, although within normal limits; BIA showed reduced lean mass in seven of ten. Parent-reported fatigue was clinically significant in two of eleven patients for general fatigue and three of eleven for sleep/rest fatigue, while children reported milder subjective fatigue.
Conclusion: Paediatric XLH patients treated with burosumab demonstrated preserved muscle strength, and static balance, with residual deficits in manual dexterity, body composition, and gait parameters. These results support the need for a multidimensional evaluation to devolp personalised rehabilitation programs and monitor treatment efficacy.
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: Outcome measures and clinimetry
PP01.380
Investigation of Dysphagia Risk and Chewing Function in SMA Type II: A Cross-Sectional Descriptive Study
Assoc. Prof. Özgü İNAL ÖZÜN1, Prof. Emre ADIGÜZEL2, Assoc. Prof. Çağlar SOYLU1, Assoc. Prof. Duygu TÜRKER1, Assoc. Prof. Didem ARDIÇLI2, Miss Ceren Şevval KARATAŞ3, Miss Merve ÖZTÜRK3, Mrs Buse BİRBİR1, Prof. Deniz YÜKSEL4
1
University of Health Sciences Turkey, Gülhane Faculty of Physiotherapy and Rehabilitation, Ankara, Turkey.
2
University of Health Sciences Turkey, Ankara Bilkent City Hospital, Ankara, Turkey.
3
University of Health Sciences Turkey, Gulhane Institute of Health Sciences, Ankara, Turkey.
4
University of Health Sciences Turkey, Ankara Etlik City Hospital, Ankara, Turkey
Background: SMA patients frequently report problems such as impaired jaw function, fatigue during chewing, difficulty swallowing solid foods, and choking. Feeding difficulties such as swallowing problems and aspiration, and chewing problems are frequently reported in SMA Type I, less frequently in SMA Type II, and occasionally in SMA Type III. More than half of SMA Type II patients have been reported to experience dysphagia, which can result in severe complications, including aspiration pneumonia and death. Patients with SMA Type II are now included in the scope of drug treatment, and the drugs used have shown positive effects on SMA Type II patients. With these new developments, problems other than motor level decline (e.g., swallowing dysfunction) have become more important in children with SMA. However, the risk of dysphagia and chewing functions in children with SMA Type II have been addressed in a limited number of studies in the literature. In this context, this study was planned to investigate the risk of dysphagia and chewing function in children with SMA Type II.
Methods: Eighteen orally fed children with SMA Type II, aged between 6.4 and 13.4 (months), were included in the study. All children were receiving Nusinersen treatment. Descriptive information and feeding information of the participants were recorded using a socio-demographic form. The Pediatric Eating Assessment Tool (PEDI-EAT-10) was used to assess dysphagia and the Karaduman Chewing Performance Scale (KÇPS) was used to assess chewing function (0-4; 0: normal chewing). The child was given a standard biscuit, and their biting and chewing behaviors during feeding were video recorded for 3-5 minutes. The videos were then reviewed and analyzed.
Results: The study evaluated 10 girls and 8 boys. The prevalence of scoliosis was 77.8% (n=14). Sucking ability was present at birth in 83.3% (n=15), and all participants were fed in a sitting position (100%, n=18). 88.9% (n=16) of participants were able to tolerate all consistencies. The average feeding time per meal was determined to be 28.06 minutes. A history of lung infection in the last year was reported in 22.2% (n=4), and a history of intensive care in 38.9% (n=7). Daytime drooling problems were observed in 55.6% (n=10) and nighttime drooling problems in 22.2% (n=4). The distribution of the KÇPS was as follows: Level 0:50% (n=9), Level 1:33.3% (n=6), Level 2:11.1% (n=2), and Level 4:5.6% (n=1). The PEDI-EAT-10 score ranged from 0 to 27, with an average of 3.17 ± 6.39.
Conclusion: In the current study, feeding problems in children with SMA Type II can manifest in different ways. During the evaluation, families reported that their children did not experience any problems with chewing or swallowing. The assessments revealed that 33.3% of participants were at risk for dysphagia, and 50% experienced chewing problems. Another notable finding of the study was the evaluation of nighttime drooling problems in addition to daytime drooling problems in children with SMA Type II.
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: Swallowing evaluation in neuromuscular diseases
PP01.381
Quantitative Muscle Ultrasound as a Clinical Correlate in Fshd: Validation of a Rapid Protocol
Dr. Nurit Birman Har-Noy1, Dr. Omer Bouzaglo1, Prof. Vivian Drory1,2, Dr. David Kravitz1, Dr. Tahani Sheikh Saker1, Assoc. Prof. Alon Abraham1,2
1
Tel Aviv Sourasky Medical Center, Tel Aviv, Israel.
2
Tel-Aviv University, Tel-Aviv, Israel
Background: Facioscapulohumeral muscular dystrophy (FSHD) requires reliable and accessible objective biomarkers for clinical monitoring and therapeutic trials. Current quantitative ultrasound (US) methods often rely on echogenicity, which frequently requires specialized software. This study aimed to validate a rapid, generic protocol focusing on muscle thickness as a clinical correlate in FSHD.
Methods: Twenty-seven genetically confirmed adult FSHD patients (mean age 45.3 ± 12.8 years) were recruited between September 2023 and April 2024. A standardized US protocol was used to measure muscle thickness in eight relaxed muscles (biceps brachii, APB, FDI, ADM, quadriceps, tibialis anterior, EDB, and AHB) on the right side. For the biceps, quadriceps, and tibialis anterior, measurements were also taken during full contraction. Hyperechogenic muscles (Heckmatt 3-4) were assigned a thickness of zero. Sum scores of relaxed and contracted thicknesses were correlated with the FSHD Clinical Score (FSHD-CS), Clinical Severity Score (FSHD-CSS), 6-minute walk test (6-MWT), and manual muscle testing.
Results: Relaxed and contracted sum muscle thickness showed strong to very strong correlations with most clinical outcomes (r = 0.61 to 0.86, p < 0.01). Specifically, relaxed sum muscle thickness demonstrated a correlation of r = -0.86 with both FSHD-CS and FSHD-CSS. Correlation with the 6-MWT was r = 0.74 for relaxed sum muscle thickness and r = 0.61 for contracted sum muscle thickness. Muscle thickness measurements did not correlate with age, BMI, or symptom duration. The entire US evaluation was completed in less than ten minutes.
Conclusion: This rapid and reproducible ultrasound protocol demonstrates a strong correlation with disease burden in FSHD across multiple clinical scales. Unlike software-dependent echogenicity methods, muscle thickness measurement is easily implementable across routine US systems and diverse clinical settings. These findings suggest that muscle thickness sum scores can serve as an objective biomarker for monitoring disease progression and assessing treatment effects in clinical trials.
Abstract Topic Groups (Submission Categories)
Topic Group 5 –Diagnostic Methods and assessment in Neuromuscular Diseases: Ultrasound
Images or Table (Optional)
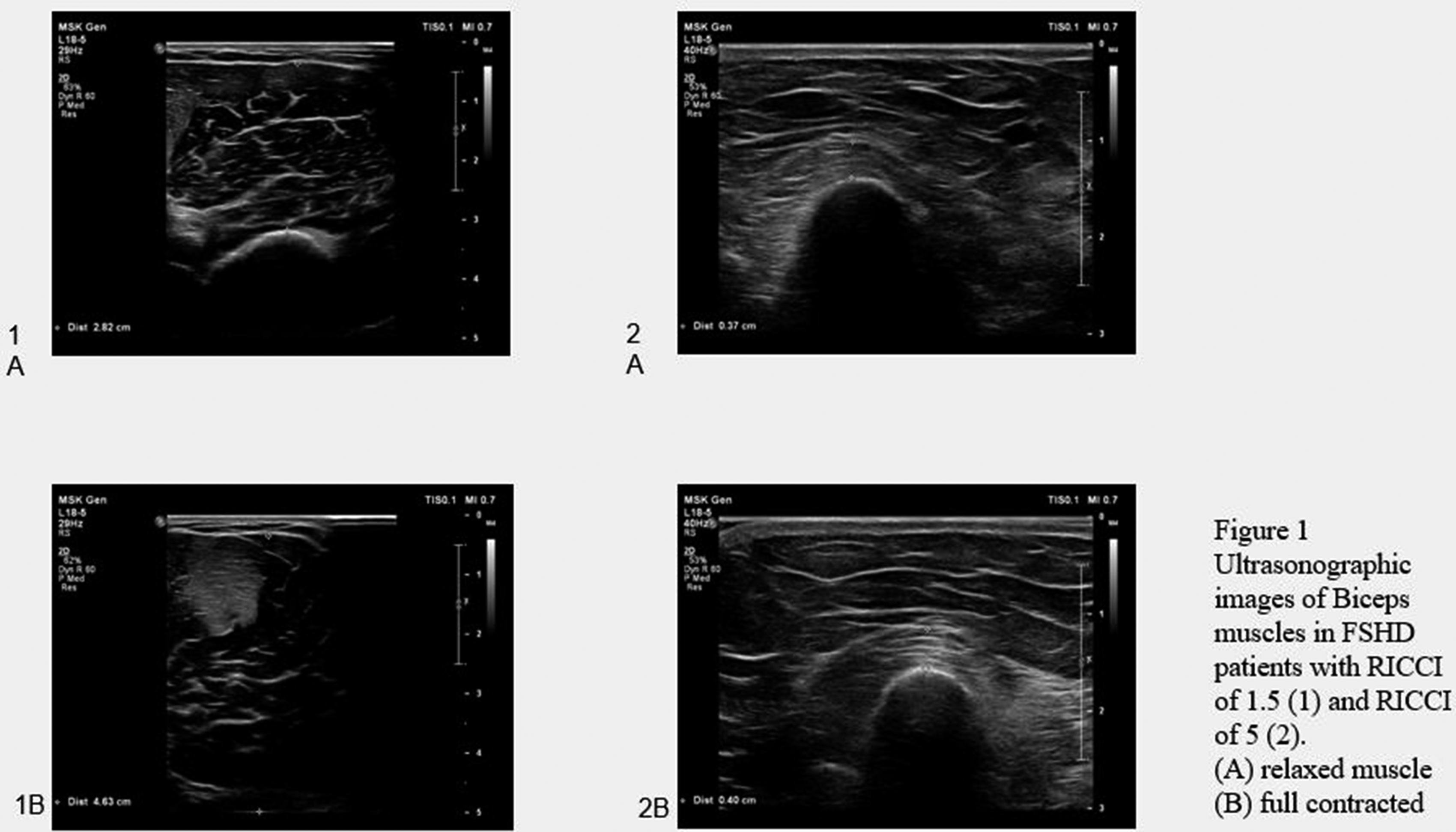
PP01.382
Neurofilament Light Chain As A Biomarker In Chronic Inflammatory Demyelinating Polyradiculoneuropathy: Insights From ADHERE
Dr. Roger Collet Vidiella1, Dr. Marta Caballero-Avila1, Dr. Tineke Casneuf2, Dr. Erik Hofman2, Dr. Geoffrey Istas2, Ms. Paula Llarch1, Dr. Lorena Martín-Aguilar1, Dr. Elba Pascual-Goñi1, Dr. Arne De Roeck2, Dr. Bianca Balbino2, Dr. Luis Querol1
1
Neuromuscular Diseases Unit, Hospital de La Santa Creu I Sant Pau, Universitat Autònoma de Barcelona, Barcelona, Spain.
2
argenx, Ghent, Belgium
Background: Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) is an immune-mediated neuropathy causing proximal and distal weakness, sensory disturbance, and possible irreversible disability. Neurofilament light chain (NfL) levels indicate axonal damage and may potentially serve as a disease activity and prognostic biomarker in inflammatory disorders, including CIDP. NfL levels were analyzed in ADHERE (NCT04281472), the largest CIDP cohort to date.
Methods: Participants with probable/definite active CIDP (demonstrated by treatment withdrawal run-in phase) received open-label, weekly subcutaneous (SC) efgartigimod PH20 1000 mg (stage A) for ≤12 weeks. Responders entered stage B and were randomized (1:1) to weekly efgartigimod PH20 SC 1000 mg or placebo for ≤48 weeks. Serum NfL (sNfL) was measured longitudinally in 214 participants, from which NfL z-score (zNfL) was determined.
Results: During stage A, sNfL levels remained stable in responders with baseline levels within healthy reference range (≤20 pg/mL, n=114); sNfL reduced by 18% over time in responders with elevated baseline levels (>20 pg/mL, n=36). In participants receiving continuous efgartigimod through stage B, sNfL reduced by 35% by week 24 (n=13). Mean (SD) sNfL levels at stage A baseline were 18.9 (22.6) pg/mL (n=214), corresponding to a mean (SD) zNfL of 0.64 (1.57). Elevated baseline zNfL levels were associated with a higher CIDP disease activity status (CDAS 5). zNfL scores at stage A baseline were the highest in samples fromCIDP-treatment-naïve participants versus those who had previously received CIDP treatment.
Conclusion: ADHERE represents the broadest CIDP dataset evaluated for NfL. Baseline NfL levels correlated with CDAS scores, with higher levels indicating more active disease. Among efgartigimod-treated responders who had elevated baseline sNfL, we observed a reduction in sNfL in both stages A and B. NfL levels may serve as a contextual biomarker, alongside clinical assessment. Further analysis will determine whether NfL can inform on disease monitoring.
Abstract Topic Groups (Submission Categories)
Topic Group 6 - Basic Sciences in Neuromuscular Diseases: Immune Mechanisms in Neuromuscular Diseases
PP01.383
Treg Cells Attenuate Neuroinflammation and Protect Neurons in a Mouse Model of Parkinson's Disease
Dr. Yan Huang
Nantong University, Nantong, China
Background: Regulatory T cells (Tregs), which secrete transforming growth factor (TGF)-β and interleukin (IL)-10, have essential role in anti-inflammatory and neurotrophic functions. Herein, we explore the neuroprotection of Tregs in Parkinson's disease (PD) by adoptive transfer of Tregs.
Methods: Tregs, isolated by magnetic sorting, were activated in vitro and then were adoptively transferred to 1-methyl-4-phenyl-1,2,3,6- tetrahydropyridine (MPTP)-treated mice. Neuroinflammation, dopaminergic neuronal loss and behavioral changes of PD mice were evaluated. Live cell imaging system detected a dynamic contact of Tregs with MN9D cells that were stained with CD45 and galectin-1, respectively.
Results: Tregs prevented MPTP-induced dopaminergic neuronal loss, behavioral changes, and attenuated the inflammatory reaction in the brain. When blockade the LFA-1 activity in Tregs or the ICAM-1 activity in endothelial cells, the percentage of Tregs in substantia nigra (SN) decreased. CD45 and galectin-1 were expressed by Tregs and MN9D cells, respectively. CD45-labeled Tregs dynamically contacted with galectin-1-labeled MN9D cells. Inhibiting CD45 in Tregs impaired the ability of Tregs to protect dopaminergic neurons against MPP+ toxicity. Similarly, galectin-1 knockdown in MN9D cells reduced the ability of Tregs neuroprotection. Adoptive transfer of Tregs protects dopaminergic neurons in PD mice by a cell-to-cell contact mechanism underlying CD45-galectin-1 interaction.
Conclusion: Treg Cells can attenuate neuroinflammation and protect dopaminergic neurons in MPTP-induced Parkinson's disease mice.
Abstract Topic Groups (Submission Categories)
Topic Group 6 - Basic Sciences in Neuromuscular Diseases: Immune Mechanisms in Neuromuscular Diseases
PP01.384
Microglia-Derived Exosomal ciRS-7 Mediates IL-17A Effect of Promoting Neurodegeneration in an Experimental Parkinson's Disease
Prof. Yi-Hua Qiu
Nantong University, Nantong, China
Background: Parkinson’ s disease (PD) is a chronic neurodegenerative disorder characterized by progressive loss of dopaminergic neurons in the substantia nigra (SN). Our research has demonstrated that the levels of interleukin (IL)-17A are elevated in the SN of rodent models of PD, and that IL-17A accelerates neurodegeneration in PD depending on microglial activation. Furthermore, existing studies indicate that exosomes released by activated microglia may play a significant role as mediators of neurodegeneration in PD.
Methods: Herein, we demonstrated that BV-2-derived exosomes were taken up by ventral mesencephalic (VM) dopaminergic neurons, and mediated IL-17A effect of promoting dopaminergic neuronal injury.
Results: IL-17A-treated BV-2-derived exosomes altered neuronal miR-7 and SNCA expression and promoted dopaminergic neuronal injury in vitro. Inhibiting BV-2 exosome formation and secretion by GW4869 alleviated dopaminergic neuronal injury. Silencing ciRS-7 in BV-2 altered neuronal miR-7 and SNCA expression and mitigated dopaminergic neuronal injury. Overexpression of ciRS-7 in VM neurons altered neuronal miR-7 and SNCA expression and promoted dopaminergic neuronal injury. Injection with exosomes derived from IL-17A-treated BV-2 altered ciRS-7, miR-7 and SNCA expression in SN in MPTP-intoxicated mice and promoted nigrostriatal dopaminergic neurodegeneration and motor impairment. However, injection with exosomes derived from IL-17A and ciRS-7-shRNA treated BV-2 attenuates the manifestations mentioned above.
Conclusion: These findings suggest that microglia-derived exosomal ciRS-7 mediates IL-17A effect of promoting neurodegeneration via miR-7 and SNCA targets and may provide a new paradigm to study the pathology of PD.
Abstract Topic Groups (Submission Categories)
Topic Group 6 - Basic Sciences in Neuromuscular Diseases: Immune Mechanisms in Neuromuscular Diseases
PP01.385
IL-17A Exacerbates Neuroinflammation and Neurodegeneration by Activatingmicroglia in Rodent Models of Parkinson's Disease
Dr. Zhan Liu
Nantong University, Nantong, China
Background: Neuroin ammation has been involved in pathogenesis of Parkinson's disease (PD), a chronic neurodegenerativedisease characterized neuropathologically by progressive dopaminergic neuronal loss in the substantia nigra(SN). We recently have shown that helper T (Th)17 cells facilitate dopaminergic neuronal loss in vitro. Herein,we demonstrated that interleukin (IL)-17A, a proin ammatory cytokine produced mainly by Th17 cells, con-tributed to PD pathogenesis depending on microglia.
Methods: Mouse and rat models for PD were prepared by intraperitoneal injection of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) or striatal injection of 1-methyl-4-phenylpyridinium (MPP+), respectively.
Results: Both in MPTP-treated mice and MPP+-treated rats, blood–brain barrier (BBB) was disrupted and IL-17A level increased in the SN but not in cortex. E ector T (Te ) cells that were adoptively transferred via tail veins in ltrated into the brain of PD mice but not into that of normal mice. The Te cell transfer aggravated nigrostriatal dopaminergic neurodegeneration, microglial activation and motor impairment. Contrarily, IL-17A de ciency alleviated BBB disruption, dopaminergic neurodegeneration, microglial activation and motor impairment. Anti-IL-17A-neutralizing antibody that was injected into lateral cerebral ventricle in PD rats ameliorated the manifestations mentioned above. IL-17A activated microglia but did not directly a ect dopaminergic neuronal survival in vitro. IL-17A exacerbated dopaminergic neuronal loss only in the presence of microglia, and silencing IL-17A receptor gene in microglia abolished the IL-17A e ect. IL-17A-treated microglial medium that contained higher concentration of tumor necrosis factor (TNF)-α facilitated dopaminergic neuronal death. Further, TNF-α-neutralizing antibody attenuated MPP+-induced neurotoxicity
Conclusion: The ndings suggest that IL-17A accelerates neurodegeneration in PD depending on microglial activation and at least partly TNF-α release.
Abstract Topic Groups (Submission Categories)
Topic Group 6 - Basic Sciences in Neuromuscular Diseases: Immune Mechanisms in Neuromuscular Diseases
PP01.386
Characterization of VCP-Interacting Proteins in VCP-iPS Cell-Derived Skeletal Muscle Cells
Dr. Fumiaki Saito1, Dr. Masato Inoue1, Ms. Miki Ikada1, Dr. Shunsuke Kobayashi1, Dr. Genta Ito2, Dr. Hidetoshi Sakurai3, Dr. Yuko Miyagoe-Suzuki1
1
Department of Neurology, School of Medicine, Teikyo University, Tokyo, Japan.
2
Department of Biomolecular Chemistry, Faculty of Pharmaceutical Sciences, Teikyo University, Tokyo, Japan.
3
Department of Clinical Application, Center for iPS Research and Application, Kyoto University, Kyoto, Japan
Background: Mutations in the valosin-containing protein (VCP) gene cause multisystem proteinopathy (MSP), manifesting as inclusion body myopathy (90%), frontotemporal dementia (30%), ALS (15%), and Paget’s disease of bone (50%). VCP is an AAA-type molecular chaperone that forms a homohexamer and collaborates with many adapter proteins to maintain proteostasis under normal, stress, and disease conditions. To clarify whether there are differences in the types and affinities of VCP-interacting proteins between normal cells and patient cells, we generated VCP-iPS cells and differentiated them into skeletal muscle cells.
Methods: We generated iPS cells from a patient carrying c.572G>A (R191Q) missense mutation in the VCP gene. Isogenic control iPS cells were generated by genome editing using homology directed repair. These iPS cells were differentiated into skeletal muscle either by overexpression of MyoD or by an improved EZ-sphere method. Stress granules were induced with sodium arsenite and detected using a G3BP1 antibody. We, further, generated iPS cells with a spot-tag inserted at the C-terminus of the endogenous VCP gene by genome editing. The tagged VCP was pulled-down by Spot-Trap magnetic agarose (Chromotek) after cross-linking with DSP, then analyzed for its interacting proteins by LC-MS.
Results: R191Q mutation did not affect the formation of stress granules induced by sodium arsenite in myoblasts differentiated from iPS cells. However, it delayed the stress granules disassembly after removal of arsenite. Spot-Tag pull-down assay identified several VCP-interacting proteins that had not been reported previously, in addition to known VCP adapters.
Conclusion: Skeletal muscle cells induced from VCP- iPS cells provide an experimental model suitable for elucidating the molecular pathogenesis of VCP-myopathy, which has been conducted mostly using non-muscle cells or over-expression systems. In fact, using VCP- iPS cells with tagged endogenous VCP, we have successfully identified VCP-interacting proteins in skeletal muscle. We aim to further analyze interacting partners of VCP in patient cells to develop therapeutic strategies.
Abstract Topic Groups (Submission Categories)
Topic Group 6 - Basic Sciences in Neuromuscular Diseases: Muscle Atrophy / Degeneration
PP01.387
Targeted Degradation of Polyglutamine-Expanded Androgen Receptor Improves Spinal and Bulbar Muscular Atrophy
Dr. Kuo-Ting Wei1, Dr. Ya-Fen Liu1, Prof. Hong-Yo Kang1,2
1
Graduate Institute of Clinical Medical Sciences, College of Medicine, Chang Gung University, Taoyuan City, Taiwan.
2
Center for Hormone and Reproductive Medicine Research, Department of Obstetrics and Gynecology, Kaohsiung Chang Gung Memorial Hospital, Kaohsiung, Taiwan
Background: Spinal and bulbar muscular atrophy (SBMA) is a rare X-linked neuromuscular disorder caused by CAG trinucleotide repeat expansion in the androgen receptor (AR) gene, resulting in an expanded polyglutamine (polyQ) tract within the AR protein. Ligand-dependent misfolding and aggregation of polyQ-AR lead to progressive lower motor neuron degeneration and muscle atrophy. Despite advances in understanding SBMA pathogenesis, the mechanisms underlying selective neuromuscular vulnerability remain incompletely defined, and disease-modifying therapies are currently lacking.This study aimed to develop and evaluate proteolysis-targeting chimeras (PROTACs) as a targeted protein degradation strategy for eliminating pathogenic polyQ-AR. We sought to assess their efficacy in cellular and in vivo SBMA models and to elucidate the molecular pathways underlying PROTAC-mediated polyQ-AR degradation.
Methods: A series of AR-targeting PROTACs were evaluated in neuron-2a cells stably expressing AR-97Q and in SBMA transgenic mice. PolyQ-AR protein levels were assessed following PROTAC treatment, with proteasome involvement examined using MG132 inhibition. Isobaric tags for relative and absolute quantitation (iTRAQ)–based proteomics were employed to identify pathways associated with protein degradation. Functional outcomes were assessed using grip strength, rotarod performance, body weight monitoring, and survival analysis. Histological analyses of skeletal muscle and spinal anterior horn were performed to evaluate neuromuscular pathology.
Results: PROTAC treatment markedly reduced aggregated AR-97Q protein levels in both cellular and tissue models, an effect reversed by proteasome inhibition with MG132. Mechanistically, PROTACs enhanced K48-linked polyubiquitination of polyQ-AR, promoting proteasomal degradation. In SBMA transgenic mice, PROTAC administration significantly improved muscle strength, motor coordination, and body mass, and prolonged survival. Histopathological analyses revealed substantial attenuation of neurogenic muscle atrophy in treated animals.
Conclusion: These findings demonstrate that PROTAC-mediated degradation of polyQ-expanded AR effectively ameliorates molecular, functional, and histopathological features of SBMA. Targeting pathogenic AR through selective ubiquitination and proteasomal degradation represents a promising therapeutic strategy for SBMA and potentially other polyglutamine-associated neurodegenerative disorders.
Abstract Topic Groups (Submission Categories)
Topic Group 6 - Basic Sciences in Neuromuscular Diseases: Muscle Atrophy / Degeneration
PP01.388
MyoScreen, a Human Skeletal Muscle Platform, Reveals Novel Targets and Muscle-Preserving Small Molecules
Dr. Mélanie Flaender, Dr. Louise Griveau, Ms. Marine Foray, Ms. Eve Duchemin-Pelletier, Ms. Caroline Roelants, Dr. Bianca Freytag, Dr. Erwann Ventre, Dr. Joanne Young
CYTOO SA, Grenoble, France
Background: Muscle wasting is associated with genetic, metabolic, and chronic disorders, including muscular dystrophy, sarcopenia, type 2 diabetes, cancer cachexia, and chronic kidney disease. Loss of muscle mass and function is also an emerging concern with the growing use of GLP-1R agonists for obesity treatment. One strategy to counter muscle loss is bimagrumab, an injectable anti-catabolic monoclonal antibody that blocks myostatin/activin type II receptors (ACTRIIA and B), key negative regulators of skeletal muscle growth. Bimagrumab has shown clinical efficacy by increasing muscle mass across diverse populations, including patients with sporadic inclusion body myositis, sarcopenia, and during weight loss. Identification of an oral small-molecule inhibitor targeting this clinically validated pathway would therefore represent a therapeutic advance, enabling simpler and more scalable manufacturing.
Methods: Experiments were conducted using the in vitro MyoScreen drug discovery platform. This human skeletal muscle system combines highly reproducible, micropatterned primary myotubes (healthy and disease-derived) in 96- or 384-well formats with high-content imaging and quantitative multiparametric readouts. The platform is specifically designed to assess clinically relevant endpoints with low variability and high sensitivity, including myotube differentiation and fusion index, myotube size, atrophy rescue, contractility, acetylcholine receptor clustering, calcium flux, glucose uptake, protein synthesis, autophagy accumulation and mitochondrial dysfunction.
Target identification and validation were performed using a siRNA-mediated knockdown strategy within a myostatin-induced muscle wasting model implemented on MyoScreen. Candidate intracellular effectors were selected based on their ability to rescue muscle size and impaired differentiation following knockdown. Small molecule inhibitors of the myostatin/activin signaling axis were screened across healthy and multiple atrophy-induced muscle models
Results: Most current therapies targeting the activin/myostatin pathway are biologics directed against soluble ligands or extracellular receptor domains. We instead prioritized small-molecule inhibitors acting against intracellular components of the signaling cascade, given the potential for improved specificity and reduced off-target effects. Target validation identified ALK5/TGFβR1 as a strong downstream mediator of myostatin-induced muscle wasting with knockdown significantly increasing fusion index and myotube size.
A panel of ten literature-reported ALK5/TGFβR1 inhibitors was subsequently evaluated for hypertrophic-inducing activity in healthy human myotubes. One compound displayed superior potency and selectivity, robustly promoting hypertrophy with no detectable cytotoxicity.
This ALK5/TGFβR1 antagonist preserved muscle size and differentiation in other MyoScreen atrophy myotube models (TGFb, TNFa, dexamethasone-induced), and was more effective than the clinical benchmark reference, bimagrumab. Further characterization revealed other positive effects such as increased myotube contractility and maturation, augmented acetylcholine receptor clustering, elevated calcium flux, and upregulation of the late myogenic marker, myosin heavy chain. These structural and functional improvements were accompanied by increased basal and insulin-stimulated glucose uptake.
Conclusion: Collectively, the data indicates that selective ALK5/TGFβR1 inhibition drives skeletal muscle hypertrophy and a transition toward a more mature and metabolically active phenotype, highlighting ALK5/TGFβR1 as a potential intracellular target for muscle-preserving therapies. These findings also establish MyoScreen as a clinically translatable human muscle platform that robustly models disease-relevant muscle states and recapitulates myofiber rescue by established standards such as bimagrumab. Importantly, this platform approach enables efficient discovery and in-depth characterization of next-generation drug candidates aimed at preserving muscle health.
Abstract Topic Groups (Submission Categories)
Topic Group 6 - Basic Sciences in Neuromuscular Diseases: Muscle Atrophy / Degeneration
Images or Table (Optional)
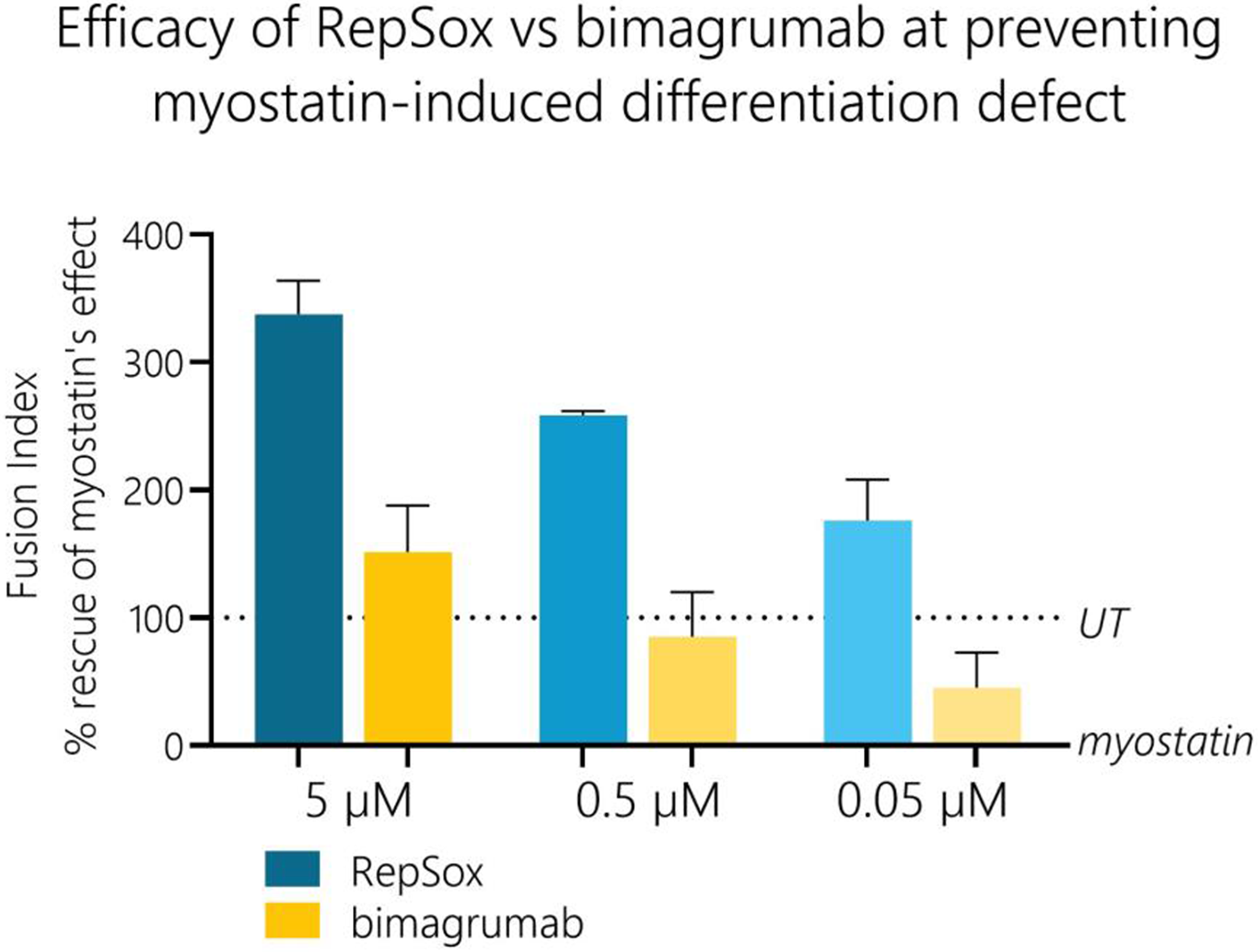
PP01.389
High-Throughput 3D Light-Sheet Imaging of Mouse Hindlimbs Enables Skeletal Muscle Morphometrics in Muscle Wasting Models
Dr. Max Hahn, Dr. Alex Addinsall, Dr. Nicolas Eskesen, Dr. Anitta Kinga Sárvári, Dr. Kasper Andersen, Dr. Alexander Hamilton, Dr. Estrid Pedersen, Dr. Marco Tozzi, Dr. Henrik Hansen, Dr. Urmas Roostalu
Gubra, Hørsholm, Denmark

Dr. Max Hahn
Background: Anatomically precise and quantitative assessment of skeletal muscle volume and morphology remains a major challenge in preclinical research of sarcopenia and related muscle disorders. Conventional readouts, such as the extrapolation of 2D stereological data originating from a single section, as well as whole-body lean mass lack spatial resolution and fail to capture changes in muscle architecture that critically determine muscle function. Rodent models of conditions involving muscle remodelling induced by pharmacological intervention (e.g.,Dexamethasone, Formoterol) aging or obesity (DIO) provide complementary and clinically relevant contexts to evaluate advanced muscle imaging approaches. Here, we characterized a scalable, AI-enabled high-resolution imaging platform based on light-sheet fluorescence microscopy (LSFM) for holistic 3D quantitative assessment of muscle volume, vascularization, innervation, and fibrosis of individual skeletal muscles in the intact mouse hindlimb.
