
Abstract
Background
A growing number of observational studies have suggested a potential association between heart rate variability (HRV) and dementia. However, the causal nature of this relationship remains unclear.
Objective
This study aims to investigate the causal relationship between HRV and dementia using two-sample Mendelian randomization (MR) analysis.
Methods
Two-sample MR analysis was conducted to examine the causal effects of HRV phenotypes, including the peak-valley respiratory sinus arrhythmia or high frequency power (pvRSA/HF), the root mean square of the successive differences of inter beat intervals (RMSSD), and normal-to-normal inter-beat intervals (SDNN), on eight dementia phenotypes (any dementia, Alzheimer's disease [AD], early-onset AD, late-onset AD, Lewy body dementia [DLB], frontotemporal dementia [FTD], Parkinson's disease dementia [PDD], and vascular dementia [VaD]).
Results
Genetically determined pvRSA/HF was associated with an elevated risk of DLB (odds ratio [OR] 1.53; 95% CI 1.04–2.25; p = 0.032) and FTD (OR 2.86; 1.06–7.66; p = 0.037). There was no significant association between HRV and the increased risks of AD, PDD, VaD, or any dementia.
Conclusions
Our study provides suggestive evidence that pvRSA/HF has a potential causal relationship with elevated risks of DLB and FTD. These findings contribute to a better understanding of the potential role of HRV in specific dementia subtypes.
Keywords
Introduction
Heart rate variability (HRV), which quantifies the fluctuation in time intervals between consecutive heartbeats, is a well-established, non-invasive measure of autonomic nervous system function. It reflects the dynamic interplay between the sympathetic (activating) and parasympathetic (resting) branches of the autonomic nervous system. 1 Higher HRV generally indicates better autonomic flexibility and adaptive capacity, whereas reduced HRV is often associated with a state of autonomic imbalance and has been linked to various adverse health outcomes, including cardiovascular diseases. 2
Accumulating evidence suggests that HRV may also be implicated in the pathogenesis of cognitive decline and dementia. A growing number of observational studies have reported that individuals with dementia, including dementia with Lewy bodies (DLB), frequently exhibit lower HRV compared to cognitively healthy individuals. 3 For instance, a recent umbrella review of meta-analyses concluded that dementia and neurocognitive disorders are associated with a significant reduction in HRV (SMD = −0.37), with the evidence grade being suggestive. 4 This systematic alteration suggests that impaired autonomic regulation may be a core feature of these conditions. The physiological interplay between the heart and the brain, often referred to as the brain-heart axis, provides a plausible basis for this connection.5,6 The central autonomic network, which includes key brain regions such as the insula, anterior cingulate cortex, and amygdala—areas notably vulnerable to pathology in various dementia subtypes—plays a critical role in regulating HRV.7–9 Moreover, growing evidence suggests that autonomic dysfunction itself may be a driving factor in dementia pathogenesis. Among physically disabled, community-dwelling women, it was found that participants with reduced HRV had a significantly higher likelihood of developing cognitive impairment than those without reduced HRV. 10 A study has demonstrated that lower HRV in mid-to-late life can independently predict subsequent cognitive decline and an increased risk of all-cause dementia. 11 Furthermore, a study that included cohorts from the United States and China found that long-term visit-to-visit resting HRV was independently associated with cognitive decline, suggesting its potential role as an early warning biomarker. 12 These findings imply that an imbalance in autonomic regulation may already be present and involved in the early pathophysiological processes of the disease, prior to the onset of clinically significant cognitive symptoms. Several key pathophysiological pathways have been proposed to explain how autonomic dysfunction might contribute to dementia pathogenesis. Impaired autonomic control can disrupt the regulation of cerebral blood flow, potentially leading to chronic cerebral hypoperfusion and impaired executive function. 13 Furthermore, HRV may impair the function of the glymphatic system, which is crucial for clearing metabolic wastes such as amyloid-β from the brain.14,15
However, observational studies are unable to definitively establish the directionality of this association. Major challenges include confounding factors, such as comorbid cardiovascular diseases, and reverse causation, whereby underlying, undetected neurodegenerative changes lead to altered HRV. Mendelian randomization (MR) provides a unique tool to address this question of causal direction. While MR has been applied to investigate the causality between certain cardiovascular metrics (e.g., blood pressure) and dementia, the specific causal roles of HRV—particularly its distinct physiological parameters—across different dementia subtypes remain largely unexplored. Therefore, this study employed a two-sample MR design to investigate the potential causal relationship between genetically predicted HRV phenotypes and the risk of major dementia subtypes, including Alzheimer's disease (AD), DLB, frontotemporal dementia (FTD), vascular dementia (VaD), and Parkinson's disease dementia (PDD). Elucidating this causality is critical, as establishing HRV as a modifiable causal risk factor would position it as a promising target for risk stratification and early intervention strategies aimed at preserving cognitive health.
