
Abstract
We postulate that 10-formyl-7,8-dihydrofolate (10-HCO-H2folate), not 10-formyl-5,6,7,8-tetrahydrofolate (10-HCO-H4folate), is the predominant in vivo substrate for mammalian aminoimidazolecarboxamide ribotide (AICAR) transformylase, an enzyme in purine nucleotide biosynthesis de novo, which introduces carbon 2 (C2) into the purine ring. 10-HCO-H2folate exists in vivo as labeled 10-formyl-folic acid (10-HCO-folic acid: an oxidation product of 10-HCO-H4folate and 10-HCO-H2folate) and is found after doses of labeled folic acid in humans or laboratory animals. The bioactivity of the unnatural isomer, [6R]-5-formyltetrahydrofolate, in humans is explained by its in vivo conversion to 10-HCO-H2folate. The structure and active site of AICAR transformylase are not consistent with other enzymes that utilize 10-HCO-H4folate. Because 10-HCO-H4folate is rapidly oxidized in vitro to 10-HCO-H2folate by cytochrome C alone and in mitochondria, it is hypothesized that this process takes place in vivo. In vitro data indicate that 10-HCO-H2folate is kinetically preferred over 10-HCO-H4folate by AICAR transformylase and that this enzyme may not have access to sufficient supplies of 10-HCO-H4folate. Methotrexate blockage of the AICAR transformylase process in patients with rheumatoid arthritis suggests that dihydrofolate (H2folate) reductase is involved and is consistent with H2folate and 10-HCO-H2folate being the product and substrate for AICAR transformylase. The labeling of purine C2 by an oral dose of [6RS]-5-H[13C]O-H4folate in a human subject is consistent with 10-H[13C]O-H2folate formation from unnatural isomer, [6R]-5-H[13C]O-H4folate, and it being a substrate for AICAR transformylase. In vitro exchange reactions of purine C2 using H4folate coenzymes are not duplicated in vivo and is consistent with H2folate coenzymes being used in vivo by AICAR transformylase.
Introduction
The vast majority of the purine ring is biosynthesized de novo rather than coming from the diet. 1 Two folate-dependent transformylases, glycinamide ribotide (GAR) and aminoimidazolecarboxamide ribotide (AICAR) transformylases, are required for purine nucleotide biosynthesis (PNB) de novo in mammals by introducing ring carbons 8 (C8) and 2 (C2), respectively. 2 It is assumed that 10-formyl-5,6,7,8-tetrahydrofolate (10-HCO-H4folate) is the in vivo substrate for both enzymes, because it has been shown that only 5,6,7,8-H4folate is capable of enzymatically acquiring a one-carbon unit. 3,4 Therefore, cellular pools of H4folate are maintained, because 7,8-dihydrofolate (H2folate) reductase (DHFR) catalyzes reduction of H2folate to H4folate. 5 However, in vitro mammalian AICAR transformylase (not GAR transformylase) is able to utilize both 10-HCO-H4folate and 10-formyl-7,8-dihydrofolate (10-HCO-H2folate) with H4folate and H2folate as products, respectively. 6 In this review, we present a hypothesis that 10-HCO-H2folate (not 10-HCO-H4folate) is the actual in vivo substrate for mammalian AICAR transformylase. To support this hypothesis, we present in vivo and in vitro findings, including our previously published data and new analyses of those as well as findings by others.