Methods: A fully automated pipeline combining optical tissue clearing, autofluorescence- and immunofluorescence-based LSFM, and AI-driven image analysis was established for intact mouse hindlimbs. In pharmacological studies, 12-week-old C57BL/6J mice were treated for 21 days with vehicle, Dexamethasone (IP, QD), or Formoterol (IP, QD). In aged-sarcopenia models, male C57BL/6J mice included young (4 months), aged (13 months), and old (23 months) lean controls, as well as aged DIO mice fed high-fat diet for 13 months. Whole hindlimbs were scanned ex vivo at micrometer resolution and downsampled for analysis. A 2D U-Net–based segmentation approach enabled volumetric quantification of individual muscles. Multidimensional LSFM readouts included muscle volume, fiber-type composition, capillary density, innervation, and fibrosis, complemented by echoMRI, muscle weights, ex vivo contractile function, and conventional histology.
Results: The LSFM pipeline enabled reproducible alignment and high-throughput, anatomically precise quantification of individual hindlimb muscles across diverse models. In pharmacological studies, Dexamethasone induced reductions in whole-body lean mass and gastrocnemius weight, whereas Formoterol increased lean mass and gastrocnemius weight, with corresponding changes in 3D muscle volume detectable at micrometer resolution. Aged DIO mice exhibited substantial increases in fat mass with only marginal lean mass gains compared with aged-matched lean controls, accompanied by pronounced alterations in muscle architecture. LSFM-derived 3D metrics bridged the gap between whole-body composition data and histological endpoints, revealing spatially resolved muscle phenotypes not captured by traditional methods.
Conclusion: We present a scalable LSFM-based platform for high-resolution, multidimensional 3D assessment of skeletal muscle remodelling across pharmacological, aging, and obesity models. This approach effectively captures dynamic changes in muscle mass, integrates structural and quality-related endpoints, and provides a mesoscopic bridge between in vivo MRI and ex vivo histology. The platform holds strong potential for preclinical drug development, mechanistic studies of muscle biology, and future translational and cross-species applications in sarcopenia and related muscle disorders.
Abstract Topic Groups (Submission Categories)
Topic Group 6 - Basic Sciences in Neuromuscular Diseases: Muscle Atrophy / Degeneration
PP01.390
Unmet Support Needs of ALS Patients Following Failed Clinical Trials
Dr. Dalisha Dalisha
National Hospital Of Neurology and Neurosurgery, London, United Kingdom
Background: Amyotrophic lateral sclerosis (ALS) is a progressive, fatal neurodegenerative disease with limited therapeutic options. Participation in clinical trials offers patients access to experimental treatments, close clinical monitoring, and often renewed hope. However, when trials fail to meet endpoints or are terminated early, patients frequently experience an abrupt loss of support, with minimal structured follow up or guidance. This transition can negatively impact well being, yet it remains underexplored in ALS research.
Patients returning to routine care after trial completion often lose access to multidisciplinary teams, regular monitoring, and communication with research staff. Trial failure can lead to emotional distress, feelings of abandonment, and declining quality of life. Caregivers experience heightened burden and uncertainty regarding future care. While informed consent addresses risks and potential lack of efficacy, it rarely prepares participants for the emotional and practical consequences of trial discontinuation. The absence of standardized post trial support highlights a gap between research obligations and patient centered care. Ethical research practice requires proactive planning for continuity of care beyond trial endpoints.
Methods: Proposing a prospective, pilot study to evaluate the feasibility, acceptability, and potential impact of a structured post-trial support program for ALS clinical trial participants. Adult patients approaching trial completion or experiencing early termination, along with their caregivers, will be invited to participate.
The intervention consists of a standardized post trial transition program initiated near trial completion and extending for six months. Core components include:
1. Structured end-of-trial consultations with research and clinical staff to communicate trial outcomes, manage expectations, and outline next steps.
2. Reintegration into multidisciplinary ALS clinic care.
3. Referral to psychological, social work, and palliative care support as indicated.
4. Written and verbal guidance regarding symptom management and potential future research opportunities.
Planned assessments include patient-reported quality of life, psychological distress, and perceived continuity of care, as well as caregiver burden and satisfaction. Semi structured interviews with patients, caregivers, and staff will explore experiences of trial transition and perceived value of post trial support. Feasibility metrics, including participation rates, referral uptake, and adherence to consultations, will inform recommendations for broader implementation.
Results: Expected Outcomes Anticipating that the program will be feasible and acceptable to patients and caregivers. Expected benefits include improved patient-reported quality of life, reduced psychological distress, enhanced perception of continuity of care, and decreased caregiver burden. Qualitative data are expected to demonstrate the importance of clear communication, coordinated care, and structured support following trial completion. Feasibility metrics will provide guidance for integrating post-trial support into future ALS clinical trial designs.
Conclusion: Addressing the unmet post trial support needs of ALS patients is critical for ethical research conduct and patient-centered care. Implementing a structured support pathway has the potential to mitigate emotional harm, preserve trust in research, and enhance the overall experience of both patients and caregivers. This approach could serve as a model for integrating patient-centered post-trial care into neurodegenerative disease research.
Abstract Topic Groups (Submission Categories)
Topic Group 8 - Patient Related Issues: Palliative Care
PP01.391
Living With TNPO3-Related Limb-Girdle Muscular Dystrophy D2: A Quality of Life Analysis
Dr. Alicia Aurora Rodríguez1, Dr. Irune García1, Miss Clara Lépée-Aragón1, Dr. Corrado Angelini2, Dr. Óscar Martínez1, Dr. Imanol Amayra1
1
University of Deusto, Bilbao, Spain.
2
University of Padua, Padua, Italy
Background: Limb–girdle muscular dystrophy (LGMD) is a group of rare genetic disorders causing progressive weakness and muscle atrophy in the proximal limbs. They are classified into autosomal dominant and recessive forms. About 10% of these dystrophies are dominant and 90% are recessive. Research on LGMD is limited, particularly for low-prevalence subtypes such as the dominant form limb–girdle muscular dystrophy D2 (LGMD-D2), which shows variable age of onset and occurs in both familiar and sporadic cases. First identified in an Italo-Spanish family, LGMD-D2 is characterized by generalized muscle atrophy and can lead to disability and dependence on others. There is currently no curative treatment, and existing therapies are symptomatic, with the disease negatively affecting life expectancy and quality of life. This study represents the first investigation into the quality of life of individuals affected by a dominant form of limb–girdle muscular dystrophy, specifically LGMD-D2.
Methods: A cross-sectional study was conducted. The study further seeks to examine patient-reported outcomes in functional performance and quality of life of patients, as well as differences between LGMD-D2 and recessive forms of LGMD. The tools employed included a sociodemographic questionnaire, the Gait-Scale-Gowers-Chair scale (GSGC), and the INQoL instrument. The sample comprised six adults diagnosed with LGMD-D2 and three patients with recessive LGMD. Participants were recruited through patient associations and a hospital in Padua. The study was approved by the Commission for Responsible Ethics (Ref: ETK-39/18-19).
Results: Individuals affected by LGMD-D2 experience multiple symptoms that result in disability and, consequently, dependence on assistance. The findings of the present study indicate that LGMD-D2 has a more pronounced impact on activities of daily living, fatigue, muscle pain, and independence compared with other LGMD subtypes. The results also suggest that age may influence quality of life, and that muscle weakness represents a particularly disabling feature of this variant.
Conclusion: People affected by LGMD-D2 have multiple symptoms that lead to disability and dependence, which ultimately lead to dependence on their environment. The present study shows that they seem to have a greater impact on the activities of daily living, independence, the emotional sphere, and body image perception than other LGMD pathologies or other NMDs, considering the limitations of the comparisons, which were only at a descriptive level. It also appears that the impact is greater if the affected person is older, and that muscle weakness is a very disabling symptom in this variant. In the context of ongoing research aimed at developing new treatments in LGMD, it is crucial to identify the aspects most significantly affected by the disease.
Abstract Topic Groups (Submission Categories)
Topic Group 8 - Patient Related Issues: Palliative Care
Invited Speaker Program – Not peer reviewed
PL01.01
Redefining Muscle Disease Management: A Roadmap to 2030
Dr. Benedikt Schoser
Friedrich-Baur-Institute dep. of Neurology LMU Clinic, Munich, Germany
Background: Neuromuscular disorder management is evolving through genomic medicine, digital health, and proactive strategies. The focus shifts from simply alleviating symptoms to implementing targeted, disease-modifying treatments, including genetic therapies and stem cell research. Precision medicine allows for personalized therapies based on genetic profiles, often combining agents like antisense oligonucleotides (ASOs) or gene vector therapy with protective or anti-inflammatory agents to enhance outcomes. Regenerative approaches involve stem cell and tissue engineering for muscle repair, driven by the development of muscle organoids, replacing earlier animal-driven strategies. AI and digital tools support diagnosis and ongoing monitoring through wearable devices and integrated electronic health data. Digital phenotyping with AI wearables enables real-world, long-term functional assessment, reducing the need for sporadic clinic visits. Diagnostic methods are advancing with multi-omic profiling to identify predictive biomarkers tailored to individuals. Standardized, multidisciplinary care pathways will ensure smooth pediatric-to-adult transitions and help reduce disparities. The use of new biomarkers in trials will speed up drug approval and access to therapies. Achieving these objectives requires collaboration among academia, industry, regulators, and patients living with neuromuscular disorders to lower barriers. Integrating technology into healthcare delivery aims to restore function and achieve remission in incurable muscle diseases.
Conclusion: By 2030, advances in molecular science, digital infrastructure, and policies should usher in a “golden age” of neuromuscular medicine, providing equitable, life-changing therapies for our patients.
PL01.02
Gene Diagnostics in 2030
Prof. Vincenzo Nigro1,2, Assoc. Prof. Annalaura Torella2
1
Telethon Institute of Genetics and Medicine, Pozzuoli, Italy.
2
Università della Campania Luigi Vanvitelli, Naples, Italy
Background: Neuromuscular disorders (NMDs) comprise a highly heterogeneous group of rare diseases in which delayed or missed molecular diagnosis still limits access to targeted therapies and appropriate genetic counselling. Experience from undiagnosed disease programs has shown that deep phenotyping combined with systematic genomic investigation can resolve a substantial proportion of long‑standing diagnostic odysseys, particularly in patients with complex neurological phenotypes. Looking toward 2030, decreasing sequencing costs, the shift from exome to whole‑genome sequencing (WGS), and large, federated rare disease initiatives in Europe are expected to make early genome‑first diagnostics the standard of care.
Methods: Building on a decade of activity of a tertiary undiagnosed disease program (TUDP) focused on pediatric and adult NMDs, we retrospectively evaluated the impact of a progressive transition from targeted panels and whole‑exome sequencing to trio‑based WGS (short and long read) as the first‑line test combined with RNA sequencing. The analysis incorporated diagnostic yield, time‑to‑diagnosis, detection of copy‑number and structural variants, reanalysis rates, and downstream clinical impact, and was used to model a 2030 scenario integrating high‑throughput short‑read WGS, improved structural variant calling, and AI‑assisted variant interpretation.
Results: In the undiagnosed program experience, the introduction of systematic short and long read WGS in trios combined with RNA sequencing increased the overall diagnostic yield for NMDs allowed the identification of pathogenic non‑coding, structural and complex alleles that had been missed by prior testing. WGS‑based copy‑number variant detection at 35× coverage proved at least comparable to chromosomal microarrays for multi‑exon events, while simultaneously providing single‑nucleotide and indel resolution in all relevant genes. Periodic reanalysis, enabled by rich phenotypic metadata and continuously expanding population reference datasets, resulted in additional diagnoses over time and facilitated participation in gene discovery consortia. By extrapolating these data to a 2030 framework—characterized by sub‑$200 genomes, harmonized data standards, and integrated European rare disease infrastructures—we estimate that genome‑first diagnostic pathways could achieve timely genetic confirmation in a clear majority of individuals with suspected inherited NMDs, while also generating high‑value longitudinal datasets for natural history and therapy development.
Conclusion: By 2030, gene diagnostics for neuromuscular disorders will likely be centred on early, trio‑based WGS with comprehensive single‑nucleotide, copy‑number and structural variant detection, embedded in undiagnosed disease networks and rare disease ecosystems. The cumulative TUDP experience underscores that the key determinants of success are not only cheaper and more powerful sequencing platforms, but also standardized deep phenotyping, interoperable data infrastructures, iterative reanalysis, and close integration between diagnostic laboratories, clinical NMD expertise and research consortia. These elements together can shorten the diagnostic odyssey, expand access to emerging disease‑modifying treatments, and provide a scalable blueprint for rare disease genomics in the next decade.
PL01.03
Towards effective low-dose gene therapy in severe muscular dystrophy models using a novel AI-designed AAV
Dr. Isabelle Richard, Mrs Eva Petat, Mrs Laurence Suel, Mrs Nathalie Bourg-Alibert, Dr. Sonia Albini, Dr. Abbass Jaber, Mr. Jérôme Poupiot, Dr. Anthony Brureau, Dr. Evelyne Gicquel, Dr. Ai Vu Hong
Genethon, Evry-Courcouronnes, France
Background: Muscular dystrophies (MDs) are debilitating genetic disorders marked by progressive muscle degeneration, with no curative treatments currently available. Adeno-associated virus (AAV)–mediated gene therapy has shown clinical promise for monogenic MDs, yet its broader application remains limited by the need for very high systemic doses, dose-related toxicities, exclusion of patients with pre-existing neutralizing antibodies, and substantial manufacturing burden. Recent advances in AAV engineering, including rational capsid design, directed evolution, and high‑throughput screening, are driving the development of vectors with improved tropism, enhanced transduction efficiency, and superior safety profiles for therapeutic applications.
Methods: A myotropic AAV capsid, LICA1, was developed to target skeletal muscle across species via binding to the conserved receptor integrin αVβ6. We further developed an AI-guided engineering strategy that generates improved αVβ6-binding of this caspid. In this process capsid sequence-structure pairs were concurrently optimized for thermostability using physics-based modeling, sequence plausibility using large protein language models, and sequence-structure compatibility using deep-learning–based protein design. Top AI-designed variants, together with AAV9 and LICA1 controls, were evaluated in a low-N barcoded library in human muscle models in vitro and in mouse biodistribution studies in vivo.
Results: We previously developed LICA1, showing that cross-species myotropism can be achieved by redirecting AAV tropism toward the conserved receptor integrin αVβ6. We further improved this capsid through an AI-guided strategy by jointly optimizing sequence-structure through thermostability modelling, use of protein language model and sequence design. Among top AI-designed variants, the lead capsid, LICA3, achieves logarithmic improvements in skeletal and cardiac muscle transduction, surpassing state-of-the-art myotropic capsids, together with strong liver-detargeting and improved manufacturability.
These myotropic capsids were tested in several models of muscular dystrophies by systemic administration at a low dose. In these experiments, while LICA1 was demonstrating superior gene transfer than AAV9, LICA3 significantly outperformed LICA1, achieving near-complete myofiber transduction, including in highly affected diaphragm, markedly reducing fibrosis and normalizing serum MYOM3 levels to near-wild-type levels, and fully restoring muscle function.
Conclusion: These results demonstrate that AI-guided design can overcome long-standing barriers in vector engineering, providing a scalable framework for the development of safe, high-potency gene therapies. Collectively, these results also establish LICA3 as a highly potent, clinically promising gene therapy vector for muscle diseases.
Improvement of capsid properties is one element to consider for ensuring higher safety and efficacy of gene therapy. Another important element is the choice of promoters and regulator elements to achieve expression at the right place, right time and right level.
PL03.02
Unravelling the Presymptomatic Phase of ALS: Clinical, Genetic, and Molecular Advances Toward Early Detection
Prof. Andrea Malaspina
UCL Queen Square MND Centre, Institute of Neurology, London, United Kingdom
Background: Capturing the presymptomatic phase of ALS through the lens of subtle clinical and molecular changes preceding phenoconversion is of critical importance. This effort is central to understanding risk, enabling early intervention and developing preventative treatment strategies. Reducing the incidence of ALS must not be underestimated, as it represents a major burden on health systems and society, requiring substantial investment in specialised end-of-life care. Apart from gene-silencing approaches benefiting a small minority of people living with ALS (plwALS), therapeutic development has recently stalled, contributing to a sense of hopelessness among patients and carers. Although significant progress has been made over the past 10–15 years in defining the clinical and molecular framework of presymptomatic disease, a complete picture of the stepwise or linear changes leading to disease onset remains elusive.
Methods: Here, we evaluate the methodological approaches used over the past two decades to study the presymptomatic stage of ALS. This has included work to understand genetic susceptibility and the broader genetic architecture of a disease with a lifetime risk of approximately 1 in 300 in the general population. Epidemiological studies of large ALS cohorts have enabled multistep modelling of disease development. In parallel, increasingly sensitive methodologies for detecting subtle molecular changes preceding clinical onset have led to the identification of biomarkers that, individually or in combination, show strong predictive value when incorporated into multivariate models of disease onset.
Results: These investigative efforts have yielded several key advances: the development of multistep population-based models; transformative progress in disease stratification and identification of the prodromal phase in mutation carriers through measurement of neurofilament light chain in biological fluids; and, more recently, the identification of biomarker panels capable of robustly predicting disease initiation. These panels are based on analyses of plasma and cerebrospinal fluid samples from presymptomatic mutation carriers, as well as large longitudinal population studies employing high-sensitivity platforms such as Simoa and Olink.
Conclusion: The future holds further promise. Continued expansion of longitudinal clinical datasets and biobanked biological samples, alongside the development of increasingly sensitive detection technologies, will enable deeper exploration of additional disease dimensions, including the proteome and metabolome. These advances, coupled with ongoing refinement of genetic and epigenetic frameworks, are expected to further elucidate the earliest stages of ALS and support the development of effective preventative strategies.
SS04.01
What we have learnt from comparative analyses?
Dr. Carlo Antozzi
Fondazione IRCCS Istituto Neurologico C. Besta, Milano, Italy
Background: Myasthenia Gravis (MG) is a chronic autoimmune disease of the neuromuscular junction characterized clinically by fluctuating weakness of voluntary muscles; the degree of fluctuations ranges from mild to severe worsening up to impending (IMC) or full myasthenic crisis (MC) with respiratory insufficiency requiring mechanical ventilation. The standard of care for clinical relapses/IMC-MC includes, apart from changes to ongoing pharmacological therapy, immunomodulation with plasmaexchange (PLEX) or high dose intravenous immunoglobulins (IVIG) effective in a proportion of patients. However, some patients may have medical contraindications to apheresis or inadequate vascular access; PLEX is also not available everywhere; IVIG are expensive and often in short supply. Moreover, high-dose steroids may carry an increased risk of infection or sepsis, which is a poor prognostic factor for patients in MC. Despite these limitations, PLEX and IVIG, together with corticosteroid, have a consolidated role in the management of MG relapses. During the last decade, considerable advances have been made in research and approval of targeted therapies such as complement or neonatal Fc (FcRn) inhibitors which have now entered clinical practice.
Methods: Literature review and experience from real world use of targeted therapies in MG
Results: Clinical experience started with eculizumab for refractory MG and efgartigimod, followed by the approval or ravulizumab and zilucoplan as complement inhibitors, and rozanolixizumab and nipocalimab as FcRn inhibitors. Apart from the different mechanisms of action, route of administraion, and treatment protocols (chronic versus cyclic regimens), targeted therapies share some features that suggest their role in the treatment of severe MG deteriorations and MC. Indeed, they provide rapid improvement (in general within two weeks in the majority of patients) and are free of the side effects of corticosteroids. Randomized clinical trials did not include MG patients in IMC/MC, hence no information is available from these studies. Nevertheless, case reports and small series are emerging in the medical literature regarding the use of efgartigimod in MC with clinical improvement observed during the first treament cycle; in this context, timing adjustments must be taken into consideration when combining FcRn inhibitors with PLEX or IVIG. Regarding complement inhibition, experience with eculizumab observed clinical improvement often within one week; in some patients improvement can be observed even within 24 hours, a condition of “super-responsiveness” observed also with ravulizumab.
Conclusion: The evidence to guide the choice between anti-complement or anti-FcRn inhibition in severe MG deterioration is still limited. Apart from issues related to autoantibody specificities (anti-acetylcholine vs MuSK), decisions must be made taking into account each patient previous history (particulary the frequency of relapses and need for rescue therapy), comorbidities, and risk of infection, as well as the need to set a strategy as a form of “bridging therapy” or as a long-term maintenance treatment. Despite the available evidence, both complement and FcRn inhibitors have been included in the recent updates of several national guidelines for the management of MG also in case of severe deterioration.
SS05.01
When drugs are available… but not for everyone
Dr. Emna Farhat, Prof. Ilhem Ben Youssef-Turki, Prof. Najoua Miladi
Tunis El Manar University, Tunis, Tunisia
Background: Neuromuscular disorders (NMD) constitute a major health care burden in the north African region (NAR) due to their continuing high frequency related to the high degree of consanguinity. The access to basic diagnostic tools such as muscle biopsy and molecular biology remains limited in the region. The discovery of innovative life-saving drugs for diseases like spinal muscular atrophy has raised hopes among clinicians. Despite continued efforts in developing regional specific diagnosis strategy, patient registries and multidisciplinary clinical management according to international standards of care, the treatment of NMD has not changed and patients continue to be treated symptomatically. The access to new targeted therapies remains limited in the NAR. Very few patients have benefited from these treatments, most within the framework of a compassionate access program, with a lack of a concrete regional strategy to improve their availability. This is probably due to the absence of structures dedicated to research/clinical trials and the lack of NMD specialists trained in this particular field, in addition to the limited economic development. The Mongi Ben Hamida National Institute of Neurology of Tunisia, founded in 1973, was the first neuromuscular centre of the NAR, with a laboratory specialized in muscle pathology and research laboratories dedicated to neurogenetics and neurosciences. In this presentation, we will discuss the difficulties and challenges of accessing NMD disease modifying treatments in the NAR, and the potential solutions for advancing a global and adapted strategy to put in place, in order to participate first of all at programs of clinical trials for these drugs, and secondary to make them accessible for all patients.
SS05.03
Crowdfunding, lottery, relocation… Treatment access inequity from a global north perspective
Prof. Laurent Servais
University of Oxford, Oxford, United Kingdom. University of Liege, Liege, Belgium
Background: This is a joined session ICNMD/WMS
Methods: We will present the perspective of treatment inequity from an European perspective
Results: We will discuss issues like relocation, lottery and crowd funding
Conclusion: To assess treatment access inequity needs the involvement of all actors, including physicians, policy makers, scientific society, patients advocacy and pharma company. It needs multimodal action including standard of care development, education to the management of rare diseases, early diagnosis to allow early treatment and of course differential costing of innovative medications.
SS06.03
Current and Future Therapies in Mitochondrial Myopathies
Assoc. Prof. Michelangelo Mancuso
University of Pisa, Pisa, Italy
Background: Mitochondrial myopathies are a heterogeneous group of disorders caused by defects in oxidative phosphorylation leading to impaired energy production. Despite advances in genetic diagnosis, effective disease-modifying therapies remain limited. This lecture will provide an overview of current management strategies, emphasizing symptomatic treatments and emerging standards of care aimed at improving quality of life.
The second part will focus on recent therapeutic developments targeting the underlying pathophysiology. These include pharmacological approaches to enhance mitochondrial biogenesis and function, such as small molecules and metabolic modulators, as well as antioxidant strategies to mitigate oxidative stress. Advances in nucleoside therapy for mitochondrial DNA depletion syndromes and the growing role of precision medicine will also be discussed.
Finally, the lecture will explore the challenges related to clinical trial design, heterogeneity of phenotypes, and outcome measures will be addressed, alongside the importance of international collaboration.
Overall, this presentation aims to provide a comprehensive and forward-looking perspective on the evolving therapeutic landscape in mitochondrial myopathies, bridging current clinical practice with cutting-edge research and offering insights into the path toward effective disease-modifying treatments.
SS08.01
AI for Redefining Natural History in Muscle Disorders
Prof. Massimiliano Filosto
University of Brescia; NeMO-Brescia Clinical Center for Neuromuscular Diseases, Brescia, Italy
Background: Muscle disorders, including hereditary myopathies, inflammatory myopathies, and neuromuscular junction disorders, exhibit heterogeneous clinical presentations and progression rates. Traditional natural history studies rely on longitudinal clinical assessments, imaging, and biomarker measurements, but are often limited by small cohort sizes, variable follow-up intervals, and the subjectivity of outcome measures. Artificial intelligence (AI) and machine learning (ML) techniques offer powerful tools to integrate multimodal data, including clinical, imaging, molecular, and wearable sensor data, to identify patterns of disease progression, predict trajectories, and refine patient stratification.
Methods: We analyze current clinical findings on the use of AI in neuromuscular disorders, focusing on its potential to improve understanding of disease progression. Key applications include combining clinical scores, imaging, genetic data, and wearable device information to identify patterns and predict functional decline. Examples from recent studies are highlighted to show how AI can complement traditional observational approaches.
Results: AI approaches can reveal previously unrecognized subgroups of patients with distinct progression patterns, which can guide individualized follow-up and management. Digital tools combined with AI allow continuous monitoring of mobility and muscle function, offering more precise and real-world information on disease trajectories. AI can also help identify early disease milestones and potential biomarkers, supporting more targeted clinical trials and personalized care.
Conclusion: AI has the potential to transform our understanding of natural history in muscle disorders, offering clinicians new tools to predict disease progression, stratify patients, and optimize trial design. While still emerging, these approaches promise to complement traditional clinical assessment and support precision medicine in neuromuscular diseases. Future work should focus on validating AI applications in clinical settings and expanding their use to rare subtypes of muscle disorders.
SS08.03
AI in histopathology?
Prof. Edoardo Malfatti
APHP, Paris, France. INSERM, Paris, France
Background: Myopathies encompass a broad spectrum of both inherited and acquired neuromuscular disorders. Interpreting muscle biopsies requires the analysis of a wide and expensive battery of histochemical and immunohistochemical techniques by experienced neuropathologists. Next-generation sequencing and serological diagnosis have changed the indication for muscle biopsies, limiting both access and interpretation skills in European Neuromuscular Reference centers (ERNC). Nevertheless, they remain indispensable to identify and characterize inflammation, to validate pathogenicity when variants of uncertain significance are identified on genetic testing, or when novel genes are discovered.
Methods: We propose an algorithm for the analysis of muscle biopsies using digital pathology. In an initial machine-learning phase, Hematoxylin–Eosin (H&E) stained whole-slide images of muscle sections were analyzed using an automated myofiber segmentation workflow that integrates QuPath, Cellpose and a home made developed pipeline to enable quantitative assessment of muscle histology.
Results: Our analysis demonstrates the efficacy of our algorythm to identify : 1)a higher fiber size variability distribution in all patterns compared to controls; 2) a significantly higher nuclear internalizations in dystrophic, inflammatory, and neurogenic patterns; 3) the presence of Inflammatory infiltrates, identified as areas of increased nuclear density, that were significantly higher in inflammatory myopathies compared to controls.
Conclusion: These preliminary results highlight the potential of artificial intelligence as a valuable tool for next-generation myopathology, supporting standardized, quantitative, and reproducible analysis of muscle biopsies. Ongoing work aims to expand and validate the approach on larger cohorts, paving the way toward machine-assisted interpretation of digitally scanned muscle sections and the development of AI-driven first-line diagnostic support in muscle pathology.
SS10.01
Painful Neuropathies: New insights/New treatment targets
Prof. Claudia Sommer
University Hospital Würzburg, Würzburg, Germany
Background: Painful neuropathies represent a major clinical challenge, often leading to chronic, debilitating pain that is difficult to treat. Neuropathic pain by definition arises from injury or dysfunction of the somatosensory nervous system and is characterized by abnormal neuronal excitability, peripheral and central sensitization, and neuroimmune interactions. The pathophysiology is complex. For example, changes in voltage-gated sodium and calcium channels can enhance ectopic neuronal firing, while microglial activation in the spinal cord can amplify pain signaling through the release of cytokines and neurotrophic factors.
Mechanistic insights have opened the door to novel therapeutic targets and more mechanism-based treatment strategies. Emerging treatments focus on modulating specific molecular pathways involved in neuronal hyperexcitability and neuroinflammation, such as selective sodium-channel blockers (e.g., targeting Nav1.7), sigma-1 receptor antagonists, and agents that interfere with glial activation or cytokine signaling. In addition, the dorsal root ganglion itself has gained attention as a promising therapeutic target, with neuromodulation techniques such as DRG stimulation and spinal cord stimulation showing potential benefits in refractory neuropathic pain.
SS10.02
Chemotherapy induced peripheral neuropathy
Dr. Paola Alberti
University of Milano-Bicocca, Monza, Italy
Background: Chemotherapy-induced peripheral neurotoxicity (CIPN) represents a significant complication arising from cancer treatment, often resulting in sensory, motor, and autonomic dysfunctions that impact patients’ quality of life. A translational approach to CIPN integrates preclinical and clinical research, bridging laboratory findings with patient care. Preclinical studies employ morpho-functional techniques to elucidate axonal damage mechanisms, focusing on ion channel dysfunctions and nerve excitability, while clinical investigations explore epidemiological trends and therapeutic interventions. This strategy facilitates the development of targeted diagnostic tools and novel therapies, ultimately improving outcomes for individuals undergoing chemotherapy.
SS10.03
Gene therapy for inherited neuropathies – how to optimally deliver to peripheral nerves
Prof. Kleopas Kleopa
The Cyprus Institute of Neurology and Genetics, Nicosia, Cyprus
Background: Emerging gene therapies for CMT inherited neuropathies include virally or non-virally mediated gene replacement, addition, silencing, or editing of genetic material. For most CMT neuropathies, gene- and disease, or even mutation-specific therapy approaches targeting lower motor and sensory neurons and their axons, or myelinating Schwann cells throughout the PNS are needed. While the efficiency of gene therapies to improve disease phenotypes has been demonstrated in cellular and in vivo rodent disease models, delivery approaches to human peripheral nerves remain to be optimized. Safety and scale-up biodistribution studies in bigger animal models, mostly non-human primates, have provided further insights into the translatability of the proposed administration routes and vector doses. Innovative delivery vehicles are currently in development, while insights from ongoing preclinical and clinical trials for other neurological and neuromuscular disorders will inform better safety strategies. Improving the risk-benefit ratio of gene therapies will facilitate future transformative treatments for CMT patients.
SS11.02
Novel Forms of LGMD
Assoc. Prof. Edmar Zanoteli
FMUSP, São Paulo, Brazil
Background: Patients with autosomal inherited limb-girdle muscular dystrophies (LGMD) present with similar clinical and histological findings, with weakness predominating in the proximal portions of the limbs. Currently, at least 34 subtypes based on protein deficiency and genetic defect are included in its classification. Some of the most common forms of the disease are caused by common pathogenic mechanisms, including sarcolemmal glycoprotein deficiency (e.g. sarcoglycan complex), defects in the extracellular matrix (e.g. collagen 6, merosin, α-dystroglycan hypoglycosylation), sarcomere remodeling dysfunction (e,g, calpain-3), and sarcolemmal repair defects (e.g. dysferlin, anoctamin-5), among others. More recently, new disease mechanisms have been associated with LGMD phenotype. For example, different genes related to the Notch signaling pathway, known to maintain the quiescence of muscle stem cells, have been recognized as being associated with both congenital myopathy (MEGF10) and LGMD (POGLUT1, JAG2). Genes involved in RNA splicing or transport of small nuclear ribonucleoproteins (snRNPs) (SNUPN) have also been associated with LGMD phenotype. Patients with LGMD phenotype and pathogenic variants in HMGCR, which is related to mevalonate pathway, were recently described. Finally, variants in POPDC genes (POPDC1 and POPDC3) that possess the evolutionarily conserved Popeye domain and are associated with protein traffic have been identified in patients with LGMD phenotype. In addition to improving the genetic diagnosis and phenotypic characterization of patients with muscular dystrophy, the identification and characterization of new pathophysiological mechanisms of LGMD may pave the way for treatments based on pathways that have therapeutic effects for multiple subtypes of the disease.
SS12.03
Trials on B Cell Depleting Agents: Success and Failure
Assoc. Prof. Raffaele Iorio
Università Cattolica del Sacro Cuore, Rome, Italy. Fondazione Policlinico Universitario Gemelli IRCCS, Rome, Italy
Background: Myasthenia gravis (MG) is an autoantibody-mediated disorder in which autoreactive B cells play a central pathogenic role by producing antibodies against acetylcholine receptors (AChR) or muscle-specific kinase (MuSK), causing fluctuating muscle weakness. B cell depletion is indeed a rational therapeutic strategy. Rituximab, an anti-CD20 monoclonal antibody, depletes mature B cells but spares plasmablasts and long-lived plasma cells that may continue producing pathogenic autoantibodies. Inebilizumab targets CD19, a marker retained across a broader B-lineage spectrum including plasmablasts and plasma cells, enabling more comprehensive depletion. Three randomized controlled trials have evaluated these approaches in generalized MG, with divergent results.
Methods: Three randomized, double-blind, placebo-controlled trials were analyzed. BeatMG (Nowak et al., 2022) was a phase 2 futility-design trial of rituximab in 52 AChR-Ab+ gMG patients on prednisone, with steroid-sparing as the primary outcome over 52 weeks. RINOMAX (Piehl et al., 2022) evaluated a single 500 mg rituximab infusion in 47 treatment-naïve patients with new-onset gMG, targeting minimal disease manifestations at 16 weeks. MINT (Nowak et al., 2025) was a phase 3 trial of inebilizumab in 238 AChR-Ab+ or MuSK-Ab+ gMG participants, with change in MG-ADL score at week 26 as the primary endpoint.
