
Abstract
Genomic imprinting is an epigenetic form of gene regulation that entails differential sex-specific methylation of the alleles of a gene. Such methylation distinguishes male and female genomes and is inherited in a parent-of-origin-specific manner. Sex-specific imprints are established in the germline during gametogenesis and remain intact throughout embryonic and postnatal development. Reprogramming of methylation patterns in gametes is essential to sex-specific inheritance of imprinted genes and assures exclusive harboring of female- and male-specific imprinted patterns in maternal and paternal gametes, respectively. The consequences of genomic imprinting are manifested by its loss, which can lead to a variety of disorders, the most prominent ones being Prader–Willi and Angelman syndromes. Although a great deal of research has been carried out to examine various imprinting mechanisms, little is known about the establishment and regulation of imprinted genes. In the present paper, we describe several epigenetic mechanisms that have relevance in imprinting and that may have impact on embryonic development, fetal growth and animal cloning.
Keywords
What is genomic imprinting?
Imprinting refers to the learning of a behavior at a critical period early in life that becomes permanent. In other words, imprinting is a type of phase-sensitive learning in which an offspring learns the characteristics of a parent and is said to be ‘behaviorally imprinted’ with those characteristics near the beginning of life. Such imprinting is exemplified by new-born geese that follow the first object with which they make visual contact, most usually their parents. Thus, the goslings survive because they learn to follow their parents at a critical stage in their lives. In addition to behavioral imprinting, the parents of mammals imprint their young ones in another way. They imprint the genome of their offspring with an epigenetic pattern, as early as the embryonic stage, and the young ones are said to be ‘genomically imprinted’. Genomic imprinting refers to a special kind of epigenetic variation that involves heritable changes in the expression of genes. Epigenetic modifications themselves are brought about primarily by chromatin remodeling, RNA interference, DNA acetylation and DNA methylation. Gene sequence per se is of little consequence. Of these four mechanisms, the most widely studied and best understood is methylation. Apparently, there is no significant alteration in the DNA sequence itself but the difference between an active and inactive gene is due to the presence of a 5′-methylcytosine, usually in those CpG dinucleotide cores that lie upstream of the gene. Methylation does not promote changes in gene activity. Rather, it maintains a gene in its silent state. The most likely explanation for such gene silencing is that the enzyme methyl transferase is biased towards daughter strands that are coupled to methylated strands which, in turn, lead to inheritance of the associated methylation patterns. Tissue-specific factors initiate the methylation process and, consequently, genes are silenced by blocking the binding of transcription factors to promoters, recruiting methyl CpG-binding proteins that compete with transcription factors or altering nucleosome formation, thus hindering transcription. 1
Methylation may be involved in two other phenomena as well, namely X chromosome inactivation and gene imprinting. 2 X chromosome inactivation is the random silencing of one of the two female X chromosomes during embryogenesis, such that the level of expression of X-linked genes is equalized in both sexes. There is a so-called ‘X inactivation center’ which includes an Xist gene. The X inactivation center is a cis-acting region located on the X chromosome that encodes RNA. The RNA, in turn, recruits methyl groups to one of the X chromosomes, leading to its subsequent silencing. Gene imprinting, on the other hand, is a process in which the maternal and paternal genomes become distinguishable from each other as a result of gamete-specific differential methylation. 3 The establishment of contrasting methylation patterns causes certain genes on one of the parental chromosomes to become active but not on the other. The consequence of such action is the presence of single active alleles of the genes involved. Because of the differences in methylation patterns between male and female pronuclei, zygotes are unable to develop from two egg or sperm pronuclei, as this would result in the absence of the active alleles of some genes. 1
Most imprinted genes studied so far are involved in placental and embryonic development and fetal growth. 4 Imprinted genes are established as sex-specific patterns in the germline during gametogenesis and are usually clustered. Once a gene is methylated and imprinted in the germline, it remains transcriptionally inactive during embryogenesis and development. 5 Owing to the existence of only one functional allele, imprinted genes are highly susceptible to lethal mutations and, because these genes are in close proximity, they often are controlled together. As such, a single mutational change can disrupt the functioning of many genes and the consequences can be deleterious. 6 Lethality may be due either to changes in the DNA sequence of the only active allele of an imprinted gene or to an ‘epimutation’ that leads to a heritable change in the gene without altering its DNA sequence. Such epimutations can alter methylation patterns in imprinted genes rendering an inactive allele active or vice versa. 7 Often referred to as ‘the battle between the sexes’, genomic imprinting and its loss can influence the precise functioning of a variety of gene networks, making it an extremely vital epigenetic mechanism in mammalian growth and development. Furthermore, there most likely is a connection between imprinted genes and the development of mental disorders such as autism and schizophrenia. 8 The reader is referred to several review articles that cover various aspects of genomic imprinting, including plants. 4,6,9,10 In this particular review, we summarize certain features of mammalian genomic imprinting, with examples, which demonstrate the significance of differential methylation in this most intriguing and mysterious phenomenon.
