
Abstract
Pulmonary embolism (PE) is a common, lethal, ischemic disease. PE-induced endothelium injury plays a critical role in the pathophysiological consequences of PE. Endothelial progenitor cells (EPCs) can be mobilized from the bone marrow to enter circulation and play important roles in repair of damaged endothelium. However, it is not yet known if EPC mobilization results from PE. The alterations of the quantity and function of bone marrow-derived EPCs were detected in acute pulmonary embolism (APE) events in mice, and the possible role of the endothelial nitric oxide synthase (eNOS)/nitric oxide (NO) pathway in those alterations was explored. APE models were established by injection of autologous thrombi into the right jugular vein of C57BL/6 mice. Mice were divided into sham and experimental groups including one hour (1H), one day (1D) and two day (2D) groups after injection. The results showed that in the APE 1D group, the thrombi were easily found in the large or medium pulmonary vessel. And CD133+ or CD34+ cells in bone marrow increased significantly, while CD133+/vascular endothelial growth factor receptor 2+ EPCs decreased. After seven days in culture, the abilities of incorporation into a vascular network, adhesion to fibronectin, migration and proliferation of bone marrow-derived EPCs in the APE 1D group increased significantly. The mRNA and protein expression levels of eNOS in EPCs increased in the APE 1D group. Treatment of EPCs with NG-nitro-L-arginine methyl ester inhibited functional alterations induced by APE. The results suggested that APE events stimulate the mobilization of EPCs from bone marrow, and enhance their functions. The eNOS/NO pathway may be involved in this process.
Keywords
Introduction
Pulmonary embolism (PE) is a common, lethal, ischemic disease with considerably high mortality and morbidity. In a community-based study, the incidence of PE was 6.0/10,000/y. 1 The case fatality rate for acute PE (APE) ranges from 7% to 11%. 2 PE is a common cause of pulmonary vascular endothelium injury. Thrombi trapping in pulmonary vessels damage the vascular endothelium and cause the release of proinflammatory mediators and secondary pathological changes. 3–6 Repeated vascular injuries of the pulmonary vessels may trigger pulmonary vascular remodeling and lead to secondary pulmonary hypertension, namely, chronic thromboembolic pulmonary hypertension. 7 Thus, PE-induced endothelium injury plays a critical role in its pathophysiological consequences.
Endothelial progenitor cells (EPCs), as an important factor in the endothelial repair, can be mobilized from bone marrow into the peripheral circulation upon the stimulus of ischemia. 8 They home to the sites of endothelial denudation, migrate and incorporate into the sites of neovascularization and improve the ischemic organs’ recovery. 8–13 EPCs can counteract endothelial cell (EC) injury induced by the ongoing risk factor, and replace dysfunctional endothelium. They may act as a predictive biomarker for certain ischemic diseases. 14–24
This study was designed to test the hypothesis that APE could lead to alterations in the quantity and function of bone marrow-derived EPCs and that the endothelial nitric oxide synthase (eNOS)/nitric oide (NO) pathway may be involved in this process.
Methods and materials
Animal model
All animal experiments were carried out in accordance with the guidelines of the local regulatory agencies and conformed to the Regulations for the Management of Laboratory Animals published by the Ministry of Science and Technology of the People's Republic of China. All the experimental protocols were approved by the Institutional Animal Care and Use Committee of Capital Medical University.
APE was induced by intravenous injection of autologous thrombi into mice, which was a modified method based on previously published protocols. 25,26 Briefly, one day before the experiment, 200 μL plasma was collected from C57BL/6 mouse tail and mixed with 50 μL thrombin CaCl2 mixture (10 U/mL bovine thrombin containing 0.5 mmol/L CaCl2). Thrombus was made by aspirating the mixture into a glass catheter which was 1.2 mm in inner diameter. The autologous thrombus was stabilized for 10 min at room temperature, then 30 min at 37°C water bath, and then stored at 4°C overnight. The thrombus was cut into 1 mm long pieces (autologous thrombi) before use.
C57BL/6 mice (20–23 g, aged 6–8 weeks, n = 193) were randomly divided into control group (n = 39), sham group (n = 62) and experimental groups (n = 92), including one hour (1H group), one day (1D group) and two day (2D group) groups after thrombi injection. After being anesthetized by intraperitoneal injection of 60 mg/kg Nembutal (pentobarbital sodium; Serva Feinbiochemica, Heidelberg, Germany), about 30 autologous thrombi with 0.4 mL saline were injected into the right jugular vein of experimental groups. The same amount of saline was injected into each sham object. After one hour, one day and two days of injection, mice were sacrificed, and the lungs, femurs and tibias were collected.
Histopathology analysis
Lungs of the mice were fixed in 10% neutral formalin and paraffin embedded. Sections (4 μm thick) were cut by an ultra-thin slicer (Leica, Wetzlar, Germany). In order to show the fibrin in the thrombi, phosphotungstic acid, hematoxylin as well as hematoxylin and eosin staining was used.
Isolation and culture of bone marrow-derived EPCs
Bone marrow cells were aspirated from tibias and femurs of mice. Mononuclear cells (MNCs) were isolated by density centrifugation over Histopaque-1083 (Sigma-Aldrich, St Louis, MO, USA). 27 In total, 1 × 106 MNCs were used for flow cytometric analysis. The remaining MNCs were plated onto fibronectin (Chemicon, Temecula, CA, USA)-coated six-well plates at a density of 3 × 106/cm2 in M199 complete medium (Invitrogen, Carlsbad, CA, USA), which is a selective medium for endothelial lineage cells, supplemented with 20% fetal bovine serum (FBS; Hyclone, Logan, UT, USA), 10 ng/mL vascular endothelial growth factor (VEGF; Chemicon) and 1 × penicillin–streptomycin (penicillin 100 U/mL, streptomycin 0.1 mg/mL; Invitrogen). After four days in culture, non-adherent cells were removed. The early outgrowth cells were used for immunochemistry identification and in vitro functional analysis after being cultured in the selective medium for seven days. 28,29
Flow cytometric analysis
CD34+, CD133+, vascular endothelial growth factor receptor 2 (VEGFR2)+ and their combinations are commonly used markers in defining EPCs. 8,30 Thus, the CD34+, CD133+, CD34+/VEGFR2+ and CD133+/VEGFR2+ cells were detected and the CD133+/VEGFR2+ cells were defined as EPCs 8,31,32 in this study. In total, 1 × 106 MNCs were stained with a three-color antibody panel: APC-anti-mouse CD133 (eBioscience, San Diego, CA, USA), PE-anti-mouse VEGFR2 (eBioscience) and fluorescein isothiocyanate (FITC)-anti-mouse CD34 (eBioscience). Antibodies were titrated to achieve working concentrations according to the manual instructions. Using a Becton-Dickinson FACS Aria cytometer (BD Biosciences, Franklin Lakes, NJ, USA), 5 × 105 MNCs were processed in each flow cytometric detection. Cell populations (VEGFR2+, CD133+, CD34+, CD133+/VEGFR2+ and CD34+/VEGFR2+) were quantified as a percentage of live events. Analysis gating criteria were set according to proper isotype controls.
