
Abstract
We report a novel PSEN1 likely pathogenic variant associated with spastic paraparesis (SP) preceding cognitive decline. A 41-year-old man with a two-year history of SP followed by cognitive impairment one year later, was found to carry a heterozygous PSEN1 c.668A > T (p.Gln223Leu) variant. Alzheimer's disease (AD) was confirmed with a combination of low CSF Aβ42 levels and amyloid positivity on [18F]Florbetaben PET. This case represents the third reported association between substitutions at PSEN1 codon 223 and AD with SP, emphasizing the need to consider PSEN1 mutations in cases of SP preceding cognitive decline.
Introduction
Alzheimer's disease (AD) is the leading cause of dementia and is neuropathologically characterized by the accumulation of amyloid plaques and neurofibrillary tangles. 1 Early-onset Alzheimer's disease (EOAD) is defined by symptom onset before the age of 65 and accounts for approximately 1–5% of all AD cases.2,3 A significant proportion of EOAD patients have a family history of the disease, with 10–15% showing an autosomal dominant inheritance pattern. Pathogenic variants in APP, PSEN1, and PSEN2 account for the majority of known genetic causes of autosomal dominant EOAD. 2
Among these genes, PSEN1 encodes a subunit of the γ-secretase complex, which plays a critical role in the proteolytic cleavage of AβPP. 4 Mutations in PSEN1 reduce γ-secretase activity and Aβ42 production 5 with lower γ-secretase activity in this genetic form of AD associated with lower Aβ42 levels, worse cognitive function, and acceleration of brain atrophy. 6 As the most frequently implicated gene in autosomal dominant AD, PSEN1 has been associated with over 300 mutations, each presenting with distinct clinical phenotypes, according to the Alzforum mutation database (accessed August 2025). In addition to AD, PSEN1 mutations have been reported in frontotemporal dementia, Lewy body disease, and spastic paraparesis (SP). 7
AD typically presents with an insidious onset of memory impairment, followed by progressive involvement of other cognitive domains. SP is characterized by progressive lower-limb spasticity and gait disturbance. Although SP is rarely observed in sporadic AD, it has been reported in approximately 10% of patients with autosomal dominant EOAD. 8
Here, we report a case of EOAD in a Korean patient carrying a novel PSEN1 c.668A > T mutation, who initially presented with SP preceding cognitive decline.
Case presentation
A 41-year-old man presented to our clinic with a two-year history of gait disturbance followed by cognitive decline one year later. His initial symptom was subjective weakness in the lower limbs. Approximately one year before his visit, he developed dysarthria and was advised to resign from his job due to slow work processing. At the time of evaluation, the patient and caregiver reported no difficulties in basic activities of daily living. Standardized assessments, however, demonstrated mild functional impairment (Barthel Index, 18/20; K-Vineland-II Daily Living Skills, 6th percentile; composite score, 0.5th percentile). He had 14 years of education, was right-handed, and was employed as an electrician. He denied medical illnesses or regular medication use. Routine laboratory tests were unremarkable, and no vascular or psychosocial risk factors for cognitive impairment were identified. His mother developed cognitive decline, SP, and parkinsonism, with symptom onset at 55 years and death at 65 years (Figure 1). Brain MRI of his mother showed numerous subcortical microbleeds and mild white-matter hyperintensities, and dopamine transporter PET demonstrated only a subtle reduction of tracer uptake in the posterior left striatum. Although dementia with Lewy bodies was considered, the overall findings favored AD with possible overlapping pathology. His maternal grandfather and uncle passed away in their 50 s, though their neurological status or genetic testing results were unknown.

Pedigree of the patient. The proband (III-1, arrow) presented at 41 years with spastic paraparesis followed by cognitive decline. His mother (II-6, gray shading) had a 10-year history of similar symptoms and died at 65 years but genetic or biomarker confirmation was unavailable. His maternal grandfather (I-3) and maternal uncle (II-7) died in their 50 s though their neurological status is unknown. Solid symbols denote clinically affected individuals with genetic confirmation; gray shading indicates clinically affected individuals without genetic confirmation.