Methylated versus imprinted genes
The establishment of imprints is a complex procedure that involves reprogramming of the entire genome. Reprogramming is of primary importance because accurate imprints must be passed on to the next generation. In other words, in males, all cells contain one set of chromosomes with male imprints (from the father) and another set with female imprints (from the mother), but when these chromosomes are passed on to the next generation, both sets in the germ cells must be reprogrammed to contain male imprints which account for paternal contribution. Likewise, in females, all cells contain one paternal set of chromosomes with male-specific imprints and a maternal set with female-specific imprints, which are later reprogrammed in the germ cells to contain female-specific imprints exclusively so that they can be passed on to the next generation with maternal methylation patterns. Reprogramming takes place in two phases and is portrayed in Figure 1. The first phase occurs in the primordial germline and involves sequential demethylation and remethylation steps to remove the existing parental methylation patterns and establish a new sex-specific pattern in the gametes. 11,12 This procedure takes place during oocyte maturation in females and at birth in males. 5 The second phase of reprogramming occurs after fertilization and involves global demethylation in the embryo prior to implantation. Global remethylation then occurs post-implantation. Most imprinted genes, however, escape the second phase of reprogramming and remain intact throughout embryonic development. 13 Studies have shown that the second erasure procedure occurs sometime between the two-cell and 16-cell stage of the embryo. 14 The primary difference between methylated genes and imprinted genes is that the latter remain inactive in all cells whereas normal methylated genes can be activated and deactivated by signals in differentiated cells.

Reprogramming and establishment of imprints. The somatic cells and germ cells of an embryo contain two homologous chromosomes: one from the egg (with female imprints) and another from the sperm (with male imprints). Primary reprogramming occurs in the primordial germline where the old imprints are erased. Subsequently, remethylation occurs in meiosis I and new sex-specific imprints are established in the new gametes
Origin of imprinting
All placental mammals examined, to date, characteristically possess imprinted genes as opposed to egg-laying mammals and birds. 6 This contrast raises interesting questions about the relevance of imprinted genes. One possibility is that imprinting is a defense mechanism for parthenogenesis. Another notion assumes that it is a protective device that inhibits tumor development in females by preventing excessive placental growth. And, yet another supposition is that imprinted genes help protect against parasites because the imprinted genes initially became inactivated due to their close positioning to foreign parasitic DNA spliced into various chromosomal regions. Perhaps, the best answer, however, lies in a rather widely accepted theory referred to as the ‘conflict hypothesis’. 6 The conflict hypothesis is based on the idea that genomic imprinting is an evolutionary response to competition between the sexes. In the situation of polyandry in which one female mates with more than one male, the male is said to possess the ability to direct an embryo that he fathers to under-express genes responsible for hindering embryonic growth. Such a condition ensures more than adequate growth of an embryo. This circumstance, however, could lead to differential maternal investment, depleting the mother of her maternal resources. Fortuitously, females possess the ability to direct an embryo to under-express genes responsible for the enhancement of embryonic growth. Thus, all embryos produced in a polyandrous relationship would have an equal chance of survival, without draining the mother of all her maternal resources. The absence of such a safeguard would bias parental genetic contributions, ultimately resulting in deterioration of the embryos. 15
Imprinting mechanisms
There is a variety of mechanisms by which genes are imprinted. The two most notable ones are summarized here. The first involves the H19 and Igf2 genes. H19 is a gene for a long non-coding RNA showing growth-repressive properties and the Igf2 gene product is an autocrine growth factor. 16 These genes are said to be ‘reciprocally imprinted’ because H19 is active on the maternal chromosome but inactive on the paternal chromosome whereas Igf2 is active on the paternal chromosome but inactive on the maternal chromosome. 3 These genes are closely linked and are a part of an imprinted gene cluster. 5 This particular situation illustrates the boundary effect of an imprinting center, also known as an insulator, which is situated between the H19 and Igf2 genes and is believed to control the boundary for enhancer activity (Figure 2). The insulator itself is a differentially methylated region whose methylation pattern is similar to that of the H19 gene, i.e., it is methylated and inactive on the paternal chromosome and unmethylated and active on the maternal chromosome. 3 When the insulator is unmethylated (maternal chromosome), a CCCTC-binding factor (CTCF) binds to it, restricting the activity of the enhancer and, thus, stimulates the expression of H19. Conversely, when the insulator is methylated (paternal chromosome), the CTCF cannot bind to it and, consequently, the enhancer acts beyond its previous boundary and promotes the expression of Igf2. 16–19 This model (Figure 2a) demonstrates the importance of methylase in the maintenance of an imprint. Studies have shown that in the absence of methylase (Figure 2b), the paternal insulator is left unmethylated, allowing CTCF to bind to it as well. 20 This action blocks the enhancer from influencing Igf2 and promotes H19 expression on both the chromosomes, leading to the complete silencing of Igf2.