Immunochemistry identification
After being cultured in vitro for seven days, the fibronectin-adherent cells were identified by using an immunochemistry method. 9,33 Cell surface lectin staining was performed using fluorescently FITC-labeled Ulexeuropaeus agglutinin 1 (UEA-1; Sigma-Aldrich) at 10 mg/L. The ability of live cells to take up fluorescently labeled acetylated-low-density lipoprotein (Dil-ac-LDL; Molecular Probes, Eugene, OR, USA) was assessed by incubating cells with Dil-ac-LDL (2.4 mg/L) for two hours at 37°C. The images were captured by Leica-SP5 confocal microscopy (Leica, Germany). EPCs were defined as both FITC-UEA-1 and Dil-ac-LDL double-stained cells. 9,33
Incorporation assay
Incorporation into a developing vascular network in vitro was used to mimic incorporation of EPCs during vasculogenesis in vivo. 34 After seven-day culture, 2 × 104 EPCs were incubated with 2.4 mg/L Dil-ac-LDL overnight and then co-plated with human umbilical vein endothelial cells (HUVECs) in a 1:2 proportion on presolidified matrigel (BD Biosciences). 29 After 4–6 h, the number of EPCs incorporated into the vascular network was counted in six high power fields (HPFs) (×200) under a fluorescence microscope (Nikon, Japan).
Adhesion assay
In total, 1 × 104 cultured EPCs were resuspended in M199 complete medium, distributed onto a 5 μg/cm2 fibronectin coated 96-well plate, and incubated for 30 min. The number of adherent cells in 10 HPFs (×200) was counted.
Migration assay
The ability of EPCs to migrate towards VEGF was evaluated by resuspending 1 × 105 EPCs in 100 μL M199 plus 0.5% FBS in the upper chamber of a modified Boyden chamber (8 μm pore size; Millipore, Billerica, MA, USA). The upper chamber was transferred into a 24-well plate containing M199 plus 20% FBS and 50 μg/L VEGF, incubated overnight at 37°C, and then fixed with 4% formaldehyde. Cell nuclei of migrated EPCs were stained with 4′,6-diamidino-2-phenylindole (1:1000, Molecular Probes) and counted in 4 HPFs (×200).
Proliferation assay
EPC proliferation was determined by MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide) assay. 35 Cells in complete medium were reseeded onto fibronectin-coated 96-well plates (2 × 104 cells/well). Each sample included three repeated wells, and blank controls were also established. After 48 h culture, the medium was removed, MTT (100 μL, 0.5 g/L) (Amersco, Solon, OH, USA) was added and incubated for 4 h, and then the supernatant was discarded and 200 μL dimethylsulfoxide was added. Samples in plates were shaken for 10 min at 37°C prior to the measurement of optical density (OD) value at 490 nm. The OD value in each group was standardized by subtracting the average value of blank controls, and cell viability percentage was calculated using standardized values by dividing the average value of normal group.
Realtime polymerase chain reaction
The eNOS mRNA levels of cultured EPCs were measured by realtime polymerase chain reaction (PCR) using the method described in detail previously. 36 The primer sequence used for eNOS was TTTGTCTGCGGCGATGT (forward) and GTGCGTATGCGGCTTGT (reverse). β-Actin was used as normalization gene transcript (forward: CTGAGAGGGAAATCGTGCGTGACA; reverse: ATACCCAAGAAGGAAGGCTGGAAAA).
eNOS immunofluorescence staining
Cultured cells were fixed with 4% paraformaldehyde (Sigma-Aldrich) for 20 min at room temperature, treated with 0.1% Triton-X-100 for 10 min and incubated in 10% normal goat serum for 20 min. After incubation with the primary antibodies (Abs) against eNOS (1:200, Santa Cruz) overnight at 4°C, EPCs were treated with secondary Abs (anti-goat IgG-Cy-3, 1:200, Sigma). Cells just exposed to the secondary Abs were used as negative controls. Images were processed by relative quantitative analysis with Image J 1.42 software (National Institutes of Health, Rockville, MD, USA).
Functional changes after NG-nitro-L-arginine methyl ester treatment in vitro
To confirm the role of eNOS/NO in the functional regulation of EPCs, cells were treated with an eNOS inhibitor NG-nitro-L-arginine methyl ester (L-NAME, 300 μmol/L) 33 for 24 h after seven days in culture. Incorporation into vascular networks, adhesion, migration and proliferation functions were assessed, and compared with the untreated groups.
Statistical analysis
Statistical analyses were performed by using GraphPad Prism 4.03 software. All data are expressed as mean ± standard error of mean. Since circulating EPC numbers were not normally distributed, data from flow cytometry were analyzed with the Wilcoxon (non-parametric) test. The Student's t-test was used to analyze EPC function in vitro. Probability values of P < 0.05 were considered statistically significant. To evaluate the role of eNOS/NO in the functional alterations of EPCs, one-way analysis of variance was used to analyze data between the three different groups.
Results
Acute PE model
As described in Figure 1, the thrombi mainly obstructed in segmental pulmonary arteries shortly after one hour of the thrombi injection (Figure 1b). In the APE 1D group, thrombi were easily found in the pulmonary vessels in 93.75% of the mice (Figure 1c). Thrombi were mainly (73.33%) found in medium or large size pulmonary arteries with a thickened lung matrix, inflammatory cell infiltration and erythrocyte aggregation. However, most of the thrombi disappeared in the APE 2D group (Figure 1d). Thus, the following functional assay experiments were primarily performed in the APE 1D group.