Results: BeatMG reached its futility endpoint (p = 0.03), with steroid-sparing rates of 60% for rituximab versus 56% for placebo, suggesting that a clinically meaningful benefit was unlikely despite an acceptable safety profile. Conversely, RINOMAX showed that early rituximab use led to minimal disease manifestations in 71% versus 29% with placebo (probability ratio 2.48; 95% CI 1.20–5.11; P = .007), with fewer rescue treatments. In the MINT trial, inebilizumab significantly improved MG-ADL scores (adjusted difference −1.9; 95% CI −2.9 to −1.0; P < 0.001) and QMG scores (adjusted difference −2.5; 95% CI −3.8 to −1.2; P < 0.001) versus placebo, with sustained benefits at 52 weeks in AChR-Ab+ patients and a favorable safety profile.
Conclusion: The efficacy of B cell depletion in MG appears influenced by disease stage, molecular target, and study design. Notable heterogeneity across trials—in patient populations (established versus new-onset disease), antibody subtypes (AChR-Ab+ only versus AChR-Ab+ and MuSK-Ab+), dosing regimens, endpoints, and sample sizes—limits direct comparisons and warrants caution. Rituximab did not demonstrate steroid-sparing superiority in established AChR-Ab+ disease but showed benefit when given early in treatment-naïve patients (RINOMAX), although the small RINOMAX sample calls for confirmatory studies. Inebilizumab, through broader CD19-mediated depletion encompassing plasmablasts and plasma cells, demonstrated significant efficacy across both antibody subtypes in the larger MINT trial, though long-term safety and durability data are needed. Collectively, these findings support the therapeutic potential of B cell depletion in MG and suggest that CD19-directed therapy may address some limitations of anti-CD20 approaches, while further research is needed to define optimal patient selection and treatment timing.
SS13.02
Hematologic related neuropathies
Prof. Chiara Briani
Department of Neurosciences, University of Padova, Padova, Italy
Background: The peripheral nervous system may be involved at any stage in the course of hematological diseases. Paraproteinemic neuropathies and those related to lymphoproliferative diseases are the most common. The different underlying mechanisms include chemotherapy neurotoxicity, direct nerve infiltration (neurolymphomatosis), infections, immune-mediated (especially IgM paraproteinemic neuropathies), paraneoplastic or metabolic processes and nutritional deficiencies. Some clinical findings, such a focal or diffuse involvement, symmetric or asymmetric pattern, presence of pain may point to the correct diagnosis. Besides a thorough medical history and neurological examination, neurophysiological studies, cerebrospinal fluid analysis, nerve biopsy (in selected patients with suspected lymphomatous infiltration) and neuroimaging techniques are needed for a proper diagnostic workup.
SS17.01
Diagnostic approach to peripheral neuropathy
Prof. Fiore Manganelli
Department of Neuroscience, Reproductive and Odontostomatological Science - University of Naples 'Federico II', Naples, Italy
Background: Peripheral neuropathies represent a heterogeneous group of disorders embracing mononeuropathies, multineuropathies and polyneuropathies, with diverse etiologies including metabolic, toxic, inflammatory, hereditary, and infectious causes. Because of their clinical variability, establishing an accurate diagnosis requires a structured and stepwise diagnostic approach integrating clinical, electrophysiological, and laboratory data.
Methods: The diagnostic approach to peripheral neuropathy is based on a systematic evaluation including detailed clinical history, neurological examination, and neurophysiological study. Nerve conduction studies and electromyography are used to define the pattern of nerve involvement (axonal vs. demyelinating, motor vs. sensory, focal vs. diffuse). Laboratory investigations are tailored according to the clinical and electrophysiological findings and may include metabolic, autoimmune, infectious, and genetic testing. Additional tools such as imaging, cerebrospinal fluid analysis, or nerve biopsy are considered in selected cases.
Results: A structured diagnostic algorithm allows the identification of the neuropathy pattern and guides targeted investigations, improving diagnostic accuracy and reducing unnecessary tests. Neurophysiology plays a central role in classifying neuropathies and identifying potentially treatable forms such as inflammatory neuropathies. The integration of clinical and laboratory data facilitates differentiation between acquired and hereditary conditions and supports appropriate therapeutic decisions.
Conclusion: A stepwise and multidisciplinary diagnostic strategy is essential for the accurate evaluation of peripheral neuropathies. Early recognition of the neuropathy pattern through clinical and neurophysiological assessment enables timely etiological diagnosis and improves patient management, particularly in treatable neuropathies.
SS18.01
Myotonic dystrophy type 1 2026 and 2030
Prof. Valeria Sansone1, Dr. Carola Ferrari Aggradi1, Dr. Alice Zanolini1, Dr. Giovanni Colacicco1, Dr. Alessandra Di Bari1, Dr. Alice Valenza1, Dr. Simone Maiorano1, Dr. Sofia Maroni1, Prof. Nicholas Johnson2
1
The NeMO Clinical Center, Neurorehabilitation Unit, University of Milan, Milan, Italy.
2
Virginia Commonwealth University, Richmond, Virginia, United States
Background: Myotonic dystrophy type 1 (DM1) is a multisystemic disorder caused by CTG repeat expansion in DMPK. Over the past decade, advances in genetic diagnosis, multisystem care, and the growing number of randomized clinical trials showing good safety profile and preliminary encouraging results, have reshaped patient outcomes. Yet substantial unmet needs remain in predicting clinical heterogeneity and patient profiling as well as quantifying response to therapy.
The talk will focus on current knowledge on DM1 as of 2026, will discuss key diagnostic and management gaps from a clinical point of view and outline priorities and opportunities to accelerate progress toward 2030 including site readiness strategies.
Methods: Clinical and demographic data from a large dataset from the NeMO Clinical Center including 321 patients with DM1 as well as results from neuromotor, respiratory, cardiac and laboratory results will be presented at baseline and over time (mean follow-up of 5 years). This data will be descriptively compared to results from the observational study where 700 patients with DM1 have been followed over a 2-year period as part of the Clinical Research Trial Network (coordinator Prof. Johnson, Virginia Commonwealth University).
Results: Patient profiles from the observational study results will be discussed - in addition preliminary results from the patient reported outcomes using the Myotonic Dystrophy Health Index as well as longitudinal assessments of swallowing studies and results from cognitive tests assessing executive functions and sleep studies will be presented.
Conclusion: The data presented will provide additional insights into the multisystem aspects of DM1 with focus on swallowing, cognition, sleep and PROs thus allowing to deep dive into domains which are critical for this population and so far not investigated in detail in large cohorts. A better understanding of the disease and its progression should be expected.
SS18.02
Myotonic dystrophy type 2 in 2026 and 2030
Prof. Nicholas Johnson
Virginia Commonwealth University, Richmond, United States
Background: Myotonic dystrophy type 2 (DM2) is a rare, multisystemic, autosomal dominant disorder characterised by proximal muscle weakness, myalgia, fatigue, and functional limitations. Despite overlapping features with myotonic dystrophy type 1 (DM1), DM2 presents a unique clinical and symptom profile, notably lacking excessive daytime sleepiness and presenting with severe myalgic pain and slower disease progression. Current clinical outcome measures inadequately capture these DM2-specific features.
Methods: Here we enrolled a pilot study seeking to adopt other composite outcome measures such as the Northstar assessment for the muscular dystrophies and assess the impact on RNA splicing in myotonic dystrophy type 2. In addition, the myotonic dystrophy clinical research network (DMCRN) has developed several composite measures that it seeks to implement to prepare this patient population for future clinical trials.
Results: Myotonic dystrophy type 2 is a slowly progressive condition that affects proximal muscles. Pilot data support the adoption of the Northstar assessment for the muscular dystrophies as an approach to capture these symptoms. Muscle biopsies revealed changes in RNA splicing consistent with those seen in myotonic dystrophy type 1. Further studies are required to assess the correlation of these splicing events and functional endpoints.
Conclusion: Myotonic dystrophy type 2 is a progressive disorder that may benefit from the development of clinical outcome assessments and biomarkers to support the development of new therapeutics. The studies proposed will prepare the field for clinical trials in the near future.
SS19.01
Closing the gap – the unsolved cohorts
Prof. Mary Reilly
Division of Neurology in UCL Queen Square Institute of Neurology, London, United Kingdom
Background: The inherited neuropathies include those neuropathies in which the neuropathy is the sole abnormality and those where the neuropathy is part of a more complex condition. The former group are known as Charcot Marie Tooth disease (CMT) and also includes the related conditions, hereditary neuropathy with liability to pressure palsy (HNPP), hereditary sensory neuropathy (HSN) and hereditary motor neuropathies (HMN).
There have been huge advances in identifying causative genes for CMT in the last two decades especially with the use of whole exome sequencing and this has been further helped by the addition of short read whole genome sequencing (SR WGS) in the last decade. We published a study in 2024 of over 1500 patients with CMT using next generation sequencing including SR WGS, in which we achieved a diagnostic rate of 77%. Although this is a marked improvement in the diagnostic rate, we still have nearly a quarter of patients who do not receive a genetic diagnosis.
Most of our unsolved cohort are patients with motor predominant axonal neuropathies often labelled as CMT2 or HMN. This talk will explore the possible reasons that these patients remain unsolved and the approaches we are taking to try to solve them. This includes the increasing use of new technologies like long read sequencing but also the recognition of the broadening of phenotypes associated with specific genes. Multidisciplinary meetings where clinicians, geneticists, clinical scientists and research scientists meet to discuss individual patients are becoming an increasingly important tool in solving the unsolved cohort.
SS20.02
Planning Pregnancy in Myasthenia Gravis (MG)
Prof. Nils Erik Gilhus
University of Bergen, Bergen, Norway. Haukeland University Hospital, Bergen, Norway
Background: Many MG women abstain from having children or have fewer children than they would have had without their MG. Main reasons are worries about MG worsening in pregnancy and after giving birth, outcome for the child because of mother's MG autoantibodies and drugs crossing the placenta, MG hereditary aspects, and long-term physical ability to care for a child. General birth rates are going down world-wide, and mother's age when giving birth is increasing. All MG women in reproductive age should receive precise information on pregnancy and puerperium, and all aspects that may lead to worries should be addressed.
Methods: The impact of MG on pregnancy, puerperium, and the child, and the impact of pregnancy and puerperium on the MG have been addressed in many studies. Registry-based studies usually include large cohorts with little selection bias, but lack detailed clinical information. Most registry-based studies are able to include relevant control groups. Single- or multicentre hospital populations are prone to selection bias, they tend to include fewer patients, and typically they do not include control populations. The Nordic countries have excellent nationwide health registries, including birth registries. The presentation will include data from various sources.
Results: Pregnancy is recommended when the MG is in a stable and well-controlled phase. Thymectomy performed before pregnancy leads to a less severe MG during pregnancy. In most MG women, the disease shows no or mild fluctuations during pregnancy. However, up to one third report a temporary deterioration. Some also improve. Myasthenic crisis during pregnancy is very rare. In puerperium, an MG deterioration is common. Therefore, relatively fast-acting therapy should be considered in this phase, either initiation or increasing the dose. Mycophenale mofetil, methotrexate, and cyclophosphamide are teratogenic and should not be used by women in reproductive age. Pyridostigmine, corticosteroids in low doses, and azathioprine can be continued during pregnancy. IVIg and plasma exchange are regarded as safe emergency treatments. Neonatal myasthenia occurs in 10-15% of children with MG mothers. This is not related to mother's MG severity. Malformations and the FARAD syndrome is rare, with 46 children reported in a large multicentre study. MG women has an increased chance for Cesarean section. This is mostly due to worries that vaginal delivery might be too demanding physically. However, ordinary vaginal birth is recommended for the large majority of MG women. Most pregnancy complications have a similar frequency with and without MG.
Conclusion: All MG women in reproductive age should receive precise information about pregnancy and giving birth. Pregnancies should be planned when the MG is stable and well-controlled. Thymectomy should usually be performed before pregnancy, and drug treatment should be adapted to a potential pregnancy. Regular follow-up of the MG mother and her baby, including ultrasound examinations, should be performed during pregnancy. Vaginal delivery is preferred in the large majority. However, all MG women should give birth in a hospital with experience in emergency neonatal care including respiratory support due to the risk of neonatal myasthenia and FARAD. MG should not be a reason for abstaining from having children.
SS21.01
Hereditary Neuropathy and myopathy : overlapping features
Dr. Tanya Stojkovic1, Dr. Marc Bitoun2
1
APHP, Pitié-Salpêtrière Hospital, Sorbonne University, Paris, France.
2
Inserm, Sorbonne University, research center of Myology, Paris, France
Background: Hereditary neuromuscular diseases include a broad range of genetic disorders affecting muscle, peripheral nerve, or both. Traditionally, hereditary myopathies (such as muscular dystrophies, congenital and metabolic myopathies) were distinguished from hereditary neuropathies (like Charcot-Marie-Tooth disease). However, increasing recognition of overlapping forms where muscle and nerve involvement coexist and intertwine is challenging this distinction. High-throughput molecular biology techniques over the last 20 years have identified mutations in the same gene causing both neuropathies and myopathie
Methods: Diagnosis involves a multidisciplinary approach including clinical evaluation,biomarkers such as creatine kinase levels, electrophysiology (EMG showing both neurogenic and myogenic patterns), muscle biopsy disclosing in some cases mixed neurogenic and myopathic features, and muscle imaging . Advances in exome and genome sequencing increasingly highlight overlaps between genes associated with myopathies and neuropathies. Temporality and spatial distribution of muscle involvement vary; neurogenic damage may appear before myopathy or vice versa, and different muscles may be differently affected.
Results: Several genes illustrate this overla:
• Mitochondrial genes (e.g., MELAS, MERRF syndromes) that cause sensory and motor neuropathies along with muscle weakness.
• LMNA (encoding nuclear lamins A/C) and DNM2 (encoding dynamin 2), where mutations can lead to either Charcot-Marie-Tooth neuropathy or myopathy depending on the mutation.
• BAG3, DES, CRYAB, mutated in both myofibrillar myopathies and isolated or combined neuropathies.
• Genes such as VCP, MATR3, SQTM1, and TIA1 involved in multisystem proteinopathies affecting nerve, brain, muscle, and bone.
• HSPB8 and SPTAN1, primarily linked to distal motor neuropathy, also implicated in distal and axial myopathies or mixed neurogenic-myogenic phenotypes.
Conclusion: Advances in high-throughput sequencing have revealed that mutations in a single gene can manifest as neuropathic, myopathic, or mixed neuro-myopathic phenotypes. This genetic overlap highlights the complexity of neuromuscular disease pathophysiology, where the same molecular defect can differently affect muscle and nerve tissues depending on factors such as cell type metabolism, energy dependence, or cytoskeletal structure.Future progress in sequencing technologies and bioinformatics will likely uncover additional overlapping genes and mechanisms, further blurring the boundaries between neuropathies and myopathies and opening new avenues for understanding and treating these complex diseases.
SS21.02
Ataxic Neuropathies
Dr. Davide Pareyson
Fondazione IRCCS Istituto Neurologico Carlo Besta,, Milan, Italy
Conclusion: Hereditary ataxic neuropathies encompass a clinically and genetically heterogeneous group of disorders predominantly characterized by sensory ataxia, either isolated or combined with motor involvement, cerebellar ataxia, or other neurological findings. This phenotype overlaps with cerebellar ataxias and related disorders and poses significant diagnostic challenges.
The discovery of biallelic intronic repeat expansions in RFC1 has revolutionized the field, identifying the most common cause of late-onset ataxia with sensory neuropathy, known as CANVAS (cerebellar ataxia, neuropathy and vestibular areflexia syndrome). Chronic cough and abnormalities of the vestibulo-ocular reflex (VOR) are strong red flags for CANVAS. The phenotypic spectrum of RFC1-related disease is broader than initially described and includes incomplete and atypical presentations. Moreover, there are CANVAS-like phenotypes not associated with RFC1 variants, including cases associated with heterozygous variant(s) in RNF170.
Several additional inherited conditions may present with sensory ataxia, either as a prominent or initial feature. These include Friedreich’s ataxia and ataxia with vitamin E deficiency (AVED), which combine cerebellar and sensory involvement, as well as mitochondrial disorders related to POLG and TWNK mutations. Recessive ataxias such as DARS2-related disease may also present with a sensory ataxic phenotype. Furthermore, certain inherited neuropathies, particularly early-onset demyelinating forms such as MPZ-related CMT1B and PRX-related CMT4F, can manifest with prominent sensory ataxia and may be misclassified. Other rare conditions to be considered are PHARC (Polyneuropathy, hearing loss, ataxia, retinitis pigmentosa, and cataract) and abetalipoproteinemia.
The differential diagnosis is broad and includes treatable metabolic disorders, such as Refsum disease, cerebrotendinous xanthomatosis and vitamin deficiencies, as well as immune-mediated sensory neuronopathies, including those associated with Sjögren syndrome or paraneoplastic conditions.
This lecture will provide a practical clinical approach to patients presenting with sensory ataxia, emphasizing key clinical features, appropriate use of neurophysiology and imaging, and a rational strategy for genetic testing. The aim is to improve diagnostic accuracy, facilitate early identification of treatable conditions, and better define the expanding spectrum of hereditary ataxic neuropathies.
SS22.02
Size does not matter - Ultrasound Parameters and Patient-Reported Outcomes after Median and Ulnar Nerve Repair
Dr. Natalie Winter1,2, Dr. Henrik Lauer3, Ms. Victoria Johnson1, Ms. Julia Wittlinger1, Dr. Stephanie Männlin1, Prof. Jonas Kolbenschlag3, Prof. Alexander Grimm1, Dr. Johannes Heinzel3
1
University Hospital Tuebingen, Tuebingen, Germany.
2
Hertie Institute for Clinical Brain Research, Tuebingen, Germany.
3
BG Klinik Tuebingen, Tuebingen, Germany
Background: Traumatic transection of the median and ulnar nerves may result in persistent functional deficits despite timely surgical repair. High-resolution nerve ultrasound enables direct visualization of fascicular integrity and morphologic nerve parameters and may complement clinical and electrophysiological assessments. The relationship between sonographic findings, clinical sensory and motor function, and long-term patient-reported outcomes remains insufficiently characterized.
Methods: Patients with traumatic transection of the median and/or ulnar nerve treated by primary end-to-end neurorrhaphy were invited for follow-up evaluation. Assessments included nerve– and muscle ultrasound, nerve conduction studies, electromyography, and standardized clinical examination including motor strength grading and two-point discrimination testing. Patient-reported outcomes were assessed using the Michigan Hand Questionnaire (MHQ) and the Short Form-36 (SF-36). Ultrasound evaluation focused on fascicular continuity and cross-sectional area (CSA) at the repair site. Associations between ultrasound, clinical, and questionnaire outcomes were analyzed. Subgroup analyses were performed for median and ulnar nerve injuries.
Results: A total of 24 patients (6 women, 18 men) with 27 nerve injuries were included (9 median nerves, 18 ulnar nerves); three patients had combined median and ulnar nerve injuries. Mean age at follow-up was 44.0 ± 17.8 years. The mean interval between surgical repair and follow-up assessment was 3.2 ± 2.1 years. Ultrasound-assessed fascicular continuity showed significant correlations with multiple patient-reported outcome measures. Higher fascicular continuity was associated with lower pain levels on the MHQ (Kendall’s tau = −0.419, p = 0.015) and with a higher MHQ total score (tau = 0.415, p = 0.011). Within the SF-36 domains, fascicular continuity correlated significantly with Energy/Fatigue (tau = 0.583, p < 0.001), Emotional Well-Being (tau = 0.427, p = 0.014), Social Functioning (tau = 0.513, p = 0.005), Pain (tau = 0.401, p = 0.024), and General Health (tau = 0.375, p = 0.033). Cross-sectional area (CSA) at the neurorrhaphy site showed no significant correlation with MHQ or SF-36 scores, muscle strength grading, or sensory function.
In a subgroup analysis of median nerve injuries, fascicular continuity showed no significant correlation with abductor pollicis brevis muscle strength or with sensory measurements; however, abductor pollicis brevis strength correlated significantly with the MHQ final score (tau = 0.569, p = 0.041). In the ulnar nerve subgroup, fascicular continuity correlated significantly with the MHQ final score (tau = 0.417, p = 0.046). Across the cohort, better two-point discrimination (minimum distance) correlated significantly with higher MHQ final scores (tau = 0.442, p = 0.038).
Conclusion: Fascicular continuity assessed by nerve ultrasound is associated with relevant patient-reported quality-of-life outcomes after median and ulnar nerve repair, whereas morphometric parameters such as CSA show limited functional relevance. Motor strength and sensory discrimination emerge as important clinical correlates of hand-related patient-reported outcomes, underscoring the complementary value of sonographic, motor, and sensory assessments in the long-term evaluation of peripheral nerve repair.
SS22.03
Nerve Ultrasound and its Role in Detecting Neuralgic Amyotrophy
Assoc. Prof. Alexander Grimm, Dr. Helene Hurth, Assoc. Prof. Martin Schumann, Dr. Katharina Kneer, Assoc. Prof. Jonas Kolbenschlag, Dr. Johannes Heinzel, Dr. Jan-Hendrik Stahl, Ms. Alexandra Hermes, Ms. Julia Wittlinger, Dr. Natalie Winter
University hospital of Neurology, Tübingen, Germany
Background: Neuralgic amyotrophy is a rare, but certainly underrecognized peripheral nerve disorder, mostly affecting the upper trunk of the brachial plexus, typically arising with severe shoulder pain followed by pareses of distinct muscles. We analyzed patients of our hospital presenting in the years 2022 to 2024.
Methods: Patients were screened clinically, electrophysiologically and by ultrasound or MRI. Most patients - depending on time of presentation - received high dosage steroids, sometimes repeated and all of them received follow-up analyses. In some patients neurosurgical steps were needed
Results: Results -1: 100 patients (7:3 male) have been included. 10% without severe pain. 92% received steroids (mostly 0,5g/die for 3 days). 5 persons had a relapsing remitting course with more than 3 episodes (no SEPT9 or PMP 22 mutation found). 81% of patients received nerve conduction studies (75% of these showed axonal damage including SNAP reduction), 80% PSA in the EMG.
Overall 64% received MRI and ultrasound, 97% revealed nerve swelling of the plexus and of several peripheral nerves (i.e. n=34 supraclavicular and axillary nerve, n=27 AIN, rarer ones were the PIN, the long thoracic and others). 15 patients did not improve with treatment and showed nerve constriction and entwinement in 24 nerves (i.e. n=10 AIN fascicle, n=3 SSC). After surgery paresis improved in 21/24 nerves
Conclusion: NA is a mostly painful acquired, probably autoimmune inflammation of the epineurium, particularly affecting the upper trunk, i.e. the SSC or the lower trunk, AIN fascicle. In about 15% of cases scar tissue building leads to nerve constriction and surgery is needed, which then improves symptoms mostly. Ultrasound helps in depicting nerve pthology in the beginning and nerve constriction in refractory disaease
SS22.04
Between neuralgic amyotrophy and torsional neuropathy – the role of surgery
Nora Florenz Dengler
Background: In recent years, a paradigm shift has occurred in the understanding of (formerly) neuralgic (shoulder) amyotrophy. It is no longer limited to shoulder nerves that are affected, and the clinical presentation can be highly heterogeneous, complicating diagnosis. In addition to genetic causes, immunological pathomechanisms (such as viral infections) are increasingly coming into focus. For some time now, nerve torsion has also been associated with neuralgic amyotrophy. Pain management and corticosteroid therapy are considered the primary treatment. Several case series on surgical treatment have been published. However, to date, there are no uniform diagnostic criteria or treatment recommendations.
Methods: Landmark publications on the topic will be shown, with a focus on surgical therapy series.Results: Following the publication of an initial review article in the German Medical Journal by Holle et al. in 2024, numerous outcome series after surgical treatment have been published (Pöschl et al. 2024, Granata et al. 2024, Campbell et al. 2025, Bang et al. 2024, Shi et al. 2024, Solaja et al. 2023). This presentation will compare and contrast different surgical treatment strategies and compare the available outcomes.
Conclusion: To date, there is no structured analysis based on comparable outcome criteria. The extent to which a multicenter study design can contribute to knowledge gain will be discussed.
SS22.05
The Thoracic Outlet Syndrome
Franz Lassner, Michael Becker
Background: The neurogenic thoracic outlet syndrome represents a nerve compression syndrome at the root and trunc level of the brachial plexus, caused by a spatial narrowness in the scalene triangle. Anatomic anomalies like hypertrophic muscles or fibrous bands have been considered as underlying pathologic mechanisms, and resection of these structures in combination with partial resection of the first rib are advocated as surgical therapy. However, a certain number of patients do not well after the procedure.
Methods: From 10/02 to 6/24, 304 operative procedures were performed in 279 patients (scalenectomy, resection of a cervical rib, limited resection of the first rib and costovertebral exarticulation of the first rib). We use a classification system with four degrees of severity:
Stage 1: Symptoms when submitted to severe physical load
Stage 2: Symptoms when submitted to moderate physical load
Stage 3: Symptoms when submitted to light physical load
Stage 4: Permanent sympoms, motorsensory deficits
The indication for surgical intervention was set for stages 3 and 4. Patient evaluation was performed at a minimum of 1 year postop, including clinical asessment and determination of the DASH score. In a retrospective clinical study, the degree of osteotomie of the first rib (limited resection vs exarticulation) was evaluated.
Conclusion: Since the variety of anatomical causes for TOS is mechanically related to the first rib, osteotomy of the first rib is considered an essential element of the surgical procedure. However, the degree of resection remains controversial. In our study, exarticulation leads to better results compared to limited resection, an average DASH score of 42 was evaluated in this group. The rate of complications in general was low in our series, with a significant higher incidence in revision cases. The causes of unfavourable results, as seen in revision surgeries, were perineural fibrosis of varying degrees of severity. Surgical trauma, lymph leakage or mechanical irritation of the posterior stump of the first rib are possible causes for these complications.
SS22.06
The potential for gain of function under direct nerve stimulation based on the long-term results of peroneal nerve stimulation
Klaus Daniel Martin
Background: The question of the extent to which electrical stimulation of a peripheral nerve achieves a functional improvement in a motor function has not yet been sufficiently clarified. We therefore examined 19 patients who received direct peroneal nerve stimulation with central foot drop with the question of the functional gain of foot lifting.
Methods: The extent and force of foot lifting and the functional gait pattern were evaluated in 19 patients who received an implantable peroneal nerve system. This was done 40 months after implantation by determining the strength level according to Janda and with the help of gait tests such as gait endurance, speed and the risk of falling.
Results: The force level of the Janda foot lift increased by a value of 2 on average. The six min gait endurance test increased from 197±39 m to 390±40 m in 78% while using the implant. Gait speed measured over 20 m increased in average in 43.6 %, the time needed decreased from 32.1±9.8 s without to 19.4±4.3 s by using electrical stimulation of a peripheral nerve. Gait steadiness improved, measured by the Timed Up and Go (TUG) test in 34.4%.
Conclusion: Electrical stimulation of a motor nerve can lead to a significant improvement in the function controlled by this muscle. This certainly depends on the complexity of the function and thus how many nerves and thus muscles have to be controlled and what range of movement and thus functionality is desired.
SS22.07
Peripheral Nerve Stimulation as a Therapeutic Option for Severe Neuropathic Pain
Walter Demmel
Background: Chronic neuropathic pain may respond inadequately to conservative treatments. For severe, therapy-refractory pain syndromes, peripheral nerve stimulation represents an effective therapeutic option, supported by recent advancements in implant technology and novel technical approaches.
SS22.08
Addressing Peripheral Neuropathic Pain from Nerve Scarring with Perforator-Based Gliding Tissue Flaps: Two Decades of Clinical Practice
Savas Tsolakidis, R Schmidhammer
Background: Peripheral neuropathic pain (PNP) is a debilitating condition affecting 7-10% of the global population, characterized by hyperalgesia, allodynia, and burning sensations that significantly impair quality of life and contribute to psychological comorbidities. Posttraumatic and postsurgical scarring of peripheral nerves represents a major causative factor, leading to loss of nerve gliding properties, local fibrosis, and compression. While pharmacological treatments remain first-line therapy, surgical intervention may be warranted when scarring is the primary etiology.
Objective: This 20-year retrospective analysis evaluates the efficacy of local perforator-based gliding tissue flaps in reducing post-neurolysis neuropathic pain in patients with peripheral nerve scarring.
Methods: A retrospective analysis was conducted examining a total of 67 patients who underwent neurolysis combined with local perforator-based gliding tissue flap reconstruction over a 20-year period. The study primarily assessed pain reduction outcomes following this surgical technique.
Results: Patients with pain duration <1 year showed VAS reduction from 7.5 to 1.25; those with pain >1 year decreased from 7.5 to 3.0. Mean gliding tissue flap size was 3,899 mm². Upper extremity nerves were most commonly affected (ulnar nerve: 21 cases; radial: 14; median: 8)
Conclusion: This study provides evidence supporting the use of local perforator-based gliding tissue flaps as a surgical treatment option for patients with post-traumatic or post surgical peripheral nerve scarring causing neuropathic pain. The technique addresses the underlying pathophysiology by restoring nerve gliding properties and reducing compression.
SS23.01
US/MRI in evaluating/diagnosing peripheral neuropathies
Dr. Stephan Goedee
Universitair Medisch Centrum Utrecht, Utrecht, Netherlands
Background: Diagnosis of peripheral neuropathies has long been based on combination of clinical evaluation and electrodiagnosis; neuromuscular imaging has been shown to be a versatile and important complementary tool that is increasingly implemented in an increasing number of neuromuscular disorders, including neuropathies.
Methods: This session wil cover the complementary role of neuromuscular imaging in the diagnosis of peripheral neuropathies, focusing on ultrasound and MRI; this includes practical examples, consideration of relevant pitfalls and implementation in routine clinical practice.
Results: Clinical assessment is an important determinant of adequate selection of ancillary testing and correct interpretation of its results, including US and MRI imaging in diagnosis of peripheral neuropathies. Each techniques has its own intrinsic strengths and limitations, and shared overlap of abnormalities found in distinct causes of peripheral neuropathies -e.g. hereditary demyelinating, chronic dysimmune and paraprotaenemic-.
Conclusion: Attendees will be able to recognize distinctive patterns of US and MRI, and consider interpretation in relevant clinical context, as complementary tool in the diagnosis of peripheral neuropathies.
SS24.03
Computer Vision and AI for the Neurological Exam
Dr. Henry Kaminski1, Dr. Gülşen Öztosun1, Dr. Quentin Lesport2, Dr. Marc Garbey2
1
George Washington University, Washington DC, United States.
2
Care Constitution, Houston TX, United States
Background: Myasthenia gravis (MG) is a fluctuating autoimmune neuromuscular disorder that presents unique challenges for both in-person and remote assessment. Clinical evaluation relies heavily on the neurological examination, which is inherently subjective and may fail to capture subtle weakness or fatigability. These limitations motivate the development of quantitative digital tools capable of augmenting traditional examination methods.
Methods: Initial development involved 52 patients with MG across six centers, generating 104 examination videos analyzed using a quantitative telemedicine platform designed to augment traditional neurological assessments. Computer vision algorithms were used to quantify ptosis, ocular motility, and limb drift, while signal processing and natural language processing methods analyzed speech-derived respiratory measures and vocalization patterns. A reproducibility analysis metric was developed to compare AI-driven quantification with expert clinician assessments. Using this system, an additional cohort of approximately 100 patients and controls has subsequently been evaluated.
Results: Initial studies demonstrated that remotely acquired video and speech data capture multiple clinically relevant features of MG, including ptosis dynamics, facial muscle weakness, dysarthria, and fatigue during sustained tasks. These digital biomarkers correlated with clinical severity measures and distinguished patients from controls. Reliability analyses revealed that variation in examiner instructions and differences in video quality significantly affected measurement reproducibility. Ongoing investigations have also identified distinctive vocal signal patterns—present even in the absence of clinically detectable dysarthria—that consistently differentiate patients with MG from controls.
Conclusion: In contrast to the categorical classifications (e.g., mild, moderate, severe) typically used in standard clinical assessments, digital examination frameworks enable continuous and quantitative measurement of neuromuscular function. These approaches have the potential to enhance assessment precision, reduce variability in neurological examination, and identify disease-related metrics not detectable during routine clinical evaluation. This scalable methodology may facilitate integration of digital biomarkers into MG clinical care and therapeutic trials.
SS25.02
Patients' Self Reported Outcomes
Dr. Umberto Manera
ALS Centre, 'Rita Levi Montalcini', Department of Neuroscience, University of Turin, Turin, Italy
Background: Amyotrophic lateral sclerosis (ALS) and motor neuron diseases (MND) are characterized by rapid progression and multidimensional burden affecting motor, respiratory, cognitive, and psychosocial domains. Traditional endpoints such as survival and clinician-administered scales (e.g., ALSFRS-R) only partially capture the patient experience. Patient-reported outcomes (PROs) and PROMs provide a complementary, patient-centered perspective, capturing quality of life, symptoms, and daily functioning. The rise of digital medicine and decentralized clinical trials (DCTs) enables remote, frequent, and real-world PRO collection, particularly relevant for patients with limited mobility. However, challenges remain in validation, integration with objective biomarkers, and ensuring reliability and adherence.
Methods: We discussed in a structured review of PROMs used in ALS, including ALSFRS-R (patient-reported versions), ROADS, ALSAQ-40, and EQ-5D, focusing on psychometric properties and suitability for remote use. Evidence from observational cohorts and clinical trials incorporating PROs was analyzed, comparing remote versus in-clinic data collection. We evaluated digital platforms (apps, web portals, telemedicine) for usability, adherence, and data quality, and reviewed the integration of PROs within decentralized and hybrid trial designs. Correlations between PROs and clinical outcomes (e.g., progression, respiratory function, survival) were assessed, along with strategies to address missing data and reporting bias. Insights from real-world clinical practice in ALS digital monitoring were also included.