Methods
Mendelian randomization design
We applied a two-sample MR design to investigate whether genetically predicted HRV exerts a causal influence on various dementia subtypes. The summary statistics used were sourced from publicly available genome-wide association study (GWAS) consortia that adhered to standard ethical guidelines. For the MR findings to be valid, three critical assumptions must hold: first, a strong association between genetic variants and HRV; second, no association between these variants and confounding factors; and third, that the variants affect dementia risk only through HRV (no horizontal pleiotropy). The study design is visually summarized in Figure 1. The reporting of the present study maintained adherence to the STROBE-MR reporting guideline. 16 All analyses were performed using publicly available summary data exclusively comprised of European ancestry populations. It is noted that all contributing original studies had received appropriate ethical approvals and collected informed consent from their respective participants.

Schematic of the study design. MR: Mendelian Randomization; pvRSA/HF: peak-valley respiratory sinus arrhythmia or high-frequency power; RMSSD: root mean square of successive differences between normal heartbeats; SDNN: standard deviation of normal-to-normal intervals; LD: linkage disequilibrium; IVs: instrumental variables; IVW: inverse variance weighted; SNPs: single-nucleotide polymorphisms; AD: Alzheimer's disease; FTD: frontotemporal dementia; DLB: dementia with Lewy bodies; PDD: Parkinson's disease dementia; VaD: vascular dementia.
Data sources for heart rate variability
The summary-statistics data for HRV measures were sourced from a large-scale GWAS conducted by Nolte et al. 17 This study rigorously quantified three key HRV phenotypes—each reflecting distinct aspects of autonomic nervous system regulation—using electrocardiogram-derived measurements in European-ancestry participants. The specific indices included peak-valley respiratory sinus arrhythmia or high-frequency power (pvRSA/HF), which captures parasympathetic (vagal) activity during respiratory cycles; root mean square of successive differences between normal heartbeats (RMSSD), representing short-term vagal-mediated oscillations; and standard deviation of normal-to-normal intervals (SDNN), an indicator of overall autonomic balance and total variability over 24-h periods. The GWAS datasets differed in sample size and genetic coverage: pvRSA/HF comprised 24,088 individuals with approximately 2.53 million single-nucleotide polymorphisms (SNPs); RMSSD included 26,523 subjects with 2.53 million SNPs; and SDNN covered 27,850 participants with 2.55 million SNPs. Further details for all HRV phenotypes are provided in Table 1.
GWAS summary statistics data utilized in Mendelian randomization (MR) analyses.