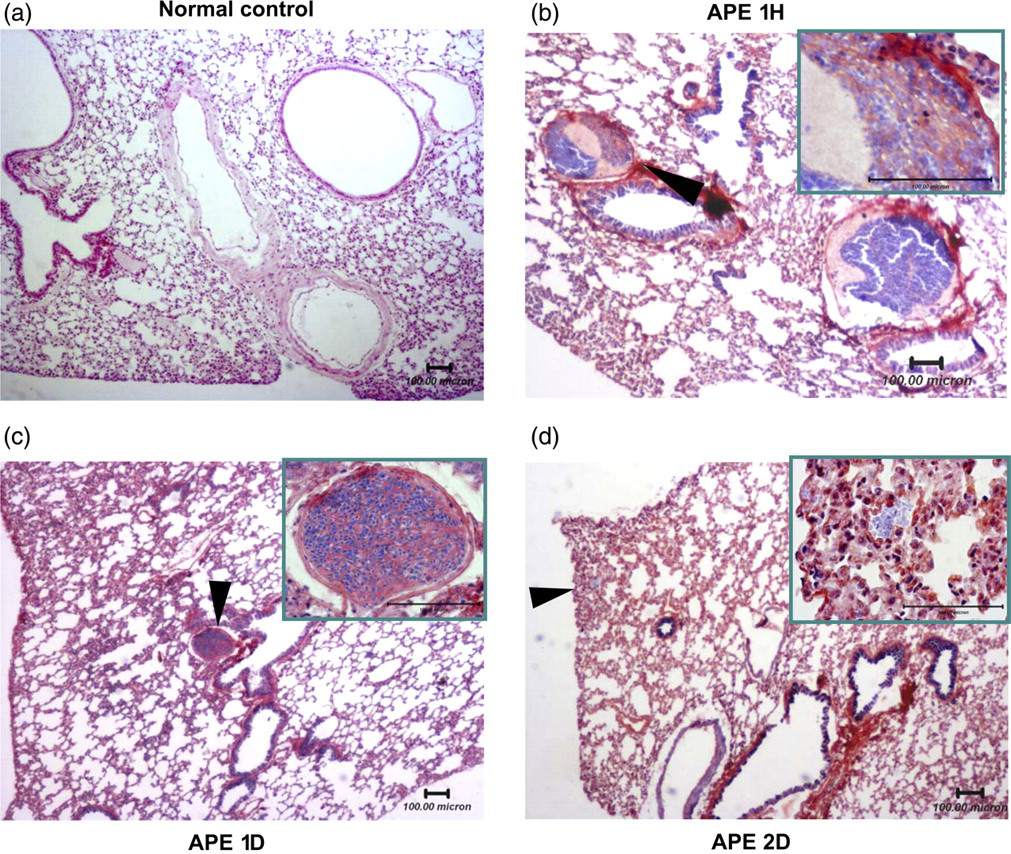
Histopathology analysis of thrombi in lung sections. (a) Photomicrograph of the normal control (HE staining; bar = 100 μm). There were no thrombi in pulmonary vessels. (b–d) Representative sections (PTAH staining; bar = 100 μm) of the APE 1H (b), APE 1D (c) and APE 2D (d) groups. In the 1H group (b), it was observed that thrombi obstruction was mainly in pulmonary segmental pulmonary arteries (arrow marked), and that the pulmonary septal thickened from pulmonary inflammatory reaction after APE. Thrombi were also easily found in medium or large pulmonary arteries with a thickened lung matrix, inflammatory cell infiltration and erythrocyte aggregation in the APE 1D group (c). Most of the thrombi disappeared in the APE 2D group (d); erythrocyte aggregation was scattered in distal vessels (arrow marked). High magnification image of the black arrow marked area is shown in the upper right corner. HE, hematoxylin and eosin; APE, acute pulmonary embolism; PTAH, phosphotungstic acid, hematoxylin; APE 1H, APE one hour; APE 1D, APE one day; APE 2D, APE two days (A color version of this figure is available in the online journal)
Changes in the quantity of bone marrow-derived EPCs following APE
CD34+, CD133+, VEGFR2+ and their combinations have been used as markers to define EPCs in human and murine in different researches. The combination phenotype of CD133+/VEGFR2+ was considered to be a suitable marker in defining EPCs. 8,31,32 Thus, in the present study, CD34+, CD133+, VEGFR2+/CD133+ as well as VEGFR2+/CD34+ cells were detected using flow cytometry and the VEGFR2+/CD133+ subpopulation was defined as EPCs. Bone marrow-derived MNCs isolated by Ficoll density gradient centrifugation were used to detect the quantity of EPCs using a fluorescence-activated sorter. The percentage of CD133+ or CD34+ cells in the APE 1D group increased significantly (versus sham group, P < 0.05), while there were no significant changes in VEGFR2+ cells percentage (Figure 2a–c). Compared with the sham group, the ratio of CD133+/VEGFR2+ cells (which were defined as EPCs 8,31,32 ) decreased significantly in the APE 1D group (P < 0.01) (Figure 2e), while no significant changes were found in the CD34+/VEGFR2+ population in this group (Figure 2d).
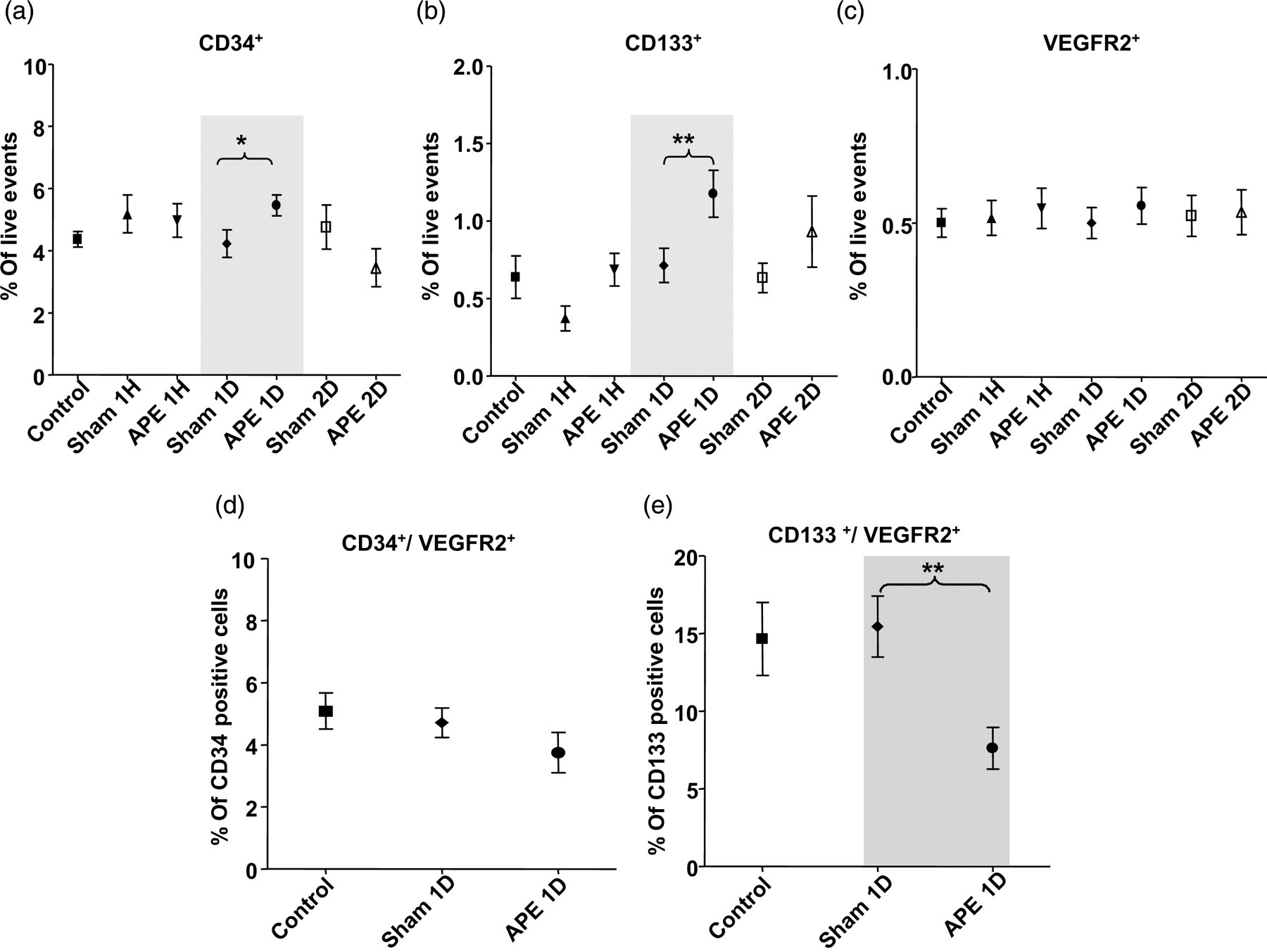
Flow cytometric analysis of bone marrow-derived mononuclear cells. Primary isolated bone marrow-derived mononuclear cells in different groups were detected by using VEGFR2, CD34 and CD133, respectively (a–c). In the APE 1D group, levels of CD133+ (**versus sham group, P < 0.01) and CD34+ (*versus sham group, P < 0.05) increased significantly, while there were no significant upregulation in VEGFR2+ expression. In CD133+ population, CD133+/VEGFR2+ level (e) descended significantly (**versus sham group, P < 0.01). The expression decline of CD34+/VEGFR2+ in CD34+ population (d) was not statistically significant. n = 8 for each group. APE, acute pulmonary embolism; VEGFR2, vascular endothelial growth factor receptor 2; APE 1D, APE one day
Incorporation capacity of bone marrow-derived EPCs in the APE model
After culture for seven days in the selective medium, EPCs in the adherent spindle-shaped MNCs cells were identified with Dil-ac-LDL and FITC-UEA-1 double labeling. 9 The results demonstrated that 86.085 ± 5.622% of adherent cells were double-positive EPCs (Figure 3), which suggested that highly purified EPCs could be selected with this culture method to use in the following functional tests.