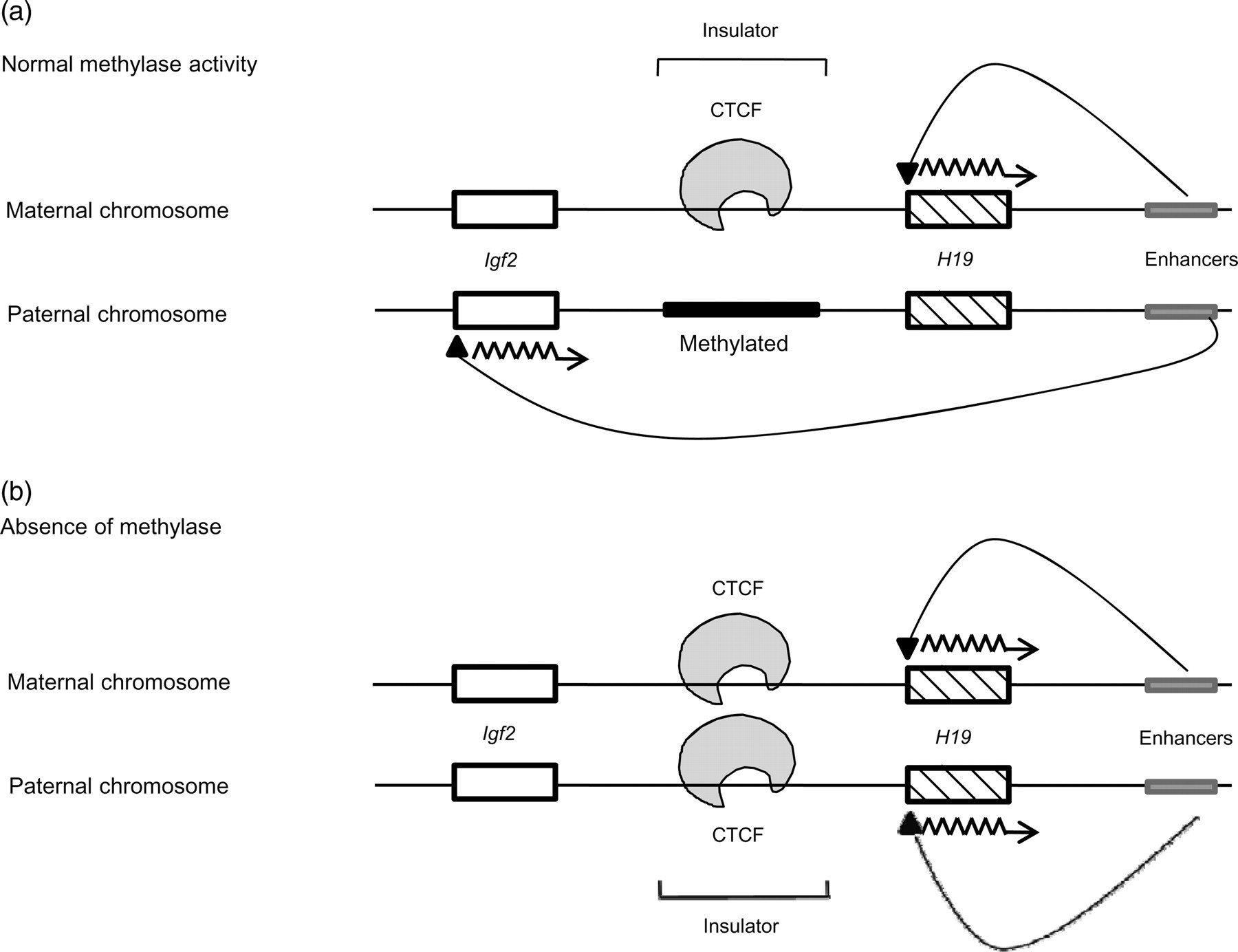
Regulation of enhancer activity by an insulator in the imprinted gene cluster H19-Igf2. (a) The maternal unmethylated insulator allows CTCF binding, thereby limiting the influence of the enhancer to the H19 gene. On the other hand, CTCF cannot bind to the paternal methylated insulator, giving the enhancer the ability to extend its influence to the Igf2 gene. (b) In the absence of methylase, the paternal insulator is left unmethylated, leading to binding of CTCF to both parental insulators, hence restricting action of the enhancer and allowing expression of only the H19 gene on both strands
Another imprinting mechanism occurs in the expression of the Igf2r gene (Figure 3). Igf2r is a maternally expressed gene and encodes the type-2 receptor for insulin-like growth factor. When in excess, it binds Igf2. 21 This system, unlike the previous one, involves the expression of the gene in the maternal strand and synthesis of an antisense strand from the paternal chromosome (Figure 3a). Apparently, a CpG island located in the second intron of the Igf2r gene helps maintain an imprinted signal. 22 The promoter for Igf2r is active on the maternal chromosome and its corresponding intronic CpG island (iCGi) is methylated. The promoter in the paternal chromosome is methylated whereas its iCGi is unmethylated. Under normal conditions, the gene in the maternal chromosome is expressed when iCGi is methylated. Coincidentally, an antisense RNA is produced in the reverse direction starting from iCGi of the paternal chromosome whose Igf2r promoter is methylated. Upon deletion of iCGi (Figure 3b), bi-allelic expression of the gene is restored and no antisense RNA forms. Hence, deletion of iCGi restores mRNA production in males but has no effect on the maternal chromosome. Most likely, iCGi is an imprinting element that acts as a cis-repressor on the Igf2r promoter. What conclusions can be drawn from such a phenomenon? First, iCGi is not required for Igf2r expression because of the bi-allelic expression in the absence of iCGi. Second, iCGi is necessary for the expression of the antisense transcript because the antisense strand is absent after deletion of iCGi. Alternatively, the promoter for the antisense transcript could lie in iCGi. In any event, methylation of iCGi leads to a loss of any inhibitory effect on the Igf2r promoter, and it is because of this negative effect that the gene is expressed in the maternal chromosome and repressed in the paternal chromosome (Figure 3; see references 21,22 ).