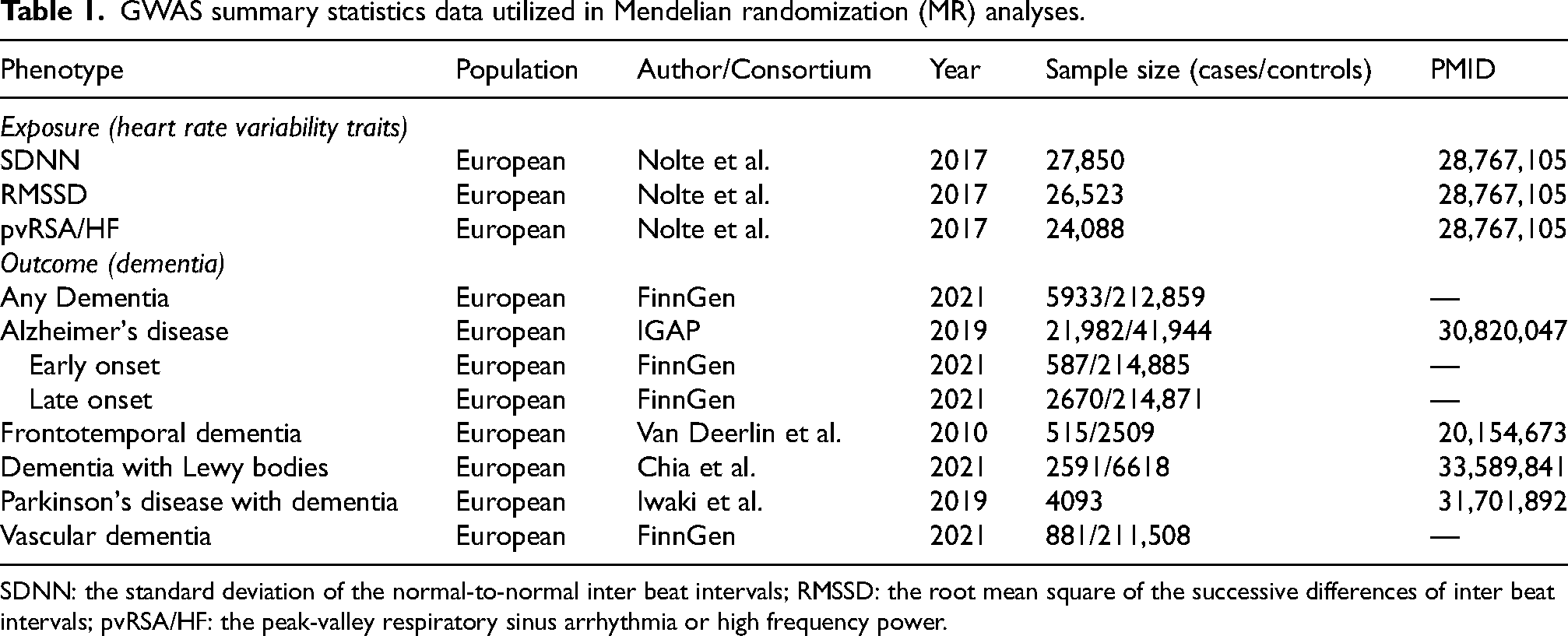
SDNN: the standard deviation of the normal-to-normal inter beat intervals; RMSSD: the root mean square of the successive differences of inter beat intervals; pvRSA/HF: the peak-valley respiratory sinus arrhythmia or high frequency power.
Data sources for dementia outcomes
We investigated eight distinct dementia phenotypes to explore potential subtype-specific causal relationships with HRV: any dementia, AD, early-onset AD (EOAD), late-onset AD (LOAD), FTD, DLB, PDD, and VaD. Summary-level genetic association estimates were obtained from publicly available GWAS conducted predominantly in European-ancestry populations. Data for any dementia, EOAD, LOAD, and VaD were accessed from the FinnGen consortium (2021 release), comprising 5933 to 881 cases and corresponding controls (ranging from ∼212,859 to ∼214,885 individuals). AD summary statistics originated from the International Genomics of Alzheimer's Project (IGAP), including 21,982 clinically diagnosed cases and 41,944 controls. 18 Genetic instruments for FTD were drawn from a study by Van Deerlin et al., which included 515 pathology-confirmed FTD with TAR DNA-binding protein (TDP 43) and 2509 controls. 19 DLB data were sourced from a multicenter GWAS across 44 consortia by Chia et al. involving 2591 cases and 6618 controls. 20 Additionally, Data for PDD was derived from a meta-analysis of 12 longitudinal cohorts of Parkinson's disease patients, totaling 4093 individuals. 21 Cognitive impairment was defined as a binary trait based on standardized neuropsychological assessments, including Mini-Mental State Examination (MMSE) scores below 27 or Montreal Cognitive Assessment (MoCA) scores below 24, evaluated during follow-up visits. Further details for all dementia phenotypes are provided in Table 1.
