
Abstract
Ketamine's ability to lift mood and spur new synapse growth has put glutamate biology at the center of modern neurotherapeutics. Yet the drug's intravenous route, monitoring requirements, and dissociative effects make it a poor candidate for long-term prevention of Alzheimer's disease (AD). This hypothesis article proposes a low-cost oral glutamatergic regimen that targets early synaptic and glutamatergic dysfunction in AD pathogenesis. Here we advance a testable hypothesis: an all-oral “synaptogenic stack” could mimic ketamine's downstream benefits—namely, the rise in brain-derived neurotrophic factor and the activation of mechanistic target of rapamycin (mTOR)—while avoiding its toxicities. The stack combines three inexpensive agents that have decades of human use. First, dextromethorphan, kept in circulation with a small dose of a CYP2D6 inhibitor, provides gentle NMDA antagonism. Second, piracetam acts as a positive modulator of AMPA receptors, boosting fast excitatory transmission. Third, oral L-glutamine replenishes presynaptic glutamate stores and buffers against excitotoxic spill-over. Working in concert, these drugs should reduce extrasynaptic NMDA stress, enhance AMPA throughput, and preserve dendritic spine density in the aging brain. If this mechanism proves sound, the regimen offers a low-cost, scalable way to delay the clinical onset of AD, particularly in people who already show prodromal biomarkers or genetic risk. Prospective trials are needed to evaluate safety, target engagement, and long-term cognitive outcomes.
Keywords
Background
Alzheimer's disease (AD) is now viewed less as a sudden loss of memory and more as a very long slide into synaptic failure that can start 20 or 30 years before a diagnosis.1,2 In the earliest phase, two things go wrong with glutamate signaling. First, extrasynaptic N-methyl-D-aspartate (NMDA) receptors, especially those that carry the NR2B sub-unit, become hyperactive. At the same time AMPA receptors, which should sit in the synapse and pass the fast excitatory message, are pulled inside the neuron and degraded.3–6 Together these shifts set off calcium overload, oxidative stress, loss of long-term potentiation, spine shrinkage and, finally, cell death. 7 Drugs on the market today arrive late in that process and work on only half of the problem—for example, memantine mostly calms the rogue NMDA channels. 8 A better preventive approach would dampen toxic NMDA activity, rescue AMPA traffic and spark new synapse growth while the network is still salvageable.
The regimen