Presence of 10-formyldihydrofolate in vivo
An important prerequisite for 10-HCO-H2folate being the in vivo substrate for AICAR transformylase in PNB is the proof indicating that this compound exists in vivo. Researchers have identified isotope-labeled or -unlabeled 10-formyl-folic acid (10-HCO-folic acid) in human bile, portal plasma and urine within a few hours after a dose of labeled folic acid or 5-formyl-5,6,7,8-tetrahydrofolate (5-HCO-H4folate). 7–9 These samples were collected in the presence of ascorbate to prevent the oxidation of reduced folates. In order to acquire a one-carbon unit, folic acid must first be reduced to H4folate. Therefore, in these studies, H4folate must have acquired a one-carbon unit to form labeled 10-HCO-H4folate that must have been oxidized to form 10-HCO-folic acid. The obligate intermediate in this two-step-oxidation process is 10-HCO-H2folate that was in fact tentatively identified in the human bile. 7 The first oxidation of 10-HCO-H4folate to 10-HCO-H2folate is more rapid than the second oxidation to 10-HCO-folic acid, 6,10 suggesting that 10-HCO-H2folate was rapidly formed after the dose of folic acid or 5-HCO-H4folate. Labeled 10-HCO-folic acid was also identified in the rat liver, bile and urine after the dose of [14C]- and [3H]-labeled folic acid. 11–13 Although labeled 10-HCO-folic acid is detected after a dose of labeled folic acid, it is in relatively small quantities compared with the pool of labeled tetrahydrofolates. Therefore, the in vivo concentration of its precursor, 10-HCO-H2folate, may also be relatively small. 7,11–13
It has been reported that an oral dose of the unnatural isomers, [6R]-5-HCO-H4folate and [6S]-5,10-methenyltetrahydrofolate (5,10-CH = H4folate), were 25% and 50% as bioactive compared with [6S]-5-HCO-H4folate, respectively, in humans. 14 As described, both [6R]-5-HCO-H4folate and [6S]-5,10-CH = H4folate are converted chemically in the gastrointestinal tract to [6S]-10-HCO-H4folate. 14 Oxidation of the pteridine ring of [6S]-10-HCO-H4folate (unnatural isomer) to the 7,8-dihydro-oxidation state (therefore, destroying the chiral center) converts this unnatural isomer to 10-HCO-H2folate. This folate is now bioactive, because it is a substrate for AICAR transformylase producing H2folate that can be enzymatically reduced to H4folate by DHFR. 6 The fact that [6S]-5,10-CH = H4folate was more bioactive than [6R]-5-HCO-H4folate is consistent with the fact that the former is an intermediate in the conversion of [6R]-5-HCO-H4folate to 10-HCO-H2folate. 14 Chemical formation of [6S]-10-HCO-H4folate and its oxidation to 10-HCO-H2folate must have been rapid because the experiments lasted only 4 h. Baggott and Tamura 14 were not the first to present data suggesting that [6R]-5-HCO-H4folate is bioactive. Although Baker et al. 15 did not report this, re-evaluation of their data using an 8-h follow-up after oral doses of [6RS]-5-HCO-H4folate (believed to be only 50% bioactive) and folic acid indicated that the unnatural isomer, [6R]-5-HCO-H4folate, was ∼80% as bioactive compared with folic acid in humans. 16,17 Devito et al. 18 also reported that an intravenous dose of [6RS]-5-HCO-H4folate produced 19% more bioactive folates than one-half that of the [6S]-5-HCO-H4folate dose in healthy subjects. Therefore, formation of [6S]-10-HCO-H4folate from [6R]-5-HCO-H4folate and its oxidation to 10-HCO-H2folate may take place in sites other than the gastrointestinal tract. In addition, using high-performance liquid chromatography with a microbiological assay, Baggott and Tamura 14 identified 10-HCO-H2folate and 10-HCO-folic acid in 2-h urine samples after doses of either [6R]- or [6S]-5-HCO-H4folate, 14 indicating that metabolites of these natural and unnatural isomers must undergo in vivo oxidation to 10-HCO-H2folate. Based on the above data, Baggott and Tamura 14 conclude that 10-HCO-H2folate exists in vivo.
In vitro data supporting our hypothesis
Unique properties of AICAR transformylase
Enzymes that utilize H2folates include thymidylate synthase, DHFR and AICAR transformylase. Thymidylate synthase catalyzes the production of H2folate from an H4folate, and DHFR reduces H2folate to H4folate. On the other hand, AICAR transformylase is unique because both substrate and product can be H2folates. Further, it is important to note that AICAR transformylase has neither structural similarity to GAR transformylase 19 nor amino acid sequence for the 10-HCO-H4folate binding site that is present in GAR transformylase, 10-HCO-H4folate synthetase and 10-HCO-H4folate dehydrogenase of a variety of species such as Escherichia coli and humans. 20,21 Site-directed mutagenesis of a suspected 10-HCO-H4folate binding site with a minor homology to the site in GAR transformylase has no effect on AICAR transformylase activity. 21 Another aspect of the uniqueness in AICAR transformylase is that it is the only folate-dependent enzyme with a homodimer quaternary structure and the active site at the dimmer interface; thus, the monomer is inactive. 19 The pterine ring of folate analogs binds differently to AICAR and GAR transformylases. The ring is more exposed to the bulk water (possibly more exposed to oxidation) in AICAR transformylase. 19 Considering the above, it would be reasonable that folate coenzyme preference by AICAR transformylase evolved with a preference for 10-HCO-H2folate in a way not similar to other 10-HCO-H4folate-utilizing enzymes.
