
Abstract
Microfluidic devices are well-suited for the study of metabolism and paracrine and autocrine signaling because they allow steady or intermittent perfusion of biological cells at cell densities that approach those in living tissue. They also enable the study of small populations of rare cells. However, it can be difficult to introduce the cells into a microfluidic device to achieve and control such densities without damaging or clumping the cells. We describe simple procedures that address the problem of efficient introduction of cells and cell culture media into microfluidic devices using small bore polyetheretherketone (PEEK) tubing and Hamilton gastight syringes. Suspension or adherent cells grown in cell culture flasks are centrifuged and extracted directly from the centrifuge pellet into the end of the PEEK tubing by aspiration. The tube end is then coupled to prepunched channels in the polydimethylsiloxane microfluidic device by friction fitting. Controlled depression of the syringe plunger expels the cells into the microfluidic device only seconds following aspiration. The gastight syringes and PEEK tubing with PEEK fittings provide a non-compliant source of pressure and suction with a rapid response time that is well suited for short-term intramicrofluidic cellular studies. The benefits of this method are its simplicity, modest expense, the short preparation time required for loading appropriate numbers of cells and the applicability of the technique to small quantities of rare or expensive cells. This should in turn lead to new applications of microfluidic devices to biology and medicine.
Introduction
Microfluidic or BioMicroElectroMechanical Systems (BioMEMS) are becoming established as precision tools for studying biological cells in highly controlled chemical environments. They enable biologists and researchers unprecedented ability to manipulate the microenvironment of individual living cells over time, while simultaneously observing and recording their responses via powerful analytical methods, including optical, electrochemical and mass spectrometry. An often cited obstacle to widespread use or commercialization of microfluidic BioMEMS devices is the ‘world-to-chip’ 1 or ‘macro-to-micro’ 2 problem of how to introduce samples from milliliter-sized volumes into the micro- and nanoliter volumes of microfluidic devices. This problem is characterized by low Reynold's number perfusion within a high surface tension device that is non-compliant, of very small volume, and resistant to flow.
The world-to-chip problem is compounded in the case of BioMEMS studies that are directed towards examining dynamic extracellular cell-to-cell (paracrine) signaling and extracellular cell-to-self (autocrine) signaling, 3,4 or nutrient consumption and metabolite production, all of which may benefit from having cells constrained in small extracellular volumes so that the concentrations of signals and metabolites are not diluted by large volumes of culture media and media can be changed rapidly for all cells. 5 In conventional cell culture, the volume occupied by the cells is on the order of one-thousandth of that of the culture media in which the cells are grown. In stark contrast, the cells in tissue may occupy 75% or more of the tissue volume, with the balance being the extracellular space that contains matrix, interstitial fluid and vascular capillaries. The challenge in BioMEMS devices for measurements of metabolism and extracellular signaling dynamics is not only to maintain the cells at near-tissue densities within an actively perfused device, but also to deliver the cells efficiently to the device at the beginning of the experiment. If these cells are obtained from either conventional suspension culture of non-adherent cells or dissociation and re-suspension of adherent ones, the cells will be at low density in the supporting media. Were cells delivered to the device along with this media, it would be necessary to incorporate a filtering or separation step to increase the cell density to tissue-like levels – a thousand-fold increase in cell concentration and a possible increase in device complexity.
There are additional technical problems with the world-to-chip interface, particularly in applications that require external, dynamic control of pressures and flows within the microfluidic device. For microfluidic circuits, the surface tensions, compliance and resistance to flow must be considered for high fidelity control of the fluid flow within the device. This problem is similar to the requirement for input impedance matching for maximum power transfer in the design of electronic equipment. 6 A driving circuit having lower or higher impedance than a load circuit will result in less than optimal power transfer. Input impedance matching of test circuitry is required for accurate control and measurement of electronic circuits. Several good solutions to this problem in microfluidics have been put forth, but most either require specialized microfabrication or are limited to specific assays and are inappropriate for cellular studies. Bings et al. 7 have connected fused silica capillary tubes to glass microfluidic devices with microdrill bits in a method similar to our approach but that is not useful for polydimethylsiloxane (PDMS) devices. Chen et al. 8 have encased the entire elastomeric chip in an epoxy resin with interconnects, a method useful for high-pressure connections, but not appropriate for low-pressure applications such as cell biology. Gray et al. 9,10 demonstrated deep reactive ion etching of mechanical and fluidic interconnections at a high spatial density, and Puntambekar and Ahn and others 1,2,11–17 have reported various types of micromachined connections that can withstand high pressure or allow high densities of connections or both, but are rather elaborate, time-consuming and expensive to implement. A recent paper by Snakenborg et al. 18 reports the performance of a patented tubing compression technique in response to varying input pressures, and briefly reviews various permanent and non-permanent connection solutions. Seo and Lean 19 and Mohanty and Beebe 20 describe techniques similar to that presented here for connecting tubing and syringes to punched holes in PDMS, but do not present results of cell studies.
