
Abstract
In non-erythroid cells, insulin stimulates a signal transduction pathway that results in the activation of phosphoinositide 3-kinase (PI3K) and subsequent phosphorylation of phosphodiesterase 3 (PDE3). Erythrocytes possess insulin receptors, PI3K and PDE3B. These cells release adenosine triphosphate (ATP) when exposed to reduced O2 tension via a signaling pathway that requires activation of the G protein, Gi, as well as increases in cAMP. Although insulin inhibits ATP release from human erythrocytes in response to Gi activation by mastoparan 7 (Mas 7), no effect on cAMP was described. Here, we investigated the hypothesis that insulin activates PDE3 in human erythrocytes via a PI3K-mediated mechanism resulting in cAMP hydrolysis and inhibition of ATP release. Incubation of human erythrocytes with Mas 7 resulted in a 62 ± 7% increase in cAMP (n = 9, P < 0.05) and a 306 ± 69% increase in ATP release (n = 9, P < 0.05), both of which were attenuated by pre-treatment with insulin. Selective inhibitors of PDE3 (cilostazol) or PI3K (LY294002) rescued these effects of insulin. These results support the hypothesis that insulin activates PDE3 in erythrocytes via a PI3K-dependent mechanism. Once activated, PDE3 limits Mas 7-induced increases in intracellular cAMP. This effect of insulin leads, ultimately, to decreased ATP release in response to Mas 7. Activation of Gi is required for reduced O
Introduction
There is strong evidence that the vascular disease of type II diabetes arises before pathological increases in blood glucose. 1–5 Indeed, microvascular dysfunction in prediabetes can be as pronounced as that in established type II diabetes. While insulin resistance is present in both prediabetes and type II diabetes, there are marked differences between these two disease states. In type II diabetes, hyperglycemia develops when insulin secretion by the pancreatic β cells is no longer sufficient to compensate for insulin resistance. This decrement in insulin release results in inadequate glucose transport into cells. In contrast, individuals with prediabetes have sufficient β-cell function and maintain near normal blood glucose levels by secreting high concentrations of insulin to overcome peripheral insulin resistance resulting in severe hyperinsulinemia. 4,6,7 In individuals with prediabetes, it was demonstrated that microvascular dysfunction correlates with plasma insulin concentration, but not with plasma glucose, β-cell function, age, body mass index, serum lipid concentration or blood pressure. 2 This correlation suggests that supraphysiological levels of insulin in prediabetes could contribute to the vascular dysfunction present in prediabetes, although the mechanism by which this occurs has not been previously defined.
One potential target for the adverse effect of insulin on the circulation is the erythrocyte. Erythrocytes deliver oxygen (O2) throughout the body to meet the metabolic needs of tissues. The precise matching of O2 supply with demand requires that blood flow be redistributed to areas of increased need. It has been suggested that erythrocytes can participate in the regulation of blood flow distribution by stimulating increases of vascular caliber via the ability of this cell to release adenosine triphosphate (ATP). 8–13 This ATP released from erythrocytes can stimulate the production of endothelium-derived vasodilators resulting in local increases in vascular caliber and, consequently, erythrocyte (O2) supply to areas of tissue under increased metabolic demand. 9,10,12 Under this hypothesis, any impairment of ATP release from the erythrocyte would hinder the matching of O2 supply with demand and could contribute to the development of microvascular dysfunction.
Recently, it has become increasingly clear that, although microvascular impairment is clearly found in type II diabetes, 3,5,14–16 vascular dysfunction is also present in the prediabetic period before the onset of increases in blood glucose, but when plasma insulin is increased. However, the mechanism(s) responsible for vascular dysfunction in prediabetes remains to be determined.
A signal transduction pathway culminating in the release of ATP from erythrocytes has been defined.
17
Stimulation of ATP release by physiological stimuli (reduced O
In adipocytes and hepatocytes, insulin stimulates the hydrolysis of cAMP by activating the PDE3 family. 24–26 We have recently identified PDE3B in rabbit and human erythrocytes and demonstrated that inhibitors of PDE3 potentiate both erythrocyte cAMP levels and ATP release. 27,28 Importantly, we have also demonstrated that insulin inhibits ATP release from erythrocytes in response to activation of Gi in these cells. 29 However, in these studies, insulin had no effect on ATP release stimulated by a non-hydrolyzable cAMP analog. Since ATP release from erythrocytes requires an accumulation of cAMP within the cell, insulin-induced activation of PDE3 within the erythrocyte could be aniticipated to stimulate cAMP hydrolysis and, thereby, limit ATP release in response to activation of Gi. It has been shown that, in adipocytes, activation of PDE3 by insulin relies on the activity of phosphoinositide-3 kinase (PI3K), 30–32 a kinase that is present in erythrocytes. 33 Here we investigated the hypothesis that insulin inhibits human erythrocyte cAMP accumulation and ATP release in a manner dependent on the activity of PI3K and PDE3.
