
Abstract
Niacin (vitamin B3) is required to form nicotinamide adenine dinucleotide (NAD) and nicotinamide adenine dinucleotide phosphate (NADP), which are involved in scores of anabolic and catabolic redox reactions throughout metabolism. It is now understood that NAD+ is also a substrate for several families of ADP-ribosylation reactions, which control processes like DNA repair, replication and transcription, the activity of G-proteins, chromatin structure and intracellular calcium signalling. Poly(ADP-ribose)polymerase-1 (PARP-1) is the most active of the PARP enzymes, and it has been implicated in both prevention and aggravation of disease processes. Inhibition of poly-ADP-ribose formation will tend to cause genomic instability and tumorigenesis in chronic models of DNA damage, but the same inhibition can prevent many acute disease processes, such as stroke, myocardial infarction and septic shock. In models of acute stress, PARP-1 inhibition may protect cellular NAD pools and prevent nuclear factor-κB-dependent inflammatory signalling, while long-term protective roles for PARP-1 include DNA repair and regulation of chromatin structure. Promising new PARP-1 inhibitors may display interactions with dietary niacin status and may have long-term deleterious effects on genomic stability, but may be extremely valuable for the treatment of acute inflammatory conditions.
Keywords
Introduction
There is a rich history in the study of niacin, leading up to our current understanding of nicotinamide adenine dinucleotide (NAD)-dependent reactions and health. Niacin deficiency in humans causes the disease pellagra, which reached epidemic proportions in many regions that adopted corn cultivation following its initial distribution from the Americas in the 1500s. 1 Pellagra resulted from an over-dependence on corn, combined with a loss of the traditional processing techniques required to improve niacin bioavailability. 2 Niacin was identified as the pellagra-preventative factor in the mid-1900s, and many roles of (NAD) and nicotinamide adenine dinucleotide phosphate (NADP) in redox reactions were established subsequently. 1 The unusual signs of pellagra, which include sun-sensitive dermatitis and profound dementia, appeared anomalous until the discovery of various forms of ADP-ribosylation reactions, which involve cleavage of NAD+, leading to the formation of poly-, mono-, cyclic- and O-acetyl-(ADP-ribose) structures. 1 It is now known that these reactions control processes such as DNA repair, chromatin structure, telomere stability, chromosome sorting, G-protein activity and neuronal calcium signalling. 3
Poly(ADP-ribose)polymerase-1 (PARP-1) is the most abundant and active enzyme in this group, and it was the first to be described. From an early stage it was appreciated that PARP-1 was capable of protecting cells from genomic instability and death as well as inducing apoptosis or necrosis. PARP-1 is an 116-kDa nuclear protein that binds to and is activated by DNA strand breaks. There are now five other confirmed PARP proteins, which are expressed in the nucleus, cytosol and vault particles. 4 PARP-2 is also activated by DNA damage, and appears to exhibit some functional redundancy with PARP-1. 5 PARP-3 also appears to participate in DNA damage responses. 6
PARP-1 the Good
Within a short time after the discovery of PARP-1, it became apparent that PARP-1 activation could damage cells, 7 but most of the early research on this enzyme focused on its beneficial effects in metabolism. 8,9 PARP-1 has two zinc-fingers that bind to DNA strand breaks, causing catalytic activation, which leads to NAD+ consumption and poly(ADP-ribose) formation. PARP-1 functions as a dimer, and much of the polymer is synthesized on the neighboring PARP-1 molecule (automodification), or on PARP-2, which will heterodimerize with PARP-1. 10 PARP-1 participates in the formation of a protein complex at the site of DNA damage, in part through direct protein:protein interactions, but also through the high-affinity binding of proteins to the poly(ADP-ribose) on automodified PARP-1. One binding partner is DNA-dependent protein kinase (DNA-PK), 11 which also binds to and is activated by DNA strand breaks. The formation of poly(ADP-ribose) and phosphorylation of protein targets by this activated complex initiates a series of events designed to control processes including cell cycle arrest, DNA repair and, if necessary, apoptosis. Part of this signalling involves regulation of p53, which is a substrate for covalent poly(ADP-ribosyl)ation, and also has a high-affinity binding site for non-covalent attachment to poly(ADP-ribose). 11 p53 is an important regulator of cell cycle arrest and apoptosis (Figure 1).
