
Abstract
Markedly elevated IgE in children is an important but nonspecific clue, particularly with early-onset atopic dermatitis, eosinophilia, and infections. We report a boy who developed generalized eczema at approximately 6 months of age and was repeatedly hospitalized during infancy for severe dermatitis, prolonged diarrhea, pneumonia, acute otitis media, and Staphylococcus aureus bacteremia. Early investigations showed eosinophilia, hypoalbuminemia, rapid IgE escalation, and lymphocyte subsets not suggestive of a clear T-, B-, or NK-cell deficiency. After 12 months, no further severe infections requiring hospitalization were documented, whereas chronic severe dermatitis, eosinophilia, markedly elevated IgE, and later asthma became dominant. Whole-exome sequencing identified two FLG variants; among these, Sanger sequencing confirmed a heterozygous FLG NM_002016.2:c.6950_6957del (p.Ser2317Ter) variant. This case highlights overlaps between severe barrier-related atopic dermatitis and hyper-IgE syndrome and emphasizes longitudinal follow-up, immunological evaluation, vaccine history, genetic interpretation, and gene–environment interactions.
Introduction
Markedly elevated IgE in children is an important clinical clue, but it is not disease specific. In practice, the combination of early-onset atopic dermatitis, eosinophilia, early bacterial infections, and very high IgE levels often raises a differential diagnosis between severe atopic dermatitis and hyper-IgE syndrome (HIES). Beyond classical HIES, early-onset severe eczematous dermatitis with marked IgE elevation and eosinophilia may also raise concern for other inborn errors of immunity, including Omenn syndrome, Wiskott–Aldrich syndrome, FOXN1 haploinsufficiency, and immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome. These disorders may share overlapping manifestations such as severe eczema, recurrent infections, eosinophilia, impaired immune responses, failure to thrive, diarrhea, or systemic immune dysregulation. Distinguishing severe atopic dermatitis from these immune disorders is particularly challenging in early life, when clinical features may substantially overlap and the more characteristic manifestations of inborn errors of immunity may not yet be fully apparent. 1 Therefore, careful longitudinal clinical assessment, immunological evaluation, and genetic interpretation are essential for accurate diagnosis.
Filaggrin, encoded by the FLG gene, is an essential structural protein of the epidermal barrier. It plays a key role in stratum corneum differentiation, maintenance of skin hydration, and protection against the penetration of allergens, microorganisms, and environmental irritants. Loss-of-function variants in FLG are among the best-established genetic risk factors for atopic dermatitis, particularly in patients with early-onset, persistent, and severe disease, often accompanied by xerosis, increased allergen sensitization, and asthma.2,3
We report a child with FLG-related early-onset atopic dermatitis who presented with markedly elevated IgE, persistent eosinophilia, and early infections, resulting in substantial clinical overlap with HIES during infancy.
Methods
This case report was prepared based on a retrospective review of the patient’s clinical records, laboratory investigations, immunological assessments, genetic testing results, treatment history, and longitudinal follow-up data. The manuscript was prepared in accordance with the principles of case-report writing, with emphasis on diagnostic reasoning, longitudinal phenotype evolution, and genotype–phenotype interpretation.
Ethical Approval
Ethical approval and informed consent details are provided in the separate title page to preserve double-anonymized peer review. Written informed consent for participation and publication was obtained from the patient’s parent/legal guardian, and all patient data were anonymized during manuscript preparation to protect patient privacy.