Materials and methods
Isolation of human erythrocytes
Twenty-five milliliters of blood were obtained from healthy human volunteers by venipuncture using a 21-gauge needle attached to a syringe containing heparin (500 units). Blood was collected from 10 females and nine males with an average age of 38 years (range 25–63 years). The protocol used to obtain blood from humans was approved by the Institutional Review Board of Saint Louis University.
After collection, whole blood was centrifuged at 500
Determination of cAMP accumulation in erythrocytes in response to incubation with pharmacological agents in the absence and presence of insulin
Washed erythrocytes diluted to a 50% hematocrit (1 mL) were pretreated with a PI3K inhibitor, LY294002 (10 μmol/L, Biomol), 32,33 or a PDE3 inhibitor, cilostazol (100 μmol/L), 34,35 for 30 min in the absence or presence of 1 nmol/L insulin (Humalog U 100®, Eli Lilly, Indianapolis, IN, USA) 29 or its vehicle, saline. Following this pretreatment protocol, mastoparan 7 (Mas 7, 30 μmol/L, Biomol), a direct activator of the G protein Gi, 18–20 was added to the cell suspension for 30 min. The reaction was stopped by the addition of 4 mL of ice-cold ethanol containing 1 mmol/L HCl. Samples were then prepared for the determination of cAMP concentration.
Measurement of cAMP
After addition of ice-cold ethanol/HCl, the samples were centrifuged at 14,000
Determination of ATP release from erythrocytes in response to incubation with pharmacological agents in the absence and presence of insulin
Washed erythrocytes diluted to a 20% hematocrit were incubated with 10 μmol/L Mas 7 (Genscript, Piscataway, NJ, USA), 18,19 following a 30-min pretreatment with LY294002 (10 μmol/L) 32,33 or cilostazol (100 μmol/L) 34,35 in the absence or presence of 1 nmol/L insulin (Humalog U 100®, Eli Lilly) 29 or its vehicle saline. The ATP concentration was determined at 5, 10 and 15 min after the addition of Mas 7. These time periods were chosen based on preliminary studies. 18,19 The maximal response to Mas 7 in the absence or presence of insulin is reported.
Measurement of ATP
ATP was measured by the luciferin–luciferase technique. 36 A 200 μL sample of erythrocyte suspension (0.04% hematocrit) was injected into a cuvette containing 100 μL of firefly lantern extract (10 mg/mL distilled water, FLE 250; Sigma, St Louis, MO, USA) 37 and 100 μL of a solution of synthetic D-luciferin (50 mg/100 mL distilled water; Sigma). The light emitted was detected using a luminometer (Turner Designs, Sunnyvale, CA, USA). A standard curve was obtained for each experiment. Cell counts were obtained from the suspension of erythrocytes and amounts of ATP measured were normalized to 4 × 108 cells/mL.
Measurement of total intracellular ATP of erythrocytes
A known number of erythrocytes were lysed in distilled water and diluted 8000 fold in wash buffer (see above). 21 ATP was measured as described above and the values were normalized to ATP concentration per erythrocyte.
Measurement of free hemoglobin
To exclude the presence of significant hemolysis in studies where the release of ATP was measured, samples were centrifuged at 500
Statisical analysis
Statistical significance between groups was determined using an analysis of variance. In the event that the F ratio indicated that a change had occurred, a Fisher's least significant difference test was performed to identify individual differences. Results were reported as means ± the standard error of the mean (SEM).
Results
Effect of insulin on Mas 7-induced increases in cAMP and ATP release from human erythrocytes
Consistent with previous studies, incubation of erythrocytes with insulin (1 nmol/L) had no effect on basal ATP release, but it attenuated Mas 7-induced ATP release from these cells (Figure 1). 29 To ensure that decreases in ATP release could not be attributed to insulin-induced decreases in erythrocyte ATP content, total intraerythrocyte ATP was measured. Total ATP levels were not different in the absence and presence of 1 nmol/L insulin (1.64 ± 0.17 and 1.52 ± 0.17 mmol/L for control and insulin treated, respectively, P < 0.05). Previously we reported that increases in cAMP are required for ATP release from human erythrocytes. 22 To begin to investigate the mechanism by which insulin inhibits ATP release, we determined the effect of insulin on Mas 7-induced increases in cAMP. Insulin (1 nmol/L) had no effect on basal cAMP levels (data not shown). However, as depicted in Figure 2, insulin did attenuate Mas 7-induced increases in cAMP. These results support the hypothesis that insulin-induced inhibition of ATP release from human erythrocytes is associated with increased cAMP hydrolysis.