Expression of the Igf2r gene and its involvement in imprinting. (a) The promoter for the Igf2r gene is active when iCGi is methylated and the Igf2r gene is expressed on the maternal chromosome whereas iCGi is unmethylated on the paternal chromosome, leading to the production of an antisense transcript. (b) Deletion of iCGi shows the restoration of biallelic expression and the absence of the antisense transcript, demonstrating that iCGi is not required for Igf2r production but is required for the synthesis of the antisense strand
Loss of imprinting
Disruption of imprinting has been identified as a causative agent in approximately 30 known diseases or disorders. The impact of genomic imprinting is manifested when an individual has one copy of a normally imprinted allele and the other active allele is either inactivated or deleted. Two striking examples of loss of imprinting (LOI) involve the Prader–Willi syndrome and the Angelman syndrome. Both conditions arise from deletions in the same region of chromosome 15, the exact location being 15q11 (Figure 4). In other words, the two diseases are associated with the same imprinting center that contains a set of three closely linked genes (Figure 4a), namely SNRPN, which codes for a small nuclear ribonucleoprotein polypeptide (SmN), the necdin-encoding gene (NDN), which lies within the centromeric portion of the Prader–Willi deletion region between the two imprinted genes ZNF127 and SNRPN, and UBE3A, which codes for an E3 ubiquitin-protein ligase. 23 ZNF127 apparently is part of the coordinately regulated imprinted domain affected in Prader–Willi patients. 24 The SNRPN and NDN genes are imprinted in females whereas the UBE3A gene is imprinted in males.
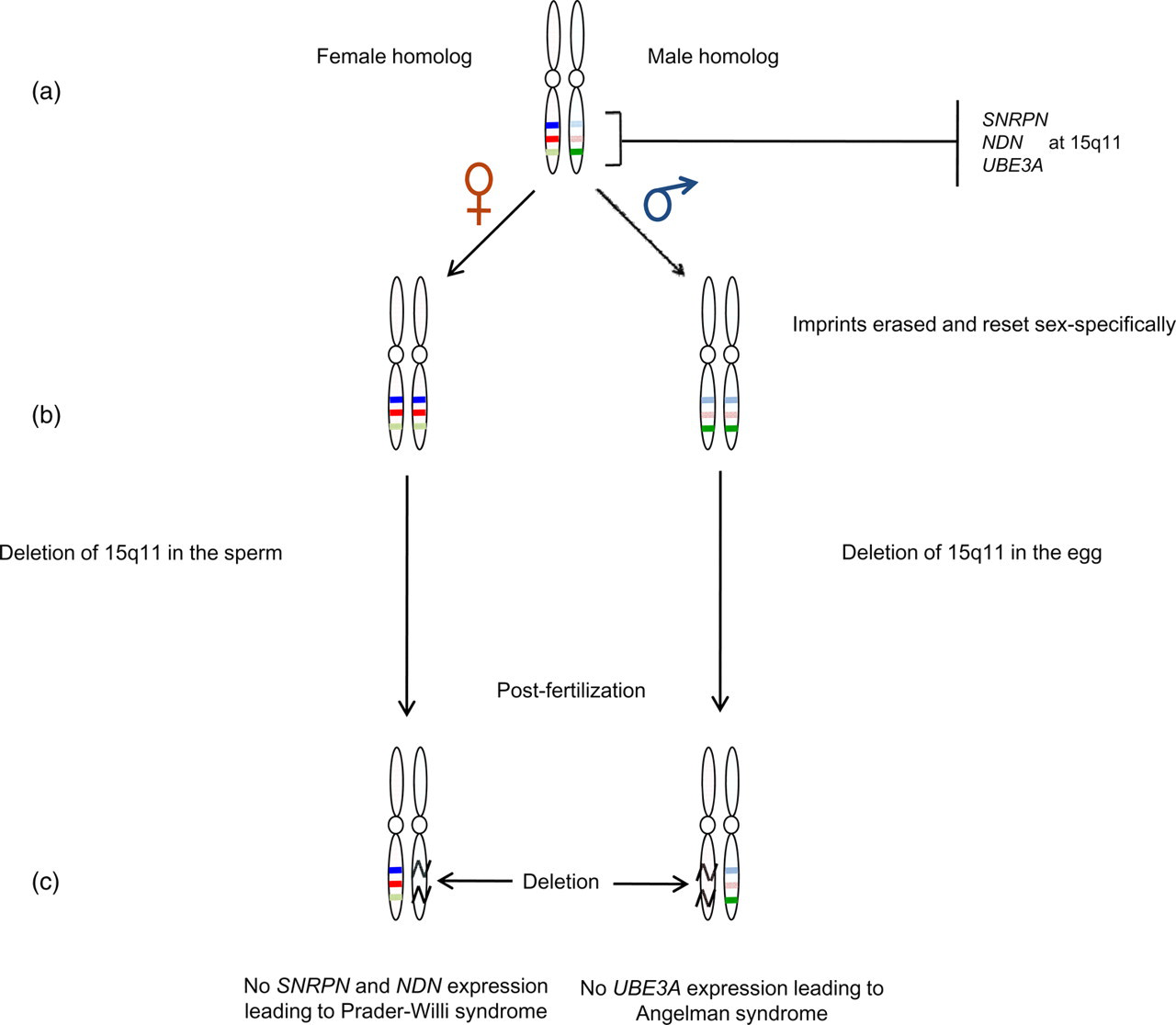
Loss of imprinting leading to Prader–Willi or Angelman syndrome. (a) Homologous chromosomes display the imprinted pattern of the genes. (b) Male and female gametes are reprogrammed to contain maternal and paternal imprints respectively. (c) Deletion in the 15q11 regions of the male and female chromosomes. Effect of the deletion: diagram on the left shows loss of SNRPN and NDN gene expression due to deletion in the sperm, which leads to Prader–Willi syndrome; diagram on the right depicts loss of UBE3A gene expression due to deletion in the egg, which leads to Angelman syndrome. The darker shaded colors represent imprinted alleles of the genes
During gametogenesis, all early imprints are erased and re-established based on the sex of the individual. Consequently, as depicted in Figure 4b, a female gamete will have SNRPN and NDN genes in the inactive state in both homologous chromosomes and the male gamete will contain an inactive UBE3A gene in both chromosomes. A deletion in the 15q11 region in either of the parental gametes creates a diseased condition. As seen in Figure 4c, a deletion in the paternal imprinting center will cause a loss in the production of SNRPN and NDN products due to the presence of the active alleles of these genes only on the deleted region of the paternal chromosome. This situation leads to manifestation of the Prader–Willi syndrome. 5,25 A deletion in the 15q11 region of the maternal chromosome shuts down the production of E3 ubiquitin-protein ligase because the active allele for this gene is found only on the maternal chromosome. The result is Angelman syndrome. 26
The Prader–Willi and Angelman syndromes both occur in approximately one out of 15,000 births and they are associated with developmental, behavioral and mental problems. Nearly all Prader–Willi patients suffer from short stature, hypogonadism, mental retardation, hyperphagia and various learning disabilities. Most Angelman syndrome patients experience ataxia, sleep disorders, seizures, hyperactivity, happy disposition with excessive laughter and motor retardation. 25 Imprinting gone awry has been associated with other serious disorders such as diabetes mellitus, autism, Alzheimer disease, schizophrenia and cancer. Although there is no experimental evidence for the role that genomic imprinting plays in mental illnesses, the fact that 40% of people suffering from Angelman syndrome develop autism and almost all children with Prader–Willi develop psychotic disorders leads scientists to believe that there are links between imprinting anomalies and certain disorders. 8
