
Abstract
Clinical trials in oncology represent a moral and scientific contract: Patients accept risk and uncertainty with the expectation that results will be reported fully, accurately, and transparently to inform future care. Despite regulatory mandates and journal policies, nonpublication, selective outcome reporting, weak linkage between protocols, registries, and manuscripts, and limited access to individual participant data remain common, particularly among investigator-initiated trials. In an era of artificial intelligence (AI)-driven evidence synthesis and precision oncology, opaque or selectively curated trial data threaten the validity of clinical evidence and risk propagating bias into downstream AI systems. This commentary argues that data transparency must be treated as foundational infrastructure for AI-ready oncology trials rather than as a discretionary compliance task. We describe how AI can function as a “transparency engine” by automating trial–publication linkage, detecting protocol-to-article discordance, and converting unstructured trial reports into standardized, machine-readable results. Embedded within journal workflows and applied with appropriate human oversight, these tools can shift transparency from episodic auditing to continuous, verifiable practice—honoring obligations to trial participants and strengthening the reliability of oncology evidence and AI-enabled care. This commentary is intended for trialists, journal editors, regulators, funders, and developers of AI systems in precision oncology.
Clinical trials in oncology embody a moral and scientific contract. Because these studies do not guarantee better outcomes for participants, patients who enroll accept uncertainty, toxicity, and logistical burden with the understanding that what is learned from their participation will be used to improve care for others—accurately, comprehensively, and transparently. That contract is only partially honored in our current system. Nonpublication, selective outcome reporting, inconsistent access to trial data, and a lack of linkage between protocols, registries, and manuscripts persist despite regulatory mandates and journal policies. In an era when artificial intelligence (AI) and machine learning (ML) are increasingly deployed to synthesize evidence and drive “precision” decisions, opaque or selectively curated trial data are not just an inconvenience; they are a structural threat to trustworthy oncology and to AI itself. For the purposes of this commentary, “AI-ready oncology trials” are defined as studies that are prospectively registered, fully reported against prespecified endpoints, linkable across protocol, registry, and publication, and accompanied by accessible, well-documented data suitable for independent verification and secondary analysis. The mechanisms described below are presented as illustrative, rather than prescriptive, examples of how AI-enabled transparency could be operationalized within existing research ecosystems.
Over the last decade, several initiatives have sought to hardwire transparency into clinical research. The International Committee of Medical Journal Editors requires prospective registration of clinical trials and, since 2018, mandates a data sharing statement for trials submitted to member journals. 1 The U.S. Food and Drug Administration Amendments Act and subsequent Final Rule define timelines and obligations for posting summary results to public registries. The U.S. National Institutes of Health Data Management and Sharing Policy, effective in 2023, requires funded investigators to plan for data sharing as a condition of support. 2 However, as with other transparency policies, the presence of formal requirements does not necessarily translate into consistent downstream compliance, enforceable access to data, or machine-actionable availability in practice. In the European Union, Regulation (EU) No. 536/2014 (Clinical Trials Regulation) establishes legally binding requirements for prospective registration, structured results reporting, and public access to trial data via the Clinical Trials Information System, representing one of the most comprehensive statutory transparency frameworks globally. At a global level, the World Health Organization’s International Clinical Trials Registry Platform aggregates primary registries worldwide, reinforcing prospective registration as a global norm and enabling cross-regional linkage of trial metadata. In parallel, the European Medicines Agency has advanced data transparency through policies enabling access to clinical study reports and, in selected contexts, patient-level data, underscoring regulatory recognition that independent scrutiny is essential to public trust in therapeutic approvals. In parallel, large-scale platforms such as Vivli, the Yale Open Data Access Project, and sponsor-specific portals have demonstrated the feasibility of controlled access to individual participant data (IPD) from completed trials. 3 On paper, the backbone for transparency in oncology and hematology exists and has been endorsed by all involved stakeholders.