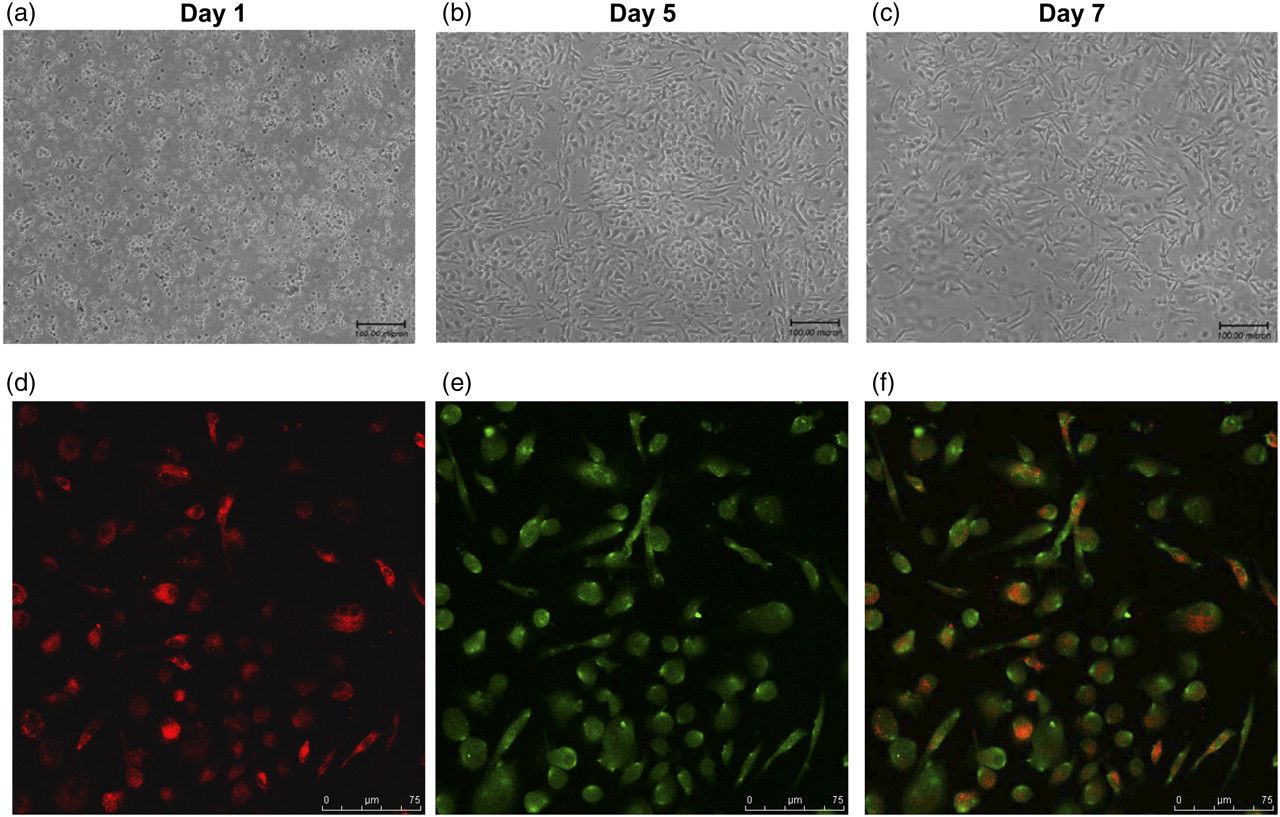
EPC culture and identification. Bone marrow-derived mononuclear cells were cultured in vitro for seven days. (a–c: representative images of cultured cells in day 1, day 5 and day 7). After seven days in culture, cells were tested for ac-LDL uptake (d) and lectin-binding (e); dual-stained positive cells (f) were recognized as EPCs. EPC, endothelial progenitor cell; ac-LDL, acetylated-low-density lipoprotein (A color version of this figure is available in the online journal)
Incorporation into a developing vascular network in vitro was used to mimic incorporation of EPCs during vasculogenesis in vivo. 34 To study the effect of APE on vasculogenic potential, the EPCs labeled DiI-ac-LDL were plated onto the matrigel in the presence of HUVECs and incorporation was determined after 4–6 h incubation. 34 As shown in Figure 4a and b, the total number of incorporated EPCs per HPF in the APE 1D group was higher than those in the sham group (35.66 ± 1.780 versus 26.93 ± 1.636 EPCs/HPF, n = 7, P < 0.01).