Neurological examination revealed dysarthria, hyperreflexia in the bilateral upper and lower limbs, and a spastic gait. However, he did not have muscle atrophy or fasciculations. During the examination, he demonstrated impairments in time orientation and right-left orientation.
The Korean Mini-Mental State Examination 2nd edition score was 16, and Clinical Dementia Rating was 1.0, with a sum of boxes score of 4.5 (memory 1, orientation 1, judgment and problem-solving 1, community affairs 1, home and hobbies 0.5, personal care 0). The Korean-Wechsler Adult Intelligence Scale 4th edition showed global cognitive impairment across all domains, with a full-scale IQ of 51 (1st percentile). Subscores included a verbal comprehension index of 66 (1st percentile), perceptual reasoning index of 59 (0.3rd percentile), working memory index of 58 (0.3rd percentile), and processing speed index of 50 (0.1st percentile). 9 Memory function was severely impaired, with composite scores for delayed recall and recognition at <0.1 percentile and 2nd percentile, respectively. 10 He had particular difficulties in sentence repetition and finger naming with preserved language comprehension. Neuropsychiatric assessment using the Neuropsychiatric Inventory revealed mild apathy (score 2) and irritability (score 4).
APOE genotype was ε2/ε3. CSF analysis showed a markedly reduced Aβ42 level of 269 pg/mL (negative if >1030 pg/mL) and borderline phosphorylated tau 181 (p-tau181) level of 27 pg/mL (negative if ≤ 27 pg/mL). [18F]Florbetaben PET demonstrated regional cortical tracer uptake grade 3 amyloid plaque deposition in bilateral parietal cortices, bilateral precuneus and posterior cingulate cortices, bilateral frontal cortices, and bilateral lateral temporal cortices (Figure 2, Supplemental Figure 1). Brain MRI was performed using a 3.0-T Siemens Magnetom Vida scanner, including axial T2-weighted, FLAIR, and GRE sequences, 3D MPR imaging with and without contrast enhancement, and diffusion-weighted imaging. Brain MRI revealed nodular hyperintensities in the subcortical white matter of the left frontoparietal region and symmetric atrophy most prominent in parietal lobe, without evidence of cerebral microbleeds (CMB) (Figure 2). No abnormalities were detected in the whole-spine MRI.

Neuroimaging of the patient. Brain MRI and [18F]Florbetaben PET images of the patient. (A) Coronal T1-weighted imaging (T1WI) showing mild bilateral hippocampal and moderate bilateral temporal lobe atrophy. (B) Sagittal T1WI revealing parieto-occipital atrophy. (C) Fluid-attenuated inversion recovery (FLAIR) image demonstrating nodular hyperintensities in the subcortical white matter of the left frontoparietal lobe, without evidence of cerebral microbleeds. (D–F) [18F]Florbetaben PET scans showing amyloid deposition in the bilateral precuneus and posterior cingulate cortex (D), bilateral frontal cortices (E), and bilateral lateral temporal cortices (F). Arrows indicate regions of increased amyloid deposition in the cortex.
Based on these findings, the patient was diagnosed with familial EOAD, 11 warranting genetic analysis. Next-generation sequencing using a targeted paraplegia panel, incorporating massively parallel sequencing and copy number analysis, identified a heterozygous missense variant in PSEN1 (NM_000021.4:c.668A > T, p.Gln223Leu) (Supplemental Table 1), classified as ‘likely pathogenic’ based on ACMG criteria. The variant was supported by 96 of 204 sequencing reads (variant allele frequency, 47.1%), consistent with a heterozygous state, and showed balanced representation across both forward and reverse strands on Integrative Genomics Viewer (Figure 3).