In vitro lability of 10-HCO-H4folate
10-HCO-H4folate is extremely labile to oxidation in vitro. It has been reported that a trace amount of iron in high-grade reagents catalyzes the oxidation of 10-HCO-H4folate to 10-HCO-H2folate. 22 Therefore, 10-HCO-H2folate cannot be prepared in vitro without being contaminated with oxidation products. 23
Oxidized cytochrome C reacts with 10-HCO-H4folate with a second-order rate constant of 1.3 × 104/mol/L × s. 10 As cytochrome C concentration is 100–200 µmol/L in the intermembrane space, and mitochondrial folate concentration is 1–5 µmol/L, the pseudo-first-order oxidation rate suggests that 10-HCO-H4folate would have a half-life of 1 s or less in the intermembrane space. 10 As the protein porin forms channels in the outer mitochondrial membrane that allow free diffusion of molecules of 10,000 Da or less, 24 10-HCO H4folate and its polyglutamates found in the cytoplasm could come in contact with cytochrome C in the intermembrane space and diffuse out to the cytoplasm as 10-HCO-H2folate to be utilized by AICAR transformylase. The above process would explain how isolated rat liver mitochondria rapidly form 10-HCO-H2folate from 10-HCO-H4folate in the solution outside the organelle. 25 Therefore, 10-HCO-H4folate must be short-lived if it is formed in the intermembrane space. However, it is possible that 10-HCO-H4folate may still be able to play a role as a one-carbon donor to AICAR transformylase before it is oxidized. In conclusion, the oxidation of 10-HCO-H4folate to 10-HCO-H2folate occurs so readily suggesting the in vivo existence of the latter.
In vitro enzyme kinetics of AICAR transformylase
There is in vitro evidence indicating that 10-HCO-H2folate is the preferred substrate for AICAR transformylase. 10-HCO-H2folate has a ∼5-fold kinetic advantage (V m/K m) over 10-HCO-H4folate for AICAR transformylases in human leukemia cell and rat bone marrow, 6 and in human recombinant AICAR transformylase. 26 This kinetic advantage is largely due to a lower K m (tighter binding) for 10-HCO-H2folate, and this is consistent with 10-HCO-H2folate, rather than 10-HCO-H4folate being the preferred in vivo substrate. However, it is not known whether polyglutamates of 10-HCO-H2folate would have the same kinetic advantage over polyglutamates of 10-HCO-H4folate.
It is likely that 10-HCO-H4folate, which is utilized by GAR transformylase, is supplied in vivo by an enzyme complex, originally described in vitro by Benkovic and colleagues. 27,28 This complex contains the trifunctional-folate-metabolizing protein (TFM), serine hydroxymethyltransferase (SHMT) and GAR transformylase. The biological purpose of the complex would be to furnish or channel the labile 10-HCO-H4folate to GAR transformylase immediately after 10-HCO-H4folate is produced.
Consistent with this channeling hypothesis, Benkovic and colleagues 29 recently reported in cultured human cancer cells that the protein with GAR transformylase activity forms clusters in the cytoplasm. In contrast, AICAR transformylase does not form such clusters, and TFM is evenly distributed in the cytoplasm. Therefore, it is likely that any 10-HCO-H4folate produced by TFM that is in close proximity to a GAR transformylase cluster must be preferentially utilized by this enzyme and stands little chance of ‘finding’ AICAR transformylase. This suggests that 10-HCO-H4folate produced by TFM must diffuse a greater distance with an increased risk of oxidation before its utilization by AICAR transformylase. In addition, Baggott et al. 6 found in human leukemia cells that 10-HCO-H4folate K m's were 4.9 and 420 µmol/L for GAR and AICAR transformylases, respectively. Thus, GAR transformylase has a higher affinity for 10-HCO-H4folate than AICAR transformylase.
