
Abstract
Synaptosomal-associated protein 29 (SNAP29) is a t-SNARE protein that is implicated in intracellular vesicle fusion. Mutations in the SNAP29 gene have been associated with cerebral dysgenesis, neuropathy, ichthyosis, and keratoderma syndrome (CEDNIK). In patients with 22q11.2 deletion syndrome, mutations in SNAP29 on the nondeleted chromosome are linked to similar ichthyotic and neurological phenotypes. Here, the authors report a patient with cerebral dysgenesis, neuropathy, ichthyosis, and keratoderma syndrome who presented with global developmental delay, polymicrogyria, dysgenesis of the corpus callosum, optic nerve dysplasia, gaze apraxia, and dysmorphic features. He has developed ichthyosis and palmoplantar keratoderma as he has grown. Exome sequencing identified a homozygous nonsense mutation in SNAP29 gene designated as c.85C>T (p.Arg29X). The authors compare the findings in the proband with previously reported cases. The previously unreported mutation in this patient and his phenotype add to the characterization of cerebral dysgenesis, neuropathy, ichthyosis, and keratoderma syndrome and the accumulating scientific evidence that implicates synaptic protein dysfunction in various neuroectodermal conditions.
Synaptosomal-associated protein 29 (SNAP29) is a 258-amino acid protein encoded by a 5-exon gene located on 22q11.21, primarily localized to intracellular membrane structures and involved in multiple membrane trafficking processes. 1 SNAP29 has 2 soluble N-ethylmaleimide-sensitive fusion protein attachment protein receptor (SNARE) domains that bind to multiple syntaxins and aid in vesicle fusion with target membranes during the processes of endocytic recycling and cell motility. 2 SNAP29 is also involved in autophagy through controlling autophagosome membrane fusion with the lysosome membrane and plays a role in ciliogenesis by regulating membrane fusions. 3,4
Loss-of-function mutations in SNAP29 have been shown to cause autosomal recessive cerebral dysgenesis, neuropathy, ichthyosis, and keratoderma syndrome (CEDNIK). 5-7 Moreover, patients with hemizygous deletions of 22q11.2 combined with mutations in the nondeleted SNAP29 display many of the features present in CEDNIK, confirming the loss-of-function mechanism. 8
Patients with CEDNIK typically present with ichthyosis (dry, scaly skin), keratoderma (marked thickening of skin, typically of the palms and soles), intellectual disability, microcephaly, facial dysmorphism, hypoplastic optic discs, sensorineural deafness, and cachexia, among other features. 5,9 Brain magnetic resonance imaging (MRI) of patients with cerebral dysgenesis, neuropathy, ichthyosis, and keratoderma shows corpus callosum dysgenesis and cortical dysplasia with pachygyria and polymicrogyria. 5-8
In line with these observations, animal studies have shown that both total SNAP29 knockout mice and keratinocyte-specific knockout mice have neonatal lethality, acanthosis, hyperkeratosis, abnormal keratinocyte differentiation, and increased proliferation. 2 Mutant mice have decreased deposition of lamellar body contents into the epidermis, as shown by their altered epidermal lipid distribution, as well as malformed lamellar bodies and an impaired epidermal barrier. They also have disturbed epidermal differentiation, as shown by their thickened stratum corneum. Similarly, Drosophila flies possessing a loss-of-function SNAP29 mutation have altered autophagy and endoplastic reticulum function that lead to impaired vesicle trafficking. 10 These findings help to explain the neurological and dermatological findings seen in cerebral dysgenesis, neuropathy, ichthyosis, and keratoderma syndrome.
Here, the authors report a patient with a novel homozygous pathogenic variant designated as c.85C>T (p.Arg29X) in SNAP29. The proband has a constellation of findings consistent with cerebral dysgenesis, neuropathy, ichthyosis, and keratoderma syndrome including neurological impairment, brain malformation, global developmental delay/intellectual disability, optic nerve dysplasia, facial dysmorphism, palmoplantar keratoderma, and ichthyosis.