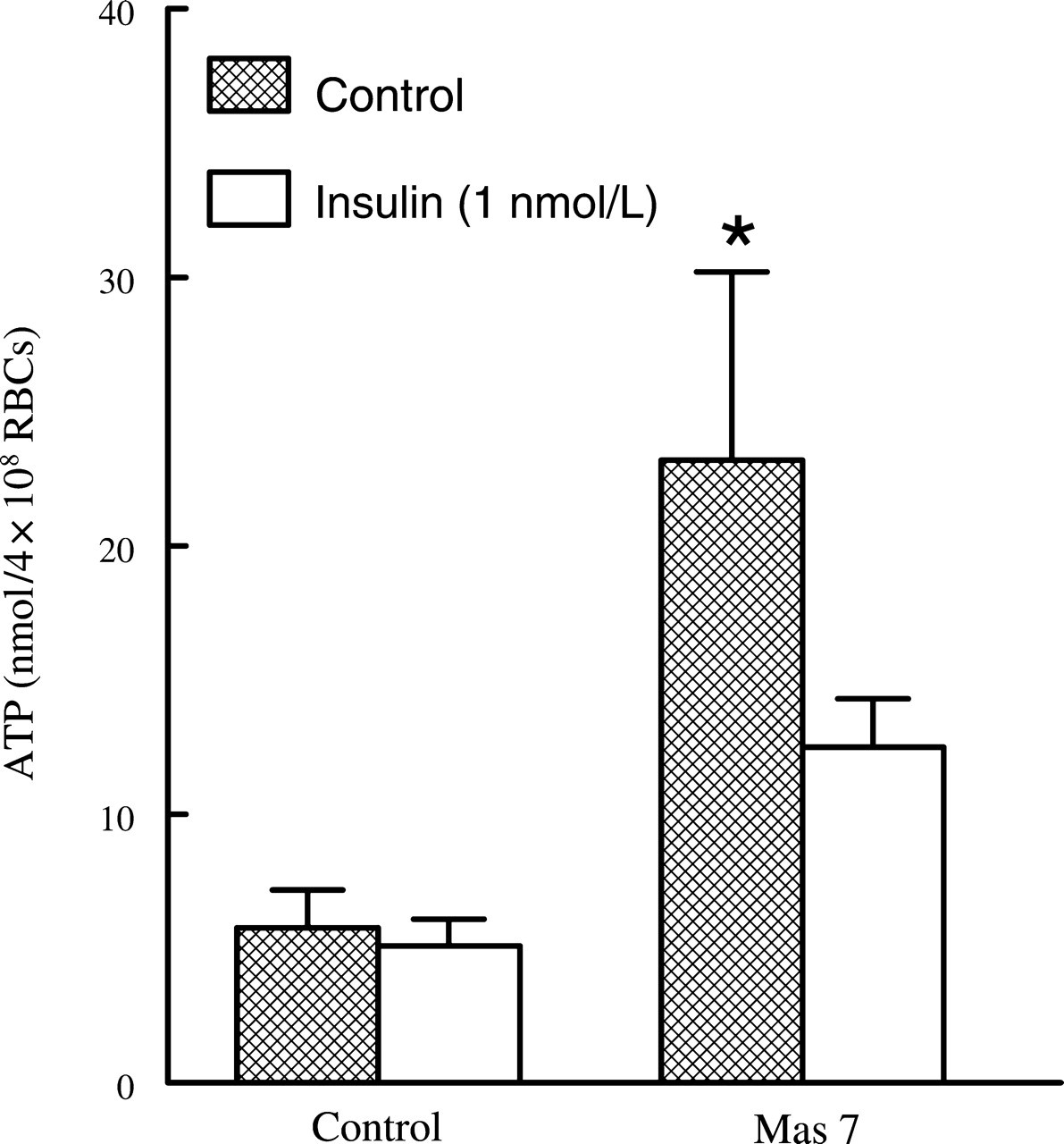
Effect of insulin on mastoparan 7 (Mas 7)-induced ATP release from erythrocytes. Washed erythrocytes were incubated with Mas 7 (10 μmol/L) 20 min after the addition of either insulin (1 nmol/L) or its vehicle (saline) (n = 9). The maximal ATP release in response to Mas 7 treatment was reported. Values are the means ± SE. *Greater than all other values (P < 0.05)
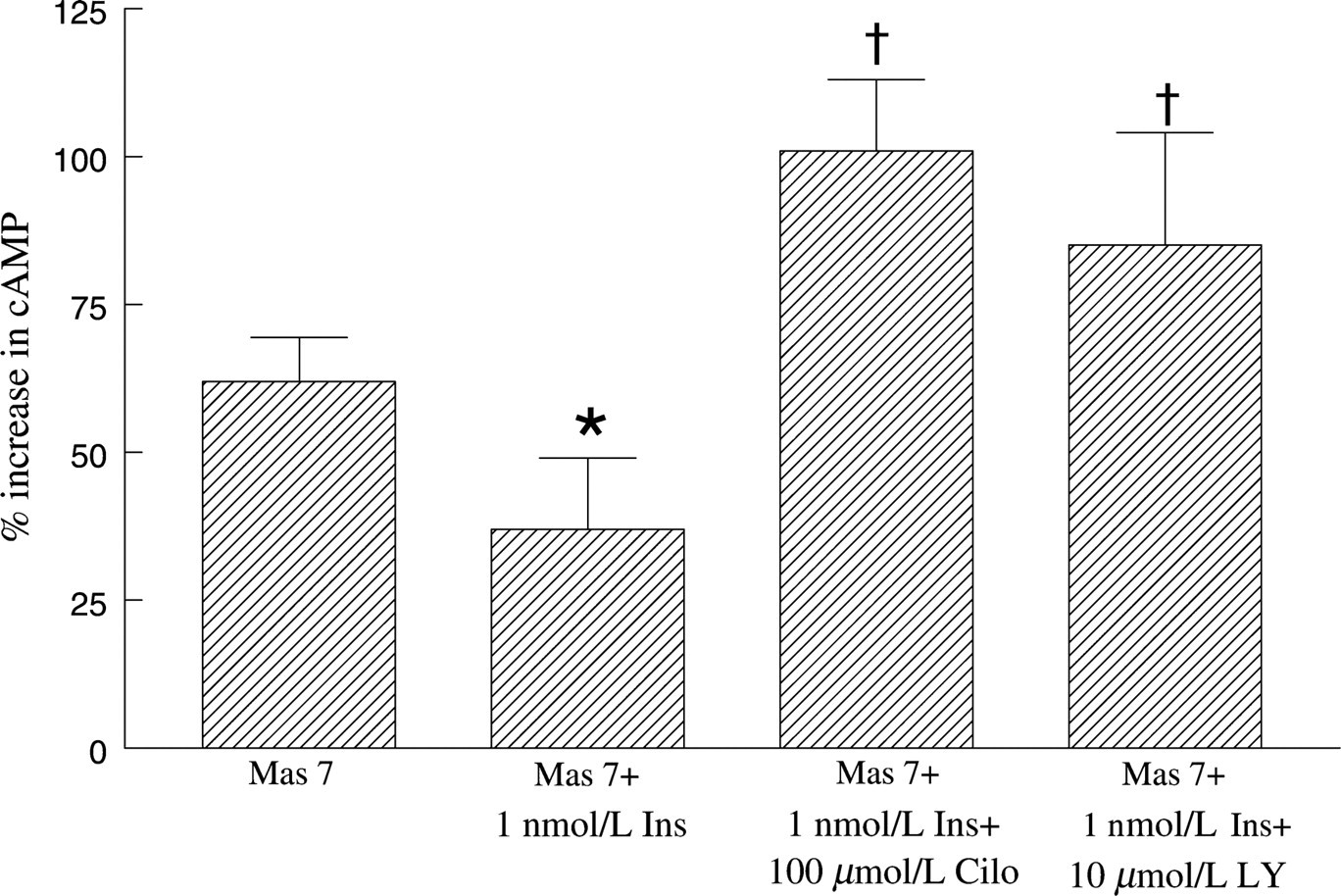
Effect of cilostazol or LY294002 on mastoparan 7 (Mas 7)-induced cAMP increases in erythrocytes in the absence and presence of insulin. Washed erythrocytes were incubated with cilostazol (Cilo, 100 μmol/L) or LY294002 (LY, 10 μmol/L) for 30 min in the absence or presence of insulin (1 nmol/L) or its vehicle (saline) and then cAMP increases were stimulated by Mas 7 (30 μmol/L) (n = 9). cAMP levels were determined 30 min after the addition of Mas 7. Values are the means ± SE. *Less than all other values; †Greater than Mas 7 and Mas 7+ 1 nmol/L Ins
Effect of a PDE3 inhibitor on insulin-induced attenuation of cAMP accumulation within erythrocytes
Insulin has been shown to antagonize the effects of cAMP by stimulating the hydrolysis of this cyclic nucleotide. 24–26 Insulin stimulates cAMP hydrolysis by activating a signal transduction pathway resulting in the phosphorylation and activation of PDE3, a PDE that has been shown to be present in human and rabbit erythrocytes. 28 To determine whether inhibition of PDE3 can rescue insulin-induced inhibition of increases in cAMP stimulated by Mas 7, erythrocytes were pre-treated with the selective PDE3 inhibitor cilostazol (100 μmol/L) in the absence and presence of 1 nmol/L insulin. 34 As shown in Table 1, 100 μmol/L cilostazol potentiated Mas 7-induced cAMP accumulation. More importantly, in the presence of cilostazol, insulin did not inhibit Mas 7-induced cAMP accumulation (Figure 2).