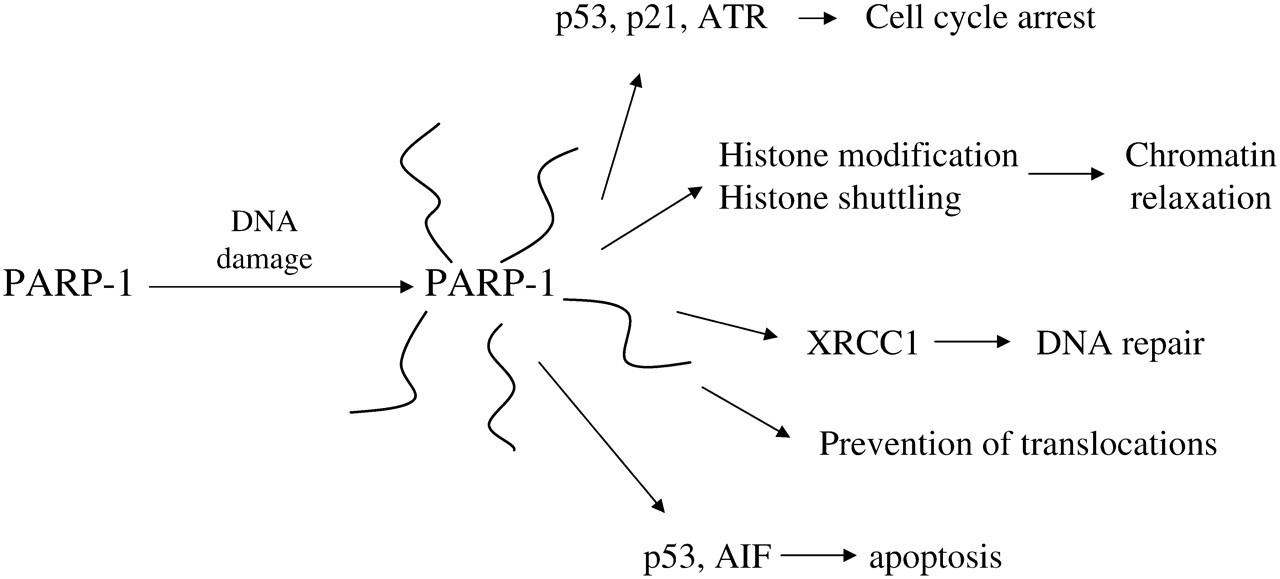
Poly(ADP-ribose)polymerase-1 (PARP-1), ‘The Good’. AIF, apoptosis-inducing factor; ATR, ATM and Rad3 related
Another main role for PARP-1 at the site of DNA damage is to induce a localized relaxation of chromatin structure, allowing DNA repair complexes to form and accomplish repair. This is also accomplished through a combination of covalent and non-covalent interactions. Activated PARP-1 attaches poly(ADP-ribose) directly to histones, and the anionic nature of the polymer causes the histones to be repelled from binding to DNA, disrupting the tightly condensed nucleosomal structure. 12 In addition, histones have high-affinity polymer binding sites that draw them out of nearby chromatin to bind to the polymer cloud around automodified PARP-1 molecules attached to strand breaks. 13 As the damage is repaired, a glycohydrolase enzyme will degrade the polymer on PARP-1 and histones, and the condensed nucleosomal structure will be restored. During the repair process, the anionic cloud of polymer around a site of damage is also thought to repel other free ends of DNA, preventing dangerous translocation events.
As mentioned above, a series of regulatory events radiate out from sites of DNA damage, and if the number of damaged sites increases, these signalling events become stronger and start to alter gene expression and cellular function. In many cases these changes will be beneficial, but they may also be harmful (see below). Cell cycle arrest may be controlled through covalent and non-covalent polymer binding to p53, which is a key regulator of short- and long-term cellular responses to DNA damage. 11 PARP-1 appears to interact in similar ways with other proteins like the checkpoint kinase ATR (ATM and Rad3 related), 14 p21, nuclear factor-κB (NF-κB), inducible nitric oxide synthase (iNOS) and DNA-PK, 11 suggesting multiple forms of regulation of cell cycle arrest. During cell cycle arrest, DNA repair reactions will work to clear the damage. PARP-1 has been shown to contribute to several different DNA repair pathways. For example, in short-patch base excision repair, automodified PARP-1 recruits XRCC1, 11 which recruits the appropriate DNA polymerase and ligase enzymes for effective repair, many of which also have polymer binding sites. If the damage levels are more significant, p53 signalling and/or regulation of apoptosis-inducing factor (AIF) 15 may cause the cell to undergo apoptosis, which is beneficial on a small scale in that it prevents the survival of cancer-prone cells. In the longer term, especially with chronic DNA damage, PARP-1 will control gene expression patterns through transcription factors such as NF-κB, p53, activator protein 1 (AP-1) and AP-2. 16 In Drosophila, PARP activity is required for the heat-shock-induced upregulation of transcription at puff loci. 17 Altered gene expression may lead to cellular resistance to further stresses, or may lead to some negative effects as discussed further down in this text.