Clinical Report
The patient is a 10-year-old Jordanian American male born at 35 weeks’ gestation via induced vaginal delivery to a gravida 3 para 3 23-year-old mother and 27-year-old father. His parents are first cousins and are both healthy. The patient has a 1-year-old sister, 6-year-old brother, and a 12-year-old sister; all are healthy with normal development. The patient has 2 paternal cousins with reportedly similar clinical features (Figure 1A). The authors have not examined these patients and their parents declined to participate in this study. The proband’s birth weight was 2.7 kg (75th percentile) and length was 50.8 cm (>97th percentile). Pregnancy was uncomplicated, and prenatal ultrasounds were normal. Neonatal course was complicated by breathing difficulties and sepsis, and the patient remained in the neonatal intensive care unit for 40 days. Between the ages of 3 months and 1 year, he was hospitalized 6 times for episodes of pneumonia.

Family tree and clinical features. A, Pedigree of the family. B, Frontal view of the proband. Note synophrys, hirsutism, low frontal hairline, bushy eyebrows, large nose, and deep-set eyes. C, Left foot heel with xerosis and hyperkeratotic plaques. D, Right hand with a hyperkeratotic plaque on hypothenar eminence. The findings in C and D are consistent with palmoplantar keratoderma.
The patient exhibited developmental delays, including deficits in gross and fine motor skills as well as sensory and visual tracking abnormalities. At 4 years, he was nonambulatory, but at 5 years, he started crawling, and at 7 years, he was able to use a stander. Currently, he is able to crawl, sit independently, and pull-to-stand. He uses a wheelchair, walker, gait trainer, and bilateral ankle–foot orthoses.
The patient did not speak his first word until 2 years. At present, he is still essentially nonverbal but has slightly more advanced receptive abilities, including communication by picture board and understanding of simple learning games. Although he has a limited appreciation of paralinguistic features (eg, eye contact, intonation, gesture, facial expression), he enjoys being with people and smiles appropriately. The patient is currently enrolled in third grade and receives physical and occupational and swimming therapy.
The patient had corrective surgery for strabismus at 2 years. A swallow study showed dysphagia and aspiration of thin liquids; therefore, he was fed only thickened liquids until 7 years. At approximately 8 years, the patient was diagnosed with kyphoscoliosis and coxa valga. Between the ages of 5 and 9 years, he had recurrent episodes of epistaxis.
His most recent examination at 10 years was notable for some dysmorphic facial features, including synophrys, hirsutism, low frontal hairline, bushy eyebrows, long eyelashes, deep-set eyes, large ears, and a bifid uvula (Figure 1B). His occipital frontal circumference was 52 cm (15th percentile), weight was 35.4 kg (56th percentile), height was 130 cm (7th percentile), and body mass index was 20.1 (88th percentile). Dermatologic examination showed palmoplantar keratoderma (Figure 1C and D) with focal areas of hyperkeratosis, eczematous dermatitis on the upper arms, and xerosis of the upper and lower extremities. Genitourinary examination demonstrated a Tanner III male with adherent prepuce and chordee. Neurologic examination revealed increased muscle tone in his lower extremities with significant spasticity in his hamstrings and adductors, decreased muscle tone in his right upper extremity, and truncal hypotonia. Deep tendon reflexes were 2/4 in upper extremities and 3/4 in lower limbs with no clonus. His muscle bulk and sensation were preserved. Musculoskeletal examination showed bilateral mild fifth finger and second and fourth toe clinodactyly. Ophthalmological assessment revealed high bilateral astigmatism and optic nerve dysplasia with reduced optic disc diameter.
Brain MRI at 3 years of age showed dysgenesis of the corpus callosum, bilateral frontoparietal polymicrogyria with abnormal cortical folding, diffuse white matter T2 hyperintensity predominately in the centrum semi ovale (Figure 2A-C), and hypoplastic intraconal optic nerves (Figure 2D and E).

Brain magnetic resonance imaging (MRI) findings in the proband. A, Sagittal T1-weighted magnetic resonance (MR) sequence demonstrates dysgenesis of the corpus callosum. Specifically, the splenium of the corpus callosum is hypoplastic (hollow arrow). B, Axial T2-weighted fluid-attenuated inversion recovery (FLAIR) MR sequence reveals marked hyperintensity in the white matter of the centrum semi ovale (yellow arrows). C, Axial T2-weighted turbo spin echo (TSE) MR sequence is notable for bilateral frontoparietal polymicrogyria and abnormal cortical folding (white arrows). D, The intraconal optic nerves are hypoplastic (black circles). E, The optic chiasm (down arrow), the intracranial optic nerves, and optic tracts are normal.