We have addressed the need for a high cellular volume fraction inside the device and efficient transfer of a compact population of cells with an inexpensive and simple method for accessing, loading and controlling micro- and nanofluidic devices for short- to long-term cellular studies. We analyze the benefits of the method in light of important experimental parameters such as cell density, sample size, flow control, flow switching and macro-to-micro transit times.
Cell density
The in vitro study of signaling and metabolism of cells requires long-term containment and nourishment of primary and/or immortalized cell lines. 11 The cells under study are usually kept in environmentally controlled incubators at bulk concentrations of approximately 1 × 106 cells/mL (1 cell/nL) or less in container sizes of 5–50 mL. For short duration experiments lasting minutes to hours, smaller volumes may be extracted from the bulk containers with pipettors into vials of approximately 1 mL. At these concentrations, a cell occupies one out of every 1000 possible positions in a suspension of cells with the balance being pure media, assuming a spherical cell with 5 μm radius and approximately 1 pL volume. Since adherent cells are often separated from their substrate and released into solution by an enzymatic digestion (i.e. trypsin) for transport or splitting, they may be assumed to exhibit roughly the same volume distribution. As a comparison, the packed cell volume (PCV) of whole blood in humans is typically 42–45%, with a cell occupying one out of every two possible positions.
Microfluidic devices for cellular studies are essentially two-dimensional flow chambers, often with vessel heights on the order of one cell diameter. The sparse cellular volume fraction of macrocell culture preparations described above leads to long loading times of microfluidic devices since every cell is not closely packed with neighboring cells. The cell–cell separation within the device is determined by the volume fraction of cells in the loaded media. A typical 650 × 870 μm2 field of view (FOV) from a wide field microscope provided by a ×20 objective will contain only 14 cells when loaded with media containing suspended cells from a macroculture vial. Integrated microfluidic filters or other kinetic separation techniques may be used to help retain or concentrate the cells that arrive early until later cells appear, but this is accomplished at the expense of time and is not always effective, since cells are deformable and tend to squeeze through small gaps in the filters and are subject to mechanical damage. In contrast to typical laboratory cell culture, whole blood virtually fills the FOV of the microscope with cells owing to its higher PCV. Many of the most simple microfluidic cell traps passively trap and retain cells by taking advantage of flow through the device. In our experience, trapping efficiency is greatly increased if the cells can be introduced at higher cellular volume fractions due to collisions between cells and their subsequent exclusion from the primary flow streams between the traps.
It is not uncommon for centrifuge pellets during cell passaging to contain 5 × 105–1 × 106 cells. We routinely load multitrap nanophysiometer (MTNP) devices from cell pellets containing 1 × 104–5 × 104 cells, which minimizes the volume of cell culture media required for an experiment. The starting cell number might be reduced further by sampling and centrifugation of culture media with capillary tubes instead of microcentrifuge tubes, perhaps to as little as 1 × 103 or 5 × 103 cells. The primary limitation one encounters as cell numbers grow very small is the ability to visualize the centrifuge pellet with the microscope.
Sample size
An important advantage of microfluidic devices for cellular studies is the ability to conduct experiments while consuming very little reagent and/or sample volumes. Centrifugation of a cell solution concentrates the cells in an accessible pellet at the bottom of the centrifuge tube. In the case of a multicomponent solution such as whole blood, centrifugation or centrifugal elutriation within a small bore capillary tube can potentially separate the components based on size and frictional drag. Centrifugation of whole blood causes stratification of platelets and white cells in a buffy coat layered on top of a condensed pellet of mostly red cells. The buffy coat is an enriched source of lymphocytes and other white cells from fresh whole blood. In our approach to cell loading, the small size of the polyetheretherketone (PEEK) tubing makes it possible to access very small centrifuge pellets in microcentrifuge tubes, as well as whole blood and buffy coat layers inside microcapillary tubes.