Selection of genetic instrumental variables
The selection process for genetic instrumental variables (IVs) is delineated in Figure 1. To construct robust genetic proxies for the HRV phenotypes, we implemented a multi-step filtering strategy based on summary-level data from the corresponding GWAS. The specific procedures were as follows: (1) Identification of Significant Variants: SNPs exhibiting a genome-wide significant association with each HRV index (p < 5 × 10–8) were initially selected as candidate IVs. (2) Linkage Disequilibrium (LD) Clumping: To ensure the independence of the IVs, candidate SNPs were clumped to exclude those in linkage disequilibrium (R2 < 0.001) within a 10,000 kb window, using the 1000 Genomes Project European population data as the reference panel. This step retains the SNP with the lowest p-value for each LD region. (3) Assessment of Instrument Strength: The strength of each selected SNP was quantified using the F-statistic, calculated as F = (βexposure/seexposure)2.22–24 SNPs with an F-statistic less than 10 were considered potential weak instruments and were removed from subsequent analyses to mitigate bias from measurement error. 25
Statistical analyses
To infer the causal effect of each HRV phenotype on the risk of dementia subtypes, we applied a suite of MR methods tailored to the number and properties of the available genetic instruments. The inverse variance weighted (IVW) method, which combines the ratio estimates of individual SNPs under a meta-analysis framework, served as the primary analytical approach when three or more independent instrumental variables were available. For analyses involving only one genetic variant, the causal estimate was derived using the Wald ratio method. 26 To ensure the robustness of the primary IVW findings and evaluate potential biases from pleiotropy, we employed several complementary MR methods where sufficient genetic instruments existed (n ≥ 3). These included the weighted median estimator, which provides consistent causal estimates even if up to 50% of the weight comes from invalid instruments 27 ; the MR-Egger regression, which can detect and adjust for directional pleiotropy via its intercept term; and the weighted mode-based approach, which identifies the causal estimate from the largest cluster (or “mode”) of similar genetic instruments, remaining consistent as long as the plurality of instruments (the largest cluster) is valid, even if the majority are invalid. 28 The presence of significant horizontal pleiotropy was formally tested using the MR-Egger intercept test. 29 Potential heterogeneity among the SNPs included in each analysis was evaluated by calculating Cochran's Q-values through the IVW and MR-Egger methods. 30 Furthermore, we implemented the MR-Pleiotropy Residual Sum and Outlier (MR-PRESSO) test to identify and remove potential outlier SNPs that might disproportionately influence the results due to pleiotropic effects. 31 The stability of the overall causal estimate was evaluated through a leave-one-out sensitivity analysis, which systematically excludes each SNP in turn to determine if the result is driven by a single influential variant.
A set of reverse MR analyses were conducted to examine whether the presence of dementia might have a causal effect on HRV levels. For these analyses, genetic instruments for each dementia subtype were selected using a genome-wide significance threshold (p < 5 × 10−8) or, when limited instruments were available, a slightly relaxed threshold (p < 5 × 10−6), after which the same MR methods were applied.
All analyses were performed in R (version 4.3.1) primarily using the TwoSampleMR package (version 0.5.7) for data harmonization and core MR analyses, and the MR-PRESSO package (version 1.0) for outlier detection.
Results
Following the selection criteria, we identified a set of robust genetic instruments for the MR analyses. Specifically, five, six, and six SNPs were selected as proxies for pvRSA/HF, RMSSD, and SDNN. All retained SNPs were robust instruments, with F-statistics ranging from 22 to 141. A complete listing of these instrumental variables is available in the accompanying Supplemental Tables 1.
Causal effects of HRV on dementia subtypes
The primary findings regarding the causal effects of HRV indices on the risks of dementia subtypes are summarized in Figure 2 and detailed in Supplemental Table 2.
