
Abstract
1q43q44 microdeletion syndrome is characterized by intellectual disability/global developmental delay, epilepsy, dysmorphic facies, stereotypic movement, language delay, recurrent infections, dental anomalies, and hand and foot anomalies. Microcephaly and corpus callosum dysplasia are present in some cases depending on gene content. 3q29 microduplication syndrome is characterized by intellectual disability, language delay, microcephaly, and dental anomalies. We report the first case with 4 de novo copy number variations with clinical features which overlap 1q43q44 microdeletion and 3q29 microduplication syndromes. Our case presented with global developmental delay, epilepsy, recurrent infections, stereotypic movements, speech delay, microcephaly, facial dysmorphism, bilateral clinodactyly, and small puffy feet with metatarsus varus; however, she had no corpus callosum dysplasia. Our case highlights the role of multiple copy number variations in the occurrence of a certain phenotype. Moreover, it supports the theory that the loss of HNRNPU gene function cannot explain the occurrence of microcephaly and abnormalities of the corpus callosum in 1q43q44 microdeletion syndrome.
Keywords
1q43q44 microdeletion syndrome (Online Mendelian Inheritance in Man 612337) is characterized by moderate to severe intellectual disability/global developmental delay, epilepsy, dysmorphic facies, hypotonia, abnormal behaviors including stereotypic movement, limited or no expressive speech, recurrent infections, dental anomalies, hand and foot anomalies, congenital heart defects, and gastroesophageal and urogenital abnormalities. 1 -4 Some patients present with microcephaly and abnormalities of the corpus callosum depending on gene content. HNRNPU gene has been proposed to be accountable for intellectual disability/global developmental delay, epilepsy, dysmorphic facies, abnormal behaviors including stereotypic movement, limited or no expressive speech, recurrent infections, dental anomalies, and hand and foot anomalies. 5 However, ZBTB18 gene has been proposed to be accountable for microcephaly and abnormalities of the corpus callosum. 6 3q29 microduplication syndrome (Online Mendelian Inheritance in Man 611936) is characterized by intellectual disability/global developmental delay, language delay, microcephaly, dental anomalies, cleft palate, palpebral fissure anomalies, congenital heart diseases, and skeletal malformations. 7,8
We report the first rare case with 4 de novo copy number variations with clinical features which overlap 1q43q44 microdeletion and 3q29 microduplication syndromes.
Case Report
The patient was 4 years old girl, born at 37 weeks gestation age through cesarean section and weighed 2850 kilograms. She was born in a nonconsanguineous family with nonremarkable family history. She developed severe global developmental delay from the age of 3 months which was accompanied by recurrent respiratory tract infections, abnormal behavior, and stereotypic movement, that is, shaking and clapping hands. On examination, she had microcephaly (-3SD), normal length, round face, short nose with broad nasal tip, ears with thin helices, hypertelorism, long and smooth philtrum, wide spaced teeth, thin vermilion borders, micrognathia, strawberry naevus in the trunk, bilateral clinodactyly, and small puffy feet with metatarsus varus (Figure 1). Ophthalmologic and audiologic examination revealed no abnormalities. She developed focal epilepsy, which was fever related from the age of 10 months and it was not controlled by valporic acid, topiramate, and levetiracetam. She was also noticed to have speech delay, and dysmorphic features became more prominent as she was growing up. Examination of the heart and kidney revealed no abnormalities, however, brain magnetic resonance imaging revealed mild brain atrophy. Electroencephalography revealed multifocal discharge arising from both hemisphere characterized by spike-wave complex. Metabolic tests were negative. Low-depth whole-genome sequencing was performed whereby she was found to have 4 de novo copy number variations: 1q44q44 deletion (4.6 Mb) spanning HNRNPU, HNRNPU-AS1, COX20, and NLRP3 genes, 3q29-q29 duplication (880 Kb) spanning CEP19, PCYT1A, RNF168, TCTEX1D2, and TFRC genes, 8p11.21-p11.1 duplication (100 Kb) spanning part of HGSNAT gene, and 11p15.4-p15.4 duplication (190 Kb) spanning PGAP2 and STIM1 genes. Low-depth whole-genome sequencing was also performed for both parents in order to identify whether they are de novo or inherited. The 4 copy number variations were not found in either of the parent, hence they were considered as de novo. The classification of the identified copy number variations was done according to the American College of Medical Genetics (ACMG) guideline for interpretation of postnatal copy number variations. 9 Two de novo copy number variations which represent 2 separate syndromes, that is, 1q43q44 microdeletion and 3q29 microduplication syndromes, were classified as pathogenic, while the other 2 de novo duplications were classified as variant of unknown significance (Table 1).