Taken together, the above data suggest that 10-HCO-H4folate is channeled to GAR transformylase by forming clusters with a binding affinity advantage over AICAR transformylase; therefore, less 10-HCO-H4folate is available to AICAR transformylase. It is likely that folate coenzymes are effectively cycled and channeled to each enzyme in the complex (TFM, SHMT and GAR transformylase) at the H4folate-oxidation state. This series of reactions are shown in Reaction Sequence 1 (Table 1).
Reaction Sequence 1. Utilization of the carbon 3 of serine by GAR transformylase
GAR, glycinamide ribotide; NADP, nicotinamide adenine dinucleotide phosphate; NADPH, reduced form of nicotinamide adenine dinucleotide phosphate; 5,10-CH = H4folate, 5,10-methenyltetrahydrofolate; TFM, trifunctional-folate-metabolizing protein; SHMT, serine hydroxymethyltransferase
No enzyme complex that generates 10-HCO-H4folate and immediately channels or furnishes it to AICAR transformylase is needed because the first oxidation product of 10-HCO-H4folate is 10-HCO-H2folate that is utilized by this transformylase. The oxidation of 10-HCO-H4folate could occur in a reaction with oxidized cytochrome C and is probably not simply an in vitro phenomenon, because the oxidation is fast enough to support respiration in isolated rat liver mitochondria. 25 The second-order rate constant for the oxidation of 10-HCO-H2folate to 10-HCO-folic acid by oxidized cytochrome C is slow. 10 Therefore, the relatively stable 10-HCO-H2folate requires less protection from oxidation than 10-HCO-H4folate and can be utilized by AICAR transformylase as shown in Reaction Sequence 2 (Table 2).
Reaction Sequence 2. Formate and 10-HCO-H2folate utilized by AICAR transformylase
10-HCO-H2folate, 10-formyl-7,8-dihydrofolate; AICAR, aminoimidazolecarboxamide ribotide; TFM, trifunctional-folate-metabolizing protein; IMP, inosine 5′-monophosphate; DHFR, 7,8-dihydrofolate (H2folate) reductase; NADP, nicotinamide adenine dinucleotide phosphate; NADPH, reduced form of nicotinamide adenine dinucleotide phosphate; OX, oxidized; RED, reduced
*This could be a reaction with cytochrome C or other compounds
†It is assumed that AICAR transformylase also possesses the IMP cyclohydrolase activity
In conclusion, the data in this section suggest that 10-HCO-H2folate, an oxidation product of 10-HCO-H4folate, is the preferred substrate for AICAR transformylase and 10-HCO-H4folate is the preferred substrate for GAR transformylase. This conclusion appears to be reasonable because it would be biologically wasteful for 10-HCO-H2folate, an intact folate molecule, to be a metabolically inert dead-end catabolite.
In vivo data in humans and animals supporting our hypothesis
Methotrexate therapy of patients with rheumatoid arthritis
Smoleńska et al. 30 measured the change in whole-blood hypoxanthine concentrations in adult patients with rheumatoid arthritis (RA) shortly before (baseline) and 2 h after an oral dose of methotrexate (MTX) (7.5 mg; patient's first MTX dose). There was a significant 36% decrease in mean hypoxanthine from baseline. The mean blood uric acid concentration was also decreased to a significant 32% from baseline, suggesting an overall decrease in PNB.
The total absorption of oral MTX is ∼80%, of which ∼70% would be absorbed in 2 h. 31 Thus, the above patients would have absorbed 4.2 mg of the 7.5-mg dose (7.5 mg × 0.8 × 0.7). Excluding urinary MTX excretion, in vivo retention would be ∼9 × 10−6 mol (4.2 mg) or in a 70-kg patient ∼1.3 × 10−7 mol/kg in 2 h. Morgan et al. 32 showed that low-dose MTX therapy significantly increases urinary aminoimidazolecarboxamide (AICA) excretion in 24 h, indicating an interference with AICAR transformylase by the drug. Therefore, it is likely that this interference is the target resulting in a decreased inosine 5′-monophosphate (IMP) biosynthesis, thus decreasing blood hypoxanthine. The question remains as to whether the interference with AICAR transformylase is directly caused by MTX. The competitive inhibition constant (K i) for MTX of AICAR transformylase is 4.0 × 10−5 mol/L, 33 and it is ∼300 times higher than the estimated in vivo MTX retention of ∼1.3 × 10−7 mol/kg, which is negligible compared with this K i. It is concluded that MTX cannot directly inhibit AICAR transformylase.
