
Abstract
Endothelial progenitor cells (EPCs), which express the CD133 marker, can differentiate into mature endothelial cells (ECs) and create new blood vessels. Normal angiogenesis is unable to repair the injured tissues that result from myocardial infarction (MI). Patients who have high cardiovascular risks have fewer EPCs and their EPCs exhibit greater in vitro senescence. Human umbilical cord blood (HUCB)-derived EPCs could be an alternative to rescue impaired stem cell function in the sick and elderly. The aim of this study was to purify HUCB-derived CD133+ cells, expand them in vitro and evaluate the efficacy of the purified and expanded cells in treating MI in rats. CD133+ cells were selected for using CD133-coupled magnetic microbeads. Purified cells stained positive for EPC markers. The cells were expanded and differentiated in media supplemented with fetal calf serum and basic fibroblast growth factor, insulin-like growth factor-I and vascular endothelial growth factor (VEGF). Differentiation was confirmed by lack of staining for EPC markers. These expanded cells exhibited increased expression of mature EC markers and formed tubule-like structures in vitro. Only the expanded cells expressed VEGF mRNA. Cells were expanded up to 70-fold during 60 days of culture, and they retained their functional activity. Finally, we evaluated the therapeutic potential of purified and expanded CD133+ cells in treating MI by intramyocardially injecting them into a rat model of MI. Rats were divided into three groups: A (purified CD133+ cells-injected); B (expanded CD133+ cells-injected) and C (saline buffer-injected). We observed a significant improvement in left ventricular ejection fraction for groups A and B. In summary, CD133+ cells can be purified from HUCB, expanded in vitro without loosing their biological activity, and both purified and expanded cells show promising results for use in cellular cardiomyoplasty. However, further pre-clinical testing should be performed to determine whether expanded CD133+ cells have any clinical advantages over purified CD133+ cells.
Introduction
Reperfusion therapy is associated with significantly reduced mortality rates and improved clinical outcomes in patients with acute myocardial infarction (MI). 1 However, normal angiogenesis is usually unable to supply the greater demand for oxygen and nutrients required following MI. In addition, it is unable to prevent hypertrophied cardiomyocyte apoptosis and ventricular remodeling. 2 Cell therapy may provide a novel therapeutic strategy to modify left ventricular remodeling processes and prevent postinfarction heart failure 3 Different cell populations have been tested in this regard in both preclinical and clinical settings. Recently, isolated CD34+ cells demonstrated increased potency and safety for therapeutic neovascularization after MI, as compared with total mononuclear cells (MNCs). 4
Endothelial progenitor cells (EPCs) are precursor cells that can differentiate into mature endothelial cells (ECs) and create new blood vessels. 5 Generally, EPCs can be identified based on their expression of CD133, CD34, KDR and/or VE-cadherin cellular markers. 6 The similar phenotype of hematopoietic and EPCs strongly supports the existence of a common precursor cell, which has been called the hemangioblast. This cell has been identified in in vitro studies using embryonic stem cells from postnatal bone marrow and cord blood. 7
EPCs represent less than 1% of all bone marrow cells and less than 0.01% of peripheral blood MNCs. 8 Human umbilical cord blood (HUCB) is a viable source of stem cells due to its large CD34 and CD133-expressing cell populations. These cells exhibit robust proliferative capacity, low immunogenicity and low infection contamination (including virions). Furthermore, the diverse representation of human leukocyte antigen (HLA) genotypes in unrelated, banked HUCB allows the use of HUCB-derived cells in allogeneic transplantations. 9 Promising results were obtained using purified HUCB stem cells as cellular therapies in animal models, and these results support their potential application in cardiovascular medicine. 10,11 Independently or in combination with biomaterials, such as collagen matrix, HUCB-derived stem cells seem to improve the efficiency of cellular cardiomyoplasty in mice. Thus, this strategy has emerged as a novel therapy. 12
Results from preclinical studies have suggested that it would take more than 10 L of autologous peripheral blood to produce a sufficient number of EPCs to induce angiogenesis in one patient. 13 Furthermore, patients with increased cardiovascular risk have fewer EPCs, as compared with healthy subjects, and their EPCs demonstrate increased in vitro senescence. 14 The use of CD133+ cells derived from purified and expanded HUCB could be used to rescue stem cell function in the sick and elderly. EPCs can be expanded in vitro to increase their potential therapeutic use. However, all current protocols involving short-term culture would not yield sufficient numbers of cells for systemic therapy in humans. On the other hand, long-term culture may introduce changes in the EPC's phenotype, which could reduce their therapeutic efficacy. 15
The aim of this study was to isolate and purify CD133+ cells from HUCB and to expand the cells in vitro by exposing them to an appropriate growth factor cocktail. We evaluated the effectiveness of the purified and expanded CD133+ cells in improving the cardiac function in a rat model of MI.
Materials and methods
This study was reviewed and approved by the Local Ethics Committee (Pontifícia Universidade Católica do Paraná: numbers 1366 and 180). Signed informed consent was obtained from each mother prior to HUCB collection. All animal experiments were carried out in accordance with: ‘The Guide for the Care and Use of Laboratory Animals’ published by the National Institute of Health (NIH publication 85-23, 7th edn, revised 1996).