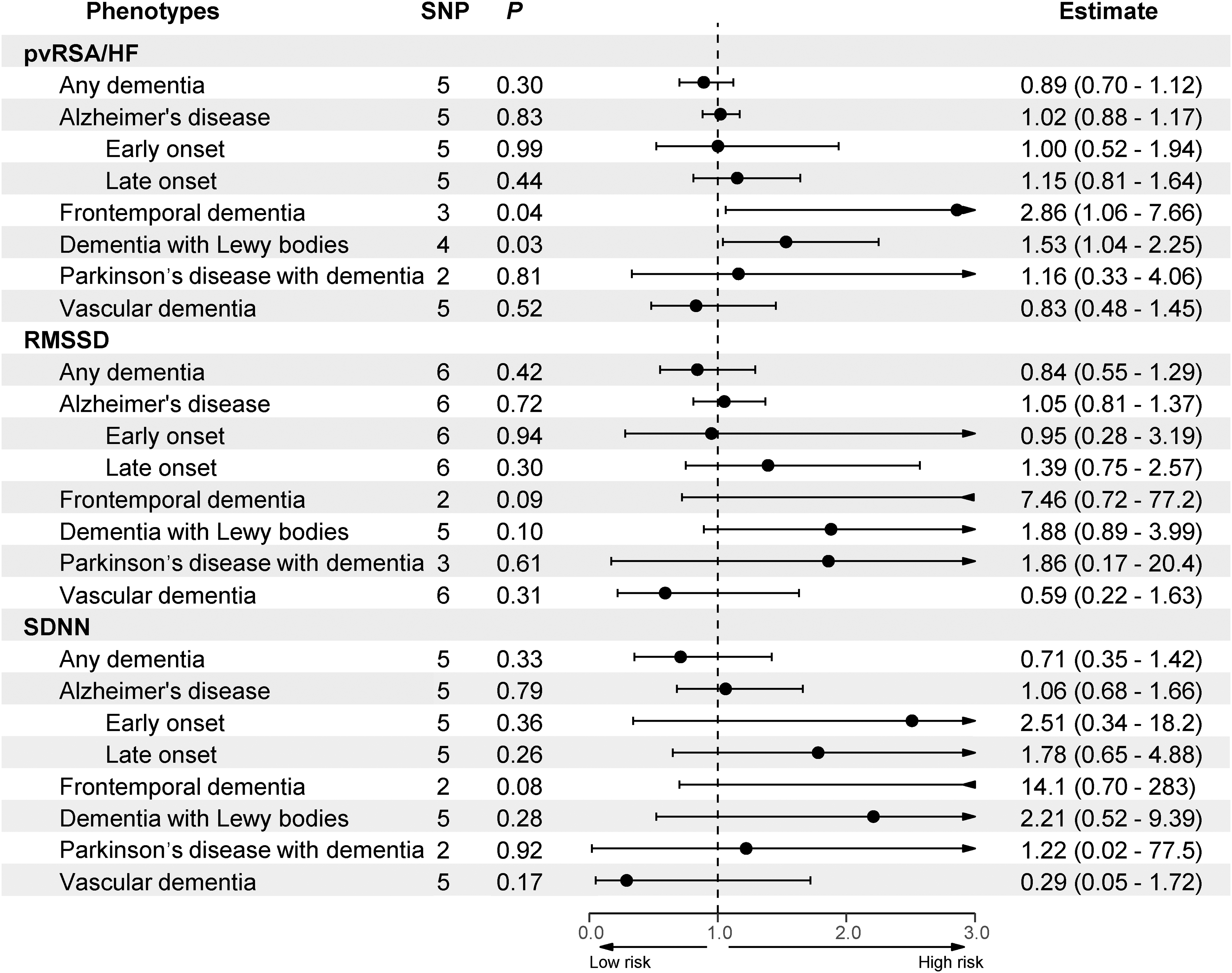
MR results of the causal effect of HRV on dementia. pvRSA/HF: peak-valley respiratory sinus arrhythmia or high-frequency power; RMSSD: root mean square of successive differences between normal heartbeats; SDNN: standard deviation of normal-to-normal intervals; SNP: single-nucleotide polymorphism.