Flow control and switching
The instantaneous fluid velocity within a microfluidic device with a chamber height of approximately one cell diameter is sensitive to seemingly mild sources of perturbation such as temperature, evaporation and mechanical jostling of supply tubing. The resistance of the device is often very high and compliance is low. Proper coupling of the fluidic resistance and compliance of the input apparatus are necessary for good flow control. Tubing, fittings and syringes that are relatively non-compliant in the macroworld must be re-examined for their ability to interface with fluidic channels of sub-nanoliter volumes. To realize the full advantage that microfluidic systems offer in terms of cellular microenvironment control, the driving system should be capable of rapid flow adjustments and switching between different flow sources as well as excellent overall flow control. This is particularly important during cell loading, when even temporarily excessive velocities can lead to cell lysis. We have found that ordinary silicone-based tubing and plastic syringes commonly used in the lab are far too compliant to supply microfluidic devices, except in cases of steady-flow and slow-flow switching. It is also advantageous to have the capability of switching between many sources of flow in a single experiment without being limited by mechanical or steric hindrances of large diameter tubing and syringes.
Transit time
Finally, transit time of reagents from the macro- to the microworld is an important consideration when conducting cellular experiments inside the microfluidic device. In many cell biology studies, it is desirable to introduce agents sequentially, such as a primary antibody, secondary antibody and fluorescent marker in an immunofluorescence experiment, or putative agents in a drug discovery screening, or when the concentrations of nutrients, metabolites or toxins must be controlled rapidly during the closed-loop control of a nanobioreactor. 5,21 When switching from normal media to media containing a bioactive agent, it becomes increasingly important for a rapid response in the cell chamber as more agents are included in a single screening experiment. Microfluidic drivers, such as the glass syringe/PEEK tube system described here, enable the conservation of expensive reagents and reduce the transit time into the chamber containing the cells, which increases data throughput by minimizing the duration of experiments. Indeed, one of the great promises of microfluidic ‘lab on a chip’ technology is high-throughput, data-dense studies.