Inhibition of an enzyme other than AICAR transformylase is likely to be responsible for decreased blood hypoxanthine 30 and increased urinary AICA excreted in these patients. 32 One logical choice of an enzyme affected by a low MTX concentration is DHFR. The K i for MTX for human DHFR is 6.1 × 10−12 mol/L, 34 and ∼1.3 × 10−7 mol/kg is ∼20,000 times higher than the K i. Even such a low MTX concentration would produce potent DHFR inhibition, which could simply reduce the H4folate-pool leading to an indirect interference with AICAR transformylase. However, this reduction in the H4folate-pool may not be the actual mechanism, because it would have to occur within 2 h. In fact, Priest and co-workers 35 found that low MTX concentrations do not reduce the H4folate-pool but increase 10-HCO-H4folate in cultured cells. Pools of other H4folate compounds were basically unchanged; therefore, there is no evidence that the intracellular H4folate-pool was depleted by MTX even though cell growth was reduced. Dervieux et al. 36 reported that erythrocyte H4folate-pools in patients with RA were decreased only 13% from baseline after ∼18 weekly doses of MTX (7.5–15 mg/week). Therefore, it is likely that the first MTX dose in patients in the investigation by Smoleńska et al. 30 resulted in a trivial reduction in the H4folate-pool in erythrocytes.
Therefore, an alternative mechanism of MTX interference with AICAR transformylase should be investigated. This may involve the utilization of 10-HCO-H2folate by the enzyme as shown in Reaction Sequence 2. This sequence is driven towards the biosynthetic direction by the DHFR-catalyzed reduction of H2folate to H4folate. The MTX inhibition of DHFR would interfere with AICAR transformylase, as the equilibrium of this step actually lies in the direction of AICAR formation. 26 Reaction Sequence 2 suggests how DHFR, 10-HCO-H2folate and H2folate are involved in the overall net reaction. The production of H2folate by AICAR transformylase would explain how low MTX concentrations interfere with the net reaction. In conclusion, 10-HCO-H2folate as the in vivo substrate for AICAR transformylase explains how low-dose MTX therapy in patients with RA affects this enzyme.
13 C enrichment at C2 and C8 of purine after a [6RS]-5-H[13C]O-H4folate dose
In this section, we attempt to explain how 10-HCO-H2folate is the in vivo substrate for AICAR transformylase by re-analyzing our published data of 13 C enrichment at C2 and C8 of purine after a [6RS]-5-H[13C]O-H4folate dose. The 13 C enrichment at C2 and C8 of urinary uric acid after a 25-mg oral dose of [6RS]-5-H[13C]O-H4folate was measured in an adult. 37,38 Figure 1 shows the relationship between the log of the smoothed C2/C8 enrichment ratios and the void number. This logarithmic transformation of the data was used to readily identify ratios <1.0 (negative numbers). Although considerable variations in the C2/C8 ratios existed, there are basically two phases after the dose. The first phase consisted of a period of enrichment ratios <1, and the second phase was a large increase of the enrichment ratio with a peak value of ∼10.

