
Abstract
Single-guide RNA (sgRNA) lentiviral infection with Cas9 protein electroporation (SLICE) enables CRISPR screening in primary cell types that require transient Cas9 expression, yet is limited by scalability and robustness. Here, we introduce dual guide RNA infection with Cas9 electroporation (DICE), which expresses two guides from the same lentiviral construct that target the same gene. In genome-wide screens, DICE outperformed SLICE in defining essential genes and modulators of PD-L1 expression in Interferon-gamma-activated THP1 cells. Collectively, these data demonstrate that DICE can be utilized for reduced-scale CRISPR screens in cell types with transient Cas9 protein expression without sacrificing screening quality.
Introduction
CRISPR-Cas systems have emerged as a potent technology to provide readily programmable and highly efficient gene perturbations on single-gene, multi-gene, and genome-wide scales. CRISPR-Cas dependent gene editing requires a guide RNA (gRNA) to recruit a Cas DNA nuclease (typically Cas9) to areas of homology in the genome where Cas9 creates a double-stranded break. 1 The imperfect repair of this lesion through nonhomologous end joining results in an insertion or deletion (indel) of bases in the genome, subsequently leading to a knockout of gene function. 2 While genome-wide CRISPR screens in immortalized cell lines have become routine, CRISPR screens in some biologically relevant models, like primary immune cells, present challenges that require modification of the standard approach.3–7 Compared with immortalized cell lines, primary cells cannot be easily engineered to express Cas9 endogenously, and cell numbers are often limited, precluding the use of standard genome-wide CRISPR libraries, which require hundreds of millions of cells for adequate gRNA coverage. To overcome the challenge of expressing Cas9 in primary immune cells, the single-gRNA (sgRNA) lentiviral infection with Cas9 protein electroporation (SLICE) method for CRISPR genome editing was recently developed. 8 SLICE enables CRISPR-mediated gene editing in cells that don’t express Cas9 protein and has been applied for genome-wide CRISPR screening in primary human T cells.8,9 While the development of the SLICE approach represents a major innovation for CRISPR screening in primary cells, to our knowledge, no head-to-head comparison of CRISPR screening with transient Cas9 (SLICE) versus constitutive Cas9 expression has been performed.
Here, we compare CRISPR screening using the Brunello library in THP1 cells with constitutive Cas9 expression or in THP1 cells requiring Cas9 protein electroporation and demonstrate that screening performance was more robust in THP1 cells constitutively expressing Cas9. 3 To improve transient Cas9 screening approaches, we developed DICE, a multiplex gRNA lentiviral system expressing two gRNAs targeting the same gene from the same construct, followed by Cas9 electroporation. We designed a miniaturized, genome-wide dual-Cas9 CRISPR library (21,034 constructs) and benchmarked it to the gold-standard Brunello library (76,441 constructs). We observe that, despite the reduction in library size, the dual-Cas9 library performs comparably to the Brunello genome-wide library in Cas9-expressing cells. Moreover, we show that the dual-Cas9 library (DICE) outperforms the Brunello library (SLICE) when Cas9 protein is introduced via electroporation. We thus present a novel approach and library that improves the outcome of genome-wide screens when Cas9 is exogenously introduced and reduces the resources required to perform genome-wide screens (i.e., cells, plasticware, sorting time, and sequencing depth) by up to 75%.
Methodology
Cell culture
THP-1 cells were maintained in RPMI 1640 (Gibco) supplemented with 10% heat inactivated fetal bovine serum (FBS, Gibco), 1% Pen/Strep (Gibco), 1%
Generation and characterization of Cas9-stable cells
Cas9-stable cells were generated by infecting parental cell lines with a lentiviral construct expressing Cas9 and blasticidin-resistance gene in 12-well plates at 1000 g for 1 h, in the presence of 8 μg/μL polybrene (Sigma-Aldrich). Plates were then returned to 37°C with 5% CO2. Cells were incubated overnight and then selected by 10 μg/mL blasticidin (Thermo Fisher Scientific). The activity of Cas9 was confirmed by flow cytometry to be greater than 75% through by introducing guides targeting CD63, CD47, and CD86. sgRNA sequences are as follows: CD86 Guide 1: TTAGAAATTGGTACTATTTC, CD86 Guide 2: GTAACCGTGTATAGATGAGC, CD63 Guide: TGTCCAGGATCGAAGCAGTG, CD47 Guide: GCACTTAAATATAGATCCGG, NTC Guide 1: GCTTTCACGGAGGTTCGACG, and NTC Guide 2: GCGAGGTATTCGGCTCCGCG.