Empirical data show, however, that practice lags behind principle. Across randomized, clinical trials (RCTs) in multiple disciplines, including oncology, a recent systematic review found that approximately 20% of trials still had no results publicly available (neither publication nor registry posting) several years after completion, most commonly in nonindustry-sponsored studies. 4 A cross-sectional analysis of registered cancer trials reported a nonpublication rate exceeding 45% in some cohorts, with cancer trials having the highest nonpublication proportion among examined disease areas. 5 Disease-focused evaluations echo this pattern; for example, in head and neck cancer RCTs, about 40% of completed trials never reached publication. 6 Publication bias compounds the problem: Tang and colleagues showed that randomized phase III oncology trials with positive results were significantly more likely to be published in high-impact factor journals, while negative trials were disproportionately underrepresented, reinforcing a systematically optimistic evidence base. 3
Even when oncology trials are published, key elements required for verification and reuse are frequently incomplete or distorted. Vera-Badillo et al. demonstrated that in breast cancer RCTs, one-third exhibited bias in reporting primary efficacy endpoints and two-thirds showed suboptimal reporting of toxicity; negative primary endpoints were often “rescued” by emphasis on favorable secondary outcomes or subgroup signals, while serious toxicities were selectively downplayed. 7 Similar concerns extend across contemporary oncology trials, where surrogate endpoints, narrow eligibility criteria, and complex designs are common, yet full protocol-concordant reporting is inconsistent. 8 From the perspective of AI in precision oncology, such selective visibility is critical: Algorithms trained or validated on biased, selectively reported, or nonreproducible trial summaries inherit those distortions, however sophisticated the modeling.
Access to IPD remains similarly constrained. In a Journal of the American Medical Association Oncology audit of 304 trials supporting FDA approvals of anticancer medicines (2011–2021), only 45% were confirmed as eligible for IPD sharing with independent researchers, despite public-facing company policies endorsing transparency. 9 A subsequent evaluation of IPD packages obtained from pharmaceutical companies for meta-analysis found that, among trials where access was granted, the usability and completeness of datasets and documentation varied substantially, limiting efficient independent re-analysis. 10 At the policy level, journal and registry requirements for data sharing statements have proliferated, but a 2024 assessment of data sharing statements in registries concluded that many remain vague, nonspecific, and difficult to interpret or implement in practice; only a minority were sufficiently clear to be machine-actionable or to guarantee real access. 11 These findings indicate that only a modest fraction of oncology trials is truly “verifiable”: prospectively registered, fully reported against their prespecified endpoints, linkable across registry–protocol–article, and accompanied by accessible IPD.
The ethical implications of this shortfall are substantial. Trial participants in hematology and oncology accept immediate and long-term risks on the premise that what is learned will be used fully and honestly to guide future patients. When results remain unpublished, when endpoints are selectively presented, or when underlying data are withheld without compelling justification, this premise is violated. Data transparency thus represents the ethical completion of the research enterprise: Patients have fulfilled their obligation, and investigators, sponsors, and journals must fulfill theirs. Scientifically, inadequate transparency inflates perceived benefit, obscures harm, enables duplicative or misdirected research, and undermines the validity of meta-analyses and guidelines that depend on a complete accounting of positive, negative, and null evidence.
Governance, Feasibility, and Implementation Boundaries
While the need for AI-enabled transparency in oncology trials is compelling, its implementation must be grounded in realistic governance structures and operational constraints. Importantly, these systems are not best deployed at the level of individual investigators or laboratories, where resources, technical expertise, and legal authority vary widely. Instead, the most practical and equitable points of implementation are journals, clinical trial registries, funders, and professional societies, where standardized workflows already exist and where transparency requirements can be applied consistently across studies. Governance structures will also be necessary to address false positives, the burden on investigator-initiated trials, and the risk of over-policing AI initiatives. These systems should operate as confidential decision-support tools, not punitive enforcement mechanisms.
A phased or incremental deployment model is likely to be most effective. Initial efforts could focus on automated trial–publication linkage and protocol–registry–article concordance checking, capabilities that are technically mature and provide immediate value with relatively low risk. More complex functions—such as structured extraction of results or verification of data-sharing claims—can be layered in over time as governance norms, validation evidence, and stakeholder trust mature.
Shared infrastructure offers an additional path to feasibility. Rather than duplicating effort across journals or institutions, professional societies (e.g., American Society of Hematology, American Society of Clinical Oncology, and American Society for Radiation Oncology) could support common transparency platforms, thereby reducing fragmentation and promoting harmonized standards across oncology and hematology. When embedded within editorial and funding workflows, the marginal cost of AI-assisted transparency can be amortized across large volumes of submissions, transforming transparency from an aspirational ideal into routine research infrastructure. Framed this way, governance is not an obstacle to AI-enabled transparency but its enabling condition.
