
Abstract
Recently, a new family of CRISPR-Cas12 endonucleases from an unexplored phylum of bacteria, Armatimonadota, was discovered. Named Cas12l, they are compact (800–900 aa), recognize a 5′ C-rich protospacer adjacent motif, and present an N-terminal domain that stretches from the beginning to the end of the ribonucleoprotein-bound DNA target site, effectively locking it in place. Here, structure-guided rational design supplemented with AI-based large protein language model predictions was used to improve rates of DNA target cleavage of a family member, Asp2Cas12l. Compared to the wild-type, engineered variants exhibited an approximately 10-fold increase in double-strand break (DSB) editing efficiency in human cells with less target-to-target variation. Moreover, frequencies of editing were comparable to those of SpCas9 at overlapping target sites, and their DSBs efficiently corrected by homology-directed repair (39–56% of editing outcomes). Altogether, this study extends our understanding of CRISPR-Cas12 protein engineering and offers a potent new alternative for DSB-mediated genome editing in human cells.
Introduction
Over the past decade, Types II and V clustered regularly interspaced short palindromic repeats (CRISPR)-associated (Cas) 9 and Cas12 endonucleases, respectively, have emerged as flexible and efficient genome editing tools.1,2 Guided by a CRISPR RNA with ∼20 bp complementary to one strand of a double-stranded (ds) DNA target adjacent to a short (1–4 bp) protospacer adjacent motif (PAM),3–5 they offer unprecedented scalability and affordability for genome editing applications. As a result, they have been widely adopted to introduce site-specific genetic edits to further the understanding of gene function, generate new sequence variation, insert desirable traits, and correct disease-related alleles.6–14
Emerging innovations have sought to perfect their use and have attempted to remove any bottleneck that may limit their application. One constraint has been the length and composition of the PAM, as it restricts the genomic sequence space that can be targeted. This is further compounded by the selection of unique targets to minimize the risk of off- or secondary target cleavage.2,15 To address this, CRISPR-Cas endonucleases with different PAM recognition and improved targeting specificity have been identified or engineered.16–22 Research to improve the intracellular delivery of CRISPR-Cas editing tools has exposed additional obstacles. For viral delivery, more compact editors with shorter coding sequences that fit within viral genomes have been coveted23,24 and have helped propel the discovery of smaller CRISPR-Cas and transposon-related RNA-guided nucleases.25–27 For methods that deliver editing reagents as short-lived transient doses of mRNA or ribonucleoprotein (RNP), the amount of RNA-guided reagent, particularly those from type V systems, needed to achieve efficient editing or the potency of the enzyme has been an underexplored area, necessitating further optimization.28–30
To help address these limitations, we engineered a Cas12l endonuclease from a bacteria species that is yet to be defined—Armatimonadota species (Asp)—as a potent DNA double-strand break (DSB) editing tool. AspCas12l proteins are compact (800–900 aa) and recognize a 5′ CCY PAM, providing a key counterbalance to the 5′ T-rich PAM recognition afforded by most other Cas12 nucleases.31,32 To improve DSB editing efficiency, the ternary RNP-DNA target structure of a family member, Asp2Cas12l, was first solved to 2.51 Å using cryo-electron microscopy (cryo-EM). Guided by the structure, arginine amino acid substitutions were next introduced to fine-tune Asp2Cas12l electrostatic binding potential at DNA and sgRNA interfaces. DSB activity, measured as the frequency of insertion and/or deletion (indel) mutations resulting from cellular nonhomologous end-joining (NHEJ) repair, was improved by approximately 7-fold over the wild-type nuclease. Next, residues suggested by artificial intelligence (AI) algorithms to improve function were introduced and evaluated. In combination with structure-guided modifications, AI predictions bolstered Asp2Cas12l DSB activity and reduced target-to-target variation. When delivered as plasmid DNA, the best variant, M82, performed comparably to SpCas9 at overlapping sites and maintained DSB activity when delivered transiently as mRNA or RNP. Satisfied with its DSB potency, its utility for homology directed repair (HDR) applications was assessed in HEK293T cells where M82 boasted efficiencies of HDR-mediated editing ranging from 39% to 56% depending on target site and edit type. Finally, the target cleavage specificity of M82 was evaluated using TEG-seq and found to be similar to other Cas12 endonucleases.
Materials and Methods
Plasmid construction
Mammalian expression vector pcDNA3.1 containing a human codon-optimized Asp2Cas12l gene was synthesized to order by GenScript (Piscataway, NJ, USA). The expression construct harbored sequence encoding a SV40 nuclear localization signal (NLS) at the N-terminus and sequence encoding a nucleoplasmin NLS at the C-terminus of the Asp2Cas12l protein.
For the Asp2Cas12l mutant expression vector construction, DNA fragments encoding respective mutation were synthesized (Twist Biosciences) and cloned into the pCDNA3.1 Asp2Cas12l expression vector by restriction cloning.
To express single-guide RNAs (sgRNAs) that direct Asp2Cas12l to target sites, plasmids were synthesized that encode a U6 polymerase III promoter sequence, Asp2Cas12l sgRNA sequence, HDV ribozyme sequence, and a U6 terminator. At the 3′ end of each sgRNA sequence, a variable spacer sequence was included. Plasmids encoding different spacer sequences of DNA were constructed by restriction cloning.