Case Presentation
The patient was a male child born at term in 2014 as the first child of a naturally conceived pregnancy and was delivered vaginally to non-consanguineous parents. There was no known family history of primary immunodeficiency, early infant death, recurrent severe infections, chronic mucocutaneous candidiasis, skeletal abnormalities, or similar severe dermatologic disease. The father was later found to carry the same heterozygous FLG variant but did not have a similarly severe atopic phenotype. The earliest manifestation was eczema, which became clearly apparent at approximately 6 months of age as generalized atopic dermatitis ( Figure 1). Between August and December 2014, he required multiple hospital admissions, mainly for severe eczema, prolonged diarrhea, and pneumonia. During the same period, he also developed acute otitis media in August 2014 and Staphylococcus aureus bacteremia in September 2014. Regarding immunization history, the patient had received only Bacillus Calmette–Guérin vaccination at birth and had not received other routine childhood vaccines. No BCG-related complication or disseminated BCG infection was documented. Vaccine-specific antibody responses were not assessed because the patient had not completed routine childhood immunizations. Early clinical presentation at approximately 6 months of age. The patient showed severe generalized eczematous dermatitis with diffuse erythema, xerosis, and widespread cutaneous involvement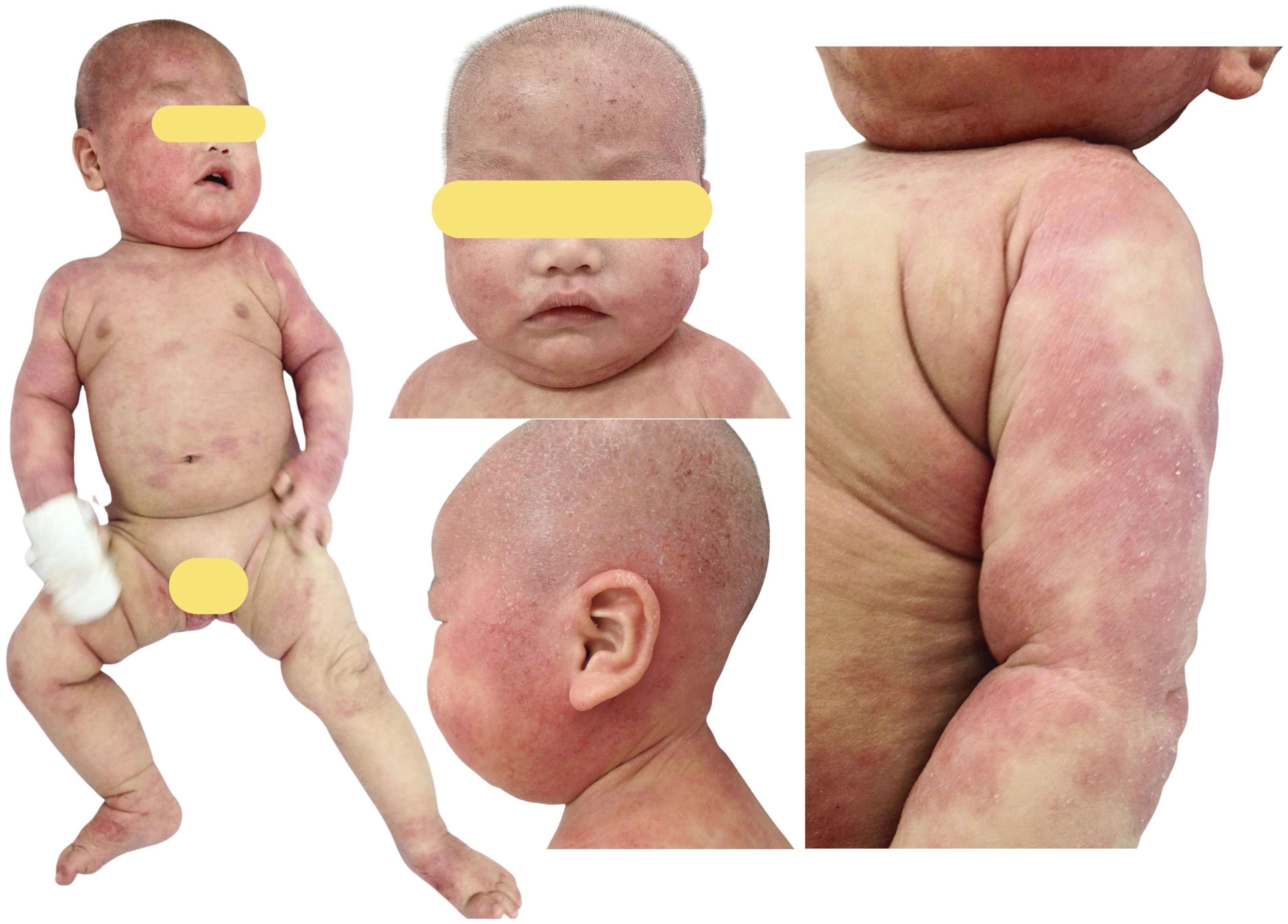
Initial management included broad-spectrum antibiotics, intravenous fluids, human albumin infusion, topical corticosteroids, and emollients. The cutaneous response remained limited. Early laboratory investigations showed leukocytosis ranging from 21 to 44 ×109/L with lymphocyte predominance, eosinophilia ranging from 8.3% to 34%, thrombocytosis ranging from 223 to 943 ×109/L, low total protein ranging from 28.6 to 54 g/L, and marked hypoalbuminemia ranging from 13.9 to 35.5 g/L. Before intravenous immunoglobulin (IVIG) administration, IgG, IgA, and IgM were generally within age-appropriate limits or only mildly decreased. Total IgE rose rapidly during the first year of life from 71.7 IU/mL to 114.3 IU/mL, 250 IU/mL, and 296 IU/mL, reaching approximately 1,150 to 1,200 IU/mL by the end of 2014. Lymphocyte subset analysis during this period showed CD3 57% to 71%, CD4 36% to 39%, CD8 20% to 30%, CD19 19% to 29%, and NK cells 7% to 9%, without evidence of a clear T-, B-, or NK-cell deficiency.