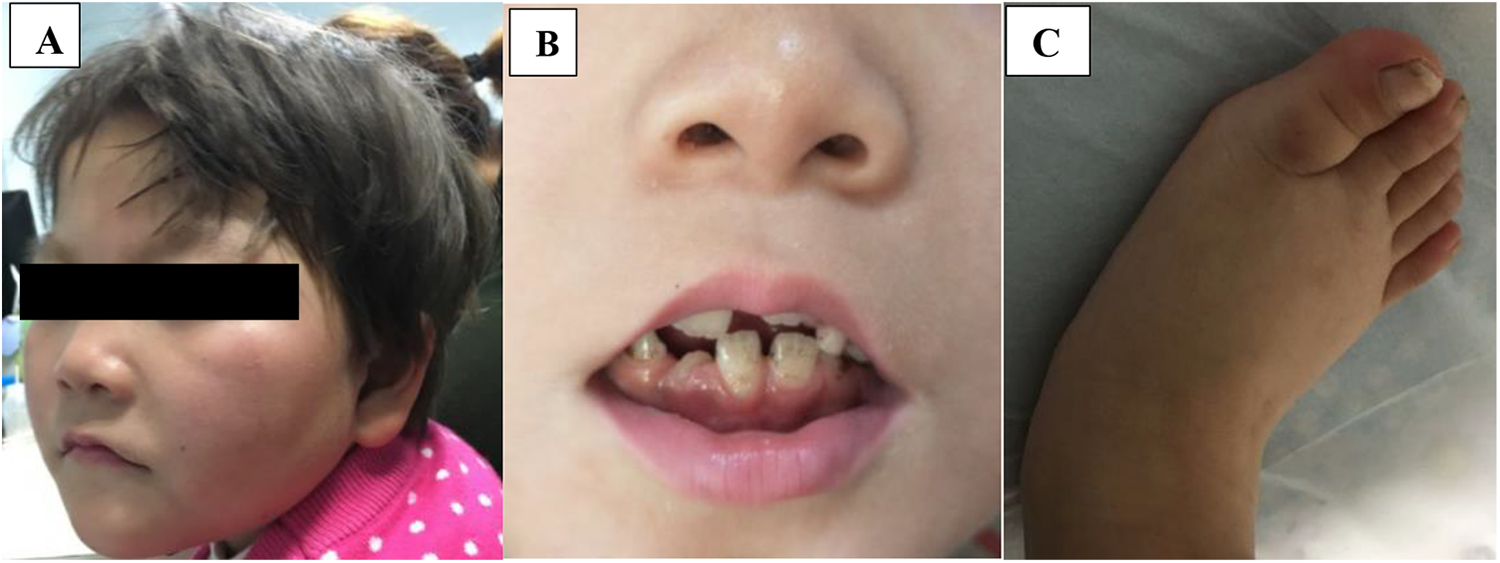
A, Microcephaly, round face, short nose with broad nasal tip, ears with thin helices, hypertelorism, and micrognathia. B, Long and smooth philtrum, wide-spaced teeth, and thin vermilion borders. C, Clinodactyly and small puffy foot with metatarsus varus.
The Identified 4 CNVs Together With Their Candidate Genes.

Abbreviations: ACMG, American College of Medical Genetics; AD, autosomal dominant; AR, autosomal recessive; CNV, copy number variation; Del, deletion; Dup, duplication; NA, not applicable; NCBI, National Center for Biotechnology Information; OMIM, Online Mendelian Inheritance in Man.