To produce mRNA in vitro transcription templates, Asp2Cas12l sequences were cloned to a plasmid vector optimized for mRNA synthesis. This optimized plasmid vector contains 5′ and 3′ human hemoglobin beta subunit (HBB) UTR sequences, T7 RNA polymerase promoter, and kanamycin resistance gene. To clone target genes into this optimized vector, Asp2Cas12l gene sequences were amplified by PCR, during which overlapping sequences from the destination vector were added; vector was also amplified by PCR, during which overlapping sequences of the target genes were added to the 5′ and 3′ ends of a linear PCR product.
mRNA production
Plasmids encoding Cas12l mRNA templates were used as PCR templates to produce in vitro transcription templates for mRNA synthesis. During this PCR step, target sequences were amplified using universal primers. Reverse primer is elongated and used for 120 nt poly(A) tail addition. After PCR, products were confirmed by gel electrophoresis and purified from PCR reactions using New England Biolabs Monarch PCR and DNA Cleanup Kit (5 µg); concentrations were measured using spectrophotometry (NanoPhotometer® NP80, IMPLEN). Approximately 1 µg of each purified PCR product was then used as a template for in vitro transcription reaction performed using HiScribe® T7 mRNA Kit with CleanCap® Reagent AG (New England Biolabs); kit-provided UTP was not used; instead, N1-Methylspeudouridine (TriLink Biotechnologies or Thermo Fisher Scientific) was added to a final concentration of 5 mM. In vitro transcription reaction was run for 1 h at 37°C after which DNaseI and nuclease-free water were added to the reaction and incubated for 30 min at 37°C to remove template DNA. After this step, the IVT reaction product was purified using the Monarch RNA Cleanup Kit (500 µg). Parts of the purified samples were set aside to assess their quality. Sample concentrations were measured using spectrophotometry (NanoPhotometer® NP80, IMPLEN) and fluorimetry (Qubit RNA Broad Range Assay Kit and Qubit 4, Thermo Fisher Scientific). mRNA integrity was assessed by performing capillary electrophoresis using 4150 TapeStation System together with RNA Screen Tape reagents (Agilent). Synthesized mRNA was aliquoted and kept at −80°C until use.
Cell culture
HEK293T and ARPE-19 cells were cultured in Dulbecco’s modified Eagle medium (DMEM) with GlutaMAX (Thermo Fisher Scientific), supplemented with 10% fetal bovine serum (Thermo Fisher Scientific) and 1% Penicillin-Streptomycin (10,000 U/mL) (Thermo Fisher Scientific) at 37°C in 5% CO2. All cells were seeded into 96-well plates (Thermo Fisher Scientific) 1 day before transfection at a density of 2 × 105/mL, 90 µL per well for HEK293T cells and of 1 × 105/mL, 100 µL per well for ARPE-19 cells.
Cell transfection
Cells were transfected using transfection reagent FuGENE® HD (Promega Corporation, Madison, WI, USA) according to the manufacturer’s protocol. For each sample, 300 of DNA containing 43 fmol of plasmid encoding Asp2Cas12l and 43 fmol of plasmid encoding guide RNA was used. A 3:1 ratio of FuGENE® HD Transfection Reagent to DNA was used (0.3 µL reagent:100 ng DNA per well). Cells were incubated at 37°C with 5% CO2 for 4 days posttransfection before cell lysis and further genome editing evaluation.
For mRNA transfection, HEK293T cells were seeded into 96-well plates (Thermo Fisher Scientific) 1 day prior to transfection at a density of 18,000 cells per well. Cells were transfected using Lipofectamine MessengerMAX Transfection Reagent (Invitrogen) following the manufacturer’s recommended protocol. Unless stated otherwise, for each well of a 96-well plate, a total amount of 100 ng RNA was used. This total amount consists of mRNA and sgRNA in a molar ratio of 1:4.
RNP electroporation (HEK293T)
Two days before the electroporation, HEK293T cells were split and maintained as described above. On the day of the experiment, cells were washed using Dulbecco’s phosphate saline buffer (DPBS) (Thermo Fisher Scientific) and trypsinized using TrypLE Express Enzyme (1X) (Thermo Fisher Scientific). Collected cells were resuspended in DPBS and diluted in SF Cell Line Nucleofector Solution (Lonza) supplemented with Supplement 1 (Lonza). RNPs were assembled in Nucleofector solution supplemented with Supplement 1 using 126 pmol of each purified nuclease and 160 pmol of sgRNA. RNP assembly reactions were incubated at room temperature for 20 min, then kept at 4°C until nucleofection. In total, 2 × 105 cells per sample were added into the RNP assembly reaction, and the reaction was transferred to 16-well Nucleocuvette Strips (Lonza). Cells were electroporated using preprogrammed protocol CM-130 using 4D-Nucleofector (Lonza). After electroporation, contents of each well were seeded into wells containing prewarmed DMEM with GlutaMAX (Thermo Fisher Scientific), supplemented with 10% fetal bovine serum (Thermo Fisher Scientific) in a 96-well plate (Thermo Fisher Scientific). Cells were then grown for 48 h at 37°C in 5% CO2 before cell lysis and further genome editing evaluation.
Asp2Cas12l-mediated HDR
HEK293T cells were electroporated with Asp2Cas12l–sgRNA or SpCas9 RNP complexes targeting the AAVS1 locus along with a dsDNA HDR template encoding the EGFP gene. 126 pmol of each purified Asp2Cas12l nuclease variant was mixed with 160 pmol of synthetic sgRNA. In the case of SpCas9, 50 pmol of EnGen® Sp Cas9 NLS (New England BioLabs) with 100 pmol sgRNA along with 500 ng of donor DNA. The mixtures were incubated for 15–20 min at room temperature. RNP electroporation performed as described earlier. The efficiency of EGFP knock-in was determined by MACSQuant® Analyzer 16 Flow Cytometer (Miltenyi Biotec); flow cytometry data were analyzed with FlowJo™ Software v10.10.0 (BD Biosciences). Single-nucleotide substitution knock-in efficiency was quantified by next-generation sequencing (NGS).