Impact of inhibition of ‘PARP-1 the Good’
It is important to define the different modes of PARP-1 inhibition before discussing their functional impact. There are three main forms of inhibition of PARP-1 function. The removal of the PARP-1 protein from the system occurs in knockout animal and cell culture models or through the use of knockdown technologies like antisense or small interfering RNA. This leads to a loss of PARP-1:DNA interactions and protein:protein interactions in addition to the loss of catalytic activity. In this respect, it may present the most complete picture of PARP-1 essentiality.
Conversely, competitive inhibitors abolish PARP-1 catalyzed polymer synthesis, but leave PARP-1 protein in the system. PARP-1 can still participate in DNA binding and protein:protein interactions. While it seems possible that this partial functionality might be a good thing, it turns out that it is generally more disruptive to the cell than complete removal of PARP-1 protein. This appears to be due to the dominant-negative effect of PARP-1 binding to strand breaks, lacking the ability for automodification required to dissociate from DNA at the appropriate stage of repair or transcription. These effects are also mimicked by the forced expression of non-catalytic DNA-binding domains of PARP-1, which compete for DNA binding, but do not undergo automodification. 18 In these models, cellular NAD pools are protected by PARP-1 inhibition, which may be one of the mechanisms of action.
The final form of inhibition is by depletion of the substrate, NAD+. When severe, this may be similar to competitive inhibition, in that it will create catalytically inactive PARP-1, which binds to DNA strand breaks inhibiting repair, replication and/or transcription processes. However, in this model NAD+ levels are decreased, with potential metabolic disruptions in other NAD+ utilizing reactions, including redox metabolism, apoptotic signalling, sirtuin function and all of the other poly-, mono- and cyclic ADP-ribosylation reactions.
Some of the earliest studies on PARP-1 used catalytic inhibitors and niacin depletion to disrupt function, 8,19 both of which delayed DNA repair and sensitized cells to genotoxicity. Many forms of genomic instability were observed with PARP inhibition, including sister chromatid exchanges, 20 supporting the idea that poly(ADP-ribose) prevents translocation events. Thus, it was surprising when the first PARP-1-null mouse was generated and it grew, reproduced and aged fairly normally. 21 With the development and characterization of two additional knockout models, it became clear that these mice suffered from basal genomic instability, and were sensitive to stress by genotoxic chemicals or radiation. 22 DNA repair was delayed, and chromosomal breaks, aberrations and sister chromatid exchanges were all increased in frequency. 22 Puzzlement over the viability of PARP-1-null mice led to the discovery of residual poly(ADP-ribose) in these tissues, followed by characterization of other members of the PARP superfamily of genes, 4 and eventually the finding that PARP-1 and -2 share functionality 5 and that double knockouts are lethal. 23 Thus, it became accepted that complete removal of PARP-1 expression did cause genomic instability, but it also led to the surprising finding that PARP-1-null mice were protected from a variety of models of tissue injury (more below).
Niacin deficiency has been used to deplete NAD+ in whole animals as well as cultured cells. While some tissues, like liver 24 and lung, 25 have high initial levels of NAD+ and are not sensitized to genotoxicity by niacin deficiency, others, like skin 26 and bone marrow, 27 are very sensitive. Niacin deficiency decreased skin NAD+ and increased UV-induced skin cancer development. Niacin deficiency dramatically decreased bone marrow poly(ADP-ribose) levels, 27 delayed base excision repair, 28 increased chromosomal aberrations, impaired cell cycle arrest and apoptosis, 29 and increased the long-term development of leukemias. 30 As mentioned earlier, these models have mechanistic distinctions; PARP inhibitors create catalytically inactive protein and preserve NAD pools, PARP-1-null models remove the protein completely and also preserve NAD pools, niacin deficiency depletes NAD pools and targets a wide range of ADP-ribosylation enzymes and redox functions, potentially creating dominant-negative forms of PARP-1, PARP-2 and other PARP enzymes that undergo automodification.