Cheung's four-part oral stack (Figure 1) was first built for refractory mood and anxiety illnesses, yet its design maps neatly onto those early AD defects.9–11 The plan is simple and inexpensive: dextromethorphan (DXM) at 30–120 mg a day; a low dose of a strong CYP2D6 blocker such as fluoxetine, paroxetine or high-dose duloxetine to keep DXM in the blood; piracetam 1.6 g a day to nudge AMPA receptors; and supplementary oral l-glutamine to refill presynaptic stores and curb excitotoxicity. Together, these agents reproduce the “NMDA-to-AMPA flip” that underlies ketamine's burst of plasticity.12–14
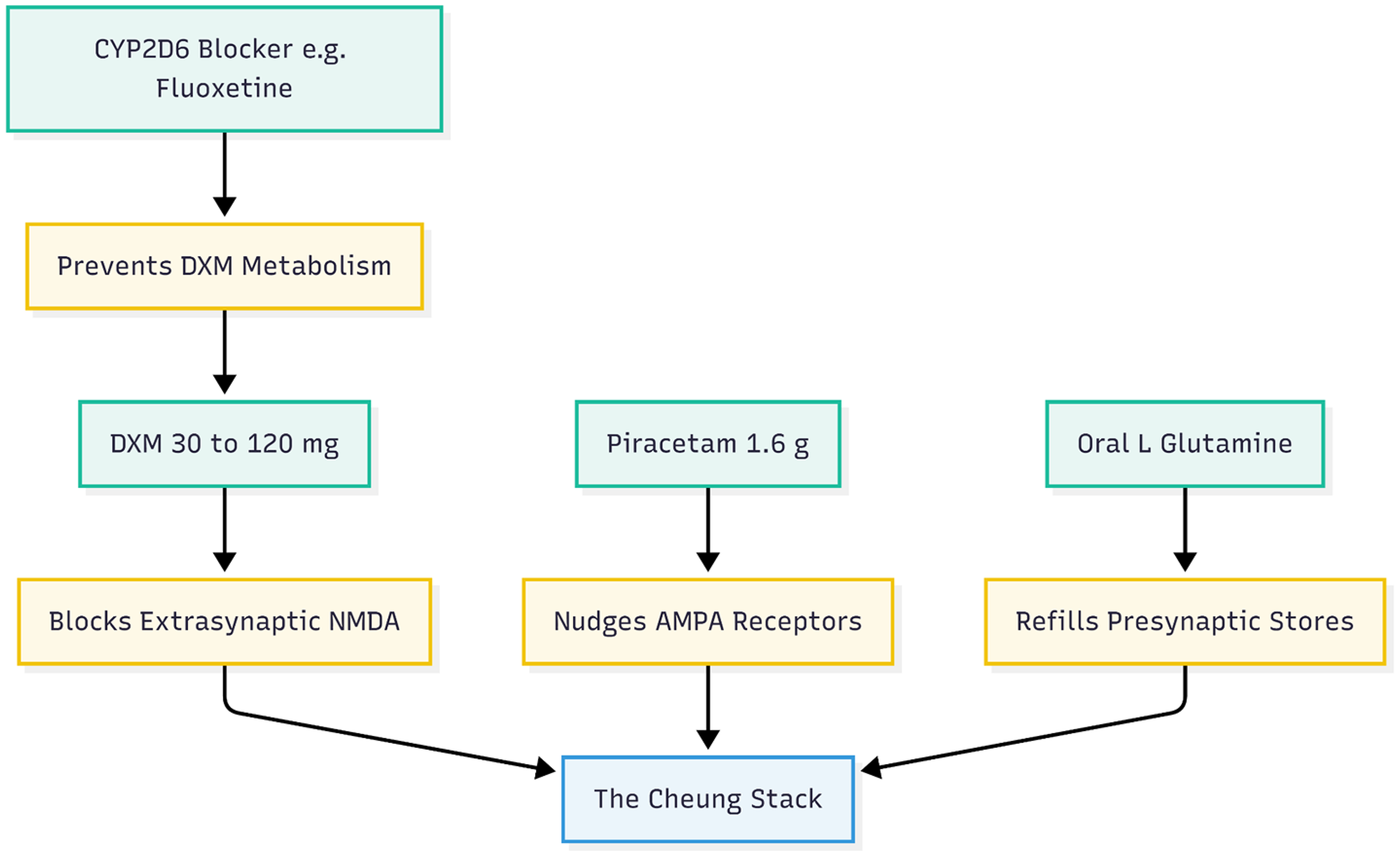
The four-part stack overview. This diagram outlines the four specific components of the regimen and their primary roles.
Mechanistic alignment with early Alzheimer's disease pathophysiology
Selective dampening of extrasynaptic NMDA noise is the first pillar (Figure 2). Like low-dose ketamine or memantine, high but steady DXM blocks open NMDA channels in a voltage-dependent way and shows a clear preference for NR2B-rich receptors. 8 Because soluble Aβ forces exactly these receptors to the cell surface and even drives damaging metabotropic signals without ion flow,7,15 DXM held in circulation by a CYP2D6 inhibitor 11 could blunt calcium overload and tau mischief long before plaques appear.

Pillar one: the NMDA brake. How DXM and CYP2D6 blockers counteract the specific NMDA defects found in early Alzheimer's disease.
The second pillar (Figure 3) is keeping AMPA receptors where they belong. Amyloid-β (Aβ) promotes rapid tagging and internalization of GluA1/GluA2 receptors, a change that tracks closely with memory loss.4–6 Piracetam reverses that trend: work in aged and amyloid-exposed systems shows more surface AMPA receptors, stronger currents and better LTP after treatment.16–18 By preserving fast transmission, piracetam may protect dendritic spines through the long pre-clinical window.

Pillar two: the AMPA accelerator. Illustrating how Piracetam fights the internalization of AMPA receptors caused by amyloid-beta.
Third, presynaptic glutamate stores need topping up (Figure 4). Stress and early amyloid lesions drain vesicular glutamate and weaken release probability.19,20 Oral l-glutamine quickly restores the glutamate–glutamine cycle, normalizes excitatory postsynaptic currents and sharpens cognition in stressed or amyloid-bearing animals.19,21 Curiously, when extracellular glutamate soars under inflammatory pressure, high-dose glutamine can actually bring it down,21,22 providing a two-way safety net against excitotoxicity.

Pillar three: the two-way safety net. How L-Glutamine acts as a buffer, helping in both low-energy states and high-inflammation states.