For the single-stranded oligo donor DNA-mediated HDR experiments, HEK293T cells were electroporated with Asp2Cas12l M82 nuclease–sgRNA RNP complex, targeting the AAVS1 or CD151 genes, along with 100 pmol of ssODN template encoding a 1 or 2 nt substitution, a 51 nt left homology arm, and 50 nt right homology arm. RNP electroporation performed as described earlier.
Evaluation of genome editing
Cells were incubated at 37°C with 5% CO2 for 4 days posttransfection before genomic DNA extraction. The cells were washed twice with 200 µL prewarmed to 37°C 1X DPBS (Thermo Fisher Scientific) and resuspended in 25 µL cell lysis solution containing 50 mM Tris-HCl, 150 mM NaCl, 0.05% Tween 20, pH 7.6 (Sigma Aldrich), and 0.2 mg/mL Proteinase K (New England Biolabs). Resuspended cells were incubated at 55°C for 1 h and 95°C for 15 min.
DNA fragments in the genomic DNA extracted by cell lysis surrounding each Cas12l target site were amplified by PCR. PCR reactions were performed using Q5 Hot Start High-Fidelity 2X Master Mix (New England Biolabs) according to the manufacturer’s instructions. The reactions were set up using 1 µL of the cell lysate and 0.5 µM of each primer in a final reaction volume of 25 µL.
For genome editing efficiency assessment by T7 Endonuclease I assay, 20 μL of each PCR reaction was combined with 3 μL NEBuffer 2 (New England Biolabs) and 7 μL of water before denaturation at 95°C for 5 min and re-annealing by temperature ramping from 95°C to 85°C at −2°C/s followed by ramping from 85°C to 25°C at −0.1°C/s. In total, 1 μL of T7 Endonuclease I (New England Biolabs) was added to each reannealed sample, and cleavage reactions were incubated at 37°C for 20 min. Fragments were analyzed by performing gel electrophoresis using E-Gel Precast Agarose Gel Electrophoresis system (Invitrogen). Overall, 8 µL of each sample was mixed with 7 µL of E-Gel Sample Loading Buffer (Invitrogen), and then the whole volume was loaded to the well. Cleaved DNA fraction was evaluated by densitometric analysis using ImageJ software.
Illumina NGS
Cell lysates of transfected HEK293T cells were used to amplify genomic target regions, and fragments were extended by Illumina sequencing that included a unique index for each sample through two rounds of PCR. A total of 4 µL of each lysate sample was used as a template in the primary PCR reaction. For the primary PCR custom primers were used that were complementary to the sequences surrounding the genomic targets and had noncomplementary “tails” with Illumina adapter sequences. Q5 HotStart 2x MasterMix (New England Biolabs) was used for the primary PCR, and the reaction was set up using 4 µL of cell lysate as template and 0.2 μM of each primer in a final volume of 25 µL. The cycling conditions used were 98°C for 2 min 30 s, 24 cycles of 98°C for 30 s, 56.5°C for 30 s, 72°C for 25 s, and final extension at 72°C for 2 min. The primary PCR product was purified using SPRIselect (Beckman Coulter) magnetic beads and used for the secondary PCR.
Secondary PCR was performed using 2X Q5 High-Fidelity Hot Start PCR Master Mix (New England Biolabs) with custom primers (synthesis ordered from Integrated DNA Technologies) containing Illumina sequences and i7 and i5 indexes (on reverse and forward primers, respectively). Cycling conditions for secondary PCR were as follows: 98°C for 2 min, 10 cycles at 98°C for 10 s, 60°C for 30 s and 72°C for 2 min, followed by final extension at 72°C for 2 min, when using 2X Q5 High-Fidelity Hot Start PCR Master Mix (New England Biolabs) and custom primers, or 98°C for 30 s, 10 cycles at 98°C for 10 s, 65°C for 1 min 15 s, followed by final extension at 65°C for 5 min. The secondary PCR products were purified using SPRISelect (Beckman Coulter) magnetic beads, and their quantity and quality were checked using spectrophotometry (NanoPhotometer® NP80, IMPLEN) and fluorimetry (Qubit 1X dsDNA HS Assay, Qubit 1X dsDNA BR Assay, and Qubit 4, Thermo Fisher Scientific). The purified samples were pooled in an equimolar ratio and size selection performed using SPRIselect (Beckman Coulter) magnetic beads. The resulting library quantified using NEBNext Library Quant Kit for Illumina (New England Biolabs). Final library pool was prepared for deep sequencing according to Illumina’s specifications. Paired-end sequencing was performed using the MiSeq Reagent Kit v2 (300 cycles) (Illumina) on the MiSeq System (Illumina) with 7% PhiX v.3 (Illumina). All sequencing data analysis was done using Geneious Prime 2025.0. Reads were trimmed and filtered using BBDuk and mapped to the reference sequence with a 50 nt quantification window set around the target site. Reads that had differences from the reference sequence in this window were included in genome editing efficiency calculations.