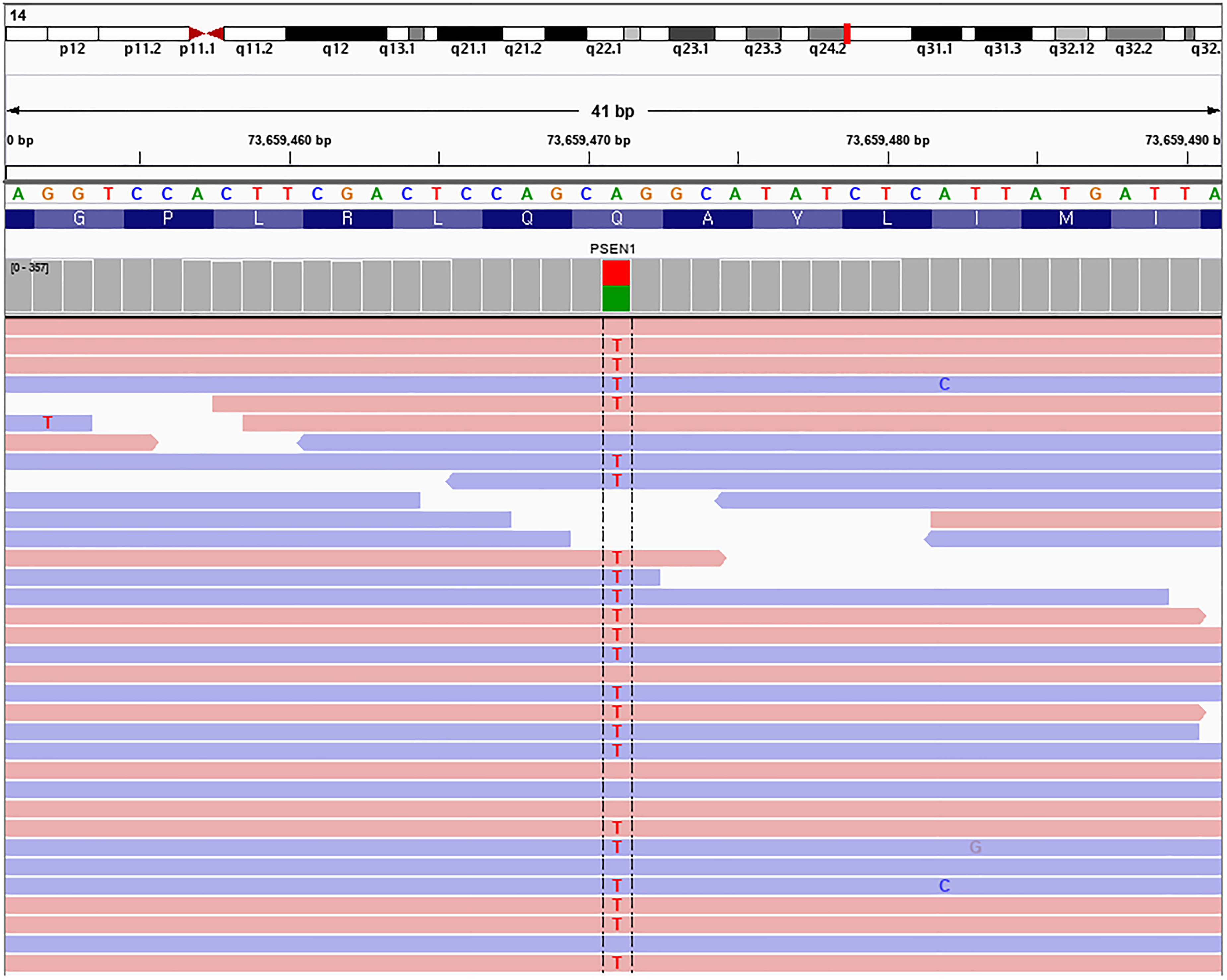
Integrative genomics viewer (IGV) visualization of the PSEN1 variant (NM_000021.4:c.668A > T, p.Gln223Leu). The heterozygous missense variant is supported by 96 of 204 sequencing reads (variant allele frequency, 47.1%), with balanced representation across forward and reverse strands and no evidence of strand bias.