Finally (Figure 5), the pairing of an NMDA brake with an AMPA accelerator triggers a well-studied plasticity cascade: brain-derived neurotrophic factor (BDNF) release and mechanistic target of rapamycin (mTOR)-dependent spine formation.12,23 Because BDNF/tropomyosin receptor kinase B (TrkB) signaling falters early in AD, repeated activation through the Cheung stack could hold synaptic density steady in people at risk. 9
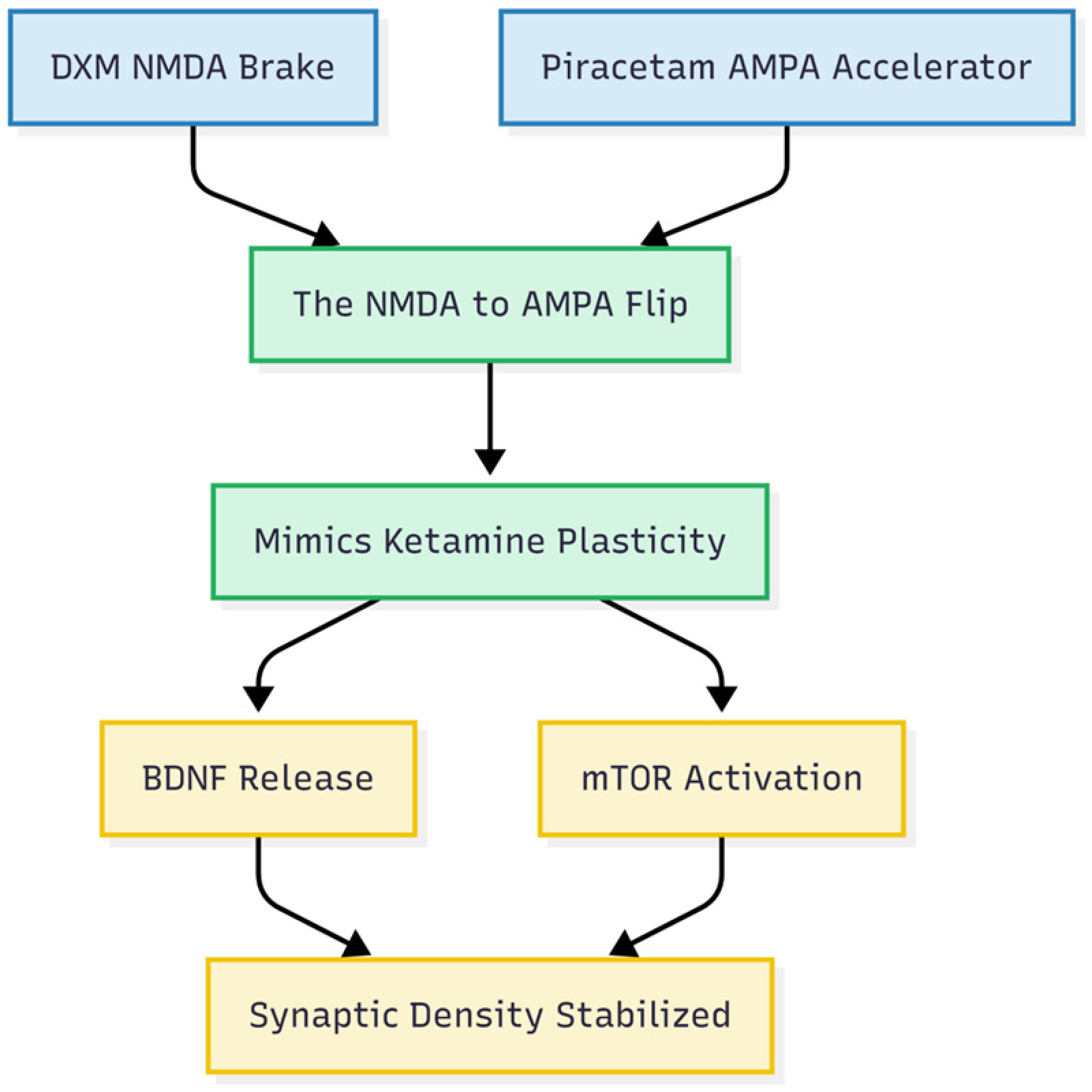
Pillar four: the plasticity cascade. The final result: How combining the “Brake” and the “Accelerator” mimics the Ketamine effect.
Nuanced effects of L-glutamine in neurodegenerative contexts
Animal work portrays L-glutamine as a “two-way safety net” for the glutamate–glutamine cycle, yet its influence in human neurodegeneration is both dose-sensitive and context-specific. After oral ingestion the amino acid is quickly absorbed, but extensive first-pass metabolism limits how much reaches the brain. 24 Rodent studies that used roughly 150 mg kg−1—about 10–20 g day−1 for people—reported synaptic benefits; higher intakes during inflammation, however, may intensify excitotoxic stress.