Flow cytometry
Cells were harvested and washed twice with fluorescence-activated cell sorting (FACS) buffer (phosphate buffer saline + 1% FBS) and resuspended in 100 µL/well of fold change-block (BioLegend, 422302) for 15 min at room temperature in the dark. Without washing, 100 μL/well of antibody is added to the FACS buffer containing Human TruStain FcX (Biolegend 422301) (1:100 final dilution). Cells were further incubated at room temperature for 15 min in the dark and then washed twice in 2× in FACS buffer. Cells were resuspended in 200 µL of FACS buffer, and fluorescence was measured using a BD LSR Fortessa Cell Analyzer. Flow cytometry antibodies used in this study are CD47 (BioLegend, 323116), CD63 (BioLegend, 353030), and CD86 (BioLegend, 374212).
Design of dual-Cas9 library
gRNAs for each human gene were designed using Broad’s Rule Set 3 and are listed in Supplementary Table S1. 10 The oligo library was synthesized at Twist Biosciences and cloned using a two-step process where the guides were cloned into pMV-AA0017 backbone, followed by the introduction of the TRACR RNA cassettes. The TRACR RNA sequences downstream of the U6 and H1 promoters are VCR1 and WCR3, respectively. 11
Genome-wide CRISPR screening
For the Brunello library transductions, 120 million THP1 cells or THP1 stably expressing Cas9 were transfected with the sgRNA lentiviral library at multiplicity of infection (MOI) = 0.3 in three biological replicates. The Brunello CRISPR-Cas9 knockout library contains 76,441 sgRNAs targeting 19,114 genes with 1000 nontargeting controls. For dual-Cas9 library transductions, 30 million THP1 cells or THP1 stably expressing Cas9 were transfected with a lentiviral library at MOI = 0.3 in three biological replicates. Following transduction, transduced cells were selected with puromycin (2 μg/mL, Thermo Fisher Scientific, Cat# A1113802) and dead cells were removed (Miltenyi Biotec, Cat# 130-090–101). For SLICE and DICE experiments, 8 μM Cas9 was nucleofected per 20E6 cells using the Expanded T cell program 2 on MaxCyte STx nucleofector. Cells were then differentiated with PMA and treated with 10 ng/mL interferon (IFN) gamma for 24 h. A total of 10 million (dual Cas9) or 40 million (Brunello) cells were collected as presort for genomic DNA extraction for each replicate. Greater than 40 or 10 million cells per replicate for Brunello or dual-Cas9 experiments, respectively, were then processed and stained for anti-Human PDL1 cell surface expression (BioLegend, Cat# A1113802) at 10 million cells/mL according to manufacturer’s recommendation. Cell populations expressing the top and bottom 10% PDL1 levels were enriched through FACS sorting using Aria Fusion (BD Biosciences). At least 1 million PDL1low and PDL1high cells were collected for genomic DNA extraction using Quick-DNA FFPE Kit (Zymo Research). PCR reactions containing up to 10 μg genomic DNA in each reaction were performed using Titanium Taq DNA Polymerase with primers to amplify the sgRNAs. Samples were then purified with SPRIselect beads and mixed and sequenced by 75-bp single-end sequencing for Brunello libraries and 2 × 150 bp paired-end sequencing for dual-Cas9 libraries.
CRISPR screen analysis
Pair-end sequencing data were demultiplexed and paired reads each with a sgRNA were quantified using custom Perl scripts. Given that the position of the sgRNA could vary per read, the primer sequences flanking the sgRNA were considered. Sequences in between the flanking primers were extracted and then compared with sequences in the sgRNA library. Only sequences with no mismatches were used in the calculation of guide-level read counts. Due to possibilities of recombination events, only pair of sgRNAs that maps to the same gene or only 1 sgRNA were identified in each read-pair and were used for downstream analysis. Overall, samples with a minimum of 75% reads mapping to a sgRNA were considered for further analysis. Through the edgeR Bioconductor package, reads were normalized between samples using the trimmed mean of M values (TMM) method. 12 Differential analysis of guide level counts was performed using the limma R Bioconductor package. 13 Gene-level effects were considered the median log fold change of the four guides associated with each gene. Statistically significant genes were identified by aggregating differential sgRNAs ranked consistently higher at gene level using a robust rank aggregation method.
Data availability
All data generated and analyzed in this study are included in the Supplementary Data.