Purification of CD133+ cells
MNCs were isolated from HUCB. Thirty-seven HUCB samples (approximately 78.53 ± 4.63 mL each) were collected from fresh placentas with the umbilical cord still attached, and acid-citrate-dextrose was used as anticoagulant. MNCs were isolated by centrifugation for 30 min at 400
Proliferation assay on CD133+ cells
A cell proliferation assay was performed to establish the optimal culture conditions for CD133+ cells. A cell suspension of 50 μL (1 × 105 cells/mL) in Iscove's Modified Dulbecco's Media (IMDM) (Invitrogen), supplemented with 10% fetal bovine serum (FBS) (Invitrogen) and 1% penicillin–streptomycin (Invitrogen), was seeded into 96-well plates (Corning, Oneonta, NY, USA). Various concentrations of growth factors were added: 0.5, 1.0 and 2.0 ng/mL of basic fibroblast growth factor (b-FGF) (Invitrogen); 1.0, 2.0 and 3.0 ng/mL of insulin-like growth factor-I (IGF-I) (Sigma-Aldrich); and 25, 50 and 75 ng/mL of vascular endothelial growth factor (VEGF) (Sigma-Aldrich). Media with a constant amount of 1 ng/mL b-FGF and 2 ng/mL IGF-I with increasing VEGF concentrations (25, 50 and 75 ng/mL) was used. The cells were incubated at 37°C with 5% CO2 in a humidified atmosphere. After five days, 1 μCi of [methyl-3H] thymidine (5.0 Ci/mmol) (GE Healthcare, Buckinghamshire, UK) was added to each well. Six hours later, the culture media and cells were removed, and the cell-associated radioactivity was measured using a scintillation counter (Beckman Coulter®, Fullerton, CA, USA). Responses to each concentration were assayed in triplicate, and results were expressed as the mean of 12 independent experiments.
Culture and expansion of CD133+ cells
After establishing the optimal culture conditions, 2 × 105 CD133+ cells in 50 μL were plated into 12-well culture plates coated with human fibronectin (Becton Dickinson) and cultured in IMDM supplemented with 50 ng/mL VEGF, 1 ng/mL b-FGF, 2 ng/mL IGF-I, 10% FBS and 1% penicillin–streptomycin. All cultures were maintained at 37°C with 5% CO2 in a humidified atmosphere. Additional feeding was performed depending on the rate of cell proliferation; the supernatant was removed by gentle pipetting and fresh medium was added. Cells were cultured to approximately 80% confluence, detached using Accutase (US Bio-Technologies, Pottstown, PA, USA) digestion and replated in T-25 tissue culture flasks for 60 d. On day 60, the cells were assessed for viability, rate of expansion and phenotype.
Phenotypic characterization of cells
Immunophenotypic analysis was performed by staining 2 × 105 of MNCs, purified and expanded CD133+ cells. MNCs and purified cells were analyzed after isolation, whereas expanded cells were analyzed after 60 days of culture. The cells were incubated with various conjugated and unconjugated monoclonal antibodies against the following human antigens: CD133 (PE conjugated; Miltenyi Biotec), CD34 (APC conjugated; BD Pharmingen), CD45 (PerCP conjugated; BD Pharmingen), CD14 (fluorescein isothiocynate [FITC] conjugated; BD Pharmingen), CD31 (BD Pharmingen), CD105 (BD Pharmingen) and von Willebrand Factor (vWF; Dako, Glostrup, Denmark). All incubations were performed at 4°C for 30 min. For CD31 and CD105 detection, cells were further incubated with an isotype-specific FITC-conjugated goat anti-mouse antibody (Caltag, Burlingame, CA, USA). For intracellular detection of vWF, cells were permeabilized using FIX & PERM cell permeabilization reagents (Caltag, Carlsbad, CA, USA), and further incubated with an isotype-specific FITC-conjugated goat anti-rabbit antibody (Sigma-Aldrich). Isotype-identical antibodies (BD Pharmingen) were served as controls. After incubation, the cells were washed with PBS containing 2% FBS and fixed with PBS containing 1% paraformaldehyde. Quantitative analyses were performed using a FACSCalibur flow cytometer and FlowJo software (Flowjo, Ashland, OR, USA).
Immunofluorescence microscopy
Expanded CD133+ cells were cultured on chamber slides (Erie Scientific Company, Portsmouth, NH, USA) to confluence. Cells were washed twice with PBS and fixed for 20 min with cold methanol. Slides were incubated for 30 min with a polyclonal rabbit anti-human vWF antibody (1:200) (Caltag), followed by an isotype-specific FITC-conjugated goat anti-rabbit antibody (1:80) (Sigma-Aldrich). Cells were washed with PBS and then stained with 4′,6-diamidino-2-phenylindole dihydrochloride (1:5) (Sigma-Aldrich). The negative control was obtained by omitting the primary antibody incubation. Slides were examined and imaged using a fluorescent microscope (Olympus, Tokyo, Japan).
Reverse transcription-polymerase chain reaction
To evaluate VEGF expression, total RNA was extracted from purified and expanded CD133+ cells using the RNeasy kit (QIAGEN, Valencia, CA, USA). RNA was treated with DNAse I (QIAGEN) in a column, according to the manufacturer's instructions. The total RNA (1 µg) was converted to cDNA by adding 10 nmol/L of oligo-dT primer (USB Corporation, Cleveland, OH, USA) and 1 µL of reverse transcriptase (RT) (IMPROM II, Promega, Fitchburg, WI, USA), according to the manufacturer's instructions. Polymerase chain reaction (PCR) reactions contained 20 ng of cDNA as a template with 20 mmol/L Tris-HCl (pH 8.4), 50 mmol/L KCl, 10 pmol of forward and reverse primers for VEGF and 5 pmol for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) primers, 2.5 mmol/L MgCl2, 62.5 µmol/L dNTPs (deoxyribonucleotide triphosphates) and 1 U Taq polymerase (Invitrogen). The oligonucleotide primers used for PCR (Midland Certified Reagent Company, Midland, TX, USA) were as follows: VEGF (Accession Number: M27281) forward (5′ CTACCTCCACCATGCCAAGTG 3′) and reverse (5′ TGCGCTGATAGACATCCATGA 3′), which yielded a 101-base pair (bp) product; and GAPDH (Accession Number 2597) forward (5′ GGCGATGCTGGCGCTGAGTAC 3′) and reverse (5′ TGGTTCACACCCATGACGA 3′), which yielded a 150-bp product. PCR conditions were: 94°C for two minutes, followed by 30 cycles of 94°C for 15 s, 62°C for VEGF and 55ºC for GAPDH for 30 s, and 72ºC for 40 s followed by a final extension step of 72°C for three minutes. All reactions were performed in a Bio-Cycler II thermocycler (Bio-Rad, Hercules, CA, USA). Amplified PCR products were separated by electrophoresis on 2% agarose gels and visualized by ethidium bromide staining. Bands were imaged using ultraviolet illumination (UV White Darkroom, UVP Bioimaging Systems, Upland, CA, USA). Relative gene expression was normalized to GAPDH.