Skin health is another area that shows clear benefits with enhanced PARP-1 function. Sun sensitivity is an obvious risk with niacin deficiency, as seen in patients with pellagra, 1 and demonstrated in cancer susceptibility in mice. 31 Supraphysiological intakes of niacin have been shown to further decrease ultraviolet radiation-induced skin cancers in mice through improved immune function. 32 Niacin supplementation of keratinocyte cultures or topical treatment of skin with lipid-soluble niacin esters have been shown to increase PARP-1 and sirtuin activities, and improve the skin differentiation process, leading to thickening of the stratum corneum and epidermis. 33 These changes improve the barrier functions of the skin layer and very likely decrease the progression towards skin cancer over time.
PARP-1 the Bad
As the PARP-1 field developed, more examples emerged showing that PARP-1 function caused cell death or tissue injury. At the cellular level this was originally noted to occur when PARP-1 was activated to such an extent that cellular NAD+ and ATP levels were lethally depleted. 7 Later, it was noted that PARP-1 is cleaved by caspases during apoptosis, and that this process played an active role in some forms of apoptosis. 34 In some models, the degree of PARP-1 activation and resulting NAD depletion determined whether cells underwent apoptotic or necrotic forms of cell death, 35 which can play an important role in the development of tissue pathologies. More recent work has shown that PARP-1 activation can lead to caspase-independent apoptosis, through the translocation of AIF, from the mitochondria to the nucleus. 15 Thus, there are a number of different forms of cell death that require PARP-1 to some degree, and these forms of cell death can be inhibited by removing PARP-1 from the system, or inhibiting its catalytic activity.
These cellular findings were soon extended to the tissue level in PARP-1 knockout mouse models. One early finding, which agreed with cell culture models, involved protection from streptozotocin-induced diabetes. Streptozotocin causes DNA damage and NAD+ depletion in pancreatic β-cells, and the development of diabetes is prevented with the use of PARP-1 inhibitors, and is also inhibited in PARP-1-null mice. 22
Tissue injury, in whole animal models, is generally more complex than the death of specific populations of cells. In most forms of tissue injury, an initial insult is followed by waves of signalling leading to an inflammatory response that significantly worsens the tissue damage. There are many examples of this, including ischemia/reperfusion conditions like stroke and myocardial infarction, respiratory distress syndromes and septic shock. PARP-1 activity is now thought to play a significant role in the development of these secondary phases of inflammatory injury, through the control of potent signalling cascades and changes in gene expression.
Many of these disease processes are caused, in whole or in part, by transient interruptions in blood flow and oxygenation (ischemia/reperfusion). Examples include myocardial infarction, stroke, circulatory shock, pulmonary embolism, thrombophlebitis, vascular surgery and organ transplantation, among others. While reperfusion is essential to survival, it induces a wave of oxidant injury due to the formation of reactive oxygen and nitrogen species, including hydroxyl radical and peroxy nitrite. This creates the first wave of cellular injury and PARP-1 activation. The extensive synthesis of poly(ADP-ribose) will deplete NAD+ and