
Abstract
Background:
The rapid adoption of digital pathology and artificial intelligence (AI) in oncology creates multiple opportunities for precision diagnostics but also creates an urgent need for evidence-based standards to ensure safe and effective implementation.
Goal:
This article, developed by the Digital Pathology Association, presents recommendations for digital pathology as well as the validation and clinical utility of AI-enabled digital pathology tools in clinical practice. This guidance addresses analytical and clinical validation, algorithm reliability, and criteria for establishing clinical utility attributed to test use. Key recommendations emphasize separate validation of scanning processes and AI algorithms, concordance studies to support interscanner generalizability, rigorous assessment of accuracy and reliability in real-world settings, and clear description of algorithmic use limitations. These recommendations further provide frameworks for when AI may replace or augment existing diagnostic approaches, such as biomarker scoring, cancer diagnosis, and prognostic risk assessment.
Conclusions:
By considering payer, regulatory, and clinical perspectives, the recommendations promote transparency, trust, and reproducibility in digital pathology while encouraging value-based care delivery. We support responsible innovation in computational pathology, ensuring that AI applications achieve not only technical performance goals but also deliver measurable clinical benefit to patients.
Executive Summary
Digital pathology and artificial intelligence (AI) are transforming anatomical pathology practice, yet their adoption has outpaced the development of comprehensive implementation standards. This recommendation statement addresses that gap by establishing evidence-based frameworks for validation, deployment, and clinical use of digital pathology systems, including AI-enabled tools, in clinical laboratories.
The Challenge: Health care institutions face critical decisions about digital pathology adoption without standardized guidance on validation requirements, appropriate clinical applications, or quality assurance measures. This uncertainty creates risks: inconsistent performance across institutions, unclear regulatory pathways, variable reimbursement, and potential patient safety concerns. Meanwhile, patients in underserved settings lack access to subspecialty pathology expertise that digital systems could provide.
Core Principles: This recommendation statement establishes that digital pathology and AI tools must meet rigorous validation standards while remaining under qualified pathologist oversight. AI augments rather than replaces pathologist expertise. Validation requirements scale appropriately with clinical risk: assistive tools that highlight features require different evidence than autonomous systems that generate diagnostic conclusions.
Key Recommendations
Health care institutions should begin planning for digital pathology adoption to enable workflow optimization, improve access to subspecialty expertise, and support artificial intelligence (AI) integration. Digital pathology is specifically indicated when traditional pathology services are unavailable, particularly for patients in Critical Access Hospitals, Rural Health Centers, and Federally Qualified Health Centers. All clinical AI applications require qualified pathologist oversight.
Slide scanning systems must be validated independently from AI algorithms. AI algorithms must demonstrate analytical and clinical validation for each scanner on which they will be used. Interscanner concordance studies can generalize performance across scanners when using validated systems as reference standards. AI validation must assess both accuracy and reliability, with quality control systems to detect batch effects and technical failures. Minimum tissue requirements and artifact limitations must be specified for each algorithm.
AI should be deployed only within validated clinical contexts where it can improve diagnostic accuracy, reliability, or efficiency. Three categories of appropriate use are defined: (1) replacing existing invasive or expensive tests when validated, (2) augmenting pathologist assessment of verifiable tasks, and (3) predicting outcomes using features that pathologists cannot readily assess. Clinical utility must be demonstrated through improved patient outcomes, either via direct evidence or validated chains of evidence.
Impact and implementation
These recommendations balance innovation with patient safety by providing clear pathways for responsible AI adoption. Our hope is to provide clarity for validation frameworks for laboratories and algorithm developers, clinical utility standards for payers, and, most importantly, improved diagnostic accuracy and expanded access to expertise to impact patient outcomes.
The recommendations are graded by evidence certainty and strength, ranging from strong recommendations backed by robust evidence to conditional guidance where further research is needed. Use cases spanning cancer detection, biomarker scoring, prognostic assessment, quality control, and case triage demonstrate current applications meeting these standards.
Stakeholders
This document is directed to a number of stakeholders that rely on, interpret, produce, pay for, or manufacture digital pathology data, devices, and software. Table 1 lays out each stakeholder, what their role is within digital pathology, and how they can use the document’s recommendations.
An Overview of the Various Stakeholders Involved in Digital Pathology and How They Can Best Utilize This Document
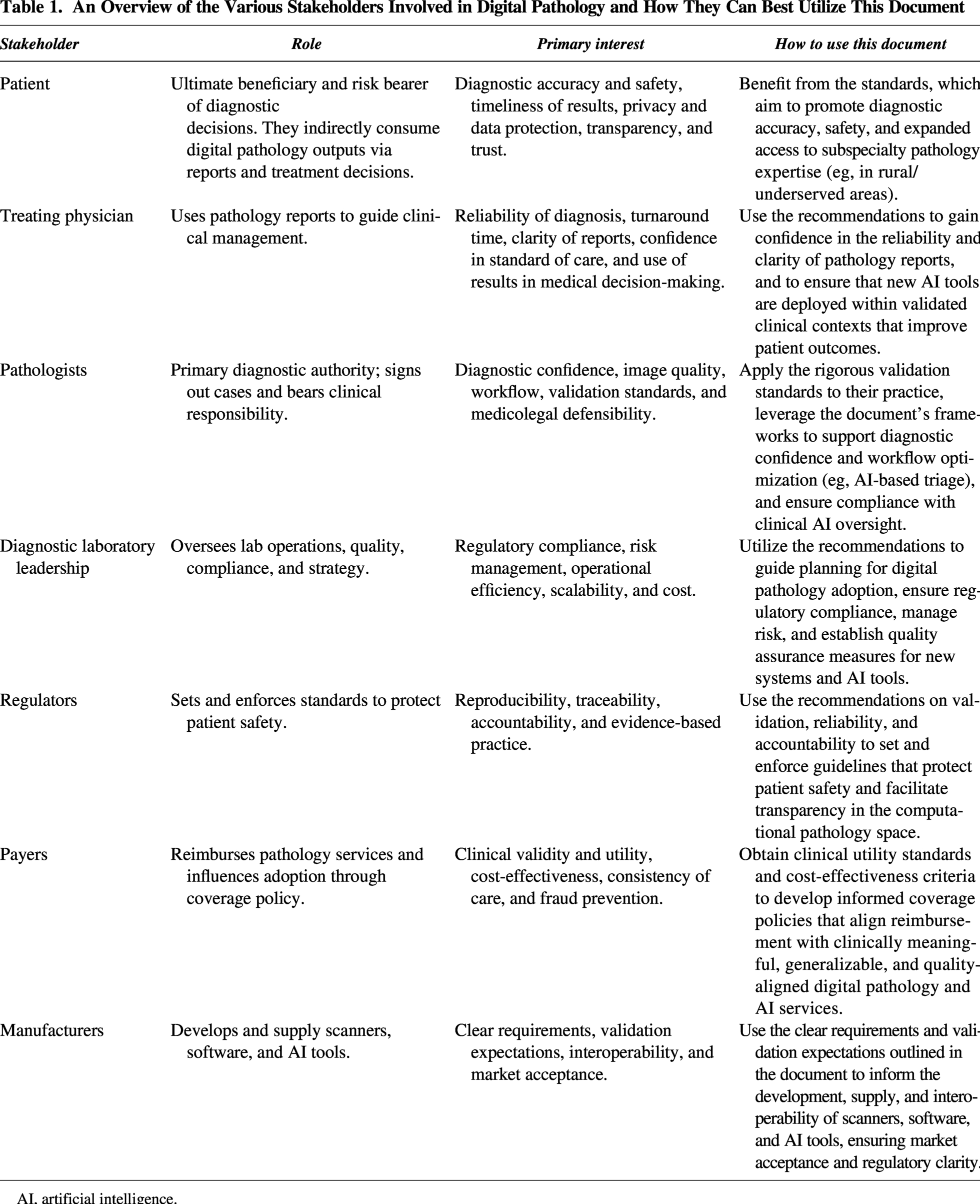
AI, artificial intelligence.
Scope
This document encompasses various elements of digital pathology including whole-slide image (WSI) scanning, image viewing, and AI algorithms for image interpretation in the clinical setting. This is inclusive of applications in solid formalin-fixed, paraffin-embedded tissue using brightfield microscopy and excludes cytology and immunofluorescence-based imaging.
Objectives
The objectives of this document are to provide clear, practical, and evidence-based recommendations for the safe, effective, and responsible use of digital pathology, inclusive of uses of AI in clinical digital pathology. Specifically, this guidance aims to:
Define key principles for evaluating and deploying digital pathology in clinical pathology workflows, with a focus on diagnostic support, quality assurance, and clinical utility. Establish best practices for the validation and performance assessment of digital pathology across varied tissue types, staining protocols, and imaging systems. Promote transparency and interpretability of digital pathology outputs to support pathologist oversight and clinical decision-making. Support consistency and reproducibility in AI-assisted outputs across institutions and settings. Facilitate payer and stakeholder confidence by outlining standards that ensure that digital pathology and its AI tools are clinically meaningful, generalizable, and aligned with quality care delivery.
These objectives are intended to support clinicians, developers, payers, institutions, and regulators in the thoughtful integration of digital pathology into pathology practice, ultimately advancing diagnostic excellence and patient care.
Definitions
Different fields use the same terms in distinct ways. This issue is particularly acute at the intersection of health care and computational technology. Indeed, some terms widely used in digital pathology or health care applications of AI either lack a clear and consistent definition or exhibit contradictory definitions in academic literature. In the absence of a clear and universally accepted definition, we provide the following definitions for the purposes of this recommendation statement:
Introduction
Digital pathology is the practice of converting glass histology slides into high-resolution digital images using specialized scanners, enabling pathologists to review and analyze tissue samples on a computer screen, mimicking the microscope but with enhanced capabilities. These digitized slides can be viewed locally or remotely, stored electronically, and analyzed using advanced software, including artificial intelligence (AI) tools. Digital pathology supports remote consultation and patient access, enhances consistency in diagnosis, and forms the foundation for computational pathology to support precision medicine and data-driven health care.
Digital pathology adoption has grown significantly in academic medical centers, integrated delivery networks, and reference laboratories. Clinical use is increasing as whole slide scanners, image management systems, and displays are used by pathologists for primary diagnosis, enabling more efficient service delivery models and improving access to subspecialty expertise through telepathology. In parallel, digitization supports downstream automation and analytics, including the use of AI-based image analysis tools.
AI in pathology has demonstrated significant promise in improving consistency1–3 and reducing diagnostic variability.4,5 Algorithms are increasingly used to assist in tumor grading,6–8 cell classification,9,10 biomarker quantification, 11 risk stratification,12–14 and triage of negative cases. The vast majority of these tools are designed to augment rather than replace the expertise of a pathologist, improving overall quality and throughput in diagnostic workflows.15–17 Importantly, AI can introduce a level of standardization that facilitates more equitable care delivery16,18 and can serve as a foundation for methodology- and performance-based reimbursement models.