After 12 months of age, no further severe infections requiring hospitalization were documented. However, the dermatitis did not resolve and instead evolved into a chronic severe disease course with seasonal fluctuation. At reassessment on December 30, 2015, CRP was 0.22 mg/L, IgG was 8.43 g/L, IgA was 0.50 g/L, IgM was 1.16 g/L, and eosinophils remained elevated at 12.4%.
From 2019 onward, the patient was diagnosed with bronchial asthma, which was well controlled with inhaled corticosteroids. In recent years, the allergic phenotype became more prominent, with total IgE reaching 12,941 IU/mL in May 2024 and 13,571 IU/mL during June to July 2025. Eosinophilia persisted, measuring 22.0% in May 2024 and 20.1% to 26.0% during June to July 2025. Albumin recovered to near-normal levels, reaching 40.7 g/L on recent assessment, while IgG remained within the age-appropriate range and CRP was not elevated.
In 2024, the patient received systemic cyclosporine for severe atopic dermatitis, but the clinical response was limited. The drug was subsequently discontinued, and he remained on intensive skin care and topical treatment. At the most recent evaluation in July 2025, at approximately 11 years and 5 months of age, he was alert and afebrile. His weight was 38.0 kg, height was 148.0 cm, and body mass index was approximately 17.4 kg/m2, corresponding to approximately the 50th, 61st, and 48th percentiles for age and sex, respectively. These anthropometric parameters were within the expected range and did not suggest persistent failure to thrive. There was no edema, purpura, or hepatosplenomegaly. The dominant findings were xerosis, generalized pruritic papules, and diffuse erythema, consistent with chronic severe atopic dermatitis (Figure 2). Clinical appearance at follow-up. Persistent xerosis, diffuse erythema, and widespread eczematous lesions with excoriations reflect chronic inflammatory skin involvement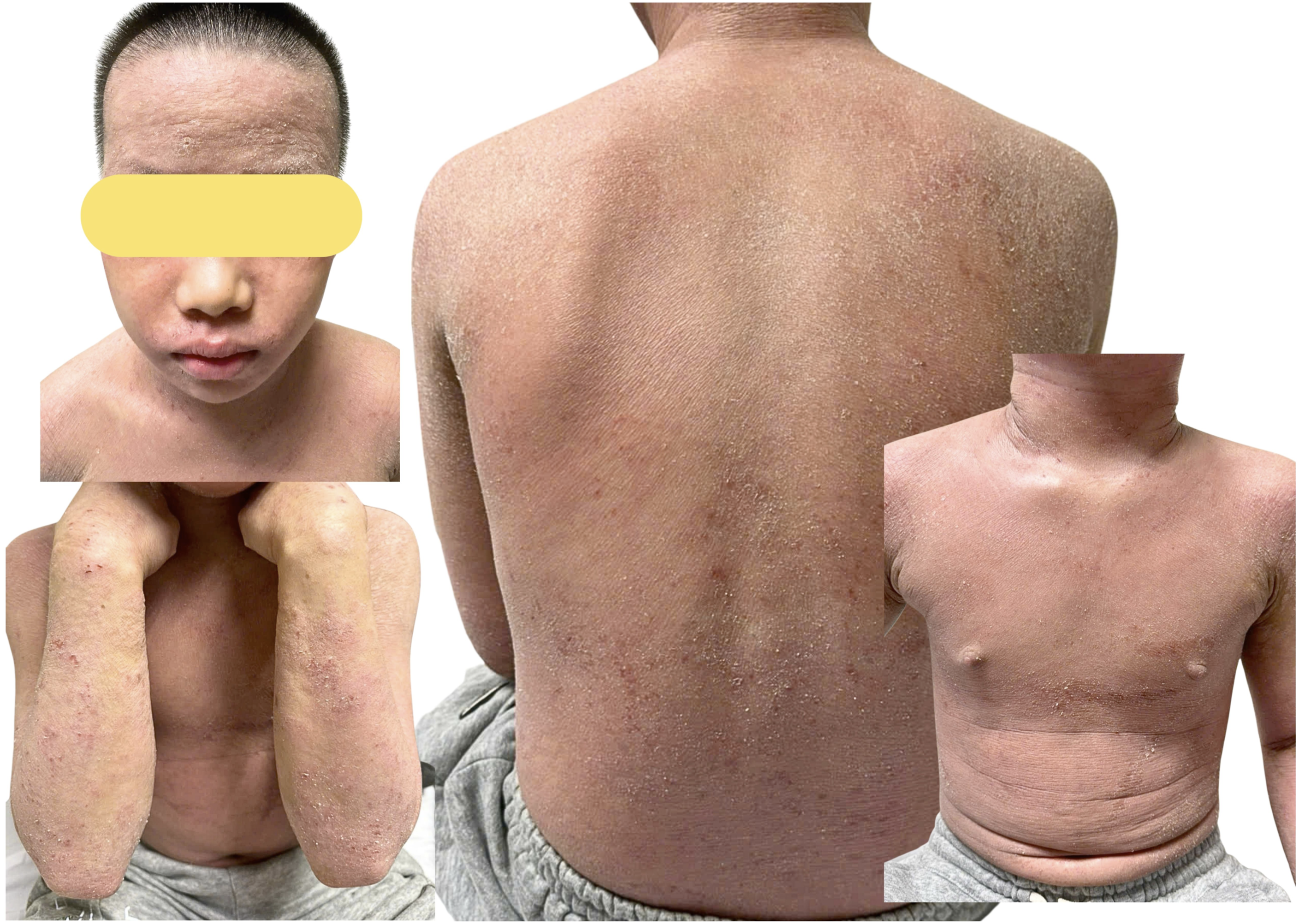