tend to induce cell death through both apoptosis and necrosis, as described in a previous paragraph. In surviving cells, important changes in signalling will be initiated. NF-κB is a redox-sensitive transcription factor that is activated by oxidant stress, leading to increased expression of inflammatory cytokines (tumor necrosis factor-α [TNF-α], interleukin-6), adhesion molecules (inter-cellular adhesion molecule) and other proinflammatory products (iNOS). PARP-1 catalytic activity is required for maximal transcription activation by NF-κB. The mechanisms involved are not clear, but PARP-1 may be part of the complex of proteins that forms with NF-κB on the promoter of target genes. 36 The formation of TNF-α can lead to significant amplification of the inflammatory response, as it causes further activation of the NF-κB pathway, attracts and activates phagocytic cells and induces circulatory shock. PARP-1 was also found to increase gene expression by other proinflammatory transcription factors, including AP-1 and hypoxia-inducible factors -α. In the final step of tissue injury, cell death in response to inflammation-induced oxidant stress may also be due to excessive activation of PARP-1, leading to necrosis or apoptosis, as discussed in the previous section. It was also found that PARP-1-null mice had low inducibility of a wide variety of NF-κB-dependent genes, 36 and that these mice were resistant in a wide range of ischemia–reperfusion and inflammatory disease models (Figure 2). 22
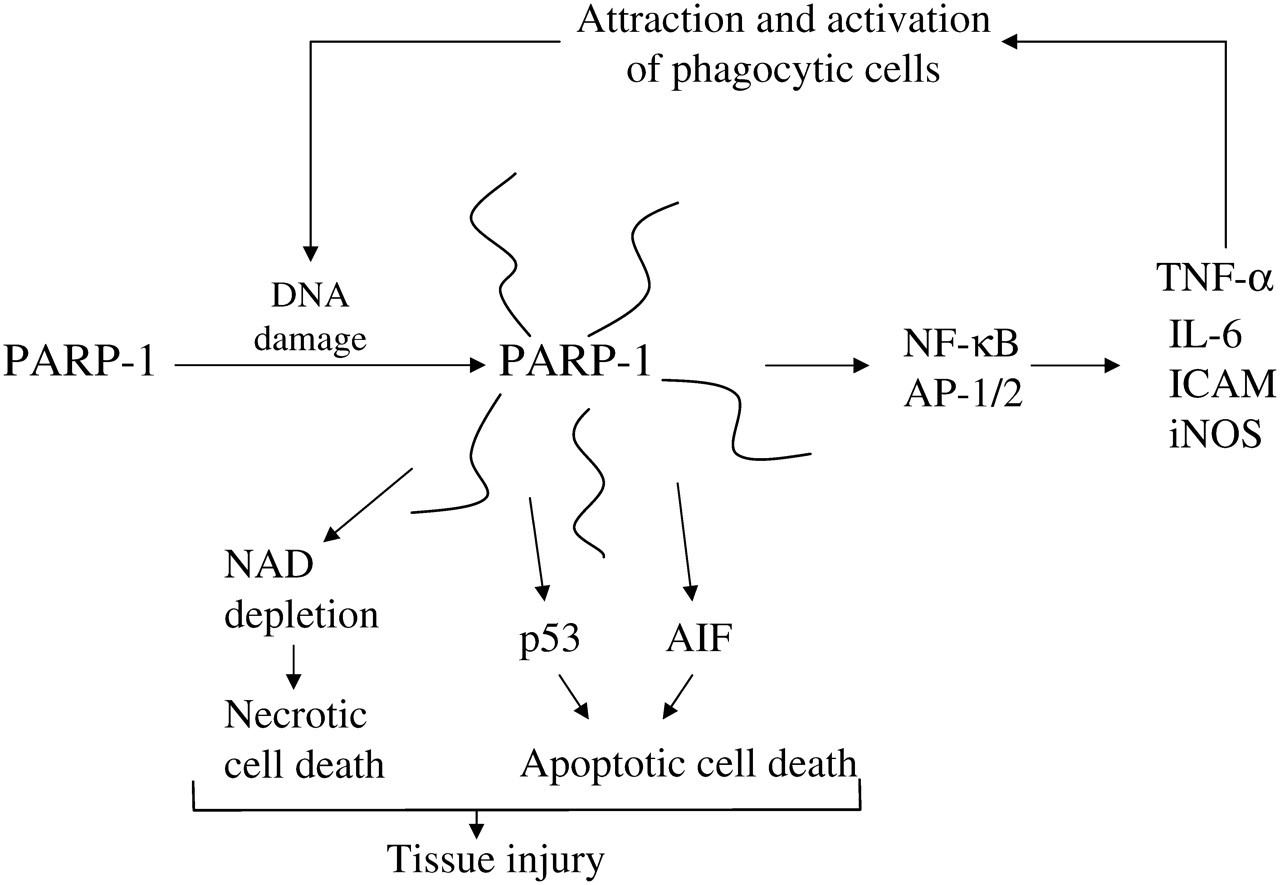
Poly(ADP-ribose)polymerase-1 (PARP-1), ‘The Bad’. TNF-α, tumor necrosis factor-α; NF-κB, nuclear factor-κB; AIF, apoptosis-inducing factor; ICAM, inter-cellular adhesion molecule; iNOS, inducible nitric oxide synthase
PARP-1 inhibitors were soon investigated for their ability to interrupt these destructive signalling cycles, and were found to produce dramatic results. The expansion of this field resulted from the development of new PARP-1 inhibitors, with improved water solubility, specificity and potency. 37,38 Compressing a large body of literature into a short space, PARP-1 inhibitors have been effective in decreasing NF-κB signalling 36 and cytokine production. 39 In various animal models, inhibitors have decreased the severity of myocardial ischemia/reperfusion injury, other acute forms of heart failure and cardiomyopathies, and ischemia/reperfusion models in other tissues, including different forms of brain injury and stroke. 37,38 PARP-1 inhibitors have also decreased tissue injury in response to circulatory shock, including models of septic and hemorrhagic shock. 37
While most of these are relatively acute injuries initiated by a significant oxidant stress, PARP-1 inhibitors have also proved effective in chronic disease models with less severe oxidant injury, including cardiovascular aging/atherosclerosis and diabetic cardiovascular complications. 37,38 This is logical, given the established roles for NF-κB signalling in the progression of endothelial dysfunction during aging, 40 but somewhat surprising given the long-term, mild nature of the injury. PARP-1 was also shown to play a deleterious role in the development of autoimmune conditions, including glomerulonephritis 41 and rheumatoid arthritis. 42
PARP-1 through natural history