Capillary-like tubule formation assay
The Matrigel® basement membrane matrix (Becton Dickinson) was used to assess EC tube formation. Matrigel was thawed overnight at 4°C and added (250 μL) to each well of a 24-well plate. The Matrigel matrix was allowed to polymerize at room temperature for 20 min. A suspension of 2 × 104 CD133+ cells/well in 250 μL of IMDM containing 10% FBS and 1% penicillin–streptomycin, and a suspension of expanded CD133+ cells/well in 250 μL of IMDM containing 50 ng/mL VEGF, 1 ng/mL b-FGF, 2 ng/mL IGF-I, 10% FBS and 1% penicillin–streptomycin were added to the wells. All assays were performed in triplicate. Cells were incubated for 24 h at 37°C in a humidified 5% CO2 incubator and observed at 2, 6, 12 and 24 h using an IX70 Olympus microscope.
Rat model of MI
Seventy male Wistar rats (200–400 g) were subjected to MI, as previously described. 16 Briefly, rats were anesthetized by intramuscularly injecting ketamine (50 mg/kg) and xylazine (10 mg/kg). The animals were intubated with a 14-gauge, 2.54-cm angiocatheter under direct vision and ventilated with a Harvard rodent ventilator model 683 (Harvard Apparatus, Holliston, MA, USA) with minute ventilations of 150 mL/min. The heart was exposed via a left anterolateral thoracotomy incision at the fourth intercostal space. The ribs were retracted to open the chest cavity. After removing the pericardium, the left anterior descending (LAD) branch of the left coronary artery was identified and then permanently ligated by passing a 7-0 polypropylene suture (Premio® Peters Surgical, Bobgny, France) under the LAD at the level of the distal margin of the retracted left atrial appendage. The chest wall, muscle layers and skin were closed with interrupted 3-0 silk sutures.
Cell transplantation
Rats with ejection fractions (EF) lower than 40% were randomly assigned to three groups: purified CD133+ cells group A; expanded CD133+ cells group B and control group C. Seven days after the MI, the animals were anesthetized and ventilated as described above. Groups A and B were treated with 2 × 105 cells in 0.3 mL of PBS injected using a tuberculin syringe with a 13 × 0.38 needle into one point of the myocardium (left ventricle [LV]), specifically in the center of the scar covering the whole infarcted and peri-infarcted area. An equal volume of PBS was injected into the heart of control animals.
Echocardiography analysis
Transthoracic echocardiography was performed in all animals five days after MI (baseline echocardiogram) and 30 days after the transplant. The evaluated variables were LV ejection fraction (LVEF), LV end-systolic volume and LV end-diastolic volume. Echocardiograms were performed using a commercially available echocardiography system equipped with a 12-MHz phased-array transducer (Hewlett-Packard Sonos 5500; Andover, MA, USA;
Statistical analysis
Continuous variables were presented as mean ± standard deviation, and categorical variables were presented as frequency and percentage. After statistical evaluation, the outliers, which were samples with values greater than 1.5 times the interquartile interval, were removed from the final analysis. Cell proliferation and immunophenotyping data were both evaluated using Friedman's non-parametric test. The normality distribution of the data was evaluated using the Shapiro–Wilks test, and the homegeneity condition was assessed by the Lévene test. Multiple comparisons were analyzed using the LSD test, and comparisons between two evaluations were assessed by the Student's t-test for paired samples. Values of P < 0.05 were considered to be statistically significant. Analysis was performed using the SPSS V.14 software (SPSS, Chicago, IL, USA).
Results
Purification and phenotypical characterization of HUCB-derived CD133+ cells
CD133+ cells were positively selected using anti-CD133-coupled magnetic microbeads. The mean number of cells obtained after magnetic CD133+ cell sorting was 0.55 × 106 ± 0.04, corresponding to 0.64% of the MNC population. As determined by flow cytometry, the mean purity of selected CD133+ cells was 74.8 ± 15.82%, and the viability was 80.87 ± 10.55%. As shown by fluorescence-activated cell sorting (FACS) analyses, the major contaminants in the CD133-enriched population were cells of the hematopoietic (CD45+) and monocytic (CD14+) lineages.
Flow cytometry analysis was performed to characterize the cell populations contributing to the MNCs, as well as the purified and expanded CD133+ cells. The staining patterns of cells for specific markers are provided in Figure 1a. The cell population profiles showed that most of the MNCs stained positive for CD45 (88.69 ± 10.19%). Some cells stained positive for CD14 (19.49 ± 10.4%), CD31 (26.09 ± 6.22%), CD105 (33.97 ± 5.88%) and vWF (34.25 ± 24.12%). A small number of MNCs stained positive for CD133 (3.37 ± 2.3%) and CD34 (3.25 ± 1.97%). After the CD133 purification, a large number of cells stained positive for CD133 (74.8% ± 15.82) and CD34 (62.15 ± 16.09%). A greater number of purified CD133+ cells co-stained for CD31 (86.27 ± 18.37%) and CD105 (69.16 ± 31.95%). A similar number of cells stained positive for vWF (35.24 ± 9.78%), and a considerably reduced number were positive for CD45 (9.03 ± 5.07%) and CD14 (3.18 ± 4.1%).
