
Abstract
Zinc is an essential nutrient for humans; however, this study demonstrated for the first time that an elevated zinc status, created by culturing cells at optimal plasma zinc concentration attainable by oral zinc supplementation, is cytotoxic for normal human bronchial epithelial (NHBE) cells. p53 plays a central role in the modulation of cell signal transduction in response to the stress from DNA damage, hypoxia and oncogene activation. The present study was designed to determine whether the previously reported increased Gadd45 expression and delayed G2/M cell cycle progression in zinc-supplemented NHBE cells is p53-dependent, and to decipher the mechanisms responsible for the regulation of Gadd45 expressions by p53, and elucidate the Gadd45 functions in impaired cell growth and cell cycle progression in NHBE cells. Cells were cultured for one passage in different concentrations of zinc: <0.4 μmol/L (ZD) as severe zinc-deficient; 4 μmol/L (ZN) as normal zinc level in culture medium; 16 μmol/L (ZA) as normal human plasma zinc level; and 32 μmol/L (ZS) as the high end of plasma zinc attainable by oral supplementation. Inhibition of cell growth and upregulation of p53 mRNA and protein expression were observed in ZS cells. Most importantly, ZS treatment also enhanced Gadd45 nuclear protein level and promoter activity, decreased CDK1-Cyclin B1 level and delayed G2/M cell cycle progression. These changes were normalized to those observed in ZN by treating ZS cells with Pifitherin, an inhibitor of p53 transactivation activity. Thus, our findings support the p53 dependency of the Gadd45-CDK1/Cyclin B1-G2/M cell cycle progression pathway in ZS NHBE cells.
Introduction
Bronchial epithelial cells are the physical barrier that separate airway connective tissue and smooth muscle from the airway luminal contents. Exposure to a high concentration of zinc in the air may cause significant health risk. 1 Zinc toxicity can cause acute respiratory tract inflammation together with bronchial hyper-responsiveness. Studies have shown that workers in mining industries had increased polymorphonuclear leukocytes and incidence of pulmonary inflammation. 2 In view of the prevalence and clinical significance of zinc deficiency in human populations, as well as extensive use of zinc supplementation in animal production and to a lesser extend in human populations, we have initiated studies designed to examine the influence of zinc status on the expression of tumor suppressor genes, p53 and the growth arrest and DNA damage-induced gene (Gadd) Gadd45, in normal human bronchial epithelial (NHBE) cells. NHBE cells have been used for this study because they are representative of a cell population capable of undergoing lung tissue transformation and are considered to be progenitor cells for human bronchial cancer.
Tumor suppressor gene p53 is the most frequently mutated gene in cancer 3 and has been implicated in maintaining genomic stability by controlling cell cycle checkpoints and apoptosis following genotoxic stress. 4–9 Stressful agents including ultraviolet (UV) radiation, alkylating agents and ionizing radiation (IR) damage DNA and activated protein. In response to DNA damage, p53 is stabilized and activated as a transcription factor. 10 Activated p53 transactivates its downstream genes, including Gadd45, p21 and Bax. 4,11,12 Subsequently, cell cycle arrest or apoptosis will prevent the cells from passing damage onto mitotic cells.
Gadd45 was originally identified as an mRNA transcript that was rapidly induced in response to UV radiation. Gadd45 is an ubiquitously expressed 21 kDa acidic protein in response to genotoxic agents, and is involved in many biological processes related to maintenance of genomic stability and apoptosis. In addition, Gadd45 has been implicated in interactions with PCNA, p21, CDK1 and MTK1. 13–16 Moreover, Gadd45 also has been shown to regulate G2/M arrest in response to genotoxic stress and to maintain genomic stability. Regulation of Gadd45 induction after DNA damage is complex and may involve both p53-dependent and -independent signaling pathways. Gadd45 induction by IR has been reported to be strictly dependent on normal cellular p53 function. However, Gadd45 induction by UV and methyl methane sulfonate (MMS) does not require p53, although p53 may contribute to the non-IR response of Gadd45. 4
The objectives of our study were to determine whether the increased Gadd45 expression with the blockage of G2/M in zinc-supplemented NHBE cells is p53-dependent or -independent, and to decipher the precise molecular mechanisms responsible for the regulation of Gadd45 expression by p53, and elucidate the Gadd45 functions in impaired cell growth and cell cycle progression in NHBE cells. By the usage of a p53 inhibitor, Pifithrin (PFT), which inhibited the transactivation of p53, this study provides evidence for the first time that the upregulation of Gadd45 is dependent on p53 in the delay in G2/M progression in NHBE cells cultured at optimal zinc concentration, which has been shown to be attainable in the plasma by oral supplementation. 17
Materials and methods
Cell culture and treatment
NHBE cells were purchased from Cambrex Bio Science (Walkersville, MD, USA). Cells were plated at 3500 cells/cm2 in tissue culture dishes containing bronchial epithelial cell growth medium (BEGM), supplemented with 0.5 μg/mL epinephrine, 10 μg/mL transferrin, 5 μg/mL insulin, 0.1 ng/mL retinoic acid, 52 μg/mL bovine pituitary extract, 0.5 μg/mL hydrocortisone, 0.5 pg/mL human recombinant epidermal growth factor and 6.5 ng/mL triiodothyronine (as growth supplements) without antibiotics, and cultured at 37°C in a 5% CO2 incubator. The cells were grown to 80% confluence for six days, and subcultured using trypsin-EDTA at a ratio of 1:8 at passage 3 for experimental zinc treatment.