Materials and Methods
The institutional review board of Washington University School of Medicine approved our genetic testing protocol. After informed consent was obtained from the patient’s parents, DNA was isolated from the proband’s and his parents’ blood samples using standard methods. Exome sequencing was performed using a trio design. Exon targets were isolated by capture using the Agilent Clinical Research Exome kit (Agilent Technologies, Santa Clara, California) and sequenced on the Illumina HiSeq 2000 system with 100 base pair paired-end reads. Sequence data were assembled using human genome build GRCh37/UCSC hg19. The mean depth of coverage was 139x, with ≥10x coverage across 97.2% of the exome. Variants were evaluated using Xome Analyzer software (GeneDx, Gaithersburg, Maryland). Kinship coefficients from exome sequencing data were calculated using kinship-based inference to control for misspecified relationships and reported consanguinity for relations as distant as second cousins. Capillary sequencing confirmed SNAP29 variant c.85C>T (p.Arg29X).
Molecular Characterization
Chromosomal microarray analysis showed multiple regions of homozygosity/segmental uniparental disomy identified on chromosomes 1, 4, 19, 21, and 22. Exome sequencing identified a homozygous pathogenic change designated as c.85C>T (p.Arg29X) in the SNAP29 gene. This variant has not been observed in approximately 6500 individuals who were sequenced as part of the National Heart, Lung, and Blood Institute exome sequence project or in ExAc database (http://exac.broadinstitute.org/). 11,12 Both parents are heterozygous for this mutation.
Discussion
The authors describe a patient with a homozygous nonsense mutation in SNAP29 associated with cerebral dysgenesis, neuropathy, ichthyosis, and keratoderma syndrome. This variant is predicted to cause loss of normal protein function either through protein truncation or nonsense-mediated messenger RNA decay. In our proband, the mutation in the SNAP29 gene is associated with global developmental delay, polymicrogyria, optic nerve dysplasia, gaze apraxia, and scoliosis. Physical abnormalities also include dysmorphic features such as synophrys, bushy eyebrows, long eyelashes, deep-set eyes, large ears, fifth finger clinodactyly bilaterally, and broad first toes bilaterally. Our patient shares many features with the patients previously reported with SNAP29 mutations (Table 1). 5-7 The neurocutaneous symptoms in cerebral dysgenesis, neuropathy, ichthyosis, and keratoderma syndrome reflect the common embryonic origin of the epidermis and neural tissues.
Clinical and Molecular Characteristics of Patients With Cerebral Dysgenesis, Neuropathy, Ichthyosis, and Keratoderma Syndrome.
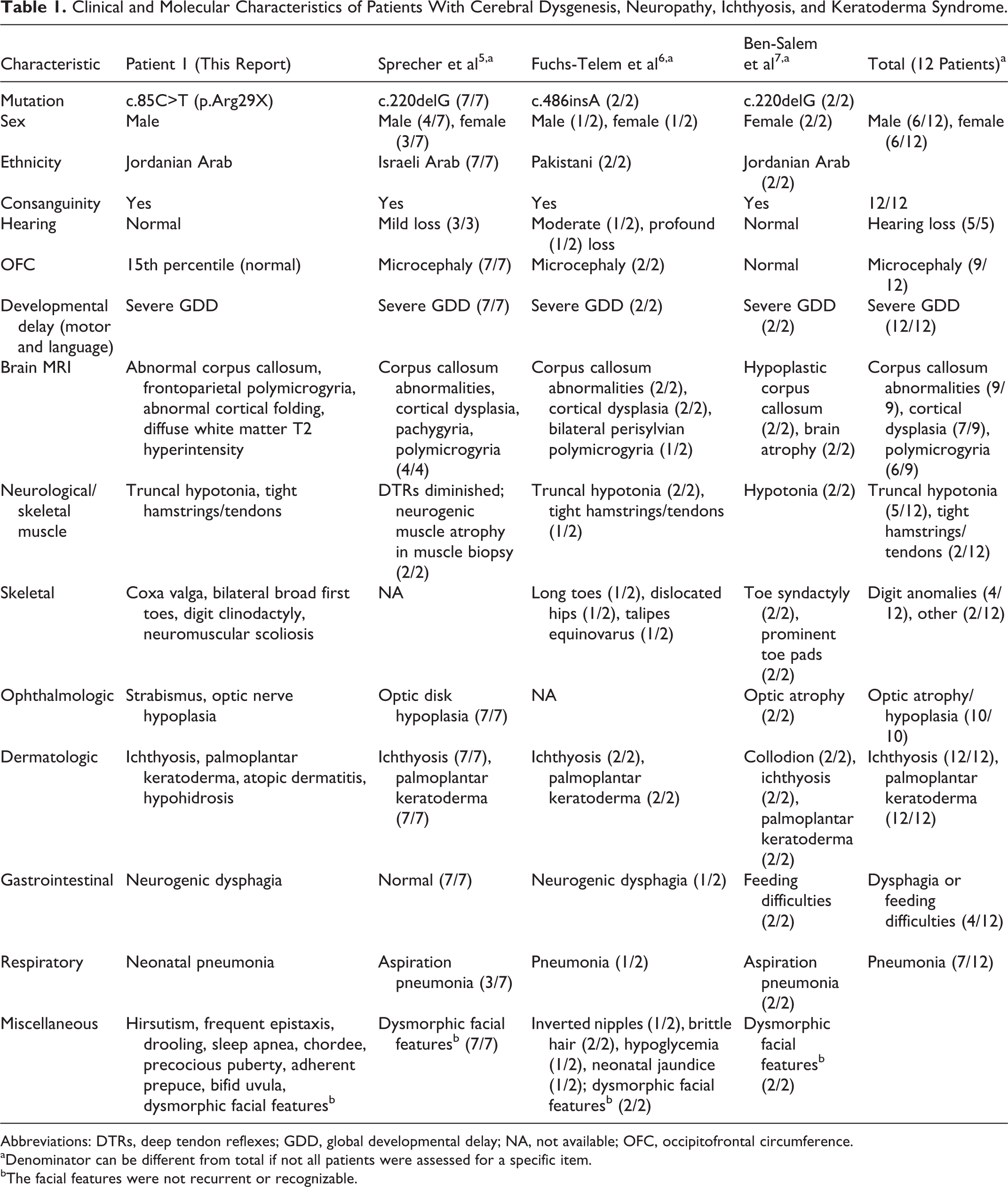