The remainder of the document provides an overview of the technologies underpinning digital pathology, including AI (Technology Background); specifies a set of concrete recommendations (Recommendations); and the methodology used to draft the recommendations (Methodology). Finally, it contains a motivating set of use cases.
Whole Slide Imaging in the Context of the Health Care System
An early hope for digital pathology was that it would enhance timely access to expert pathology interpretations via remote image review and interpretation. In the late 1990s, early telepathology efforts focused on expanding access to subspecialty pathology expertise. The Mediterranean Institute for Transplantation and Advanced Specialized Therapies in Palermo, Italy, collaborated with University of Pittsburgh Medical Center (UPMC) to provide continuous (24-h) expert evaluation of transplant allografts, including both primary and second-opinion interpretations by UPMC transplant pathologists. 19 In a series of 78 second-opinion cases via telepathology, the UPMC pathologist disagreed with the primary pathologist 11 times, of which 3 were considered clinically significant. 20 This collaboration demonstrated the feasibility and clinical value of around-the-clock remote subspecialty transplant pathology services.
More recently, the National Academies of Sciences, Engineering, and Medicine convened a 2018 workshop on improving cancer diagnosis and care. 21 A common issue raised was the increasing complexity of diagnostic assessment in cancer and the need for expertise along with patient access to that expertise. 22 Digital pathology was raised as a potential solution to address this need. 22
In addition to the need for specialized pathologist opinions, some pathological interpretations also call for timely critical reviews and reports to treating clinicians. In general, conditions in which there is a need for urgent communication of results tend to be those in which there is a potentially urgent medical condition associated with the finding.
The College of American Pathologists (CAP) calls on laboratories and institutions to establish protocols and criteria for identifying and reporting urgent and significant diagnoses to the treating clinician. 23 A survey 24 of 1,130 CAP-accredited pathology laboratories’ policies regarding significant, unexpected, and critical diagnoses in surgical pathology found that the following specific conditions were often included in written policies: findings not expected by clinical history, malignancy, life-threatening infections, organ rejection or graft-versus-host disease, inflammatory or immunological processes, and no chorionic villi in products of conception.
As the workload for each pathologist continues to increase, 25 driven by expanding diagnostic criteria required for precision medicine, regulatory demands, and concomitant understaffing in many pathology departments, individual pathologists must take on more responsibilities to keep up with clinical demand. In traditional glass slide workflows, critical cases are often “hidden” among hundreds of other slides, awaiting their turn. Digital pathology introduces the possibility of AI-based triage, allowing cases with potentially critical findings to be prioritized within the pathologist’s worklist. This prioritization improves the likelihood of timely reporting and adherence to recommended turnaround times. AI systems designed to triage cancer in pathology images have already been reported, demonstrating the feasibility of identifying and prioritizing cases with critical findings for earlier review. 26 In radiology, for example, AI has shown the value of effective worklist prioritization: intracranial hemorrhage on CT scans can be automatically flagged, ensuring that cases with critical findings are reviewed sooner and patients receive faster care. 27
Access to care and specialized centers
The increasing complexity of diagnostic assessments has increasingly required greater levels of specialization and expertise in pathology, like much of the rest of medicine. While this affects all health care institutions to some degree, it disproportionately burdens those serving regions and populations for whom access to care is already limited. Care settings that are explicitly designed to serve underserved populations or communities include Rural Health Clinics (RHCs), Critical Access Hospitals (CAHs), and Federally Qualified Health Centers (FQHCs), 28 with Medicare criteria as described in the State Operations Manual. 28 Many health care institutions that do not have one of these designations may also serve those with limited access to care.
Limited access to specialty care in rural areas has long been recognized as a persistent challenge. Analyses of cancer incidence and mortality have shown that, despite declining cancer incidence rates in rural populations, cancer-related mortality has increased. 29 This disparity has been attributed in part to barriers in accessing care, including the geographic concentration of oncologists in densely populated urban centers rather than in rural areas. 30 Unger and colleagues attempted to more closely study this issue by examining outcomes from rural versus nonrural patients in SWOG studies for which protocol-directed care regardless of geography (ie, patients had access to care) would mitigate differences in rural versus urban patients. 31 In line with this hypothesis, they found that within the SWOG cohort, outcomes were similar among the rural and nonrural patients, suggesting that improved access to care for rural patients may help mitigate the poorer cancer outcomes observed in rural populations.
FQHCs serve patients regardless of their ability to pay, rather than based on location, 28 but all of these facility types are recognized as a matter of definition as entities providing care to a population that has limited health care access. Health care providers may be recognized by Centers for Medicare & Medicaid Services (CMS) as an FQHC or RHC. In 2022, such community health centers served over 30 million Americans, including 1 in 9 children, 9.6 million rural residents, and 395,000 veterans. 32 These health care centers continue to serve a wide range of Medicare beneficiaries, many of whom have significant comorbid illnesses such as cancer, heart disease, and lung disease. 33 A Mathematica Policy Research Report 34 prepared for the HHS Office of the Assistant Secretary for Planning and Evaluation noted that providing patient access to specialty care has been an important challenge among federally funded health centers, and one of the strategies being attempted to address this is the use of electronic consultations.
WSI and digital pathology open a new opportunity to give patients access to specialized pathology interpretations even if those pathologists are geographically far removed from the site of care.
Multimedia reports for treating clinicians and patients
Medicine has developed an extensive vocabulary to describe anatomical regions, tissue architecture, and pathology. This vocabulary is helpful to facilitate communication between health care professionals and patients regarding what is seen in diagnostic imaging. However, jargon is often confusing to nonclinicians, and patients may find images of their own disease easier to understand than medical language that attempts to translate those images into verbal descriptions. In addition, seeing images may give patients a sense of engagement and better ownership of their own health and care.
Availability of digital images enables treating clinicians to better understand their diagnostic findings and share those images with patients in discussions regarding the disease to provide both education and assist in informed decision-making. It is a very common practice in the United States for treating clinicians, especially specialists, to review their own images and even show them to patients since the advent of digital radiology. A study by Nyak 35 and colleagues of 160 US physicians sought to understand the role of images accompanying radiology text reports. The overwhelming majority (91%) of respondents indicated that access to images helps to understand the text report, and 60% of clinicians felt strongly or very strongly that access to images accompanying text would significantly improve patient care and outcomes. Not only has this result been consistently demonstrated in radiology,36,37 but the same trends are occurring in pathology. An increasing number of patients are relying on online patient portals to access their pathology reports, 38 and patients prefer to augment the text reports with the images necessary to understand them. 39 Satisfying this patient need ultimately can only be done via the reliance on digital pathology. This benefit of digital pathology goes beyond even the patient as research suggests that the review of digital pathology slides with patients at the clinic can reduce pathologist burnout. 40
In addition, in a study of patient satisfaction, evidence from digital radiology suggests that patients prefer to see the images themselves, 41 and visualization of pulmonary nodules by patients 42 provides context to the patients to assist with understanding an ambiguous diagnosis. To date, there has not been much opportunity for clinicians to review pathology images with patients. Prior to digital pathology, the practice of pathology relied on fragile glass slides that required expensive microscopes to review, which also required maintenance. This contrasts with radiology, which could be reviewed with patients using robust silver halide film that could readily be viewed on cheap light boxes even prior to the emergence of digital radiology. Because digital pathology obviates the need to transport fragile slides and enables the viewing of slides on digital screens, the technical capability for clinicians to begin reviewing pathology images with patients is now present. While not yet widely practiced, patient-facing pathology clinics where pathology images are reviewed with patients are emerging, an encouraging practice that would be impossible without digital pathology.
Technology Background
The Technology Background section provides a high-level overview of the foundational technologies underpinning the digital transformation of pathology. It first describes brightfield imaging for creating high-resolution WSIs. This is followed by an explanation of specialized image viewing systems, which are essential software for the smooth visualization, navigation, and clinical review of WSI files, often with integration into laboratory systems. Finally, the section delves into AI, highlighting its basis in deep learning and neural networks (eg, convolutional neural networks and Transformers) as a set of computational tools for automated image interpretation, designed to augment pathologist expertise in tasks such as classification, segmentation, and detection.
Brightfield imaging
The foundational imaging technique in clinical digital pathology is brightfield scanning, which uses transmitted white light and high-resolution color cameras to create images from stained tissue sections. These scanners are capable of handling large slide volumes, offering fast scan times, automated focus, and integration into laboratory information systems (LIS). The ability to digitize slides not only supports remote review and centralized workflows but also creates a standardized, reproducible dataset that serves as the foundation for AI-driven analysis.
Image viewing
Digital pathology viewers are software applications that allow for the visualization, navigation, and manipulation of WSIs—digitized representations of traditional glass pathology slides scanned at high magnification (typically 20× or 40×). These viewers are purpose-built to handle the extremely large file sizes and resolutions associated with WSIs, often several gigabytes per image. To ensure smooth performance, viewers commonly use dynamic image tiling and streaming, loading only the portion of the image currently in view at the desired resolution, which minimizes memory usage and latency. This allows pathologists to pan and zoom through digital slides fluidly, mimicking the experience of a traditional light microscope.
Modern digital pathology viewers often support additional functionality that enhances diagnostic accuracy and efficiency. These features can include measurement tools, annotations, side-by-side comparison of serial sections or stains, and synchronized viewing of multiple images. Many platforms are integrated with LIS, electronic health records (EHR), and picture archiving and communication systems (PACS), allowing for streamlined case review and reporting. Increasingly, viewers also incorporate or interface with, AI algorithms, enabling automated quantification (eg, Ki-67, HER2), feature detection (eg, mitotic figures, tumor margins), and quality control (QC) checks (eg, blur, stain variation).
For clinical deployment, digital pathology viewers must meet high standards for usability, reliability, and regulatory compliance. Clinical-grade viewers often support audit trails, user access controls, and secure data transmission, which are critical for HIPAA and GDPR compliance and other data protection regulations. Regulatory body-approved (such as the Food and Drug Administration in the US, etc) systems may be required for use in primary diagnosis, depending on the region. Viewers used in regulated environments may undergo formal validation processes to ensure diagnostic equivalence to traditional microscopy. Overall, digital pathology viewers are a core technology underpinning the broader digital transformation of pathology, enabling remote diagnosis, collaboration, education, and integration of computational pathology tools into routine practice.
Artificial intelligence
AI refers to a set of computational techniques that enable machines to perform tasks typically associated with human cognition, such as visual perception, language understanding, and decision-making. In digital pathology, AI is most commonly applied through a subfield known as computer vision, which allows computers to interpret and extract information from high-resolution histopathological images. These AI systems are not intended to replace pathologists or their human cognition, but to assist them in improving diagnostic accuracy, efficiency, and consistency.