For these reasons we have examined the use of small-volume glass body Hamilton Gastight syringes (80920, Hamilton Company, Reno, NV, USA) and PEEK tubing and fittings (Upchurch Scientific, Oak Harbor, WA, USA) as flow drivers for many of our microfluidic structures. We developed the techniques described here for loading cells into microfluidic cell-trapping devices of approximately 100 nL overall volume from the condensed cell pellet. Our goal was to decrease reagent waste and increase experimental throughput by minimizing delays due to sparseness of cells while efficiently accessing small-volume samples such as 10–50 K cell pellets and whole blood from finger sticks.
Methods
Primary human T cells were obtained from healthy donors using separation methods described elsewhere.
22
The T cells were stored in liquid nitrogen in aliquots of approximately 1 × 106 cells/tube. For experiments, the cells were thawed rapidly, washed and re-suspended in Roswell Park Memorial Institute (RPMI) complete media in a 100 mL flat-bottom culture flask and maintained at 37°C and 5% CO2 in an incubator for 4–5 days. The Jurkat T-cell line was maintained in suspension cell culture at approximately 1 × 106 cells/mL in RPMI complete media. An overview of the aspiration method is shown in Figure 1. Precise volumes of media and cells were removed by sterile pipetting from the culture flask and dispensed into 1.6 mL microcentrifuge tubes. The tubes were centrifugated at 1000
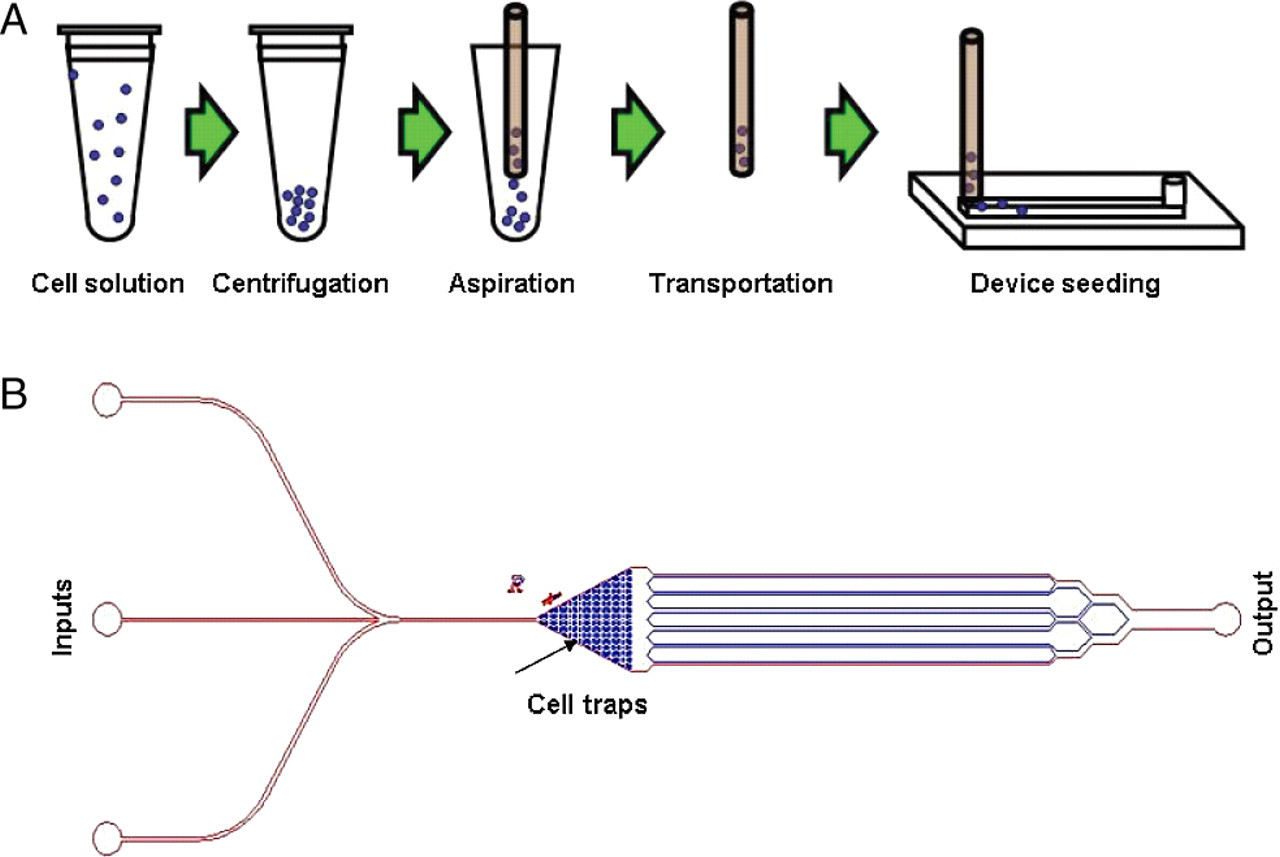
(A) Overview of the technique for transporting cells from macrocell culture to nanoliter-volume microculture devices. Beginning in the upper left, cells are pipetted from a large flask into a microcentrifuge tube using standard sterile technique. The cells are centrifuged to the bottom of the microcentrifuge tube and aspirated into the PEEK tubing while being visualized on a microscope (Figure 2). The PEEK tubing