Results: PROs capture key aspects of disease burden often missed by clinician-based measures, including fatigue, emotional well-being, and social impact. Patient-reported functional scales show strong correlation with clinician-administered assessments, supporting their reliability. Frequent digital PRO collection improves resolution of disease trajectories and patient stratification. In DCTs, PROs enable remote endpoint collection, increasing accessibility and inclusivity while reducing patient burden. Adherence is generally good but declines in advanced disease, requiring caregiver support and adaptive tools. Limitations include variability in reporting, missing data, and digital access disparities. Integration with objective biomarkers enhances interpretability. Regulatory acceptance of PRO-based endpoints is growing but still evolving.
Conclusion: PROs are central to a more patient-centered approach in ALS and MND research and care. Combined with digital medicine, they enable continuous, real-world monitoring and support decentralized trial models, improving inclusion and engagement. Their integration with multimodal data (wearables, biomarkers) can enhance disease modeling and personalized care. Future priorities include validation of digital PROMs, standardization, and improved accessibility across disease stages. Collaboration among stakeholders will be essential to establish PROs as robust endpoints in clinical trials. Overall, PROs bridge the gap between clinical measures and lived experience, offering a critical tool to advance research and improve outcomes in ALS.
SS26.03
ACCESS ALS
Dr. Angela Genge
McGill University Health Center Research Institute, Montreal, Canada
Background: ACCESS ALS is a new initiative in Canada based upon the success of Dr Genge's site at McGill university and now at the McGill University Health Center Research Institute. Access ALS will develop phase 1 clinical trials sites across Canada based upon the success of the Mcgill efforts. The coordinating center will partner with academic ALS sites across Canada to develop, train and support the staff tasked with executing Phase 1 trials. This model can/will be extended to other neuromuscular diseases to take advantage of the favorable regulatory environment in Canada for innovative Phase 1 drug development.
Methods: Access ALS has developed a model coordinating center for the execution of Phase 1 trials in ALS and ultimately other neurological conditions. It includes an operations manager, network coordinator, nurse navigator, pharmacist, contracts specialist, ethics and regulatory manager and phase 1 supervisor. This coordinating center will network with sites across the country to increase the expertise and capabilities across a large geographic region.
Results: Access ALS is currently being established at the McGill University Health Center Research Institute and partners with ALS CAPTURE and the CALS network
Conclusion: The network will immediately enable a more efficient approach to phase 1 drug development in ALS/MND
SS27.02
Diabetic Lumbosacral Radiculoplexus Neuropathy: Recent Updates and Unmet Needs
Asst. Prof. Marcus Pinto
Mayo Clinic, Rochester, United States
Background: Diabetic Lumbosacral radiculoplexus neuropathy (DLRPN) is a rapidly progressive, painful lower-limb neuropathy. This disease usually begins focally and unilaterally with pain but evolves into widespread often bilateral neuropathy with weakness. Although it is usually monophasic, patients have prolonged morbidity from pain and weakness, may have relapses and many patients become wheelchair-dependent. DLRPN is usually a motor-predominant disease but there is unequivocal evidence of autonomic and sensory involvement. Cutaneous nerves from patients with DLRPN show pathologic evidence of ischemic injury (multifocal fiber loss, perineurial thickening neovascularization, and injury neuroma) and microvasculitis (mural and perivascular inflammation and fragmentation of smooth muscle layers of microvessels).
Methods: This lecture examines the clinical and pathological characteristics of DLRPN, evaluates current management and outcomes, and highlights the significant unmet needs in patient care.
Results: The pathophysiology of DLRPN remains under investigation, but pathological studies have demonstrated evidence of ischemic injury, microvasculitis, and an upregulation of inflammatory mediators within the nerves. Risk factors associated with an increased risk of development include a history of autoimmune disorders, stroke, and a higher BMI. Potential triggers—which may occur in up to two-thirds of cases—include infections, vaccinations, surgery, and rapid glycemic control. Ultimately, altered metabolism and immune dysfunction appear to be the most influential factors in the development of this immune-mediated neuropathy. In Olmsted County, MN, the incidence of DLRPN is 2.7/100,000 persons per year, fulfilling the definition of a rare disease. Despite its rarity, it is the most common immune-mediated neuropathy in that population. While the disorder is typically considered monophasic, most patients are left with significant disability, and approximately 20% experience a progressive disease course. Consequently, DLRPN is an inflammatory neuropathy characterized by high morbidity and immense unmet medical needs.
Recent observations have shown a frequent association between DLRPN and the use of GLP1-RA drugs. A case-control study demonstrated that GLP1-RA use increases the risk of DLRPN by 51%. While the exact mechanisms are not yet clear, the profound metabolic shift induced by these drugs appears to be a primary trigger. Research also indicates that nerve pathology features are very similar between patients with treatment-induced neuropathy of diabetes (TIND) and those with DLRPN. This suggests that DLRPN can be treatment-induced and exists as part of the broader TIND spectrum.
Currently, DLRPN is classified as a variant of nonsystemic vasculitic neuropathy. However, there remains no international consensus regarding its clinical management, and there are currently no FDA or EMA-approved therapies available.
Conclusion: DLRPN is a rare but highly debilitating immune-mediated neuropathy, identified as the most common of its kind in Olmsted County. Its pathophysiology involves ischemic injury and microvasculitis, often triggered by rapid metabolic shifts from glycemic control or GLP1-RA use. This places TI-DLRPN within the TIND spectrum, characterized by severe morbidity and long-term disability. Despite its classification as a nonsystemic vasculitic neuropathy, the lack of FDAor EMA-approved therapies and clinical consensus highlights an urgent need for targeted research and standardized treatment strategies.
SS27.03
GLP1 agonists and neuropathy
Dr. Vincenza Spallone
University of Rome Tor Vergata, Rome, Italy
Background: GLP1 receptor agonists (GLP1-RAs) represent a new class of drugs with multi-faceted beneficial effects on glycaemic control, overweight, cardiovascular and renal outcomes of diabetes. The advent of double or triple incretin agonists represents another strong opportunity to achieve almost unthinkable therapeutic goals in people with diabetes. Given the unmet need of a pathogenetic treatment for diabetic polyneuropathy (DPN), there is growing interest in exploring a potential place of GLP1-RAs in DPN, in addition to their potential effects on central neurological diseases. Preclinical studies have documented neuroprotective and significant antinociceptive effects of GLP1-RAs, thus providing the premises for clinical trials with the primary outcome of DPN or neuropathic pain.
In preclinical studies GLP1-RAs provide a multimodal neuroprotection (by limiting inflammation through inhibition of NF-κB and MAPK pathways and reduction of pro-inflammatory cytokines), oxidative stress, and mitochondrial dysfunction, promoting Schwann cell survival and neurite growth, and reducing astrocyte and microglial activity. They may also activate μ-opioid receptors via increased IL-10 and β-endorphin in microglia thus suppressing pain-related synaptic transmission in the spinal dorsal horn.
Prospective observational studies documenting a reduction in incidence of DPN in parallel with the increased adoption of GLP1-RAs in people with diabetes are lacking, though these drugs are linked with favourable changes in oxidative stress biomarkers and proteomic signatures of neuropathic pain. Moreover, few small controlled clinical trials of 6-18 months duration — including even fewer double-blind, placebo-controlled studies — have investigated the effects of GLP1-RAs on DPN measures but none demonstrated clear superiority over comparators. Therefore, current evidence from these small trials does not support a disease-modifying effect of GLP1-RAs on DPN.
This presentation will briefly review preclinical findings in animal models of DPN and highlight clinical trial results, including ongoing research on dual and triple incretin agonists and combinations of GLP1-RAs with amylin agonists. Both GIP and amylin receptors are expressed in the peripheral nervous system, with limited preclinical evidence suggesting benefits for nerve repair and nociception regulation. Reduced relative risk of DPN development has been described with the GLP-1R/GIP dual agonist tirzepatide, and a trial with GLP1-RA semaglutide and amylin receptor agonist cagrilintide is currently underway for patients with type 2 diabetes and painful DPN.
On the other hand, some cases of peripheral neuropathies have been reported in individuals with diabetes or obesity as possible consequences of GLP1-RAs-associated significant weight loss and rapid improvement in glycaemic control. These neuropathies resemble lumbosacral radiculoplexus neuropathy or treatment induced diabetic neuropathy, with inflammation and microvasculitis as possible causes. Although rare, these comorbidities of GLP1-RAs warrant awareness and surveillance.
SS28.01
Looking forward: Duchenne muscular dystrophy in 2030
Prof. Francesco Muntoni
Paediatric Neurology and the Director of the Dubowitz Neuromuscular Centre, at the UCL Great Ormond Street Institute of Child Health and Great Ormond Street Hospital for Children, and Director of the Genetic Therapy Accelerator at the UCL Institute of Neurology, London, United Kingdom
Background: In the last decade there have been consistent advances in different approaches aimed at restoring dystrophin production in skeletal and cardiac muscle in patients with Duchenne muscular dystrophy (DMD). RNA therapies paved the way, with a number of first generation splice-switching antisense oligonucleotides (ASOs), several having received conditional FDA approval, targeting specific “skippable” mutations. We are now in the era of second generation ASOs, with several pivotal trials underway that are likely to become the next standard of care. The path of adeno-associated virus (AAV) gene therapy has also demonstrated success of the first generation vectors; and many competitors are currently building on the experience of the first studies to try to develop more potent therapies. Parallel but different efforts have also been devoted to a growing list of interventions which deal with other aspects of muscle pathology clearly delineating a path of multiple combinatorial therapies that will represent the standards of care in the year to come.
In my presentation I will provide an overview of what could be the hypothetical therapeutic landscape in DMD, and also which are bottlenecks that will need to be overcome to have the maximal impact for the life of people living with DMD
SS30.01
Challenges in supportive and palliative neuromuscular care in developing countries
Prof. Jorge A. Bevilacqua
Universidad de Chile, Santiago, Chile
Background: Palliative and supportive care for neuromuscular disorders (NMDs) faces complex, structural, cultural, and clinical-scientific challenges in developing countries. Some of them are universal; however, unlike in high-income countries, where services are relatively integrated, a lack of visibility persists in low-and middle-income countries, leaving many patients without any specific diagnosis, treatment, or support.
These challenges encompass at least five main critical areas: health infrastructure and public policies; late and misdiagnosis; access to treatments and symptom management; training of healthcare personnel and fragmentation of general and specialized care; caregiver burden; and psychosocial support. The lack of prioritization of rare diseases, particularly NMDs, on most public health agendas often leads to a lack of registries, preventing us from assessing the magnitude of the problem and planning appropriate healthcare resource allocation and strategies accordingly. The existing palliative care infrastructure, often basic and insufficient, typically focuses on more common, urgent, or visible illnesses, such as cancer, or multiple sclerosis, neglecting rare and progressive NMDs. In most developing countries, timely diagnosis presents an enormous clinical and logistical challenge. NMDs are often mistaken for more common conditions. The lack of specialists and the absence of specific diagnostic tools leads to long diagnostic odysseys. Once diagnosed, the disease are often at an advanced stage, and access to therapy is limited, hindering timely management and treatment. Disease-modifying therapies are often inaccessible or unavailable in the public sector, requiring legal action to obtain them, resulting in delays and loss of the therapeutic windows for prescription, additionally to the administrative burden put on the medical team's shoulders. Taken together, this forces palliative care teams to focus exclusively on symptom management with basic resources.
Furthermore, a significant knowledge gap exists between both general practitioners and specialists, who do not associate NMDs with palliative care, perceiving them solely as “end-of-life care.” Additionally, multidisciplinary care is only accessible in large cities and is virtually nonexistent in rural areas. In contexts where institutionalized care is unavailable, the responsibility falls almost exclusively on the family, often on women or children. The absence of universal health insurance systems or government subsidies on the other hand, forces families to cover prohibitive costs with their own scarce resources.
Conclusion: Palliative and supportive care for patients with NMDs faces challenges that are exacerbated in low-income developing countries. Several strategies have been identified to address these challenges effectively. Integrated home-based palliative care has emerged as a viable and effective model. Civil society organizations constitute another strategy that not only helps raise awareness about these diseases but also provides social support networks and community education. There is a clear and urgent need for a paradigm shift: to move from considering palliative care as a luxury of high-income countries to reading it as an essential component of the treatment of neuromuscular diseases at a universal level; secondly, to move from medicine centralized in medical specialties to one centered on the patient's health problem as the central axis of the healthcare team's strategy to treat it through the implementation of integrated multidisciplinary approaches including transition.
SS33.01
Epidemiological Changes in MG
Prof. Valentina Damato
Department of Neuroscience, University of Florence, Florence, Italy. SOD Neurologia d'urgenza, Careggi University Hospital, Florence, Italy
Background: Myasthenia gravis (MG) is a rare autoimmune neuromuscular junction disorder with marked heterogeneity in clinical phenotype and immunopathogenesis. Over recent decades, its epidemiology has evolved substantially, offering insights into disease mechanisms and healthcare needs. Contemporary population-based studies report an annual incidence of approximately 10–30 per million person-years and a prevalence ranging from 10 to over 30 per 100,000 population, with consistent evidence of increasing disease frequency worldwide. This rise is attributed to population ageing, improved diagnostic accuracy, and reduced mortality. Notably, shifts in MG subtypes, particularly the growing predominance of late-onset and very-late-onset MG, suggest changing interactions between genetic susceptibility, environmental exposures, and immune ageing.
Methods: This presentation synthesizes data from recent population-based epidemiological studies, systematic reviews, and longitudinal cohort analyses spanning the past five decades. Key metrics include incidence, prevalence, demographic distribution, and subtype-specific trends. Special emphasis is placed on large registry-based studies and retrospective cohort analyses examining temporal changes in age at onset, antibody status, and clinical outcomes. Comparative evaluation of methodological approaches (registry-based vs clinical cohorts) and subgroup stratification (AChR, MuSK, seronegative MG; early vs late onset) is performed to contextualize observed epidemiological variability.
Results: Global MG incidence ranges from approximately 2.7 to 61 cases per million person-years, with more robust estimates clustering between 10 and 30 per million, while prevalence has increased up to 25–50 per 100,000 in recent large-scale studies . A consistent temporal increase in prevalence is driven by improved survival and ageing populations. Demographically, MG shows a bimodal age distribution, with early-onset predominance in females and increasing incidence in older males . Recent longitudinal data demonstrate a marked shift toward very-late-onset MG (≥65 years), with median age at onset increasing from ∼35 years in the 1970s to ∼65 years in recent decades. This shift is accompanied by a higher proportion of AChR-antibody-positive disease and increased comorbidities. Subtype variation remains geographically distinct: MuSK-MG is more prevalent in Mediterranean and African populations, whereas juvenile MG is more common in East Asia . Despite increasing prevalence, mortality has declined, with only modestly elevated standardized mortality ratios in treated populations.
Conclusion: The epidemiology of MG is undergoing a significant transition characterized by increasing prevalence, ageing patient populations, and evolving subtype distribution. The rise of late- and very-late-onset MG has important implications for disease management, given higher comorbidity burden and treatment-related risks in older patients. These trends likely reflect both improved healthcare systems and true biological changes, including immunosenescence and environmental influences. Future epidemiological research should prioritize standardized methodologies and detailed subgroup analyses to better elucidate etiological mechanisms and guide precision medicine strategies.
SS33.03
MG Prognosis in Very Late Onset Patients
Dr. Elena Cortes-Vicente
Hospital de la Santa Creu i Sant Pau, Barcelona, Spain
Background: Myasthenia Gravis (MG) was initially considered a disorder in women under 40 years of age. Patients have been generally classified into 2 subgroups according to age at onset: early-onset MG, when they are under 50 at disease onset, and late-onset MG, when they are aged 50 or older at onset. However, in recent decades the incidence has increased in both men and women over 65. This group have been defined as very-late-onset MG patients.
Methods: We designed an observational cross-sectional multicenter study based on information in the neurologist-driven Spanish Registry of Neuromuscular Diseases (NMD-ES). All patients were >18 years of age at onset of MG and onset occurred between 2000 and 2016 in all cases. Patients were classified into 3 age subgroups: early-onset MG (age at onset <50 years), late onset MG (onset ≥50 and <65 years), and very-late-onset MG (onset ≥65 years). Demographic, immunologic, clinical, and therapeutic data were reviewed.
Results: This study was published in Neurology in 2020 (Cortés-Vicente E, et al. Neurology 2019;00:1-10. doi:10.1212/WNL.0000000000008903). We studied a total of 939 patients from 15 hospitals: 288 (30.7%) had early-onset MG, 227 (24.2%) late-onset MG, and 424 (45.2%) very-late-onset MG. The mean follow-up was 9.1 years (SD 4.3). Patients with late onset and very late onset were more frequently men (p < 0.0001). Compared to the early-onset and late-onset groups, in the very-late-onset group, the presence of anti-acetylcholine receptor (anti-AChR) antibodies (p < 0.0001) was higher and fewer patients had thymoma (p < 0.0001). Late-onset MG and very-late-onset MG groups more frequently had ocular MG, both at onset (<0.0001) and at maximal worsening (p = 0.001). Although the very-late-onset group presented more life-threatening events (Myasthenia Gravis Foundation of America IVB and V) at onset (p = 0.002), they required fewer drugs (p < 0.0001) and were less frequently drug-refractory (p < 0.0001).
Conclusion: Patients with MG are primarily very-late-onset with anti-AChR antibodies and no thymoma. Although patients with very-late-onset MG may present life-threatening events at onset, their outcome is usually good when diagnosed and treated properly.
SS34.02
Dysfunctional autonomic disorders
Prof. Paola Sandroni
Mayo Clinic, Rochester, United States
Background: As symptoms referable to autonomic dysfunction are relatively common, it is a frequent occurrence to diagnose patients as having a primary autonomic failure or dysfunction when the actual pathology may be elsewhere.
Particularly conditions affecting cardiovascular and gastrointestinal systems could be extremely difficult to differentiate from a primary autonomic problem. One of the most common mimickers that we encounter these days is probably persistent postural perceptual dizziness as well as pervasive conditions such as myalgic encephalomyelitis.
Rheumatologic and immune mediated conditions can also contribute to the patient is suffering from autonomic symptoms and other conditions such as pelvic floor dyssynergia could mimic gastrointestinal dysmotility.
There is certainly a subset of patients that appears to suffer from a constellation of symptoms characterized by orthostatic intolerance, pain, fatigue, cognitive dysfunction (“brain fog”), functional dyspepsia, interstitial cystitis, sleep disturbances, widespread pain and other symptoms such as difficulty thermoregulating, which may suggest predisposition to electrical instability at the level of autonomic pathways resulting in dysregulation across central and peripheral autonomic networks generating multiple symptoms that Individually are non‑dangerous; however, their cumulative effect can be profoundly disabling.
There is little value in addressing these symptoms separately. Multiple studies have indicated that a more global strategy with a multidisciplinary team approach yields a much better outcome than when care is fragmented among different specialists that do not coordinate care in a cohesive manner. But there is still a lot of discussion on what these strategies should be.
Addressing symptoms in isolation has limited clinical value. Fragmented care commonly results in conflicting treatment recommendations, redundant diagnostic testing, poor symptom control, patients’ disengagement and providers’ dissatisfaction.
Many physicians report discomfort managing these patients due to diagnostic ambiguity, lack of standardized care pathways, resource and coordination challenges.
Unified diagnostic formulation, patient education focused on symptom coherence and coordinated therapeutic interventions are likely to be the best strategy to improve functional status, reduce symptom burden and enhance patient and provider confidence.
SS36.03
Antibody Discovery in Immune Neuropathies
Dr. Divyanshu Dubey
Mayo Clinic, Rochester, United States
Background: Immune-mediated neuromuscular disorders comprise a heterogeneous group of peripheral nervous system disorders in which autoantibodies play key diagnostic and mechanistic roles. While established antibodies have refined disease classification, a substantial proportion of patients remain seronegative, limiting diagnostic precision and targeted therapy.
Methods: We employ a multimodal discovery platform integrating tissue-based indirect immunofluorescence assays, phage immunoprecipitation sequencing (PhIP-Seq), immunoprecipitation coupled with mass spectrometry, and protein microarray. Serum and CSF samples from well-phenotyped patients with suspected autoimmune neuropathies are systematically evaluated through a biomarker discovery pipeline, followed by antigen identification and orthogonal confirmation.
Results: This approach has enabled the identification and validation of novel antibody targets associated with distinct clinical phenotypes. Some of the new characterized antibodies demonstrate specific staining patterns, reproducible antigen binding, and clinicopathologic correlations.
Conclusion: Advanced antibody discovery platforms are expanding the spectrum of immune neuropathies and improving diagnostic yield. Identification of novel autoantibodies enhances disease classification, informs prognosis, and may guide therapeutic decision-making, underscoring the importance of continued translational efforts in neuroimmunology.
SS37.02
Whole RNA Profile of Response to Thymectomy
Dr. Henry Kaminski, Prof. Linda Kusner
George Washington University, Washington DC, United States
Background: Thymectomy provides durable clinical benefit in myasthenia gravis (MG), yet the systemic immune mechanisms underlying its efficacy remain incompletely understood.
Methods: We performed longitudinal whole-blood RNA sequencing and serum proteomic profiling in participants from a randomized clinical trial comparing prednisone alone with prednisone plus thymectomy. Analyses were designed to identify treatment-associated transcriptional signatures linked to clinical severity over time.
Results: Global clustering and compound Poisson mixed-effects modeling showed that baseline gene expression had limited predictive value for short-term clinical improvement. In contrast, longitudinal modeling identified robust treatment-by-severity interactions involving 238 genes. Early disease severity was associated with increased interferon/STAT1 signaling, enhanced antigen-loading machinery, NF-κB regulatory networks, and elevated immunometabolic demand. Although baseline features did not predict outcomes, long-term clinical improvement was associated with coordinated modulation of pathways regulating STING activity (SKIL), dendritic cell tolerance and antigen presentation (DCSTAMP, AIRE), cytokine negative feedback (SOCS3), inflammasome-associated regulatory programs (AIM2, IL18R), and B-cell receptor and plasma-cell survival pathways (IGLV5-45, TNFRSF17/BCMA), together with epigenetic regulators of HLA expression (BRD2, HCG27).
Conclusion: Thymectomy was associated with sustained attenuation of immune activation, reduced cellular metabolic demand, and remodeling of intracellular transport and cytoskeletal pathways, consistent with transition toward a less inflammatory immune state. These findings demonstrate that dynamic transcriptional trajectories, rather than baseline molecular profiles, capture treatment response and provide mechanistic insight into the durable therapeutic benefit of thymectomy in MG.
SS39.02
Interventional respiratory training
Asst. Prof. Stephan Wenninger
Friedrich-Baur-Institute, Munich, Germany
Background: Respiratory muscle weakness (RMW) is a major contributor to morbidity and mortality in many neuromuscular disorders (NMD). Progressive weakness of inspiratory and expiratory muscles leads to restrictive ventilatory insufficiency, impaired cough, sleep-disordered breathing, and ultimately chronic respiratory failure. Current respiratory management in NMD primarily focuses on supportive interventions such as non-invasive ventilation and airway clearance techniques. However, strategies aimed at directly improving respiratory muscle performance have received comparatively little attention in clinical practice.
Respiratory muscle training (RMT), particularly inspiratory muscle training (IMT), has emerged as a potentially valuable and feasable therapeutic approach to enhance respiratory strength, ventilatory efficiency, and exercise tolerance. Small interventional studies in conditions such as Duchenne muscular dystrophy, late-onset Pompe disease, and myotonic dystrophy type 1 suggest that structured respiratory training programs may improve maximal inspiratory pressure and other pulmonary function parameters. Nevertheless, evidence remains heterogeneous with regard to training protocols, duration, and clinical endpoints.
Methods: The presentation is based on a narrative literature review which was conducted to identify interventional studies evaluating respiratory muscle training in patients with neuromuscular disorders. Particular emphasis was placed on studies assessing inspiratory muscle training using threshold resistance devices, which represent the most widely applied and validated training modality. Outcome measures of interest included maximal inspiratory pressure (MIP), forced vital capacity (FVC), maximal voluntary ventilation (MVV), and patient-reported outcomes.
Results: Across published studies as well as based on own experience in daily routine care, IMT demonstrated consistent improvements in respiratory muscle strength, most commonly reflected by increases in maximal inspiratory pressure. Improvements in ventilatory parameters such as FVC and MVV were less frequently reported in some cohorts, particularly when endurance-focused training protocols were applied.
Evidence from randomized and controlled studies remains limited but suggests that structured respiratory training can lead to clinically meaningful physiological adaptations. Across different studies, IMT was well tolerated and no serious adverse events attributed to IMT were observed in any trial. Moreover, adherence rates were generally acceptable when training was performed in structured home-based programs with regular follow-up. Importantly, greater adherence to the training protocol was associated with larger improvements in respiratory muscle strength, highlighting the relevance of sustained patient engagement.
Conclusion: IMT represents a feasible, safe, and potentially effective therapeutic strategy for patients with NMD and respiratory involvement. Available clinical evidence indicates that structured inspiratory training programs can improve respiratory muscle strength and may enhance ventilatory efficiency in selected patient populations. Both strength-oriented and endurance-oriented training protocols appear beneficial, although endurance-based approaches may provide broader physiological effects on ventilation.
Despite these encouraging findings, the overall evidence base remains limited by small sample sizes, heterogeneous study designs, and variability in training protocols and outcome measures.
Within multidisciplinary neuromuscular care, respiratory muscle training may represent a valuable proactive intervention, particularly in early stages of respiratory involvement before the development of advanced ventilatory insufficiency. Future research should focus on standardized training protocols, integration with telemedicine-supported care models, and the evaluation of clinically meaningful outcomes in neuromuscular disease cohorts.
SS40.01
Update on Lambert-Eaton myasthenic syndrome
Prof. Jan Verschuuren
Leiden University Medical Centre, Leiden, Netherlands
Background: Lambert-Eaton myasthenic syndrome (LEMS) is an ultrarare autoimmune disease caused by antibodies to presynaptic voltage-gated calcium channels. It is characterized by proximal muscle weakness, most predominantly in the legs, and loss of tendon reflexes and autonomic dysfunction. The incidence is less than 1 per million. LEMS has a strong HLA association in the predominantly female, early-onset group. Aside from this idiopathic form, in about half of the patients the disease is associated by small cell lung cancer, more often in the older population.
LEMS is very likely to be underdiagnosed, as the most predominant symptom of proximal leg weakness is not very specific and can be explained by other disorders, especially at an older age. LEMS is a treatable condition. Effective symptomatic and immunosuppressive treatments are available. The diagnosis of LEMS is relatively easy, if the patient is tested for serum VGCC antibodies or electromyography with repetitive nerve stimulation is performed.
Conclusion: Current treatment and the newly evolving therapies for autoimmune neuromuscular synapse disorders will be discussed.
SS40.02
Autoreactome of Thymoma-Associated Myasthenia Gravis
Dr. Linda Kusner, Dr. Henry Kaminski
George Washington University, Washington, United States
Background: Thymomas are unique among neoplasms for their strong association with autoantibody-mediated diseases with the highest occurrence of acetylcholine receptor antibody-positive myasthenia gravis (thymoma associated MG, TAMG). A major contributor to this pathogenesis is disruption of thymic epithelial cell function, which impairs T cell development and central immune tolerance through a deficiency in the autoimmune regulator (AIRE) protein. While autoantibodies targeting striated muscle proteins, the ryanodine receptor, and potassium channels have been described, the full scope of autoreactivity remains incompletely defined.
Methods: To address this, we applied a broad, unbiased profiling approach. Serum samples from 43 patients with TAMG and 18 healthy, non-autoimmune controls were analyzed by molecular indexing by self-assembly to characterize the autoreactome, assessing over 16,000 full-length proteins and nearly 400,000 peptide sequences for autoreactive antibodies.
Results: Both TAMG subjects and controls demonstrated substantial autoreactivity, ranging from 62 to 623 protein-targeted autoantibodies per individual. Although no consistent pattern of autoreactivity distinguished the TAMG cohort, a subset of patients exhibited high levels of antibodies directed against multiple type I interferons. Within this subgroup, all patients had antibodies against TRIM46 and P2X4 which have not previously been identified in TAMG. Analysis of the subpopulation did not reveal association with thymoma WHO classification or clinical characteristics.
Conclusion: These findings support widespread disruption of immune tolerance in TAMG, particularly affecting antigen processing and regulatory T-cell function. Further investigation into the functional significance of these autoantibodies may provide insight into disease mechanisms.
SS40.03
Thymoma-Associated Non-MG Syndromes
Dr. Gil Wolfe
University at Buffalo/SUNY, Buffalo, United States
Background: Although relatively rare, thymomas are associated with a variety of autoimmune and paraneoplastic phenomena, with the neuromuscular transmission disorder myasthenia gravis (MG) being the most common. However, a variety of other peripheral and central nervous system disorders have been described in the context of thymoma.
Methods: Case series literature was reviewed based on a PubMed search that merged autoimmune and paraneoplastic neurologic disorders with thymoma. Autoantibody associations and therapeutic approaches beyond thymoma resection and systemic tumor management were also reviewed.
Results: From the standpoint of neurologic disorders linked to thymoma, MG predominates, representing one-third to one-half of all associations. B2 thymomas are the most common histologic type, and autoantibodies to the acetylcholine receptor are present in nearly all these MG cases. The second most common association is most likely the central nervous system disorder of limbic encephalitis/ encephalomyelitis, associated with autoantibodies to ANNA-1 and Ma2. Cognitive, neuropsychiatric, spinal cord and even peripheral nerve involvement can occur with this syndrome, with immunosuppressive therapy (IST), plasma exchange (PE), intravenous immunoglobulin (IVIg), and B-cell depletion representing the avenues of therapy. Other central nervous system disorders include brainstem encephalitis (ANNA-2 and Ma2 autoantibodies), opsoclonus-myoclonus (ANNA-1 or 2, Ma1 or 2, or Yo autoantibodies), cerebellar degeneration (Yo or ANNA-1 autoantibodies), and neuromyelitis optica (NMO; AQPR, MOG, or ANNA-1 autoantibodies). Management of these disorders is similar to the approach for limbic encephalitis with the exception that C5 complement inhibitors, and anti-IL6 receptor and anti-CD19 agents are approved for NMO.
The spectrum of peripheral nervous system disorders associated with thymoma is wide. In addition, to MG, Lambert Eaton myasthenic syndrome (LEMS) associated with the typical autoantibodies to the P/Q voltage-gated calcium channel has been reported with thymoma. Other conditions include cranial and polyneuropathies (GAD, amphiphysin, glycine receptor autoantibodies), autonomic neuropathy (neuronal acetylcholine receptor and CRMP5 autoantibodies), rippling muscle disease (neuronal acetylcholine receptor autoantibodies if thymomatous MG also present), neuromyotonia in the form of either Isaacs’ or Morvan’s syndrome (LGI1/CASPR2 autoantibodies), and inflammatory myopathies including granulomatous myositis. Of note, rippling muscle disease can also be inherited, with caveolin-3 gene mutations, and both stiff person and Morvan’s syndromes do cross over to involve the central nervous system. Beyond the specialized symptomatic and immunotherapy used for MG and LEMS, management of these other disorders typically involves IST, PE and IVIg. Stiff person syndrome can respond to benzodiazepines and anti-spasticity agents such as baclofen, while acetylcholinesterase inhibitors and agents used for orthostatic hypotension such as midodrine can help with autonomic neuropathies. Anti-epileptic agents such as phenytoin and carbamazepine have been used for many years to ameliorate symptoms of muscle stiffness in neuromyotonia.
Conclusion: Although thymomas are an uncommon form of neoplasia, they are frequently associated with a wide variety of autoimmune disorders and paraneoplastic syndromes, with the peripheral and central nervous systems often involved. It is important for clinicians to be aware of these disorders so that appropriate chest imaging can be ordered.
Although the data on prognosis remains mixed, earlier diagnosis and management of thymoma in general can positively influence survival.
SS41.02
Treatment options in newly diagnosed patients and refractory patients
Dr. Julie Paik
Johns Hopkins University, Baltimore, United States
Background: Idiopathic inflammatory myopathies (IIM) are heterogeneous autoimmune diseases with variable clinical manifestations and outcomes. Early diagnosis and timely initiation of therapy are critical to prevent irreversible muscle damage and systemic complications. However, treatment selection remains challenging due to disease heterogeneity, limited comparative data, and the absence of standardized treatment pathways. A subset of patients develops refractory disease, underscoring the need for more effective and targeted therapies.
Methods: This presentation provides a state-of-the-art review of treatment strategies for IIM, with a focus on dermatomyositis and immune-mediated necrotizing myopathy. Evidence is drawn from randomized controlled trials, prospective cohort studies, and emerging data on targeted therapeutics. Practical treatment approaches for newly diagnosed and refractory patients are discussed, incorporating clinical phenotype, autoantibody profiles, and organ involvement.
Results: In newly diagnosed IIM, glucocorticoids in combination with steroid-sparing immunosuppressive agents remain the foundation of therapy, with intravenous immunoglobulin and other agents used in selected patients. Recent advances in targeted therapies are reshaping the treatment landscape, particularly in dermatomyositis. Recently reported phase 3 trial data evaluating JAK/STAT pathway inhibition demonstrate clinically meaningful improvements in disease activity and provide new options for patients with refractory disease. In this setting, biologic and targeted therapies are increasingly used following inadequate response to conventional immunosuppression. Integration of clinical features with serologic and imaging data is improving patient stratification and guiding therapeutic decisions.
Conclusion: The treatment landscape of IIM is rapidly evolving with the emergence of targeted therapies and increasing availability of high-quality clinical trial data. While therapeutic decision-making remains individualized, recent advances offer new opportunities to improve outcomes, particularly in refractory disease. Ongoing multicenter studies will be critical to define optimal treatment strategies and support a more precision-based approach to care.
