
Abstract
Due to the genetic and clinical heterogeneity of Rett syndrome, patients with nonclassic phenotypes are classified as an atypical Rett syndrome, that is, preserved speech variant, early seizure variant, and congenital variant. Respectively, MECP2, CDKL5, and FOXG1 have been found to be the causative genes, but FOXG1 variants are the rarest and least studied. We performed mutational analyses for FOXG1 on 11 unrelated patients without MECP2 and CDKL5 mutations, who were diagnosed with atypical Rett syndrome. One patient, who suffered from severe early-onset mental retardation and multiple-type intractable seizures, carried a novel, de novo FOXG1 mutation (p.Gln70Pro). This case concurs with previous studies that have reported yields of ∼10%. FOXG1-related atypical Rett syndrome is rare in Korean population, but screening of this gene in patients with severe mental retardation, microcephaly, and early-onset multiple seizure types without specific genetic causes can help broaden the phenotypic spectrum of the distinct FOXG1-related syndrome.
Keywords
Rett syndrome, first described by Andreas Rett in the 1960s, is a severe neurodevelopmental disorder characterized by psychomotor regression with the development of distinctive hand stereotypies. 1 It is one of the most common genetic causes of mental retardation in females, affecting approximately 1 in 10 000 live births. 2 Common clinical features include microcephaly, autism, seizure, breathing abnormalities, growth retardation, and gait apraxia. 3,4 Clinical diagnosis is based on the assessment of these phenotypes according to the Rett syndrome inclusion criteria, which have been modified minimally over time to retain key clinical elements of classic or, typical, Rett syndrome. 2 However, patients without some of these clinical characteristics have been recognized. These patients form distinct subgroups classified as “variant” or “atypical” Rett syndrome, which includes preserved speech variant, early seizure variant, and congenital variant Rett syndrome. 5
The genetic and clinical heterogeneity of Rett syndrome is notable: 95% to 97% of individuals with typical Rett syndrome have mutations in the MECP2 gene, compared to 50% to 70% in atypical cases. 6,7 The early seizure variant has been attributed to the CDKL5 gene after 2 unrelated female patients displaying characteristics of atypical Rett syndrome were found to have disruption in the CDKL5 gene. 8 –10 The congenital variant remains the least studied. In 2008, the FOXG1 gene was implicated in the congenital variant based on 2 cases. 11 By 2011, only 18 intragenic sequence changes in FOXG1 had been reported, including 14 null mutations and 4 missense mutations. 12,13 FOXG1 mutation has been found to be especially rare in the Asian population. Takahashi et al 14 identified 2 cases with FOXG1 mutations, one of which was novel, whereas Zhang et al 15 found no FOXG1 mutations in a large cohort of 365 patients in the same year. Here, we report 1 case with a novel mutation of FOXG1 and describe the clinical features in detail with the previous literature review.
Methods
Eleven unrelated Korean patients (9 females and 2 males) who fit the criteria for typical and atypical Rett syndrome based on the revised criteria 2 were examined at Seoul National University Children’s Hospital. We had previously analyzed the MECP2 gene and the CDKL5 gene by direct sequencing and multiple ligand probe amplification for these 11 patients and discovered no mutations in either gene in each patient.
Mutational Analysis
Blood samples were obtained from enrolled patients who provided informed consent. Genomic DNA was extracted from peripheral blood leukocytes using a QIAamp DNA Blood Midi Kit according to the manufacturer instructions (Qiagen, Valencia, California). Direct sequencing of all coding exons and flanking intronic sequences of the FOXG1 gene was performed using primer pairs designed by the authors. The primers are available on request. Polymerase chain reaction amplification was performed in a thermal cycler (Model 9700; Applied Biosystems, Foster City, California) and cycle sequencing was performed on an ABI Prism 3730xl DNA Analyzer using the BigDye Terminator Sequencing Ready Reaction Kit (Applied Biosystems). Sequence variations were analyzed via comparison to the wild-type sequence. The mutation nomenclature followed the recommendations of the Human Genome Variation Society. Parental DNA was also collected and tested for the presence of the identified variant.
Results
Molecular Analysis
A novel point mutation in the FOXG1 gene was identified in 1 of the 11 patients who were clinically diagnosed with atypical Rett syndrome (Figure 1). The de novo origin of the mutation was confirmed with maternal and paternal DNA samples. The missense variant c.209A>C, p.Gln70Pro was analyzed with PolyPhen-2 (Polymorphism Phenotyping v2) 16 and Sorting Intolerant From Tolerant 17 programs. The variant was predicted as “possibly damaging” with a pph prob score of 0.797 and “tolerated” with Sorting Intolerant From Tolerant score of 0.35.

The novel FOXG1 variant. A, Direct sequencing of FOXG1 exon 1 shows c.209A>C, p.Gln70Pro mutation in the patient DNA. The position is indicated by a red asterisk. B, Protein sequence alignment shows allele conservation for Gln70 among closely and remotely related species.