is friction-fitted into a prepunched hole in the PDMS and cells are ejected into the device at a velocity that does not cause cell lysing. The cells normally remain in the PEEK tubing for seconds to minutes, although we have demonstrated that they remain viable in the tubing for relatively long periods of time. (B) Schematic of the multitrap nanophysiometer showing multiple inputs on the left, a chamber containing over 400 individual cell traps and six parallel exit channels converging into a single output on the left. PEEK, polyetheretherketone; PDMS, polydimethylsiloxane
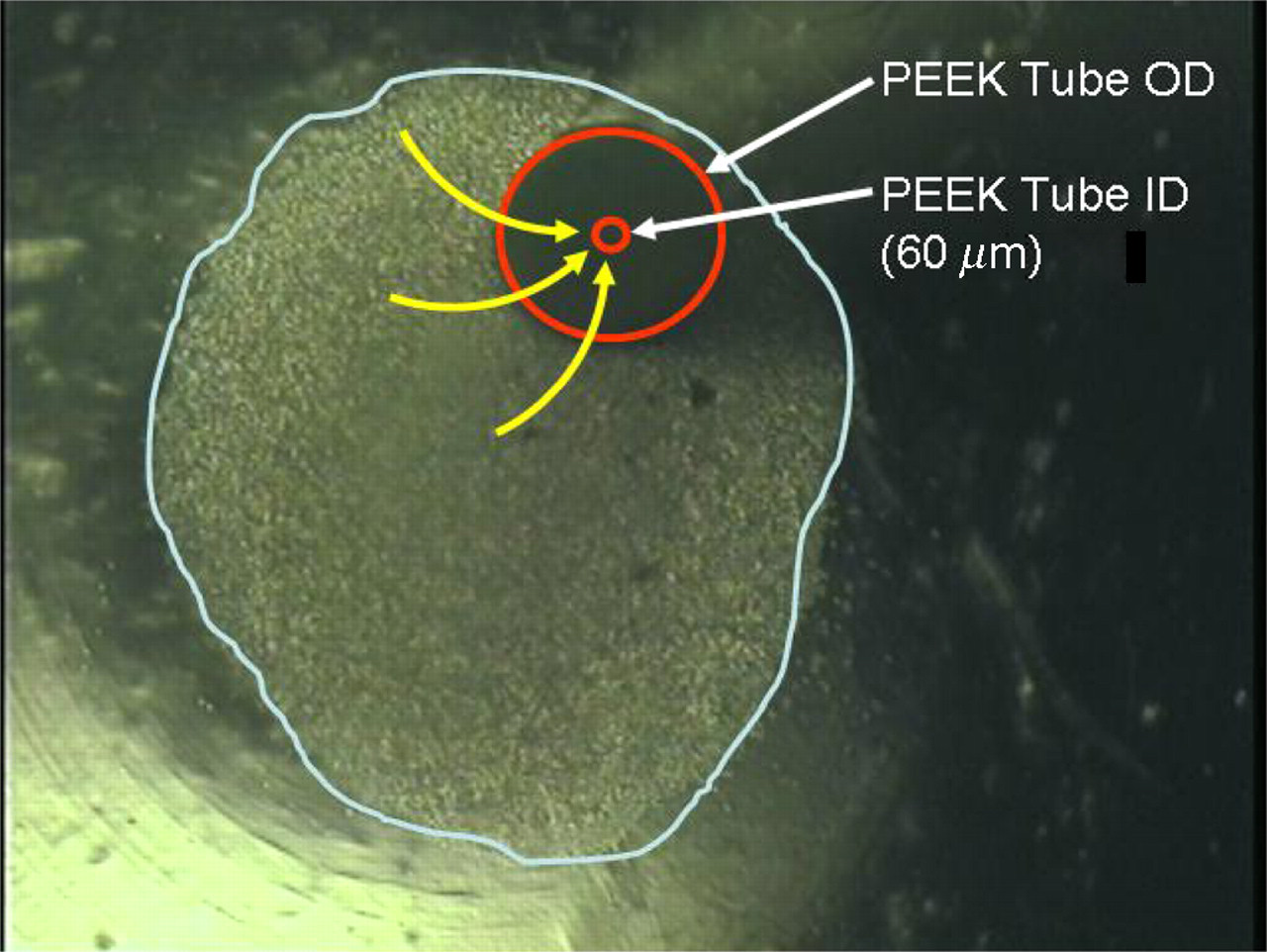
An inverted microscope image of a 500,000 cell pellet (white overlay) in the bottom of a 1.6 mL microcentrifuge tube during aspiration. Red overlay: PEEK tube end. Yellow overlay: direction of cell motion into tube. PEEK, polyetheretherketone

Various cell types loading into a microfluidic device. (Upper left) Primary human CD4+ T cells flowing from left to right. (Upper right) Primary human dendritic cells. (Middle left) Cells from 12 L whole blood (mostly erythrocytes, photo courtesy Eric Chung). Erythrocytes escaping through trap gaps. (Middle right) Murine mesenchymal stem cells (photo courtesy Susan Opalenik). (Bottom left) Saccharomyces cerevisiae loading at very high density. Several distributaries cut through a densely packed double layer of yeast cells. (Bottom right) S. cerevisiae after approximately 10 h in the device (yeast photos courtesy Erik Schneibel). Red dashed lines indicate paths of trapped cells. Scale bar is approximately 40 μm
When we fabricate our devices, we punch the entrance port with sharpened 0.25-gauge needle stock (#75165A686, McMaster-Carr Supply Company, Santa Fe Springs, CA, USA). Typically our PDMS structures are 2–5 mm thick, leading to 2–5 mm of contact between the outer surface of the PEEK tube and the inner PDMS surface of the punched hole. We have found this friction fit is more than adequate to support flow rates of 0–3000 nL/min. Due to the low dead volume and low compliance of the system, cells begin appearing in the channels of the device immediately upon the coupling of the tube to the device, or at time delays selectable by aspiration of a leading plug of pure media.