SS42.03
Small Molecule Mediated Therapies
Dr. Sally Spendiff
CHEO Research Institute, Ottawa, Canada
Background: Congenital myasthenic syndromes (CMS) are rare inherited disorders of the neuromuscular junction (NMJ) caused by mutations in one of more than 35 genes. These defects impair neuromuscular signalling through diverse molecular mechanisms. CMS typically presents with fatigable weakness, and in severe cases, life threatening respiratory crisis. Treatment outcomes vary widely by genetic subtype. Acetylcholinesterase inhibitors (AChEi) are first line treatments for many forms but have limitations, including short half-life, muscarinic side effects, and variable efficacy. They may worsen symptoms in common subtypes such as COLQ- and DOK7-CMS.
Recent advances have identified promising mechanistic directed therapies. MuSK agonist antibodies show strong benefit in DOK7-CMS mice models by restoring MuSK phosphorylation, NMJ morphology, and motor performance. These findings suggest potential use in additional CMS forms involving defects in AGRIN-LRP4-MuSK signaling or in proteins that interact with the pathway. In parallel, chloride channel (ClC-1) inhibitors, which enhance muscle excitability, have produced functional gains in NMD models and early human studies.
Methods: MuSK agonist antibody experiments used differentiated mouse or human myotubes to measure MuSK phosphorylation, acetylcholine receptor (AChR) clustering, and downstream signalling activation. In vivo experiments assed therapeutic benefit in mouse models with Dok7, Agrn, or ColQ mutations. Outcomes included survival, body weight, motor performance, and NMJ morphology and function. Studies of ClC‑1 chloride channel inhibitors used a combination of electrophysiology, pharmacokinetics, and functional testing. Motor performance was evaluated using grip strength, hindlimb suspension, inverted screen assays, and measures of fatigability. Electrophysiological approaches such as repetitive nerve stimulation were used to assess neuromuscular transmission. Tissues were collected for histological and biochemical analyses, including fibre-type immunolabelling, quantification of fibre size distributions, and measurement of protein expression.
Results: Both strategies demonstrated measurable improvements in NMJ function, motor performance, and survival of mutant mice. MuSK agonist antibodies consistently restored AGRIN-LRP4-MuSK signalling, increasing MuSK phosphorylation, and AChR clustering in Dok7- and Agrn-CMS models. Treatment produced robust gains in body weight, grip strength, and fatigue resistance, with some models showing full rescue of survival. However, therapeutic response was gene-dependent, with no benefit observed ColQ-CMS models. ClC-1 inhibitors also yielded encouraging results, increasing muscle excitability, grip force, and overall motor endurance. Treatment was associated with enlarged muscle fibre size, improved neuromuscular transmission, and partial correction of aberrant fibre-type composition.
Conclusion: Together, these results demonstrate opportunities to improve neuromuscular transmission across multiple forms of CMS, highlighting the therapeutic promise of pathway targeted interventions. They also underscore the importance of matching treatment strategy to genetic subtype. As both mechanistic understanding and therapeutic toolkits expand, the future of CMS care appears increasingly precise, personalised, and hopeful.
SS45.01
Europe
Ms. Evy Reviers
EUpALS, Leuven, Belgium
Background: The European Organisation for Professionals and People with ALS (EUpALS) unites 30 national ALS Assiociations from 22 European countries to collaborate better together. As a non-profit European umbrella organisation, we take the interests of European people with ALS (pALS) to heart. In particular, we facilitate access to ALS clinical trials and provide information about it.
Methods: EUpALS stimulates ALS clinical research by engaging in partnerships with industry and academic consortia, to bring in and safegarding the patient perspective.
Results: Gathered information is disseminated via the EUpALS website, social media, and quaterly Newsletters. In addition, we organise Round Tables satellite to scientific meetings, and are partner in a number of EU Horizon projects.
Conclusion: Several examples of the aforementioned EUpALS activities will be presented and discussed during this session.
SS45.02
North America
Prof. Senda Ajroud Driss
Northwestern Univeristy, Chicago, United States
Background: US patient organizations have made transformative contributions to neuromuscular disease research, therapy development, and clinical care over the past 20 years, fundamentally reshaping the landscape from basic research through FDA approval and clinical implementation.
US patients’ organizations have been active on many fronts, they interact with legislators to change policies that directly affect neuromuscular patients, they engage with the FDA, they fund research and clinical infrastructure and play a role in care standardization and access to care not just locally but also globally.
During the presentation, I will review the history of patients and patients’ organizations in the US their direct role in fundamentally changing the way we care for our patients and how they propelled us in the era of genetic therapies and precision medicine
SS46.01
Cellular Senescence in Tissue Repair and Dysfunction
Dr. Mikolaj Ogrodnik
Ludwig Boltzmann Institute for Traumatology, Vienna, Austria
Background: Cellular senescence is a state of stable cell cycle arrest accompanied by characteristic morphological and molecular alterations. The accumulation of senescent cells (SCs) in various tissues, including the nervous system and skeletal muscle, has been shown to promote inflammation and impair tissue function. Numerous studies have demonstrated a causal relationship between SC accumulation and age-related tissue dysfunction, with the introduction of SCs accelerating aging phenotypes and their clearance attenuating them. In aging muscle, an increased burden of SCs correlates with a decline in muscle performance.
This presentation will summarize recent advances in the reliable identification and study of cellular senescence in animal tissues, as outlined in the Guidelines for Minimal Information on Cellular Senescence Experimentation in vivo (MICSE). Furthermore, I will discuss key research directions and findings concerning the role of cellular senescence in the neuromuscular system. Finally, I will present some of our most recent results on cellular senescence in other tissues, highlighting their potential implications for future research on the neuromuscular system and aging.
SS46.02
Ageing of peripheral nerves
Prof. Claudia Sommer
University Hospital Würzburg, Würzburg, Germany
Background: The prevalence of peripheral neuropathy is greatly increased in elderly people. Various factors may be involved in this finding. Cumulative effects of adversive lifestyle factors and environmental toxins have been implied. Furthermore, senescent Schwann cells are induced by aging and are associated with impaired axonal regeneration and inflammation. These senescent Schwann cells may be a negative prognostic factor in patients with neuropathies, especially in older patients and may be a therapeutic target. Age-related mediators like sirtuins and neprilysin are supposed to form an important link between aging and inflammation. SIRT1 a is a NAD+-dependent deacetylase that participates in various biological processes. Decreased SIRT1 activity may be associated with autoimmune diseases, and neuropathic pain. Neprilysin also decreases with aging, and rare variants in the encoding gene are associated with a late-onset neuropathy, indicating that decreased neprilysin activity may promote the development of chronic nerve damage.
SS46.03
Molecular insights of ageing sarcopenia
Prof. Marco Sandri
University of Padova, Padua, Italy
Background: Maintaining the integrity of organelles and proteins despite the cellular stresses that arise from environmental pressure is essential for life. The time-dependent accumulation of cellular damage is widely considered to be the general cause of cellular degeneration and disease onset. Alteration of the protein quality control, called the loss of proteostasis, contributes to impairment of macromolecule and organelle function and is considered among the primary hallmark of aging, morbidity and mortality. Postmitotic tissues such as brain and striated muscles are particularly vulnerable to changes of proteostasis.
Methods: By using genetic approaches for gain and loss of function experiments as well as transgenic mice we have shown that the regulation of protein and organelles turnover is under the control of transcription factors belonging to different signalling pathways. Moreover, to identify novel players in ageing sarcopenia we screened transcriptomic data sets and identified a novel autophagy regulator gene that is required for a healthy ageing.
Results: Our findings showed that the the shape of mitochondrial network and impaired proteostasis in muscles regulates signalling pathways that not only promote sarcopenia but also a secretory pattern to induce sterile systemic inflammation, tissue senescence and organism premature ageing. These alterations in bioenergetics and proteostasis are linked to life styles and I will describe why physical activity and exercise are beneficial for human health, while sedentary life and inactivity are one of the major detrimental behaviours of industrialized societies that contribute to mortality and morbidity
Conclusion: We have started to dissect the molecular insights that promote sarcopenia. These results enable us to identify blood biomarkers that predict the risk of sarcopenia onset and unhealthy ageing
TC03.03
CIDP and Autoimmune Nodopathies
Asst. Prof. Marcus Vinicius Pinto
Mayo Clinic, Rochester, United States
Background: Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) is frequently misdiagnosed, particularly in its atypical clinical phenotypes. Accurate identification is critical as delays in recognizing inflammatory neuropathies increase patient morbidity and mortality. Recent clinical advancements have led to the classification of autoimmune nodopathies (e.g., targeting Neurofascin-155, Contactin-1) as a distinct group of disorders separate from CIDP, requiring unique therapeutic approaches due to their poor response to standard intravenous immunoglobulin (IVIg) therapy
Methods: This lecture will review the diagnosis and management of CIDP and autoimmune nodopathies.
Results: CIDP is diagnosed through a combination of clinical phenotypes (typical vs. variants) and rigorous nerve conduction studies that identify non-uniform demyelination, including multifocal slowing, conduction block, and temporal dispersion. Autoimmune nodopathies (targeting proteins like NF155 and CNTN1) are now classified as separate clinical entities and are no longer considered CIDP variants. While IVIg remains the established first-line therapy for CIDP, autoimmune nodopathies are typically resistant to IVIg. There have been no randomized clinical trials specifically for autoimmune nodopathies to date, but clinical experience indicates promising results with the use of corticosteroids and rituximab
Conclusion: Successful management of demyelinating neuropathies relies on integrating clinical phenotypes with rigorous EAN/PNS diagnostic guidelines. Clinicians must remain vigilant for “red flags” and CIDP mimics—such as POEMS syndrome, amyloidosis, and vasculitic neuropathy—especially in cases refractory to initial immunotherapy. Early differentiation of autoimmune nodopathies is essential to implementing effective, targeted B-cell depletion therapies.
HO01.01
Cranial Nerves and Trunk
Dr. Stefan Meng
Radiology, Hanusch Hospital, Vienna, Austria. Center for Anatomy and Cell Biology, Medical University of Vienna, Vienna, Austria
Background: This part of the nerve ultrasound workshop focuses on landmark-based localization and tracking of clinically important nerves at the head, neck, and trunk. Pitfalls and technical implications will be discussed.
Methods: As ultrasound is based on the combination of knowledge and manual skills, this workshop will be hands-on.
Results: Ideally, the participants take home practical skills and experience for nerve ultrasound.
Conclusion: Ultrasound is an important imaging modality in the diagnosis of neuropathies. Physicians should be aware of the strengths and weaknesses.
AUTHOR INDEX
A. Lewis, Richard PP01.261
Ab Ghapar, Amirul Asyraf OS07.01
Abati, Elena OS08.04
Abbasi, Maria PP01.137
Abdel-Hamid, Hoda Z. PP01.46
Abdelhak, Sihem PP01.69, PP01.70
Abdelkrim, Abdelmadjid PP01.69, PP01.70
Abe, Tatsuya PP01.334, PP01.356
Abedi, Mehrdad PP01.171
Abicht, Angela PP01.66
Abraham, Alon PP01.381
Abrahao, Agessandro PP01.305
Abuzinadah, Ahmad PP01.328
Acar Arslan, Elif PP01.109
Acerra, Gabriella Maria PP01.204
Acsadi, Gyula PP01.67
Addinsall, Alex PP01.389
Adetoro, Nikki PP01.130
ADIGÜZEL, Emre PP01.380
Aggarwal, Ayush PP01.220
Aggarwal, Rohit PP01.103, PP01.262, PP01.90
Agircan, Dilek PP01.181
Aguennouz, Mhammed PP01.120
Aguirre, Maria Adela PP01.153
Ahmadiharchegani, Fatemeh PP01.42
Ahmed, Bouhouche PP01.06, PP01.07
Ahn, Sohyun PP01.227, PP01.302
Ahn, Tae-Beom PP01.330
Ain, Quratul PP01.362
Ait Allali, Nesaiba OS02.03, PP01.03, PP01.323
Ait-Tihyaty, Maria PP01.206, PP01.211
Ajroud Driss, Senda SS45.02
Akbeyaz, Hakki PP01.109
AKCADAG CAMAN, Tugce PP01.190
Akhavanfar, Mohammad H. PP01.372
Akpınar, Ahmet PP01.77
Al Chalabi, Ammar SS32.02
Al-Khalili Szigyarto, Cristina PP01.73
Al-Khalili, Yasir PP01.287
Al-Muhaizea, Mohammad PP01.343
Al-Rubayie, Karnavaal PP01.367
Ala, Pierpaolo PP01.44
Albano, Noemi OS01.04
Alberti, Claudia OS08.04, PP01.104
Alberti, Paola SS10.02
Albini, Sonia PL01.03
Alboini, Paolo Emilio OS04.03, OS04.05, PP01.214, PP01.231
Aldridge, Jamie PP01.215
Alecu, Iulian PP01.52
Alemañ, Jose PP01.203
Alessi, Federica OS02.05, PP01.67
Alfaidi, Nouf PP01.305
Alfredsson, Lars PP01.205
Ali Pacha, Lamia PP01.241
Allegra, Cosimo OS07.04
Allegri, Isabella OS01.04
Allen, David PP01.213
Allen, Jeffrey PP01.268, PP01.88
Allen, Jeffrey A PP01.267
Allen, Jeffrey A. PP01.279, PP01.282, PP01.284
Allenbach, Yves PL04.03, PP01.103
Almeida, Jaqueline PP01.43
Alnajjar, Sara PP01.305
Alonso Alonso, Miguel PP01.267
Alonso-Alonso, Miguel PP01.261, PP01.265
Alonso-Jiménez, Alicia PP01.172
Alpaydın Baslo, Sezin PP01.355
Alroughani, Raed PP01.173
Alshareef, Aysha PP01.328
Alshehri, Ali PP01.193
Alsulami, Manal PP01.328
Altamura, Concetta PP01.25
Amato, Anthony OS01.01, TC14.01
Amato, Anthony A. PP01.262
Amayra, Imanol PP01.391
Ambati, Ravi PP01.245
Amornvit, Jakkrit PP01.371
An Haack, Kristina OS03.06
An, Jae-Young PP01.306
Andersen, Henning OS07.03
Andersen, Kasper PP01.389
Anderson, Ashley PP01.218, PP01.219
Andzeva, Elisa PP01.275
Angadi, sanjana PP01.139
Angelelli, Alessia PP01.35
Angelini, Corrado OS03.01, PP01.107, PP01.111, PP01.391
Anijs, Thomas PP01.61, PP01.62
Annoni, Karin PP01.214, PP01.231
Anokhina, Katerina PP01.278
Antonini, Giovanni OS01.04, PP01.145, PP01.187, PP01.232
Antozzi, Carlo PP01.175, PP01.176, PP01.177, PP01.178, PP01.189, PP01.235, PP01.236, SS04.01
Aoyagi, Ryohei PP01.191
Appelqvist, Hanna PP01.128
Araújo, Alexandra PQC PP01.343
Araúzo-Bravo, Marcos J PP01.362
Arbogast, Thomas OS06.06
ARDIÇLI, Didem PP01.380
Arena, Ignazio PP01.127
Arena, Ignazio Giuseppe PP01.119, PP01.120
Arici, Sehnaz PP01.291
Arman, Inbar PP01.199
Armani, Andrea PP01.152
Arnaoutoglou, Marianthi PP01.110
Arndt, Raquel Cristina OS05.02
Arnold, W. David PP01.329
Aronica, Eleanora OS06.03
Aronica, Eleonora PP01.103
Arpaia, Chiara PP01.55
Arslan, Doruk OS07.06
Arsov, Todor PP01.357
Arteaga Rodriguez, Carlos PP01.254
ARTERO, RUBÉN OS01.03
Arunachal, Gautham PP01.138, PP01.150
Arustamyan, Kristine PP01.217
Arvin-Berod, Clémence PP01.272
Arvin-Bérod, Clémence PP01.279
Ascherman, Dana OS01.01
Ashraghi, Mohammad PP01.188, PP01.192
Assis, Carolina OS07.05
Astrea, Guja PP01.11
Atchayaram, Nalini PP01.138, PP01.139
Atiya, Yael PP01.199
Attarian, Shahram PP01.169, PP01.194, PP01.265, PP01.282
Audhya, Ivana F. PP01.48
Auinger, Peggy PP01.367
Ayanoglu, Cigdem OS01.06
Aydin, Gozde PP01.237
Azcona, Carolina PP01.153
Azuma, Hiroto OS03.04
Azzimonti, Matteo PP01.117
Babu, Merly PP01.65
Bachman, Eric OS04.06
Bae, Jisun PP01.304
Bae, Jong Seok PP01.293
Baek, Seol-Hee PP01.333
Baets, Jonathan PP01.172
Bagg, Matthew PP01.99
Baima, Jader PP01.366
Bakircioglu Duman, Ezgi PP01.188, PP01.192
Bakircioglu-Duman, Ezgi OS09.01
Bal, Mehmet PP01.181
Balasubramaniam, Shanti PP01.65
Balbino, Bianca PP01.382
Baldassari, Simona PP01.09
Baldissera, Martina PP01.316
Baldwin, Joel OS04.04, PP01.79
Baldwin, Tomas PP01.44
Balique-Laplanche, Aurore PP01.132
Ballarini, Nicolas PP01.346, PP01.347
Bamps, Kobe OS08.06, PP01.359
Banerjee, Tanya PP01.290
Bansal, Prashant PP01.59
Baptista, Linda PP01.286
Baranello, Giovanni OS01.06, PP01.33, PP01.44, PP01.46, TC09.04
Baratto, Serena PP01.34
Barbaccia, Adele OS01.04
Barbosa, Guilherme PP01.165
Barbov, Ivan PP01.157
BARGIELA, ARIADNA OS01.03
Baric, Ivo PP01.129
Barnett-Tapia, Carolina PP01.178, PP01.238, PP01.305
Baronchelli, Carla PP01.04, PP01.05
Barone, Carolina OS05.06
Barone, Paolo OS04.03, PP01.155, PP01.204
Barp, Andrea PP01.373
Barrera-Sierra, Sergio PP01.289
Bartels, Bart PP01.17
Barthelemy, Florian PP01.28
Bartolomejovava, Pavla Bortolomejovava PP01.40
Basa, Mihail PP01.13
Baskar, Dipti PP01.139, PP01.150
BASSE FAYE, ANNA MODJI PP01.354
Bauer-Ventura, Iazsmin PP01.90
Baumann, Matthias PP01.325
Beange, Kristen H.E. PP01.372
Bearden, David PP01.259
Beatrice, Francesca OS08.01
Beazley, Bethany PP01.268
Becker, Michael SS22.05
Bedir, Ali Zeki PP01.08
Beer, Kelly PP01.99
Beermann, Mary Lou PP01.37
Beggs, Simon PP01.45
Behin, Anthony PP01.113
Bekoeva, Zemfira PP01.298
Belachew, Shibeshish PP01.368
Belimezi, Maria PP01.182
Bellanti, Roberto OS06.04
Bello, Luca OS01.02, PP01.42
Beltran, Sergi PP01.129
BEN OMRAN, TAWFEG PP01.72
Ben Youssef-Turki, Ilhem SS05.01
Benatar, Michael PP01.170, PP01.199, PP01.80, PP01.81
Benchaabi, Ouissem PP01.321, PP01.69, PP01.70
Benedetti, Luana PP01.370
Benguerba, Kamal PP01.52
Benkirane, Nada PP01.126
Benmhammed, Noor PP01.369
Bennetts, Bruce PP01.65
Benussi, Alberto PP01.316
Benveniste, Olivier OS01.01
Beretta, Francesca PP01.212, PP01.86
Bergin, Cathleen PP01.218, PP01.219
Bergstrom Johnson, Claire OS06.04
Berling, Edouard PP01.132, PP01.151
Bernardo, Katrina PP01.249
Bernert, Guenther PP01.40
Bernert, Günther PP01.325
Bertella, Enrica OS02.03, PP01.04, PP01.05
Berti, Beatrice PP01.55
Berti, Gianna OS03.05
Bertini, Enrico PP01.11, PP01.343
Bertolasi, Laura OS07.03
Bertolasi, Letizia PP01.104, PP01.105
Bestetti, Ilaria OS08.04
Bettica, Paolo OS02.05, PP01.67, PP01.74
Bevilacqua, Jorge A. SS30.01
Bevilacqua, Liliana OS04.03, PP01.155, PP01.204
Beydoun, Said OS01.01, PP01.206
Beytia, Maria de los Angeles PP01.58
Bhatia, Rohit PP01.220
Bhavaraju-Sanka, Ratna PP01.176, PP01.269
Biddeci, Roberta PP01.10
Bieuvelet, Séverine PP01.133
Bikmazer, Bilgihan PP01.109
Bilgraer, Raphael PP01.223
Bini, Paola PP01.86
Biran, Yifat PP01.237
Birman Har-Noy, Nurit PP01.373, PP01.381
Birouk, Nazha PP01.124, PP01.125, PP01.126
Bischoff, Gert PP01.135
Bischoff, Monika PP01.135
Bisecco, Alvino OS04.03
Bisson, Camille OS08.02, PP01.369
Bitoun, Marc SS21.01
BİRBİR, Buse PP01.380
Blaauw, Bert PP01.152
Blanco, Rocio PP01.153
Blank, Bert PP01.364
Blankenbiller, Tricia PP01.108
Blaschek, Astrid OS02.04
Blasini, Romina OS09.04
Bleecker, Jan De PP01.175
Blein, Cécile PP01.191, PP01.194, PP01.272
Bloemers, Jos PP01.179, PP01.186, PP01.195, PP01.201
Blum, Stefan PP01.173, PP01.221, PP01.237, PP01.238
Bobadilla, Edna Julieth PP01.40
Boccanegra, Brigida PP01.29, PP01.30
Boemia, Virginia PP01.352
Boespflug-Tanguy, Odile PP01.67, PP01.74
Boffa, Laura PP01.144, PP01.247, PP01.250, PP01.251, PP01.318
Bogaards, Johannes OS01.05
Bogatyreva, Anna PP01.01
Boggs, Kirsten PP01.65
Boluk, Ebru PP01.291
Bonaccorso, Rosa OS02.02, PP01.19
Bonanno, Silvia OS01.04, OS04.03, PP01.96
Bonar, Kerina PP01.214, PP01.231
Bonci, Tecla PP01.368
Bongiolatti, Stefano PP01.212
Bonofiglio, Federico PP01.283
Bonventre, Joseph V. PP01.46
Boonyapisit, Kanokwan PP01.174
Boonyuen, Wanicha PP01.371
Borghs, Herman PP01.61, PP01.62
Bormann, Daniel PP01.325, PP01.326
Boroojerdi, Babak PP01.179
Bortolani, Sara OS08.03
Boschi, Silvia PP01.47, PP01.84
Bossuyt, Patrick PP01.103
Botrous, Mina PP01.238
Botta, Annalisa PP01.145
Bougamra, Sirine PP01.169
Bouman, Karlijn PP01.14
Bourg-Alibert, Nathalie PL01.03
Bouzaglo, Omer PP01.381
Bovy, Nicolas PP01.369
Boyer, Stéphane PP01.286
Boz, Cavit PP01.173
Braen, Angélique PP01.329
Brais, Bernard PP01.154
Brajuskovic, Goran PP01.143
Brajušković, Goran PP01.358
Branas, Maria PP01.57
Brandsema, John F. PP01.67
Brannagan, Thomas OS01.01
Brannagan, Thomas H. PP01.284
Bratina, Alessio PP01.316
Braun, Jessica PP01.54
Brauner, Susanna OS02.06, OS04.02, OS06.05, OS09.03, PP01.197, PP01.205
Bray, Sarah PP01.170
Breiner, Ari PP01.193
Brennan, Christopher M. PP01.37
Briani, Chiara PP01.297, PP01.370, SS13.02
Brierley, Russell PP01.130
Briggs, Caitlin PP01.159, PP01.206, PP01.211
Brighina, Filippo OS04.03, PP01.370
Bril, Vera OS04.04, PP01.175, PP01.186, PP01.195, PP01.201, PP01.218, PP01.219, PP01.228, PP01.305, PP01.78, PP01.79
Brkusanin, Milos PP01.143, PP01.27, PP01.358
Brockt, Alicia PP01.367
Brogna, Claudia PP01.55
Brokamp, Katharina PP01.180
Brolatti, Noemi OS07.04, PP01.35, PP01.36
Brooks, Gabriel PP01.68
Brown, Thomas PP01.339
Bruex, Hannah PP01.363
Brulc, Erika PP01.153
Bruno, Claudio OS07.04, PP01.11, PP01.34, PP01.35, PP01.36
Brureau, Anthony PL01.03
Brusa, Chiara PP01.33
Bubb, Vivien OS06.01
Bucci, Elisabetta PP01.145
Budding, Kevin PP01.284
Budikayanti, Astri PP01.308
Buffel, Connor PP01.168
Bugatti, Mattia PP01.04, PP01.05
Bukacova, Katerina PP01.40
Buliņa, Inita PP01.98
Burcklen, Michel PP01.175
Burden, Steven PP01.158
Burden, Steven J. PP01.156, PP01.166
Businaro, Pietro OS04.05
Butler, Lesley PP01.222
Butzkueven, Helmut PP01.238
Buzzard, Katherine PP01.173, PP01.237, PP01.238
Bähr, Mathias PP01.363
Bönnemann, Carsten G. OS02.04
Caballero-Avila, Marta PP01.382
Cabraal, Isobel PP01.188, PP01.192
Cacchio', Gabbriella PP01.297
Cadour, Stéphanie PP01.279, PP01.282
Cadri, Silvi PP01.373
Cai, Na PP01.46
Cakar, Arman PP01.118
Calabresi, Paolo OS05.05
Calderon Castro, Andrea del Pilar PP01.40
Calma, Aicee Dawn OS07.01, OS07.02
Calvo, Andrea TC13.03
Camacho, Ana PP01.57
CAMAN, Mahmut Bilal PP01.190
Cambier, Audrey PP01.113
Camdessanché, Jean-Philippe PP01.194
Camelo, Clara PP01.40
Cameron, Donnie PP01.14
Camizaru, Alexander PP01.11
Campbell, Craig PP01.46
Campbell, Nolan PP01.209
Campo, Sofia OS04.03
Camprubi, Sandra OS07.03
Canale, Fabrizio PP01.370
Canales, Blanca PP01.80
Canali, Elena OS01.04
Candrea, Elisabeta PP01.106
Canning, Jake OS09.01
Cano, Robert PP01.329
Cantisani, Teresa PP01.370
Cao, Weihua PP01.228, PP01.78
Capasso, Anna PP01.55
Cappellari, Ornella PP01.29, PP01.30, PP01.31
Carapella, Nicola PP01.04, PP01.100
Cardoso, Igor Baroni PP01.257
Caress, James OS01.01
Caria, Filomena OS02.03, PP01.04, PP01.05, PP01.323, PP01.373
Carment, Loïc PP01.132, PP01.133
Carmona-Martinez, Ricardo PP01.129
Carnazzi, Alessandra PP01.21, PP01.22, PP01.96
Carpo, Marinella PP01.370
Carrera-García, Laura PP01.341
Carrillo, Nuria PP01.268
Carugati, Roberto OS02.03, PP01.03