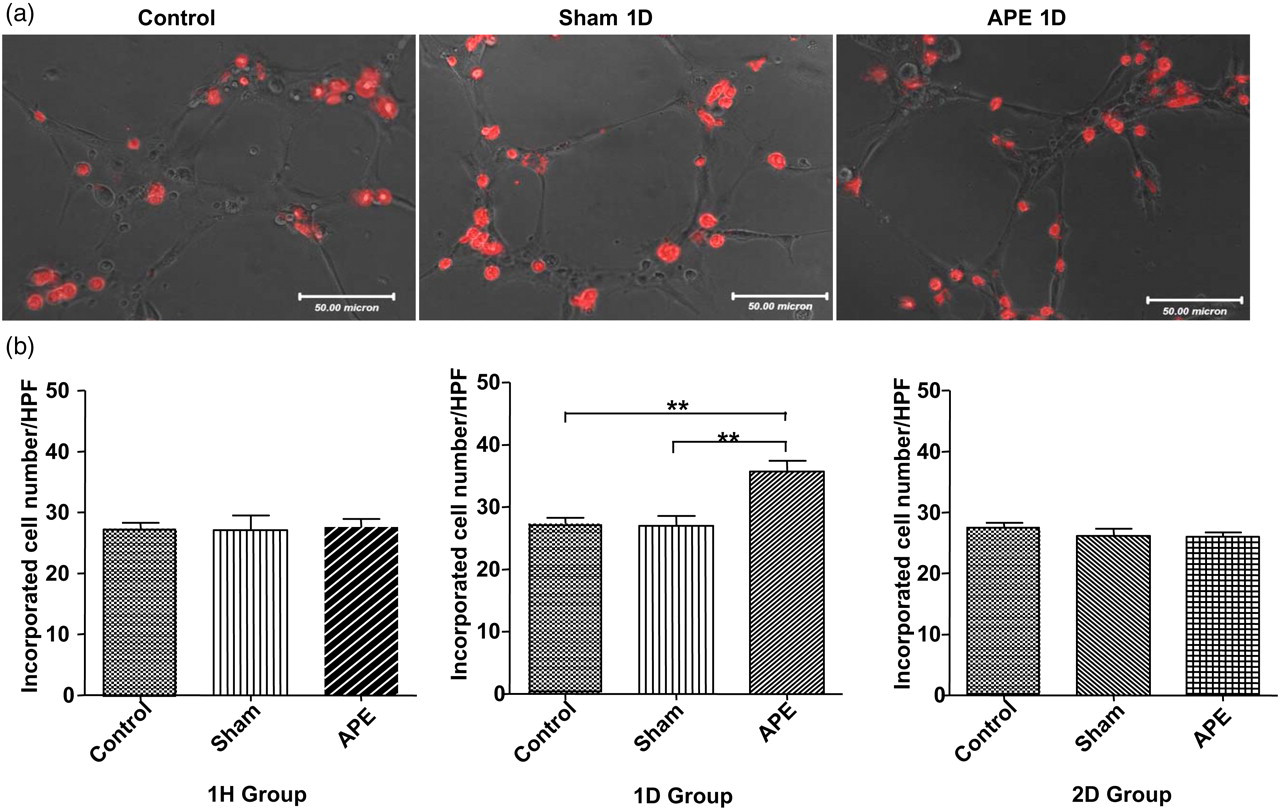
Effects of APE on EPC incorporation capacity. (a) Tube formation with DiI-labeled EPCs that were co-plated with HUVECs on matrigel. (b) Summarized cell numbers of incorporation into vascular network in different groups (**P < 0.01, n = 7 for each group). APE, acute pulmonary embolism; EPC, endothelial progenitor cell; HUVEC, human umbilical vein endothelial cell (A color version of this figure is available in the online journal)
Adhesive capacity of bone marrow-derived EPCs in the APE model
Adhesion to the extracellular matrix is believed to be an important step during new blood vessel growth. The results showed that the adhesive capacity of EPCs significantly increased in the APE 1D group compared with the sham group (37.04 ± 5.090 versus 18.36 ± 2.080 EPCs/HPF n = 7, P < 0.01) as shown in Figure 5a and b.
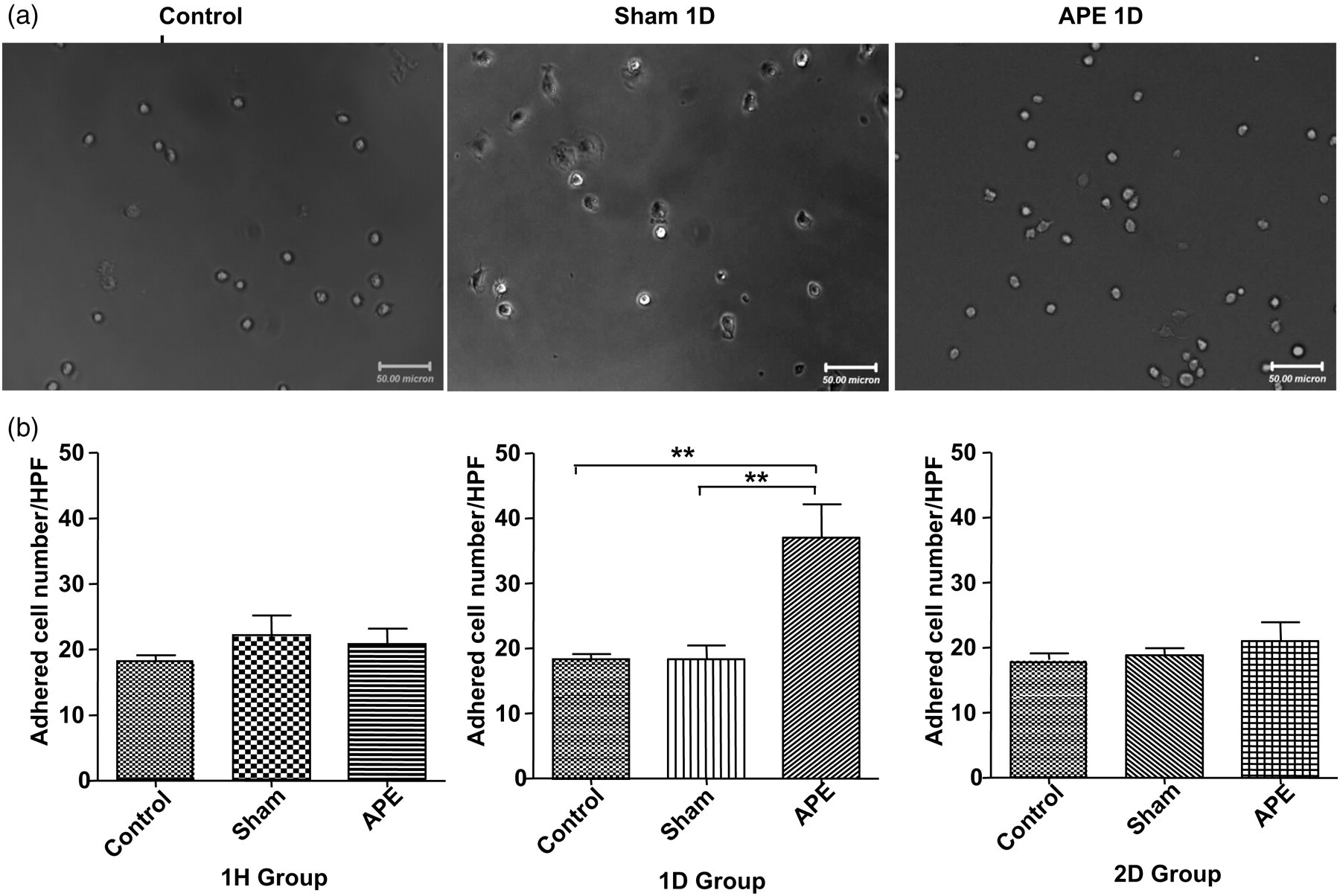
Effects of APE on EPC adhesion capacity. (a) Adhesive capacity of EPCs on fibronectin-coated plate. (b) Summarized numbers of adhered EPCs to fibronectin (**P < 0.01, n = 7 for each group). APE, acute pulmonary embolism; EPC, endothelial progenitor cell
Migratory capacity of bone marrow-derived EPCs in the APE model
Migration of isolated EPCs in response to VEGF was determined using a modified Boyden chamber assay. As illustrated in Figure 6, the migratory capacity of EPCs from APE 1D mice was markedly increased (28.09 ± 4.357 versus the shame group of 17.24 ± 2.234 migrating EPCs/HPF, n = 7 for each group, P < 0.01).