A genetically determined increase in the pvRSA/HF index was significantly associated with a higher risk of DLB, as evidenced by the primary IVW method (odds ratio [OR] = 1.53; 95% confidence interval [CI] = 1.04–2.25; p = 0.032). The robustness of this finding was supported by consistent estimates from complementary methods, including the weighted median (OR = 1.58; 95% CI = 1.003–2.47; p = 0.048). Sensitivity analyses reinforced the validity of this causal inference. Cochran's Q statistic indicated no significant heterogeneity among the instrumental variants (Q-value = 2.38; p = 0.498; Table 2 and Supplemental Table 3). Furthermore, both the MR-Egger intercept test (intercept = −0.04; p = 0.436; Table 2 and Supplemental Table 4) and the MR-PRESSO global test (p = 0.677; Table 2 and Supplemental Table 5) found no evidence of substantial horizontal pleiotropy. A leave-one-out sensitivity analysis, displayed in Figure 3, confirmed that the overall association was not driven by any single influential SNP.

Scatter plot and leave-one-out result of the causal effect of pvRSA/HF on DLB and FTD risk. pvRSA/HF: peak-valley respiratory sinus arrhythmia or high-frequency power; FTD: frontotemporal dementia; DLB: dementia with Lewy bodies.
The results of sensitivity analyses for the causal links between heart rate variability and dementia (FTD and DLB).

FTD: frontotemporal dementia; DLB: dementia with Lewy bodies; MR: Mendelian Randomization; pvRSA/HF: peak-valley respiratory sinus arrhythmia or high-frequency power; RMSSD: root mean square of successive differences between normal heartbeats; SDNN: standard deviation of normal-to-normal intervals; MR-PRESSO: MR-Pleiotropy Residual Sum and Outlier.
For FTD, the MR analysis also revealed a significant causal effect of pvRSA/HF. The IVW estimate indicated that a genetic predisposition to higher pvRSA/HF was associated with an increased risk of FTD (OR = 2.86; 95% CI = 1.06–7.66; p = 0.037). A suite of sensitivity analyses affirmed the robustness of this finding. There was no significant heterogeneity detected among the genetic instruments (Cochran's Q = 0.11; p = 0.948; Table 2 and Supplemental Table 3). Moreover, the MR-Egger intercept test (intercept = 0.05; p = 0.810; Table 2 and Supplemental Table 4) provided no indication of horizontal pleiotropy. The leave-one-out analysis, displayed in Figure 3, confirmed that the observed causal effect was not attributable to any single outlier SNP, as the significance remained stable upon the iterative removal of each variant.
The reverse MR analysis, which assessed the causal effect of genetic liability for each dementia subtype on HRV phenotypes, yielded no statistically significant associations (Table 2 and Supplemental Table 6). This result indicates that reverse causality is unlikely to explain the observed significant effects of pvRSA/HF on DLB (β = −0.019; SE = 0.029; p = 0.528) and FTD (β = 0.006; SE = 0.021; p = 0.772) risk. No genetically predicted levels of RMSSD, SDNN, or pvRSA/HF showed statistically significant causal effects on AD, VaD, PDD, or any dementia. However, it should be noted that the number of valid instrumental SNPs was limited, particularly for rarer subtypes such as FTD and DLB. Therefore, these null findings do not definitively exclude a potential causal effect of dementia on HRV.
Discussion
In this large-scale two-sample MR study, we investigated the causal relationships between genetically predicted HRV phenotypes and the risk of major dementia subtypes. Our primary analysis revealed that genetically determined higher pvRSA/HF—an index predominantly reflecting parasympathetic (vagal) activity—exerts a suggestive causal effect on increasing the risk of both DLB and FTD. Conversely, we observed no significant causal associations for any of the three examined HRV phenotypes with AD, VaD, PDD, or the broad phenotype of ‘any dementia’. The robustness of these primary findings is substantiated by consistent results across complementary MR methods, comprehensive sensitivity analyses indicating no significant heterogeneity or horizontal pleiotropy, and the absence of potential evidence for reverse causality in our bidirectional MR analyses.