Carvalho, Alzira PP01.112, PP01.116, PP01.163, PP01.164, PP01.165, PP01.18, PP01.91, PP01.92, PP01.93, PP01.94, PP01.95
Carvalho, Diana PP01.40
Carvalho, Nara OS07.05
Carver, Aleks PP01.335, PP01.336, PP01.337
Casiraghi, Jacopo Luca PP01.373
Casneuf, Tineke PP01.382
Cassano, Emanuele PP01.370
Castellar-Leones, Sandra Milena OS03.03, PP01.246, PP01.258
Castiglioni, Claudia PP01.58
Castillo, Laura PP01.368
Castro, Ana Laura OS03.03, PP01.246, PP01.258
Castro, Paz PP01.340, PP01.341
Cataldi, Matteo PP01.09
Cataldo, Stefania PP01.317
Catapano, Francesco OS02.02
Catteruccia, Michela PP01.11
Cavalcante França Jr, Marcondes PP01.344
Cavalcante, Paola PP01.96
Cavka, Mislav PP01.129
Cazzaniga, Sara OS02.05
Ceccanti, Marco OS05.05
Ceccherini, Elisa PP01.32
Cella, Ricardo Rojo PP01.177
Cenacchi, Giovanna PP01.107
Cendron, Laura PP01.152
Centonze, Diego PP01.144, PP01.247, PP01.250, PP01.251, PP01.318
Cepele, Alba OS04.05, PP01.233
Cerchiara, Alessandro Giovanni PP01.29
Cerovac, Natasa PP01.27
Cetin, Hakan OS08.05, PP01.325, PP01.326
Chacon, Aline PP01.43
Chae, Juhee PP01.294
Chahin, Nizar PP01.88, PP01.90
Chakraborty, Eesha PP01.89
Chambers, Darren PP01.45
CHAN, Irene Heung Lan PP01.315
Chan, Sophelia PP01.40
Chan, Sophelia Hoi Shan PP01.345
Chan, Stephen Wing Wai PP01.345
Chang, B. Kalei PP01.270
Chang, David PP01.171
Chang, David J PP01.90
Chang, Hee Jin PP01.330
Chang, Herng-Hua PP01.311
Chang, Kai Chieh PP01.365
Chao, Chi-Chao OS05.03, PP01.311
Chao, Kenneth PP01.249
Chary, Sowmya PP01.142
Chaudhari, Umesh OS04.06
Cheli, Marta OS04.03, PP01.96
Chen, Lei PP01.243
Cheng, Mei-Fang OS05.03
Cheng, Sue PP01.170, PP01.80, PP01.81
Cheng-Zhang, Jia Qi PP01.37
Cherney, David PP01.305
Chernova, Natalia PP01.298
Cherubino, Christabella PP01.339
Chesmore, Kevin PP01.28
Cheung, Mei Wun PP01.345
Cheva, Aggeliki PP01.110
Chiang, Ming-Chang OS05.03
Chiarella, Lorenzo PP01.36
Chien, Yin-Hsiu OS03.06
CHING, Wai Kin PP01.315
Chinoy, Hector PP01.262
Chitimus, Diana PP01.115
Chiyonobu, Tomohiro PP01.348
Chloca, Fernando PP01.41
cho, eun bin PP01.324
Cho, Jeong Hee PP01.167, PP01.20, PP01.23, PP01.24
Cho, Jin Whan PP01.330
Cho, Joong-Yang PP01.277
Choi, Kyomin PP01.330
Choi, Young-Chul PP01.167, PP01.20, PP01.23, PP01.24
Choi, Yunjung PP01.167, PP01.20, PP01.23, PP01.24
Chollet, Grégory PP01.214, PP01.231
Choo, Seung Ho PP01.324
Choudhary, Gaurav PP01.90
Choudhry, Zia PP01.177, PP01.210
Chourpiliadis, Charilaos OS06.05
Christiansen, Silvia PP01.153
Christopher-Stine, Lisa OS01.01
Chroni, Elisabeth PP01.173, PP01.193, PP01.238
Chu, Binh PP01.329
Chu, Wing Sum PP01.45
Chuang, Tzu-I PP01.311
Chávez, Daniel Cesar PP01.246
Chávez, Daniel César OS03.03
Ciafaloni, Emma PP01.170, PP01.367, PP01.41, PP01.80, PP01.81
Ciaiola, Francesca PP01.318
Cicala, Gianpaolo PP01.55, TC04.01
Cinnante, Claudia Maria PP01.105
Cintas, Pascal PP01.132, PP01.214
Ciscato, Patrizia PP01.104, PP01.105
Ciumas, Mariana PP01.272
Ciurea, Stephan PP01.171
Claeys, Kristl PP01.130, PP01.359
Claeys, Kristl G. OS08.06, PP01.169, PP01.178, PP01.193, PP01.202, PP01.279, PP01.61
Claeys, Kristl. G. PP01.62
Clarke, Andrew PP01.264
Clarkson, Kristen A. PP01.81
Claudione, Letizia PP01.30
Clay, Ieuan PP01.156
Clemens, Paula OS01.02, PP01.42
Clemens, Paula R. PP01.51
Cluzeau, Céline OS08.02, PP01.369
Coarelli, Giulia PP01.154
Coats, Julie OS02.04
Coccia, Michela PP01.373
Cocito, Dario PP01.370
Cogell, Ashley PP01.285
Colacicco, Giovanni SS18.01
Cole, Suzanne OS09.06
Colitta, Alessandro PP01.233
Collet Vidiella, Roger PP01.382
Collins, Michael OS01.01
Colombi, Chiara PP01.03
Comi, Giacomo OS08.01, OS08.04
Comi, Giacomo Pietro PP01.100, PP01.104, PP01.105, PP01.117
Conte, Elena PP01.30
Conte, Giorgio PP01.117
Cooper, Ian PP01.99
Coppejans, Rani PP01.166
Coratti, Giorgia PP01.55
Corazzini, Roseli PP01.112, PP01.116, PP01.163, PP01.164, PP01.165, PP01.92, PP01.93, PP01.94, PP01.95
Corbo, Michela Maria PP01.212
Cordani, Ramona PP01.36
Cordiglieri, Chiara OS08.01
Cornblath, David PP01.268
Cornell, Nikki PP01.44
Correa-Arrieta, Cristian OS03.03, PP01.246, PP01.258
Correia, Carolina PP01.164
Corse, Andrea PP01.215
Cortes-Vicente, Elena SS33.03
Cortese, Andrea PL02.02
Corti, Stefania OS03.05, OS08.01, OS08.04, PP01.100, PP01.104, PP01.105, PP01.117
Cortés Vicente, Elena PP01.186, PP01.201
Cortés-Vicente, Elena PP01.169, PP01.236
Cosentino, Giuseppe PP01.244, PP01.370
Costanza, Massimo PP01.96
Cotti Piccinelli, Stefano OS02.03
Coudyzer, Walter PP01.61, PP01.62
Crisan, Ioana PP01.106
Cristiano, Enrica PP01.31
Croce, Michele Giovanni PP01.100, PP01.244
Crockett, Lucy PP01.364
Cross, Jennifer PP01.160
Cruse, Belinda PP01.173, PP01.237, PP01.238
Cunha, Keyla OS07.05
Cuoghi Costantini, Riccardo OS01.04
Curtin, Julie PP01.65
Cutter, Gary PP01.238, PP01.89
Cuveele, Eline PP01.38, PP01.39, PP01.40
César-Chávez, Daniel PP01.258
D' Angelo, Andrea OS08.01
D'Alvano, Giulia OS04.03
D'Amico, Adele PP01.11
D'Andrea, Francesca PP01.374, PP01.375, PP01.378, PP01.379
D'Angelo, Maria Grazia OS01.04
D'Errico, Eustachio PP01.317
Da Silva Cardoso, Juliana PP01.40
Dadali, Elena PP01.342
Dal-Pra Ducci Cirino, Renata PP01.252, PP01.255, PP01.327
Dalakas, Marinos C. OS07.03
Dalisha, Dalisha PP01.390
Dalla Zanna, Gianmarco PP01.154
Dallorso, Stefania PP01.09
Damasio, Joana PP01.154
Damato, Valentina OS04.05, PP01.212, PP01.86, SS33.01
Damioli, Simona OS02.03, PP01.03, PP01.05
Damsker, Jesse OS01.02
Dang, Utkarsh OS01.02, PP01.42, PP01.73
Dang, Utkarsh J. PP01.51
Daniel Martin, Klaus SS22.06
Danielsson, Olof PP01.128
Daron, Aurore OS08.02
Das, Animesh PP01.220
Dash, Suravi PP01.138
Davis, Mark PP01.65
Day, John W. PP01.52
De Baets, Marc PP01.199
De Bleecker, Jan OS01.01
De Bleecker, Jan L. PP01.176
De Clercq, Lieselot PP01.166
De Cuyper, Tine PP01.62
de Cuyper, Tine PP01.61
De Felice, Sofia PP01.152
de Laat, Elisabeth C.M. PP01.17
de Laat, Ilse PP01.14
De Lorenzo, Alberto PP01.370
De Luca, Annamaria PP01.29, PP01.30, PP01.31
De Luca, Marcella OS06.02
De Maria, Martina PP01.352
De Merlier, Martijn PP01.202
De Mori, Roberta PP01.318
de Paepe, Els PP01.289
De Ridder, Willem PP01.172
de Rivera Garrido, Francisco JR PP01.133
De Roeck, Arne PP01.269, PP01.278, PP01.382
De Spirito, Marco OS08.03
De Stefano, Nicola OS03.05, OS05.05
de Toledo Justo, João Guilherme PP01.253
De Valle, Katy PP01.40
de Visser, Marianne PP01.103
De Vivo, Darryl C. PP01.52
De Waele, Liesbeth PP01.38, PP01.39, PP01.40, PP01.46, PP01.53, PP01.63, PP01.64
Decio, Alessandra PP01.30
Deconinck, Nicolas PP01.41, PP01.46, PP01.53, PP01.71
Deepracha, Tharadon PP01.371
Degan, Chiara PP01.73
Dejaeger, Marian PP01.61, PP01.62
Del Bo, Roberto OS08.04, PP01.104
Del Vecchio, Guido PP01.117
Del-Águila-Mejía, Javier PP01.230
DeLasHeras, Virginia PP01.173
Della Bella, Eleonora PP01.370
Della Marina, Adela OS02.01
Della Toffola, Jacopo PP01.316
Delmont, Emilien PP01.272
Delmonte, Valeria PP01.317
Delstanche, Stephanie PP01.116
Delstanche, Stéphanie PP01.369
Demeret, Sophie PP01.214
Demmel, Walter SS22.07
Demsar, Nina PP01.40
Dengler, Nora Florenz SS22.04
Denkberg, Galit PP01.199
Denlinger, Katherine OS03.06
Desai, Soaham PP01.173
Desaphy, Jean-François PP01.25
Desguerre, Isabelle PP01.52
DeVeaux, Michelle OS04.06
DeWaele, Liesbeth PP01.71
Dhall, Aishwarya PP01.361
Dhanabal, Mohanraj PP01.37
Di Bari, Alessandra SS18.01
Di Feo, Maria Francesca OS01.04
Di Iorgi, Natascia PP01.35
Di Marco, Carmine OS08.03
Di Martino, Giuseppe OS04.03
Di Muzio, Antonio PP01.55
Di Stefano, Vincenzo OS04.03, PP01.214, PP01.370
DIAGNE, NGOR PP01.354
Diamanti, Luca PP01.244
Diaz Manera, Jordi PP01.73
Diaz-Ruiz, Jorge Arturo OS03.03, PP01.246, PP01.258
Diefes, Alexander PP01.88
DiFeo, Mariafrancesca PP01.11
Difonzo, Gianni PP01.317
Dimachkie, Mazen OS01.01, PP01.211, PP01.88, TC06.02
Dimachkie, Mazen M PP01.133
Dimachkie, Mazen M. PP01.283
Dimova, Kalina PP01.53
Dinoi, Giorgia PP01.30
Dionne, Annie PP01.261
DIOP SENE, MARIEME SODA PP01.354
Dittmayer, Carsten PP01.180
Dixit, Vineet PP01.310
Dixon, Stacy OS01.01
Dmitrienko, Alex OS01.01
do V.P. Rodrigues, Paula Raquel PP01.327
do Vale Pascoal Rodrigues, Paula Raquel PP01.252, PP01.255
Dobeddu, Pietro PL02.03
Dobelmann, Vera PP01.129
Dobrescu, Amelia PP01.11
Dohring, Orlando PP01.346, PP01.347
Dohrn, Maike PP01.243
Doksani, Paolo OS02.01, OS08.05, PP01.180
Dolanska, Anezka PP01.40
DOLEK, Turgay PP01.190
Domanska, Barbara PP01.229
Donadio, Vincenzo TC12.03
Doneddu, Pietro E. PP01.282
Doneddu, Pietro Emiliano PP01.283, PP01.284, PP01.285, PP01.370
Dong, Yin PP01.166
Dos Santos Souza, Cleide PP01.329
dos Santos, Thalita Aparecida PP01.252
Dosi, Claudia PP01.40
Douglas, Lyndal PP01.65
Doverty, Althea PP01.99
Dowery, Reem PP01.199
Dowling, James J. OS02.04
Doykov, Ivan PP01.44
Drago, Selene PP01.119, PP01.127
Drago, Selene Francesca Anna PP01.120
Drakou, Eleni PP01.129
Dreezen, Jente PP01.39
Dreyer, Meret PP01.363
Drory, Vivian PP01.381, TC07.01
Drudge, Chris PP01.346, PP01.347
Drummond, Kerri PP01.41
Druzhinina, Eugenia PP01.296
Drużdż, Artur OS04.04, PP01.201
Dube, Bright PP01.222
Dubey, Divyanshu SS36.03
Dubois, Charline PP01.369
Dubuisson, Nicolas OS06.04
Ducci, Renata Dal-Pra OS05.02, PP01.257
Duchemin-Pelletier, Eve PP01.388
Duchesne, Elise PP01.136
Dufva, Ann Eriksson PP01.197
Dugas, Marc-Olivier PP01.136
Dulak, Józef PP01.26
Duncombe, Paul PP01.262
Duong, Monica PP01.346, PP01.347
Duong, Tina PP01.82
Dupont, Melissa PP01.285
Dupont, Patrick OS08.06, PP01.359, PP01.61, PP01.62
Durmus, Hacer PP01.118
Durr, Alexandra PP01.154
Dusemund, Carla OS02.01, OS08.05
Dutta, Bhaskar PP01.132
Dyck, P James SS23.02
Dyck, P. James PP01.270
Dyck, P. James B. Dyck TC12.02
Dysgaard, Tina PP01.268
D’Hooghe, Lena OS09.06
Ebong, Ima PP01.132
Echaniz-Laguna, Andoni PP01.194, PP01.272
Echeverry, Eduardo OS03.03, PP01.246, PP01.258
Edens, Coughi PP01.90
Edmundson, Christyn PP01.218, PP01.219
Edwards, Robert PP01.177, PP01.189
Eftimov, Filip OS01.05, PP01.103, PP01.268
Eggenspieler, Damien OS08.02, PP01.369
Eggers, Christian PP01.269, PP01.325
Eisenblätter, Luca PP01.363
Ejarque, Vanessa PP01.341
Ekinci, Aysen Suzen PP01.291
Ekstrom, Anne-Berit PP01.40
Elavarasi, Arunmozhimaran PP01.220
Elgsås, Tonje PP01.40
Ellor, Susan PP01.284
Elsouda, Dina PP01.366
Engebrecht, Charlotte PP01.367
Engelstad, JaNean PP01.270
Engvall, Martin PP01.128
Erasmus, Corrie PP01.14, PP01.40
Erasmus, Corrie. E. PP01.17
Erbaş, Yasemin PP01.52
Erdem-Ozdamar, Sevim OS07.06
Erdler, Marcus PP01.325, PP01.56
Erez, Shir PP01.199
Eriksson-Dufva, Ann OS04.02
Erra, Carmen OS04.05, PP01.155, PP01.214
Eskesen, Nicolas PP01.389
ESPINONSA-ESPINOSA, JORGE OS01.03
Esposito, Gabriella OS03.05
Esposito, Martina PP01.152
Espíndola, Gisele PP01.116
Etemadifar, Masoud PP01.173
Eura, Nobuyuki PP01.102, PP01.123
Europa, Tarin PP01.259
Evangelista, Teresinha TC12.01
Evers, Sanne OS01.05, PP01.103
Evoli, Amelia TS10.03
Ewenczyk, Claire PP01.154
Eymard, Bruno PP01.241
Fadli, Nurul PP01.308
Faedo, Elena PP01.34
Faelens, Ruben PP01.208
Fajardo, Val PP01.54
Falso, Siliva PP01.86
Falso, Silvia OS04.03, OS04.05, PP01.231
Falzone, Yuri OS04.03, PP01.370
Fan, Dongsheng SS32.01
Fang, Fang OS04.02, OS06.05, PP01.197, 611
Fang, Yuan OS01.02
Farhat, Emna SS05.01
Faria, Nathalia PP01.327
Farmakidis, Constantine PP01.175
Farrar, Michelle OS07.02, PP01.65
Farrar, Michelle A PP01.343
Farrugia, Emily PP01.313, PP01.314
Farwell, Wildon PP01.75
Feder, David PP01.163, PP01.164, PP01.165, PP01.92, PP01.93, PP01.94, PP01.95
Feitoza, Pablo PP01.116
Ferguson, Francesca PP01.188, PP01.192
Fernandez, David OS01.01
Fernandez, Joaquin Alejandro PP01.57
Fernández-Torrón, Roberto PP01.147
Ferraiuolo, Laura PP01.329
Ferrari Aggradi, Carola SS18.01
Ferrari, Elisabetta PP01.03
Ferri, Carolina PP01.32
Ferullo, Lucia OS02.03, PP01.04, PP01.05, PP01.323
Feticu, Marian PP01.115
Filosto, Massimiliano OS01.04, OS02.03, OS03.05, PP01.03, PP01.04, PP01.05, PP01.100, PP01.323, PP01.370, PP01.373, SS08.01
Finanger, Erika L. PP01.67
Finelli, Palma OS08.04
Finkel, Richard S PP01.343
Finkel, Richard S. PP01.52
Finsterer, Josef PP01.121, PP01.122
Fiogbé, Elie PP01.136
Fionda, Laura OS04.03, OS04.05, PP01.187, PP01.201, PP01.214, PP01.86
Fiorillo, Chiara PP01.09, PP01.10, PP01.11, PP01.34
Firholz, Monika PP01.40
Fitzgibbon, Marie PP01.175, PP01.177, PP01.189, PP01.208, PP01.209, PP01.210, PP01.235, PP01.236
Flaender, Mélanie PP01.388
Flanigan, Kevin PP01.129, PP01.68, PP01.71
Florczyk-Soluch, Urszula PP01.26
Florea, Laura PP01.115
Florio, Lucia OS04.05
Floudiotis, Niki PP01.259
Folz, Mary PP01.360
Fontana, Pedro PP01.164
Fontanelli, Lorenzo PP01.32
Foong, Yi Chao PP01.173, PP01.238
Foray, Marine PP01.388
Forcina, Francesca OS05.05, PP01.232, PP01.370, PP01.86
Fortes, Clarisse PP01.43
Foschi, Matteo PP01.173
Fossati, Gianluca PP01.30
Fox, Ashley PP01.329
Fraddosio, Angela PP01.317
Fraga, Luiza PP01.327
Francavilla, Teresa PP01.317
Franchi, Ester PP01.03, PP01.323
Franco da Graça, Felipe PP01.344
Frangiamore, Rita OS04.03, PP01.231
Fratini, Paula PP01.91
Freimer, Miriam OS01.01, PP01.179, PP01.229
Freytag, Bianca PP01.388
Frezatti, Rodrigo PP01.18
Frezza, Erica PP01.144, PP01.145, PP01.247, PP01.250, PP01.251, PP01.318
Frieler, Ryan PP01.82
Fu, Lijun OS03.06
Fujino, Masahiro PP01.02, PP01.76
Fujita, Syugo PP01.356
Fukumoto, Yuta PP01.240
Fukushima, Naoshi OS09.03
Fulop, Krisztina PP01.312
Furmanak, Thomas PP01.171, PP01.90
Furness, Kate PP01.314
Fustes, Otto Jesus Hernandez OS05.02, PP01.257
Gable, Karissa PP01.283, PP01.285, PP01.88
Gable, Karissa L. PP01.284, PP01.372
Gadaleta, Giulio PP01.47, PP01.84
Gaegan, Chloe PP01.40
Gaffney, Daniel PP01.137
Gafson, Arie PP01.269, PP01.278
Gagliardi, Delia OS08.01, OS08.04, PP01.104, PP01.105
Gagnon, Cynthia PP01.136
Gaignard, Pauline PP01.113
Gaio, Nikolas PP01.31
Gaki, Eleni PP01.343
Galatolo, Daniele PP01.11
Gallus, Gian Nicola OS03.05
Gana, Simone PP01.244
Gandhi, Kavita PP01.177, PP01.209, PP01.210, PP01.235, PP01.236
Garai, Nemanja PP01.358
Garassini, Marina SS43.03
Garay-Albízuri, Patricia PP01.147
Garbey, Marc SS24.03
Garcia Dominguez, Carlota OS03.06
Garcia, Beatriz OS07.03
García, Irune PP01.391
García-López, Sofía PP01.341
GARCÍA-MARTÍN, ALBERTO OS01.03
Garg, Ajay PP01.220
Garibaldi, Matteo PP01.187, PP01.232, PP01.86
Garletti, Giorgia PP01.03
Garofali, Francesca OS02.03, PP01.03
Garrido, Cristina PP01.40
Garrido-Hernández, Tania PP01.230
Gastaldi, Matteo OS04.05, PP01.231
Gauthier, Remiche PP01.130
Gavanji, Roya PP01.346, PP01.347
Gavrilaki, Maria PP01.110
GAYE, ARAME PP01.354
Gaëlle-Dosne, Anne PP01.223
Geckinli, Bilge Bilgen PP01.109
Gedikbası, Asuman PP01.118
Geiger, Christopher OS01.01
Gelinas, Deborah PP01.218, PP01.219
Gemelli, Chiara OS05.06
Gemma, Marco OS04.03
Genge, Angela SS26.03
Gentile, Luca PP01.370
Gentyala, Rahul OS02.04
George, Kelly OS03.06
Georgiadou, Elissavet PP01.185
Gerischer, Lea OS02.01, PP01.133
Gerovska, Daniela PP01.362
Geuens, Sam PP01.38, PP01.39, PP01.63, PP01.64
Gevorgyan, Edgar PP01.217
Ghati, Chetan PP01.138
Ghizoni Teive, Hélio PP01.254
Giacobbe, Juliette PP01.01
Giacopuzzi Grigioli, Eleonora OS04.03
Giagnorio, Eleonora PP01.21
Giampá, Sara PP01.253
Gibertini, Sara PP01.21, PP01.22, PP01.25, PP01.96
Gicquel, Evelyne PL01.03
Gil Garzon, Monica Rebeca PP01.45
Gilberti, Giulia PP01.03, PP01.323
Gilhus, Nils Erik PP01.206, PP01.222, SS20.02
Gillebert, Céline PP01.38, PP01.39, PP01.40
Gillenstrand, Jonas PP01.40
Gilmour, Kimberly PP01.44
Giouroukos, Sotiris PP01.185
Giovanelli, Giorgia OS02.03, PP01.03, PP01.04, PP01.05
Giramé-Rizzo, Lídia PP01.203
Girgenrath, Mahasweta PP01.37
Gistelinck, Fien PP01.178
Giunta, Giada PP01.96
Gkougka, Dionysia PP01.207
Glaubitz, Stefanie PP01.363
Gleeson, Frank PP01.75
Glāzere, Ieva PP01.274, PP01.98
Godoy, Lara PP01.253
Goedee, Stephan PP01.289, SS23.01
Goedeker, Natalie PP01.74
Goel, Abeer PP01.290, PP01.310
Goglia, Mariangela PP01.144, PP01.145, PP01.247, PP01.250, PP01.251, PP01.318
Gokhale, Sankalp PP01.159
Gold, Valeriu PP01.325
Gomez-Figueroa, Enrique PP01.173
Gonorazky, Hernan PP01.68
Gontika, Maria PP01.207
Gonzalez, Patrick PP01.68
Gonzalez-Perez, Paloma OS01.01
Gooch, Katherine L. PP01.48
Goodwin, Robert OS03.05
Goosens, Veerle OS08.06, PP01.359, PP01.61, PP01.62
Gordon, Robert PP01.177
Goret, Marie OS06.06
Gorin, Clarissa PP01.132
Gorni, Ksenija PP01.343
Gosk-Tomek, Magdalena PP01.325
Govaarts, Rosanne PP01.63, PP01.64
Govaarts, Rossane PP01.40
Goyal, Namita OS01.01
Goyal, Neelam OS01.01
Gozke, Eren PP01.77
Graham, Ryan B. PP01.372
Grandis, Marina OS01.04, OS03.05, OS04.03, OS05.06, PP01.34
Grandone, Anna PP01.379
Grazia, Devigili SS44.01
Graziosi, Bruno PP01.112, PP01.163, PP01.164, PP01.165
Greco, Giulia PP01.144, PP01.145, PP01.247, PP01.250, PP01.251, PP01.318
Greenslade, Annie PP01.364
Griedl, Theresa Antonia PP01.325
Grigoryan, Mariam PP01.217
Grimm, Alexander PP01.248, PP01.260, SS22.02, SS22.03
Grinzinger, Susanne PP01.325
Griveau, Louise PP01.388
Groepenhoff, Floor OS01.05
Groothuis, Jan PP01.14
Groothuis, Jan T. PP01.17
Grote, Daniela PP01.135
Gruber, Felix PP01.325, PP01.326
Grudniak, Mariusz PP01.175
Grumati, Paolo PP01.152
Gruosso, Francesco PP01.144, PP01.247, PP01.250, PP01.251, PP01.318
Gräßl, Michael PP01.325
Gröhl, Janek PP01.363
Gueye, Mouhamed PP01.339
Guglielmi, Marco PP01.317
Guglielmino, Valeria OS05.05
Guglieri, Michaela PP01.71
Guglieri, Michela OS01.02, PP01.40, PP01.42, PP01.51, PP01.67, PP01.73
Guha, Shashwat PP01.45
Guida, Melania OS04.03, OS04.05, PP01.231, PP01.233
Guillen, Elisa PP01.57
Guite, Kaitey PP01.51
Gulati, Sheffali PP01.41
Guliani, Gaurav K PP01.132
Gunawardane, Shivi PP01.287
Gundogan, Sevgin PP01.291
Guntrum, Debra PP01.367
Gupta, Anu PP01.220
Guptill, Jeff PP01.223
Guptill, Jeffrey PP01.169
Guptill, Jeffrey T. PP01.193
Gurgel Giannetti, Juliana OS07.05
Gurgel-Giannetti, Juliana PP01.344
GURSOY, Melik PP01.190
Gutierrez, Haydee PP01.329
Guye, Sabrina PP01.222
Guémy, Clément PP01.151, PP01.351
Gwathmey, Kelly OS01.01
Gómez Andrés, David PP01.74
Gómez-Andrés, David PP01.340, PP01.53
Ha, Sue young PP01.149
Haack, Tobias PP01.248
Haarmann, Axel PP01.216
Haberlova, Jana PP01.40
Habetswallner, Francesco OS04.05, PP01.155, PP01.370
Habib, Ali OS04.06, PP01.170, PP01.171, PP01.186, PP01.215
Habib, Ali A. OS04.04, PP01.169, PP01.195, PP01.201, PP01.79
Hachisuka, Akiko PP01.334
Haddad, Hafedh PP01.284
Hagenacker, Tim PP01.216
Hagerty, Laura OS01.02
Haghikia, Aiden OS04.01
Hahn, Andreas OS03.06
Hahn, Katrin TC08.01
Hahn, Max PP01.389
Hahn, Sihoun OS03.06
Hajdú, Nándor PP01.141
Hajok, Kalina PP01.26
Hakim, Manfaluthy PP01.308
Hallal, Sihem PP01.69
hallal, Sihem PP01.70
Haltner, Anja PP01.346, PP01.347
Hamedani, Mehrnaz OS05.06
Hamilton, Alexander PP01.389
Hamilton, Tara PP01.301
Hamir, Muhammed PP01.364
Hamstra, Sophie PP01.54
Han, Kyungdo PP01.324
Han, Mitchell PP01.31
Hansen, Henrik PP01.389
Harbo, Thomas PP01.268, PP01.282
Harel, Ofer PP01.199
Harmelink, Matthew PP01.68
Harsono, Adrian Ridski PP01.308
Hartung, Hans-Peter PP01.265
Hashimoto, Atsuki OS02.04
Hasuike, Yuhei PP01.85
Hathout, Yetrib OS01.02, PP01.73
Haugh, Joy PP01.46
Hawley, Brien PP01.283
Hayashi, Shinichiro OS03.04, PP01.140
Hayat, Ghazala PP01.177
Hayat, Ghazala S. PP01.189
Hazotte, Aurélie PP01.286
Heatwole, Chad PP01.367
Hebestreit, Katja PP01.137
Heckmann, Jeannine PP01.173, PP01.238, PP01.259
Hedberg-Oldfors, Carola PP01.08, PP01.349
Heerlein, Kristin PP01.218, PP01.219, PP01.223
Hegelmaier, Tobias OS04.01, PP01.216
Heindl, Felix PP01.154
Heinzel, Johannes SS22.02, SS22.03
Heinzmann, Anna PP01.154
Heitzman, Daragh OS01.01
Hejas, Reka PP01.312
Helmlinger, Gabriel PP01.59
Henderson, Robert OS01.01, PP01.108
Heng, Hazel PP01.313
Henriquez, Alicia PP01.68
Henshaw, Joshua PP01.364
Hentschel, Andreas PP01.129, PP01.180, PP01.243
Henzi, Bettina PP01.40
Heo, Namjin OS05.04
Herczegfalvi, Agnes PP01.129
Herdick, Meret OS02.01, OS08.05
Hermes, Alexandra SS22.03
Hernandez Fustes, Otto PP01.252, PP01.253, PP01.254, PP01.255
Hernandez Fustes, Otto Jesus PP01.327
Hernandez, Ana Luisa PP01.92
Hewamadduma, Channa PP01.179, PP01.279, PP01.368
Heywood, Wendy PP01.44
Hickey, Luke PP01.159, PP01.206, PP01.211, PP01.268
Hideki Kawamura Junior, Edson PP01.252
Hiebeler, Miriam PP01.56, PP01.66
Higashimoto, Yuji PP01.320, PP01.322
Hill-Smith, Emilie PP01.40
HIRANO, MAKITO PP01.322
Hirano, Makito PP01.240, PP01.320
Hjartarson, Helgi OS02.06
Ho, Gladys PP01.65
Ho, Kelly PP01.166
Hobson-Webb, Lisa OS01.01
Hockey, Briana PP01.54
Hoffman, Eric OS01.02, PP01.42
Hoffman, Eric P. PP01.51
Hoffman, Sarah PP01.223
Hoffmann, Sarah OS02.01, OS08.05, PP01.175, PP01.180, PP01.193
Hofman, Erik PP01.382
Hogan, Jonathan PP01.171
Holla, Elisa PP01.129
Holliday, Nicholas OS09.06
Hong, Seokchan PP01.97
Horber, Veronka PP01.248
Horeau, Mathieu PP01.60
Horlings, Corinne PP01.325
Horvath, Rita PP01.129, PP01.243
Hoshino, Genki PP01.240
Hotter, Anna PP01.325
Hou, Can OS06.05
Houwen - van Opstal, Saskia PP01.14
Houwen-Van Opstal, Saskia PP01.40
Howard Jr, James F OS04.06
Howard Jr, James F. PP01.176, PP01.178, PP01.193, PP01.223
Howard Jr., James F. PP01.170, PP01.80, PP01.81
Howard, James PP01.179, PP01.215, PP01.228, PP01.78
Howe, B. Matthew PP01.270
Hsieh, Sung-Tsang OS05.03, PP01.311
Hsueh, Hsueh-Wen OS05.03, PP01.311
Hsueh, Sung-Ju PP01.365
Hu, Yihan OS06.05
Huang, Wan-Yi PP01.176, PP01.193
Huang, Yan PP01.383
Hughes, Tom PP01.218, PP01.219
Huijbers, Maartje PP01.158
Hummelgen, Eduardo PP01.253
Huntemann, Niklas PP01.216
Hurley, Erin PP01.329
Hurth, Helene PP01.260, SS22.03
Hussain, Yessar OS01.01, OS04.06, PP01.169
Hussain, Yessar M. PP01.278, PP01.282
Hustinx, Manon PP01.116, PP01.369
Huynh-Ba, Olivier OS03.06
Huysmans, Lotte OS08.06, PP01.359
Hwang, Eunbyol PP01.271
Héjas, Réka PP01.141
Höftberger, Romana OS08.05
Hübner, Miriam OS09.04
Hüpper, Anna PP01.325
Iadarola, Antonella PP01.36
Iaffaldano, Antonio PP01.317
Iannibelli, Eliana PP01.21, PP01.22, PP01.96
Idoux, Romane PP01.129
Iguchi, Naohiko PP01.102
Ikada, Miki PP01.386
Ilagan, Janine PP01.137
Ilderton, Charlotte PP01.40
Ilic, Nikola PP01.13
Im, Sun PP01.304
Imai, Tomihiro PP01.356
Imbrici, Paola PP01.29
Inagaki, Sho PP01.191
Indrawati, Luh Ari PP01.308
Inghilleri, Maurizio OS05.05, PP01.370
Ingre, Caroline OS06.05
Inkhao, Peerakan PP01.371
Inoue, Masato PP01.386
Iolascon, Giovanni PP01.374, PP01.375, PP01.376, PP01.377, PP01.378, PP01.379
Iorio, Raffaele OS04.03, OS04.05, PP01.133, PP01.214, PP01.218, PP01.219, PP01.86, SS12.03
Iovino, Aniello PP01.204
Iovino, Veronica OS04.05, PP01.233
Iruzubieta, Pablo PP01.147, PP01.154
Irving, Adam PP01.237
Irving, Kathryn PP01.40
Irwin, Charlotte PP01.367
Ishigaki, Keiko PP01.40
Ishiguro, Kumiko PP01.40
Ishizuchi, Kei PP01.198
ISONO, CHIHARU PP01.322
Isono, Chiharu PP01.320
Istas, Geoffrey PP01.382
Iterbeke, Louise PP01.359
Ito, Genta PP01.386
Ito, Yasuo PP01.356
Ivanova, Kristīne PP01.98
Iwabe, Cristina PP01.344
Iyisenyurek, Seyme PP01.109
Izumi, Yuishin PP01.320
Iñarra Velasco, María José PP01.147
İNAL ÖZÜN, Özgü PP01.380
J Hopkin, Robert OS03.06
Jaber, Abbass PL01.03
Jackson, Louis PP01.210
Jacob, Saiju OS04.06
Jaffe, Linda PP01.307
Jahr, Kristen PP01.364
Jaiswal, Jyoti OS01.02
Jakobsen, Johannes OS07.03
Jakovčevic, Antonia PP01.129
Jakubczak, Ewa PP01.26
Jakubiec, Alexander PP01.368
Jalali, Neda OS04.06
James, Lucy OS02.04
Januario, Nelio OS07.05
Januzi, Donjetë PP01.362
Jaque, Carlos PP01.58
Jayaseelan, Dipa PP01.130
Jeon, Hara PP01.242
Jerez, Fátima PP01.340
Jevtic, Svetlana PP01.228, PP01.78
Jih, Kang-Yang OS03.02
Jimenez, Rosa H. PP01.169, PP01.193
Jimenez, Rosa Hermina PP01.176, PP01.178
Jiménez, Belén Valenzuela PP01.208
Jin, Lee-Way PP01.239
Jo, Geun Yeol PP01.200
Joe, Sangwon PP01.167
Jofré, Javiera PP01.58
Johnson, Nicholas PP01.82, SS18.01, SS18.02