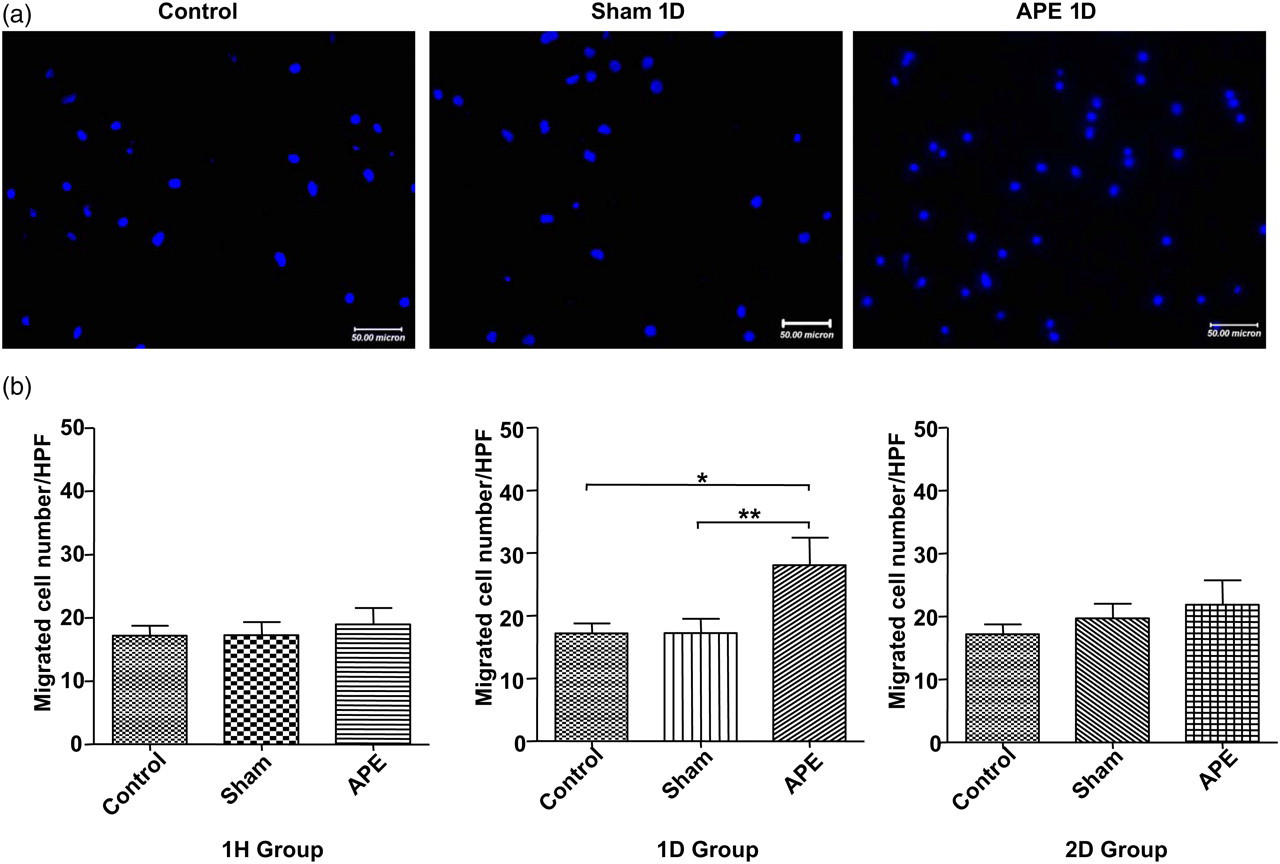
Effects of APE on EPC migration capacity. (a) Representative images of migrated cells stained with DAPI in normal (left panel), Sham 1D (middle panel) or APE 1D group (right panel). (b) Summarized numbers of migrated EPCs in different groups (*P < 0.05; **P < 0.01, n = 7 for each group). APE, acute pulmonary embolism; EPC, endothelial progenitor cell; DAPI, 4′,6-diamidino-2-phenylindole; Sham 1D, sham one day; APE 1D, APE one day (A color version of this figure is available in the online journal)
Proliferation ability of bone marrow-derived EPCs in the APE model
The effect of APE on the proliferation of bone marrow-derived EPCs was assayed by the MTT technique. The proliferation ability of EPCs was significantly enhanced in APE 1D mice (versus the sham group, n = 6 for each group, P < 0.05) (Figure 7).
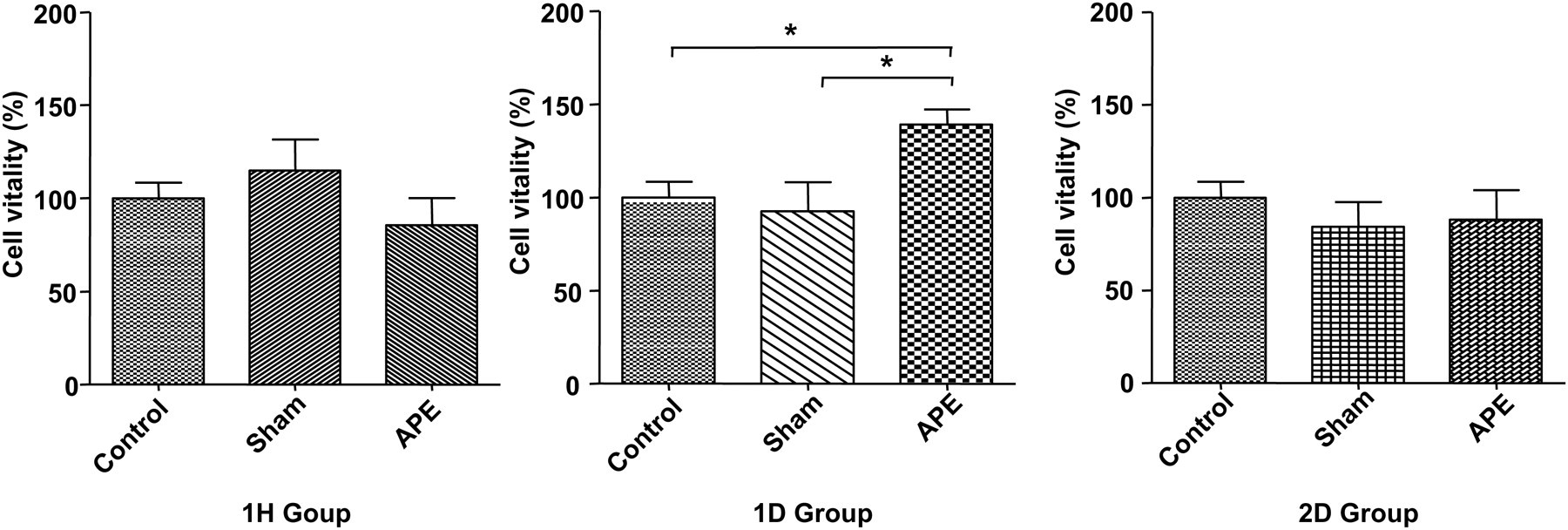
Effects of APE on EPC proliferation. The proliferation ability of EPCs was significantly enhanced in APE 1D mice versus sham operation mice (*APE 1D versus sham 1D group, P < 0.05, n = 6 for each group). EPC proliferations were detected using the MTT technique. APE, acute pulmonary embolism; EPC, endothelial progenitor cell; APE 1D, APE one day; MTT, 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide
Effect of the eNOS/NO pathway on APE-induced functional alterations in bone marrow-derived EPCs
Realtime PCR was employed to detect the eNOS mRNA expression level in EPCs. As shown in Figure 8, eNOS mRNA level increased significantly in APE 1D mice compared with the sham and control mice (n = 4, P < 0.01). Consistent with the mRNA detection data, eNOS protein expression measured by using immunofluorescence staining was also increased in the APE 1D group (versus the sham group, n = 4 for each group, P < 0.001).