The earliest approaches to AI relied on rule-based systems in which explicit instructions were written by humans (eg, “if a cell is larger than 50 microns and has an irregular nucleus, flag it as abnormal”). These approaches were displaced by data-driven algorithms, collectively referred to as machine learning, in which algorithms automatically learned from data rather than relying on hard-coded rules. In machine learning, a clinician might label thousands of slides (eg, benign vs malignant) and indicate which features a machine should rely on (nucleus size, shape, perimeter, staining intensity, etc), and the machine would learn from the data which features were important and how to combine them to produce a correct result.
The vast majority of modern AI approaches in digital pathology are based on deep learning, a subfield of machine learning that takes this approach one step further. Deep learning can either rely on human-specific features or it can automatically discover which features are most useful for performing the requested task from the raw images, without needing a human to hand-pick them. For example, instead of a human specifying that an algorithm should focus on cell count or cell shape, a deep learning algorithm will discover that these are useful features by itself through the process of reviewing millions of image patches.
Deep learning algorithms rely on structures called neural networks, which interpret inputs via a series of layers of computational processing. Each layer in a neural network processes and transforms information provided to it by an earlier layer. This approach is loosely modeled on the human visual cortex in which layers of biological neurons in the brain, referred to as V1, V2, etc, process and transform visual signals from the previous layer. Like the human visual cortex, the layers of a neural network extract and represent different types of information at different stages of processing. For example, the initial layers might identify features such as edges, corners, or lines; intermediate layers might identify coarser structures such as morphological patterns; and the final layers identify more abstract concepts such as biomarkers, risk factors, and diagnoses.
In human brains, neurons are only connected to a relatively small subset of other neurons. This topology or map of connections defines how information flows. Similarly, deep learning algorithms have a topology that defines which digital neurons are connected. This map of connections is referred to as the “architecture” of the deep learning algorithm. Perhaps the most common architecture for image-based tasks is the convolutional neural network (CNN), which is specifically designed to process and analyze image data. More recently, transformer-based architectures 42 have begun to replace CNNs in many cutting-edge models. Originally developed for natural language processing, transformers excel at capturing long-range dependencies and global context and have been adapted to vision tasks with remarkable success. This architectural shift is also evident in multiple pathology foundation models,43–54 where vision transformers are increasingly serving as the backbone for large-scale image understanding, marking a major advance in the field.
A digital pathology image—such as a WSI—is represented mathematically as a 3D tensor, or multidimensional array. Each image can be thought of as a grid of pixels, where each pixel has associated intensity values for color channels (typically red, green, and blue), resulting in an image tensor of shape [height, width, channels]. For example, an image that is 512 pixels tall, 512 pixels wide, and has 3 color channels would be represented as a tensor of shape [512, 512, 3]. This tensor becomes the input to the AI model.
Depending on the task, an AI model may produce different types of outputs including, but not limited to, the following:
For For For For
Due to the extremely large size of WSIs, often containing gigapixels of data, it is computationally infeasible for most AI models to process an entire image at once. Instead, a common strategy is to divide the image into smaller subregions called Combining patch-level classifications to generate a heatmap showing spatial distributions of tumor and nontumor areas. Summing detected features (eg, mitoses) across all patches to compute a total count. Averaging or voting across patch-level predictions to generate a final diagnostic classification.
In some cases, models are trained using
To make these outputs useful in clinical settings, the model’s predictions are typically presented as overlays (eg, false-color segmentation masks), visual heatmaps, or structured outputs (eg, scores or classifications) integrated into digital pathology platforms. The goal is to offer interpretable, reproducible results that complement the pathologist’s expertise.
By understanding how AI models process, interpret, and aggregate image data, clinicians and regulators are better equipped to assess their reliability, clinical value, and appropriate use in practice. Transparency in how models function—and how they arrive at their predictions—is essential for building trust and ensuring safe integration into diagnostic workflows.
Categories of AI applications
There are multiple ways to categorize AI applications. We discuss two that are particularly relevant in the context of validation, regulation, and payment for AI algorithms applied to digital pathology.
Regulatory status of AI laboratory tests in pathology
Regulatory agencies across countries and regions rely on distinct approaches to regulating medical software. These are rapidly changing as AI applications continue to evolve and mature. However, broadly speaking, software (with or without AI) is categorized based on its intended use.
In particular, software is considered a medical device if it is intended to diagnose, treat, or drive or inform clinical decisions for an individual patient. Digital pathology software that would clearly fall into this bucket includes software intended to diagnose cancer detection, produce automatic cancer grading, or automatically select patients for the use of a drug as a companion diagnostic.
Software that stores, transfers, displays, or manages data without interpretation is generally not a medical device. Examples in digital pathology of nonmedical device software include PACS/LIS integration software, image compression or caching software, software intended to segment regions of interest but not diagnose, and WSI viewers specifically being used for nondiagnostic purposes.
United States
In the United States, regulatory oversight of AI laboratory tests in pathology involves a combination of federal agencies and legislative frameworks.
First, the Food and Drug Administration (FDA) regulates AI software and devices that meet the definition of a medical device. The FDA defines Software as a Medical Device (SaMD) as “software intended to be used for one or more medical purposes that perform these purposes without being part of a hardware medical device.” 55 The FDA has been actively developing policies for SaMD, including AI/ML-based technologies, emphasizing a total product lifecycle approach that accommodates iterative learning and updates. The agency’s framework distinguishes between locked algorithms (fixed behavior) and adaptive algorithms that change with real-world data. AI tools that aid in diagnosis or inform clinical decisions typically require premarket review and clearance or approval, depending on risk classification.
The FDA does not normally regulate a laboratory’s use of a device. Consequently, many laboratory tests are not FDA-cleared, even though the devices used to perform those tests may be. This distinction proved crucial in the 2025 Texas federal court decision American Clinical Laboratory Association v FDA, 56 which vacated the FDA’s attempt to regulate laboratory-developed tests (LDTs) as medical devices. The court’s reasoning centered on the interpretation of “test system,” holding that while physical devices are subject to FDA jurisdiction, the professional laboratory service using those devices is not. We should note that we are unaware of the use of the term “laboratory-developed test” within any regulations that are currently in force, so we are unable to provide a formal definition. However, this term is generally used to describe laboratory services using devices that have not been FDA-cleared or that use devices in a way that does not align with the FDA labeling that perform LDTs. This remains an evolving area of law, and future legislative or regulatory developments may alter this framework.
Second, the CMS plays a dual role in laboratory oversight: it administers the Clinical Laboratory Improvement Amendments (CLIA) program that regulates laboratories nationwide, and it operates Medicare, the largest health payment program in the United States. Clinical laboratories in the United States must obtain a CLIA certificate in order to operate, and some states have additional licensing requirements. While not required, many laboratories in the United States seek additional accreditation by the CAP. 57
Under Medicare, CMS distinguishes between physician pathology services and clinical diagnostic laboratory tests. Physician services must be both performed by a physician and ordinarily require a physician. 58 Clinical diagnostic laboratory tests, such as genetic tests and serum chemistry panels, do not ordinarily require a physician to perform the service. AI applications in digital pathology may fall into either category depending on its use case. When a physician pathologist provides the work, the AI serves as part of a physician pathology service. When no physician work is required to generate the test result, the AI functions as a clinical laboratory test.
Europe and the United Kingdom
In the European Union, in vitro diagnostics (IVDs) are regulated under the In Vitro Diagnostic Regulation (IVDR), 59 which replaced the earlier IVDD 60 and substantially strengthened requirements for clinical evidence, risk classification, postmarket surveillance, and oversight of higher-risk devices. Unlike the centralized FDA model in the United States, EU device oversight relies on notified bodies, independent organizations designated by member states, to conduct conformity assessments. Manufacturers must demonstrate compliance with the IVDR’s general safety and performance requirements, implement a quality management system (typically ISO 13485 61 ), and obtain CE marking to place a device on the market. In the United Kingdom, most of Great Britain continues to operate under a transitional IVDD-based framework overseen by the Medicines and Health care products Regulatory Agency, while Northern Ireland aligns with the EU IVDR.
Software, including AI algorithms, is regulated as a medical device in Europe when its intended use involves diagnosis, prevention, monitoring, prediction, prognosis, or treatment of disease. 61 AI tools used in laboratory medicine, such as diagnostic classifiers or risk-stratification algorithms, therefore qualify as IVD medical devices when they generate clinical outputs based on biological samples. Under the IVDR, such systems are risk-classified (Classes A–D), with most clinically impactful AI systems falling into higher-risk categories requiring notified body review, robust performance evaluation (scientific validity, analytical performance, and clinical performance), and ongoing postmarket performance follow-up.
Clinical laboratories themselves are regulated separately from devices. While the IVDR governs commercially marketed IVDs and imposes conditions on in-house tests, laboratories are primarily overseen through national licensing and accreditation systems. Most European medical laboratories are accredited under ISO 15189, 62 which sets standards for quality management, personnel competence, assay validation, and external quality assessment. Thus, in Europe, AI algorithms used in laboratory medicine are regulated as medical devices under IVDR (or UK medical device law), whereas laboratories operate under parallel accreditation and health system regulatory frameworks.
Other jurisdictions
In Canada, IVDs are regulated as medical devices under the Food and Drugs Act and the Medical Devices Regulations, administered by Health Canada. 63 IVDs are classified into Classes I–IV based on risk, with most clinically significant diagnostic assays and AI-driven diagnostic software falling into Classes II–IV. Higher-risk devices require a Medical Device Licence 64 supported by evidence of safety and effectiveness, quality system certification under ISO 13485 through the Medical Device Single Audit Program, and ongoing postmarket surveillance. Software intended for diagnostic or clinical decision-making purposes, including AI-based tools, is regulated as SaMD. In parallel, clinical laboratories are regulated at the provincial level and are typically accredited under ISO 15189 or equivalent provincial accreditation frameworks (eg, Accreditation Canada Diagnostics), creating a dual structure in which devices are federally regulated while laboratories are provincially overseen.
Across Asia, regulatory systems vary considerably but generally treat IVDs—including AI-based diagnostic software—as medical devices subject to national regulatory authority oversight. In Japan, IVDs are regulated under the Pharmaceuticals and Medical Devices Act (PMD Act) by the Pharmaceuticals and Medical Devices Agency (PMDA) 65 and the Ministry of Health, Labour and Welfare (MHLW), with risk-based classifications and premarket review for higher-risk devices. In China, the National Medical Products Administration (NMPA) regulates IVDs under a three-class risk system, requiring local type testing and, for higher-risk products, clinical evaluation. Singapore’s Health Sciences Authority (HSA) and South Korea’s Ministry of Food and Drug Safety (MFDS) operate similar risk-based frameworks. AI algorithms intended for diagnostic use are typically regulated as SaMD and must demonstrate analytical and clinical performance. Laboratory oversight, however, is generally handled separately through national accreditation schemes, frequently aligned with ISO 15189 standards.