Effect of cilostazol (100 μmol/L) or LY294002 (10 μmol/L) on mastoparan 7 (30 μmol/L)-induced cAMP increase in human erythrocytes
Mas 7, mastoparan 7; Values are mean ± SEM
*Significantly different from Mas 7 alone (P < 0.05)
Effect of a PI3K inhibitor on insulin-induced attenuation of cAMP accumulation within erythrocytes
Many of the cellular effects of insulin are mediated through its activation of PI3K. 38 Indeed, activation of PDE3 by insulin and subsequent cAMP hydrolysis has been shown to require the activity of PI3K. 31,32 To determine whether insulin attenuates Mas 7-induced increases in cAMP via a mechanism that requires activation of PI3K, erythrocytes were incubated with Mas 7 or Mas 7 plus insulin (1 nmol/L) in the absence and presence of the selective PI3K inhibitor LY294002 (10 μmol/L). As shown in Table 1, 10 μmol/L LY294002 potentiated Mas 7-induced cAMP accumulation in the absence of insulin. More importantly, in the presence of LY294002, insulin had no effect on Mas 7-induced cAMP accumulation (Figure 2).
Effect of a PDE3 or PI3K inhibitor on insulin-induced attenuation of ATP release from erythrocytes
To determine whether inhibitors of PDE3 or PI3K prevent the effects of insulin on Mas 7-induced ATP release from erythrocytes, cells were incubated with insulin in the absence and presence of either cilostazol (100 μmol/L) or LY294002 (10 μmol/L). ATP release was then stimulated by Mas 7. Cilostazol and LY294002 alone had no effect on Mas 7-induced ATP release in the absence of insulin (data not shown). However, both cilostazol (Figure 3) and LY294002 (Figure 4) prevented the insulin-induced inhibition of ATP release produced by Mas 7. These results provide strong support for the hypothesis that insulin stimulates the hydrolysis of cAMP within the erythrocyte via PI3K-mediated PDE3 activation resulting, ultimately, in inhibition of ATP release from these cells.
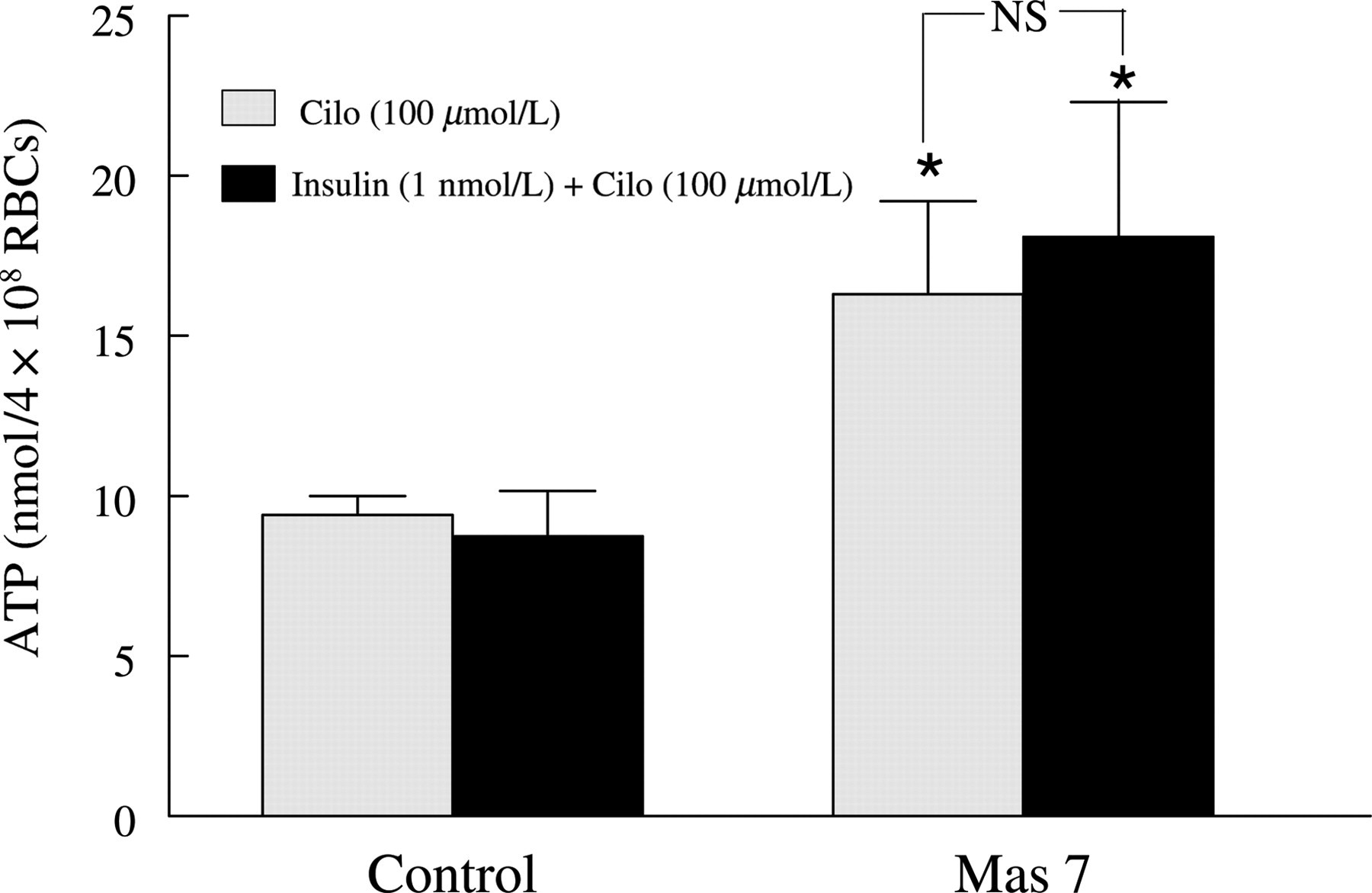
Effect of cilostazol on mastoparan 7 (Mas 7)-induced ATP release from human erythrocytes in the absence and presence of insulin. Washed erythrocytes were incubated with cilostazol (Cilo, 100 μmol/L) for 30 min in the absence or presence of insulin (1 nmol/L) or its vehicle (saline) and then ATP release was induced by Mas 7 (10 μmol/L) (n = 8). The maximal ATP release in response to Mas 7 treatment was reported. Values are the means ± SE. *Greater than all other values (P < 0.05), NS, no significant difference between values