Clinical Features
The patient with a novel FOXG1 mutation is the first child of a nonconsanguineous couple. She was born at 37 weeks by cesarean section after an uneventful pregnancy. At birth, she weighed 2.03 kg and had breathing difficulties with germinal matrix hemorrhage and cystic change; these issues resolved after 6 months. She had severe mental retardation and no head control until her third year of age. Her development slowly improved after her third year, and she began walking, although unsteadily, at age 7. Her head circumference had decelerated from borderline microcephaly at birth to severe postnatal microcephaly: It measured 42 cm at 8 months of age (3-10 percentile) and 47 cm at 8 years of age (<3 percentile). She had multiple-type intractable seizures: At 8 months of age, dialeptic seizures gradually developed into multiple seizure types such as generalized tonic, tonic–clonic, and atonic seizures, which were unsuccessfully treated with multiple combined antiepileptic drugs. By the age of 7 years, the seizures were gradually controlled with 3 new antiepileptic drugs, namely, valproic acid, levetiracetam, and lacosamide. Video electroencephalography monitoring taken at 2 years of age revealed atypical absence seizures with consistent ictal changes, and further electroencephalography revealed frequent generalized slow spike and wave and paroxysmal fast activity. On last examination at 8 years of age, the patient was seizure free and intermittently showed purposeful use of her hands but also presented with abnormal movements such as generalized fine tremulous movements and choreoathetoid movements in the head and trunk. She never acquired speech or receptive language besides the use of the word “mama.” Her brain magnetic resonance imaging revealed no abnormalities. The clinical features of the other 10 patients, who had no identifiable genetic causes, are summarized in Table 1.
Clinical Features of Patients With Atypical Rett Syndrome in This Study.

Abbreviations: +, present; −, absent; N/A, not available; F, female; M, male.
Discussion
We describe here a novel, heterozygous FOXG1 mutation in a patient with atypical Rett syndrome with early-onset developmental delay and multiple seizure types. According to the FOXG1 variant database maintained by the International Rett Syndrome Foundation, 18 only 20 pathogenic variants have been found to date (Figure 2). A small number of pathogenic missense variants identified in FOXG1 all affect highly conserved amino acid residues in the fork head domain that serves a crucial role in DNA binding. 19 –21 The novel variant lies 300 bp before the fork head domain in a region in which no deleterious missense variants have been reported. Although there is an insufficient number of reported cases to infer genotype–phenotype correlations or mutation hot spots, 13 further investigation of this novel variant can reveal critical information on the yet unknown roles of the N-terminal domain of FOXG1 gene.
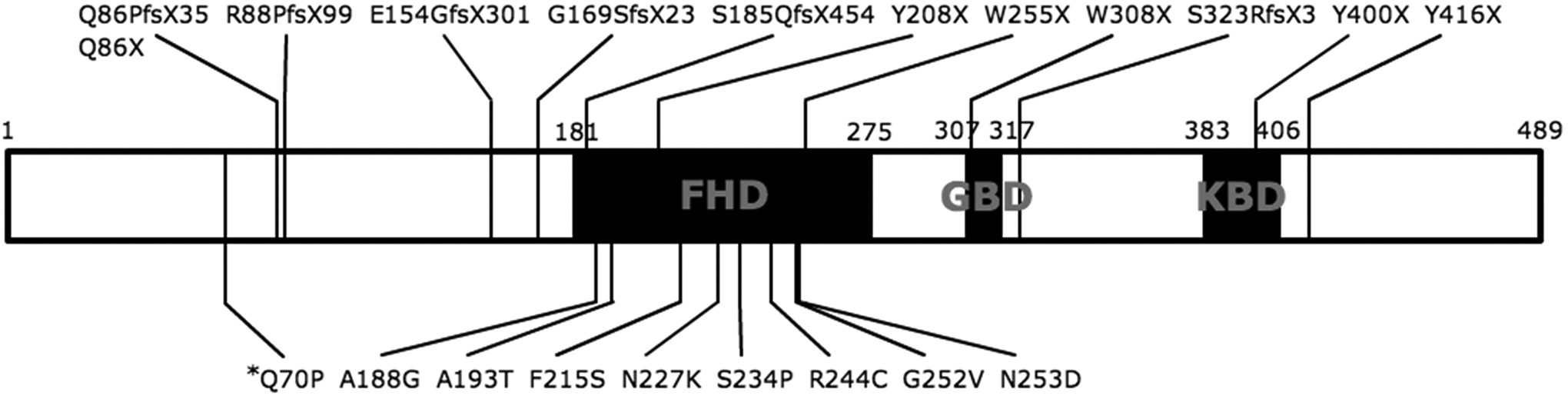
Schematic gene structure of FOXG1. The 3 main functional domains are shown, namely, the DNA binding fork-head domain (FHD), the Groucho binding domain (GBD), and the JARID1B binding domain (JBD). Twenty reported pathogenic variants and their positions are indicated. 18
The clinical phenotype of the patient described in this study would initially suggest the congenital variant, that is, delayed initial development, postnatal microcephaly, seizures, absence of characteristic eye gaze, hand stereotypies, and regression. However, there are growing doubts regarding the validity of an all-inclusive Rett syndrome classification system, and there have been continuous attempts to revise the classification system according to new findings. 13,22 In 1988, Trevathan and Moser removed female sex from the Rett syndrome criteria to include possible male patients with Rett syndrome. Yet despite this early attempt at generalization, Rett syndrome, associated with MECP2 and CDKL5, was a syndrome that exclusively affected females until the recent discovery of the FOXG1 gene. 20,21,23 The key clinical features remain, that is, profound cognitive disability, period of regression followed by stabilization, hand stereotypies, loss of spoken language, and gait abnormalities. However, as Rett syndrome remains a clinical diagnosis, amassing more data on atypical Rett syndrome will allow us to challenge the breadth and integrity of the classic Rett syndrome diagnostic criteria. 2,24 Recent reports now suggest FOXG1 syndrome to be a distinct form of developmental encephalopathy with detailed and consistent clinical data to support the association. 12,25,26