In Australia, IVDs are regulated by the TGA 66 under the Therapeutic Goods Act 1989 and associated medical device regulations. IVDs are classified into Classes 1–4 (low to high risk), with most clinically significant diagnostic tests and AI-based diagnostic software falling into higher classes requiring conformity assessment and inclusion in the Australian Register of Therapeutic Goods (ARTG). Australia recognizes conformity assessment evidence from comparable jurisdictions in certain circumstances but maintains its own regulatory oversight. Laboratories are regulated separately through the National Pathology Accreditation Advisory Council standards and accreditation by the National Association of Testing Authorities, typically to ISO 15189. As in Canada and much of Asia, this creates a bifurcated system in which AI diagnostic devices are regulated as medical devices at the national level, while laboratories operate under accreditation and professional regulatory frameworks.
AMA classification
The American Medical Association’s (AMA) CPT Manual classifies AI into “assistive,” “augmentative,” or “autonomous.” Autonomous AI is further classified into Levels I, II, or III based on the extent to which the AI initiates treatment. While this taxonomy was designed to fit all specialties, some specialty-specific nuance must be considered when applying this framework to a particular medical specialty. 67
The AMA’s taxonomy may be appropriate for coding but does not meet the needs of these recommendations for three reasons. First, broadly speaking, the AMA’s taxonomy focuses on the level of human interpretive effort that the provider billing for the service must provide. Second, these recommendations represent an approach to evaluating the role of AI within a patient’s entire care journey, rather than limiting it to the nature of the specific procedure provided by a billing entity, which is out of the scope of the AMA’s CPT process. Third, the AMA classification is still being very actively edited as of the writing of this article.
Validation
Validation of digital pathology systems, and more broadly, software and hardware systems that perform a clinical function, is a very broad space encompassing aspects of clinical validation, usability, information quality, accuracy, repeatability, reproducibility, robustness, and compliance. The scope of these recommendations is limited to the three areas of validation described below.
Analytical validation of AI
In the context of digital pathology, what constitutes analytical validation has been outlined by various regulatory and professional bodies, which include:
Clinical and Laboratory Standards Institute (CLSI): Provides detailed protocols in documents such as CLSI EP05-A3 (precision),
68
EP17-A2 (detection limits),
69
and EP15-A3 (verification of precision and accuracy).
70
U.S. FDA: Outlines expectations for analytical validation in the context of SaMD.
55
CMS under CLIA: Requires laboratories to verify or establish performance specifications for nonwaived tests.
71
CAP: Offers accreditation checklists that require evidence of analytical validation for new or modified tests.
72
In the context of these recommendations, a summary of Analytical Validation inclusive of the aforementioned definitions would be the process of systematically evaluating and documenting a test or system’s technical performance to ensure it reliably and accurately measures the intended analyte or output under defined conditions. In other words, does the test accurately and reliably measure what it is supposed to under defined conditions, regardless of clinical meaning.
While we are not proposing an entirely new approach to analytical validation, these guidelines do propose adjustments and applications of the aforementioned principles that are particular to AI.
Clinical validation of AI
Several regulatory and standards organizations provide guidance on clinical validation, especially for SaMD, LDTs, and IVDs:
U.S. FDA: FDA defines
55
clinical validation as the process of establishing that the test output is clinically meaningful for its intended use. This is outlined in guidance such as: “Clinical performance must be demonstrated through studies that measure how accurately the test predicts a clinical condition.” CLSI: CLSI provides
73
protocols for clinical performance studies and test evaluation in documents like EP24-A2. CAP: CAP’s accreditation program
72
requires laboratories to document clinical validation for tests, especially LDTs and modified assays, in accordance with stated clinical purpose.
In the context of these recommendations, a summary of Clinical Validation inclusive of the aforementioned definitions would be the process of systematically evaluating that a test or system is clinically meaningful, that it accurately and reliably predicts or correlates with a clinical condition, risk, or outcome in the intended population and setting.
Clinical utility
Clinical utility is a critical aspect of evidence generation for test adoption and reimbursement. Major sources defining clinical utility include:
CMS and Medicare Administrative Contractors: CMS requires evidence of clinical utility for coverage decisions under the Medicare program. National Academy of Medicine (formerly IOM): Emphasizes that clinical utility is a cornerstone of evidence-based medicine, particularly for genomic and personalized diagnostics.
In the context of these recommendations, a summary of Clinical Utility inclusive of the aforementioned definitions would be the evidence that improved patient outcomes can be attributed to the use of the test either directly or via a chain-of-evidence approach.
Digital Pathology Association’s approach to clinical utility
Clinical utility is a complex and often contested concept, representing one of the greatest sources of interpretation and disagreement in diagnostic testing. Accordingly, the Digital Pathology Association (DPA) seeks not only to define clinical utility and establish associated standards but also to articulate a structured process for evaluating whether clinical utility has been demonstrated.
Before we propose a formal framework to guide determinations of clinical utility, we first review four sources that appear to use differing language to address similar notions.
First, the National Cancer Institute defines 74 clinical utility as: “A term that refers to the likelihood that a test will, by prompting an intervention, result in an improved health outcome. The clinical utility of a genetic test is based on the health benefits related to the interventions offered to individuals with positive test results.” Second, the National Academy of Medicine has adopted 75 the definition of the EGAPP working group of clinical utility 76 : “Simply stated, clinical utility is defined as the use of a clinical test’s result to make a treatment decision that positively changes the outcome of a patient.” Third, Medicare does not explicitly provide a definition for clinical utility but instead uses clinical utility as part of the evidentiary standard to determine whether a test is “reasonable and necessary.”77,78 Specifically, Medicare’s criteria require that medical intervention be safe and effective, not experimental, appropriate in duration and frequency, consistent with accepted standards of medical practice, delivered in an appropriate setting by qualified personnel, tailored to meet the patient’s medical needs, and “it should be at least as beneficial as any existing, medically appropriate alternative.” Lastly, the Agency for Healthcare Research and Quality uses a framework 79 for assessing clinical utility based on attaching patient outcomes to a particular diagnostic technology and outlining particular evidentiary questions.
A common theme among all of these approaches is the tying of a particular medical intervention (service or test) to an improved health outcome (reduced morbidity, mortality). In certain cases, the improvement in patient outcomes can be based on very direct evidence (mortality benefit of statins being a prime example 80 ). However, most diagnostics in routine clinical use have not used this evidentiary approach. Even FDA-approved companion claims for diagnostics81,82 are supported by a chain of evidence via concordance to a clinical trial assay. Indeed, the chain of evidence forms the typical approach for establishing clinical utility in diagnostics, in which concordance studies to well-accepted diagnostic approaches and noninferiority studies to accepted diagnostic approaches are expected to be among the approaches to evidence generation. Figure 1 illustrates the two approaches to evidence generation.
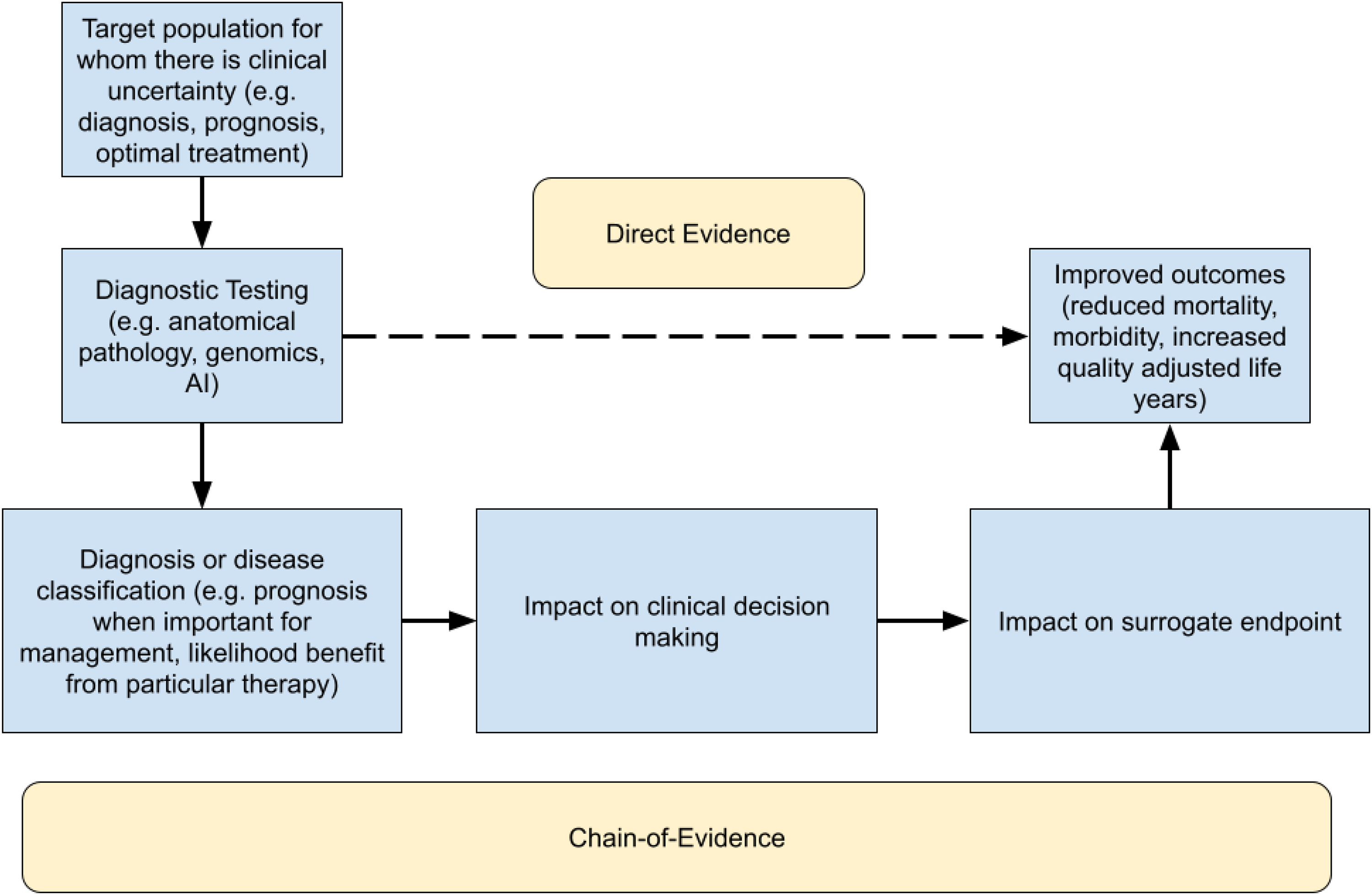
A flowchart illustrating the manner in which evidence for clinical utility can be obtained either directly or via chain-of-evidence methods.