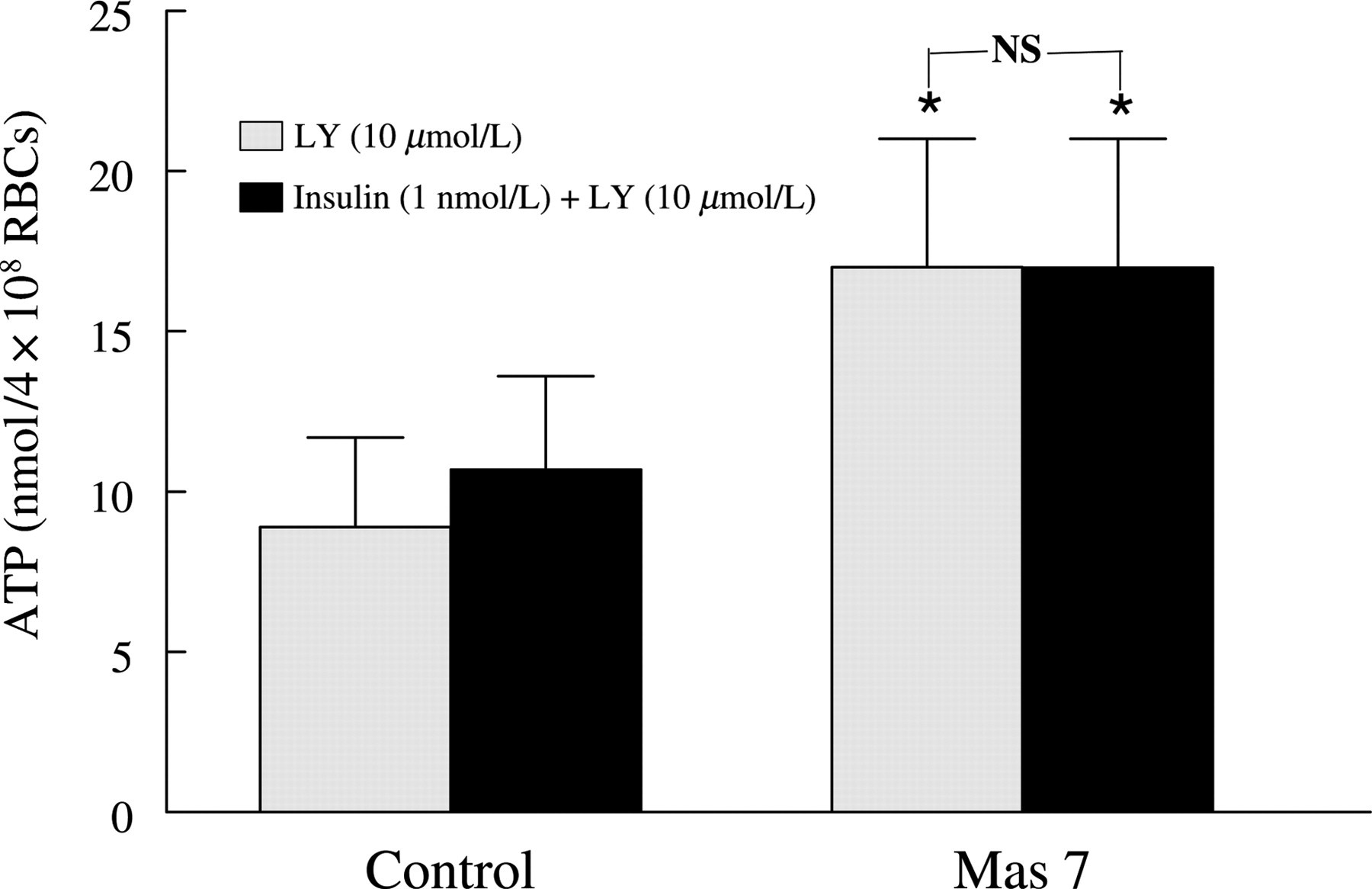
Effect of LY294002 on mastoparan 7 (Mas 7)-induced ATP release from human erythrocytes in the absence and presence of insulin. Washed erythrocytes were incubated with LY294002 (LY, 10 μmol/L) for 30 min in the absence or presence of insulin (1 nmol/L) or its vehicle (saline) and then ATP release was induced by Mas 7 (10 μmol/L) (n = 8). The maximal ATP release in response to Mas 7 treatment was reported. Values are the means ± SE. *Greater than all other values (P < 0.05), NS, no significant difference between values
Discussion
Adequate O
We, and others, have suggested that the erythrocyte plays a critical role in the regulation of vascular caliber permitting that cell to participate in the matching of O
A signal transduction pathway that results in ATP release from erythrocytes in response to both physiological (reduced O
A critical control point in the pathway stimulating erythrocyte ATP release is the intraerythrocyte concentration of cAMP. 16 PDEs, via their ability to hydrolyze cAMP, not only regulate the intracellular concentrations of this second messenger but also ultimately regulate biological processes that involve this cyclic nucleotide. Although PDE activity has been described in erythrocytes, the ability of these enzymes to regulate erythrocyte cAMP levels and ATP release has not been extensively studied. Recently, we identified PDE3B in rabbit and human erythrocytes and have also demonstrated that inhibitors of this PDE family potentiate agonist-induced cAMP increases within these cells as well as ATP release. 27,28
We demonstrated previously that 1 nmol/L insulin, a concentration of insulin that reflects postprandial levels in humans with prediabetes,
44–47
inhibits ATP release from human erythrocytes stimulated by exposure to reduced O
We first confirmed that insulin inhibits Mas 7-induced ATP release (Figure 1). Additionally, we demonstrate that insulin inhibits Mas 7-induced increases in cAMP within these cells (Figure 2). In other cell types, insulin stimulates cAMP hydrolysis via a signaling pathway that results in the phosphorylation and activation of PDE3. 24–26 To determine whether PDE3 activity is required for insulin-induced inhibition of cAMP increases in erythrocytes, cells were preincubated with 1 nmol/L insulin followed by Mas 7 in the presence or absence of the selective PDE3 inhibitor cilostazol. As shown in Figure 2, cilostazol prevented insulin-induced inhibition of increases of cAMP produced by Mas 7. This suggests that PDE3 regulates cAMP accumulation stimulated by activation of Gi and that PDE3 activity is required for insulin to inhibit cAMP increases in erythrocytes.