Results
Cryo-EM structure of Asp2Cas12l-sgRNA-dsDNA complex
The cryo-EM structure of wild-type Asp2Cas12l in complex with sgRNA and its DNA substrate was determined at a resolution of 2.51 Å (see Supplementary Data S1). To maximize R-loop formation, a 16 nt long nontarget strand without complementarity to the target strand was utilized (Fig. 1A–C, Supplementary Fig. S1, Supplementary Table S1). The structure revealed extensive similarity to a related nuclease, Asp3Cas12l, which was published during the preparation of this study (labeled CasPi-2 in the referred publication) (Fig. 1D).31,32 At the sequence level, these proteins shared 43.6% amino acid identity, however, the ternary complexes superimposed with an root-mean-square deviation (RMSD) distance of 1.022 Å over 600 pruned residue pairs in ChimeraX. 33 Protein structure comparison via the Dali server confirmed the high structural similarity with a Z-score of 38.3. 34 In general, functional domains of both proteins were arranged similarly. Minor differences included several flexible and thus unresolved stretches of amino acids in the Asp2Cas12l structure, a slightly different path of the proline-rich string (PRS) as it encircled the ternary complex, and a segment containing two antiparallel beta-strands in the helical and NTSB chimera (HNC) domain of Asp3Cas12l, which was not present in Asp2Cas12l (Fig. 1D). Therefore, we adopted the same naming convention for Asp2Cas12l as reported earlier (Fig. 1A–B). 32

The structure of the Asp2Cas12l-sgRNA-DNA ternary complex.
The sgRNA of Asp2Cas12l and Asp3Cas12l also formed similar architecture motifs with a difference observed in the positioning of the crRNA repeat and tracrRNA antirepeat duplex relative to the rest of the complex (Fig. 1D, Supplementary Fig. S2a). Defined by a tetrapartite structure, the sgRNA for Asp2Cas12l comprised a junction region that linked the other RNA folds, a large scaffold stem that interfaces with the HC domain, a duplex stem rising from the crRNA repeat and tracrRNA antirepeat interactions, and an RNA pseudoknot structure that orients the spacer into the DNA binding pocket for R-loop formation (Fig. 1E, Supplementary Fig. S2b).
Rational engineering of CRISPR-Cas12l
DSB-stimulated indel editing with Asp2Cas12l and its relatives was previously shown to be less efficient and more target site dependent compared to SpCas9 in human cells. 32 To ameliorate this, the Asp2Cas12l-sgRNA-DNA structure was first analyzed for amino acids whose atoms were less than 0.4 Å away from the negatively charged phosphodiester backbone of the DNA target. Each position was then substituted with a positively charged arginine residue (if not already arginine) to increase its electrostatic attraction to the target site. To improve RNP complex formation, positions shown to be near the sgRNA were similarly modified. In all, 34 positions were altered (Fig. 2A).
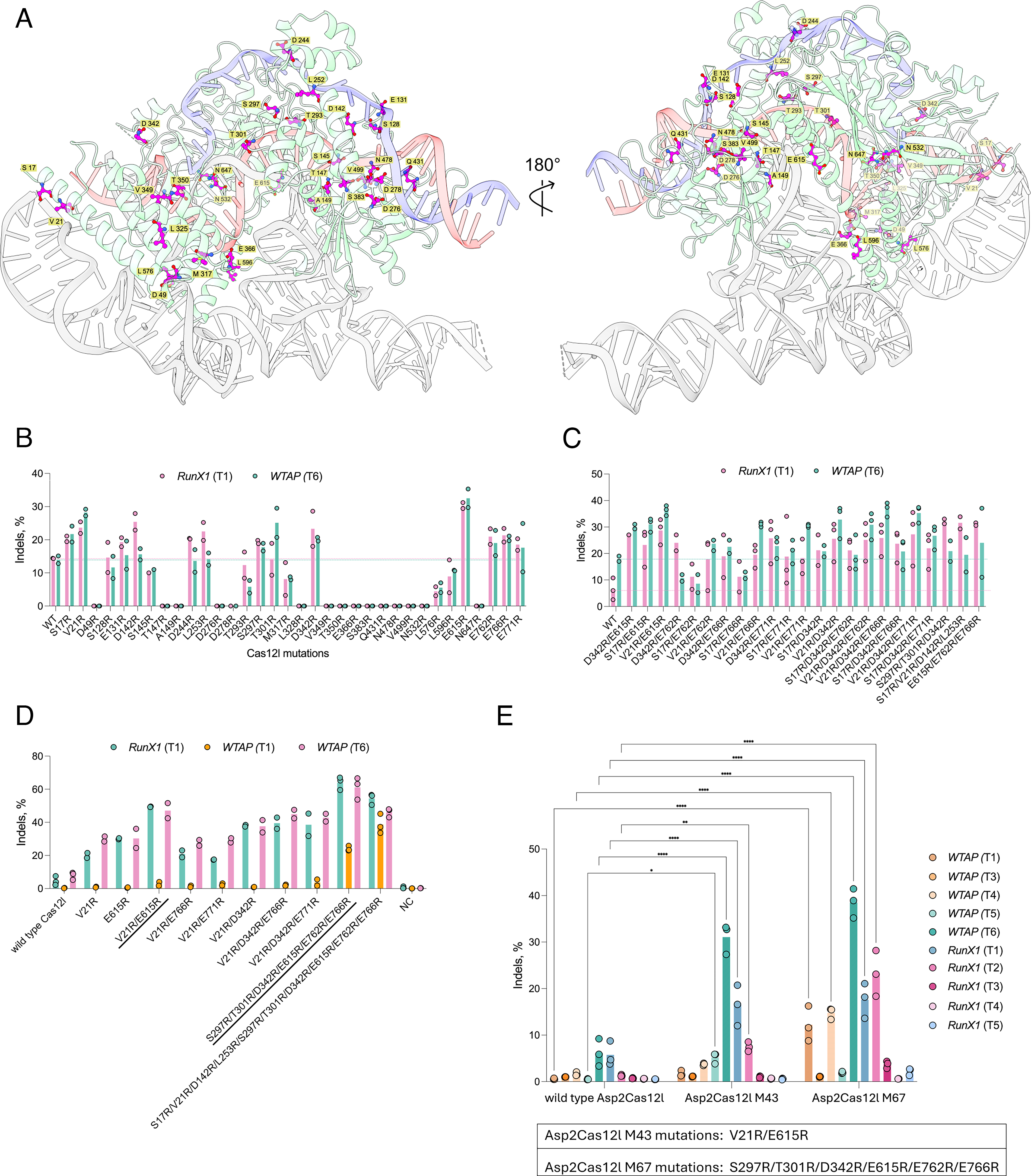
Asp2Cas12l mutagenesis and HEK293T DNA editing.