Abdominal ultrasound and abdominal radiography showed no abnormalities. CMV, EBV, and SARS-CoV-2 testing were negative. Clinically, despite markedly elevated IgE, persistent eosinophilia, and early bacterial infections, longitudinal follow-up did not identify features strongly supportive of classical hyper-IgE syndrome, such as recurrent deep abscesses, recurrent severe sinopulmonary infections, chronic mucocutaneous candidiasis, pathologic fractures, retained primary teeth, skeletal or joint abnormalities, or characteristic facies.
Because of the combination of early-onset severe dermatitis, persistent eosinophilia, markedly elevated IgE, and early bacterial infections, genetic evaluation was undertaken. Whole-exome sequencing initially identified two FLG variants; among these, Sanger sequencing confirmed a heterozygous FLG NM_002016.2:c.6950_6957del (p.Ser2317Ter) variant, a predicted loss-of-function change resulting in a premature termination codon.
Family testing showed that the father carried the same heterozygous variant, whereas the mother was negative for the tested variant. In the available genetic interpretation, no additional pathogenic or likely pathogenic variants were reported that could establish a defined monogenic inborn error of immunity, including syndromic hyper-IgE phenotypes, Wiskott–Aldrich syndrome, IPEX syndrome, Omenn syndrome, or severe combined immunodeficiency. Therefore, the genetic findings were interpreted in conjunction with the longitudinal clinical course, immunological profile, and family segregation results.