Insulin-induced activation of PDE3 has been reported to require the activity of PI3K. 30–32 PI3K is present in erythrocytes, 33 but this protein has not been related to PDE activity within this cell. To determine whether PI3K is involved in the insulin-induced inhibition of cAMP accumulation in erythrocytes, cells were preincubated with insulin and cAMP increases were stimulated by Mas 7 in the presence and absence of the PI3K inhibitor LY294002. LY294002 prevented insulin-induced inhibition of increases of cAMP produced by Mas 7 (Figure 2). This demonstrates that PI3K is involved in the activation of PDE3 by insulin in human erythrocytes. Taken together, these results support the hypothesis that insulin stimulates a signal transduction pathway within the erythrocyte involving PI3K that results in activation of PDE3 and, ultimately, increased cAMP hydrolysis.
Since insulin also inhibits Mas 7-induced ATP release from erythrocytes (Figure 1), 29 we next determined whether inhibition of PDE3 or PI3K rescues Mas 7-induced ATP release. We found that inhibitors of either PDE3 or PI3K prevented insulin-induced inhibition of ATP release from these cells (Figures 3 and 4). These data provide additional support for the hypothesis that, in human erythrocytes, insulin-induced activation of PI3K and, subsequently, PDE3 stimulates the hydrolysis of intracellular cAMP resulting in inhibition of ATP release.
In summary, this study demonstrates that, in human erythrocytes: (1) insulin attenuates Mas 7-induced cAMP accumulation and ATP release and (2) inhibition of PI3K or PDE3 prevents these effects of insulin. These results support the hypothesis that insulin inhibits erythrocyte cAMP accumulation and ATP release by activating PDE3 in this cell. Since activation of Gi and increases in cAMP levels in human erythrocytes are required for ATP release when these cells are exposed to reduced O2 tension, such an action of insulin could hinder the ability of erythrocytes to stimulate vasodilation and, thereby, attenuate increases in erythrocyte (O2) supply to areas of skeletal muscle with increased O2 demand.