Finally, we can propose an approach to clinical utility that applies the definitions above to the context of digital pathology in a manner sensitive to how evidence is practically generated. In particular, clinical utility can be defined as the following: The use of a clinical test’s result to make a treatment decision that positively changes the outcome of a patient, whether demonstrated via a direct or chain-of-evidence approach.
Recommendations
The following are a set of comprehensive recommendations that we hope will assist providers, patients, algorithm developers, and payers to ensure that digital pathology systems, inclusive of uses of AI in digital pathology, are properly validated and deployed in a safe and effective manner and that they have the intended effect of ultimately improving patient outcomes. Our recommendations are subdivided into three sections.
The first section covers recommendations on how digital pathology systems should be deployed. We hope that these will assist pathology laboratories and large health care organizations in planning for, and increasing adoption of, digital pathology.
The second section covers recommendations on how digital pathology algorithms are validated. We hope that these will assist pathology laboratories in verification and validation of AI systems and device manufacturers in communicating the performance characteristics of their models.
The third section covers recommendations for how AI for digital pathology algorithms are clinically used to maximize safety and efficacy. We hope that these will assist providers in adopting digital pathology, and payers and regulators to evaluate proper uses of digital pathology.
Recommendations for the deployment of digital pathology
R1: Pathology labs and health care institutions should begin planning for digital pathology
Digital pathology already enables significant improvement to patient care and will continue to expand opportunities in the future. The benefits of adoption of digital pathology include workflow optimization and efficiency,83,84 reproducible image analysis,85,86 remote access for consultation, research, and education,85–88 the ability to align and review slides across slide stains, 89 prevention of slide loss or degradation, 89 and the ability to use AI algorithms.83,89 Furthermore, adoption of AI algorithms on top of digital pathology alone includes further increases in workflow efficiency,12,90,91 diagnostic objectivity,4,91,92 and more informed decision support. 12
Unfortunately, individual physicians are often unable to implement digital pathology recommendations in their own practice until their institutions adopt digital pathology. By adopting digital pathology, institutions will enable pathologists to practice in manner contemporary with current standards of excellence and enable better communication and coordination of care.
A critical barrier to the adoption of digital pathology is that it relies on not only equipment and software purchases but also institutional changes in both human processes and information technology (IT) infrastructure, which require significant financial and operational planning. Consequently, we recommend beginning this planning early so as to ensure that laboratories and health care institutions have adequate financial, human, and IT resources to successfully implement digital pathology.
R2: Access to digital pathology is specifically indicated for cases where traditional pathology is not readily available
Digital pathology, independent of AI, is recognized as a medical necessity in settings where traditional pathology services are limited or unavailable, enabling timely and accurate diagnostic support through remote access and consultation. The CAP and the American Telemedicine Association have endorsed digital pathology for remote interpretation, particularly in underserved or rural areas, to bridge gaps in access to subspecialty expertise. 93 In global health contexts, the World Health Organization has also highlighted digital pathology as a tool to reduce diagnostic delays and improve cancer care equity where pathology infrastructure is lacking. 94 Studies have demonstrated that WSI is noninferior to traditional glass slide review for primary diagnosis, with concordance rates exceeding 95%, thereby supporting its use for routine clinical interpretation. 95 As such, digital pathology is not merely a convenience but a critical infrastructure element that ensures diagnostic continuity and timely patient care in scenarios where in-person pathology is not feasible due to geographic, logistical, or personnel constraints.
Consequently, we recommend that if a specimen being reviewed is from a patient seen in one of the following settings, documented in the patient record, then the use of digital pathology should be seen as particularly indicated in the following settings:
CAHs Rural Health Center FQHC Independent Laboratory The service is provided for a patient of one of the facility types above as part of a contractual arrangement with the facility.
R3: Clinical AI used in digital pathology should be overseen by a qualified pathologist and does not replace a pathologist
Pathologists are central to the practice of anatomical pathology and laboratory medicine, not only for their diagnostic skills but also for overseeing and ensuring high-quality results from a laboratory. Within the United States, there are varying laws governing the practice of medicine and the oversight of laboratories. This recommendation generally assumes that a pathologist is a licensed medical doctor who specializes in the diagnosis of disease primarily from in vitro specimens and who engages in the oversight of laboratories or laboratory tests. As part of training, pathologists learn to recognize a wide range of diseases and tissue types from a wide variety of specimen types.
Current AI systems may be very good at addressing specific tasks for which they were trained with well-defined inputs and outputs. However, high-quality health care demands that health care providers identify even very rare diseases and accommodate the diagnosis of patients with rare presentations of common diseases. This requires experts who are able to assess and interpret a wide range of potentially relevant information. Therefore, clinical AI systems used in pathology require pathologist oversight, even if the systems provide information that does not require pathologist interpretation. Clinical AI systems complement the skills of a pathologist rather than replace them.
Recommendations for algorithm validation
R4: The digital pathology scanning process should be validated separately from an AI algorithm
The process of scanning a physical histology slide to produce a digital image file is a prerequisite for both human and AI interpretation of digital pathology images. For AI interpretation of digital pathology images, we recommend that the digital pathology scanning mechanism (typically a combination of staining protocol and a digital slide scanner) be validated separately from any subsequent image analysis component. CAP has already provided guidelines 96 for validating WSIs independently of whether the images are consumed by humans or algorithms. In cases where the digital slide scanner is not part of a regulatory-approved (FDA, CE, etc) system, then it should be validated in the lab in which it is being used in accordance with a consensus guideline. 96 Once a digital pathology scanner is validated, any AI algorithm intended to be used with the scanner must be subsequently validated with digital images produced from that scanner (see R5).
R5: Analytical and clinical validation of AI algorithms must be demonstrated for each scanner model in which the algorithm is to be clinically used
AI algorithms in digital pathology rely on analyzing WSIs, and the performance of these algorithms can vary significantly depending on the slide scanner used to digitize the pathology specimens. Differences in scanner hardware, optics, resolution, color calibration, compression algorithms, and file formats can subtly alter the appearance of histological features in ways that AI models may not be robust, especially when models are trained on a narrow set of devices.
Multiple peer-reviewed studies have demonstrated that even state-of-the-art AI models show performance degradation when applied to images captured on a different set of scanners than those used during AI algorithm training. In a study by Aubreville et al., 97 an AI algorithm is trained to detect mitotic figures. When the algorithm is applied to the exact same slides scanned on three different digital slide scanners, they observe a 20–40% difference in F1 scores between scanners. In Swiderska-Chadaj et al, 98 the authors develop an algorithm for cancer detection that is tested via a multicenter, multiscanner protocol. They find that the algorithm’s accuracy varies between 5% and 15% depending on the digital slide scanner used for testing.
Regulators have recognized this risk. The FDA, in its 2021 clearance of Paige Prostate, 99 explicitly limited the algorithm’s use to images generated by the Philips Ultra Fast Scanner, reflecting that performance data had only been validated on that system. This highlights the agency’s position that AI tools must be evaluated within the specific ecosystem in which they are deployed, including scanner hardware. The VENTANA® TROP2 (EPR20043) RxDx Device achieved breakthrough FDA status as the first computational pathology companion diagnostic. 100 This significant achievement clears the way for the use of AstraZeneca’s Quantitative Continuous Scoring (QCS) AI algorithm to be used to objectively quantify TROP2 expression and subsequently support treatment decision-making for targeted therapies based on TROP2 quantification. This algorithm is validated exclusively for Roche’s DP 200 and DP 600 scanners.
Without scanner-specific validation, AI algorithms are at risk of reduced accuracy, false negatives, or overcalls, particularly in edge cases or rare morphologies. To mitigate these risks, the CAP statement for AI in pathology recommends validating AI tools on representative scanner platforms and ensuring ongoing performance monitoring as hardware evolves. 101
Note that many commonly used digital slide scanners are not yet FDA-approved. In these cases, in order to reliably validate and utilize AI algorithms on such scanners, images from these scanners must first be validated for each indication separately from the AI algorithm (see R4), and second, each algorithm must be shown to validate on images from these scanners.
R6: Interscanner concordance studies are acceptable to generalize a validated AI algorithm’s performance from one slide scanner to another when using the original digital slide scanner as the gold standard
When the same physical slide is scanned using two different slide scanners, the resulting pair of digital slide images can be significantly distinct in appearance. This raises the natural question: if an algorithm is validated on slide scanner A, can it be reliably used on slide scanner B? To be clear, this is not a question of comparing the algorithm’s performance on slide scanner B to human performance but rather ensuring that an algorithm’s performance on slide scanner B is effectively equivalent to its performance on slide scanner A.
This question has become central to regulatory, clinical, and operational decision-making. Sufficient evidence97,102,103 suggests that we cannot assume that an algorithm validated on scanner A will necessarily also validate on scanner B. Furthermore, while many color normalization techniques have been introduced and may improve cross-scanner performance, they do not necessarily close the performance gap.104,105 Consequently, a scientifically grounded and pragmatic approach is to conduct concordance studies, using the original validated scanner as the reference standard to assess performance on alternate scanners.
Interscanner concordance studies aim to determine whether an algorithm, when applied to slides digitized on a new scanner, produces output values that are statistically and clinically equivalent to those it generates from its originally validated scanner. These studies typically compare diagnostic outputs, such as detection sites, scores, or classifications, from images of the same slide scanned on different systems. If high agreement is demonstrated, it can be reasonably concluded that the algorithm performs similarly on both scanners for the intended use. As an illustrative example, a study might demonstrate that Cohen’s kappa >0.8, or that categorical concordance exceeds ≥95. Ultimately, the choice of both the metric and threshold are task-dependent and ultimately require higher thresholds for higher-risk tasks.
A critical component of such interscanner concordance studies is the use of a set of slides, typically referred to as reference slides or quality assurance slides, which are scanned on each of the digital slide scanners in the study. The number of slides involved and their content should reflect the typical variability of samples that one would expect the algorithm to be applied to.
This approach mirrors established practices in pathology and radiology. For example, assay bridging studies in laboratory medicine routinely use a reference standard as the comparator to validate performance on alternative platforms, and the FDA has accepted such bridging methodologies 106 for companion diagnostics and digital imaging systems. Moreover, CAP’s statement on AI validation in pathology 101 recognizes that “analytical validation using digital concordance across systems can be sufficient if it is performed rigorously and includes pathologist review or clinical outcome linkage.”
Scientific studies support this method of concordance. A paper by Lu et al 107 demonstrated that AI-based Gleason scoring models maintained >95% concordance when applied to prostate biopsies scanned on three different WSI systems—after modest color normalization was applied.