Whole blood from a finger prick was loaded into a 10 μL heparinized glass capillary tube (Drummond Scientific, Broomall, PA, USA), and the tube end was plugged with sealing clay. The capillary tube was centrifugated at low revolutions per minute for approximately one minute to pack the red blood cells and separate the buffy coat layer. The PEEK tubing was gently inserted into the glass capillary tube on the microscope stage until the bore was in contact with the buffy coat. The white cells, platelets and some red cells were then aspirated into the PEEK tubing and loaded into the device as described above. The aspherical shape and deformability of the red cells often enabled them to escape the microfluidic traps. Spherical cells from the buffy coat were thus preferentially trapped in the device. We have used an alternate method, in which the whole blood is aspirated directly without centrifugation and pumped through the microfluidic device, and noted a similar escape of erythrocytes from the microfluidic traps.
Residence time within the tubing during ordinary experiments is kept to a minimum and typically does not exceed five minutes. To gauge the effects of aspiration and residence time in the PEEK tubing on cell viability, controlled studies were performed comparing residence times from zero minutes (no residence in the PEEK tubing) to 90 min. As shown in Figure 4, no appreciable decrease in cell viability was noted in samples that were resident in the tubing up to 30 min. Furthermore, long-term microfluidic experiments performed using cells loaded in this manner have shown that both Jurkat and primary CD4+ T cells remain viable for more than 24 h, indicating that PEEK tubing aspiration does not induce apoptosis (data not shown). Taken together, this suggests that this aspiration method is well-suited for cellular applications.
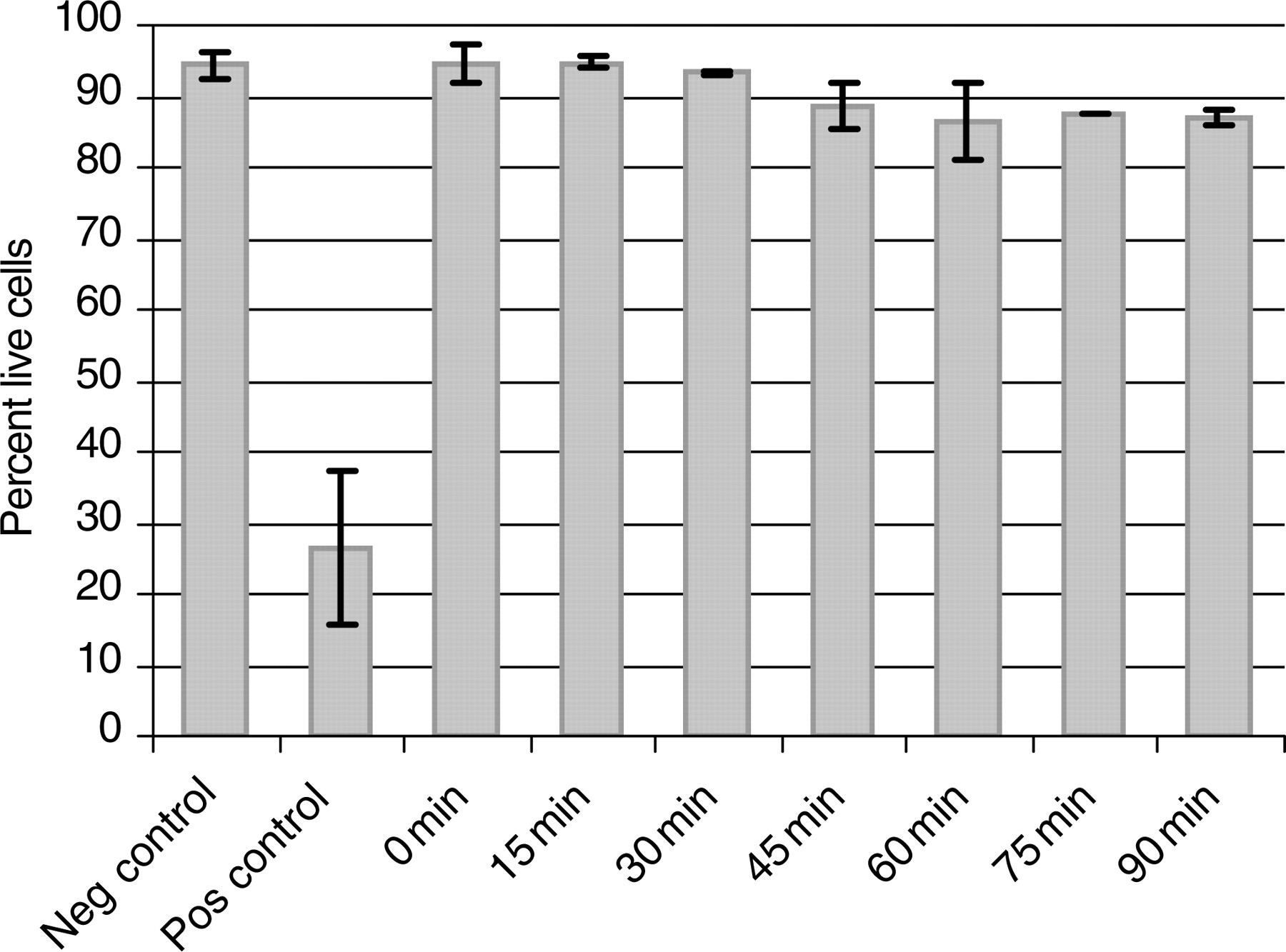
Cell viability versus PEEK residence time. The graph depicts the percentage of live Jurkat T cells following aspiration and residence within PEEK tubing for various times. Negative control is samples taken straight from cell pellet following centrifugation. Positive control is cells incubated in 70% water/media solution for five minutes. Viability was estimated with yopro-1 staining and microscopic imaging. No appreciable decrease in viability occurs prior to 30 min, while more than 85% of cells remain viable following 90-min incubation. N = 3, error bars indicate ±one standard deviation. PEEK, polyetheretherketone