Results
Genome-wide SLICE has limited performance relative to traditional screening
Genome-wide CRISPR screens are routinely executed in immortalized cell lines, as these cells can be engineered to express Cas9 protein and expanded to essentially unlimited numbers; however, screening in cell lines does not always yield disease relevant targets. Therefore, there is an emphasis on developing approaches to enable CRISPR screening in primary cell models that are more likely to reflect disease biology and donor diversity. A hybrid CRISPR approach termed “single-guide RNA lentiviral infection with Cas9 protein electroporation” (SLICE) was recently developed for pooled screening in primary human T cells, wherein cells are transduced with a lentiviral CRISPR sgRNA library and Cas9 is subsequently introduced by electroporation. 8 Given that Cas9 activity within a cell persists for only 24–48 h14,15 when Cas9 is introduced via electroporation and that most published screens allow 1+ weeks for genetic perturbation in Cas9-expressing cells, we sought to evaluate the global effect of Cas9 expression modes on CRISPR screen performance.
To evaluate the contribution of Cas9 expression on screening performance, we executed genome-wide screens in THP-1 macrophages and evaluated two independent phenotypic readouts: (1) gene essentiality and (2) IFN-gamma-dependent regulators of PD-L1 surface expression in cells expressing constitutive Cas9 and in parental cells electroporated with Cas9 protein (Fig. 1A–C). Briefly, we transduced Cas9-expressing or parental THP-1 cells with the Brunello genome-wide library at 500× coverage with an MOI of 0.3 and selected for cells transduced with gRNAs. THP-1 parental cells expressing gRNAs were electroporated with 8 µM Cas9 protein, a dose of Cas9 that provides near 100% activity, 8 to induce genome editing following the SLICE protocol. Edited cells were then differentiated into macrophages with PMA and treated with IFN gamma to induce PD-L1 surface expression. Cells were FACS sorted for the top and bottom 10% of PD-L1 expressors. Thus, our screening workflow allowed us to assess library performance using both a viability and FACS-based readouts.
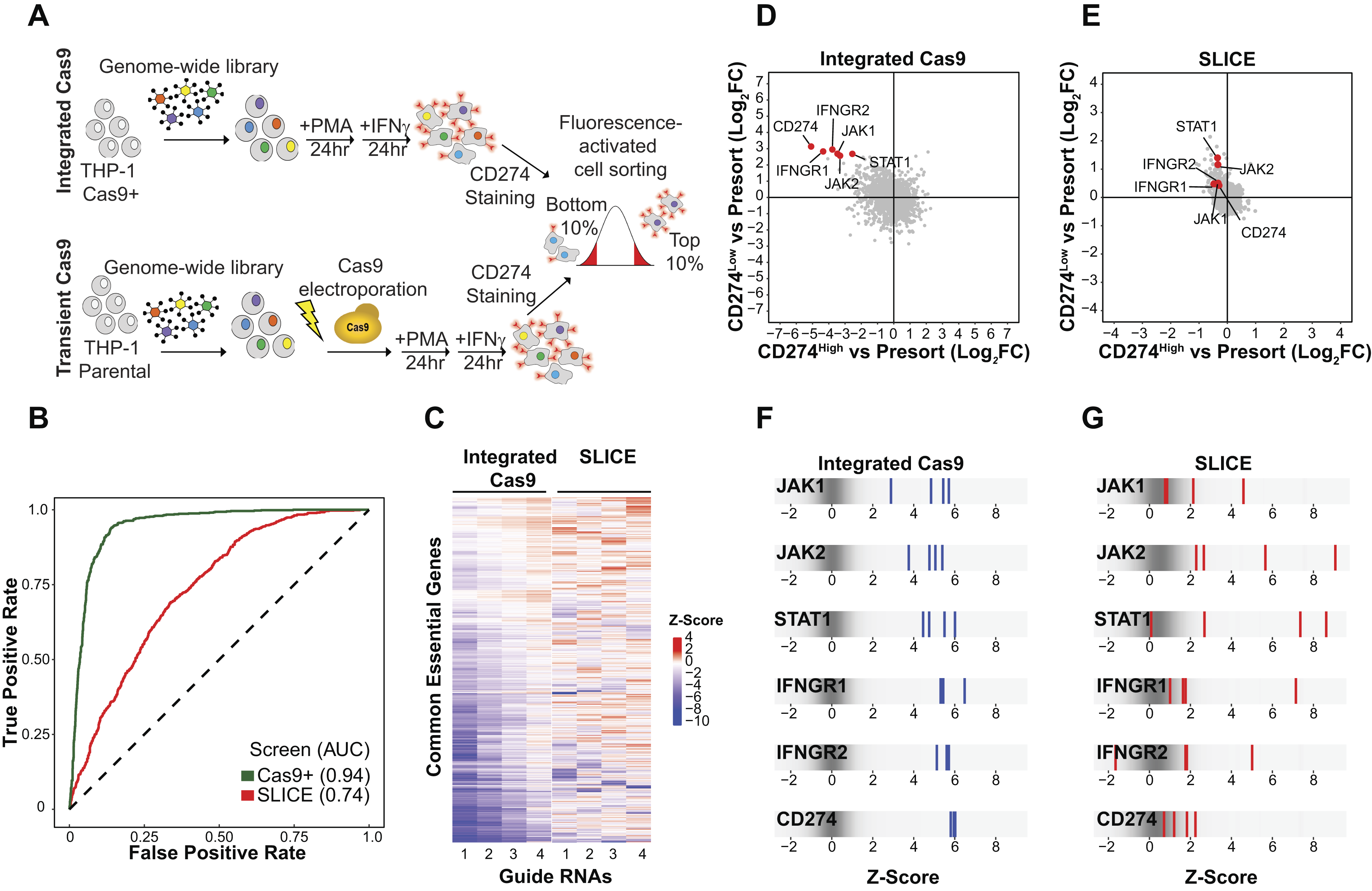
Performance of Brunello genome-wide CRISPR library with constitutive and transient Cas9 expression.