Summary of Key Clinical, Immunological, and Genetic Findings

Discussion
This case illustrates that markedly elevated IgE in childhood should not be interpreted in isolation as evidence of hyper-IgE syndrome. In infancy, our patient presented with generalized severe dermatitis, eosinophilia, pneumonia, acute otitis media, and Staphylococcus aureus bacteremia, creating substantial clinical overlap with HIES. However, severe atopic dermatitis can mimic HIES, especially early in life when eczema, eosinophilia, and very high IgE coexist. In our patient, the later course was less consistent with classical HIES: after 12 months of age, no further severe infections requiring hospitalization were documented, and there were no recurrent deep abscesses, chronic mucocutaneous candidiasis, retained primary teeth, pathologic fractures, skeletal abnormalities, or characteristic facies.4,5
The differential diagnosis in early life was broader than classical HIES alone. Omenn syndrome, Wiskott–Aldrich syndrome, FOXN1 haploinsufficiency, and IPEX syndrome may present with severe eczema or erythroderma, eosinophilia, elevated IgE, infections, diarrhea, growth impairment, autoimmunity, or immune dysregulation. In the present case, the absence of persistent severe infections after infancy, chronic mucocutaneous candidiasis, thrombocytopenia with small platelets, severe enteropathy, endocrinopathy, persistent lymphopenia, syndromic skeletal features, or craniofacial abnormalities made these diagnoses less likely during longitudinal follow-up. Nevertheless, continued monitoring remains important because some inborn errors of immunity may evolve over time.
A barrier-related atopic disorder associated with FLG provides a more coherent explanation for the overall phenotype. Filaggrin is central to epidermal differentiation, skin hydration, and protection against environmental allergens, irritants, and microbes. Loss-of-function FLG variants are among the best-established genetic risk factors for early-onset, persistent, and more severe atopic dermatitis, and are also associated with broader atopic phenotypes, including allergen sensitization and asthma.2,3 This framework closely fits our patient, whose disease began in infancy with severe generalized dermatitis and later evolved into a persistent atopic phenotype marked by chronic severe eczema, eosinophilia, markedly elevated IgE, and asthma. The severe phenotype in this patient is likely multifactorial rather than attributable to the FLG variant alone. The heterozygous FLG loss-of-function variant may contribute to epidermal barrier impairment, increased transepidermal water loss, enhanced allergen penetration, microbial colonization, and amplification of type 2 inflammation. These mechanisms may help explain the combination of early-onset severe dermatitis, persistent eosinophilia, markedly elevated IgE, and later asthma. However, the discordance between the patient and his father, who carried the same heterozygous variant but did not have a similarly severe phenotype, suggests that additional modifiers may be involved. Potential contributors include the broader atopic background, environmental allergen exposure, skin microbiome interactions, recurrent early-life inflammation, and other genetic or epigenetic factors not fully captured by routine variant interpretation.
The laboratory course further supports this interpretation. During the early severe phase, the patient had profound hypoalbuminemia and low total protein, together with marked eosinophilia and extensive skin disease, suggesting systemic consequences of severe barrier disruption and inflammation. Over time, albumin recovered to near-normal levels, whereas total IgE remained markedly elevated. Serial immunoglobulin measurements further showed that IgA, IgG, and IgM remained relatively stable over time, without evidence of a clear evolving humoral immunodeficiency (Figure 3). Taken together, these findings are more consistent with ongoing barrier-related atopic inflammation than with progressive immunodeficiency or persistent severe infection.2,3,6 Longitudinal trends in serum immunoglobulin levels during follow-up. Serial measurements of IgA, IgG, IgM, and total IgE demonstrate persistently elevated total IgE, while IgA, IgG, and IgM remained relatively stable over time
The genetic findings further support this interpretation. Whole-exome sequencing initially identified two FLG variants; among these, Sanger sequencing confirmed a heterozygous FLG NM_002016.2:c.6950_6957del (p.Ser2317Ter) variant, predicted to result in premature truncation. An important feature of this case is the intrafamilial phenotypic discordance: the father carried the same heterozygous variant but did not have a similarly severe phenotype. Rather than excluding the pathogenic relevance of the variant, this finding is compatible with incomplete penetrance and variable expressivity in FLG-related disease, in which some carriers may remain asymptomatic or only mildly affected, whereas others develop early-onset and severe eczema.6,7 This discordance also suggests that the clinical impact of an FLG loss-of-function variant may be modified by additional factors, including the surrounding atopic milieu, type 2 inflammation, environmental exposures, skin microbiome interactions, and possibly other genetic or epigenetic contributors.