It seems odd that PARP-1 is conserved across animal species and expressed constitutively at high levels, given that mice are viable when it is deleted, animals can be treated beneficially with potent inhibitors for extended periods, and that PARP-1 activation causes mortality from many acute conditions that would appear to decrease fitness in a competitive environment. However, there are not any currently known examples of natural familial deletion of PARP-1 function, suggesting that it is beneficial enough that complete loss is not competitive in the long term. It should be remembered that even small changes in fitness, undetectable in a laboratory animal environment, could prevent any loss of function in a natural environment. This could include small changes in immunity or fertility or changes in learning or social behaviors that are not required for survival in laboratory colonies.
There have been some interesting findings on the association of polymorphisms in the human PARP-1 gene with the risk of various disease processes. A common variation in the PARP-1 gene is a valine to alanine shift in position 762, which has an allelic frequency of 0.2, 0.4 and 0.6 in Caucasians, Asians and African-Americans, respectively. 43 The polymorphism is thought to decrease PARP-1 activity by about 40%, depending on substrate conditions. 44 One might hypothesize that this would decrease the incidence of autoimmune and inflammatory conditions, but increase the risk of some cancers due to problems in DNA repair. Increased risk was observed for some cancers, including gastric and lung. 43 Also fitting the hypothesis, this polymorphism was associated with decreased risk of asthma in a Turkish population, 45 but was associated with an increased risk of diabetic polyneuropathy 46 and glomerulonephritis 47 in Russian populations. As additional PARP-1 polymorphisms are identified and functionally characterized, this field should generate some interesting findings.
In addition to genetic variation, there are a number of environmental factors that can modulate PARP-1 function, and these may have changed over time, potentially leading to more harmful outcomes from PARP-1 activation. In the last several thousand years, there have been significant changes in dietary fat type and quantity, caloric intake, physical activity and body composition. The ratio of omega-6 to omega-3 fatty acids has changed from an estimated 1:1 during historical times to 15:1 or higher in modern westernized countries. 48 Greater dietary fat content and caloric intake have combined with lower physical activity to cause dramatic increases in obesity, metabolic syndrome and type 2 diabetes. 49 Together, these changes lead to increased formation of proinflammatory prostaglandins and leukotrienes, cytokines and reactive oxygen species. 48,49 It is likely that the negative impact of PARP-1 function has been exaggerated by this modern metabolic pattern, given the similarity in the proinflammatory signalling pathways involved.
At the same time, there has been a decreased intake of natural plant products, leading to decreased exposure to non-nutrient phytochemicals, such as flavonoids. Different classes of these polyphenolic compounds are found in fruits, vegetables, wines and teas. Some individual flavonoids (quercitin, tricetin, fisetin) have been found to inhibit the polymer-synthesizing activity of PARP-1 and to inhibit the resulting induction of inflammatory gene expression in cultured cells. 50 In a mouse model, fisetin was shown to reduce lipopolysaccharide-induced lung inflammation dramatically. 51 Flavonoids have been used in traditional medicine as anti-inflammatory agents, and it appears that their efficacy may be due in part to modulation of the PARP-1/NF-κB/TNF-α signalling axis.