Receiver operating characteristic (ROC)–area under the curve (AUC) analysis of a logistic regression classifier that was trained with reference sets containing essential and nonessential genes was performed to estimate true-positive and false-positive rates for the Cas9-expressing cells and SLICE. This analysis revealed that the screen executed in Cas9-expressing cells (AUC = 0.94) outperformed the screen with transient Cas9 expression (AUC = 0.74) (Fig. 1B). To further understand the difference in library performance between the stable and transient Cas9 screens, we compared the performance of the four gRNAs for each common essential gene in both screens. While the Z-score for guides targeting the same gene was well-correlated in the integrated Cas9 screen, this correlation was markedly reduced in the SLICE screen, indicating that the consistency of some individual guides is dependent on the mode and duration of Cas9 expression (Fig. 1C).
FACS-based screening identified key regulators of IFNγ-induced PD-L1 expression (Fig. 1D–E, Supplementary Fig. S1).16–18 As expected, in Cas9-expressing cells, the core IFNγ signaling pathway genes (JAK1/2, STAT1, IFNGR1/2) and PD-L1 (CD274) were identified as top-enriched genes in the PD-L1low population (Fig. 1D, Supplementary Fig. S1A). In the SLICE screen, some of the positive controls were identified as significant hits, including STAT1 (Log fold change [LFC] = 1.4) and JAK2 (LFC = 1.2); however, strikingly, several core IFNγ signaling pathway genes, including IFNGR1 (LFC = 0.479), IFNGR2 (LFC = 0.504) and CD274 (LFC = 0.429), were not significantly enriched in the PD-L1low population (Fig. 1E, Supplementary Fig. S1C). The Z-scores of each of the four gRNA constructs for the known PD-L1 regulator genes were enriched to similar levels in the integrated Cas9 screen (Fig. 1F). In contrast, in the SLICE screen, the variability in Z-scores for guides targeting the same gene increased dramatically, with some guides showing no enrichment (Fig. 1G). Taken together, these results indicate gRNA enrichment within genome-wide screens is highly dependent on the mode of Cas9 expression and delivery.
Dual-guide RNA lentiviral infection followed by Cas9 electroporation (DICE) improves editing efficiency compared with SLICE
Multiplex Cas9 systems have emerged, which express multiple sgRNAs that target the same gene or multiple genes from the same lentiviral construct.11,19–21 These approaches improve gene editing efficiencies compared with single-guide systems. 22 Therefore, we reasoned that we could increase gene editing efficiencies in experimental contexts requiring nucleofection of Cas9 protein by leveraging a recently developed spCas9/dual gRNA approach wherein each lentiviral construct harbors two gRNAs targeting separate genes. This dual gRNA Cas9 (dual-Cas9) system features lentiviral constructs designed with separate U6-driven and H1-driven gRNA expression cassettes in opposing orientations. U6-VCR1 and H1-WCR3 promoter-tracr pairings were chosen based on superior efficacy and reduced recombination rate as previously reported. 11 Using this system, we designed lentiviral constructs with guides targeting both CD47 and CD63 expressed from both proximal and distal positions (Fig. 2A). We transduced cells with both lentiviral constructs and then introduced Cas9 by electroporation. We observed greater than 75% knockout of both CD47 and CD63 with both construct orientations, indicating that the U6 and H1 promoters express sgRNAs for similar gene editing efficiencies with transient Cas9 (Fig. 2A).