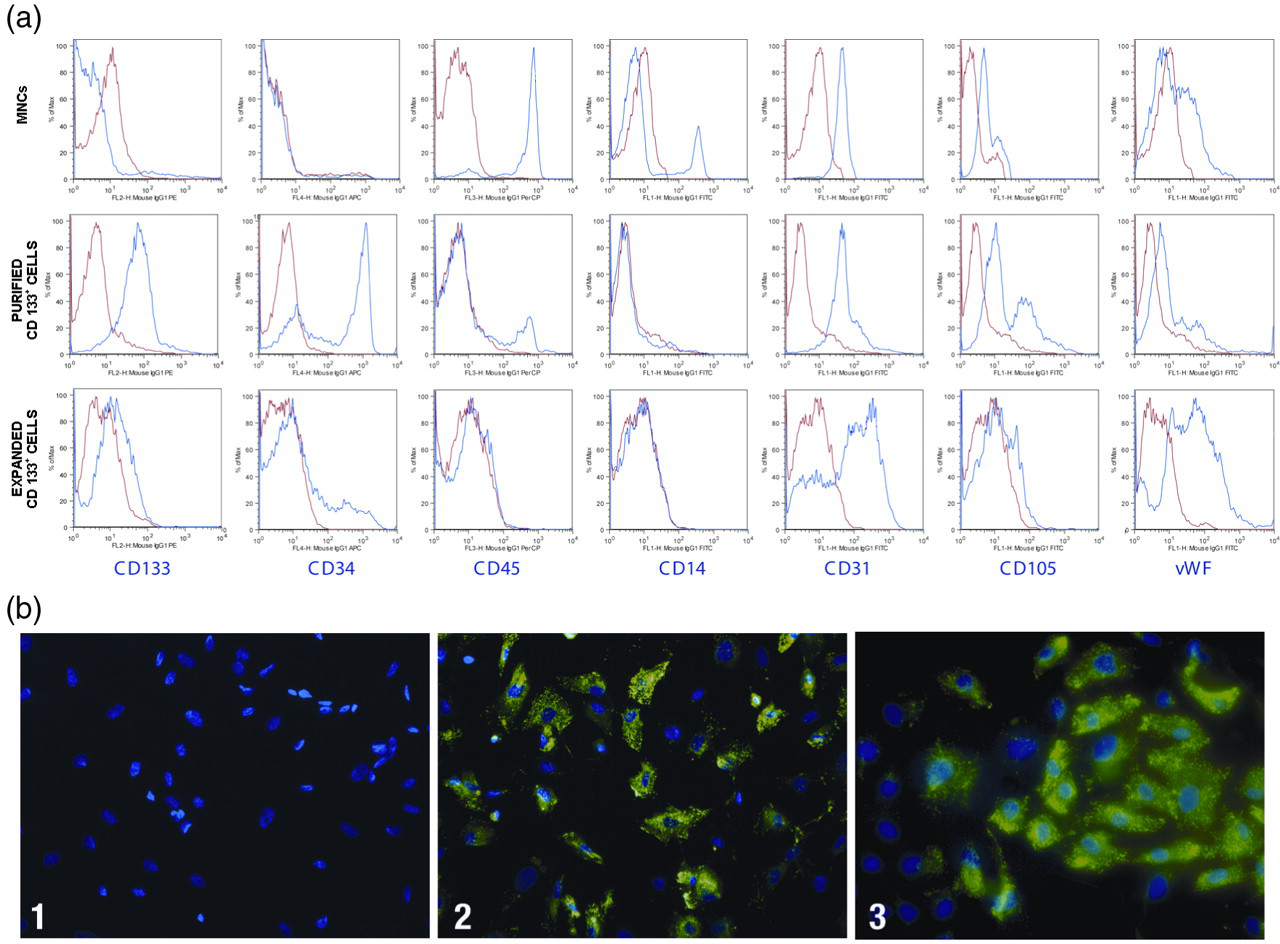
Phenotypic characterization by flow cytometry and fluorescence microscopy. (a) Human umbilical cord blood (HUCB)-derived mononuclear cells, purified and expanded CD133+ cells were labeled with fluorescent human antibodies against CD133, CD34, CD45, CD14, CD31, CD105 and von Willebrand factor (vWF). Blue histograms indicate the percentage of positive cell populations for each antibody, whereas red histograms indicate isotype-control antibodies. Histograms represent one experiment. The X-axis represents the fluorescence intensity. (b) Fluorescent images (×400) labeled with anti-human vWF isotype-specific fluorescein isothiocynate (FITC)-conjugated goat anti-rabbit antibody (green) and 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) (blue). 1: Negative control, expanded CD133+ cells (labeled with FITC-conjugated secondary antibody and DAPI). 2: Positive control; coronary artery-derived endothelial cells. 3: HUCB-derived expanded CD133+ cells after 60 days of culture
Proliferation assays on growth factor-treated CD133+ cells
We evaluated the effects of using different concentrations of three growth factors (b-FGF, IGF-I and VEGF) on CD133+ cell proliferation. Proliferation was assessed after five days of treatment, which included thymidine incorporation for six hours. Three concentrations of each growth factor were independently used, in addition to media with a constant concentration of b-FGF and IGF-I and varying VEGF concentrations. The proliferation rate was calculated by dividing the mean cell count at each growth factor concentration by the mean cell count of the control. The only statistically relevant effect for b-FGF was observed when 1.0 ng/mL was added to the cultures (P = 0.04). There were no significant differences in proliferation observed when various concentrations of IGF-I or VEGF were independently used. Constant concentrations of b-FGF (1.0 ng/mL) and IGF-I (2.0 ng/mL) were added along with varying concentrations of VEGF. Incubating the cells with 50 ng/mL VEGF increased their proliferation (P = 0.004).
CD133+ cells actively proliferate and differentiate into endothelial-like cells
CD133+ cells were grown on fibronectin-coated chamber slides in the presence of VEGF, b-FGF and IGF-I to induce the differentiation of putative purified EPCs into endothelial-like cells. Adherent cells formed a monolayer, predominantly consisting of small-sized cells, during the first 10 days of culture. No significant proliferation was noted during this culture period. Cells began to form clusters in some areas of the chamber on day 14 of culture. After this, several cells (increased on a daily basis) became spindle-shaped and tended to form cell lines. We also observed an increased population density, suggesting that the culture was actively proliferating. Adherent cells were still actively growing between days 30 and 60; therefore, cultures needed to be expanded on average every five days. At this point, morphological analysis of the cells revealed large flat cells with a cobblestone appearance, which is characteristic of ECs (Figure 2). Growth curves exhibited up to a 17-fold increase in the cell number by day 30 (passage 1) and up to a 70-fold increase after 60 days of culture (passage 4) and a viability of 85.32 ± 8.36% (Figure 3).