In the case of cancer, two types of genes are important – tumor suppressor genes and oncogenes – both of which are critical in maintaining the cell cycle. Tumor suppressor genes protect a cell from entering the path to cancer by limiting the number of divisions a cell undergoes. Loss or reduction in the function of tumor suppressor genes can promote cancer. Oncogenes also can contribute to the production of cancer when they are mutated or expressed at high levels (Figure 5). Under normal conditions, two working copies of a tumor suppressor gene help maintain proper functioning of the cell cycle. If a mutation occurs in one of the tumor suppressor genes or if one of the copies is imprinted (Figure 5a), only one functional copy remains in either case, rendering an individual more vulnerable to cancer caused by any additional mutation in the remaining copy. Imprinting plays the same role as a pre-existing mutation. As shown in Figure 5b, an imprint on one copy of an oncogene prevents the cell from being driven uncontrollably through the cell cycle, thereby keeping a check on cancer. A loss of such an imprint will cause acceleration of cell division which, in turn, leads to cancer. 6,27

Effect of loss of an imprint on tumor suppressor genes and oncogenes. (a) The first example shows two working copies resulting in normal cell function. The second and third examples demonstrate how a mutation or an imprint on one of the copies can lead to a state of increased cancer risk. The fourth example demonstrates how the combination of an imprint and mutation can be lethal. (b) The first example shows how an imprint on one copy can maintain a check on over-proliferation of cells, which does not lead to cancer. The second example shows how a loss of the imprint drives the cell into uncontrollable division, leading to cancer
Perhaps, the most intriguing disorder caused by LOI is the Beckwith–Wiedemann syndrome. 15 This syndrome is a maternally transmitted disorder, which leads to fetal and postnatal overgrowth as well as to a proclivity for tumor formation. Manifestation of these conditions is a result of perturbation of a complex network of genes. The Beckwith–Wiedemann syndrome locus is situated at 11p15.5 and includes many imprinted genes. It consists mainly of two independently regulated domains, H19-Igf2 and KCNQ1-KCNQ1OT1 (Figure 6). Functionality of the latter domain is similar to that of Igf2r (Figure 3) wherein the KCNQ1 gene (which has an imprint pattern similar to the Igf2r gene) is maternally expressed and codes for a potassium voltage-gated channel. The KCNQ1OT1 gene, which is imprinted similar to iCGi (Figure 3), is paternally expressed and produces an antisense transcript. This region also contains a maternally expressed gene CDKN1C, which controls growth of the fetus (Figure 6). Each domain contains an imprinting control region (ICR) with a CTCF binding site responsible for its regulation. The ICRs control methylation of CDKN1C, H19, Igf2 and KCNQ1OT1, among other genes involved in normal growth. As can be seen in Figure 6, the paternal allele of ICR1 and the maternal allele of ICR2 are methylated in normal cells and are responsible for the regulation of H19-Igf2 and KCNQ1-KCNQ1OT1 genes, respectively. Abnormal methylation disrupts the regulation of these genes, rendering overgrowth and other distinguishing features of the Beckwith–Wiedemann syndrome.