Donepezil 5 mg was prescribed and titrated to 10 mg and baclofen 30 mg per day and clonazepam 1 mg per day were also prescribed. His father reported that attention improved but without changes to the spastic gait. Approximately 10 months later, he became unable to walk.
Discussion
We report a novel PSEN1 variant, c.668A > T (p.Gln223Leu) associated with EOAD, initially presented with SP. This case contributes to the expanding clinical and genetic spectrum of PSEN1-associated phenotypes and highlights the diagnostic challenges of atypical EOAD presentations, particularly those with SP preceding cognitive decline.
PSEN1, a subunit of the γ-secretase complex, is a transmembrane protein which plays a crucial role in γ-secretase cleavage and AβPP fragment transport. Impairment of PSEN1 function can alter the amount and length of amyloid peptides, contributing to AD pathogenesis. 3 PSEN1 is also involved in multiple cellular pathways, contributing to the phenotypic heterogeneity. 7
The pathogenicity classification was determined according to ACMG guidelines. p.Gln223Leu maps to the fifth transmembrane helix (TM5) of PSEN1 within the TM2–TM5 four-helix bundle of γ-secretase, a well-established functional domain that constitutes a mutational hot spot in which PSEN1 substitutions cluster and pathogenic/likely pathogenic classifications are enriched relative to extramembranous regions.12,13 The p.Gln223Leu mutation is absent from gnomAD v4.1.0 and has a high conservation score of 9.163 according to PhyloP100way. Multiple in silico prediction tools, including SIFT, PolyPhen-2, Mutationtaster, indicate pathogenic impact, with a CADD score of 26. The variant satisfies three moderate-level evidence criteria—PM1 (mutational hot spot), PM2 (absent from 1000Genome, ExAC, KRGDB), and PM5 (novel missense change at a known pathogenic residue)—as well as one supporting criteria (PP3: computational evidence), thereby warranting classification as likely pathogenic. 14 Two previously reported cases involving p.Gln223Arg substitution at the same codon also presented with EOAD and prominent SP.15,16 Both cases involved male patients in their 30 s–one presented with SP followed by cognitive decline, while the other initially exhibited cognitive impairment. Our patient carried a p.Gln223Leu substitution at the identical amino acid residue and presented with overlapping clinical presentations.
EOAD with SP is a rare but increasingly recognized phenotype, accounting for approximately 14% of known PSEN1 pathogenic variants. 17 Although most EOAD-SP cases are linked to PSEN1 mutations, rare exceptions involving PSEN2 (e.g., M239 V) 18 and APP (e.g., V717I) 19 have been reported. Cotton wool plaques have been reported but are not specific to EOAD with SP. 20 Furthermore, these cases exhibit prominent microgliosis and amyloid deposition in motor cortices, which, while severe, appear insufficient to fully explain the extent of neuronal loss observed. 21
Notably, fewer than 10% of reported PSEN1 variants present with SP preceding cognitive decline. 17 The studies of patients clinically suspected to have hereditary spastic paraplegia (HSP) have found that approximately 1% harbor pathogenic PSEN1 variants,17,22 most of whom develop cognitive symptoms subsequently. As a result, diagnosing autosomal dominant AD is particularly challenging when patients initially present with isolated SP. In the present case, the patient underwent extensive evaluation for gait disturbance, including workup for ataxia, without an identified etiology. It was only after the emergence of cognitive symptoms that a diagnosis of autosomal dominant AD with coexisting SP was considered.
One potential distinguishing feature is the later age at onset of SP in individuals with PSEN1 mutations compared to those with common HSP genes. Patients with pathogenic PSEN1 variants typically report symptom onset around the age of 40, 17 whereas the average age at onset for individuals with mutations in SPAST, SPG11, or CYP7B1—the most frequent genetic causes of HSP—is generally in the second decade of life or earlier.23,24
The presence of the APOE ε2 allele may have contributed to a relatively later age at onset in our patient compared with the two previously reported cases involving substitutions at the same codon. APOE ε2 is considered protective against AD-related pathology and has been associated with slower cognitive decline,25,26 with less consistent effects in ε2/ε3 carriers. 27 It may therefore have partially influenced symptom onset.