CD133+ cell culture in the presence of growth factors (basic fibroblast growth factor, insulin-like growth factor-I and vascular endothelial growth factor). (1) Small-sized round cells at day 5 (×100). (2) Small-sized round and flat elongated cells at day 14 (×200). (3) Cluster formation at day 15 (×200). (4) Cell lines at day 21 (×100). (5) Proliferation enhanced at day 21 (×200). (6) Confluent cells with cobblestone appearance at day 27 (×200)
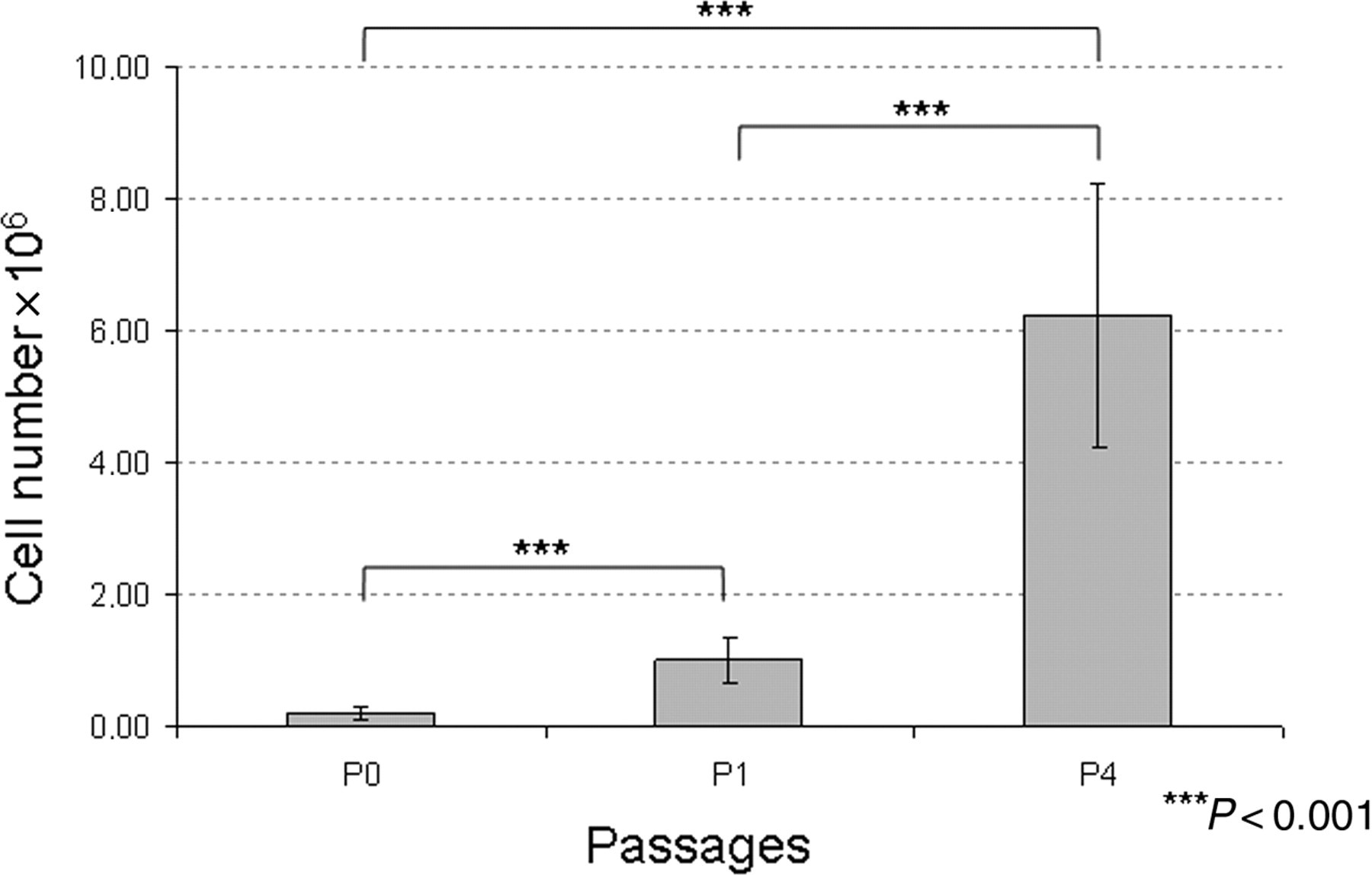
Proliferation rates. Eleven samples were used to evaluate the increase in cell number at three time points. P0, day 0: purified CD133+ cells were grown on fibronectin-coated chamber slides (mean = 0.19 × 106). P1, day 30: first passage; cells reached 80% confluence, and the morphological analysis showed spindle-shaped cells without a differentiated phenotype. The cell number increased up to 17-fold (mean = 1 × 106). P4, day 60: fourth passage; the morphological analysis revealed large flat cells with a cobblestone appearance, which is characteristic of expanded endothelial cells, and the cell number increased up to 70-fold (mean = 6.23 × 106)
We analyzed the expanded CD133+ cells after 60 days of culture. Very few cells stained positive for CD133 (2.21 ± 1.65%), CD45 (3.36 ± 2.1%) and CD14 (2.03 ± 1.36%). The number of cells expressing CD31 (63.58 ± 16.3%) decreased over time but still represented a large population. The CD105 (14.14 ± 14.14%) and CD34 (11.52 ± 7.09%) positive populations decreased, and most of these cells were also positive for vWF (79.59 ± 8.33%). The mean viability for MNCs, purified CD133+ cells and expanded CD133+ cells was greater than 92%, 80% and 87%, respectively.