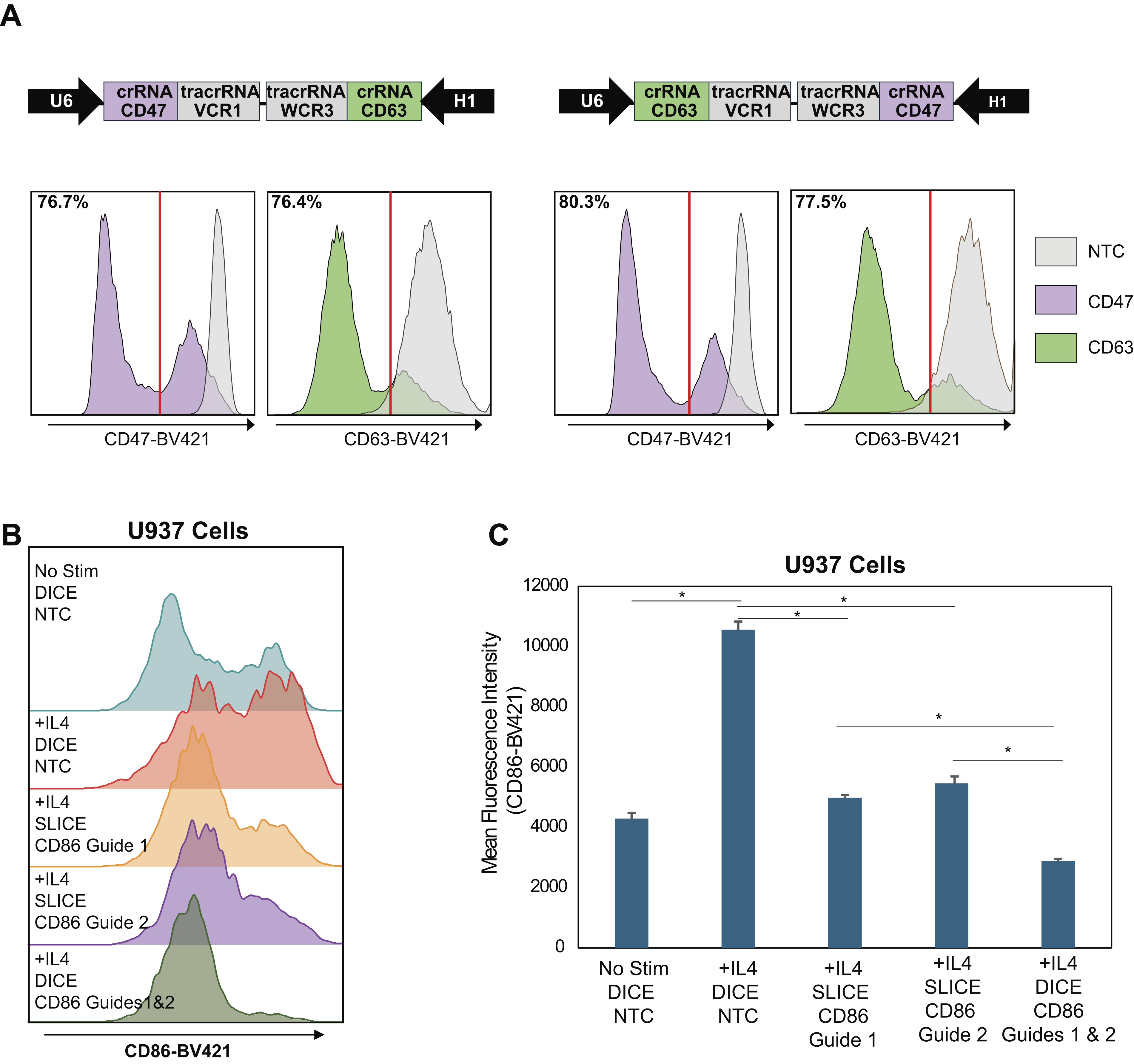
Development of DICE as a strategy for multiplex gene editing.