C2/C8 13C enrichment ratios of urinary uric acid after an oral dose of 25 mg [6RS]-5-H[13C]O-H4folate in a human. The smoothed 1:2:3:2:1 weighted running average of the % 13C enrichments of urinary uric acid at the C2 and C8 positions were calculated after an oral dose of 25 mg [6RS]-5-H[13C]O-H4folate.38 Closed circles represent the log of the 13C enrichment ratio (C2/C8) that was plotted against the void number. The data from the first two and last two voids were lost in this process. Open circles represent data that were negative numbers, thus presumed to be low ratios (i.e. negative log C2/C8 enrichment). The line is a best-fit spine curve. C2, carbon 2; C8, carbon 8
The previously held notion that an equal utilization by both GAR (C8) and AICAR (C2) transformylases, of [6R]-10-H[13C]O-H4folate (formed by rapid enzymatic reactions from [6S]-5-H[13C]O-H4folate), is not consistent with the data shown in Figure 1. If an equal utilization were true, the C2/C8 ratios should have been 1.0 (log = 0) for all voids. To explain these data, we postulate the following hypotheses. The first phase is the result of the rapid enzymatic formation of [6R]-10-H[13C]O-H4folate that is utilized primarily by GAR transformylase. This folate is channeled to and preferentially utilized by GAR transformylase (see the section ‘In vitro lability of 10-HCO-H4folate’); hence, C2/C8 ratios are <1.0. In the second phase, a slow non-enzymatic formation of 10-H[13C]O-H2folate from the unnatural isomer [6R]-5-H[13C]O-H4folate predominates, 14 resulting in high C2/C8 ratios, since 10-H[13C]O-H2folate can only be utilized by AICAR transformylase. 6,14 These data resemble what one would predict, if [6R]-10-H[13C]O-H4folate is rapidly formed, channeled to and utilized only by GAR transformylase, and 10-H[13C]O-H2folate is slowly formed and utilized only by AICAR transformylase. Human data reported by Baggott and Tamura 14 are also consistent with 10-HCO-H2folate being the in vivo substrate for AICAR transformylase.
Carbon exchange at the second position of the purine ring
A C2 to C8 exchange in the purine ring was reported by Warren et al. 39 When an avian liver preparation containing GAR and AICAR transformylases was incubated with IMP, GAR and H4folate, both AICAR and formyl-GAR were readily formed (Table 3, Reaction Sequence 3). The C2 from IMP (in equilibrium with formyl-AICAR) was transferred to H4folate, and the resulting 10-HCO-H4folate was utilized to form formyl-GAR. Reaction Sequence 3 shows these reactions for which the apparent equilibrium lies in favor of formyl-GAR and AICAR formation because the GAR transformylase reaction is irreversible. When this formyl-GAR is metabolized to IMP, the former carbon at the C2 position now appears in the C8 position. Because GAR transformylase cannot utilize 10-HCO-H2folate, this exchange requires that H4folate and 10-HCO-H4folate are the coenzymes for the AICAR transformylase reaction. 6 In theory, this exchange shown in Reaction Sequence 3 can occur in vivo.
Reaction Sequence 3. Exchange of carbon 2 of IMP to carbon 8 of IMP
IMP, inosine 5′-monophosphate; AICAR, aminoimidazolecarboxamide ribotide; 10-HCO-H2folate, 10-formyl-7,8-dihydrofolate; GAR, glycinamide ribotide
Using an avian liver preparation, Flaks et al. 40 reported an exchange reaction of C2 of IMP with the carbon 3 of serine (Reaction Sequence 4, Table 4). This exchange involves TFM, SHMT and H4folate coenzymes (not H2folate coenzymes). In theory, this exchange can occur in vivo, and the serine formed by this reaction would be readily oxidized to CO2.
Reaction Sequence 4. Exchange of carbon 2 of IMP to carbon 3 of serine
IMP, inosine 5′-monophosphate; AICAR, aminoimidazolecarboxamide ribotide; 10-HCO-H2folate, 10-formyl-7,8-dihydrofolate; TFM, trifunctional-folate-metabolizing protein; 5,10-CH = H4folate, 5,10-methenyltetrahydrofolate; NADP, nicotinamide adenine dinucleotide phosphate; NADPH, reduced form of nicotinamide adenine dinucleotide phosphate; SHMT, serine hydroxymethyltransferase
It should be emphasized that exchange reactions (Sequences 3 and 4) require that H4folate coenzymes function in the AICAR transformylase process. However, it remained to be seen whether these exchange reactions actually occur in vivo. Using adenine labeled with 13 C at the C2 position and 14C at the C8 position, Abrams 41 evaluated the possibility of these exchanges in rats. Adenine is readily metabolized to IMP. 2 The C2/C8 labeling ratio was not changed compared with the starting material in liver RNA adenine and guanine days after the dose of the double-labeled adenine. Abrams 41 concluded that C2 exchanges described above do not occur in vivo. However, the fact that no exchange reactions are detected indicates that H2folate coenzymes are utilized by AICAR transformylase.