Discussion
We report the first rare case with 4 de novo copy number variations with clinical features which overlap 1q43q44 microdeletion and 3q29 microduplication syndromes. This case highlights the role of multiple copy number variations in the occurrence of a certain phenotype. Our case was identified to have de novo 1q44q44 deletion (4.6 Mb) spanning HNRNPU, HNRNPU-AS1, COX20, and NLRP3 genes, de novo 3q29-q29 duplication (880 Kb) spanning CEP19, PCYT1A, RNF168, TCTEX1D2, and TFRC genes, de novo 8p11.21-p11.1 duplication (100 Kb) spanning part of HGSNAT gene, and de novo 11p15.4-p15.4 duplication (190 Kb) spanning PGAP2 and STIM1 genes. 1q44q44 deletion and 3q29-q29 duplication represent 1q43q44 microdeletion syndrome and 3q29 microduplication syndrome, respectively.
1q43q44 microdeletion syndrome is characterized by moderate to severe intellectual disability/global developmental delay, epilepsy, dysmorphic facies, hypotonia, abnormal behaviors including stereotypic movement, limited or no expressive speech, recurrent infections, dental anomalies, hand and foot anomalies, congenital heart defects, and gastroesophageal and urogenital abnormalities. 1 -4 Some patients present with microcephaly and abnormalities of the corpus callosum depending on gene content. HNRNPU, ZBTB18, and AKT3 have been proposed as possible candidate genes for this syndrome. HNRNPU gene could be accountable for intellectual disability/global developmental delay, epilepsy, and dysmorphic features, 2,5 while ZBTB18 gene and AKT3 could explain abnormalities of the corpus callosum and microcephaly. 2,6 Thierry et al studied 11 unrelated patients with 1q44 microdeletion in which all patients presented with moderate to severe intellectual disability, epilepsy, and nonspecific craniofacial anomalies. 2 Two out of 11 cases had microcephaly and their copy number variations spanned AKT3 gene. 2 The other 9 cases without microcephaly had copy number variations which spanned HNRNPU gene (Figure 2). Therefore, they suggested HNRNPU gene as a good candidate for intellectual disability/global developmental delay and epilepsy. 2 3q29 microduplication syndrome is characterized by intellectual disability/global developmental delay, language delay, dental anomalies, and microcephaly. 7,8 The genes responsible for the phenotype are not clear currently; however, the copy number variation ranges from 1.6 to 2.3 Mb, spanning from TFRC to BDH1 genes.

A map of the deletions in chromosomal band 1q44. Comparison of our case with 9 reported cases that presented with intellectual disability (ID)/global developmental delay (GDD), epilepsy (EP), and dysmorphic features without microcephaly. A proposed critical region for ID, EP, and dysmorphic features encompasses HNRNPU gene.
Our case presented with severe global developmental delay, focal epilepsy, recurrent respiratory tract infections, abnormal behavior, stereotypic movements, speech delay, microcephaly, round face, short nose with broad nasal tip, ears with thin helices, hypertelorism, long and smooth philtrum, wide spaced teeth, thin vermilion borders, micrognathia, strawberry naevus in the trunk, bilateral clinodactyly and small puffy feet with metatarsus varus, and mild brain atrophy. Most of the clinical features of our case overlap with those of the reported cases with 1q43q44 microdeletion syndrome. 1 -4 1q44q44 deletion which was identified in our case spans HNRNPU gene which explains majority of the clinical features except microcephaly (Figure 2). HNRNPU encodes for a heterogeneous nuclear ribonucleoprotein and is expressed ubiquitously in humans and mice and has been proposed to contribute to brain development. Nevertheless, 3q29-q29 duplication (880 Kb) spanning CEP19, PCYT1A, RNF168, TCTEX1D2, and TFRC could also explain global developmental delay, dental anomalies, speech delay, and microcephaly. Importantly, RNF168 gene mutation has been reported to associate with microcephaly. 10 Ballif et al, 8 Lisi et al, 11 and Goobie et al 12 reported 3 cases diagnosed with 3q29-q29 microduplication syndrome which presented with microcephaly. Their copy number variations overlap with that of our case since they encompass RNF168 PCYT1A, TCTEX102, and CEP19 genes (Figure 3). Consequently, we think the phenotype of our case was contributed by these 2 copy number variations: deletion at 1q44q44 and duplication at 3q29-q29. We also think RNF168 gene can explain microcephaly in 3q29-q29 microduplication syndrome. However, more similar cases are needed for comparison.