Importantly, full analytical revalidation of AI tools on every scanner is impractical and unnecessarily resource-intensive, especially when high-quality reference slides and rigorous concordance protocols can demonstrate equivalency. Requiring full clinical validation on every new scanner would create significant regulatory and adoption bottlenecks, slowing innovation and access to AI-enabled care without clear gains in safety.
Consequently, interscanner concordance studies are an efficient, evidence-supported, and clinically meaningful mechanism to generalize AI performance across digital pathology scanners.
R7: When scanner settings can be adjusted, AI algorithm validation using a specific scanner should specify scanner settings and conditions
Some WSI systems are not locked with specific settings. For example, changing focus parameters, image compression, or z-stacking parameters may be sources of variability that can impact the results of an AI model. This is less of a concern on WSI systems with locked settings, but laboratories or manufacturers should ensure that they specify the relevant settings and parameters for use of AI algorithms when scanner settings are adjustable. It may be reasonable to specify a range of parameters or settings, but in such a case, validation should be done across that range.
When a WSI system has locked settings, or always runs using clearly specified settings, then the validation may simply reference the default settings, so long as these remain the default settings.
R8: Analytical validation should include measures of both accuracy and reliability
Accuracy refers to how closely an AI tool’s output matches the reference standard (eg, expert pathologist consensus or established clinical outcomes). Without a quantification of accuracy, it is impossible to assess the model’s ability to detect or classify pathology correctly. Numerous studies have shown that measuring diagnostic accuracy is critical in determining the clinical value of AI algorithms. For example, Campanella et al 108 evaluate deep learning models at diagnosing patients with prostate, breast, and lung cancer using the area under the ROC curve (AUC) in order to determine the trade-off between sensitivity and specificity. Steiner et al 109 similarly use AUC to evaluate AI-assisted reads in breast cancer. Coudray et al 110 evaluate their AI algorithms for predicting lung cancer mutations using Cohen’s kappa to measure the concordance between the algorithm and the reference standard of expert pathologists.
Reliability, also sometimes referred to as precision, is often operationalized as intra- and inter-run reproducibility or consistency across varying conditions (eg, different scanners, staining batches, or image artifacts) and is equally important. AI tools that are accurate in a controlled setting but yield inconsistent results when deployed in the real-world pose significant clinical risk. Lin et al 105 demonstrate the degree to which AI models trained on a particular batch of images failed to generalize to images in a different batch. Miranda Ruiz et al 111 demonstrate that different scanner types affect a trained AI model’s outputs in the context of Amyloid-β detection. Ochi et al 112 demonstrate that AI models are sensitive to variations in the hematoxylin and eosin (H&E) staining protocol. These examples underscore the need for reliability assessments in validation studies.
The FDA and CAP both emphasize the need for both accuracy and reliability in analytical validation. The FDA’s guidance 55 on SaMD explicitly recommends verification and validation procedures that include performance characteristics like accuracy and reproducibility. Likewise, CAP’s recommendations 101 on validating AI in pathology recommend evaluation of both “diagnostic performance (accuracy)” and “consistency across relevant input and operating conditions (reliability).”
Excluding either metric introduces clinical risk: an AI tool that is not accurate may produce misleading outputs, while one that is not reliable may produce unpredictable results across cases, patients, or environments.
R9: AI predictions that can be verified by pathologists should rely on a cohort whose size should be determined by the intended use
Many AI algorithms produce outputs that a pathologist can verify. These include AI algorithms that highlight regions of interest in WSIs, such as mitotic figures, tumor areas, or areas suspicious for metastasis in lymph nodes 109 ; segmenting and classifying nuclei into categories (tumor cells, stromal cells, and lymphocytes); computing cell-specific features (nuclear size, shape, density); counting lymphocytes and mapping their spatial proximity to tumor cells; and performing automated QC of digital pathology slides by identifying blur, pen markings, bubbles, and other artifacts. The typical approach to validation of such algorithms would be to compare an AI-generated prediction to that of a human pathologist.
The question of how many pathologists to compare with naturally arises because interobserver variability is well-documented in pathology (and diagnostic decision-making more generally as well), particularly in the interpretation of complex or borderline cases. Relying on a single pathologist’s diagnosis risks encoding individual bias into the evaluation process, while a consensus among three or more provides a more stable and representative ground truth. Such individual bias is an inherent part of medical practice. Consequently, AI that mimics the bias of its sole user would be noninferior to that user. However, if the AI is intended to reduce bias, then validation on a consensus using multiple pathologists may be necessary.
As a general standard, we would recommend reliance on a minimum of three pathologists. Studies across tumor types, including prostate,4,113–117 breast,118–122 colon,123–127 and lung cancer,128,129 have shown that diagnostic agreement improves significantly when using multipathologist adjudication panels. Furthermore, using three pathologists enables majority voting in the absence of full consensus, offering a statistically stronger comparator and reducing the likelihood of idiosyncratic error influencing algorithm validation. This approach aligns with practices used in regulatory submissions (though regulatory bodies may demand more depending on the context) and high-quality AI development frameworks where multireader, multicase studies are the standard for establishing reference truth.
That being said, there are several exceptions to this standard.
First, even once validated by the device manufacturer, individual labs may want to add their own validation to ensure that their users interact with the AI tool in a manner acceptable to the performance within the lab. In this case, a single pathologist may suffice, and it is often infeasible for individual labs to rely on greater numbers.
Second, deployments that are research-use only may rely on a single pathologist, but AI developers and clinical collaborators should be made aware of the aforementioned limitations of reliance on single experts, and the use of less than three should be adequately justified.
Third, while the intended use of many algorithms is meant to be user agnostic, other tools might be customized to assist a single individual pathologist. In this case, that single pathologist may provide the gold standard for validation. Such AI would not be used to represent a consensus diagnosis and therefore not validated for such a purpose. Examples of such algorithms include tools that might draw contours of regions of interest on digital slides or language models that help craft pathology reports in the style of a single pathologist.
R10: AI predictions that are distinct from pathologist interpretations should use a reference standard that corresponds with the output being predicted
For many tasks that AI algorithms perform (cell segmentation, artifact detection), the natural reference for clinical validation performance is human pathologists. However, what if the AI algorithm is performing a task that pathologists cannot easily visually verify, such as therapeutic response?
AI tasks that pathologists cannot easily visually verify are those tasks that a pathologist cannot simply examine the contents of one or more slides to produce the same output. Examples include quantifying molecular biomarkers when there is insufficient tissue for performing immunohistochemical (IHC) staining, quantifying the likelihood that a particular patient’s tumor exhibits high microsatellite instability (MSI), or quantifying the likelihood that a particular patient’s tumor is characterized by HRD. A growing number of products have been released or FDA-cleared in recent years. The FDA granted Breakthrough Device Designation to the TROP2-QCS AI 100 biomarker, which calculates a normalized membrane ratio of TROP2 IHC staining to predict response to Datopotamab deruxtecan that a human pathologist cannot visualize with precision.130–132 The Artera AI test 12 produces predictive outputs, such as whether a patient is likely to benefit from adding androgen deprivation therapy to radiation in order to guide decisions about treatment intensification versus de-escalation. A number of vendors have commercialized AI algorithms that produce prognostic risk scores12,133–135 by analyzing digitized H&E pathology slides from a patient’s biopsy along with clinical variables. AI algorithms have also demonstrated the ability to predict molecular biomarkers from H&E136–139 and even simulate IHC staining from H&E alone. 140
With this in mind, for tasks that pathologists can visually verify, one or more human pathologists should serve as the reference standard. For tasks that pathologists cannot visually verify, an appropriate reference standard must be obtained for sufficient clinical validation of the algorithm. Table 2 provides a noncomprehensive list of examples of the references that might be used to clinically validate AI algorithms that are not easily visually verifiable.
Examples of Reference Standards for Digital-Pathology-Based Tasks That Are Not Verifiable by a Pathologist

H&E, hematoxylin and eosin; IHC, immunohistochemical; MSI, microsatellite instability.
R11: As with any laboratory test, analytical quality systems and processes should be established so as to ensure that invalid results are not reported due to laboratory errors
Responsible lab operators are already accustomed to putting into place laboratory-level countermeasures to protect patients from device-generated medical errors. In this respect, AI software should be no different. Indeed, many problems can arise within the performance of AI testing, so it is critical that systems be put in place so as to detect problems and address them. QC should be implemented to address the full end-to-end processes in the testing system to detect any problems that may arise with the test processes that could threaten the validity of results reported on patients. A growing body of evidence97,102,103,105,111,112 demonstrates the degree to which AI models in particular are sensitive to batch effects, changes in H&E staining protocols, and scanner changes.
QC methods and acceptance criteria should be developed based on the intended use of the AI test and any established criteria on the appropriate specimen types as well as the reportable range of test output. QC may be performed using methods such as intermittent testing of control specimens or within-batch controls. To develop appropriate QC processes, laboratories implementing AI should systematically identify potential sources of variability within the end-to-end testing process and ensure that they have adequate QC methods to identify all such sources of potential test failure.
R12: Laboratories and manufacturers should specify minimum usable sample requirements for AI algorithms
AI algorithms applied to pathology slides must specify limits of use, such as minimum usable tissue area and minimum cancer content, because their performance is highly dependent on input quality and context.
Algorithms trained on datasets with adequate tumor representation may produce unreliable outputs when applied to small biopsies or slides with sparse cancerous regions, leading to false negatives or clinically misleading quantification. For instance, FDA-cleared tools like Paige Prostate 99 explicitly limit use to prostate needle biopsies with sufficient tumor presence, noting that performance degrades in benign or low-tumor-content samples. 99 Similarly, studies evaluating PD-L1 scoring algorithms emphasize that accurate quantification requires a minimum number of tumor cells, often recommending thresholds such as ≥100 viable tumor cells to ensure reliability.141,142 Without enforcing these boundaries, AI tools risk being misapplied outside of their validated domain, undermining diagnostic accuracy and patient safety. Thus, clear specification of use parameters is essential for clinical integration and regulatory compliance.
Labs and manufacturers should consider how to establish what constitutes usable tissue. Artifacts within tissue may impact the extent to which the tissue can be used for inferential purposes by AI algorithms. Tissue containing artifacts to which the algorithm is robust may be usable, while tissue containing artifacts that adversely impact algorithmic validity or containing artifacts with an unknown impact on algorithmic validity should not be considered usable.
Research has shown that artifacts can confound deep learning models, introducing false positives or negatives, particularly in tasks like tumor detection or cell quantification. 143 For example, a study by Tizhoosh and Pantanowitz 144 highlights how AI performance degrades in the presence of common slide defects unless explicitly trained on artifact-diverse datasets or equipped with preprocessing artifact detection modules. Moreover, the FDA and regulatory bodies increasingly expect manufacturers to characterize performance in the presence of real-world variability, including artifacts, to ensure safe clinical deployment. 145 Therefore, transparent documentation of algorithm robustness—or limitations—in artifact-laden conditions is essential to guide clinical users and avoid misapplication in suboptimal imaging scenarios.