Johnson, Victoria SS22.02
Jones, Kristi J PP01.65
Jones, Maxwell PP01.346, PP01.347
Jonson, Per-Harald PP01.21
JOUBAILY, ZEINA PP01.354
Jovanovic, Vladimir PP01.143
Joy, Shiny PP01.220
Ju, Hyunjin PP01.148
Juang, Jyh-Ming Jimmy OS05.03
Judit Molnar, Maria PP01.41
Juel, Vern C. PP01.178
Julazadeh, Hana PP01.329
Junek, Rosie PP01.65
Jung, Hee-Jae PP01.97
Jung, Ye Joon PP01.226
Jungbluth, Heinz PP01.17, TC09.02
Juntas, Raúl PP01.203
Juntas-Morales, Raul PP01.214
Juraver, Adrien PP01.368
Järvinen, Elina PP01.133
Júnior, Euldes PP01.116
Kabli, Bouchra PP01.125
Kably, Bouchra PP01.124
Kaffe, Korina PP01.110
Kaibin Kuan, Kelvi PP01.292
Kalcev, Goce PP01.157
Kaltsa, Argiro PP01.183
Kalvāne-Brokāne, Sintija PP01.98
Kaminski, Henry PP01.199, SS24.03, SS37.02, SS40.02
Kamoi Kay, Claudia Suemi PP01.327
Kamperman, Renske OS01.05, PP01.103
Kamsteeg, Erik-Jan PP01.129, PP01.154
Kan, Hermien E. PP01.63, PP01.64
Kanagaiah, Preshetha PP01.367
Kang, Heung-Won PP01.97
Kang, Hong-Yo PP01.387
Kang, Min PP01.171
Kang, Sa-Yoon PP01.224, PP01.225
Kao, Yi-Hui PP01.311
Kapetanovic Garcia, Solange PP01.231
Kapoor, Mahima PP01.238
Karali, Sukru PP01.229
Karam, Chafic PP01.282, PP01.88, PP01.89
KARATAŞ, Ceren Şevval PP01.380
Karcagi, Veronika PP01.129
Kariyawasam, Didu PP01.65
Karkkainen, Elena PP01.335, PP01.336, PP01.337
Karlsson, Markus PP01.360
Karmous, Wisam PP01.210, PP01.235, PP01.236
Karydi, Eirini-Anastasia PP01.207
Kaspar, Allan PP01.329
Kaspar, Brian PP01.329
Katsalouli, Marina PP01.183, PP01.184, PP01.185
Katz, Jon PP01.268
Katz, Sonja OS02.01
Katzberg, Hans PP01.268, PP01.282, PP01.305
Kavanagh, Alex PP01.45
Kawasaki, Hitoshi PP01.356
Kawka, Aleksandra OS03.05
Kay, Claudia Suemi Kamoi OS05.02, PP01.257
Kecelioglu Binnetoglu, Kiymet PP01.109
Kediha, Mohamed Islam PP01.241
Keerthipriya, Muddassu PP01.139
Keerthipriya, Muddasu Suhasini PP01.150
Kenis, Vladimir PP01.342
Kennelly, Laura PP01.280
Kerasidis, Anastassia PP01.287
Keritam, Omar PP01.325, PP01.326
Kermode, Allan PP01.173
Kerr, Douglas PP01.142, PP01.59, PP01.71
Keyzor, Ian PP01.366
Khachatryan, Lili PP01.281
Khan, Farid PP01.173
Khan, Waleed PP01.88
Khosla, Tanvir PP01.288
Khuram, Hassan PP01.287
Kief, Katharina PP01.363
Kieseier, Bernd PP01.78
Kihara, Yuki PP01.40
Kilinc, Muhammed OS07.06
Kim, Ahwon PP01.149
Kim, Bongseong PP01.324
Kim, Byoung Joon OS05.04, PP01.330
Kim, Byung-Jo PP01.332, PP01.333
Kim, Dayoung PP01.330
Kim, Ho Cheol PP01.97
Kim, Hyun Gi PP01.273
Kim, Hyunjin OS06.03, PP01.97
Kim, Jee-Eun PP01.276
Kim, Jin-Ah PP01.331
Kim, Jinhyeong OS06.03
Kim, Jong-Il PP01.331
Kim, Junho OS06.03
Kim, MinGi PP01.273
Kim, Sang Beom PP01.167, PP01.20, PP01.23, PP01.24, PP01.309
Kim, Sang-Beom PP01.350
Kim, Seung Woo PP01.196, PP01.226
Kim, Sooyoung PP01.330
Kim, Sunyoung PP01.299
Kim, Won PP01.97
Kim, Woo-Kyung PP01.167, PP01.20, PP01.23, PP01.24
Kim, Young Chul PP01.97
Kimiskidis, Vasilios PP01.110
Kimura, Yosuke PP01.348
Kinga Sárvári, Anitta PP01.389
Kiriyama, Takao PP01.102, PP01.123
Kirk, Edwin PP01.65
Kirschner, Janbernd PP01.40, PP01.52
Kishnani, Priya OS03.06
Kiss, Christian PP01.325
Klabsook, Ratiporn PP01.174
Klein, Andrea PP01.40
Kleinveld, Vera PP01.325
Kleopa, Kleopas SS10.03
Kletzl, Heidemarie PP01.343
Klopčič, Matic PP01.83
Kluczka, Eugénie PP01.366
Kneer, Katharina PP01.260, SS22.03
Knipp, Florian PP01.325
Knopp, Rachel PP01.295
Ko, Vivian PP01.329
Kobayashi, Shunsuke PP01.386
Kok, Jannigje PP01.329
Kokkinou, Eleftheria PP01.183, PP01.184, PP01.185
Koks, Sulev OS06.01
Kolb, Stephen J. PP01.329
Kolbenschlag, Jonas SS22.02, SS22.03
Komaki, Hirofumi PP01.40
Komori, Tetsuo PP01.334
Koneczny, Inga OS08.05
Korngut, Lawrence OS01.01
Korobko, Denis PP01.176
Kosac, Ana PP01.27
Kostera-Pruszczyk, Anna PP01.01, PP01.176
Kotambail, Ananthapadmanabha PP01.150
Koutsioumpa, Charalampia PP01.88
Kovacevic, Gordana PP01.13
Kovalchuk, Maria PP01.281, PP01.296
Kravitz, David PP01.381
Krenn, Martin OS08.05, PP01.325, PP01.326
Kretz, Alexandra PP01.362
Krittanupong, Supaporn PP01.371
Kroeksattayaporn, Apinya PP01.174
Krutovs, Vladimirs PP01.98
Krämer, Heidrun H. PP01.216
Krümmer, Norma PP01.216
Kteyan, Armine PP01.217
Kubo, Hiroshi OS09.03
Kueh, Kelly PP01.173
Kumutpongpanich, Theerawat PP01.174
Kunsztler, Luca PP01.55
Kuntz, Nancy L. OS02.04, PP01.01, PP01.156
Kurt, Can Ebru OS07.06
Kusner, Linda SS37.02, SS40.02
Kusunoki, Susumu PP01.240
Kutch, Michael PP01.189, PP01.235, PP01.236
Kuthiala, Manni PP01.343
KUTLU, Gulnihal PP01.190
Kuwahara, Motoi PP01.240
Kvalsund, Michelle PP01.259
Kvarnung, Malin PP01.12
Kwon, Patrick PP01.269
Kwon, Soonwook PP01.324
Kölbel, Heike PP01.243
Küpper, Hanna PP01.248
Küsters, Benno PP01.129
Kıyan, Esen PP01.118
La Rosa, Elena OS07.04
Labella, Beatrice OS02.03, PP01.05, PP01.113
Lacueille, Clémentine PP01.272
Ladisa, Alberto PP01.30
Laforet, Pascal PP01.115, PP01.351, TC11.01
Laforêt, Pascal PP01.132, PP01.133, PP01.151
Laghetti, Paola PP01.25
Lagrange, Emmeline PP01.132, PP01.214
Lalla, Marianna PP01.160, PP01.206, PP01.211
Lambert, Phil PP01.75
Lan, Lan PP01.01
Land, Natalie PP01.339
Landrieu, Blandine PP01.133
Lange, Dale J. OS04.04
Langenscheidt, Dieter PP01.325
Lanzi, Gaetana PP01.04, PP01.05
laporte, jocelyn OS06.06
Larimore, Kevin PP01.137, PP01.364
Lasorella, Piergiorgio PP01.352
Lassner, Franz SS22.05
Lauer, Henrik SS22.02
Lauletta, Antonio PP01.187, PP01.232, PP01.86
Laura, Fionda PP01.232
Lauria Pinter, Giuseppe PP01.370
Lauria, Giuseppe TC08.02
Laurie, Steven PP01.129
Lauro, Rita OS07.04
Laverty, Chamindra PP01.71
Lavrov, Andreea PP01.222
Lavrov, Arseniy PP01.78
Law, Eric PP01.65
Le Lam, Thao Nguyen PP01.369
Le Masson, Gwendal PP01.272
Leandri, Massimo OS05.06
Lee, Ada PP01.108
Lee, Do-Yeon PP01.149, PP01.331
Lee, Eun Kyoung PP01.330
Lee, Eun-Jae PP01.97
Lee, Han PP01.239
Lee, Hyung-Soo PP01.353
Lee, Jeehun PP01.71
Lee, Jeong Ho OS06.03
LEE, Jhee PP01.319
Lee, Jin Yong PP01.309
Lee, Jung Hwang PP01.271
Lee, Seungkeun PP01.276
Lee, Sun PP01.215
LEE, William Kah Howe PP01.87
Lee, Yi-Chung OS03.02
Lefeuvre, Claire PP01.151
Lefever, Kristen PP01.221
Lehnerer, Sophie OS02.01, PP01.133
Leila, Tamaoui PP01.06
Leite, M Isabel PP01.192
Leite, M. Isabel PP01.170, PP01.179, PP01.188, PP01.80, PP01.81
Lenti, Roberta PP01.30
Leonardi, Luca OS05.05, PP01.187, PP01.232, PP01.370, PP01.86
Leonardis, Lea PP01.83
Leone, Daniela PP01.55
Leopizzi, Eleonora PP01.47, PP01.84
Lerario, Alberto PP01.104, PP01.105
Lerman, Andrew PP01.278
Lesport, Quentin SS24.03
Leu, Jocelyn H. PP01.208, PP01.209
Leung, Rebecca PP01.221
Levine, Todd OS04.06
Lewis, Richard PP01.268
León, Ana María PP01.341
Li, Jenny PP01.53
Li, Lingyun PP01.215, PP01.218, PP01.219
Li, Mingming OS09.03
Li, Qing PP01.80
Li, Tianhong PP01.239
Li, Xiang PP01.37
Li, Yuebing PP01.210
Lian, Wenlong PP01.37
Liang, Christina OS01.01
Liantonio, Antonella PP01.29
Licandro, Simonetta Andrea PP01.30
Lignos, Dimitris PP01.183, PP01.184
Ligouri, Rocco TC05.02
Liguori, Sara PP01.374, PP01.375, PP01.376, PP01.377, PP01.378, PP01.379
Lilleker, James OS01.01
Lim, Jamie PP01.158
Lim, Young-Min PP01.97
Lima Nogueira Tolentino, Alessandra PP01.116
Lin, Boya PP01.218, PP01.219
Lin, Cheng-Chen PP01.311
Lin, Jie PP01.261
Lin, Yen-Hung OS05.03
Lindgren, Ulrika PP01.128
Lindroos, Jenny PP01.222
Litchy, William PP01.270, TC05.03
Little, Courtney PP01.90
Liu, Hai OS01.06
Liu, Haoming PP01.37
Liu, Jingyi PP01.218, PP01.219
Liu, Li PP01.169, PP01.176
Liu, Ling PP01.137
Liu, Nanjun PP01.53
Liu, Weijian PP01.41
Liu, Ya-Fen PP01.387
Liu, Zhan PP01.385
Lizio, Andrea PP01.373
Llarch, Paula PP01.382
Llauradó, Arnau PP01.203
Lledó-Garcia, Rocio OS09.06
Lloyd, Thomas OS01.01
Lo Iacono, Aurora OS04.05, PP01.233
Lo, Daryl Yin Keong PP01.292
Lochmuller, Hanns PL04.01
Lochmüller, Hanns PP01.129
Lochmülller, Hanns PP01.156, PP01.166
Lodato, Simona OS08.01
Logullo, Francesco PP01.297
Long, Davalos PP01.88
Long, Nick PP01.37, PP01.53
Longone, Patrizia PP01.318
Longoni, Natalia OS07.04
Lopergolo, Diego OS03.05
Lopez, Cristina PP01.214, PP01.231
Lopez, Juan David OS03.03, PP01.246, PP01.258
Lorentzos, Michelle PP01.65
Lorenzoni, Paulo Jose PP01.252, PP01.255, PP01.327
Lorenzoni, Paulo José OS05.02, PP01.257
Lotti, Antonio PP01.86
Lovas, Gabor PP01.238
Lozi, Claudia PP01.370
Lu, Huifang PP01.90
Lu, Jessica PP01.65
Lu, Yi PP01.261, PP01.265, PP01.267
Lucchetta, Marta PP01.370
Lucchiari, Sabrina OS08.04
Luigetti, Marco OS05.05, PP01.370
Luiza Romanelli Tavares, Vanessa PP01.344
Lukash, Oleg PP01.178
Lundberg, Ingrid E. PP01.262
Luo, Jiangyuan PP01.191
Luo, Sushan PP01.193
Lure-Berregi, María PP01.147
Luzuriaga-Carpio, Diana OS03.03, PP01.246, PP01.258
Lyden, Kate PP01.156
LYECE, YARGUI PP01.338
Lykopoulou, Evangelia PP01.185
Lymperatos, Kosmas PP01.183, PP01.184
Lynch, Karen PP01.283, PP01.285
Lynch, Karen M. PP01.267, PP01.372
Lépée-Aragón, Clara PP01.391
Löscher, Wolfgang PP01.325
Lünemann, Jan D. OS02.01
Macaione, Vincenzo PP01.120
Maccarrone, Amanda PP01.36
Maccarrone, Giulia PP01.47
Macedo-Silva, André PP01.92
Machado, Pedro OS01.01
Mackay, Lori PP01.221
MacNally, Meghan PP01.53
MacPherson, Rebecca PP01.54
Madić, Sanja PP01.358
Madruga-Garrido, Marcos OS01.06
Maes, Frederik OS08.06, PP01.359
Maestri Tassoni, Michelangelo OS04.05, PP01.233
Maestri, Michelangelo PP01.214
Maggi, Lorenzo OS01.04, OS04.03, PP01.156, PP01.21, PP01.22, PP01.25, PP01.96, TC11.03
Maghnie, Mohamad PP01.35
Magistrado-Coxen, Pamela PP01.41
Magri, Francesca OS03.05, OS08.04, PP01.104, PP01.117
Mahajerin, Arash PP01.215
Mahal, Simone PP01.325, PP01.40
Maharramov, Eshgin OS07.06
Mahesh, karthik Vinay PP01.310
Mahoney, Paul PP01.179, PP01.186, PP01.195, PP01.201
Mahuwala, Zabeen PP01.186, PP01.195, PP01.208, PP01.79
Mahuwala, Zabeen K. PP01.201
Maiorano, Simone SS18.01
Maisenbacher, Mathias PP01.131
Major, Kinga Andrea PP01.106
Major, Zoltan Zsigmond PP01.106
Makuch, Mateusz OS06.04
Makunina, Eleonora PP01.298
Malaichamy, Sivasankar PP01.129
Malandrini, Alessandro OS03.05
Malaspina, Andrea PL03.02
Malatesta, Lindsay PP01.132
Malfatti, Edoardo PP01.11, SS08.03, TC09.03
Malm, Eva PP01.135, PP01.66
Mammen, Andrew L. PP01.262
Mancardi, Margherita PP01.10
Manco, Carlo OS05.05
Mancuso, Michelangelo SS06.03
Manera, Umberto SS25.02
Manfrini, Marianna PP01.343
Manganelli, Fiore OS05.05, PP01.370, SS17.01
Manganotti, Paolo PP01.316
Maniaci, Marisa OS07.04
Maniaol, Angelina PP01.179
Mannila, Maria PP01.12
Mantegazza, Renato OS04.06, PP01.169, PP01.195, PP01.201, PP01.79
Mantero, Vittorio OS04.03
Mantuano, Paola PP01.29, PP01.30
Manzur, Adnan PP01.33
Manzur, Adnar PP01.11
MAO, Shanshan PP01.161, PP01.162
Marando, Demetrio PP01.232, PP01.86
Marantz, Jing PP01.82
Maravic, Melissa PP01.339
Marchese, Davide OS08.03
Marchi, Margheria SS44.02
Marchioni, Enrico PP01.86
Marciano E Ortolano, Anita PP01.352
Marcotte, Robert T. PP01.156
Marcuzzo, Stefania PP01.21, PP01.96
Marfia, Girolama Alessandra PP01.370
Margollicci, Giuseppina OS02.03
Maric, Nina PP01.13
Marigliano, Davide OS04.03
Marina, Anna OS07.05
Marinelli, Manuel PP01.31
Marini Bettolo, Chiara PP01.335, PP01.336, PP01.337
Marini, Sofia OS04.05, PP01.86
Mariën, Lore PP01.166
Markandeya, Yogananda PP01.138
Markov, Martin PP01.284
Maroni, Sofia SS18.01
Marques, Wilson PP01.18
Martinovic, Jelena PP01.27
Martins, Bruno PP01.218, PP01.219, PP01.48
Martorell, Loreto PP01.147
Martín-Aguilar, Lorena PP01.382
Martínez, Óscar PP01.391
Martínez-Salmerón, María del Mar PP01.230
Marx, Almuth PP01.346, PP01.347
MARZOUK, BASMA PP01.124
MARZOUK, Basma PP01.126
Marzouk, Basma PP01.125
Masciocchi, Stefano PP01.86
Maselli, Ricardo PP01.239
Masingue, Marion PP01.113
Mason, Stefanie PP01.46
Massa, Roberto OS03.05, PP01.144, PP01.145, PP01.247, PP01.250, PP01.251, PP01.318
Massacesi, Luca PP01.212
Masschaele, Delphine PP01.169, PP01.193
Masson, Riccardo PP01.40
Massucco, Sara OS05.06, PP01.34
Masud, Ameneh PP01.41
Matalonga, Leslie PP01.129
Mataluni, Giorgia PP01.370
Materia, Roberto OS07.04
Mathews, Katherine PP01.108, PP01.46
Matijašević Joković, Suzana PP01.358
Matrone, Federica OS04.05
Matsubara, Tomoyasu PP01.320
Matsumura, Tsuyoshi PP01.85
Matsuoka, Taro PP01.348
Matsuura, Amane PP01.348
Matà, Sabrina PP01.370
Mauermann, Michelle PP01.270, SS13.02
Maurino, Jorge PP01.57
Mauriño, Jorge PP01.340, PP01.341
Mavor, Matthew P. PP01.372
Mazor, Roei D. PP01.199
Mazurkiewicz-Beldzinska, Maria PP01.41
Mazurkiewicz-Bełdzińska, Maria PP01.343
Mazzeo, Anna OS06.02, PP01.370
Mazzà, Claudia PP01.368
Maçaneiro, Bruna PP01.327
McAdam, Laura PP01.40
McAlonis-Downes, Melissa PP01.329
McCarthy, Grace PP01.346, PP01.347
McCombe, Pamela PP01.173
McConnon, Aine PP01.173
McDonald, Craig PP01.41, PP01.68
MCFARLANE, Robert PP01.87
MCKINTY, Ailie PP01.87
McLaughlin, Laurie PP01.221, PP01.237, PP01.238
McMillan, Hugh PP01.40, PP01.71
Meagher, Karoline A OS04.06
Medeiros Mota dos Reis, Tertuliana PP01.344
Mehrabyan, Anahit PP01.134, PP01.217
Meilleur, Katy PP01.59
Meisel, Andreas OS02.01, OS04.06, OS08.05, PP01.169, PP01.176, PP01.178, PP01.180
Melnik, Evgeniya PP01.342
Melton, Andrew PP01.364
Mendoza, Meg PP01.305
Menezes, Flavia PP01.01
Meng, Stefan HO01.01
Menichella, Daniela PL02.01
Menon, Parvathi OS07.01, OS07.02
Menon, Sarath SS29.02
Mercelis, Rudy PP01.172
Mercuri, Eugenio OS02.05, PP01.41, PP01.46, PP01.68, PP01.74
Mercuri, Eugenio M. PP01.52
Mercuri, Eugenio Maria PP01.40, PP01.55
Mertens, Johan PP01.62
Messer, Jordan PP01.59
Messina, Sonia OS06.02, OS07.04, PP01.55
Metay, Corinne PP01.113
Meyer, Stefanie PP01.363
Meyer, Thomas TC13.02
Meznaric, Marija PP01.111
Mi-Young, Mi-Young OS05.04
Miceli, Carrie PP01.28
Michael, Eva PP01.08
Michaud, Maud PP01.132
Michelassi, Francesco OS03.05
Michiura, Toru PP01.240
Miguet, Lauren PP01.202
Mihara, Masahito PP01.02
Miladi, Najoua SS05.01
Milanez, Lucina OS07.05
Milic Rasic, Vedrana PP01.27
Mills, Kevin PP01.44
Min, Ju-Hong PP01.324
Minetti, Carlo PP01.36
Minks, Eduard PP01.175
Minn, Yang-Ki PP01.20, PP01.23, PP01.24, PP01.309
Minn, Yangki PP01.227, PP01.302
Miotto, Matteo OS08.01
Miranda, Rubén PP01.63
Mironovs, Staņislavs PP01.274
Misheva, Mariya OS06.04
Missaglia, Sara OS03.01
Mitchell, Ryan PP01.75
Mitsui, Yoshiyuki PP01.240
Miyagoe-Suzuki, Yuko PP01.386
Miyamoto, Yosuke PP01.348
Mladenovic, Jelena PP01.27
Moat, Dionne PP01.336, PP01.337
Modi, Manish PP01.290
Modi, Tanish PP01.300
Moggio, Maurizio OS01.04
Mohassel, Payam OS01.01
Molitierno, Nicola OS08.04, PP01.104, PP01.105
Mondou, Elsa OS07.03
Monforte, Mauro OS08.03
Mongini, Tiziana PP01.47, PP01.84
Mongini, Tiziana Enrica OS01.04
Monickaraj, Judith PP01.367
Monif, Mastura PP01.238
Montolio, Marisol PP01.57
Moon, Sookin PP01.148
Moraes, Ana PP01.91
Morando, Simone PP01.36
Moreno, Cristiane PP01.40
Moret, Federica PP01.370
Moretti, Antimo PP01.374, PP01.375, PP01.376, PP01.377, PP01.378, PP01.379
Morgan, Jennifer PP01.19
Morgenroth, Lauren OS01.02
Mori, Eiichiro PP01.123
Morimoto, Masafumi PP01.348
Morino, Stefania PP01.187, PP01.232
morino, stefania PP01.86
Moroni, Isabella PP01.21, PP01.22, PP01.25
Morrison, Alexander PP01.88
Moschou, Maria PP01.110
Mou, Yongchao PP01.37
Mouly, Vincent PP01.31
Moura, Ana Carolina OS07.05
Mousser, Souheila PP01.69
Moustafellou, Anna PP01.183, PP01.184, PP01.185
Mouzis, Nikolaos PP01.183, PP01.184
Moynihan, Meghan PP01.222
Mozaffar, Tahseen PP01.108, PP01.90
Muaremoska Kanzoska, Ljelja PP01.357
Mucha, Olga PP01.26
Muhmann, David PP01.243
Muni Lofra, Robert PP01.335, PP01.336, PP01.337
Munot, Pinki PP01.11, PP01.33
Muntean Firanescu, Cristina PP01.12
Muntoni, Francesco OS02.02, PP01.15, PP01.19, PP01.33, PP01.40, PP01.41, PP01.44, PP01.45, PP01.52, SS28.01
Muppidi, Srikanth PP01.228, PP01.78, PP01.88
Murai, Hiroyuki OS04.06, PP01.210
Murayama, Shigeo PP01.320
Musumeci, Olimpia PP01.119, PP01.120, PP01.127, SS39.03
Muñoz, Juan Pablo OS03.03, PP01.246, PP01.258
Myo, Arkar PP01.87
Myszka, Małgorzata PP01.26
Männlin, Stephanie SS22.02
Müller, Petra PP01.325
Müller-Felber, Wolfgang OS02.04
Nadaj-Pakleza, Aleksandra PP01.132
Naddaf, Elie OS01.01, PP01.90
Nag, Heidi Elisabeth PP01.40
Nagai, Yoshitaka PP01.240, PP01.320, PP01.322
Nagano, Mamoru PP01.320
Nagaraju, Kanneboyina OS01.02
Nagy, Sara PP01.154
Naito, Hiroyuki OS04.04
Najem, Catherine PP01.170
Nakahara, Jin PP01.198
Nakamura, Harumasa PP01.85
Nakamura, Hiroto PP01.240
Nakamura, Shuhei PP01.123
Nalepa, Anna PP01.26
Nalini, Atchayaram PP01.150, SS31.02
Namura, Hitoshi PP01.240
Nanaura, Hitoki PP01.102, PP01.123
Napoli, Laura PP01.104, PP01.105
Narayanaswami, Pushpa PP01.159
Nardes dos Santos, Flávia PP01.344
Nardes, Flávia PP01.43
Narrayanaswami, Pushpa TS10.02
Nascimento Osorio, Andrés PP01.41
Nascimento, Andres PP01.46
Nascimento, Andrés PP01.341
Nashi, Saraswati PP01.139, PP01.150
Nathani, Dev PP01.245
Nathorst-Böös, Kristofer OS02.06
Natoli, Silvia OS04.05
Naylor, Maria PP01.59, PP01.71
Nazha, Birouk PP01.06, PP01.07
NDOYE SALL, NDEYE FATOU PP01.354
Nectoux, Juliette PP01.113
Needham, Merilee OS01.01
Needham, Merrilee PP01.99
Neil, David OS01.06
Nel, Melissa PP01.259
Nelson, Leslie PP01.339, PP01.343, PP01.82
Nelson, Stanley PP01.28
Nennesmo, Inger PP01.128
Neyens, Martine PP01.208
Ng, Joanne PP01.45
Nicolas, Guillaume PP01.132, PP01.151, PP01.351
Nicolini De Gaetano, Lucia PP01.22, PP01.96
Nicolis, Filippo PP01.03
Nicolosi, Silvia PP01.100
Nigro, Vincenzo PL01.02
Nikitin, Sergei PP01.342
Niks, Erik OS01.06, PP01.40
Niks, Erik H. PP01.01, PP01.64
Nilsson, Emil PP01.128
Nilsson, Malin PP01.349
Nishimatsu, Shin-ichiro PP01.02, PP01.76
Nishimori, Yukako PP01.102, PP01.123
Nishino, Ichizo OS03.04, PP01.123, PP01.140, PP01.85, TC07.02
Niu, Dau-Ming PP01.114
Nobile-Orazio, Eduardo PP01.268, PP01.269, PP01.282, PP01.370
Nobili, Lino PP01.09, PP01.10, PP01.36
Noel, Wim PP01.177, PP01.210, PP01.235, PP01.236
Noer, Julie B PP01.362
Nogara, Leonardo PP01.152
Noguchi, Satoru OS03.04, PP01.140
Noioso, Ciro Maria PP01.204
Nollet, Fran OS07.03
Norcia, Giulia PP01.55
Norris, Sarah PP01.65
Notturno, Francesca PP01.370
Nouioua, Sonia PP01.321, PP01.69, PP01.70
Noukens, Jan PP01.01
Noushad, Muhammed Ameen PP01.213
Novelli, Giuseppe PP01.145
Nowacek, Dustin PP01.215
Nowak, Richard J. PP01.170, PP01.175, PP01.80, PP01.81
Nuccetelli, Marzia PP01.144, PP01.145, PP01.250
Nucifora, Elsa PP01.153
Nunez, Daniel PP01.171
Nunez, Manuel PP01.267
Nuredini, Andi OS01.04
Nyougui, Elisabeth OS09.04
Nümann, Astrid PP01.154
O'Toole, Orna PP01.280, PP01.301
Obadović, Ksenija PP01.358
Obadović, Vanja PP01.358
Obrochta Moss, Kristin PP01.137
Obrochta-Moss, Kristin PP01.364
Octaviana, Fitri PP01.308
Oezturk, Menekse OS02.01
Ogrodnik, Mikolaj SS46.01
Oh, Eungseok PP01.330
Oh, Jeeyoung PP01.330
Oh, Seong-il PP01.330
Oh, Sun-Young PP01.294
Ohagen, Patrik PP01.214, PP01.231
Ohashi, Tomohito PP01.102, PP01.123
Ohsawa, Yutaka PP01.02, PP01.76
Okada, Kensuke PP01.198
Okuba, Kei PP01.356
Oldfors, Anders PP01.08, PP01.128, PP01.349
Oldfors, Carola PP01.128
Oldham, Andrew PP01.366
Oliveira, Thiago PP01.116
Oliveri, Emanuele PP01.05
Olivieri, Emanuele OS02.03
Olulana, Oluwaseyi PP01.287
Opsomer, Matthias OS08.06, PP01.61, PP01.62
Orban, Nikolett PP01.312
Ortez, Carlos PP01.57
Ortiz-Corredor, Fernando OS03.03, PP01.246, PP01.258
Osredkar, Damjan OS08.02, PP01.40
Ostojic, Slavica PP01.13
Ostrovskiy, Denis PP01.132
Ottelli, Elisa PP01.03
Ottoboni, Linda OS08.01
Ouaja, Rabye PP01.286
Ouchkat, Fatima PP01.124, PP01.125
OUISSEM, BENCHAABI PP01.338
Oury, Julien PP01.166
Oved, Kfir PP01.199
Oyama, Genko PP01.356
Ozonoff, Alexander PP01.89
Ozturk, Gulten PP01.109
Ozyurt Kose, Selen OS09.05
O’Neil, Daniel PP01.129
Packnett, Elizabeth PP01.222
Pacucci, Eleonora PP01.317
Padovani, Alessandro OS02.03, PP01.03, PP01.04, PP01.05, PP01.323
Page, Jess PP01.335, PP01.336, PP01.337
Pagliarani, Serena OS08.04
Paik, Julie SS41.02
Pajot, François PP01.168
Paker, Asif PP01.261, PP01.265, PP01.267
Pal, Endre PP01.312
Palace, Jacqueline PP01.156
Palombo, Flavia OS03.05
Paltiel, A. David PP01.89
Pamfil, Cristina PP01.106
Pandit, Awadh Kishor PP01.220
Pane, Marika OS01.06, PP01.55
Panicucci, Chiara PP01.11, PP01.34, PP01.35, PP01.36
Panos-Basterra, Paula PP01.113
Paoletta, Marco PP01.374, PP01.375, PP01.376, PP01.377, PP01.378, PP01.379
Paolicelli, Damiano PP01.317
Paolucci, Sophia PP01.55
Papadaki, Iliana PP01.183
Papadaki, Ilianna PP01.184
Papanikolaou, Aikaterini PP01.183
Parasrampuria, Ridhi PP01.53
Paravisi, Elena PP01.61, PP01.62
Pareyson, Davide SS21.02, TC11.02
Parihar, Jasmine PP01.220
Parinello, Guillaume OS08.02, PP01.369
Parisi, Mosè OS08.04, PP01.104, PP01.105
Park, Geun-Young PP01.304
Park, Hyung Jun PP01.167, PP01.20, PP01.23, PP01.24
Park, Jeongju PP01.196
Park, Jin-Mo PP01.146
Park, Jin-Sung PP01.146, PP01.175, PP01.97
Park, Jin-Woo PP01.333
Park, Jinse PP01.330
Park, Jinwoo PP01.332, SS34.03
Parlindungan, Faisal PP01.308
Parman, Yesim PP01.118
Parodi, Andrea PP01.74
Parravicini, Stefano PP01.40
Pascual-Goñi, Elba PP01.382
Pascuzzi, Robert OS04.04, PP01.186, PP01.79
Pascuzzi, Robert M. PP01.195, PP01.201
Patel, Reena PP01.284
Patel, Vihar PP01.239
Patki, Kiran PP01.53
Paul, David PP01.108
Pavani, Rodrigo OS04.06
Pavey, Nathan OS07.01, OS07.02
Pavlou, Miranta PP01.183, PP01.184
Pawaskar, Dipti OS04.06
Pechmann, Astrid PP01.40
Pedemonte, Marina OS07.04, PP01.35, PP01.36
Pedersen, Estrid PP01.389
Pedroso, José PP01.116
Peer, Cody J. PP01.81
Peeters, Herman PP01.61
Peeters, Ronald OS08.06, PP01.359
Pegoraro, Elena OS01.02, OS01.04, PP01.42
Pellacani, Enzo PP01.112, PP01.163, PP01.164, PP01.165, PP01.18, PP01.91, PP01.92, PP01.93, PP01.94, PP01.95
Pellerin, David PP01.154
Pensrisirikul, Benjamat PP01.371
Pepe, Georgis PP01.110
Perez de Arenaza, Diego PP01.153
Perez Ruixo, Juan-Jose PP01.208
Peric, Stojan PP01.143, PP01.159, PP01.268, PP01.279, PP01.282
Peris-Moreno, Dulce OS01.03
Perlee, Lorah OS04.06
Perlman, Seth J. PP01.46
Perna, Alessia PP01.145
Persson, Emma PP01.279
Persson, Emma K PP01.289
Persson, Emma K. PP01.282
Pesic-Heuvrard, Natacha PP01.133
Pesovic, Jovan PP01.143, PP01.27
Petat, Eva PL01.03
Petersson, Malin OS04.02, PP01.205
Petitta, Ilaria PP01.144
Petri, Susanne OS07.03
Petrie, Dennis PP01.237
Petrucci, Antonio PP01.145, PP01.247, PP01.86
Petschke, Kurt PP01.89
Petterson, Malin PP01.197
Pezzini, Camilla OS09.02, PP01.60
Peña Padilla, Christian PP01.129
Pešović, Jovan PP01.358
Pfaff, Abigail OS06.01
Pham, Tan P. PP01.366
Phan, Han PP01.46
Philips, Glenn PP01.215
Phillips, Glenn PP01.218, PP01.219
Pianto, Samuele PP01.03
Piasecka–Stryczyńska, Karolina OS07.03
Piccione, Ezequiel OS01.01
Piccolo, Gwenaelle OS06.06
Pichiecchio, Anna PP01.100
Piehl, Fredrik OS04.02, OS06.05, OS09.03, PP01.197, PP01.205
Pierno, Sabata PP01.30
Piga, Daniela OS08.04, PP01.117
Pikus, Liubov PP01.298
Pini, Veronica OS02.02, PP01.15, PP01.19
Pinto, Ashwin PP01.213
Pinto, Marcus PP01.295, PP01.300, SS27.02
Pinto, Marcus Vinicius TC03.03
Pinzur, Lena PP01.199
Pirreca, Shelley PP01.65
Piscosquito, Giuseppe PP01.204, PP01.370
Plantone, Domenico OS05.05
Plecko, Barbara PP01.325
Podhorna, Jana PP01.194
Pogosean, Artor PP01.234
Polavarapu, Kiran PP01.129, PP01.139, PP01.150
Poleur, Margaux OS08.02, PP01.369
Poli, Loris OS02.03, PP01.04, PP01.05, PP01.323, PP01.370
Pons, Roser PP01.185
Poon, Ming Chung PP01.345
Porco, Dania M.D. PP01.53
Porrello, Emanuela PP01.19
Poulidou, Vasiliki PP01.110
Poupiot, Jérôme PL01.03
Povedano, Monica SS31.01
Povedano, Mónica PP01.340
Power, Robert PP01.137
Poza, Juan José PP01.147
Press, Rayomand PP01.12
Preusse, Corinna PP01.180
Previtali, Stefano OS01.04, PP01.71
Previtali, Stefano Carlo OS02.02, OS04.03, PP01.15, PP01.19
Prigent, Hélène PP01.132, PP01.151
Primiano, Guido OS05.05, PP01.130
Prior, Robert OS06.04
Privolizzi, Riccardo PP01.45
Proud, Crystal PP01.68