Abbreviations: DTRs, deep tendon reflexes; GDD, global developmental delay; NA, not available; OFC, occipitofrontal circumference.
aDenominator can be different from total if not all patients were assessed for a specific item.
bThe facial features were not recurrent or recognizable.
All previously reported patients with cerebral dysgenesis, neuropathy, ichthyosis, and keratoderma syndrome, including the patient in this report, presented with severe global developmental delay and intellectual disability, which are probably related to SNAP29’s function as a negative modulator of neurotransmitter release. 13 Specifically, SNAP29 has been shown to play an important role in the recycling of clathrin-dependent and clathrin-independent ligands in fibroblasts. Fibroblasts of human patients with cerebral dysgenesis, neuropathy, ichthyosis, and keratoderma syndrome show impaired endocytic recycling due to reduced expression of transferrin and B1-integrin and altered destruction of focal adhesion complexes. 9 A decreased amount of SNAP29 may slow the recycling of vesicle fusion proteins, leading to increased neurotransmitter release and eventually the neurological symptoms seen in patients with cerebral dysgenesis, neuropathy, ichthyosis, and keratoderma syndrome.
Furthermore, all patients with cerebral dysgenesis, neuropathy, ichthyosis, and keratoderma syndrome have developed palmoplantar keratoderma and generalized ichthyosis. These dermatological manifestations demonstrate SNAP29’s integral role in proper cell motility and migration, as well as cell spreading and wound healing. 9 During normal development, the epidermis is formed when keratinocytes migrate upward from the basal cell layer and transform into spinous, granular, and cornified cells. 5 The epidermal barrier of the skin is made of a cornified cell envelope, extracellular lipid layers, and keratin filaments. Lamellar bodies, which are secretory organelles found in the Golgi complex of keratinocytes, are found in granular cells and typically release glucosylceramides, kallikrein-5, and kallikrein-7 into the extracellular space beneath the stratum corneum to aid the cells in desquamation and reinforce the epidermal barrier by forming an lipid membrane impermeable to water. SNAP29 is necessary for epidermal differentiation and barrier formation because it allows lamellar bodies to secrete their contents into the extracellular space. In cerebral dysgenesis, neuropathy, ichthyosis, and keratoderma syndrome, lamellar granules are abnormally retained in the cornified layer, making them unable to secrete their contents into the extracellular space. 5 This prevents the cells separating from each other and the epidermal layer thickens considerably as a response to this impaired barrier formation process.