Inadequate matching of O
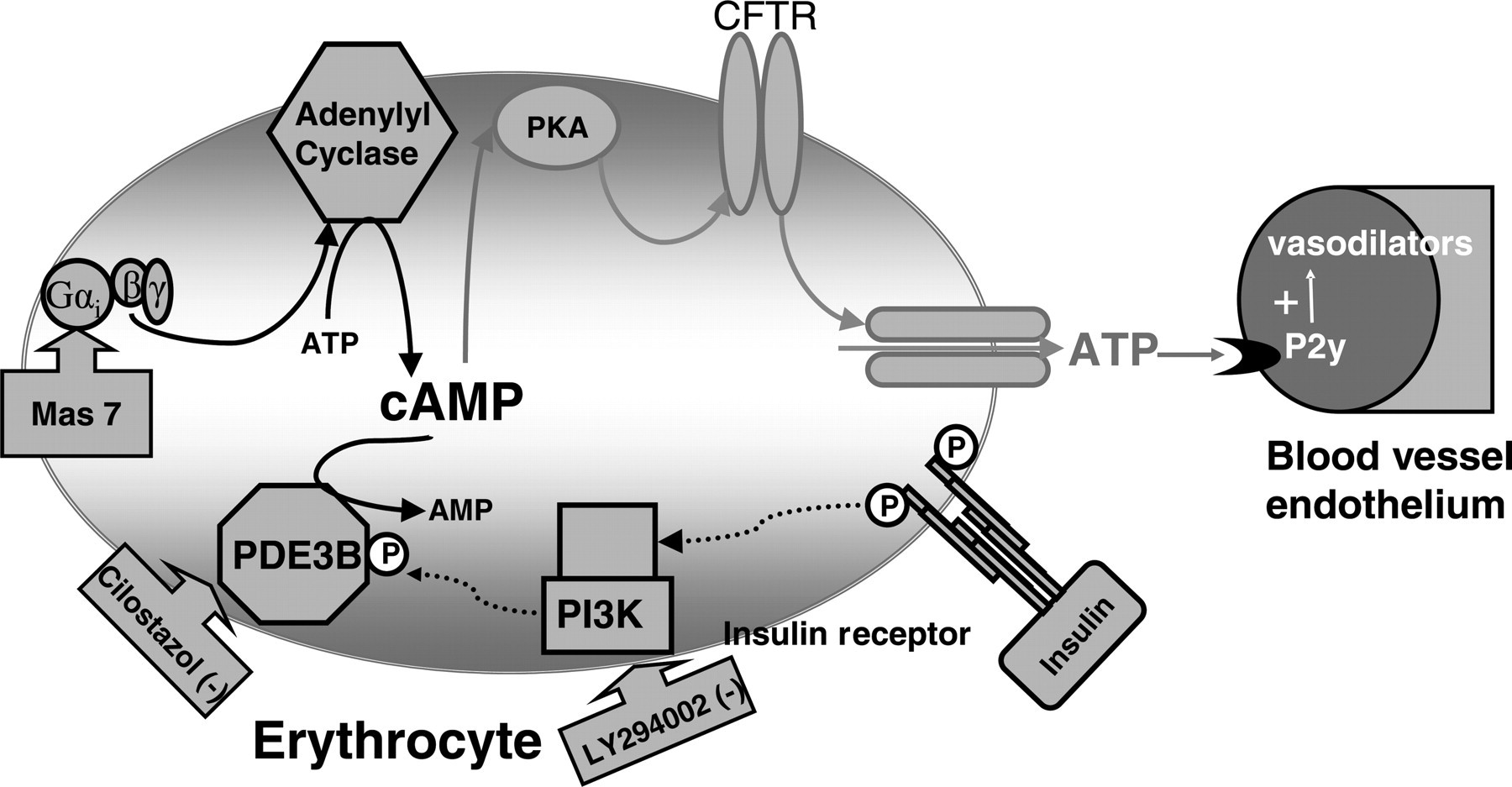
Proposed mechanism by which insulin activates PDE3 in erythrocytes, inhibiting ATP release from these cells. Gαi, heterotrimeric G protein Gi; Mas 7, mastoparan 7; ATP, adenosine triphosphate; cAMP, cyclic adenosine monophosphate; PKA, protein kinase A; CFTR, cystic fibrosis transmembrane conductance regulator; PI3K, phosphoinositide-3 kinase; PDE3B, phosphdiesterase 3B; AMP, adenosine monophosphate
Footnotes
Acknowledgements
The authors extend their gratitude to Dr Mary L Ellsworth for her editorial assistance. The authors also would like to thank JL Sprague for inspiration. This work was supported by National Institutes of Health grants HL-64180 and HL-89094, American Diabetes Association grant RA-133 and an American Heart Association Fellowship (MSH).