Our findings indicate that elevated pvRSA/HF may increase the risk of DLB. The potential causal link between pvRSA/HF and DLB is biologically plausible. DLB is characterized by the early accumulation of alpha-synuclein pathology, which frequently affects key brainstem nuclei integral to the central autonomic network, such as the dorsal motor nucleus of the vagus nerve.32,33 This direct neuroanatomical overlap provides a compelling mechanism whereby autonomic dysfunction, quantified by HRV, could be intrinsically linked to DLB pathogenesis. Furthermore, elevated pvRSA/HF might reflect heightened vagal activity. The vagus nerve not only regulates cardiac function but also connects the gut and the brain via the gut-brain axis. Research suggests that abnormal aggregation of alpha-synuclein might originate in the gut and subsequently undergo retrograde transport to the brainstem (e.g., the dorsal motor nucleus of the vagus) via vagal pathways, ultimately spreading to the cerebral cortex.34–36 Intriguingly, a meta-analysis comparing HRV across dementia subtypes found significantly lower HRV in DLB patients compared to those with AD (p = 0.0381), 3 highlighting the potential diagnostic value of HRV assessment in patients suspected of having DLB.
Moreover, our study identified that higher pvRSA/HF may increase the risk of FTD. The behavioral variant FTD typically involves early and prominent pathology in brain regions critical for autonomic and emotional regulation, including the insula, anterior cingulate cortex, and orbitofrontal cortex.37,38 Neurodegeneration in these higher-order cortical regions could disrupt top-down control of autonomic function. Additionally, patients with FTD can exhibit reduced HRV and blood pressure dysregulation, patterns that align with regional brain atrophy, as various frontal subregions contribute to the modulation of HRV and blood pressure.39,40 Our findings suggest that autonomic dysregulation may constitute a component of the FTD pathological cascade.
Autonomic dysfunction may serve as a potential early biomarker for neurodegeneration. 41 The temporal sequence implied by our MR estimates—where genetic predisposition to autonomic dysfunction precedes clinical dementia—aligns with this concept. It is particularly noteworthy that HRV indices have also been proposed as potential markers for specific neuropsychiatric symptoms in dementia, such as agitation propensity in AD, 42 indicating that autonomic metrics might reflect both disease risk and phenotypic expression. It is important to note, however, that the relationship between neurodegenerative diseases and autonomic function is likely bidirectional. 40 On the one hand, neurodegenerative changes may lead to autonomic dysfunction. On the other hand, age-related decline in autonomic function could also accelerate the progression of neurodegenerative pathology. Our findings suggest a potential genetic pathway from parasympathetic activity to the onset of specific dementia subtypes, yet these results should be interpreted with caution. Although reverse-causality MR analyses did not support an effect of dementia on HRV alterations, we cannot rule out the possibility that neurodegenerative changes may contribute to autonomic impairment.
The null finding for AD in our primary analysis is intriguing but does not preclude autonomic involvement at different disease stages. A meta-analysis showed that among various dementia subtypes, HRV was significantly reduced in patients with mild cognitive impairment rather than in those with AD. 3 The relationship between cortical Aβ accumulation and autonomic cardiac function represents an early phenomenon in AD pathogenesis. 43 It is plausible that our genetic instruments for HRV, which reflect lifelong autonomic tone, may not capture the dynamic autonomic changes coinciding with initial Aβ deposition in preclinical AD. This potential early interaction is mechanistically supported by interventions like HRV biofeedback and emotion regulation training, which have been shown to modulate plasma Aβ40 and Aβ42 levels. 44 This suggests that non-pharmacological improvement of autonomic function could potentially influence the amyloid pathway, offering a novel therapeutic avenue.