Recommendations for realizing clinical utility
R13: A clinical AI algorithm should be used per its intended use
Analogous to immunohistochemistry or special stains, the ability to use AI in the laboratory does not imply that it should be universally used. AI should be used when it can improve the interpretation of the pathologist or provide new information that the pathologist is not able to provide in the absence of AI.
For example, clear-cut cases of prostate cancer are unlikely to require AI to determine if cancer is present. Conversely, if there is a question on whether the cancer should be classified as a Gleason 3 + 4 or a Gleason 4 + 3, it may be appropriate to apply AI to address this question so long as the AI has been appropriately validated. Similarly, a liver biopsy showing clear cirrhosis is unlikely to benefit from AI assessment of fibrosis status. However, when the biopsy shows fibrosis, AI that can provide a histological grade may be appropriate for use if the patient’s treatment is dependent upon accurate fibrosis measurement.
Some evidence from inside4,146 and outside147,148 of digital pathology suggests that while AI algorithms have demonstrated improved efficiency and accuracy at diagnostic tasks, clinicians may end up relying on the AI algorithm in a manner inconsistent with its intended use. For example, one can imagine a “second read” algorithm being overly relied on in a manner that results in the algorithm effectively being trusted as the primary read.
Ultimately, clinicians should be familiar with each algorithm’s intended use and realize their adoption of such algorithms in a manner consistent with their intended uses.
R14: AI algorithms are appropriate for use when they have been validated to complement or replace an existing test
AI algorithms have emerged as a laboratory tool that are appropriate for clinical use when they are reliable complements, screens, or substitutes for more invasive, expensive, or time-consuming tests, such as molecular or genomic assays, by extracting equivalent or superior prognostic or predictive information directly from histopathology slides. Indeed, in resource-constrained settings, or when tissue quantity is limited, molecular testing may be intentionally limited.
While AI algorithms that predict molecular signatures are still emerging, an increasing number of studies have demonstrated that AI models can indeed infer molecular features like MSI, tumor mutational burden, or even specific mutations (eg, IDH1, EGFR) from routine H&E-stained slides with clinically relevant accuracy.149,150 For example, Echle et al 149 showed that MSI could be predicted from colorectal cancer histology with an AUC of up to 0.89, suggesting the potential to triage cases for confirmatory molecular testing.
Commercially available AI algorithms are also emerging that produce similar outputs to existing molecular tests. For example, the ArteraAI Prostate test 151 combines pathology image data and clinical variables to stratify patients by risk and guide treatment decisions based on digital pathology images and clinical variables. Comparable molecular tests include Decipher Prostate 152 and MDX Health’s GPS. 153 However, CMS’s price 154 for the AI-driven test, $706.26, is markedly different than the price for the molecular tests, $3,873.
In addition, AI IHC quantification algorithms that do not require additional reflex testing (eg, in situ hybridization [ISH]) are also medically necessary. For example, a CE-marked HER2 algorithm 155 has demonstrated a reduction in the need for ISH testing by reclassifying HER2 IHC from 2+ to either 0/1+ or 3+. This reclassification of IHC was validated using FISH. When the algorithm downgrades an IHC 2+ (human read 2+) to a 0/1+, the FISH score is 0. When the algorithm upgrades the IHC score to 3+, the FISH is very positive.
The ability to replicate such insights from routinely acquired data such as H&E-stained pathology slides not only streamlines diagnostics and reduces costs but also shortens time to treatment decisions. However, clinical use in this context requires rigorous validation and transparent reporting of model performance to ensure AI can match or exceed the reliability of the replaced test, as recommended by recent best-practice guidelines. 156
R15: AI algorithms are recommended to assist physicians when sufficiently validated and medically necessary
AI algorithms have already had a significant effect in digital pathology in several respects and, provided they are sufficiently validated and medically necessary, we recommend their use in assisting physicians.
In particular, a number of AI algorithms already assist clinicians in their workflows by detecting and surfacing clinically relevant data without generating conclusions. Examples include AI algorithms that highlight regions of interest in WSIs. 109 FDA-approved devices such as Paige Prostate Detect 99 assist pathologists by flagging prostate biopsy regions that may contain cancerous features, requiring expert review for final diagnosis. Similarly, algorithms can prescreen colorectal biopsies to identify slides unlikely to contain malignant findings, allowing for more efficient case triaging. 108 These systems have already been shown to improve case review efficiency,4,157 reduce oversight-related errors, and support pathologists in managing rising case volumes.144,158 While these particular algorithms do not make clinical decisions themselves, they enhance the diagnostic process by focusing attention, reducing fatigue-related error, and enabling faster, more consistent interpretations—ultimately contributing to improved patient care 159 with increased potential in community hospital settings. 160
Furthermore, a number of AI algorithms produce diagnostically relevant information that can be used to improve patient care. The TROP2 QCS algorithm developed by AstraZeneca objectively quantifies TROP2 expression on IHC WSIs by identifying tumor regions and TROP2-positive membranes to generate a continuous score reflecting staining intensity and tumor cell prevalence,130–132 reducing interobserver variability and enabling patient stratification in trials of TROP2-targeted therapies. As the first AI-driven H&E companion diagnostic, 161 TROP2 demonstrates that clinically actionable biomarker information can be inferred directly from routine H&E slides, reducing tissue use, cost, and turnaround time while capturing intratumoral heterogeneity and establishing AI as a regulated, treatment-enabling diagnostic modality. Another example is the algorithm used in the FDA-approved Artera AI test. 151 The algorithm analyzes digitized H&E pathology slides from a patient’s biopsy along with clinical variables such as PSA, age, and tumor stage and produces prognostic outputs (eg, risk of metastasis or prostate cancer-specific mortality) as well as predictive outputs, such as whether a patient is likely to benefit from adding androgen deprivation therapy to radiation. Clinically, it is used to help stratify patients across low-, intermediate-, and high-risk disease and to guide decisions about treatment intensification versus de-escalation.
With this in mind, we propose that AI algorithms are considered sufficiently validated when:
Peer-reviewed published research shows that the AI algorithm improves the accuracy, reliability, or throughput of diagnostic assessments by pathologists. Analytical validation and clinical validation follow Recommendations 4–12. The AI has been verified in the laboratory and implemented with appropriate processes for QC.
In addition, AI algorithms should be considered medically necessary when:
There is an order for an anatomical pathology service by the treating clinician. A board-eligible or board-certified pathologist or dermatopathologist is using the AI to perform a medically necessary assessment of the tissue.
Methodology
The DPA assembled a working group of pathologists and technologists with a mix of backgrounds in academia and industry. The goal of the cohort was to provide guidance to the aforementioned stakeholders in a manner that would ensure that digital pathology continues its rapid adoption in a manner that ultimately yields improved patient outcomes, based on rigorous approaches to validation and clinical utility.
To inform the development of these guidelines, a comprehensive search of publicly available sources was conducted to identify relevant evidence pertaining to digital pathology and AI applications. Sources included peer-reviewed journal articles, conference proceedings, publicly available preprints, regulatory guidance documents, professional society statements, and other authoritative materials.
The evidence collection process was guided by the principles of comprehensiveness and relevance. Searches were performed using a combination of key terms related to digital pathology, computational pathology, AI, machine learning, clinical and analytical validation, regulatory frameworks, and diagnostic performance. Identified sources were evaluated for applicability to the guideline topics, with priority given to materials that provided empirical data, systematic reviews, or expert consensus relevant to the safe and effective implementation of DP and AI technologies.
All evidence sources were documented and cited within the guideline to ensure transparency. While this approach did not employ a formal systematic review methodology, it aimed to capture the breadth of current literature and guidance to support evidence-informed recommendations.
For each recommendation, the group adapted the GRADE system 162 as we excluded any recommendations with “Very Low” confidence. Table 3 illustrates the levels of evidence categories and the definitions we relied on.
The Levels of Evidence Used to Evaluate Each Recommendation

Furthermore, we not only provide a description of our strength of recommendations in Table 4 but also make clear, for an abbreviated set of stakeholders, how they should interpret each corresponding guideline based on the strength.
The Strength of Each Recommendation as Well as an Abbreviated Guide for How Various Stakeholders Should Interpret Each Guideline Based on the Strength

AI, artificial intelligence.
Use cases/examples
The preceding sections established evidence-based recommendations and standards for the validation and implementation of digital pathology and AI-enabled tools. The following examples, spanning applications such as cancer detection and prognostic risk assessment, illustrate specific use cases and examples where these guidelines would be applied.
Cancer detection
AI-powered cancer detection is one of the most mature and widely adopted applications of AI in digital pathology, demonstrating clear value in diagnostic accuracy, efficiency, and QC. Solutions from companies such as Paige, Ibex Medical Analytics, PathAI, and Aiforia are being deployed in clinical settings to detect malignancy in WSIs, assist in tumor grading, and enhance safety-net functions through diagnostic discrepancy detection.
A landmark example is Paige Prostate, which in 2021 became the first AI tool for pathology to receive FDA de novo clearance 99 for clinical use in prostate cancer detection. In a prospective multireader study, Paige Prostate improved the diagnostic sensitivity of general pathologists from 88.7% to 96.6% when used as an assistive tool, without a significant decrease in specificity. 4
Similarly, Ibex’s Galen™ Prostate and Galen™ Breast platforms are CE-marked 163 and used across Europe and the Middle East, and Ibex Prostate Detect received FDA clearance. 164 Pantanowitz et al 85 demonstrated Galen Prostate’s ability to achieve 99.0% sensitivity and 97.6% specificity in identifying prostate cancer in core needle biopsies.
Though reimbursement mechanisms are still emerging, particularly in the United States, many of these tools are deployed under LDT frameworks or embedded in institutional workflows. Their value is evident: multiple studies report that AI-assisted triage and detection can reduce review time, 157 improve interobserver agreement, 3 and help ensure rare or subtle cancers are not overlooked. 165
As validation studies continue and regulatory pathways expand, AI-driven cancer detection is transitioning from an innovative add-on to a foundational element of modern digital pathology practice.
Prognostic risk assessment
AI-driven prognostic risk assessment in digital pathology is rapidly emerging as a powerful clinical tool, enabling more precise and individualized predictions about cancer progression, treatment response, and patient outcomes. Unlike traditional diagnostic AI tools that focus on detecting the presence of malignancy, prognostic models extract quantitative histological features from digitized slides—often imperceptible to the human eye—and use them to predict disease aggressiveness, likelihood of recurrence, or survival.
A leading example is Artera, which developed the first multimodal prognostic and predictive biomarker platform for localized prostate cancer. 12 Artera’s test combines pathology image data and clinical variables to stratify patients by risk and guide treatment decisions. In a validation study, 166 the model successfully identified which patients benefited from intensified therapy, such as long-term androgen deprivation, and which could safely avoid it—demonstrating clinical utility beyond existing genomic or clinical risk models.