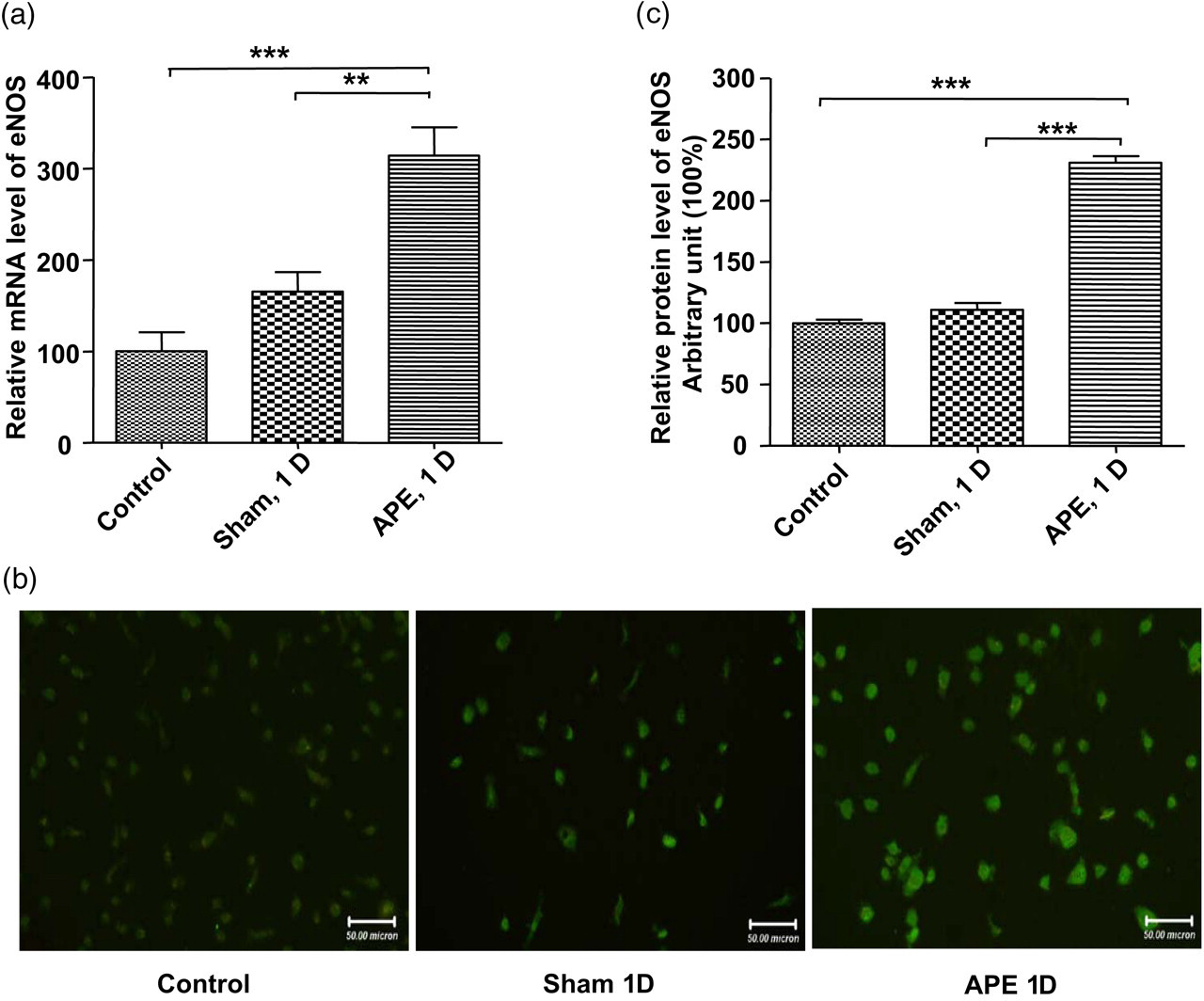
Changes of eNOS/NO expression level in EPCs. (a) The mRNA level of eNOS in EPCs detected with realtime PCR was enhanced in the APE 1D group compared with that of sham and control group (**P < 0.01; ***P < 0.001, n = 4). (b and c) The protein level of eNOS detected by immunofluorescence staining increased in the APE 1D group (***P < 0.001, n = 4). EPC, endothelial progenitor cell; APE, acute pulmonary embolism; eNOS, endothelial nitric oxide synthase; APE 1D, APE one day (A color version of this figure is available in the online journal)
Following treatment of EPCs with an eNOS inhibitor, L-NAME, the functional alterations in EPCs induced by APE (including proliferation, incorporation, adhesive capacity and migratory capacity) were suppressed close to the control levels (Figure 9). These results indicated that the eNOS/NO pathway maybe partially responsible for the APE-induced functional alterations in EPCs.