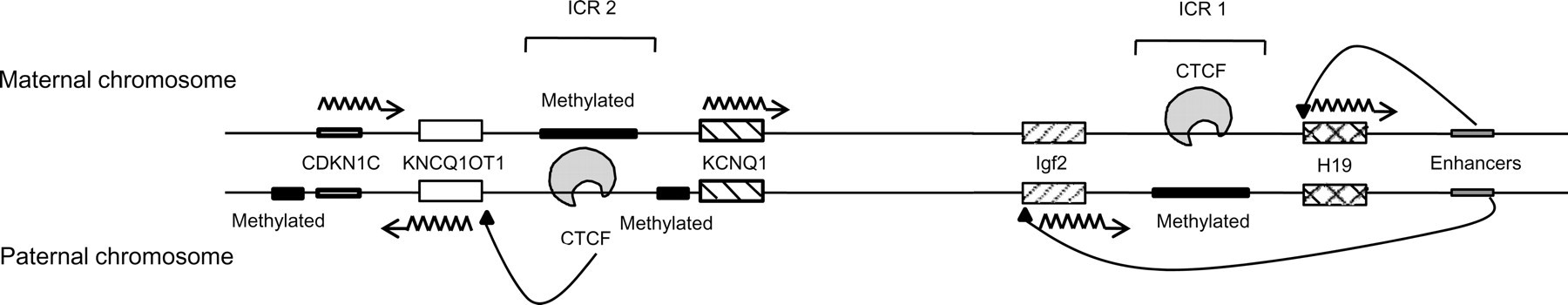
Locus 11p15.5 with ICR1 and ICR2 responsible for the Beckwith–Wiedemann syndrome. ICR1 is situated at the 3′ end of the DNA segment and regulates functioning of the H19-Igf2 cluster. ICR2 is located towards the 5′ end of the DNA segment and is responsible for the regulation of the KCNQ1-KCNQ1OT1 cluster. Both ICRs require CTCF for efficient functioning. The upper half of the diagram represents the pattern of imprinting in the female genome and the lower half represents the imprinting pattern in the male genome. A loss in the function of ICR2 and ICR1 leads to the manifestation of the Beckwith–Wiedemann syndrome and tumorigenesis, respectively
Additional disruptions in the system can cause severe effects as well. About 20% of all clinical cases are the result of uniparental disomy in which progenies inherit both copies of a gene from one parent and none from the other. Uniparental disomy can also result from a trisomic embryo in which some cells lose the extra chromosome, leaving two homologs from one parent. Inheritance of two paternal copies of a region within a chromosome in which there is non-disjunction of the chromosome in sperm and oocytes can lead to over-expression of paternally active genes such as Igf2 and reduced expression of maternally active genes such as H19 and CDKN1C (Figure 6). Uniparental disomy also may cause problems if it removes the active allele of an imprinted gene. Considering such a phenomenon, a mutation would result in LOI at ICR2 or ICR1. Errors in imprinting in the ICR2 generally give rise to the Beckwith–Wiedemann syndrome phenotype whereas abnormalities in the ICR1 lead to the over-expression of certain maternally silenced genes that eventually can cause tumorigenesis. 28
Effect of erroneous reprogramming on mammalian clones
A special area of interest that has attracted a considerable amount of attention is the production of healthy clones by somatic cell nuclear transfer (SCNT). The success rate of this technique is not very high owing to the significantly large number of deaths of cloned embryos during post-implantation development. Embryonic fatality is due to the occurrence of abnormalities such as large offspring syndrome, 29 elevated levels of ammonia and lactate dehydrogenase in the blood, 19 reduced antibody production 30 and reprogramming errors, 31 to name a few. SCNT involves the transfer of a nucleus from an adult somatic cell into an enucleated donor egg, which then divides and is transferred into a surrogate uterus. As mentioned earlier (see the section on Methylated versus imprinted genes), a fertilized egg undergoes secondary reprogramming that involves global demethylation prior to implantation (Figure 7). For embryos resulting from natural fertilization, the extent of methylation in the male gamete is actively reduced within four hours of fertilization in the absence of DNA replication. 13 The female methylation pattern however undergoes passive demethylation after the embryo reaches the two-cell stage. Passive demethylation occurs without any involvement of DNA methyltransferases, and the daughter DNA strands are not methylated upon replication. This process of gradual demethylation continues until the embryo has reached the morula (16-cell) stage (Figure 7a). At this point, cells of the morula differentiate into blastomeres that become the inner cell mass and blastomeres that are flattened on the surface become the trophoblast. The embryo or blastocyst ultimately is implanted in the uterus. Methylation patterns are set in the inner cell mass from this point onward while the trophectoderm, from the trophoblast, remains hypomethylated and progresses toward formation of the placenta 14 (see Figure 7a for the normal flow of events).