Environmental exposures were also considered as potential disease modifiers, although detailed environmental assessment was limited in this case. In patients with FLG-related epidermal barrier dysfunction, exposure to house dust mites, animal dander, molds, irritants, and microbial colonization may promote allergen penetration through the impaired skin barrier and amplify type 2 inflammation. Therefore, the severe and persistent phenotype in this patient may reflect a complex gene–environment interaction involving FLG-associated barrier impairment, recurrent early-life inflammation, environmental exposures, skin microbiome interactions, and sustained type 2 immune activation. The absence of a comprehensive environmental exposure assessment is acknowledged as a limitation of this case report.
From a practical perspective, this case emphasizes that management of severe FLG-associated dermatitis should not be viewed only through the lens of nonspecific immunosuppression. In our patient, asthma responded well to inhaled corticosteroids, whereas the skin response to systemic cyclosporine was limited. This supports a mechanism-based view in which intensive barrier care and appropriate anti-inflammatory treatment remain central. Recent evidence also suggests that targeted therapy such as dupilumab is not necessarily less effective in patients with pathogenic FLG variants, although dryness-related symptoms may remain more prominent in some genetically affected patients. 8 Overall, this case underscores the diagnostic complexity of severe allergic phenotypes in early childhood and illustrates how a barrier-related disorder may clinically overlap with hyper-IgE syndrome during infancy.
This case report has several limitations. First, functional validation of the FLG variant was not performed. Second, vaccine-specific antibody responses could not be assessed because the patient had received only BCG vaccination and had not completed routine childhood immunizations; therefore, assessment of antigen-specific humoral responses to vaccines such as tetanus, diphtheria, pneumococcus, or Haemophilus influenzae type b was not feasible. This limitation should be considered when interpreting the humoral immune evaluation, although serial IgG, IgA, and IgM levels did not show a clear pattern of progressive humoral immunodeficiency. Third, detailed environmental exposure assessment, including standardized evaluation of house dust mite exposure, animal exposure, molds, and other irritants, was limited. Fourth, although the available genetic interpretation did not identify additional pathogenic or likely pathogenic variants sufficient to establish a defined inborn error of immunity, non-coding variants, epigenetic mechanisms, polygenic risk, or gene–environment interactions cannot be completely excluded. Longitudinal follow-up remains necessary to monitor for recurrent infections, immune dysregulation, and evolution toward a more clearly defined immunological phenotype.
Conclusion
This case describes severe early-onset atopic dermatitis associated with a heterozygous FLGloss-of-function variant, presenting with persistent eosinophilia, markedly elevated IgE, and early clinical overlap with hyper-IgE syndrome. Longitudinal evolution, the absence of defining HIES features, and genetic confirmation supported a barrier-related atopic disorder rather than classical HIES.
Footnotes
Acknowledgments
The authors sincerely thank the patient and his family for their cooperation and for consenting to the publication of this case report. The authors also acknowledge the treating clinicians and laboratory staff involved in the patient’s clinical care and diagnostic evaluation.
ORCID iDs
Ethical Considerations
This case report was reviewed and approved by the Institutional Review Board of the Vietnam University of Traditional Medicine, Hanoi, Vietnam, under approval number IRB-VN01.014. The report was conducted in accordance with the principles of the Declaration of Helsinki.
Consent to Participate
Written informed consent to participate was obtained from the patient’s parent/legal guardian.
Consent for Publication
Written informed consent for publication of the patient’s clinical information, laboratory findings, genetic results, and accompanying clinical images was obtained from the patient’s parent/legal guardian.
Author Contributions
TVC and KVN conceived and designed the case report. TVC, ATVN, MTPN and LHP collected and interpreted the clinical data. TMH contributed to manuscript development and critical revision for important intellectual content. KVN supervised the work. All authors reviewed and approved the final manuscript and agree to be accountable for all aspects of the work.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
The data supporting this case report are not publicly available because they contain information that could compromise patient privacy. De-identified data may be available from the corresponding author on reasonable request and subject to institutional approval.
Use of Artificial Intelligence
Artificial intelligence was used only for language and grammar editing. It was not used to generate data, analysis, or conclusions, and no identifiable patient information was entered into any artificial intelligence tool. All authors reviewed and approved the final manuscript and take full responsibility for its content.
Patient Privacy Statement
All images and clinical details included in this report were reviewed to minimize the risk of patient identification.