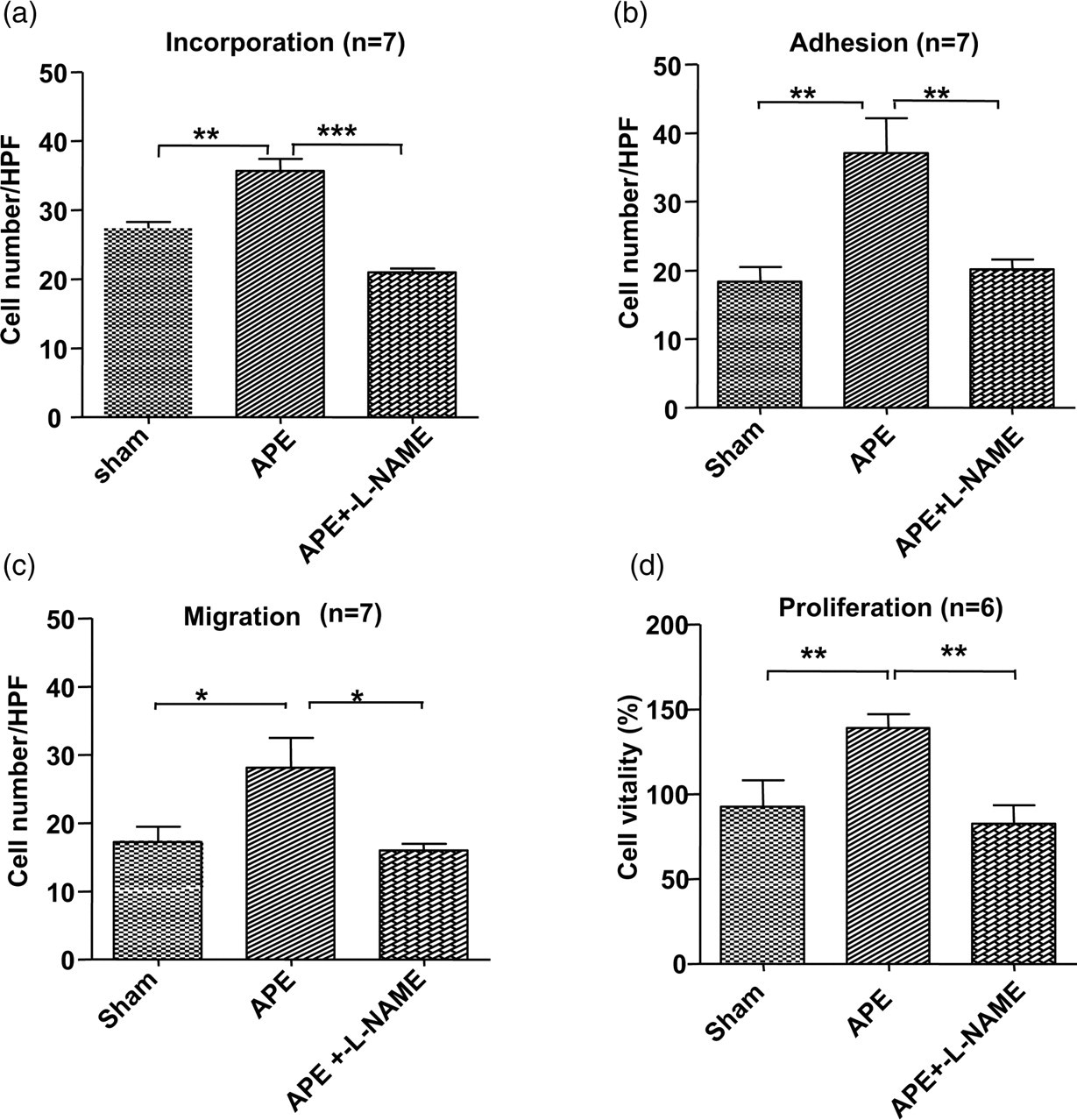
L-NAME treatment blocked APE-induced EPC functional changes. After treatment with L-NAME, the incorporation (a), adhesion (b), migration (c) and proliferation (d) functions of EPCs in the APE 1D group were significantly suppressed and dropped close to the levels of the control group (*P < 0.05; **P < 0.01; ***P < 0.001). APE, acute pulmonary embolism; EPC, endothelial progenitor cell; APE 1D, APE one day; L-NAME, NG-nitro-L-arginine methyl ester
Discussion
In this study, the autologous thrombi injection was used to create a mouse APE model. Autologous thrombi injection avoided the variant thrombi-induced rejective effect in the body. The APE model used in this study was shown to be stable and feasible.
Endothelial injury is a fundamental process in PE. Healing the injured endothelium requires rapid endothelialization. Two possible sources of endothelialization are as follows: (1) migration and co-option of pre-existing vascular wall ECs or (2) recruitment of EPCs from the stem cell reservoirs such as bone marrow. 8 Many previous studies demonstrated alterations of peripheral circulating EPCs during different ischemic conditions, 10,15,17,19–24 but the steps preceding alterations of peripheral circulating EPCs, namely what the effect of ischemia attack is on the bone marrow EPCs, are not known. Using our APE mouse model, we found that the quantity and function of bone marrow-derived EPCs increased at the early stage of an APE event. This study, to the best of our knowledge, is the first one to examine the effect of APE stress on the quantity and function of EPCs in bone marrow.
EPCs, as precursors of ECs, are considered to originate from hematopoietic stem cells that are positive for CD34 or the more immature marker protein CD133. In 1997, Asahara and colleagues reported that purified CD34+ hematopoietic progenitor cells can differentiate ex vivo to an endothelial phenotype. These cells were named ‘EPCs’ and both showed expression of various endothelial markers, and incorporation into neovessels at sites of ischemia. 9 CD133 is a highly conserved antigen which is expressed by hematopoietic stem cells but is absent from mature ECs and monocytic cells. 37 Bone marrow CD133+ progenitor cells can be activated by inflammatory cytokines and give rise to highly purified ECs with rapid downregulation of CD133 and acquisition of some mature endothelial markers. 38,39 Friedrich et al. 40 reported that the CD133+/34− EPC subpopulation was recognized as a precursor of the CD133+/34+ EPCs, and functionally more potent than the CD133+/34+ EPCs with respect to homing and vascular repair. To differentiate EPCs from hematopoietic stem/progenitor cells, VEGFR2, VE cadherin, or E-selectin were used in subsequent studies. 41 Recently, the combination phenotype of CD133+/VEGFR2+ was considered to be a good marker for defining EPCs. 8,31,32 In the present study, the ratio of CD133+ or CD34+ cells increased in APE. This may imply that APE events could induce proliferative promotion of bone marrow stem cells. The much more striking increase in CD133+ cells in bone marrow suggested that it could be more sensitive and potent than CD34+ cells in APE. However, the quantity of CD133+/VEGFR2+ EPCs decreased significantly in the APE 1D group, which may be the result of APE events inducing acute mobilization of EPCs from bone marrow.
In ischemic tissue, the recruitment and incorporation of EPCs requires a coordinated sequence of multistep events including migration, adhesion and finally differentiation to mature ECs, 8 which reflect their reparative capacity. In this study, after culture in selective medium, 29 the capacity to incorporate into a vascular network, adhesion, migration and the proliferation abilities of EPCs were significantly enhanced in the APE 1D group. The upregulation of EPC functions could be adaptations to the needs of post-APE vascular repair.
By using the eNOS inhibitor, L-NAME, APE-induced EPCs functional changes were blocked. This result suggested that the eNOS/NO pathway may be involved in APE-induced functional alterations in EPCs. This was in agreement with the role of eNOS/NO as a key mediator in stimulating cell proliferation and other functions through activating the phosphoinositide 3-kinase/Akt pathway and as a downstream effecter in VEGF-induced angiogenesis. 42–44
In conclusion, the main contributions of this study were as follows: (1) establishment of an acute PE mouse model; (2) the demonstration that APE events changed the number and functions of bone-marrow derived EPCs; and (3) the demonstration that the eNOS/NO pathway may participate in APE-induced EPCs changes. These findings provide potential insight into the pathophysiological mechanisms of PE. However, further studies are needed to define the mechanisms that underlie the APE-induced changes in the number and activity of EPCs.
Footnotes
ACKNOWLEDGEMENTS
This study was supported by the National Nature Science Foundation of China (30770940 and 30971162), Fund of China 973 program (2009CB522107), the Major International Joint Research Project of Natural Science Foundation of China (30810103904), Fund of Scientific Research Common Program of Beijing Municipal Commission of Education (KM200710025002), Fund of Natural Science Foundation of Beijing (7082012) and Fund for Key Project of Ministry of Education (208002).