A map of the duplications in chromosomal band 3q29. Comparison of our case with 3 reported cases of 3q29-q29 microduplication syndrome. A proposed critical region contains the 4 genes: PCYT1A, TCTEX102, RNF168, and CEP19.
Some of the reported cases of 1q43q44 microdeletion syndrome had corpus callosum dysplasia which was absent in our case. This could be due to the fact that our case’s copy number variation does not span ZBTB18 gene which has been proposed to be accountable for microcephaly and abnormalities of the corpus callosum. 6,13 ZBTB18 (zinc finger and BTB domain-containing protein 18) encodes a transcriptional repressor that plays an important role in cortical and cerebellar development. 14 Animal studies have revealed that Zbtb18 deficiency results in abnormal growth, differentiation, and maturation of cortical and cerebellar neurons, leading to a phenotype of microcephaly, cerebellar vermis hypoplasia, and corpus callosum dysplasia. 15 Moreover, AKT3 gene, which has also been proposed to associate with microcephaly, is not within our case’s copy number variation. Therefore, microcephaly in our case could be part of 3q29 microduplication syndrome and not 1q43q44 microdeletion syndrome. Thus, our case adds evidence that both the microcephaly and abnormalities of the corpus callosum phenotypes may not be explained by a loss of HNRNPU function but may be due to a loss of function of AKT3 and ZBTB18 genes.
The pathogenic role of de novo duplications at 8p11.21-p11.1 and 11p15.4-p15.4 is unknown, hence they have been classified as variant of unknown significance. These 2 copy number variations span genes which are associated with autosomal recessive disorders so it is difficult to understand their contribution to the patient’s phenotype. Additionally, in DECIPHER (Database of Chromosomal Imbalance and Phenotype in Humans using Ensemble Resources (https://decipher.sanger.ac.uk) and DGV (Database of Genomic Variants, http://dgv.tcag.ca/gb2/gbrowse/dgv) databases, there are few copy number variations with duplication in these regions that associate with a wide range of phenotypes, while others have no phenotype. As a result, they have discordant definitions ranging from likely benign to variant of unknown significance. Therefore, despite the fact that they are de novo aberrations of small size, it is difficult to understand their contribution in our case’s phenotype.
In conclusion, we have reported the first rare case with 4 de novo copy number variations in which 2 of them represent 2 different syndromes with overlapping clinical features: 1q43q44 microdeletion syndrome and 3q29 microduplication syndrome. Our case highlights the role of multiple copy number variations in the occurrence of a certain phenotype. Moreover, it supports the theory that a loss of HNRNPU function cannot explain the occurrence of microcephaly and abnormalities of the corpus callosum in 1q43q44 microdeletion syndrome.
Footnotes
Authors’ Note
The authors confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Acknowledgments
The authors thank the patient and her parents for their cooperation and participation in this report.
Author’s Contribution
MK contributed to conception and design; contributed to acquisition, analysis, and interpretation; drafted the manuscript; critically revised the manuscript; gave final approval. JP contributed to conception and design; contributed to acquisition, analysis, and interpretation; drafted the manuscript; critically revised the manuscript; and gave final approval. LY contributed to conception and design; contributed to acquisition, analysis, and interpretation; drafted the manuscript; critically revised the manuscript, and gave final approval. HD contributed to conception and design; contributed to acquisition, analysis, and interpretation; drafted the manuscript; critically revised the manuscript, and gave final approval. YT contributed to conception and design; contributed to acquisition, analysis, and interpretation; drafted the manuscript; critically revised the manuscript, and gave final approval. FY contributed to conception and design; contributed to acquisition, analysis, and interpretation; drafted the manuscript; critically revised the manuscript, and gave final approval. All authors agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Ethical Approval
This case report was reviewed and approved by the ethic committee of the Xiangya Hospital of Central South University.