Immunofluorescence microscopy analysis using anti-vWF antibody was performed to characterize the expanded CD133+ cells in culture and to confirm our flow cytometry data. At day 60, most cells exhibited strong staining and clear cytoplasmic localization of vWF, which is a characteristic of ECs (Figure 1b).
Expanded CD133+ cells express VEGF mRNA and form capillary-like tubes
VEGF mRNA was not detected in purified CD133+ cells by RT-PCR; however, high VEGF mRNA levels were observed in all five biological replicates of cells induced to differentiate. Thus far, the data suggest that CD133+ cells are indeed EPCs, and expanded CD133+ cells are endothelial-like cells. Based on these conclusions, we characterized the function of expanded CD133+ cells by studying their ability to form tubule-like structures on a Matrigel layer. Only expanded CD133+ cells, not purified CD133+ cells, formed a branched network of tubule-like structures after 24 h of culture on Matrigel (Figure 4). Thus, the in vitro data indicated that expanded CD133+ cells clearly exhibit EC-like characteristics.

Capillary-like tubule formation assay of human umbilical cord blood-derived CD133+ cells cultured for 24 h on a Matrigel® layer. (1) Purified cells after 24 h of culture (×40). Expanded CD133+ cells formed tubules in response to basic fibroblast growth factor, insulin-like growth factor-I and vascular endothelial growth factor after (2) 2 h (×40); (3) 6 h (×200); (4) 12 h (×200); (5) 24 h (×40) and (6) 24 h (×100)
Purified and in vitro expanded CD133+ cells improved LVEF in a rat model of MI
To confirm the in vivo functional activity of purified and expanded HUCB-derived CD133+ cells and their potential use as a cell therapy, the cells were implanted in a rat model of MI. Seventy rats were infarcted and 11 animals died after this procedure. Twenty-seven rats showed left ventricular dysfunction (EF below 40%) in the first echocardiographic evaluation. Four rats died during or immediately following cell transplantation. The remaining 23 rats were divided into three groups: group A, purified CD133+ cells-injected rats (7 rats); group B, expanded CD133+ cells-injected rats (6 rats), and group C, saline buffer-injected rats (10 rats). Three rats in group C died after surgery (12, 16 and 24 days). There were no deaths in groups A and B. After statistical evaluation, the outliers in each group were discarded. There was one outlier in group A, one in group B and two in group C.
Echocardiography measurements were applied to evaluate whether purified and/or in vitro expanded HUCB-derived CD133+ cells could restore cardiac function in a rat model of MI 30 days after cell injection. Five days after MI, the LVEF measurements for the groups A, B and C were 22.51 ± 7.48%, 20.75 ± 5.42% and 25.99 ± 5.03%, respectively. In comparison to the second echocardiography (30 days after cell injection), the LVEF had increased by 10.16% for group A and by 6.8% for group B. The control group (group C) exhibited a 7.09% reduction in the LVEF. However, these data were not statistically significant. At this point, the LVEF values for groups A, B and C were 32.67 ± 14.37%, 27.55 ± 5.42% and 20.46 ± 4.05%, respectively. When we compared the difference between the first and second echocardiographic evaluations, we observed an improvement in the LVEF for the cell injection groups (A and B). The LVEF for group A (purified CD133+ cell injection) was improved, as compared with the control group (P = 0.001). Group B (expanded CD133+ cell injection) also exhibited a significant improvement in the LVEF, as compared with the control group (P = 0,011). There was no statistical difference observed between groups A and B (Figure 5).
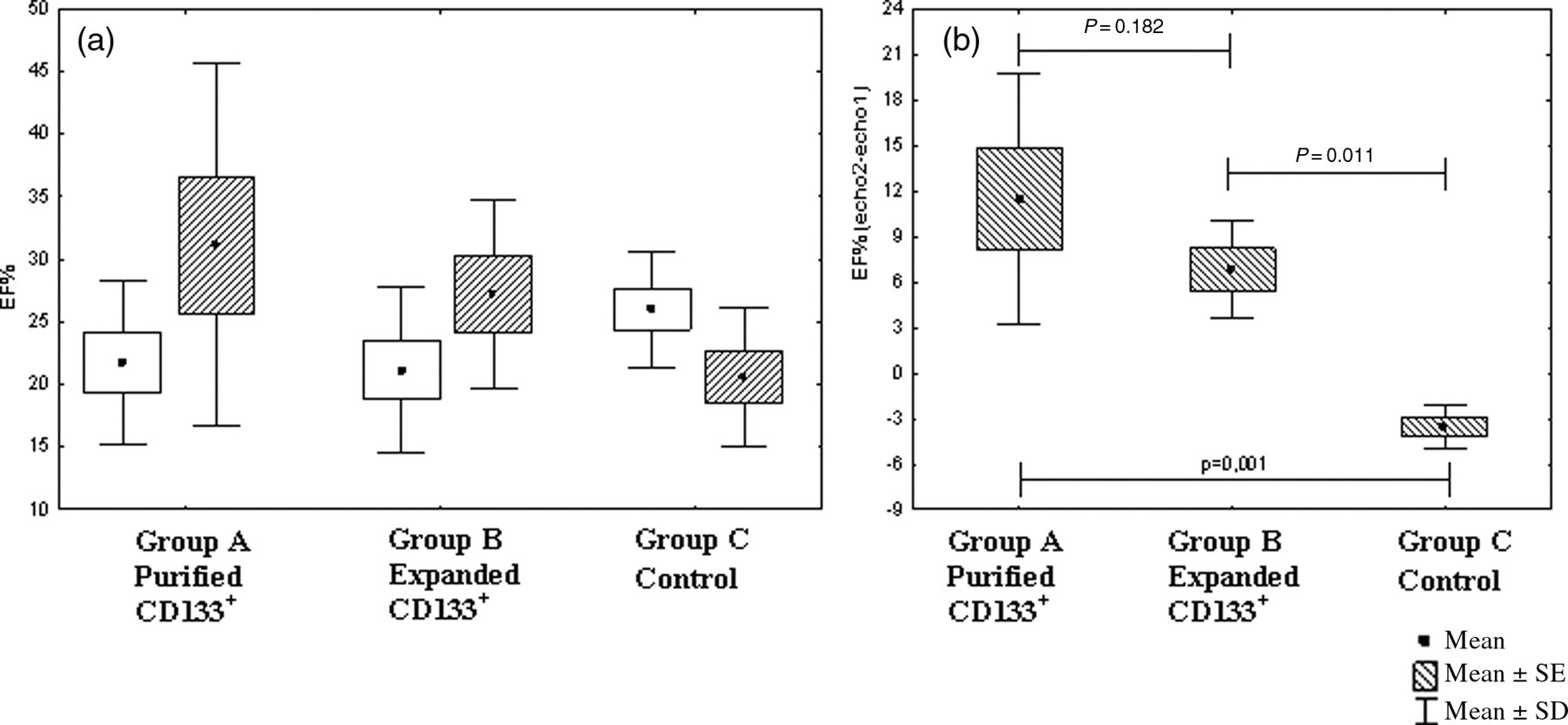
Echocardiographic evaluation. (a) Comparison of the left ventricle ejection fraction (LVEF) in two evaluations: echo 1, five days after myocardial infarction (white box); and echo 2, 30 d after cell transplantation (striped box). (b) Differences in the LVEF between echocardiography evaluations 1 and 2, comparing the groups in pairs and all together
Discussion
Neoangiogenesis within the infarcted tissue is an integral component of the remodeling process; however, the capillary network is unable to support the increased requirements of the hypertrophied myocardium, resulting in a progressive loss of viable tissue, infarct extension and fibrous replacement. 17 Limitations of the current strategy for inducing angiogenesis in ischemic tissues has led to the exploration of cell therapy as a means of stimulating neovascularization. 18–20 Most clinical assays use autologous bone marrow-derived MNCs for revascularization cell therapy. 21–24 In spite of bone marrow-derived MNCs showing positive effects in the treatment of heart disease in humans, 2,25 it has been suggested that using a specific fraction of progenitor cells (such as EPCs) could be more effective in rescuing the ischemic-induced injured areas of the heart. 26,27 Overall, the available clinical data indicate that cell therapy with EPCs is safe, feasible and associated with improved heart function. 28,29 The outcomes of clinical trials have been mixed and not as robust as they were in the animal models, partly because of the conflicts regarding the definition of human EPCs and the heterogeneity of the cell populations used in the treatments. 30