Another example is Paige’s AI-based grading and quantification tools, 99 which are being developed to not only detect cancer but also provide risk stratification information based on tumor architecture, gland morphology, and spatial features. These features are often used to refine Gleason grading or supplement prognostic indices.
Studies using open datasets such as The Cancer Genome Atlas (TCGA) and CPTAC have shown that deep learning models trained on pathology slides can predict overall survival, recurrence-free survival, and molecular subtypes in multiple cancers, including breast, lung, colorectal, and glioma.167–169 These models often rival or surpass traditional molecular classifiers, while being significantly more scalable and cost-effective since they use routine H&E slides.
Biomarker scoring
AI-driven biomarker scoring is transforming how IHC and ISH biomarkers are quantified in digital pathology, enabling faster, more standardized assessments that support precision oncology. These tools analyze stained tissue sections—such as PD-L1, HER2, ER/PR, and Ki-67—on WSIs to provide objective, reproducible scoring aligned with clinical guidelines or companion diagnostics (CDx).
Multiple companies now offer AI solutions that aid biomarker quantification. For example, PathAI, Aiforia, Visiopharm, and Roche have developed algorithms for PD-L1 scoring in non-small cell lung cancer (NSCLC), triple-negative breast cancer, and gastric cancers. These tools assist in calculating tumor proportion score or combined positive score, improving consistency in determining patient eligibility for immunotherapies such as pembrolizumab or atezolizumab.
PathAI’s AISight platform has supported studies in PD-L1 and Ki-67 scoring, including partnerships with pharmaceutical sponsors to enhance companion diagnostic reproducibility. In one study involving PD-L1 scoring for gastric cancer, AI-assisted pathologists showed greater concordance with expert consensus than unaided readers. 170
Biomarker scoring with AI also supports HER2 status determination, a critical factor in selecting HER2-targeted therapies in breast and gastric cancers. Tools from Visiopharm and Paige are being explored for HER2 quantification and HER2-low classification, which has emerged as a new clinical category.155,171
While reimbursement for AI biomarker tools is often embedded within broader digital pathology services or companion diagnostic workflows, they are increasingly being recognized in regulatory filings. The AMA’s CPT Editorial Panel has introduced taxonomy guidance to classify autonomous AI scoring tools (CPT Panel, 2023), and CMS has begun to consider coverage pathways for software that enhances FDA-approved CDx tests.
Ultimately, AI-powered biomarker scoring provides a scalable way to reduce variability, shorten turnaround times, and ensure more equitable access to precision medicine. By enabling consistent interpretation of complex markers, these tools help ensure the right patients get the right therapy—especially in contexts where biomarker scoring is known to be subjective or variable.
Quality control
AI-enabled QC in tissue slide preparation is an emerging yet essential application in digital pathology, addressing a persistent and often underrecognized contributor to diagnostic error: preanalytic variability. From tissue processing and sectioning to staining and slide digitization, suboptimal slide quality can compromise diagnosis, delay turnaround, and reduce the utility of downstream AI tools. AI-driven QC solutions help flag poor-quality slides in real time, enabling reprocessing before diagnostic review or AI inference.
A number of different efforts have illustrated the degree to which AI algorithms can perform various types of QC in an automated fashion commensurate with human pathologists. An early attempt was the work of Janowczyk et al, 172 which illustrated that hand-crafted machine learning features could be used to identify artifactless regions of interest in digital slides and demonstrated strong agreement (94%) with human experts. Using a larger dataset from TCGA, 173 Haghighat et al 174 demonstrate a correlation of 0.89 with human pathologists at slide-level overall diagnostic usability using a multistage deep learning algorithm. Weng et al 175 and Jabar et al 176 demonstrate high pixel-level accuracy (Dice score of ∼94%) for segmentation of artifactless tumor pixels from a variety of institutions and digital slide scanners.
While these efforts have demonstrated the viability of automated QC, a number of companies have commercialized AI tools for automated quality assurance of H&E and IHC slides. Algorithms such as Conflux’s QC suite, 177 Aiosyn’s AiosynQC, 178 Leica’s SlideQC BF, 179 and PathAI’s Artifact Detect 180 identify common artifacts in WSIs, such as air bubbles, folds, debris, blurry/out-of-focus regions, and pen marks, and can highlight affected areas to flag slides that may need rescan or manual review.
While most QC tools have not yet undergone formal FDA review, they are increasingly integrated into LDT pipelines and digital pathology platforms. For example, PathAI’s Artifact Detect can be added to their AISight Image Viewer, 181 and AiosynQC has been integrated into Sectra’s image viewer via their app marketplace. 182 Some vendors offer QC modules as part of broader whole-slide imaging systems, such as the Leica Aperio iQC Software 183 or Proscia’s Automated QC 184 in Concentriq LS, which monitors scan fidelity and alerts technicians in real time.
Triage
AI-powered triage systems in digital pathology can improve efficiency by automatically identifying and prioritizing slide cases based on clinical urgency and diagnostic complexity. Slides most likely to be benign or low-risk—such as benign prostate or breast biopsies—can be flagged for later review, allowing pathologists to focus first on complex or high-likelihood-of-disease cases. By ensuring that critical cases are reviewed earlier, these systems can facilitate earlier diagnosis, which in turn enables timely treatment initiation and more efficient scheduling of additional testing. This targeted prioritization helps reduce diagnostic backlog, mitigate fatigue, and streamline workflows in high-volume laboratories. It also supports pathologists who must balance multiple responsibilities, such as intraoperative consultations, fine needle aspiration procedures, and tumor boards, by ensuring that critical cases rise to the top of the worklist.
The impact of triage has been demonstrated in radiology, where AI-based prioritization has led to measurable clinical benefits. At Cedars-Sinai, the implementation of AI triage software for intracranial hemorrhage and pulmonary embolism reduced hospital length of stay by 1.3 days (11.9%) and 2.07 days (26.3%), respectively, compared with pre-AI periods. These reductions were attributed to faster case flagging, earlier radiologist notification, and quicker clinical intervention, all of which are critical for patient outcomes. 185 Similar AI-driven triage in digital pathology could enable earlier diagnosis of high-risk cases, faster communication with clinical teams, and more efficient use of pathologists’ time, ultimately improving patient care while maintaining safety for low-risk cases.
One of the most established examples is Paige Prostate Detect, 99 which is FDA-cleared to assist in the identification of prostate cancer in WSIs. While its core function is cancer detection, the tool has been shown to triage out benign slides with high negative predictive value, enabling pathologists to spend up to 65% less time on negative cases. 109 The tool achieved an AUC of 0.99 for differentiating between benign and malignant cases in multireader studies.
Similarly, Ibex’s Galen™ Prostate platform, CE-marked and in routine clinical use in Europe, provides triage and prioritization functionality, highlighting suspicious regions and enabling lab workflows that automatically escalate slides with AI-flagged abnormalities. 186 In real-world deployments, this has led to reductions in diagnostic turnaround times and improved case prioritization for subspecialty review.
Academic studies have replicated this utility across other cancer types. For example, AI models trained on breast cancer biopsies can triage out benign cases with >95% accuracy, reducing review burden while maintaining diagnostic safety nets. Triage models have also been evaluated for colorectal polyps, cervical cytology, and lung nodules, with high performance in excluding normal or low-risk findings.
Tools like Paige PanCancer Detect, 99 a foundation model–based system recognized by the U.S. FDA as a Breakthrough Device, are designed to flag slides suspicious for malignancy across multiple tissue and organ types. In real-world testing, Paige PanCancer achieved high sensitivity (93–95%) and 90% overall accuracy, even identifying previously missed small carcinoma foci. 26
While these tools are not typically reimbursed as standalone services, their operational impact is substantial. Pathology departments using triage AI report shortened average case turnaround times, lower review variability, and reduced subspecialty bottlenecks—especially valuable in under-resourced or high-volume labs.
Triage AI is increasingly seen not just as a convenience but as a critical component of safe and scalable digital pathology—automating what pathologists already do intuitively and ensuring that scarce human expertise is focused where it matters most.
Future Directions
The landscape of digital pathology continues to evolve at an unprecedented pace, driven by technological innovations in both hardware and AI systems. As we look toward the future, several key developments will likely reshape the field and present new opportunities and challenges for pathology practice.
Emerging technologies in slide scanning
The next generation of slide scanning technologies promises significant advances in imaging capabilities, throughput, and accessibility. We anticipate developments in ultra-high-resolution imaging systems that may surpass current 40× magnification standards, potentially enabling visualization of subcellular structures with greater clarity. Multispectral and hyperspectral imaging technologies 187 are emerging that could provide enhanced tissue characterization beyond traditional brightfield microscopy, offering new diagnostic capabilities through spectral analysis of tissue components. For example, quantification of HER2 using multispectral imaging provides superior prognostic information for invasive breast cancer compared with conventional RGB imaging, demonstrating higher predictive accuracy for 5-year disease-free survival and stronger association with patient outcomes. 188
Real-time scanning technologies may eliminate current batch-processing limitations, allowing for immediate digitization as slides are prepared. In addition, portable and point-of-care scanning solutions are being developed that could democratize access to digital pathology in resource-limited settings and enable rapid consultation in surgical and clinical environments.
Advances in optical technologies, including computational imaging and deep learning-enhanced image reconstruction, may also improve image quality while reducing scanning time and storage requirements. These developments could make digital pathology more efficient and cost-effective for widespread adoption.
Artificial intelligence
The integration of AI in digital pathology represents one of the most transformative aspects of the field’s future. The vast majority of AI tools in clinical use are ultimately relied on by physicians to obtain a better outcome while maintaining pathologist oversight and final decision-making authority.
At present, there are no widely deployed Autonomous Level II (systems that can make independent diagnostic decisions with pathologist review) or Autonomous Level III (fully autonomous diagnostic systems) solutions in routine clinical practice. However, the rapid pace of AI development suggests this landscape will continue to evolve significantly.
We anticipate continued expansion of AI tools that help pathologists identify regions of interest, quantify biomarkers, and detect potential diagnostic pitfalls. Augmentative systems will likely become more sophisticated in their ability to enhance pathologist capabilities through advanced image analysis, pattern recognition, and integration of multimodal data, including genomic, clinical, and imaging information. Autonomous Level I applications may expand to cover more specialized areas of pathology, potentially including complex morphological assessments and prognostic and predictive scoring systems. As these systems mature and demonstrate consistent performance across diverse populations and institutions, the regulatory and clinical pathways for higher levels of autonomy may begin to emerge.
The development trajectory toward higher autonomy levels (Level II and Level III) remains to be seen, not just in digital pathology but across other areas of radiological imaging and health care in general.