A zinc-free BEGM baseline media, in which Cambrex omitted the addition of ZnSO4, was used as the zinc-depleted media. This media consisted of bronchial epithelial basal media (BEBM) supplemented with growth components, and contained residue amounts of zinc (<0.4 μmol/L), as detected by flame atomic absorption spectrophotometry. The zinc-free basal medium of <0.4 μmol/L was used as the zinc-depleted medium (ZD). For the other four treatment groups, zinc was added to the media in the form of ZnSO4 so that the only difference between these media was the zinc concentration. For the other media: the zinc-normal medium (ZN) contained 4 μmol/L of ZnSO4; the zinc-adequate medium (ZA) contained 16 μmol/L of ZnSO4; and the zinc-supplemented medium (ZS) contained 32 μmol/L ZnSO4. The ZN medium was used as a comparison to standard culture media and was used as the control group for experiments. The ZA treatment was used as a representative of human plasma zinc levels, and the ZS group was used to represent plasma zinc levels attainable by oral supplementation in humans. After NHBE cells were subcultured and assigned into one of the four corresponding groups, the cells were cultured overnight in ZN media before changing to their respective medium. Cells were then cultured in ZD, ZN, ZA and ZS for six days. PFT was purchased from Sigma-Aldrich (St Louis, MO, USA), and was used to prepare stock solutions in dimethylsulfoxide. PFT was administrated to the cells at the working concentration of 2 μmol/L for 48 h.
RNase protection assay
Total RNA was isolated from NHBE cells by using the RNAqueous Kit (Qiagen, Valencia, CA, USA), according to the manufacturer's instruction. The integrity of the RNA was verified by electrophoresis and quantified by spectrophotometry. The mRNA levels of human genes including PARP, NF-κB, TNF-β, p53, Gadd45, p38, p21, Brca1, PCNA, MDM2 were measured by non-radioactive RNase Protection Assay (RPA) (Pharmingen, San Diego, CA, USA). The human GAPDH probe was also included in the multiprobe and was used as house-keeping gene for normalization. Labeled riboprobes were synthesized using the Non-Radioactive In Vitro Transcription kit with T7 RNA polymerase (Pharmingen), and biotin-dUTP (Roche, Alameda, CA, USA).
RPA was performed using the Pharmingen RPA kit. Each sample contained 10 μg of total RNA from NHBE cells, and 2 μg of the multiriboprobe. The RNA and labeled probes were co-precipitated with ammonium acetate and ethanol, and resuspended in hybridization buffer at 56°C for 16 h. The RNase digestion was performed at 30°C for 45 min, followed by inactivation with proteinase K cocktail, and subsequent precipitation. Protected fragments were separated by tris-urea polyacrylamide gel electrophoresis (PAGE) with precasted gel from BioRad (Hercules, CA, USA). Control sample were processed without RNase digestion and only full-length probes were applied. No protected bands appeared in controls, in which yeast RNA replaced NHBE RNA indicating that digestion was complete. The PAGE gel resolved protected-probes were transferred to nylon membrane and subjected to UV cross-linking. The biotin-labeled protected cDNA transferred onto the membrane were detected by chemiluminescent signal and visualized by X-ray film exposure.
Gadd45 promoter activity assay
Preparation of Gadd45-luciferase
The Gadd45 promoter construct was a kind gift provided by Dr Towia Libermann. The promoter region was isolated and inserted into the plasmid pGL3-basic (Promega, Madison, WI, USA) to generate construct pGL3-Gadd45-Luci plasmid. The plasmid was transfected into Escherichia coli DH5α competent cells (Invitrogen, Grand Island, NY, USA) by standard protocol for transformation. The plasmid was prepared by using Wizard PureFection Plasmid DNA purification system from Promega.
Transient transfection and luciferase assay
NHBE cells were transfected by using Tfx-20 reagent according to the protocol provided by the manufacturer (Promega). NHBE cells were seeded at a density of 2 × 105 cells/well in 24-well plates and cultured for four days in complete medium containing 0, 4.0 or 32.0 μmol/L zinc. Just before transfection, the medium was removed and co-transfection was performed in accordance with Dual-Luciferase reporter Kit protocol from Promega. Briefly, 500 ng of Gadd45 promoter reporter plasmid, which expresses firefly luciferase, were mixed with 10 ng of an internal control plasmid, pRL-SV40, which expresses Renilla luciferase. The mixture of plasmids was combined with transfection reagent and used to transfect the cells. One hour after transfection, the transfection medium was changed to their respective zinc concentration media and continuously cultured for two additional days. Firefly and Renilla luciferase expression signals were measured using the Luminometer TD-20/20 (Turner Designs, Sunnyvale, CA, USA) following the Dual-Luciferase reporter Kit protocol. Firefly luciferase signals, which represent Gadd45 promoter activity, were normalized with the internal control Renilla luciferase signals to overcome the variations of transfection efficiency in different samples.
