
Abstract
Autism spectrum disorders are a group of neurodevelopmental disorders with a strong genetic etiology. Cytogenetic abnormalities have been detected in 5% to 10% of the patients with autism spectrum disorders. In this study, the authors present the clinical and array-based comparative genomic hybridization evaluation of a 4-year-old male with autism spectrum disorder and mental retardation. The patient was found to carry a de novo duplication of chromosome 8p22-21.3 of 1.0 Mb as ascertained by quantitative polymerase chain reaction, and this region encompassed 3 genes including Pleckstrin and Sec7 domains-containing protein 3 (PSD3), SH2 domain-containing 4A (SH2D4A), and Chondroitin Sulfate N-Acetylgalactosaminyltransferase 1 (CSGALNACT1). This represents the smallest rearrangement of chromosome 8p as yet found in a patient with autism spectrum disorder, but the significance of this mutation is still ambiguous.
Autism spectrum disorders are neurodevelopmental disorders with complex etiology and strong genetic basis, characterized by impaired communication, reduced social interaction, and stereotyped and/or repetitive behavior. 1 –3 Autism spectrum disorder is the most common among these disorders with an average estimated global prevalence of 62 cases per 10 000 children, 4 and a male to female ratio of 4:1. 5 The prevalence of mental retardation in autism spectrum disorder has been reported to range between 40% and 70%. 6,7 The heritability of autism spectrum disorder was reported to be approximately 90%. 8 Concordance rate in monozygotic twins is as high as 70%, 2 while the recurrence rate in siblings is nearly 20%. 9 So genetics play a major role in autism spectrum disorder. Until now, more than 100 disease genes and over 40 genomic loci were identified in autism spectrum disorder probands. 10 Endorsed by the American College of Medical Genetics, analysis of chromosomal copy number variations using array-based comparative genomic hybridization or chromosomal microarray is now routinely performed in clinical genetics laboratories. A consensus statement, published in 2010, recommended chromosomal microarray as the first tier clinical diagnostic test for individuals with autism spectrum disorder. 11 Four reports on patients with autism spectrum disorder and chromosome 8p duplications have been published. 12 –15 The clinical diagnostic procedures and precision of the molecular cytogenetic analyses in these cases are extremely variable.
Here, the authors describe clinical and molecular cytogenetic findings in a patient with autism spectrum disorder and mental retardation but not growth retardation, self-mutilation, dysmorphic feature, congenital defects, or epilepsy. Using array-based comparative genomic hybridization and quantitative polymerase chain reaction (PCR), the authors identified a 1.0 Mb de novo duplication in chromosome 8p22-21.3 in our patient. To our knowledge, no patient having autism spectrum disorder with 1.0 Mb duplication of chromosome 8p22-21.3 has been described in the medical literature to date.
Methods
In order to detect a wide range of genome imbalance in children diagnosed with autism spectrum disorder, patients diagnosed with autism spectrum disorder using Diagnostic and Statistical Manual of Mental Disorders (Fourth Edition) 16 criteria at the Children’s Hospital of Fudan University were tested. One boy was identified with a 1.0 Mb duplication of chromosome 8p22-21.3 (chr8:18,549,078-19,570,474). The result was further confirmed using quantitative PCR and showed that this duplication is de novo.
Array-Based Comparative Genomic Hybridization
The DNA sample from the proband was analyzed using an Agilent SurePrint G3 custom comparative genomic hybridization and SNP Microarray 4x180 k (Agilent Technologies, USA). Following the manufacturer’s instructions, CytoGenomics were subsequently used for data normalization, quality evaluation, and data visualization. A minimum of 5 probes were required to meet the size cutoff. The chromosomal copy number variations were checked against the Database of Genomic Variants (http://dgv.tcag.ca/dgv/app/home) to exclude those present in healthy individuals. Nucleotide positions are as in human genome GRCh37/hg19 assembly.
Quantitative PCR
For chromosomal copy number variations identified in this study, the authors performed quantitative PCR to confirm for probands and also their parents to investigate the genetic status. The primer pairs of each gene for quantitative PCR were designed using the Primer3 (http://flypush.imgen.bcm.tmc.edu/primer/primer3_www.cgi; Supplemental Table S1). The PCR reactions were performed in volume of 10 μL contained 5 μL SybrGreen I Master mix (Toyota, China), 0.4 mmol/L primers, and ∼25 ng template complementary DNA. Hydroxymethylbilane synthase (HMBS) of Housekeeping genes was employed as endogenous controls for normalization. Final working volume of 10 μL contained 5 μL SybrGreen I Master mix (Toyota, China), 0.4 mmol/L primers, and ∼25 ng template genomic DNA. Each sample was conducted in triplicate. The runs were performed on the StepOnePlus Real-Time PCR Systems (Applied Biosystems, USA). The thermal profile for quantitative PCR was preincubation for 20 seconds at 95°C, followed by 40 cycles for 3 seconds at 95°C, and 30 seconds at 60°C, melting curve including 15 seconds at 95°C, 1 minute at 60°C, and 15 seconds at 95°C. Raw data were analyzed by the StepOne Software v2.1. Amplification level was calculated using the ΔΔCt method.