Regarding prognosis, reported disease duration in PSEN1-associated AD with SP is heterogeneous but is typically within a decade after symptom onset, although longer courses have been described.20,21,28 In contrast, hereditary spastic paraplegia generally does not shorten life expectancy, despite poorer outcomes in selected subtype. 29
In addition to the clinical course, neuroimaging findings provided further insight into the underlying pathology. Brain MRI demonstrated white matter hyperintensities in the absence of conventional vascular risk factors. Emerging evidence suggests that such white matter changes in AD may reflect amyloid-related vascular pathology rather than small vessel cerebrovascular disease.30,31 Accordingly, the cerebrovascular changes observed in our patient may be interpreted as amyloid-related vascular pathology, despite the absence of CMBs on MRI.
Our study had several limitations, including the lack of segregation analysis and functional validation of this variant. In addition, histopathological confirmation would provide further insight into the pathophysiology of SP preceding cognitive impairment in PSEN1-associated disease. These investigations may be particularly informative given that our patient's CSF profile revealed a normal p-tau181 level. While p-tau181 elevation typically precedes the development of tau pathology, 32 which is linked to clinical expression in AD, reduced soluble Aβ42 level is increasingly recognized as playing a critical role in disease progression. 33 Notably, CSF p-tau181 is not uniformly elevated in all cases of autosomal dominant AD. 34 Taken together, further functional and histopathological studies would clarify the mechanisms underlying neurodegeneration in PSEN1-associated disease.
Despite these limitations, this report expands the clinical spectrum of PSEN1-associated EOAD by describing a novel variant with an atypical presentation. Since the seminal report of a Finnish pedigree, 35 subsequent studies have demonstrated considerable heterogeneity in the clinical presentation of PSEN1-related disease. The identification of a previously unreported PSEN1 c.668A > T (p.Gln223Leu) variant supports a potential genotype–phenotype association at codon 223. In addition, the diagnosis was supported by contemporary biomarker evidence, including reduced CSF Aβ42 and amyloid PET positivity, and by a detailed longitudinal characterization showing SP preceding cognitive decline, with intrafamilial comparison.
Conclusion
This case illustrates the importance of early consideration of genetic testing in EOAD patients who present with non-amnestic features such as spasticity or dysarthria. Recognizing atypical presentations may facilitate timely diagnosis, inform family counseling, and support enrollment in precision medicine trials. Future investigations, including protein modeling and functional assays, will be needed to validate the pathogenicity of the p.Gln223Leu variant and clarify its mechanistic role in neurodegeneration.
Supplemental Material
sj-docx-1-alr-10.1177_25424823261419068 - Supplemental material for A novel PSEN1 (p.Gln223Leu) variant associated with spastic paraparesis and early-onset Alzheimer's disease
Supplemental material, sj-docx-1-alr-10.1177_25424823261419068 for A novel PSEN1 (p.Gln223Leu) variant associated with spastic paraparesis and early-onset Alzheimer's disease by Hyuk-je Lee, Hansol Im, Bora Yoon, YongSoo Shim, Dongju Won and Jung Hwan Lee in Journal of Alzheimer's Disease Reports
Footnotes
Acknowledgements
The authors have no acknowledgments to report.
Ethical considerations
The study is exempt from approval of Institutional Review Board, The Catholic University of Korea, Seoul St Mary's Hospital (KC25ZASE0331). Requirement for informed consent to participate has been waived by the Institutional Review Board.
Consent to participate
Not applicable
Consent for publication
A consent-to-disclose form was written by the patient.
Author contribution(s)
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
The data not published within this article may be available by request from any qualified investigator.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.