Next, the indel editing efficiency of each of the 34 variants was evaluated. For this, plasmid DNA encoding each variant and a sgRNA targeting the RunX1 (T1) and WTAP (T6) sites were co-delivered into HEK293T cells. Using T7 Endonuclease I to rapidly read out the target site mutation frequency, 13 of the 34 arginine substitutions enhanced indel rates for at least one of the tested target loci (Fig. 2B). These were next evaluated for additive effects by stacking substitutions located in different Asp2Cas12l domains in sets of two, three, or four (Fig. 2C). Exceptions included variants that combined S17R/V21R, S297R/T301R, or E762R/E766R (Fig. 2C). Based on the magnitude of improvement at the RunX1 (T1) target, S17R/V21R/D142R/L253R, S297R/T301R/D342R, and E615R/E762R/E766R were selected for a third round of stacking (Fig. 2C). For this, S297R/T301R/D342R and E615R/E762R/E766R were combined as well as one variant that united all three (Fig. 2D). When compared with the wild-type and other best-performing variants from the first and second rounds of engineering using Ampli-Seq, both enzymes outperformed all others (Fig. 2D). Between the two, S297R/T301R/D342R/E615R/E762R/E766R exhibited the highest rates of editing at the RunX1 (T1) and WTAP (T6) targets, while the one with 10 arginine substitutions had an advantage at the WTAP (T1) site (Fig. 2D). Given this, S297R/T301R/D342R/E615R/E762R/E766R (M67) was selected and evaluated on a larger collection of target sequences in WTAP and RunX1 loci in HEK293T cells (Fig. 2E). The potent improvement afforded by just two alterations, V21R/E615R (M43), at the RunX1 (T1) and WTAP (T6) targets also prompted its testing at additional sites. As averaged across ten targets, M43 and M67 produced a 3.5- and 7.1-fold improvement in indel editing, respectively, compared to wild-type Asp2Cas12l (Fig. 2E).
Close inspection of the amino acid changes in M67 revealed that three of the six arginine substitutions, S297R, T301R, and D342R, resided close to the DNA backbone of the target strand and were hypothesized to enhance target DNA engagement (Supplementary Fig. S3a,b). E615R was situated near the sgRNA pseudoknot, potentially stabilizing it (Supplementary Fig. S3c). Lastly, E762 and E766 were found in a disordered stretch of the C-terminal domain (Supplementary Fig. 3d) and posited to help orient the RuvC domain for optimal DNA target cleavage.
Mutagenesis via large language model predictions
While Asp2Cas12l M67 enhanced rates of targeted indels at some sites, several remained recalcitrant to editing (Fig. 2E). To improve further, M67 was subjected to in silico mutagenesis, and the fitness of the resulting variants was predicted using the 15B parameter ESM-2 protein large language model paired with zero-shot learning.35,36 For this, variant fitness was measured via a masked marginal scoring function, which outputs the log odds ratio of the mutated position, reflecting how likely that amino acid can be naturally found in a similar sequence context. 35 Assuming that evolution tends to preserve function, a higher score increases the probability that the mutation will have a positive effect. 37 Based on this, the nine highest scoring zero-shot predictions were selected, individually introduced into M67, and tested for improvements in HEK293T cells (Fig. 3A). Q572R exhibited around a 2-fold higher mean indel frequency compared to the parental M67 variant at the RunX1 (T5) target and retained activity at WTAP (T6) and RunX1 (T1) sites (Fig. 3B). Since F607S maintained similar editing efficiencies on all targets, it and Q572R were selected for testing across a wider collection of targets (Fig. 3C). The combination of Q572R and F607S retained similar indel efficiencies on target sites previously shown to be efficiently (>20%) edited (W4, W6, R1, R2) and increased indel rates at those that were not (W1, W2, W3, R3, R4, R5) (Fig. 3C). Q572R had a stronger impact than F607S, but in tandem and when averaged across targets, they outperformed Q572R alone, resulting in the Asp2Cas12l M82 variant containing the combined mutations S297R/T301R/D342R/Q572R/F607S/E615R/E762R/E766R (Fig. 3C). Positioned in the bridge-helix domain and close to the phosphodiester backbone of the sgRNA spacer and PRS, Q572R was reasoned to stabilize sgRNA-mediated DNA target recognition similar to that posited for S297R, T301R, and D342R and/or promote PRS wrapping (Supplementary Fig. S4a). Located in the RuvC domain and pointing into the sgRNA pseudoknot, F607S was thought to modulate sgRNA binding (Supplementary Fig. S4b).

LLM-based Asp2Cas12l M67 mutagenesis.