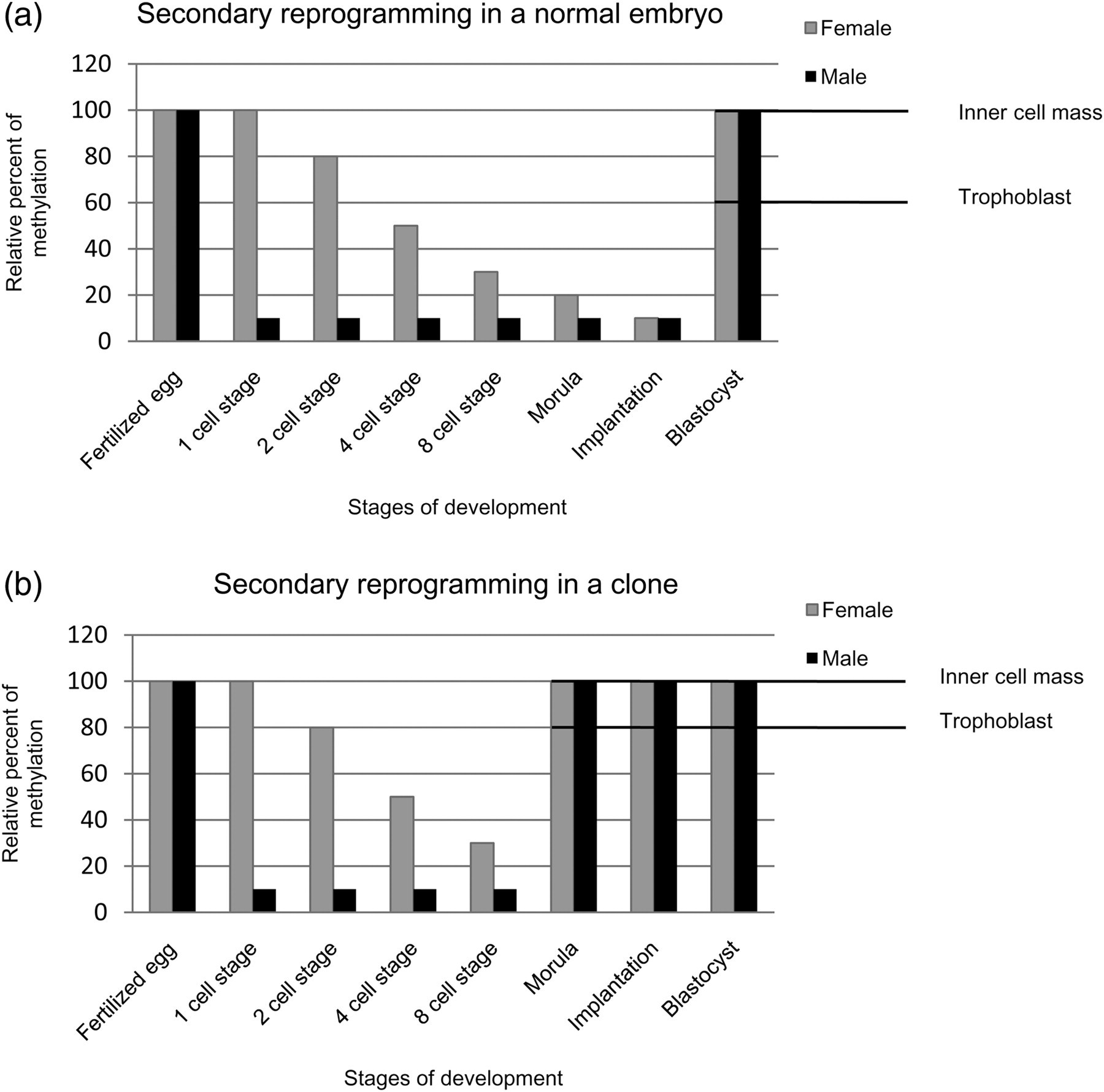
Secondary reprogramming in normal and cloned embryos. The graphs depict the demethylation and remethylation steps that take place in an embryo between fertilization and formation of the blastocyst. The grey bars represent maternal methylation and the black bars represent paternal methylation. The differences in the levels of methylation between the inner cell mass and trophoblast are seen in the blastocyst stage. (a) Reprogramming in an embryo that has undergone natural fertilization in which the paternal genome undergoes immediate active demethylation and the maternal genome undergoes gradual passive demethylation until implantation. Methylation patterns are set post-implantation. (b) Reprogramming in a cloned bovine embryo in which remethylation is initiated prior to implantation in the morula, leading to abnormalities. Obviously, the degree of methylation in the trophoblast is significantly more in a clone at the blastocyst stage than in a normal embryo
Interestingly, cloned embryos initiate demethylation actively but, unlike normal embryos, demethylation after the two-cell stage is minimal if not lacking altogether. 32 Cells of cloned embryos undergo de novo methylation earlier than usual, starting at the four- or eight-cell stages. As a result, the trophoblastic layer of the blastocyst, which eventually must be hypomethylated, does not have adequate time to undergo complete demethylation, leading to an unusual state of hypermethylation. By the time the embryo has reached the 32- or 64-cell stage, there is little difference between the trophectoderm and the inner cell mass with respect to methylation (Figure 7b). Hence, disturbance in the normal timing of events results in potentially grave changes in epigenetic reprogramming and subsequent development of the extra-embryonic lineage, which consequently can be detrimental to the development of the cloned embryo. 29 Therefore, it is safe to say that mammalian cloning as a reproductive tool is impractical until scientists find a way to control the timing of events taking place during fertilization and embryonic development in vitro.