Discussion
We have loaded microfluidic devices with both adherent and suspension cells and whole blood using our technique for aspirating cells into small bore PEEK tubing. The tubing enables the suction of high-cell-fraction media from the pellet and immediate loading into a perfused microfluidic device. We have used this aspiration method with a variety of cells, including the Jurkat T-cell line, primary T and B cells, dendritic cells and adherent mouse mesenchymal stem cells and Dictyostelium and yeast.
The low-compliance PEEK tubing and glass body syringes with syringe pumps allow us to control absolute flow and switch between many flow sources very rapidly with short delays. Figure 5 shows a fluorescent image montage of the trap chamber of a microfluidic device during a switch from water to water with fluorescein isothiocyanate at three flow rates (50, 150 and 250 nL/min). The data illustrate that switching occurs very rapidly and repeatedly when using the methods described here. At the higher flow rates trapped cells experience a very rapid change in their microenvironment. The figure illustrates a subtle but critical advantage of microfluidic experiments: the ability to know with great precision the timing of cellular microenvironment changes. The small bore of the PEEK tubing and the ease with which it can be cut and bent allow the access to the high-density cell centrifugation pellet. Aspiration of cells from the pellet with this setup leads to very rapid transition from cell culture volumes and concentrations to microfluidic volumes and concentrations, with the whole technique (including centrifugation) being accomplished in 3–5 min. The small outside diameter and 50 μm inside diameter and the easy deformability of the PEEK tubing allow interconnection of microfluidic devices, with the output from one device providing input to a downstream device, etc. 23 Using these methods, we have routinely supplied devices with 3–5 different perfusion sources and can switch rapidly between perfusion media or any combination or mixture of media. We could potentially achieve a dozen or more supply lines in a single device using these small, deformable tubes. Finally, we have extracted cells from microfluidic devices back into the PEEK tubing, suggesting the possibility for return to long-term traditional culture in incubators after experiments or treatments in the microfluidic device, as might be required prior to a monoclonal expansion of the cell population.