To further validate potency, the indel editing efficiency of Asp2Cas12l M67 and M82 was compared when delivered transiently as either RNP or mRNA into HEK293T cells. Here, indel rates were evaluated at WTAP (T6), as well as three previously untested target sites, either CD34, CD151, and AAVS1 or CD34, CD151, and VEGFA for RNP and mRNA delivery experiments, respectively (Supplementary Fig. S5a, b). When nucleofected as RNP, M82 outperformed M67 and exhibited high rates of targeted indels (74–99%) at all four target sites (Supplementary Fig. S5a). Co-lipofection of M67 and M82 mRNA and sgRNA produced a similar outcome with M82, resulting in higher indel rates than M67 (Supplementary Fig. S5b).
Benchmarking Asp2Cas12l M82 for DSB-mediated genome editing applications
To gauge the effectiveness of Asp2Cas12l M82 to introduce targeted indels, it was compared with SpCas9 at overlapping targets. For this, seven sites sharing the same PAM region (3′ RGG for SpCas9 and 5′ CCY for Asp2Cas12l) with each enzyme’s sgRNA base pairing on opposite strands of the same target were selected. When delivered as plasmid DNA into HEK293T cells, three of the seven targets showed comparable indel rates between M82 and SpCas9 (Fig. 4A). Two of the remaining sites, WTAP T6 and VEGFA T10, were edited more efficiently with M82, while WTAP T4 and VEGFA T9 showed higher indel rates with SpCas9 (Fig. 4A). Altogether, the average rate of indel editing observed with Asp2Cas12l M82 (67.4%) was nearly identical to SpCas9 (64.8%) (Fig. 4A).
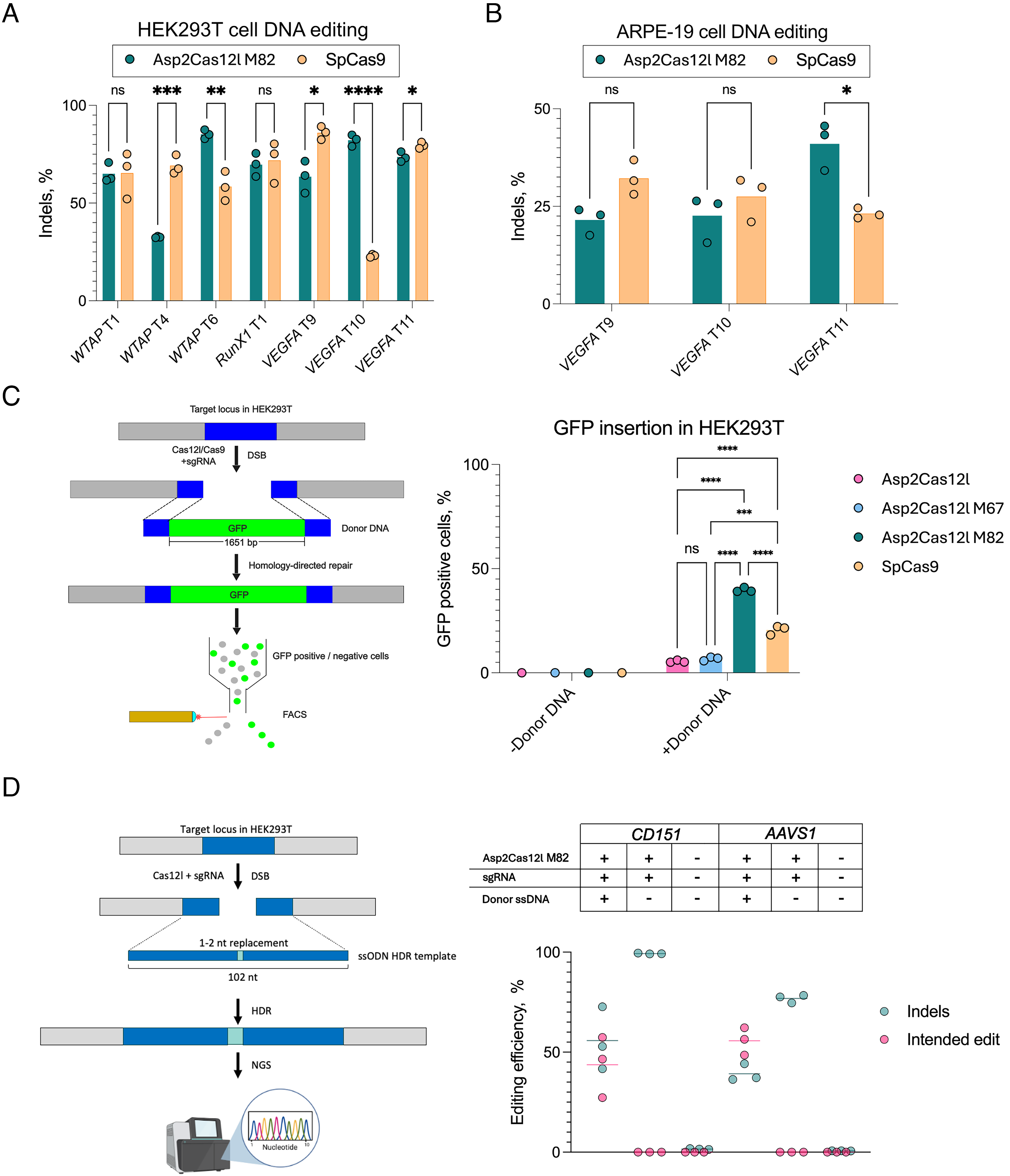
Asp2Cas12l M82 DNA editing validation and benchmarking.
To support therapeutic applicability, the VEGFA gene associated with age-related macular degradation37,38 in the human retinal cell line ARPE-19 was edited. In this case, mRNA encoding Asp2Cas12l M82 or SpCas9 was electroporated into cells, alongside the respective sgRNA. In ARPE-19 cells, Asp2Cas12l M82 exhibited similar editing efficiencies to SpCas9, with up to 40% indel editing as measured by T7 Endonuclease I (Fig. 4B).