What does the future hold?
Obviously, imprinted genes are vital to gene expression, gene regulation and development. They are present in clusters and function within complex and intricate networks. Imprinting is indispensable to embryonic growth and is considered to be nature's check on inequality between the sexes. A large number of imprinting studies has been conducted and more than 80 imprinted genes have been discovered and catalogued to date. 33 However, a significant amount of information is still needed to fully understand the functionality and the purpose of these genes.
An area of particular interest is the origin of mutations that cause numerous human disorders. The National Institutes of Health (NIH) is focusing on the identification of structural characteristics of DNA at breakpoint sites to provide clues about the origin of chromosomal deletions and non-disjunction as they relate to the manifestation of the Prader–Willi and Angelman syndromes. 34 Many researchers are in quest of more imprinted genes and other genes that are associated with various defects in an attempt to better understand them mechanistically. One approach involves genome-wide screening and knockout studies of individual genes. 35 For example, a research group at the University of Southampton has developed a unique approach for the identification of candidate imprinted genes by studying consistent differences between the sequences of imprinted and control genes. 36 These differences include reduced occurrence of short interspersed transposable elements, members of the Alu family and mammalian-wide interspersed repeats in imprinted genes. 25 Research is also being undertaken to discern the impact of environmental factors on imprinted genes to gain insight into the kind of mutations that lead directly to particular disease symptoms.
In addition to studies that concentrate on single genes or imprinted gene clusters, investigations are underway to decipher and assess epigenetic patterns that span the entire genome – a field of study termed ‘epigenomics’. The main objective of epigenomic analysis is to gain better understanding of those systems involved in the establishment and maintenance of epigenetic blueprints and signatures. Support for this approach is being provided by another NIH venture called the NIH Roadmap Epigenomics Project, 37 which purports that epigenomic regulation of the genome accounts for vulnerability to a disease. This NIH project funds development of technologies and methods for epigenomic analysis and imaging of epigenetic activities. There are also public/private collaborations, one of which is called the Human Epigenome Project. 38 This particular project is spearheaded by the Human Epigenome Consortium, consisting of various members such as the Sanger Institute, Epigenomics AG and the Centre National de Génotypage. The overall aim of the Consortium is to identify, catalog and analyze global methylation patterns across the human genome. It is believed that distinct methylation patterns can serve as biomarkers of a certain tissue type or diseased state and that these markers holds promise for quicker and more accurate diagnosis through early recognition of predisposition to diseases and disorders, which are more often than not associated with epigenetic changes rather than genetic changes. Identification of methylation patterns on a genome-wide basis is being made easier with the help of commercially available DNA methylation kits, specific anti-5′-methylcystosine antibodies and methylated DNA immunoprecipitation reagents.
What lies ahead in genomic imprinting? One perplexing issue is the identification of the trigger(s) that initiate the establishment of imprints. As reviewed herein, a number of studies have revealed the involvement of methylation in maintenance of imprinted patterns from generation to generation but the precise mechanism(s) that prompts sex-specificity of an imprint remains elusive. To approach this issue, knockout studies of genes responsible for differences between the two sexes might identify which genes are responsible for the initiation of an imprint. An example worthy of consideration is the SRY gene, found on the Y chromosome, which is responsible for determining ‘maleness’ of an individual. Knockout of this gene most likely would yield virtually identical male and female genomes. Imprinting patterns in the resulting genomes most likely would lend better understanding of the regulatory mechanisms that influence methylation of imprinted genes. As for the origin of imprinting, comparative analyses of genome imprinting in the mouse and human coupled with additional mammal studies will provide evolutionary evidence not only for the basis of imprinting but also for important evolutionary relationships as well. Comparative studies that concentrate on homologous genes would reduce the number of genes to be screened and examined. And finally, unveiling the intricacies of those mechanisms that regulate imprinting per se and elucidating the role of epigenetics in the manifestation of disease should go a long way toward development of newer and better diagnostic tools and therapeutic procedures.