AI Accountability, Auditability, and Validation
AI-enabled transparency systems must themselves be accountable, auditable, and subject to validation if they are to enhance, rather than undermine, trust in oncology research. At a minimum, such systems should rely on versioned models, with clearly documented training data sources, update histories, and intended use cases. Performance metrics—including sensitivity for detecting reporting discrepancies and false-positive rates—should be reported and periodically reassessed across diverse trial designs, disease areas, and publication formats.
Bias testing is particularly important. Algorithms trained primarily on large, industry-sponsored trials or high-resource journal formats may perform less reliably when applied to investigator-initiated studies or trials conducted in lower-resource settings. Without explicit monitoring, such disparities risk amplifying inequities rather than correcting them. Transparent reporting of known failure modes—such as difficulty interpreting complex adaptive designs, unconventional endpoints, or poorly structured supplements—should therefore be considered a core requirement.
Crucially, AI outputs must never be treated as final adjudications. Human override and expert review should be explicitly embedded into all workflows, with AI functioning as a decision-support layer that surfaces potential inconsistencies rather than enforcing compliance. When designed with these safeguards, AI systems can strengthen transparency while preserving scientific judgment, contextual interpretation, and ethical responsibility.
Achieving Transparency
AI is often discussed in oncology as a tool for prediction and classification. Equally important is its role as infrastructure for verifiable evidence. Several capabilities are already technically mature enough to be deployed (Fig. 1).
First, AI can automate linkage between trial registrations and publications at scale. Tools such as the web-based system developed by Smalheiser et al. use ML to match ClinicalTrials.gov entries to PubMed articles with high accuracy, even when explicit identifiers are missing. 12 Deployed as background infrastructure by registries, journals, or professional societies, such systems can continuously monitor oncology and hematology trials, determine whether corresponding results have appeared either as registry postings or peer-reviewed articles, and flag apparent gaps. This transforms sporadic, labor-intensive audits into routine surveillance in real-time: An oncology trial that passes its reporting deadline without results becomes an automatically visible exception, not a needle in a haystack.
Second, natural language processing can be used to check concordance between protocols, registry records, and manuscripts. ML systems such as RobotReviewer have already shown that automated extraction of trial features and risk-of-bias assessments from PDFs is feasible and reasonably concordant with human review. 13,14 Extending these methods, an AI engine can parse the prespecified primary and secondary endpoints, planned sample size, eligibility criteria, and analysis plans from protocols and registries and compare them directly with what is reported in submitted or published articles. Unacknowledged changes—such as endpoint switching, unexplained shortfalls in sample size, selective omission of negative secondary endpoints, or materially altered inclusion criteria—can be highlighted for editors, reviewers, and, ultimately, readers. When used appropriately, AI is acting as a consistency checker that forces deviations into the open where they can be explained or corrected.
Third, AI can help convert unstructured oncology trial reports into structured, machine-readable data layers. Information extraction models can identify all reported endpoints, per-arm denominators, event counts, hazard ratios, confidence intervals, and adverse event frequencies and organize them into standardized tables aligned with CONSORT and related guidelines. 15 These same models can track flows of participants from enrollment to analysis to quantify attrition and missing data, verify that totals are internally consistent, and detect when key elements—such as grades 3–4 toxicities or serious adverse events—are summarized inadequately compared with expectations for the disease and intervention class. For endpoints prespecified but never reported, the absence itself becomes visible: One can algorithmically answer whether each primary and secondary endpoint listed in the registry appears in the publication, and, if not, whether an explanation is provided. In the context of precision oncology, this structured layer enables robust evidence synthesis, cross-trial comparisons, and training of AI models on comprehensive rather than cherry-picked results.