Next, the utility of Asp2Cas12l for HDR was evaluated. Initially, the safe harbor AAVS1 locus in HEK293T cells was targeted for the site-specific insertion of a GFP expression cassette at overlapping SpCas9 and Asp2Cas12l sites (Fig. 4C). The exogenously supplied dsDNA repair template consisted of two 500 bp AAVS1 homologous sequences flanking the DSB site for SpCas9 with the GFP gene nestled in between (Fig. 4C). To induce the DSB, cells were electroporated with either Asp2Cas12l or SpCas9 RNP and donor DNA. After 5 days, fluorescence-activated cell sorting (FACS) was performed to measure the frequency of HDR (Supplementary Fig. S6). Asp2Cas12l M82 facilitated the highest mean percentage of GFP-positive cells (39.2%) and thus site-specific gene insertion compared to wild-type Asp2Cas12l (6.08%), M67 (6.99%), and SpCas9 (22.2%) (Fig. 4C).
To confirm these observations, the efficiency of HDR was examined at two additional target sites for Asp2Cas12l M82. In these experiments, single-stranded (ss) DNA donor templates with 50 bp of homologous sequence flanking both sides of the target cut site containing 1–2 nt replacement(s) were used (Fig. 4D). HEK293T cells were electroporated with Asp2Cas12l M82 RNPs and donor DNA, and HDR efficiency was quantified with Ampli-Seq. Depending on the target, 44–56% of sequence reads showed the seamless incorporation of 1–2 nt polymorphism(s) while indel editing outcomes ranged from 39% to 56% (Fig. 4D).
In vitro characterization of AspCas12l engineered variants
The biochemical attributes of Asp2Cas12l M43, M67, and M82 dsDNA target cleavage were next characterized in vitro (see Supplementary Data S1). Initially, this included the rate of linear dsDNA target cleavage at 37°C in the buffers established earlier for the wild-type enzyme (Supplementary Fig. S7). Surprisingly, the best DSB editor, Asp2Cas12l M82, did not exhibit the highest cleavage activity in vitro (Supplementary Fig. S7). To explore further, the effect of temperature, ionic concentration, and pH on linear dsDNA target cleavage were evaluated (Supplementary Figs. S8, S9). In doing so, it was found that the wild-type enzyme exhibited the broadest thermal profile (Supplementary Fig. S8), the engineered variants showed the greatest tolerance to increasing ionic concentrations (NaCl) (Supplementary Fig. S9a), and all variants functioned at pHs ranging from 5.5 to 8.0 (Supplementary Fig. S9b). Since the intracellular ionic composition of a HEK293 cell has been estimated to be ∼150 mM, 39 it was presumed to be a key factor behind the increased activity of the engineered variants.
Asp2Cas12l dsDNA target cleavage specificity
Next, dsDNA target cleavage specificity of Asp2Cas12l M82 was examined. Unbiased and with a sensitivity level equivalent to 0.0002–1% mutant reads by Ampli-Seq, TEG-seq was used to assess Asp2Cas12l M82 specificity (see Supplementary Data S1). 40 Briefly, HEK293T cells were electroporated with the corresponding Asp2Cas12l M82 RNP along with a DNA oligo duplex tag that was destined to be incorporated into intended and secondary target site DSBs. Next, genomic DNA was extracted, processed to enrich fragments containing the integrated tag via PCR, and sequenced using Ion Torrent chemistry. A custom analysis pipeline was then used to map sequence reads to the reference genome. Overall, four HEK293T sites were targeted by Asp2Cas12l M82 nuclease, and the potential for off or secondary target cleavage was evaluated (Fig. 5A, Table 1). Sequencing read depth, associated with putative secondary targets, varied between 0.06% and 3.7% across 48–149 loci, depending on the site (Table 1). Arising from low sequence read counts relative to the on-target, most of these were near or below the lower limit of TEG-Seq detection (Fig. 5A). As a control, TEG-seq was also performed with SpCas9 at a previously characterized target prone to secondary site cleavage in human cells, AAVS1 (Fig. 5B, Table 1). 41 In line with previous results, a much larger proportion of sequencing reads could be attributed to SpCas9 secondary versus on-target cleavage, 62.31% and 37.69% of reads mapped to secondary sites and the on-target across 90 loci, respectively (Fig. 5B, Table 1). Secondary targets with highest read counts also matched sites that were more heavily edited in the earlier study (Fig. 5). 41

Asp2Cas12l M82 specificity assessment in HEK293T. Nuclease specificity was evaluated using TEG-seq methodology.
Summary of Asp2Cas12l M82 secondary target activity evaluation via TEG-seq

To confirm these findings with an orthogonal approach, Cas-OFFinder was used to identify 29 putative secondary sites containing between three and nine mismatches for four on-targets, three in the CD5 locus and one in the VEGFA gene. CD5-targeting sgRNAs were individually co-transfected along with a mRNA encoding M82 into HEK293T cells. The VEGFA-targeting sgRNA and M82 were delivered similarly into ARPE-19 cells. Ampli-Seq was then used to measure indel rates at the respective on-sites and secondary targets. While on-targets showed detectable frequencies of indels, none of the secondary sites accumulated indels at frequencies above the negative control (DNA amplicons from untransfected cells) (Supplementary Fig. S10). Taken together, this suggests that the risk of Asp2Cas12l secondary target cleavage is low.