However, EPCs isolated from patients with increased cardiovascular risk factors such as hypertension, atherosclerosis, smoking, family history and diabetes mellitus, as compared with those isolated from healthy subjects, are reduced in number and demonstrate increased in vitro senescence. 14,31–33 In patients either at high risk for future cardiovascular events or who have documented coronary disease, the body's endogenous stem cell supply and bone marrow response may be inadequate. 9 Impairment may be due to age-related diminution of vascular EC number and function. This may limit the efficacy of patient-derived progenitor cells in mediating neovascularization; 14 therefore, the use of stem cells for myocardial tissue repair might be limited among the elderly and sick because their cell supply is depleted and/or exhausted. 34
Cord blood-derived EPCs may have a potentially powerful therapeutic advantage in ischemic diseases, especially MI and stroke. 35 HUCB has been previously described as a potential source of EPCs, 36 but little is known about the optimal culture conditions and differentiation of these cells. 15 Previous studies have demonstrated that circulating CD34+ cells co-expressing the CD133 maker can be isolated from MNCs. 6,37 Using a similar methodology, we purified CD133+ cells from HUCB, resulting in a cellular fraction of approximately 75% purity. The frequency of EPCs in bone marrow-derived MNCs is 0.01%. 38 In this study, we obtained approximately 0.64% EPCs from HUCB-derived MNCs, confirming that HUCB is a better source for EPCs. 15
After deciding on an EPC source, we attempted to establish the optimal conditions for cell expansion. VEGF was one of the growth factors chosen for cultivation, because it has been established to be involved in the first steps of hemoangioblast differentiation and is associated with faster endothelium growth. 20 VEGF could also be important in stimulating growth in relation to neovascularization, 39,40 as it is considered to be an important mitogen for ECs. 41 FGF is a regulator of proliferation; it influences EC growth, differentiation and migration both in vivo and in vitro. 39 ECs cultivated without b-FGF undergo apoptosis. 42 In clinical trials, co-administration of VEGF and b-FGF led to a faster and more efficient neovascularization than if either growth factor was independently used. It has been proposed that these growth factors promote the differentiation of EPCs into EC-like cells in vitro. IGF-I was also chosen, as it is a mitogen that regulates cellular proliferation and differentiation. 43
There is no consensus regarding the optimal dose for stem cell administration. 44 Extrapolating from the data available on the injected EPC number required to obtain therapeutic efficacy in mice, 37 the number of EPCs required for clinical therapy in humans would exceed the output of most published protocols. 15 We expanded CD133+ cells in the presence of three growth factors, b-FGF, IGF-I and VEGF. Cellular proliferation assays obtained with each growth factor showed that the optimal and most statistically significant concentration of b-FGF was 1.0 ng/mL. Different IGF-I and VEGF concentrations were not statistically significant when individually used. When VEGF was analyzed with co-administration of b-FGF and IGF-I (at a constant concentration), a VEGF dose of 50 ng/mL was found to be the most effective and statistically significant. Eighty milliliters of cord blood, a volume that can be routinely extracted from one cord, yields about 6.23 × 106 EPCs after expansion. According to Forraz et al., 45 HUCB-derived EPCs exhibit a high proliferative capacity and also demonstrate rapid self-renewal. We found this same result in our study. Growth curves showed up to a 17-fold increase on day 30, and cells had the typical morphology of undifferentiated EPCs. After this point and until the fourth passage (approximately 60 days), the cell population increased up to 70-fold, and they acquired similar morphological and functional phenotypes to ECs, suggesting that the cells lost expression of EPC markers. At this point, the cells continued to proliferate at least until the fourth passage, suggesting that a few undifferentiated cells were still present. The cell number increased by 17-fold at P1, so at this point, these cells could be clinically used, since there is a reasonable number of undifferentiated EPCs that have been passaged for a relatively short period of time.