Vitamin D status is also thought to have changed over time in developed countries due to a decrease in sun exposure with modern work and lifestyles. 52 Many recent studies show significant and widespread health benefits with vitamin D supplementation, including decreased cardiovascular disease, diabetes and cancer incidence, decreased severity of autoimmune conditions and improved bone and muscle strength and nerve function. 52 Many of these conditions also benefit from inhibition of PARP-1 activity, and a recent paper has suggested a mechanistic connection with the finding that 1,25-dihydroxy-vitamin D (calcitriol) is an effective PARP-1 inhibitor in cultured cells. 53 If this interaction occurs in vivo, it could explain the link between low vitamin D status and increased incidence of conditions like diabetes, lupus, rheumatoid arthritis, multiple sclerosis and psoriasis, given that PARP-1 inhibitors have been found to decrease the development of autoimmune conditions in experimental models. 41,42 Inhibition of PARP-1 by calcitriol could also decrease the inflammatory component of chronic diseases like cardiovascular disease, cancer and diabetes.
To develop a more complete understanding of the function of PARP-1 in normal development and healthy tissue function, we will have to learn more about the activation of this enzyme in the absence of DNA strand breaks. There are a number of interesting experimental models in this poorly appreciated area. PARP-1 activation is required for the neurite outgrowth and synaptic plasticity required for the development of long-term memory. 54 In related work, PARP-1 activation in hippocampal neurons was induced by cholinergic signals and mediated by inositol-1,4,5-trisphosphate and calcium release. 55 PARP-1 is known to play multiple roles in transcriptional activation, through non-catalytic and catalytic mechanisms. The formation of puff-loci around actively transcribed Drosophila genes is dependent on poly(ADP-ribosyl)ation, 17 and it is now known that PARP-1 and histone H1 compete for specific binding sites in the promoters of actively transcribed genes. 56 PARP-1 binds to and is catalytically activated by hairpin or cruciform structures in DNA, in the absence of strand breaks. 57 These observations likely represent a small proportion of what we will eventually learn about the non-strand break regulation of PARP-1 and the associated roles in health and disease.
Current clinical use and future potential of PARP-1 inhibitors
A recent review listed eight PARP-1 inhibitors in phase 1 and 2 clinical trials 58 targeting advanced tumor chemotherapy, with one study on prevention of cardiovascular surgery complications. 37 There are several other medical conditions where the evidence from animal models is very strong and clinical trials will likely appear in the near future. These areas are discussed below with consideration of potential benefits and risks of PARP-1 inhibition:
(i) Critical care medicine: Experimental models of circulatory shock and sepsis represent the constant challenges faced by patients and medical practitioners in intensive care units. Many different injuries, such as burns and broken bones, can lead to a degenerative series of events, involving excessive inflammatory signalling, neutrophil margination and activation, and tissue hypoxia leading to circulatory shock and multiple organ failure. At the same time, immune barriers falter and sepsis is initiated. Anti-inflammatory drugs are immune suppressive and may cause a worsening of sepsis. This series of events has a severe prognosis, with up to 60% mortality, 59 and it often occurs in otherwise healthy patients with an expectation of full recovery from the initial injury. If PARP-1 inhibitors effectively block the inflammatory cascade without causing severe immune suppression, this patient population would benefit tremendously, and the animal models are very supportive. 37,38 The duration of treatment would be brief, and the risks of PARP-1 inhibition would likely be minimal.
(ii) Ischemia/reperfusion events: Many patients who experience a stroke or myocardial infarction receive initial treatment in time to prevent some of the downstream events that exaggerate the tissue injury. Reperfusion injury following transplantation or cardiovascular surgeries can be treated before and after the ischemic event, and all of these conditions could benefit from effective use of PARP-1 inhibitors. The treatment duration would be brief, and the risk/reward balance should be favorable.
(iii) Chronic inflammatory conditions: Using PARP-1 inhibitors to minimize the complications of chronic conditions, like type 2 diabetes, would require extended periods of drug use. It is interesting that animal models have shown that PARP-1 inhibitors can be quite effective in this type of regimen, 37,38 without an obvious increase in cancer risk, although the experimental models were not necessarily designed for that end point. It may turn out that certain doses of PARP-1 inhibitors may allow a low level of polymer synthesis that is compatible with DNA repair processes, while still protecting NAD pools and/or inhibiting inflammatory signalling. Interestingly, the protection provided by PARP-1 inhibitors in this model may be similar to what would be expected from a combination of improved vitamin D status, increased dietary omega-3 fatty acid intake and increased intake of fruit and vegetable phytochemicals (as discussed earlier).