Previous reports have demonstrated that utilizing multiple guides targeting the same gene increases the activity for CRISPR-dependent gene modulation.23,24 To increase the editing efficiency of the single-gene perturbation Cas9 system, we designed dual-Cas9 constructs containing either single or two gRNAs targeting the cell surface protein CD86 (Fig. 2B–C). We assessed CD86 editing in cells transduced with lentiviral single- or dual-sgCD86 constructs followed by electroporation of Cas9 and IL4 stimulation. IL4 treatment increased the mean fluorescence intensity of CD86 by 2.5-fold (Fig. 2B–C) in cells containing the nontargeting control gRNAs. Single-guide expression (SLICE) for CD86 resulted in a significant reduction of CD86 surface expression compared with nontargeting containing cells. Cell surface expression of CD86 was further reduced in cells transduced with the dual guide expression construct (DICE). We thus present a dual gRNA lentiviral infection followed by Cas9 electroporation (DICE) approach, which shows improved editing efficiency compared with SLICE.
Design and benchmark of dual-Cas9 genome-wide library against the gold-standard Brunello library
To apply the DICE approach for genome-wide CRISPR screening, we designed a dual-Cas9 genome-wide CRISPR library (Fig. 3A). Given the observed increase in editing efficiency with two guides per perturbation as opposed to one, we reasoned that we could efficiently target each gene with one dual-Cas9 construct as opposed to the four separate single-guide constructs per gene in the standard Brunello genome-wide library, reducing the total library size by 75%. gRNA sequences for the dual-Cas9 library were chosen based on the top two gRNAs for each target gene as predicted by Rule Set 3, 25 which incorporates variations in tracrRNA sequences and improves accuracy in predicting on-target efficiency over previous models. As the Brunello library was generated prior to the development of Rule Set 3, ∼70% of guides in the dual-Cas9 library are unique, while ∼30% are also found in the Brunello library (Fig. 3A).

Miniature dual-Cas9 library provides comparable performance to the Brunello library.
Dual-Cas9 and Brunello libraries perform comparably to identify common essential genes and PD-L1 regulators in Cas9-expressing THP-1 macrophages
We first compared the dual-Cas9 and Brunello library screening performance in THP-1 macrophages with stable Cas9 expression. The Pearson correlation for essential genes between the dual-Cas9 and Brunello was 0.806, and the AUC score from ROC curve analyses indicated high and comparable performance between the two libraries in the detection of essential genes (Brunello, AUC 0.94; dual-Cas9, AUC 0.93) (Fig. 3B and C). In addition to common essential genes, we assessed whether our dual-Cas9 library was able to identify known regulators of PD-L1 expression in response to IFNγ treatment (Fig. 1A and 3D). We plotted the median LFC of genes in our sorted PD-L1low versus our PD-L1high populations and found that all five of the known regulators, as well as PD-L1 (CD274), were enriched in the PD-L1low population and depleted in PD-L1high population in dual-Cas9 screen (Fig. 3D, Supplementary Fig. S1B). When we compared the Z-score of each sgRNA construct in the Brunello (red) and dual-Cas9 (blue) libraries for five known regulator hits in our PD-L1low population, we observed high enrichment and tight distribution of all constructs targeting each gene (Fig. 3E). Collectively, these results demonstrate that the novel dual-Cas9 library identifies regulators of PDL1 expression at a similar robustness as the gold-standard Brunello library.
DICE outperforms SLICE for genome-wide CRISPR screening
Given that our dual-Cas9 library performed comparably to the Brunello library in cells that stably express Cas9 and that gene editing efficiency is higher with the DICE approach compared with SLICE, we hypothesized that the dual-Cas9 library would outperform the Brunello library in screens where Cas9 expression is transient. The DICE platform would thus provide more robust gene editing to improve screening quality as well as enable genome-wide scale screens in cell models where scale is limited. We compared the performance of SLICE (Brunello) and DICE (dual-Cas9) genome-wide screens in our viability and FACS-based screens (Fig. 1A). Depletion of essential genes was more robust in the DICE screen compared with SLICE, indicating improved editing efficiency with the dual-Cas9 library compared with Brunello (Fig. 4A). As the sgRNA composition of the two libraries is not identical, performance of each library could depend on the repertoire of sgRNAs in each library and/or the dual compared with the single sgRNA library format. The overlap of the guides between the two libraries allowed for comparison of the performance on a subsample of essential genes. Essential genes where both guides in the dual-Cas9 library are also present in the Brunello library show similar dropout rates in THP1-Cas9+ cells (Fig. 3B); however, in cells requiring nucleofection of Cas9 protein, dropout of essential genes was more robust using the DICE approach (Fig. 4A). Similarly, the DICE approach significantly outperformed the SLICE approach as shown by ROC–AUC analysis (Fig. 4B). We next evaluated whether the DICE screening platform was better able to identify known regulators of PD-L1 compared with SLICE. In the FACS-based screens, DICE identified all five known regulators of PD-L1 as significantly enriched in the PD-L1low population, whereas LFCs for known PD-L1 regulators were lower in the SLICE screen, and PD-L1 itself was not identified as a statistically significant hit (Figs. 1E, Fig. 4C, and Supplementary Fig. S1D). Moreover, the Z-score for each sgRNA construct in the Brunello (red) were generally lower than the guides in the dual-Cas9 (blue) library for known regulators of our PD-L1 (Fig. 4D). Both guides present in the dual-Cas9 library for JAK1 are also represented in the Brunello library. While the dual-Cas9 construct and shared guides for JAK1 performed comparably in the integrated Cas9 screens (Fig. 3E), neither individual guide construct in the Brunello library performed as well as the dual guide construct in the transient Cas9 screens (Fig. 4D). Collectively, these data demonstrate that the DICE approach significantly outperforms the SLICE in viability and FACS-based CRISPR screens.