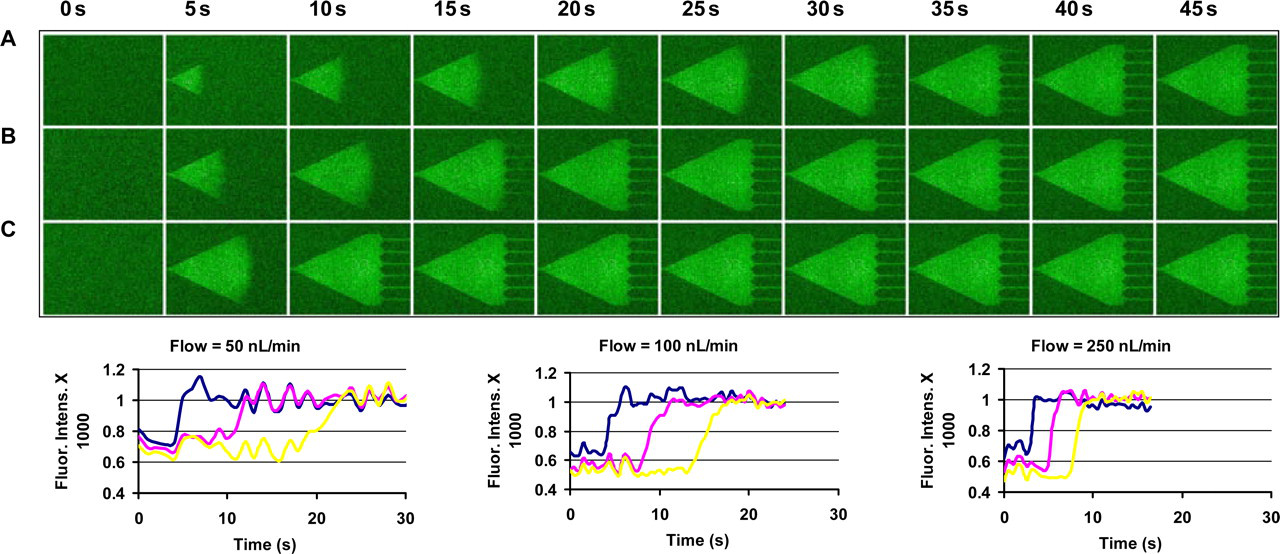
Response and transit time of the MTNP supplied with PEEK tubing and Hamilton syringes. (Top panel) Fluorescent image montage of fluorescein isothiocyanate (FITC) solution entering the MTNP after switching from water at three flow rates (50, 100 and 250 nL/min). (Bottom panel) Graphs of the fluorescence at upstream, midstream and downstream (blue, pink and yellow lines, respectively) positions in the trap chamber for the three different flow rates. At 250 nL/min the advancing wave of FITC propagates from the front to the back of the device in about five seconds, and a cell anywhere in the device will experience a complete microenvironment switch in one second or less. PEEK, polyetheretherketone; MTNP, multitrap nanophysiometer
The MTNP used in the images of Figure 3 is designed to study individual cells, especially non-adherent cells such as cells of the blood, or small numbers of yeast cells. The MTNP is configurable with up to tens of thousands of traps, so the number of cells needed to load every trap varies depending on the specific device being used. Calcium transient experiments like those described in Faley et al. 3 can be conducted on as few as one cell to as many cells as the device can hold. The methods described here for loading cells give a researcher access to a few hundred (possibly fewer) or several million cells, which is flexible enough to load MTNP devices with adequate numbers of cells in a reasonable amount of time.
Conclusions
Microfluidic devices are developing into powerful tools in cell biology and medicine, but the interface of the device to conventional cell culture techniques is often inconvenient and inefficient. The methods described here offer four advantages over other methods for conducting cellular studies within microfluidic devices: (1) small-volume flow drivers conserve samples and experiment time; (2) non-compliant drivers lead to fast flow response and rapid switching; (3) small tubing bore and outside diameter facilitate the aspiration of cells from centrifuge pellets and hematocrit tubes; (4) the low expense, small size, flexibility and non-compliant walls of the PEEK tubing facilitate the interfacing of microfluidic devices with many different perfusion sources.
Rigid, low-compliance, low-dead-volume perfusion systems reduce the time required to set up cell studies in microfluidic devices. Aspiration from the centrifuge pellet or hematocrit tube as demonstrated here enables rapid access to sparse cells in macrocell culture dishes. Many independent perfusion mixtures may access the same microfluidic chip and any combination of switching or mixing may be rapidly accomplished during an experiment. Interconnection of separate microfluidic devices is easily achieved with PEEK tubing with very low transit time between devices. Cells that have been studied or treated within the device can also be easily off-loaded from the device and may possibly be returned to conventional cell culture flasks or multiwell plates using this system if sterility is maintained throughout the experiment. This cell handling technique is hence a key first step in applying simple, low-cost microfluidic devices to challenging problems in biology and medicine.
Footnotes
Acknowledgements
This work was supported in part by grants from the Defense Advanced Research Projects Agency (DARPA), the Air Force Office of Scientific Research (AFOSR), the National Institutes of Health grant U01 A1061223, the Vanderbilt Institute for Integrative Biosystems Research and Education (VIIBRE) and the Searle Systems Biology and Bioengineering Undergraduate Research Experience (Searle SyBBURE).