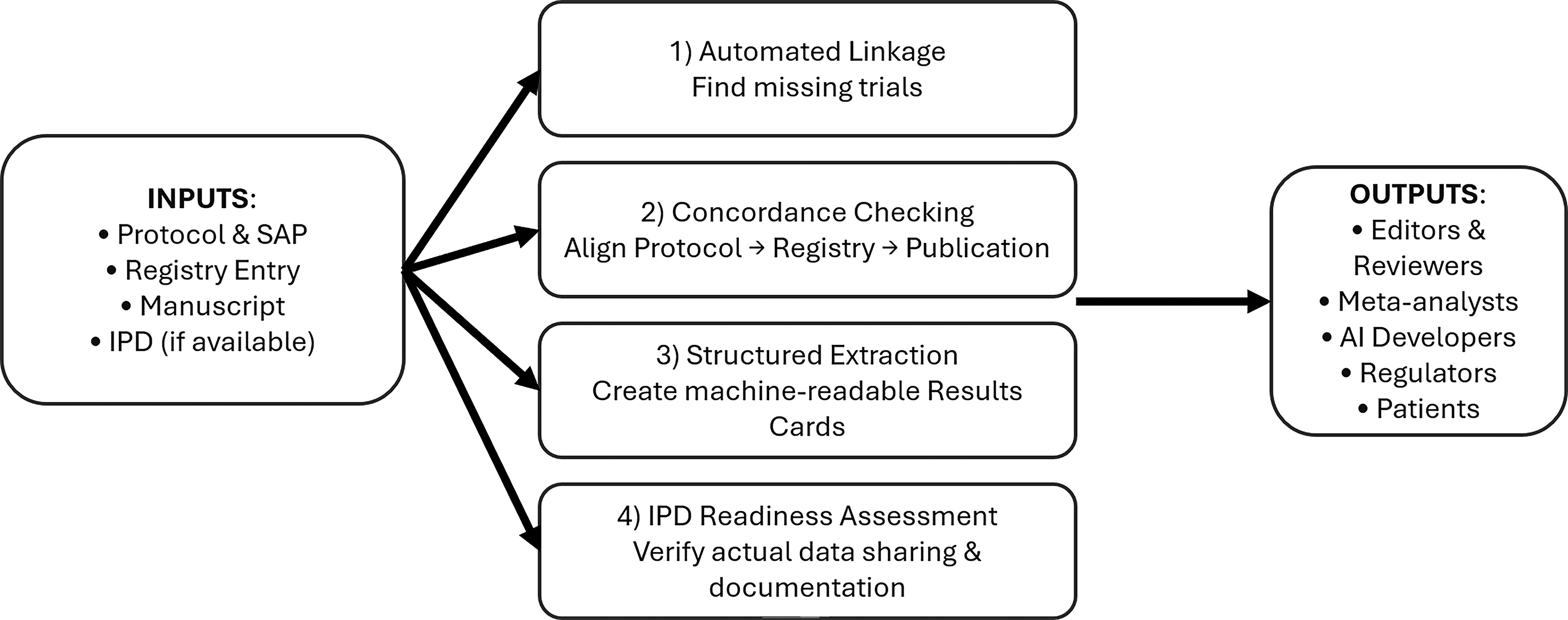
AI-augmented transparency workflow for oncology trials. This figure depicts a left-to-right pipeline. On the left, trial inputs include the preregistered protocol and statistical analysis plan, the registry entry, and the final article (and supplementary materials). In the center, an “AI Transparency Engine” comprises four modules:
While this commentary does not advocate for a single enforcement model, several illustrative mechanisms merit consideration as potential levers to improve transparency. Funding agencies could link grant renewal or future funding eligibility to timely and complete reporting of trial results, reinforcing transparency as a core component of research stewardship. Journals may explore conditional acceptance models, in which final publication is contingent on verified registry–article concordance or completion of structured results reporting. Automated checks at submission—analogous to plagiarism screening—could further normalize transparency expectations without singling out individual investigators. Finally, public transparency dashboards, framed nonpunitively, could provide aggregate reporting and data-sharing metrics at the institutional or sponsor level, promoting accountability through visibility rather than sanction. Together, these emerging approaches suggest that transparency can be incentivized through routine infrastructure rather than episodic enforcement.
When framed in these ways, AI serves as critical decision support: Their outputs should trigger confidential human review, not public accusation; their algorithms and performance characteristics should be transparent and subject to peer scrutiny; and their deployment should explicitly safeguard against penalizing studies from under-resourced settings where formatting or metadata may be imperfect. Although many examples draw from U.S. and EU frameworks, the transparency gaps and AI-related risks described here are global, particularly in regions where regulatory enforcement, registry interoperability, and journal infrastructure vary widely.