Bennett and Karlsson 42 studied the exchange of 2-[14C]adenine and 8-[14C]adenine in mice. Measuring hepatic RNA adenine- and guanine-specific activities, they found no difference comparing C2 to C8 in experiments that lasted for 2 weeks, and concluded that C2 was not involved in exchange reactions described above. Of particular interest is the measurement of expired 14CO2 from the C2- and C8-labeled compounds in mice. After 20 h, 14CO2 production from both compounds was small, and there was no marked difference in 14CO2 production from 2-[14C]adenine compared with 8-[14C]adenine. These findings are also evidence indicating that the exchange reactions discussed above do not occur in vivo in mice.
In humans, however, data are limited. Baggott et al. 43 conducted a human study to measure incorporation of [13C]formate into C2 and C8 of urinary uric acid. In three-day experiments, two subjects had little incorporation into C8, but C2 incorporation was significant. Therefore, the C2 to C8 exchange (Reaction Sequence 3) was small. However, a subject incorporated significant amounts into the C8 position as well as into C2. Therefore, the data in this subject could be used to test the possible C2 to C8 exchange. If C8 incorporation (all or part) was the result of the exchange from C2, more enrichment at C8 should occur in a time-dependent manner as C2 becomes more enriched. Thus, both C8 enrichment and the C8/C2 enrichment ratio should have increased with time. Figure 2 shows both of these values plotted over a three-day period after the [13C]formate dose, and linear regression of the data in both plots indicates negative slopes. Thus, there was no evidence of C2 to C8 exchange in this subject. The findings in humans by Baggott et al. 43 are, therefore, in agreement with the animal studies. 41,42 It is possible that in vitro exchange reactions are not operable in vivo, because enzymes are not in close proximity and do not exchange folate coenzymes or other factors. However, if H2folate coenzymes are the in vivo substrate for AICAR transformylase, these exchanges should not occur in vivo.
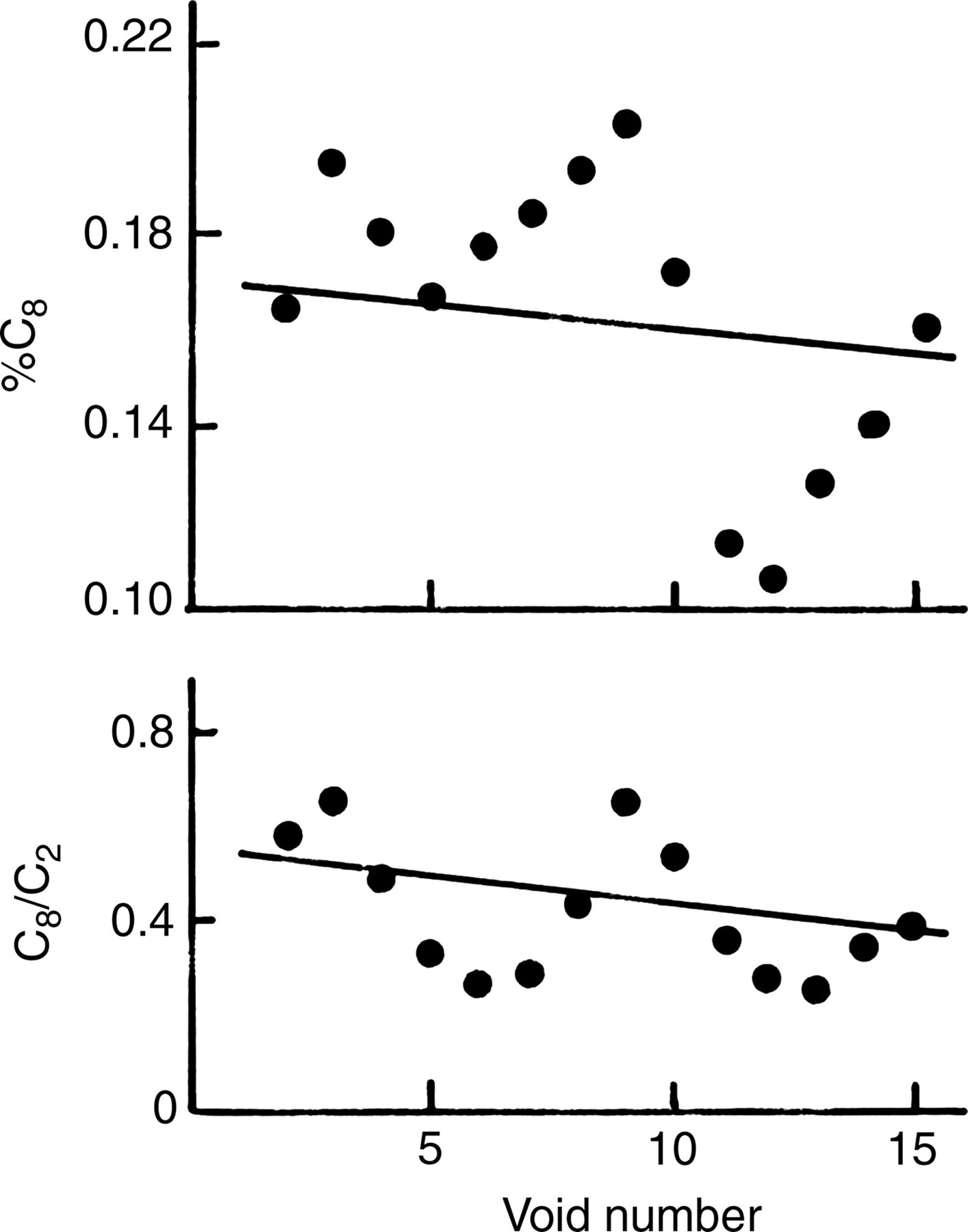
C8/C2 13C enrichment ratios and % 13C enrichment at the C8 position of urinary uric acid after an oral dose of 1.0 g of sodium [13C]formate in a human. The smoothed 1:2:1 weighted running averages of % enrichment at the C2 and C8 positions (%C8) and the enrichment ratio (C8/C2) of urinary uric acid after an oral dose of 1.0 g of sodium [13C]formate were plotted against the void number for subject C.38 The data from the first and last void were lost in this process. Linear regression lines are negative, but are not significant (P > 0.05)