Results
Cytogenetic Findings
Whole-genome comparative genomic hybridization analysis showed the patient has a 1.0 Mb duplication in chromosome 8p22-21.3 (chr8:18,549,078-19,570,474; Figure 1). The duplication was further confirmed by quantitative PCR (Figure 2), which was not found in parents, indicating that the duplication is a de novo mutation. And this region encompassed 3 genes including PSD3, SH2D4A, and CSGALNACT1.
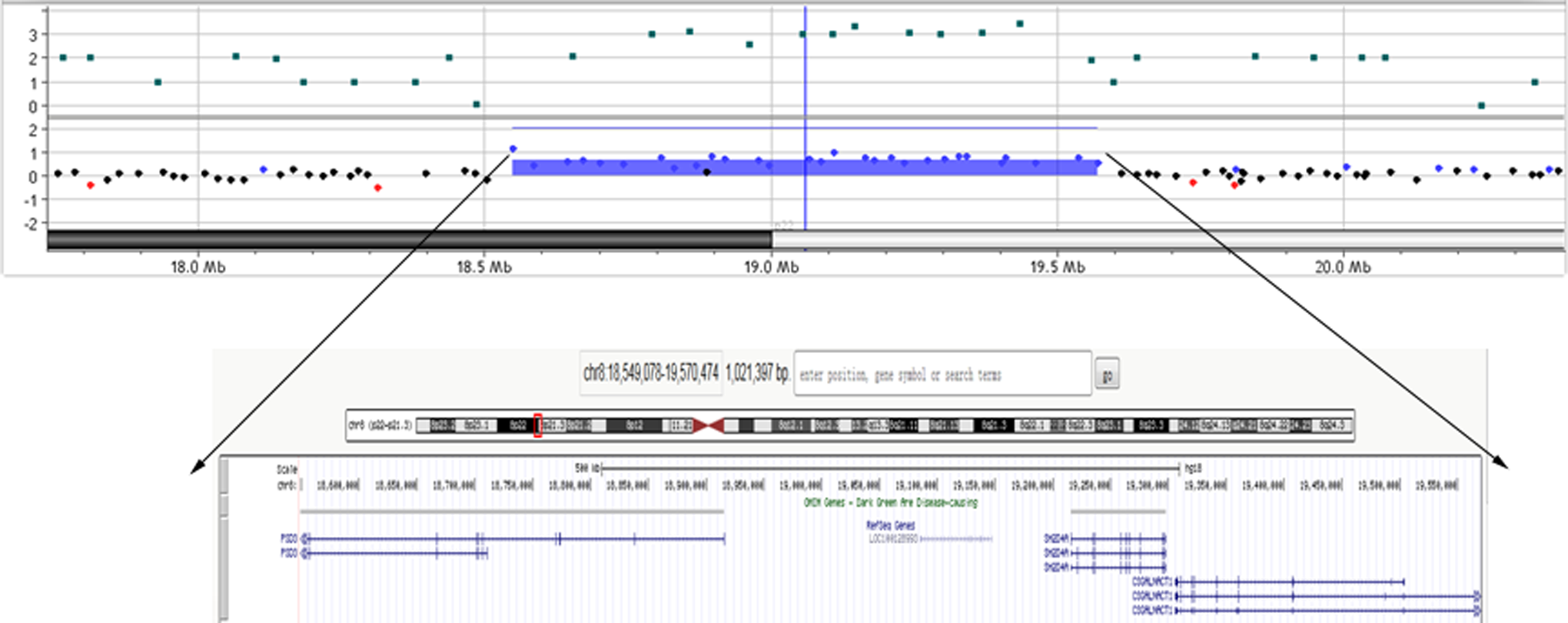
Identification of a 1.0 Mb duplication in chromosome 8p22-21.3 (chr8:18, 549,078-19,570,474) by array-based comparative genomic hybridization. Dots represent relative intensities in log2 ratio and genomic locations of the oligonucleotide probes employed in array-based comparative genomic hybridization assay. For patient, red (loss), black (no change), and blue (gain) dots represent log2 ratio deviation from the horizontal line of 0. Regions with copy number gains are indicated with blue horizontal bars.

Identification of a de novo duplication of chromosome 8p22-21.3 in a patient with autism spectrum disorder by quantitative polymerase chain reaction (PCR). De novo duplication of chromosome 8p22-21.3 in a patient with autism spectrum disorder. Confirmation of results using quantitative PCR. Mother and father were found to have single PSD3 and SH2D4A gene dosage, while patient had 2 copies of PSD3 and SH2D4A gene dosage.