EPCs are subdivided into two groups: early and late EPCs. Both can induce neovascularization in animals and can synergistically interact with each other. 46 Early EPCs appear within 4–7 days of in vitro culture; they are spindle-shaped and express CD14. 14,47 Late EPCs develop after 2–3 weeks of culture; they have a cobblestone pattern, are very proliferative and do not express CD14. 48 Our culture was maintained for 60 days, and the cells continued to significantly expand without any signs of senescence. EC-like colonies appeared around the third week, and they had the appearance of a cobblestone monolayer. These results were consistent with those reported by Ingram and colleagues, 49 i.e. the expanded cell population was characteristic of late EPCs.
We used flow cytometry to characterize the cell phenotype. Immunophenotyping of MNCs showed the typical phenotype for the hematopoietic lineage, i.e. cells were positive for CD45 50 and CD14, which is characteristic of monocytes. 51 There were practically no cells positive for CD133 52–54 and CD34. 55 Some cells expressed both endothelial and hematopoietic markers 56 and were CD31+ . 57 We also observed the presence of CD105-positive cells, which is characteristic of HUCB-derived cells and proliferating ECs. 58 Purified EPCs expressed predominantly CD133, CD34, CD31 and CD105. There was a small percentage of contamination from hematopoietic lineage cells (CD45+). The cells reached confluence after four weeks of culture and displayed a typical EC phenotype, including vWF expression 59 and CD31 enhancement. We observed a reduction in CD34+ and CD105+ cells in the population; however, some cells remained, which was consistent with the previous study by Gross and Herbrig. 38 Our results presented a similar loss of CD133 cells, as previously described by Shmelkov and colleagues.60 EPCs adopted a more mature endothelial phenotype during their in vitro culture and expansion, which was consistent with a previous study.15
We evaluated the functional phenotype of CD133+ cells. One important finding is that CD133+ cells only form capillary tubes and express VEGF after induction of differentiation, suggesting that only these cells have the potential to induce vascular regeneration. Samadja and colleagues61 have demonstrated that bone morphogenetic proteins 2 and 4, which play a key role in endothelial colony-forming cell commitment and outgrowth during neovascularization, were selectively expressed by late EPCs but not by early and circulating EPCs.
During in vitro expansion, stem cells experience a long replicative history; thus, they are subject to damage from intracellular and extracellular insults.62 Therefore, expanded and purified CD133+ cells could show different biological properties when transplanted in vivo. We decided to test both populations in terms of their ability to repair myocardial tissue after inducing MI in a rat model.
The rats that received purified CD133+ cells (Group A) and expanded CD133+ cells (Group B) showed a significant improvement in their LVEF, as compared with the control group. The smaller improvement observed for Group B, as compared with Group A, may be related to the lower proliferation rate of these differentiated cells. The high proliferation rate of purified CD133+ cells suggests that these cells would continue to proliferate after injection into the heart. Despite the ventricular enlargement, the results suggest that LV systolic dilation is reduced. Leor and colleagues11 obtained similar results using 1.2–2 × 106 purified HUCB-derived CD133+ cells intravenously injected seven days after permanent coronary artery ligation in athymic nude rats. The cells showed functional recovery as there was abrogation of scar thinning and LV systolic dilation. We used approximately 6–10 times less cells in our experiments and obtained the same improvement. The direct infusion of the cells into the myocardial scar could explain our results.
The therapeutic effects of EPCs have been recently demonstrated in preclinical studies of hindlimb ischemia,15,63 strokes64 and MI.11 One disadvantage of using HUCB-derived EPCs in clinical trials is the inherent risk of rejection. Previous studies suggest that HUCB-derived cells are not very immunogenic,65–67 so these cells may be a useful source for cell therapy. In addition, a previous Eurocord study demonstrated that unrelated HLA-mismatched umbilical cord blood transplantation in children with acute leukemia produces comparable results to HLA-matched transplantation with other stem cell sources.68 However, the reduced immunogenicity of HUCB cells is unproven and warrants further investigation. According to Cho and colleagues,69 umbilical cord tissue derived cells are major histocompatibility complex (MHC) class I dim and negative for MHC class II. A single injection of MHC-mismatched UTC-derived cells did not induce a detectable immune response. However, when these cells were injected into an inflamed region, repeatedly injected into the same region, or stimulated with interferon γ prior to injection, they were immunogenic. The reported successes of CD133+ and mesenchymal cell allograft-xenografts in animal models encouraged further experiments to test the efficacy of clinical-grade HUCB CD133+ cells in promoting cardiac regeneration using more accurate immunogenicity studies.70 In summary, the cells from HUCB-compatible MHC donors in cord blood banks have the potential for therapeutic application.
In conclusion, we purified and expanded CD133+ cells in vitro up to 70-fold. After 60 days of cultivation, the cells were functionally active in vitro and expressed cellular differentiation markers. We observed a significant improvement in the LVEF and attenuation of ventricular remodeling after transplantation of the purified and expanded HUCB-derived CD133+ cells, suggesting that both purified and expanded cells are promising candidates for use in cellular cardiomyoplasty. However, further pre-clinical trials should be performed to elucidate whether expanded CD133+ cells have any clinical advantages over purified CD133+ cells.
Footnotes
Acknowledgements
This work was supported by grants from the Ministry of Health of Brazil and CNPq (552233/2005-6). We would like to thank Marco Stimamiglio for assisting us with the statistical analysis and Dr Vivian Ferreira do Amaral for supervising the harvesting of human umbilical cord bloods. The authors have no competing financial interests.