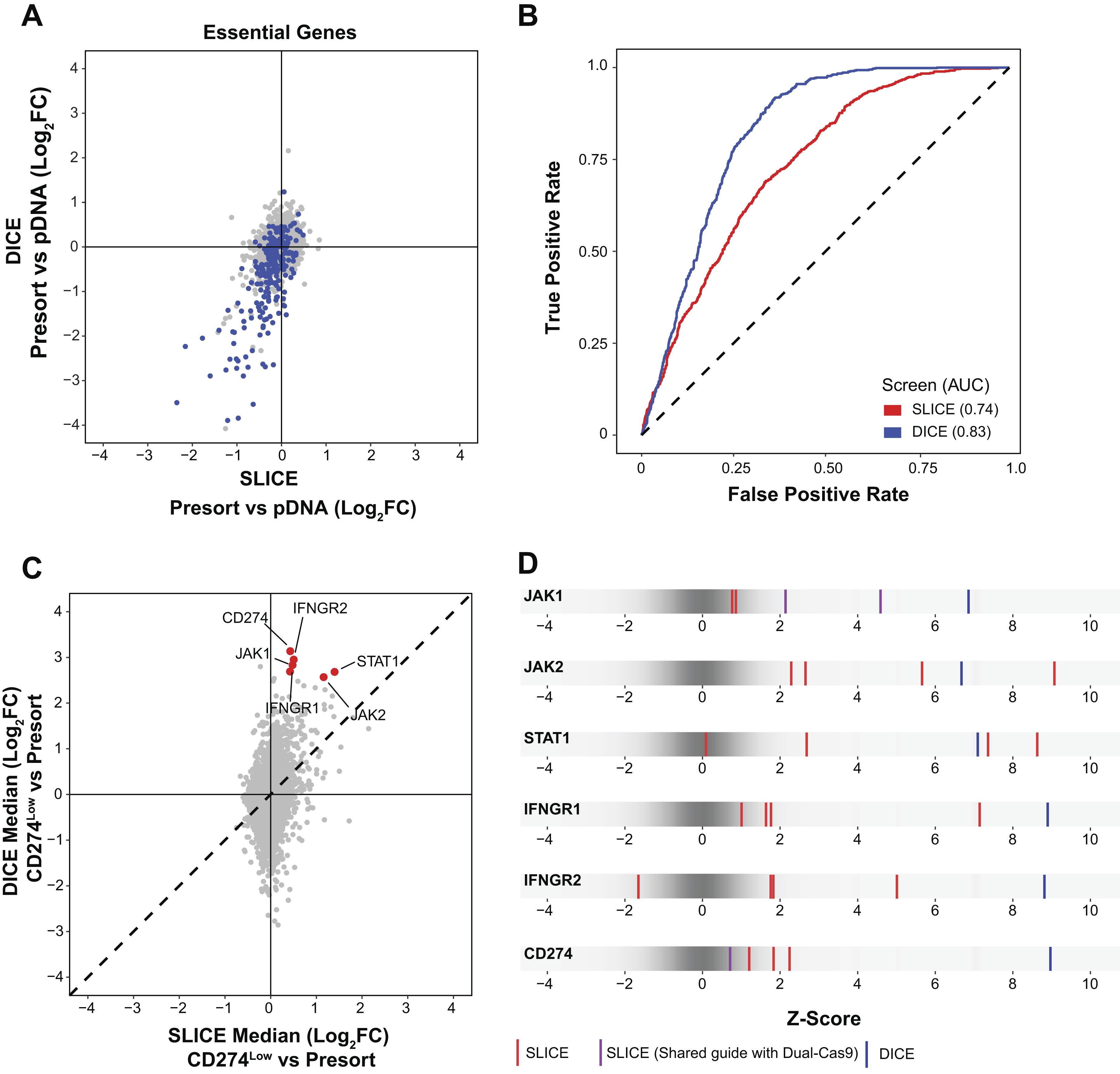
Dual-Cas9 outperforms Brunello to identify essential genes and PD-L1 regulators in THP-1 macrophages when Cas9 is introduced transiently.