Other mechanisms might also explain the causal role of autonomic dysfunction in these dementias. Impaired HRV may contribute to cerebral hypoperfusion and disrupt glymphatic clearance of neurotoxic proteins.13–15 Future studies are warranted to explore these and other potential mediating pathways.
Our study has several strengths, including the MR design that minimizes confounding and reverse causation, the use of robust genetic instruments, and comprehensive analyses across multiple dementia subtypes and HRV parameters. However, limitations must be acknowledged. First, the sample sizes for certain dementia outcomes, particularly FTD and DLB, remain limited, potentially constraining statistical power to detect weaker associations. It should be noted that the sample sizes for FTD and DLB, as well as the number of SNPs obtained, were relatively limited, highlighting the need to expand the cohort for further validation of the findings from this study. Second, all included GWAS participants were of European ancestry, cautioning against generalizing our findings to other populations. Third, although we employed multiple methods to assess pleiotropy, residual influence cannot be entirely ruled out. Fourth, as our study is exploratory in nature, our analysis examined multiple exposure-outcome combinations without applying multiplicity correction. Therefore, the possibility of false-positive findings should be considered. Accordingly, the MR results for pvRSA/HF in relation to DLB and FTD should be interpreted with caution, and the level of evidence should be regarded as suggestive. Fifthly, each HRV phenotype was instrumented by a relatively small set of SNPs. Although all retained SNPs were strong instruments (F > 10), the limited number of IVs may reduce the sensitivity of pleiotropy-robust methods to detect pleiotropy. Therefore, the absence of significant pleiotropy in our sensitivity analyses should be interpreted with caution, and residual pleiotropy cannot be entirely ruled out. Finally, the HRV parameters assessed do not fully capture the entire spectrum of autonomic function, necessitating future validation with GWAS data on a broader range of phenotypic indicators. It should be noted that due to the limited number of valid instrumental SNPs, the reverse MR analyses may not have had sufficient power to detect a causal effect of dementia on HRV. Therefore, these results should be interpreted with caution and cannot fully rule out the possibility of bidirectional effects.
Conclusions
In conclusion, our MR study provides suggestive genetic evidence supporting a role for autonomic dysfunction in the pathogenesis of DLB and FTD. These findings refine our understanding of the brain-heart axis in neurodegeneration. Our results suggest the potential of HRV as a biomarker for identifying individuals at high risk for specific dementias and position the autonomic nervous system as a plausible target for preventive strategies aimed at mitigating dementia risk.
Supplemental Material
sj-xlsx-1-alr-10.1177_25424823261431319 - Supplemental material for Unraveling the brain-heart axis: Mendelian randomization study on the causal relationship between heart rate variability and dementia
Supplemental material, sj-xlsx-1-alr-10.1177_25424823261431319 for Unraveling the brain-heart axis: Mendelian randomization study on the causal relationship between heart rate variability and dementia by Songbiao Li, Yan Zhang, Li Kou and Jinfeng Deng in Journal of Alzheimer's Disease Reports
Footnotes
Acknowledgements
The authors thank Nolte et al., Van Deerlin et al., Chia et al., Iwaki et al., FinnGen, and IGAP for providing GWAS summary data.
Ethical considerations
This study used only published summary statistics and no individual-level data, thus obviating the need for new ethical board review. All source studies and consortia had secured prior ethical approvals and informed consent from their participants.
Consent to participate
The analysis presented in this study was confined to publicly accessible summary statistics derived from previously published genome-wide association studies. As no individual-level data were utilized, obtaining additional informed consent was not required.
Consent for publication
Not applicable
Author contribution(s)
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
The publicly available GWAS data employed in this study (summarized in Table 1) were compiled from two repositories. The OpenGWAS platform (![]() ) provided the data for HRV, AD, Early-onset AD, Late-onset AD, FTD, and DLB. The FinnGen consortium served as the source for the data on any dementia and vascular dementia.
) provided the data for HRV, AD, Early-onset AD, Late-onset AD, FTD, and DLB. The FinnGen consortium served as the source for the data on any dementia and vascular dementia.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.