Summary
We conclude that 10-HCO-H2folate is the in vivo predominant substrate for mammalian AICAR transformylase, and summarize as follows. The in vivo existence of 10-HCO-H2folate is likely due to the lability to oxidation of 10-HCO-H4folate, suggesting that 10-HCO-H2folate must have a metabolic role rather than being a dead-end catabolite. AICAR transformylase does not have a 10-HCO-H4folate binding site that is present in other 10-HCO-H4folate-utilizing enzymes and has a unique active site formed by a homodimer. Enzyme-kinetic data suggest the utilization of 10-HCO-H2folate by AICAR transformylase and utilization of only 10-HCO-H4folate by GAR transformylase. The labile 10-HCO-H4folate is likely channeled to GAR transformylase in vivo and the relatively stable 10-HCO-H2folate is utilized by AICAR transformylase. These findings are consistent with labeling patterns in the purine ring after a dose of [6RS]-5-H[13C]O-H4folate in a human. The unique sensitivity of AICAR transformylase to the low-dose MTX therapy in patients suggests that formation of H2folate from 10-HCO-H2folate and inhibition of DHFR are involved in the transformylation process. The facile in vitro exchanges of the C2 position of purine in the presence of H4folates and enzymes are not duplicated in vivo, suggesting that H2folates (which cannot produce facile in vitro exchanges), not H4folates, are involved in the in vivo AICAR transformylase (C2) process.
Finally, it may be difficult, if not impossible, to prove that 10-HCO-H2folate is the in vivo substrate for mammalian AICAR transformylase; however, we can turn the table on this argument. Simply stated, at present, we believe that it is difficult (if not impossible) to design experiments proving that 10-HCO-H4folate is the one and only substrate for AICAR transformylase in vivo and we are not aware of any experiments that attempted to prove this. It would be biologically wasteful for 10-HCO-H2folate to be a metabolically inert dead-end catabolite. It is possible that mammalian AICAR transformylase evolved with metabolic flexibility to utilize both 10-HCO-H2folate and 10-HCO-H4folate in order to metabolically ‘salvage’ former utilizing the PNB pathway. Changing the relatively in vivo amounts of 10-HCO-H4folate and 10-HCO-H2folate could be a regulatory mechanism in the PNB pathway.