(iv) Autoimmune conditions: PARP-1 inhibitors have proved to be effective in preventing animal models of various autoimmune diseases, including type 1 diabetes, 22,60 glomerulonephritis 41 and rheumatoid arthritis. 42 Streptozotocin-induced DNA damage is sometimes used to model the autoimmune destruction of pancreatic β-cells. PARP-1 inhibitors prevent diabetes in this rather severe model, but also illustrate the potential risks; 100% of surviving animals develop β-cells tumors due to the survival of damaged cells combined with genomic instability associated with PARP-1 inhibition. 61 More physiological animal models of type 1 diabetes paint a more optimistic picture, and suggest that PARP-1 inhibition prevents the initial immune attack on the pancreatic β-cells, 60 without the risk of initiating pancreatic tumors. The difficulty in transferring these models into human clinical practice is that autoimmune conditions need to be predicted to be prevented. This has actually been attempted in some large human clinical trials on type 1 diabetes in which high-risk family members were screened for the early appearance of β-cell antibodies and placed on high-dose nicotinamide treatment. 62 These trials failed to demonstrate a benefit of nicotinamide, but nicotinamide is also not a very effective PARP-1 inhibitor. 63 As expected, there is a large onus for safety in the design of disease prevention trials, especially those conducted in children. It may be some time before clinical trials are launched to study the efficacy of newer PARP-1 inhibitors in the prevention of autoimmune conditions. It is possible that supplementation with flavonoids and vitamin D would provide safe and effective prevention of some autoimmune conditions through modulation of PARP-1 function.
(v) Cancer therapy: Most of the current clinical trials of PARP-1 inhibitors involve treatment of advanced cancers. 37 The mechanisms involved in cancer therapy differ fundamentally from the other treatment models described in this paper. The approach is most effectively targeted towards tumor cells with BRCA1 and BRCA2 deficiencies (breast, ovarian), since they are deficient in double-strand break repair capacity. Inhibition of PARP-1 blocks the repair of spontaneous single-strand lesions, leading to persistent single-strand breaks, which advance to double-strand breaks through mechanisms like stalled replication forks. These double-strand breaks become lethal due to the BRCA deficiencies. There is exciting potential for monotherapy with PARP-1 inhibitors to combat cancers with this genotype, as shown by positive results in several recent clinical trials. 58
Given the severity of the disease, significant risks are accepted in most forms of chemotherapy. However, rapidly dividing bone marrow cells are very sensitive to most chemotherapy drugs. Clinical trials treating non-BRCA cancers with combinations of PARP-1 inhibitors and genotoxic drugs have found persistent dose-limiting myelosuppression. 58 Animal models have shown that low niacin status and impaired PARP-1 functionality cause an increased incidence of acute myelosuppression and the long-term development of leukemias. 64 The risk of secondary cancers may need to be considered if there are other effective treatments for the same cancer, especially in younger patient populations. Nicotinamide has also been used in chemotherapy regimens with the goal of inhibiting PARP-1 activity, but it is not very effective, especially in whole animal models, where it is readily cleared and converted to NAD+. 63 In most cases, nicotinamide treatment probably increases PARP-1 activity, but it also prevents lethal levels of NAD+ depletion, 63 which may be the main benefit of nicotinamide supplementation in most in vivo models. It would be of interest to fully characterize the mechanisms of action of nicotinamide therapy in acute inflammatory models, given that this treatment will likely prevent NAD+ depletion, but enhance poly(ADP-ribose) synthesis, providing insight into the relative importance of these two actions of PARP-1 ‘The Bad’.
Summary
PARP-1 function is critical for the long-term maintenance of genomic stability through the regulation of chromatin structure, cell cycle arrest, DNA repair and apoptosis. However, under certain conditions, PARP-1 activity can amplify tissue damage by participating directly in necrotic or apoptotic cell death, or through enhanced inflammatory signalling and secondary damage. Potent new PARP-1 inhibitors may play significant roles in the treatment or prevention of a variety of diseases in the near future. Lifestyle and dietary changes may also prove effective in modulating PARP-1 function to improve health.
Footnotes
ACKNOWLEDGEMENTS
This work was supported by funds from the Natural Sciences and Engineering Research Council of Canada