Clinical Summary
The proband is the first-born child of nonconsanguineous parents of Chinese origin. He was born after an otherwise uncomplicated pregnancy by normal delivery at term with a birth weight of 3020 g. Concerns about his development arose from 21 months of age because of delayed speech. He can unconsciously say “mom” and “dad” at 7 to 8 months old and walk alone at 15 months old. He knew no language but could passively understand simple sentences as spoken by his mother when he was 21 months. At that time, eye-to-eye contact was good and no repetitive behavior was observed. He was diagnosed with developmental delay. When followed up at 4½ years old, his expressive language was limited (3 to 5 words), and his eye contact with the parents was poor. He occasionally ran around with other children but could not engage in interactive games. He showed repetitive behaviors such as frequently jumping, running back and forth, playing with strings, and shaking his hands. He wasn’t found to have impulsivity. He was diagnosed with autism spectrum disorder using Diagnostic and Statistical Manual of Mental Disorders (Fourth Edition) 16 at 4 years and 7 months, had an Autism Diagnostic Observation Schedule score (Table 1) 17 of 22 at the time of this examination (4 years and 11 months), well above the cutoff for autistic disorder. His head circumference was on 10th to 25th centile as well as height and weight on the 25th to 50th centile. He had no facial anomalies, kyphoscoliosis, spastic paraparesis, cerebral atrophy, or epilepsy. Cognitive/developmental evaluation using the Gesell Developmental Scale 18 showed that he had cognitive and language skills as well as motor skills well below that for his age (Table 1). The magnetic resonance imaging of brain, electroencephalogram, hearing screening, and karyotype analysis were normal.
Quantification of Developmental, Cognitive, and Autism Phenotypes of the Proband.

a The normal value of developmental quotient is ≥85.
Discussion
Duplication of 8p has been recognized since the early 1970s. 19 Most cases involve duplication of much of 8p, often as a duplication with distal breakpoints in 8p22 or 8p23 and proximal breakpoints in 8p11.2, 8p12, or 8p21. 20,21 Papanikolaou et al reviewed several cases associating chromosomal interstitial 8p with autism spectrum disorder. 14 The clinical picture of patients with duplication of much of 8p was described as a recognizable multiple congenital anomalies such as facial anomalies, hypotonia, structural brain abnormalities, and mental retardation syndrome while profound orthopedic problems, spastic paraplegia, frequently develop in elder patients. 22 –24 In the database of DECIPHER v8.7, there were 2 cases having a chromosomal aberration mostly analogous to us. One case (Patient 249443) had a 3.41 Mb duplication at 8p22 to 8p21.3 (chr8:18403254-21816649, encompassing 11 genes [ATP6V1B2 (ATPase, H+ transporting, lysosomal, V1 subunit B2), CSGALNACT1, DOK2 (Docking protein 2), GFRA2 (GDNF family receptor alpha 2), INTS10 (Integrator complex subunit 10), LPL (Lipoprotein lipase), LZTS1 (Leucine zipper, putative tumor suppressor 1), PSD3, SH2D4A, SLC18A1 (Solute carrier family 18 member 1), and XPO7 (Exportin 7)], inheritance unknown), with the phenotypes such as autism, inguinal hernia, intellectual disability, long face, macrocephaly, macrotia, proportionate short stature, and seizures; the other (Patient 249579) also had a 3.41 Mb duplication at 8p22 to 8p21.3 (chr8:18403338-21816628, encompassing genes the same as above, inheritance unknown), with a clinical picture of autism, brachycephaly, cryptorchidism, intellectual disability, proptosis, short philtrum, short toe, spasticity, and upslanted palpebral fissure. Although breakpoints were different, the duplication of region 8p21 to p22 was a consistent finding in all cases, meaning that these bands indicate the critical region for social and communication deficits. Patients with autism having abnormalities in these bands often have unrecognized autism spectrum disorder until 3 or more years and with a negative family history of autism spectrum disorder, 14,25 so the autistic phenotypes were milder than those with abnormalities in other chromosomes. Here the authors reported the patient had the smallest rearrangement of chromosome 8p as yet found in a patient with autism spectrum disorder. And our patient lacked many features of severe congenital anomalies seen in patients described earlier. He didn’t have encephalodysplasia, facial anomalies, or seizures. He was diagnosed with mental retardation at the age of 2, while he developed autistic phenotypes at about 4 years old. His symptoms were comparatively mild. The factor that may contribute to the attenuated phenotype in our patient is a relatively small size of the duplicated segment compared to many other patients. On the other hand, it might also be that in many cases of chromosome abnormalities, autistic spectrum symptoms go unrecognized because of a more general diagnosis of mental retardation.