The core FOXG1 syndrome phenotype consists of severe mental retardation, hypotonia, absent language, postnatal microcephaly, dyskinesia, and corpus callosum hypogenesis. 12,27 Severe microcephaly along with delayed myelination, gyral simplification, and corpus callosum hypogenesis is frequently visible on magnetic resonance imaging scan. However, the imaging results published in the literature for FOXG1 syndrome 27 are nonspecific and similar to magnetic resonance imaging abnormalities in patients with other causes of developmental delay, including typical and atypical Rett syndrome cases. 26 Epilepsy is also common with generalized tonic–clonic and myoclonic seizures, and most seizures are easily controlled unlike CDKL5-related disorders. 11,25 Additionally, 10 patients with FOXG1 point mutations all presented with intense hyperkinetic movement disorders with polymorphic midline stereotypies and jerky movements. 28 Although the patient presented in this article showed normal magnetic resonance imaging without gyral simplification or corpus callosum hypogenesis, she showed all other main features of the FOXG1 syndrome, which are also characteristic of the typical Rett syndrome, that is, abnormal development, postnatal microcephaly, dyskinesia, and presence of hand stereotypies. 13
Notably, the patient had intractable seizures, initially presenting as dialeptic seizures starting in her eighth month. Over time, she had further multiple seizure types, such as drop attacks, generalized tonic–clonic seizures, atonic seizures, and generalized tonic seizures, followed by severe retardation with microcephaly. Her early clinical features led to a tentative diagnosis of Lennox-Gastaut syndrome, but as the patient began to show intermittent abnormal jerking hand movements and other autistic behavior, along with small, cold hands and feet, the investigators decided to analyze the genes associated with Rett syndrome and other Rett-like syndromes.
The yield for genotyping FOXG1 variants can be as low as 1.5% 28 or as high as 13% 14 depending on the breadth of inclusion criteria but has been found to be potentially as high as 15%: Zhang et al 15 and Mencarelli et al 19 screened 27 MECP2 and CDKL5 mutation negative patients with atypical variant Rett syndrome or FOXG1-related phenotype and found no mutation in 14 patients and 4 patients, respectively. Our study moderately concurs with these findings, with a yield of 9% (1 of 11).
Conclusively, we report low but significant incidence of FOXG1 mutation in patients with atypical Rett syndrome in the Korean population. This index case supports recent studies that associate distinctive and clinically recognizable phenotypes such as congenital developmental delay, early microcephaly, and dyskinesia to a FOXG1-related syndrome. We also note early-onset, multiple-type, intractable seizures that have not been observed in other reported cases. Considering the broad spectrum of FOXG1-related phenotypes and consistent yield in gene analysis, FOXG1 mutation assay would be helpful in determining genetic causes in patients who present with severe mental retardation and microcephaly at a very early age with negative MECP2 and CDKL5 mutation screening.
Footnotes
Acknowledgment
We would like to thank Sung-Wook J. Bang for English revision of this article.
Author Contributions
CKB contributed to data acquisition and prepared the first draft of the manuscript, editing the manuscript drafts. JSL, BCL, KJK, and YSH made substantial contributions to data acquisition, interpretation, and revised the manuscript. J-HC provided the case and edited the manuscript drafts until the final draft was produced and mentored CKB through the process as a correspondent.
Declaration of Conflicting Interests
The authors declare no conflicts of interest with respect to the research, authorship, or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by a grant from the Korea Healthcare Technology R&D project, Ministry for Health, Welfare and Family Affairs, Republic of Korea (Grant No. A120099).
Ethical Approval
This study was approved by the local Institution Review Board (IRB).