Discussion
In this study, we present a new approach for improved CRISPR editing using single-target, dual sgRNA expressing lentiviral constructs and Cas9 protein electroporation. We designed a genome-wide dual sgRNA (dual-Cas9) library wherein each gene is targeted by two separate sgRNAs expressed from a single construct, reducing the library size by 75% compared with the gold standard Brunello genome-wide library. We compared the performance of the dual-Cas9 genome-wide CRISPR library to the Brunello library using both viability- and FACS-based screening readouts. We confirm that the dual-Cas9 library performs comparably to the Brunello library in cells expressing Cas9 but significantly outperforms the Brunello library in screens where Cas9 is delivered exogenously. The observation that both libraries perform similarly in the context of constitutive Cas9 suggests that genome-wide screens can be scaled down by 75% without missing valuable biological information. Miniature libraries can enable genome-wide screening in cell types where cell numbers are limited, such as primary immune and neuronal cells. At the same time, the dual-Cas9 library, which expresses two sgRNAs targeting the same gene, provides a performance advantage over the Brunello library in screens where Cas9 must be introduced by electroporation. A potential explanation for improved screening quality with the dual-Cas9 despite reduced library size is that under transient expression, Cas9 protein has twice as many opportunities to edit the genome in each cell with the dual-Cas9 system compared with a single-guide library. This explanation is supported by the tight distribution of LFCs for Brunello and dual-Cas9 library constructs targeting known PD-L1 regulators in screens in THP-1 Cas9+ cells, but relative loss of enrichment of many Brunello constructs compared with dual-Cas9 constructs in the SLICE screens. In SLICE experiments, key members of the JAK/STAT pathway were not identified as significant hits when using the Brunello library, suggesting that single-guide libraries have the potential to miss important regulators of the targeted biology. While dual sgRNA systems provide more robust gene editing than single-guide approaches, generating multiple double-stranded DNA breaks may increase the presence of off-target effects, increase the probability of both deletions and translocations, and may induce p53-dependent cell death.26,27 Therefore, careful evaluation of sgRNA expression is warranted prior to phenotypic screening to prevent the potential deleterious effects of multiple guides mentioned above. Despite these potential liabilities, our dual-Cas9 platform fills critical gaps in CRISPR screening capabilities to enable improved screen performance when Cas9 is transient and reduces genome-wide screening scale, both of which are critical for screening in primary human immune and neuronal cells.
Conclusion
This study presents a novel dual-Cas9 genome-wide CRISPR library consisting of one gRNA construct per gene and compares its performance to the four-construct-per-gene Brunello library. In both viability- and FACS-based screens, the dual-Cas9 library performs similarly to Brunello when Cas9 is constitutively expressed and outperforms Brunello when Cas9 is delivered exogenously. The dual-Cas9 platform enables screening with 75% fewer resources without compromising quality, making it ideal for primary cells with limited numbers. Moreover, its superior performance in contexts with exogenous Cas9 highlights the potential of the dual-Cas9 library to capture biologically valuable information missed by single-guide libraries.
Footnotes
Acknowledgments
The authors thank David Root, Olivia Bare, John Doench, and other members of the Genetic Perturbation Platform of The Broad Institute (Cambridge, MA) for the Brunello library as well as the design and cloning of the dual-Cas9 sgRNA library. The authors thank Jing Wang, Si Wu, and Fedik Rahimov for the critical review of the article.
Authors’ Contributions
C.P., C.L., M.J.F., and J.D.S. conceived, designed, and led the study and drafted the article; C.P. and V.V. performed experiments; C.L., A.M., C.P., and J.D.S. analyzed the data; S.K., M.J.F., and A.I.d.H. provided resources and critical feedback. All authors reviewed the article, provided feedback, and approved the final version of the article.
Author Disclosure Statement
All authors are or were employees of AbbVie at the time of the study. The design, study conduct, and financial support for this research were provided by AbbVie. AbbVie participated in the interpretation of data, review, and approval of the publication. No honoraria or payments were made for authorship.
Funding Information
No funding was received for this article.
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.