Several studies have identified numerous candidate genes for autism spectrum disorder. 26 Thus far, alterations in genes involved in pathways such as chromatin remodeling and gene regulation; cytoskeleton dynamics and synaptic scaffolding; cell adhesion; second messenger systems; and genes encoding secreted proteins, receptors, and transporters have been associated with autism spectrum disorder. 26 This by no means excludes the possibility that other genetic pathways may be involved in autism spectrum disorder. And all genes of relevance to autism spectrum disorder share expression in the central nervous system as a common feature. Therefore, of the 3 genes with known function in the duplicated region in our patient, the authors consider those which show clear transcript expression in the central nervous system and with apparent functions in the pathways mentioned as primary candidates for autism spectrum disorder. Based on this criterion, the gene encoding human PSD3 represents the most plausible gene-dosage sensitive candidate gene for development of autism spectrum disorder in our patient. PSD3 protein is a guanine nucleotide exchange factor for adenosine diphosphate-ribosylation factor 6, 27 which regulates the membrane trafficking of small G proteins. 28 PSD3 also regulates Ras-related C3 botulinum toxin substrate 1, which is a Rho guanosine triphosphatase. 27 Consequently, PSD3 protein coordinates endocytosis with cytoskeletal rearrangement by catalyzing nucleotide exchange on adenosine diphosphate-ribosylation factor 6 and regulating Ras-related C3 botulinum toxin substrate 1 activation. 27 Thus, altered gene dosage and consequential elevated expression levels of PSD3 can interfere with cytoskeleton dynamics, a pathway related to autism spectrum disorder. And so far, the aberrant expression of PSD3 has been related to some intracranial tumors such as astrocytoma progression 29 and the damage of cognitive function in the aged individuals. 30 Another gene showing transcript expression in the central nervous system in our study is SH2D4A. The SH2D4A gene encodes SH2 domain-containing protein 4A. Using exon trapping and exon linking at chromosome 8p22, Dai et al 31 cloned SH2D4A, which they called SH2A. The deduced 454-amino acid protein contains a single SH2 domain. Reverse transcription polymerase chain reaction (RT-PCR) and Northern blot analysis detected ubiquitous expression of 3 SH2A transcripts as well as aberrant expression in some cancer cell lines. Dai et al 31 concluded that SH2A is a docking protein that may be involved in signal transduction. Lapinski et al 32 noted that SH2D4A shares a similar SH2 domain and other structural features with the T-cell adapter proteins TSAD (SH2D2A; 604514) and ALX (608349). However, they found that knockdown of SH2D4A expression in human T cells had no impact on T-cell function. And this gene has never been implicated before in any brain-related disorders. The CSGALNACT1 gene encodes chondroitin sulfate N-acetylgalactosaminyltransferase 1, which is a key glycosyltransferase of chondroitin sulfate biosynthesis. It catalyzes the transfer of an N-acetylgalactosamine residue onto the nonreducing end of sulfated glucuronic acid and has been shown to play a key role in chondroitin sulfate chain initiation, possibly in the elongation process. 33 And it has not yet shown an expression in the central nervous system.
Similar duplications have also been reported in the Database of Genomic Variants (http://dgv.tcag.ca/dgv/app/home; nssv694958, 8p22-8p21.3, 1.0 Mb duplication).
In summary, the authors first reported a patient with autism spectrum disorder having a 1.0 Mb de novo duplication at 8p22-21.3 with clear breakpoints (chr8:18,549,078-19,570,474), but the significance of mutations is still ambiguous. Our report highlights both the diagnostic value of array-based comparative genomic hybridization in children with autistic phenotypes and emphasizes the usefulness of the thus generated precise molecular cytogenetic information for identifying candidate genes for autism spectrum disorder and other neurodevelopmental disorders. Screening of larger population of patients with autism together with healthy controls, as well as functional studies and resequencing of the PSD3 gene, can aid to elucidate the possible involvement of this gene in autism spectrum disorder.
Footnotes
Acknowledgments
The authors thank the children and their families for participating in this study.
Author Contributions
XX and QX conceived the study and participated in its design and coordination; PD, QX, BZ, PL, and XX identified the patient and carried out the clinical characterizations; PD, QX, YA, and RL carried out the molecular genetics studies; PD and QX wrote the manuscript; all authors read and approved the final manuscript.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by grants 81371270 from the National Science Foundation of China (to Xiu Xu).
Ethical Approval
This study was approved by the Ethics Committee of the Children’s Hospital of Fudan University (Approval number: Children’s Hospital of Fudan University Ethics. Protocol 2011–040). Written informed consent for the collection of peripheral blood samples and subsequent analyses was obtained from the participating families, with the parents giving consent for themselves and on behalf of their minor children.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.