Proud, Crystal M. PP01.52
Prufer, Alexandra PP01.43
Prunetti, Paolo PP01.244
Psarra, Aggeliki PP01.207
Psichari, Iliana PP01.183
Psichari, Ilianna PP01.184
Pugliese, Alessia OS04.05
Puig, Cristina PP01.341
Pulley, Michael T PP01.132
Pumberger, Matthias PP01.180
Punga, Anna Rostedt PP01.234
Pupillo, Elisabetta SS29.01
Putko, Brendan PP01.300
Puzzi, Mattia PP01.03
Pyko, Andrei OS06.05
Pál, Endre PP01.141
Péréon, Yann PP01.132
Qi, Cynthia PP01.215, PP01.218, PP01.219
Qi, Yulan PP01.364
Qin, Yating PP01.362
Qiu, Yi-Hua PP01.384
Quagliotto, Magda PP01.316
Quarta, Raffaella PP01.30, PP01.31
Quasthoff, Stefan PP01.325
Querol, Luis PP01.261, PP01.267, PP01.268, PP01.279, PP01.282, PP01.284, PP01.382
Quick, Stephen PP01.313
Quijano-Roy, Susana PP01.52
Quinn, Colin OS01.01
Quinn, John OS06.01
Quintiens, Jilmen PP01.61, PP01.62
Raaphorst, Joost OS01.05, PP01.103
Rabinowicz, Shira PP01.33
Radenkovic, Lana PP01.143
Radenković, Lana PP01.358
Radovanovic, Nemanja PP01.143
Radovanović, Nemanja PP01.358
Ragole, Thomas PP01.132
Rainey, Amy PP01.108
Raja, Shruti M PP01.132
Rajabally, Yusuf A. PP01.268
Rajasingham, Thulashitha PP01.108
Raju, Dheeraj PP01.52
Rakocevic-Stojanovic, Vidosava PP01.143
Ramadža, Danijela Petković PP01.129
Ramchandren, Sindhu PP01.175, PP01.177, PP01.189, PP01.208, PP01.209, PP01.210, PP01.235, PP01.236
Ramdane Cherif, Ferroudja PP01.69, PP01.70
Ramdane-Cherif, Farroudja PP01.321
Ramdas, Sithara PP01.01, TC04.03
Ramirez-Montaño, Diana OS03.03, PP01.246, PP01.258
Ramos Masa, Maria PP01.223
Ramprasad, V L PP01.139
Randhawa, Simrat PP01.159
Ranieri, Enzo PP01.65
Ranum, Laura P. W. PP01.320
Rappold, Mika PP01.325
Rath, Jakob PP01.325, PP01.326
Ravaglia, Sabrina OS01.04, PP01.100, PP01.244
Ravera, Beatrice OS08.03
Ray, Soma PP01.142, PP01.59, PP01.71
Rayner, Chris PP01.137
Raza, Syed PP01.168
Rebel, Marcos PP01.43
Recchia, Fabio Anastasio PP01.32
Reddel, Stephen PP01.173, PP01.228, PP01.237, PP01.238, PP01.78
Reddel, Stephen W. PP01.178
Reddy, Divya PP01.53
Reef, Sharon PP01.199
Rehaume, Linda PP01.160
REID, Alice PP01.87
Reilly, Mary SS19.01
Rejdak, Konrad OS07.03
Remmerie, Anneleen PP01.269, PP01.278
Renaud, Mathilde PP01.154
Renegarajan, Sophie PP01.28
Repetto, Agnese PP01.35
Reshef, Ran PP01.171
Restrepo, Juan Luis PP01.203
Retailleau, Emilie PP01.351
Reviers, Evy SS45.01
Reyes, Óscar PP01.368
Reyna, Sandra P. PP01.52
Rezania, Fatemeh OS09.01, PP01.303
Ribera Armengol, Mar OS09.06
Ribolla, Fulvia PP01.84
Ricci, Edoardo PP01.316
Ricci, Enzo OS08.03
Ricci, Federica Silvia PP01.40
Ricci, Giulia OS01.04
Ricciardi, Dario OS04.03, PP01.155, PP01.370
Richard, Isabelle PL01.03
Richman, David PP01.171, PP01.239
RIEDEL, NATALIA OS01.03
Rigamonti, Andrea OS04.03
Righi, Delia OS05.05
Rigler, Nicole PP01.101
Riley, Nicholas PP01.346, PP01.347
Rinaldi, Simon OS06.04, PP01.282, PP01.284
Rini, Nicasio OS04.03, PP01.231
Riolo, Giorgia PP01.96
Ripolone, Michela PP01.104, PP01.105
Risi, Barbara OS02.03, OS03.05, PP01.03, PP01.04, PP01.05, PP01.100, PP01.323, PP01.370
Riso, Vittorio PP01.145
Riyanto, Dinda Larastika PP01.308
Rizzo, Federica OS08.01
Robb, Stephanie PP01.33
Roberts, Lauren PP01.314
Roberts, Leslie OS09.01, PP01.303
Roberts, Mark PP01.366
Rocchi, Camilla PP01.247, PP01.251, PP01.318
Rocchiccioli, Silvia PP01.32
Roddate, Marija PP01.274, PP01.98
Rodolico, Carmelo OS01.04, OS04.05, OS06.02, PP01.119, PP01.120, PP01.127, PP01.231
Rodrigues da Silva, Ádria PP01.255
Rodrigues, Gonçalo M.C. PP01.189
Rodrigues, Paula Raquel do Vale Pascoal OS05.02, PP01.257
Rodrigues, Thaina Louise PP01.112, PP01.164
Rodrigues, Thainá Louise PP01.163, PP01.18
Rodriguez, Alicia PP01.107
Rodriguez, Sebastian PP01.262
Rodríguez, Alicia Aurora PP01.391
Rodríguez-Camacho, Marta PP01.230
Roelants, Caroline PP01.388
Rohonczi, Mirtill PP01.312
Rolle, Enrica PP01.47, PP01.84
Romano, Angela OS05.05
Romano, Carmela PP01.86
Ronchi, Dario OS08.04, PP01.104, PP01.117
Roos, Andreas PP01.129, PP01.180, PP01.243
Roostalu, Urmas PP01.389
Rosen, Orli OS01.06
Rosenberger, Andreas Dybesland PP01.40
Rosenfeld, Marie PP01.53
Rosero, Spencer PP01.367
Rosewich, Hendrik PP01.248
Rosignoli, Paula Silvia PP01.327
Rosow, Laura OS01.01
Ross, Lauren OS09.01, PP01.303
Ross, Ryan PP01.222
Rossi, Salvatore PP01.145
Rossi, Simone PP01.86
Rossini, Elena OS04.03, OS04.05, PP01.187, PP01.231, PP01.232, PP01.86
Rosso, Tiziana PP01.370
Rostedt Punga, Anna TC05.01
Rouyer, Alice PP01.151
Roveta, Edoardo OS05.06
Roy, Bhaskar OS01.01, PP01.234, PP01.279, PP01.88, PP01.89
Rozsa, Csilla PP01.238
Ruas, Erica PP01.43
Ruck, Tobias OS01.01, PP01.129
Rudnicki, Michael PP01.75
Rueckert, Jens-Carsten PP01.180
Ruffin, Nicolas OS09.03
Ruggero, Susanna PP01.297
Ruggieri, Alessandra PP01.21, PP01.22, PP01.25, PP01.96
Ruggieri, Martino PP01.36
Ruggiero, Lucia OS01.04, OS03.05, PP01.352
Rugginini, Bianca PP01.244
Rugiero, Marcelo PP01.153
Ruiter, Annabel M OS02.01
Ruiz-Ospina, Edicson OS03.03, PP01.246, PP01.258
Russell, Jacqu PP01.65
Russo, Anna PP01.352
Russo, Massimo OS06.02
Russo, Rosario PP01.352
Ruszin-Perecz, Brigitta PP01.141
Rzepiński, Łukasz PP01.178, PP01.193
Röttger, Richard OS09.04
Sabbatini, Daniele OS01.02, PP01.42
Sabino, Andrea OS05.05
Sacca, Francesco PP01.211
Sacconi, Sabrina OS04.04, PP01.132, PP01.195, PP01.201, PP01.214, PP01.231, PP01.79, TC07.03
Sadjadi, Reza PP01.88
Saeki, Satoru PP01.334
Safri, Ahmad Yanuar PP01.308
Sahagian, Gregory PP01.169
Said, Ibrahim OS07.04
Saigoh, Kazumasa PP01.240, PP01.322
Saito, Fumiaki PP01.386
Saito, Kayoko PP01.52
Saito, Yoshihiko PP01.85
Saito, Yuko PP01.320
Sakata, Hanami PP01.240
Sakurai, Hidetoshi PP01.386
Salerno, Franco PP01.22, PP01.96
Saltarella, Ilaria PP01.25
Salvadó, Maria PP01.203
Salvalaggio, Alessandro PP01.297, PP01.370
Salvi, Erika PP01.22
Sam, Geuens PP01.40
Sambruni, Irene PP01.22
Samuelsson, Kristin PP01.12
SAMUKAWA, MAKOTO PP01.322
Samukawa, Makoto PP01.240, PP01.320
Sandri, Marco OS09.02, PP01.152, PP01.60, SS46.03
Sandroni, Paola SS34.02
Sanga, Panna PP01.175
Sangrut, Phuettha PP01.371
Sansone, Valeria OS01.06, PP01.74, SS18.01
Sansone, Valeria Ada PP01.373
Sansone, Valeria Ada Maria PP01.55
Santilli, Ashley PP01.101
Santorelli, Filippo PP01.154
Santorelli, Filippo Maria OS03.05
Santos Benetti, Webert Alex PP01.252
Santos Silva, Claudia OS07.02
Santos, Schwend PP01.364
Sarajlija, Adrijan PP01.13
Sareidaki, Doxa-Eleni PP01.183, PP01.184, PP01.185
Sarkozy, Anna PP01.11, PP01.33
Sarparanta, Jaakko PP01.21
SARR, MAMADOU MOUSTAPHA PP01.354
Sartori, Roberta OS09.02, PP01.60
Savarese, Marco OS03.05, PP01.21, TC14.02
Savic Pavicevic, Dusanka PP01.27
Savic, Natasa PP01.179, PP01.214, PP01.229
Savic-Pavicevic, Dusanka PP01.143
Savić Pavićević, Dušanka PP01.358
Saxon, David PP01.287
Scannell Bryan, Molly PP01.285
Schaarschmidt, Marco OS09.04
Schara, Ulrike PP01.243
Schara-Schmidt, Ulrike PP01.129, PP01.40, TC09.01
Scheiner, Christophe A PP01.132
Schelfhout, Matthias PP01.63, PP01.64
Schenone, Angelo OS05.06, PP01.370
Schenone, Cristina OS05.06
Schepers, Josef OS09.04
Schipani, Elke PP01.270
Schirinzi, Erika PP01.370
Schlader-Ratzinger, Michaela PP01.170
Schmidhammer, R SS22.08
Schmidt, Jens OS01.01
Schneider-Gold, Christiane PP01.216
Schoser, Benedikt PL01.01
Schouten, Meyke PP01.129
Schroeter, Michael OS02.01
Schuelke, Markus PP01.180
Schuhmann, Martin PP01.260
Schuller, Jan C. PP01.285, PP01.372
Schumann, Martin SS22.03
Schutze, Katherine PP01.99
Schwab, Matthias PP01.362
Schwabe, Paula PP01.363
Schwaede, Abigail N. PP01.01
Schwerin-Nagl, Anette PP01.325
Schänzer, Anne PP01.129
Schömig, Friederik PP01.180
Sciacco, Monica OS08.04, PP01.104
Sciarrone, Maria Ausilia OS05.05
Scola, Rosana Herminia OS05.02, PP01.252, PP01.257, PP01.327
Scola, Rosana Hermínia PP01.255
Scoto, Mariacristina PP01.33, PP01.44
Secil, Yaprak PP01.291
SECK, LALA BOUNA PP01.354
Seferian, Andrea OS02.04
Segall, Hailey PP01.134
Seghers, Ineke PP01.193
Sehinovych, Ihor PP01.41
Seiderer, Linda OS09.01
Seier, Mara PP01.256
Sejersen, Thomas OS02.06, PP01.40
Sellami, Noura PP01.132, PP01.133
Seluzhytsky, Alex PP01.285, PP01.372
Semciw, Adam PP01.313
Sen, Annabel PP01.28
SENGHOR, HENRIETTE PP01.354
Seo, Jung-Hwa PP01.266
Seok, Jin Myoung PP01.330
Servais, Aude PP01.151
Servais, Laurent OS08.02, PP01.343, PP01.369, PP01.52, PP01.53, SS05.03, 659
Seshadri, Madhav PP01.171
Seth, Arjun OS01.01, PP01.169, PP01.278, PP01.283, PP01.285
Sethuraman, Natarajan PP01.53
Sevilla, Teresa PP01.175
Sframeli, Maria OS07.04
Shah, Hemant PP01.305
Shah, Jaimin OS01.01
Shaibani, Aziz OS01.01, OS04.06
Shane, Olivia PP01.48
SHARMA, MEHAR PP01.361
Shaughnessy, Laura PP01.222
Sheehan, Jennie TC04.02
Sheffield, James PP01.268
Shehatha, Ruby PP01.99
Sheikh Saker, Tahani PP01.381
Shelly, Shahar OS05.01, PP01.159, PP01.160, TS10.01
Shen, Yan PP01.128
Sherman, Steven OS04.06
Shieh, Perry OS01.01, PP01.68, PP01.71
Shieh, Perry B. OS02.04, PP01.52
Shimazaki, Rui PP01.123, PP01.140
Shimizu, Hironori PP01.102
Shimizu-Motohashi, Yuko PP01.40
Shin, Dong Wook PP01.324
Shin, Ha Young PP01.173, PP01.226, PP01.273
Shin, Kyong Jin PP01.200, PP01.330
Shin, Sarah PP01.65
Shiota, Tomo PP01.102, PP01.123
Shock, Anthony OS09.06
Shouman, Kamal PP01.300
Shree, Ritu PP01.290
Shupe, Christina PP01.367
Sian, Veronica PP01.21
Siciliano, Gabriele OS01.04, PP01.32, PP01.370, TC14.03
Siddiqui, Uzma PP01.159, PP01.206, PP01.211
Sidhu, Ishnoor PP01.137
Signore, Giovanni PP01.32
Signori, Alessio PP01.370
SIHAM, HALLAL PP01.338
Sikorski, Patricia SS43.01
Silva, Ana Marina PP01.112, PP01.116, PP01.163, PP01.164, PP01.18, PP01.92, PP01.93, PP01.94, PP01.95
Silva, Erika Christina PP01.255
Silvestri, Gabriella PP01.145
Silvestri, Nicholas PP01.168, PP01.215
Simmons, Zachary OS01.01
Simms, Laura PP01.335, PP01.336, PP01.337
Singh, Rajesh kumar PP01.220
Singhvi-Hanns, Laura OS09.06
Sinha, Uma PP01.108
Sipos, Andrea PP01.141, PP01.312
Sirbu, Carmen Adella PP01.115
Sirina, Julija OS09.06
Sivakumar, Kumaraswamy OS01.01, PP01.175
Skopek, Jiri OS07.03
Skripuletz, Thomas PP01.269
Slader, Cassandra PP01.214, PP01.231
Slowik, Agnieszka PP01.159
Sluzevich, Jason PP01.90
Smilowski, Marek PP01.159, PP01.175, PP01.206, PP01.211
Smith, Barbara K. OS02.04
Smith, Gordon PP01.215
Smith, Tanya PP01.99
Smithuis, Frank OS01.05
So, Jungmin PP01.353
Soekhradj, Jayant A. PP01.63, PP01.64
Sohn, Eunhee PP01.330
Solidoro, Paolo PP01.84
Solé, Guilhem PP01.194, PP01.231
Somma, Teresa PP01.352
Sommer, Claudia PP01.267, SS10.01, SS46.02
Song, Guochen PP01.82
Song, Hee-Jung PP01.309
Songsliph, Nicha PP01.45
SONIA, NOUIOUA PP01.338
Soontrapa, Pannathat PP01.174
Sorgente, Annamaria PP01.03
Sorrenti, Benedetta OS04.03
Souttou, Amal OS04.06
SOYLU, Çağlar PP01.380
Soysal, Aysun PP01.355
Spagni, Gregorio PP01.212, PP01.86
Spallone, Vincenza SS27.03
Spalloni, Alida PP01.318
Spelman, Tim PP01.173
Spendiff, Sally PP01.129, PP01.166, SS42.03
Spies, Judith TC08.03
Spin, Paul PP01.346, PP01.347
Spinner, Robert PP01.270, SS23.03
Spitali, Pietro PP01.73
Sproule, Douglas PP01.108
Squillaci, Angelica OS06.02
Srikanth, Nikethana PP01.346, PP01.347
Srivastava, Achal kumar PP01.220
St. Andre, Michael PP01.37
Stadheim, Terrance A. PP01.37
Stahl, Jan-Hendrik PP01.260, SS22.03
Stahl, Mark PP01.329
Stamatakis, Iasonas-George PP01.183, PP01.184, PP01.185
Starling, Ana Lucia OS07.05
Staropoli, John PP01.82
Stascheit, Frauke OS02.01
Staskus, Lexa PP01.137
Statland, Jeffrey PP01.82
Stavropoulou- De Lorenzo, Sotiria PP01.207
Steeland, Sophie PP01.223
Steffan, Davide PP01.152
Stein, Maike OS02.01
Steiner, Leonie PP01.40
Steinkühler, Christian PP01.30
Stenzel, Werner PP01.180
Sternberg, Damien PP01.241
Stettner, Mark PP01.282, PP01.284
Stinghel Pellacani, Enzo PP01.116
Stobbe, Alica PP01.363
Stocker, Grace PP01.129
Stoddart, Leigh OS09.06
Stojkovic, Tanya PP01.113, SS21.01
Stokke, Simen PP01.40
Strano, Camilla PP01.370
Strataki, Eleni PP01.182
Straub, Volker PP01.108, PP01.40
Street, Jonathan PP01.368
Streicher, Nicholas PP01.287
Striano, Pasquale PP01.36
Strijbos, Paul OS08.02
Stromillo, Maria Laura OS05.05
Stroppi, Marco PP01.117
Strupp, Michael PP01.154
Study Group, MGBase PP01.173
Stufano, Angela OS05.05
Stögmann, Eva PP01.325
Su, Mao-Yuan OS05.03
Su, Wendy PP01.228
Suboh, Amani OS02.01, OS08.05
Suel, Laurence PL01.03
Suemi Kamoi Kay, Cláudia PP01.252, PP01.255
Sugata, Mayu PP01.102
Sugie, Kazuma OS03.04, PP01.102, PP01.123, PP01.140
Suh, Bum Chun PP01.167, PP01.20, PP01.23, PP01.24, PP01.309, PP01.350
Suh, Jee Hyun PP01.49, PP01.50
Suk, Jungim PP01.16
Suman, Adrian Florentin PP01.32
Summers, Dave PP01.343
Sun, Fang PP01.169
Sun, Yuyao PP01.283, PP01.285
Suna, Inga PP01.275
Sunada, Yoshihide PP01.02, PP01.76
Sunebo, Sofie PP01.128
Sung, Jung-Joon OS06.03, PP01.149, PP01.331
Sunnegardh, Oskar OS02.06
suri, vaishali PP01.361
Suárez, Bernardita PP01.58
Swijsen, Ann PP01.158
SY, MAMADOU PP01.354
Synofzik, Matthis PP01.154
Szabo, Lena OS08.02
Szabolcs, Szatmari PP01.115
Szczałuba, Krzysztof OS03.05
Szmulewicz, David OS09.01, PP01.303
Sáiz, Pilar OS07.03
Sánchez-Tejerina, Daniel PP01.203
Tae, Woo-Suk PP01.333
Takacs, Sara PP01.132
Takada, Rei PP01.348
Takahashi, Masanori P. PP01.191
Takatoshi, Sato PP01.40
Takeshita, Erit PP01.40
Takizawa, Hotake PP01.85
Taliani, Rebecca PP01.212
Talloen, Willem PP01.156
Tamaoui, Leila PP01.124, PP01.124, PP01.125, PP01.126
Tan, Elizabeth Ming Jing PP01.292
Tan, Ersin OS07.06
Tan, Pauline PP01.37
Tan, Zhibin PP01.292
Tanaka, Akito PP01.102
Tanaka, Hiroaki PP01.102
Tang, Fengming PP01.80, PP01.81
Tannemaat, Martijn R OS02.01
Tanner, Stephanie PP01.336
Tanriverdi, Zeynep PP01.291
Tarancón, Thais OS04.04
Tarancón, Thaïs PP01.186, PP01.195, PP01.201, PP01.231, PP01.79
Tard, Céline PP01.132
Tasca, Giorgio OS08.03
Taskin, Seyhan PP01.181
Tavian, Daniela OS03.01
Teng, Sophie PP01.239, PP01.288
Teng, Yee Sean PP01.292
Teran, Nataša PP01.83
Teranishi, Hirofumi PP01.191, PP01.194
Terrancle, Angeles PP01.340, PP01.57
Tetorou, Konstantina PP01.45
Tetunashvili, Kristine PP01.56
Thaisetthawatkul, Pariwat PP01.131, PP01.256
Thams, Sebastian PP01.12
Thenral, S G PP01.139
Thiele, Simone PP01.135, PP01.56, PP01.66
Thomas-Ahner, Jennifer M. PP01.46
Thompson, Ben OS06.03
Thompson, Rachel PP01.129
Thomsen, Gretchen PP01.329
Tierro, Benedetta PP01.297
Tiet, May PP01.243
Tikhomirova, Evgeniya Anatolyevna PP01.296
Tizzano, Eduardo PP01.52
Tobin, Rebecca PP01.73
Tolentino, Alessandra PP01.112, PP01.163, PP01.164, PP01.92, PP01.93, PP01.94, PP01.95
Tomaselli, Pedro PP01.18
Tonholo, Thiago PP01.116
Tonin, Paola PP01.107
Topakian, Raffi PP01.325
Topaloglu, Haluk OS01.06
Torchia, Eleonora OS08.03
Torella, Annalaura PL01.02
Toriello, Antonella PP01.204
Toscano, Antonio OS06.02, PP01.119, PP01.120, PP01.127
Tozawa, Takenori PP01.348
Tozza, Stefano OS05.05
Tozzi, Marco PP01.389
Trakman, Gina PP01.314
Traum, Avram Z. PP01.46
Trauschütz, Andreas PP01.154
Traverso, Monica PP01.09, PP01.10, PP01.11, PP01.34
Tripathi, Manjari PP01.220
Troger, Johannes PP01.325
Trojan, Daria A. OS07.03
Trucco, Federica OS07.04, PP01.11, PP01.34, PP01.35, PP01.36
Truini, Andrea SS44.03
Truman, Matt PP01.159
Tsai, I-Ching PP01.210
Tsargush, Vadim PP01.342
Tsimakidi, Chrysanthi PP01.207
Tsolakidis, Savas SS22.08
Tsonaka, Roula PP01.73
Tsuda, Koichi PP01.191
Tsuji, Yukiko OS07.01, OS07.02
Tuccillo, Francesco PP01.155
Tucht, Calvin PP01.129
Tufano, Laura OS04.05, PP01.145, PP01.187, PP01.232, PP01.86
Tulimiero, Lisamaura PP01.30
Tummala, Raj PP01.171, PP01.90
Tung, Angel Wing Lam PP01.345
Tupler, Rossella OS01.04
Turkdogan, Dilsad PP01.109
Turkoz, Ibrahim PP01.175, PP01.189, PP01.209, PP01.210, PP01.235, PP01.236
Turner, Catherine PP01.40
Tuttle, Edward PP01.48
Tuzova, Elizaveta PP01.281
Tzartos, John PP01.173
Tzavella, Dimitra PP01.182
Tzeng, Shiou-Ru OS05.03
TÜRKER, Duygu PP01.380
Ueda, Reoto OS03.04
Ulane, Christina PP01.171
Unver, Olcay PP01.109
Uppara Kowthalam, Madhurima OS03.06
Urbano, Guido PP01.47, PP01.84
Urbano, Isabella PP01.03
Usbergo, Cristian PP01.119, PP01.120, PP01.127
Utsugisawa, Kimiaki OS04.04, PP01.170, PP01.176, PP01.179, PP01.201, PP01.228, PP01.78, PP01.80, PP01.81
Uzawa, Akiyuki PP01.178, PP01.194
Vakrakou, Aigli G PP01.182
Valente, Enza Maria PP01.244
Valentino, Maria Lucia OS01.04, OS03.05
Valenza, Alice SS18.01
Vallefuoco, Francesca PP01.352
Vallve Maine, Camila PP01.45
van Alfen, Nens PP01.17
van Bragt, Tonke PP01.01
van de Camp, Sanne A.J.H. PP01.17
Van de Steen, Olivier PP01.279, PP01.284, PP01.289
Van de Walle, Inge PP01.284
van de Warrenburg, Bart PP01.154
van den Ameele3, Jelle PP01.243
van den Anker, Johannes OS01.02
van den Bos, Mehdi A.J OS07.01
Van den Bos, Mehdi A.J. OS07.02
van der Burgt, Yuri PP01.73
van der Kooi, Anneke OS01.05, PP01.103
van der Pol, Ludo PP01.289
van der Pol, W. Ludo PP01.17, PP01.282
Van der Walt, Anneke PP01.173, PP01.237
van der Walt, Anneke PP01.238
van der Woning, Bas PP01.262
van Doorn, Jeroen L.M. PP01.17
van Doorn, Pieter PP01.269
Van Hoorick, Benjamin PP01.156, PP01.168
Van Huynegem, Karolien PP01.158
van Leeuwen, Esther PP01.103
van Lenthe, Harry PP01.61, PP01.62
Van Lieshout, Marloes PP01.40
van Prooije, Teije PP01.154
van Schaik, Ivo OS01.05, PP01.103
Van Schil, Paul PP01.172
Van Vleet, Jeremy PP01.364
Vandenborne, Krista OS02.05
Vangeneugden, Tony PP01.223
Vanhauwaert, Roeland PP01.156, PP01.158, PP01.166
Vankerckhoven, Bernhardt PP01.166
Vanoli, Fiammetta OS04.03, OS04.04, PP01.214
Varga, David PP01.312
Varga, Dávid PP01.141
Varma, Anika PP01.367
Vartianen, Jenny PP01.45
Vashishta, Prashanth PP01.138
Vasquez, Edison OS03.03, PP01.246, PP01.258
Vattemi, Gaetano OS01.04, PP01.107
Veerapandiyan, Aravindhan PP01.68
Vegezzi, Elisa PP01.244, PP01.370
Velardo, Daniele OS01.04, OS08.04, PP01.100, PP01.104, PP01.105
Velazquez, Peter OS01.06
Veltsista, Dimitra PP01.238
Vencovský, Jiri PP01.262
Venditti, Romina PP01.55
Veneruso, Marco PP01.36
Vengalil, Seena PP01.138, PP01.150
vengalil, seena PP01.139
Ventre, Erwann PP01.388
Vera, Valentina PP01.187, PP01.232, PP01.86
Vercelli, Liliana PP01.47, PP01.84
Verebi, Camille PP01.113
Verhamme, Camiel PP01.103
Verhamme, Fien M. PP01.176, PP01.178
Verovnik, Barbara PP01.40
Verschuuren, Jan PP01.158, SS40.01
Verschuuren, Jan J. G. M. PP01.176
Verza, Massimiliano Ugo OS04.05
Vettori, Katia PP01.325
Vibha, Deepti PP01.220
Vicart, Savine PP01.130
Vicino, Alex PP01.96
Vietri, Giovanni PP01.144, PP01.247, PP01.250, PP01.251, PP01.318
Villa, Marianna PP01.55
Villano, Daniella PP01.40
Villar-Quiles, Rocío-Nur PP01.130
Villella, Chiara PP01.104
Vinay Mahesh, Karthik PP01.290
Vincenten, Sanne PP01.14
Vinciguerra, Claudia OS04.03, PP01.155, PP01.204, PP01.214
Vinçonneau, Gilles PP01.229
Visconti, Virginia Veronica PP01.145
Visentin, Andrea PP01.297
Vissing, John OS04.04, OS04.06, PP01.108, PP01.133, PP01.159, PP01.170, PP01.186, PP01.195, PP01.201, PP01.228, PP01.78, PP01.79, PP01.80, PP01.81, PP01.82, TC06.01
Visuttijai, Kittichate PP01.08
Vitali, Francesca OS05.05
Vlodavets, Dmitry PP01.343
Voermans, Nicol PP01.129, PP01.14
Voermans, Nicol C. PP01.17
Voet, Nicoline B.M. PP01.17
Vogrig, Alberto PP01.86
Volkov, Jenell PP01.171, PP01.90
Voltolini, Luca PP01.212
Voorn, Eric Lukas PP01.136
Vorobeva, Sofya PP01.296
Vovk, Andrej PP01.40
Vrščaj, Eva OS08.02
Vu Hong, Ai PL01.03
Vu, Tuan OS04.04, OS04.06, PP01.159, PP01.160, PP01.175, PP01.176, PP01.186, PP01.193, PP01.201, PP01.209, PP01.215, PP01.79
Vucic, Steve OS07.01, OS07.02
Vucinic, Dragana PP01.27
Vujcic, Miodrag PP01.282, PP01.289
VY, Vishnu PP01.220
Vélez-Gómez, Beatriz PP01.230
Wa Somwe, Somwe PP01.259
Waddington, Simon PP01.45
Wadman, Renske I. PP01.17
Walsh, Sarah PP01.346, PP01.347
Walter, Dr. Maggie C. PP01.56
Walter, Farah J. PP01.135, PP01.56
Walter, Maggie PP01.66
Walter, Maggie C. PP01.135
WAN, Lok Man PP01.315
Wang, Dazhe PP01.71
Wang, Jin PP01.363
Wang, Leo OS01.01
Wang, Te-Wei OS05.03
Wang, Yong Lin PP01.238
Wanschitz, Julia PP01.325
Warren, Francis PP01.343
Wasilewicz, Luc PP01.54
Waters, Kate PP01.33
Waters, Patrick OS09.01
Waśniowska, Urszula PP01.26
Weber, Markus TC13.01
Webster, Richard PP01.166
Wee, Yong Kiat PP01.65
Wei, Kuo-Ting PP01.387
Weigl, Lukas OS08.05
Weihl, Conrad OS01.01
Weinstein, Jennifer PP01.367
Weis, Joachim PP01.243
Weisburd, Ben OS02.02
Weisman, Mia PP01.88
Weiss, Michael D. PP01.179
Weiss, Ryan PP01.329
Wendel, Caterina PP01.66
Wendel, Caterina M. PP01.135
Wenninger, Stephan PP01.135, SS39.02
Werneck, Lucas Cesar PP01.257
White, Yvonne PP01.171
Whyms, Dermot OS07.03
Widholm, Per PP01.360
Wiendl, Heinz PP01.176, PP01.228, PP01.78
Wierenga, Lara M. PP01.63
Wiesenhofer, Anna PP01.325
Wilfong, Erin Marie PP01.90
Wilkins, H. Jeffrey OS01.01
Willcocks, Rebecca OS02.05
Willems, Natasha OS09.01
Willen, Joanna PP01.38, PP01.39
Willi, Roman PP01.228
Willis, Tracey PP01.74
Winkel, Antony PP01.173, PP01.263, PP01.264
Winter, Natalie SS22.02, SS22.03
Winter, Nathalie PP01.260
Winthrop, Kevin L. PP01.177
Wiratman, Winnugroho PP01.308
Withana, Ayana PP01.45
Witt, Evan PP01.137
Witte, Otto W PP01.362
Wittebols, Kim PP01.38, PP01.39
Witters, Peter OS03.06
Wittlinger, Julia SS22.02, SS22.03
Wolfe, Gil PP01.199, PP01.215, PP01.218, PP01.219, SS40.03
Wolff, David PP01.52
Wolfsgruber, Marlene OS08.05
Wong, Sui H. PP01.178
Woodcock, Ian PP01.40, PP01.71
Worner, Murray OS09.01, PP01.303
Wotton, Tiffany PP01.65
Wu, Jing OS02.06, OS04.02, OS06.05, PP01.197, PP01.205
Wu, Ning PP01.239, PP01.288
Wu, Wanqing OS02.06, OS04.02, PP01.197
Wu, Yao-Yu PP01.311
Wu, Yujie PP01.215
Wurm, Raphael PP01.325, PP01.326
Xi, Jianying PP01.178
Xiang, Cheryl PP01.218, PP01.219
Xiong, Ge PP01.239, PP01.288
Xiong, Li PP01.267
Yamada, Nanami PP01.102
Yamagishi, Yuko PP01.240
Yamanaka, Ai OS03.04, PP01.102, PP01.123
Yamaoka, Minako PP01.102, PP01.123
Yang, Chia-Feng PP01.114
Yang, Hongbo PP01.215
Yang, Huan PP01.175
Yang, Min PP01.218, PP01.219, PP01.48
Ye, Dongni PP01.215
Yee, Patrick PP01.40
Yeghiazaryan, Nune PP01.217
Yeh, Jiann-Horng OS04.04
Yeh, Ti-Yen PP01.311
Yilmaz, Zeynep PP01.109
Yin, Xiaoyan PP01.59
Yiu, Eppie PP01.40
Yoo, Dallah PP01.330
Yoon, Byeol-A PP01.266
Yoon, Seo Yeon PP01.49
Yoon, Won Tae PP01.350
Yoshida, Hideki PP01.348
Yoshikawa, Keisuke PP01.240
Yoshioka, Wakako PP01.85
Youn, Jinyoung PP01.330
Young, Joanne PP01.388
Yu, Ara PP01.364
Yu, Shuli PP01.142
Yuan, Julie Yue PP01.15
Yuan, Yue Julie PP01.19
Yungher, Benjamin PP01.132
YÜKSEL, Deniz PP01.380
Zabudskaya, Kseniya PP01.342
Zaidman, Craig PP01.68
Zambrano-Vera, Dario OS03.03, PP01.246, PP01.258
Zanolini, Alice SS18.01
Zanoteli, Edmar PP01.343, PP01.40, PP01.92, SS11.02
Zanotti, Simona PP01.104, PP01.105
Zhai, Yaya PP01.156
Zhang, WenWen PP01.238
Zhang, Xin PP01.237
Zhao, Chongbo PP01.178, PP01.193
Zhao, Xin PP01.168
Zhao, Yang PP01.160
Zheng, Yunlong PP01.191
Zhong, Wayne PP01.234
Zhou, Haiyan PP01.44
Zhou, Huiyu PP01.364
Zhou, Junguo PP01.329
Zhou, Tianyue OS03.06
Zhu, Chao PP01.238
Zhu, Yaowei PP01.175, PP01.208, PP01.209
Zimba, Stanley PP01.259
Zimprich, Fritz OS08.05, PP01.325, PP01.326
Zinaï, Saad PP01.132, PP01.133
Zinman, Lorne PP01.305
Zoppi, Dario OS01.04, OS03.05, PP01.352
Zouvelou, Vasiliki PP01.182, PP01.185
Zozulya-Weidenfeller, Alla PP01.130
Zschuentzsch, Jana OS02.01, OS09.04
Zschüntzsch, Jana PP01.130, PP01.195, PP01.363, PP01.79
Zuccarino, Riccardo PP01.373
Zulehner, Gudrun PP01.325, PP01.326
Zupan, Amadea PP01.83
Álvarez, Carmen PP01.341
Álvarez-Velasco, Rodrigo PP01.178
Ávila-Smirnov, Daniela PP01.58
Özbezen Kızıltan, Ebru PP01.355
Özdemir, Sadıka PP01.355
Özkaynar, Pinar OS01.05, PP01.103
Öztosun, Gülşen SS24.03
ÖZTÜRK, Merve PP01.380
Čopič, Alenka PP01.21
Ķauķe, Gundega PP01.274, PP01.98
Ķēniņa, Viktorija PP01.274, PP01.98
Łoboda, Agnieszka PP01.26
Šikic, Katarina PP01.129
Šuput, Dušan PP01.40
Žigman, Tamara PP01.129