Human findings diverge. Some cerebrospinal fluid (CSF) studies show higher glutamate and glutamine in AD, perhaps an early compensatory response, whereas magnetic-resonance spectroscopy often records lower cortical glutamate.25,26 In 3×Tg-AD mice, oral glutamine preserved medial-prefrontal synaptic currents, postponed memory loss, and lowered oxidative markers with no toxicity. 27 Mendelian randomization work supports causality: each standard-deviation rise in circulating glutamine reduced AD odds by 17%. 28 Nonetheless, under severe excitotoxicity glutamine may drive extra astrocytic glutamate release, so any future trials must titrate dose and monitor metabolites. 29
Balanced evidence on individual components
Pre-clinical data are strongest. DXM blocks NR2B-rich NMDA receptors and limits neuronal loss in ischemia and amyloid models. Piracetam boosts AMPA trafficking and mitochondrial function in aged neurons.30,31 Glutamine supplementation lessens tau phosphorylation and inflammation in transgenic mice.27,32
Clinical signals are weaker. A Taiwanese cohort (≈199,000 adults, 16 years) linked DXM use to a 43% lower dementia risk, with the largest benefit at ≥91 defined-daily-doses year−1. 33 The DXM–bupropion tablet AXS-05 delayed relapse of AD agitation in phase 3 trials and was well tolerated. 34 Meta-analyses of piracetam (19 randomized controlled trials (RCTs), n = 1489) show modest overall improvement but memory-specific gains remain inconsistent.35,36 No sizeable AD trial has tested glutamine, although it is widely judged safe in other settings. Taken together, a mechanistic rationale exists, yet randomized studies—especially in biomarker-positive, prodromal AD—remain essential.
Emerging clinical signals relevant to cognition and neuroprotection
A proprietary tablet that pairs dextromethorphan with bupropion (AXS-05) keeps dextromethorphan levels high by blocking CYP2D6 and has delivered encouraging Phase 3 results in AD agitation: two randomized-withdrawal trials (ACCORD-2, ACCORD-1) cut the risk of relapse (hazard ratio 0.276; p ≤ 0.014) and slowed both agitation and overall disease progression, the parallel ADVANCE-1 trial reduced agitation acutely, and although ADVANCE-2 missed significance, most outcomes still favored the drug 37–39. Across studies and up to a year of follow-up, side-effect rates matched placebo, falls and discontinuations stayed under 3% and 1% respectively, no deaths or extra sedation occurred, and cognition remained stable, supporting long-term use.37,38 On this record the FDA granted Breakthrough Therapy status and has now accepted a priority-review submission, setting 30 April 2026 as the action date. 39
On the other hand, case reports already hint at brisk cognitive gains with the full or partial regimen. Patients describe clearer thinking, faster processing and stronger working memory within weeks—improvements that mirror the subtle executive losses seen years before an AD diagnosis.11,40–42 One young man with schizoaffective disorder said the stack “cleared decades of mental sludge” after everything else had failed. 41 Although anecdotal, such stories match the known importance of healthy AMPA throughput for high-order cognition. Table 1 summarizes the preclinical and clinical evidence for glutamatergic modulation in neurodegeneration.
Summary of preclinical and clinical evidence for glutamatergic modulation in neurodegeneration.

Feasibility for long-term prevention
All four ingredients are off-patent, cheap (well under two dollars a day), taken by mouth and backed by decades of safety data. Side-effects tend to be mild—occasional insomnia, jitters or stomach upset—and usually ease with dose adjustment.9,10 Because many clinicians already prescribe the stack off-label, it is uniquely positioned for pragmatic prevention trials in APOE ε4 carriers, people with subjective cognitive decline or biomarker-positive preclinical AD.
Limitations, interactions, and safety
Ease of access is counter-balanced by untested drug–drug interactions. Long-term CYP2D6 inhibition by fluoxetine, paroxetine, or duloxetine can persist for weeks, raising the risk of serotonin toxicity, QT prolongation, and falls in older adults.44,45 How glutamine might modify CYP2D6 kinetics or glutamate flux is unknown. Piracetam is renally cleared but can cause insomnia or agitation at high doses; glutamine, usually tolerated up to 30 g day−1, requires caution in renal impairment or hyperammonemia.
Early-phase trials should track electrocardiograms, falls, serum electrolytes, and genotype (APOE, CYP2D6). Imaging or fluid biomarkers, such as BDNF, spine density, or phospho-tau, would confirm target engagement. Off-label use offers informal reassurance, yet systematic safety data in frail populations are lacking. 46
Conclusion
The earliest biology of AD looks like a tilt from a balanced NMDA–AMPA partnership toward runaway extrasynaptic NMDA activity and dwindling AMPA support. The Cheung Glutamatergic Regimen addresses both faults at once: sustained DXM plus a CYP2D6 blocker reins in toxic NMDA signaling; piracetam revives AMPA strength; glutamine refuels presynaptic stores and cushions against spill-over damage. Collectively, bench studies, genetic evidence, and preliminary human findings justify a staged clinical program. Phase 1/2 work in prodromal AD should establish safety, optimal dosing, and biological engagement. If successful, larger randomized trials in high-risk cohorts could test whether this low-cost, orally available regimen slows synaptic failure and preserves cognition.
Footnotes
Acknowledgements
The author has no acknowledgments to report.
Author contribution(s)
Funding
The author received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.