Several lines of evidence support the pathogenicity of the SNAP29 mutations and their association with the previously mentioned phenotypes. A previous report documented the 220delG SNAP29 mutation in 2 unrelated, consanguineous Arab families with cerebral dysgenesis, neuropathy, ichthyosis, and keratoderma syndrome features. This mutation in SNAP29 was expected to cause loss of the protein function. 5 All 7 affected children from these 2 families had failure to thrive, roving eye movements, and poor head and trunk control in the first 4 months of life, similar to our patient. They also had progressive microcephaly, facial dysmorphisms including elongated faces, antimongoloid eye slant, slight hypertelorism, and a flat broad nasal root, as well as palmoplantar keratoderma and ichthyosis. By 15 months of age, all patients demonstrated severe intellectual disability and were unable to sit or walk without assistance. Brain MRIs in 4 patients showed corpus callosum abnormalities, as well as cortical dysplasia with pachygyria and polymicrogyria, as seen in our patient as well. Three of the patients died of aspiration pneumonia between 5 and 12 years of age. A subsequent study further elucidated the pathogenesis of cerebral dysgenesis, neuropathy, ichthyosis, and keratoderma syndrome by identifying a novel homozygous insertion in SNAP29 at complementary DNA position 486, which caused a frameshift mutation and a truncated protein due to premature termination of translation 5 amino acids downstream to the mutation. 6 Transfection of wild-type and complementary DNA constructs with the same insertion in SNAP29 seen in these patients resulted in the production of truncated and mislocated protein. An additional report described 2 female siblings with the 220delG mutation causing cerebral dysgenesis, neuropathy, ichthyosis, and keratoderma without polymicrogyria. 7 Finally, mutations in SNAP29 have been shown to unmask an autosomal recessive condition in patients with 22q11.2 deletion syndromes similar to cerebral dysgenesis, neuropathy, ichthyosis, and keratoderma. 8 Patients who have both hemizygous deletions of 22q11.2 and mutations in SNAP29 exhibit neurological and cutaneous features of cerebral dysgenesis, neuropathy, ichthyosis, and keratoderma syndrome. The neurological phenotypes and neuroradiological findings are more severe than in patients with the deletion only.
In summary, the authors report a fourth family with cerebral dysgenesis, neuropathy, ichthyosis, and keratoderma syndrome caused by a novel homozygous nonsense SNAP29 mutation. SNAP29 haploinsufficiency causes a unique neurodevelopmental phenotype encompassing global developmental delay, polymicrogyria, cerebral dysgenesis, optic nerve dysplasia/hypoplasia, some dysmorphic features, ichthyosis, and keratoderma. Additional reports are needed to better define the molecular and phenotypic spectrum associated with abnormal intracellular transport processes.
Footnotes
Acknowledgments
The authors thank the family of the patient for participating in this study.
Author Contributions
TH contributed to conception and design, contributed to acquisition and analysis, drafted the manuscript, and gave final approval. CCC contributed to acquisition, analysis, and interpretation, critically revised the manuscript, and gave final approval. KGM contributed to acquisition, analysis, and interpretation, critically revised the manuscript, and gave final approval. EF contributed to acquisition and interpretation, critically revised the manuscript, and gave final approval. RCM contributed to acquisition, analysis, and interpretation, critically revised the manuscript, and gave final approval. ARP contributed to acquisition, critically revised the manuscript, and gave final approval. MS contributed to conception and design, contributed to acquisition, analysis, and interpretation, critically revised the manuscript, and gave final approval. TH, CC, KM, EF, RCM, ARP, and MS agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Ethical Approval
The institutional review board of Washington University School of Medicine approved our genetic testing protocol after informed consent was obtained from the patient’s parents.