Discussion
Here, structure-guided rational engineering, zero-shot predictions from AI-based large language models trained on protein folding and stability, and the screening of multiple target sites in each round of engineering were applied to improve the DSB editing of Asp2Cas12l. In doing so, an improved variant, M82, was developed without laborious and expensive alternatives, like protein-directed evolution. 42 It contained eight amino acid substitutions compared to the wild-type parent, six from the structure-based rational engineering and two from AI-based suggestions. In combination, the alterations resulted in consistent and specific indel editing that rivaled those produced by SpCas9, independent of delivery method and human cell line used, with low target-to-target variability (Figs. 2E, 3C, 4A, B, 5; Supplementary Fig. S5a, b).
Close examination of the amino acid changes underlying Asp2Cas12l M82 were also suggestive of methods that may be used to streamline the improvement of other Cas12l orthologs or even Cas proteins. First, the observation that arginine substitutions enhanced DSB editing efficiency indicates that native Cas12l proteins may be deficient in sgRNA and DNA binding and that approaches aimed at improving these aspects may generally improve editing activity. Second, when M82 alterations were mapped (either through multiple sequence alignment of AlphaFold predicted structures) onto related Cas12l proteins, they were strongly conserved and therefore might represent evolutionary hotspots for the improvement of orthologs (Supplementary Fig. S11). Indeed, some Cas12l family members already exhibited positionally equivalent amino acid substitutions to those found in M82 (e.g., S297R, Q572R, E615R, E766R) (Supplementary Fig. S11). Finally, the masked marginal scoring from the 15B parameter ESM-2 protein large language model predictions lacked strong correlation with DSB editing efficiency in human cells (Fig. 3A, B). Since the model in essence predicts amino acid sequence plausibility based on orthologous sequence patterns rather than actual function, the small and relatively divergent evolutionary branch of known Cas12l nuclease family members may present a challenge for zero-shot predictions, and methods that remove this limitation may help future engineering campaigns.
In our study, it was observed that Asp2Cas12l M82 DSBs were also efficiently corrected by HDR (Fig. 4D, E; Supplementary Fig. S6). While this warrants further investigation, there may be several features inherent to M82 that may contribute to this outcome. As initially hypothesized for the Cas12a nuclease from Lachnospiraceae bacterium and later demonstrated with SpCas9, the continued or recurrent cleavage of target site indel mutations can produce a 2- to 3-fold improvement in SpCas9 HDR with frequencies exceeding 50% in rapidly dividing human cells.43–45 For Cas12 family members, this seems to occur naturally as part of their PAM-distal target cleavage mechanism, while for Cas9 one or more gRNA(s) need to be delivered to direct cleavage of the most prevalent NHEJ repair outcome(s).5,25,44–47 In addition, the 5–9 nt 5′ staggered cut produced by Asp2Cas12l target cleavage 31 may also enhance HDR, although other studies using paired SpCas9 nickases suggest that its contribution may be minimal.48,49 Furthermore, the unique framework by which Cas12l enzymes encircle their dsDNA targets via a pair of interlocking helical bundles and a PRS of charged (e.g., arginine and lysine) residues 32 may also bias repair toward HDR, and additional studies are needed to understand its impact (if any) and underlying mechanism(s).
Conclusions
Asp2Cas12l M82 presents itself as a potent new option for DSB-based genome editing applications. As highlighted herein, it can be delivered in a variety of different molecular formats, potentially enabling its introduction as RNP or mRNA either directly to cells or through engineered lipid nanoparticles (LNPs) or virus-like particles.50–52 Depending on application, this has several benefits that include further reducing risk for secondary target cleavage, decreasing cellular toxicity, and alleviating potential regulatory concerns.53–56 Moreover, it is compact in size (867 aa) making it compatible with many viral delivery systems, including adeno-associated viruses (AAVs).57,58 Finally, its targets are efficiently corrected by HDR, offering a fresh perspective on traditional DSB-mediated genome editing.
Authors’ Contributions
T.U., J.K.Y., and G.G. conceived the study. T.U., L.T., I.L., M.P., R.K., M.S., and M.S. performed the experiments. T.U., L.T., I.L., M.P., R.K., M.S., M.S., and G.G. analyzed the data. G.S. and G.T. determined and refined the cryo-EM structure. T.U. and G.G. wrote the article with contributions and guidance from J.K.Y.
Footnotes
Acknowledgments
The authors would like to thank David Papp and Mudra Hegde (Thermo Fisher Scientific) for the assistance in setting up the TEG-seq experiments and performing the data analysis. Also, they thank Ieva Narkeviciute and Greta Busmaite (Vilnius University Hospital Santaros Klinikos) for performing the cell sorting procedures.
Author Disclosure Statement
T.U., J.K.Y., and G.G. have filed patent applications related to the article. T.U., L.T., I.L., M.P., R.K., and G.G. are employees of Caszyme. M. Stitilyte and M. Sabaliauskas were employees of Caszyme at the time of the study. Current affiliation of M. Stitilyte—Gensinta, Vilnius, Lithuania; M. Sabaliauskas—Thermo Fisher Scientific Baltics, Vilnius, Lithuania J.K.Y. is an employee of Corteva Agriscience. V.S. is the chairman of Caszyme. V.S. and G.G. have a financial interest in Caszyme. The remaining authors declare that they have no conflicts of interest.
Funding Information
Research was conducted as part of the execution of Project “Mission-driven Implementation of Science and Innovation Programmes” (No.
Supplemental Material
Supplemental Material
Supplemental Material
Supplemental Material
Supplemental Material
Supplemental Material
Supplemental Material
Supplemental Material
Supplemental Material
Supplemental Material
Supplemental Material
Supplemental Material
Supplemental Material
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.