Cell cycle analysis
DNA contents of cells were assayed by fluorescence-activated cell sorting (FACS). NHBE cells were cultured in ZD, ZN, ZA and ZS media for one passage, trypsinized, washed in PBS (Ca2+, Mg2+ free) and fixed in 70% cold ethanol. Cells were stored at 4°C. For staining, cells were collected by centrifugation, and pellets were suspended in 1.0 mL propidium iodide staining solution (20 μg/mL propidium iodide, 100 U/mL RNase in PBS), and incubated at room temperature for one hour. Staining was quantitated with a FACSCalibur cytometer (Becton Dickinson, San Jose, CA, USA). The cellular events (10,000) were acquired with CELLQuestPro software program (Becton Dickinson). Cell cycle distribution percentages of stained nuclei were calculated by using Modfit LT software (Verity Software House, Topsham, ME, USA). The calibration standard LinearFlow Green and the DNA QC Particle kit were purchased from Molecular Probes (Eugene, OR, USA) and Becton Dickinson, respectively, and were used for verification of instrument performance.
Nuclear and cytoplasmic extract preparation
The NE-PER nuclear and cytoplasmic extraction reagents and the Halt Protease Inhibitor Cocktail Kits (Pierce Biotechnology, Rockford, IL, USA) were used for nuclear and cytoplasmic extract preparation according to the manufacturer's instructions. Nuclear and cytoplasmic extracts were then stored in aliquots at −80°C. Protein concentrations were determined by using the BCA Protein Assay Reagent kit (Pierce). Contaminations of nuclear extracts by cytoplasmic proteins or contaminations of cytoplasmic extracts by nuclear proteins, detected by Western blot analysis of Hsp90 or Oct-1, respectively, were routinely found to be less than 5% in our lab.
Western blot analysis
Nuclear and cytoplasmic protein concentrations were determined by using the BCA kit (Pierce). Forty micrograms of protein were resolved on a 10% sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis and transferred to Hybond ECL nitrocellulose membrane (Amersham Pharmacia Biotech, Germany) by using a mini-transfer system (Bio-Rad, Hercules, CA, USA). Membranes were blocked with 5% non-fat dry milk in PBS-T (10 mmol/L phosphate buffer pH 7.3, 137 mmol/L NaCl, 2.7 mmol/L KCL and 0.1% Tween-20) for one hour at room temperature, prior to incubation with 1 μg/mL of primary antibody from Santa Cruz Biotechnology (Santa Cruz, CA, USA), in PBS-T containing 5% non-fat milk at 4°C overnight. Membranes were then washed three times with PBS-T and blotted with a secondary antibody conjugated with horseradish peroxidase (Santa Cruz Biotechnology) at room temperature for one hour, followed by three washes in PBS-T. The protein was visualized by using the SuperSignal West Pico Chemiluminescent Substrate (Pierce). Anti-Gadd45, anti-p53, anti-Cyclin B1, anti-actin and anti-histone H1 were purchased from Santa Cruz Biotechnology.
Immunoprecipitation of CDK1/CyclinB1
Cells that reached 70% confluency were harvested, and washed with 10 mL of PBS. After washing, the cells were completely resuspended in 1 mL of cold Lysis Buffer containing 1× Protease Inhibitor Cocktail (Pierce). Cells were then placed on ice for 30 min and centrifuged at 10,000
Statistical analyses
Each experiment was repeated at least three times, with each experiment yielding essentially identical results. Data were expressed as means ± SEM. Statistical comparisons were carried out by one-way analysis of variance. Means were examined by the least significant difference post hoc analysis (SPSS Inc, Chicago, IL, USA). P < 0.05 was considered statistically significant.
Results
Zinc supplementation markedly reduced cell growth in NHBE cells
In ZD, ZA and ZS cells, cell growth as measured by cell number per plate, was found to be reduced to 79 ± 1.7%, 84 ± 3.9% and 68 ± 5.0%, respectively, of the ZN cells (Figure 1). The culture of cells in ZD resulted in significant 18% reduction of cellular zinc as compared with control ZN cells (Figure 2). Moreover, cellular zinc level in ZA and ZS cells was 150% and 300% of that of ZN control cells. Thus, cell growth was reduced both by the low-zinc and by the high-zinc status, particularly in ZS cells. Furthermore, a dose-dependent elevation in cellular zinc content was observed as the zinc concentration in the media was increased.

Zinc-deficient and -supplemented cells decreased in cell number. Cell number per plate in normal human bronchial epithelial (NHBE) cells after cultured in zinc-deficient (ZD, <0.4 μmol/L zinc), zinc-normal (ZN, 4.0 μmol/L zinc), zinc-adequate (ZA, 16.0 μmol/L zinc) and zinc-supplemented (ZS, 32.0 μmol/L zinc) media for one passage. Cell number was determined using a hemacytometer. Similar results were obtained from experiments with cells from another subject
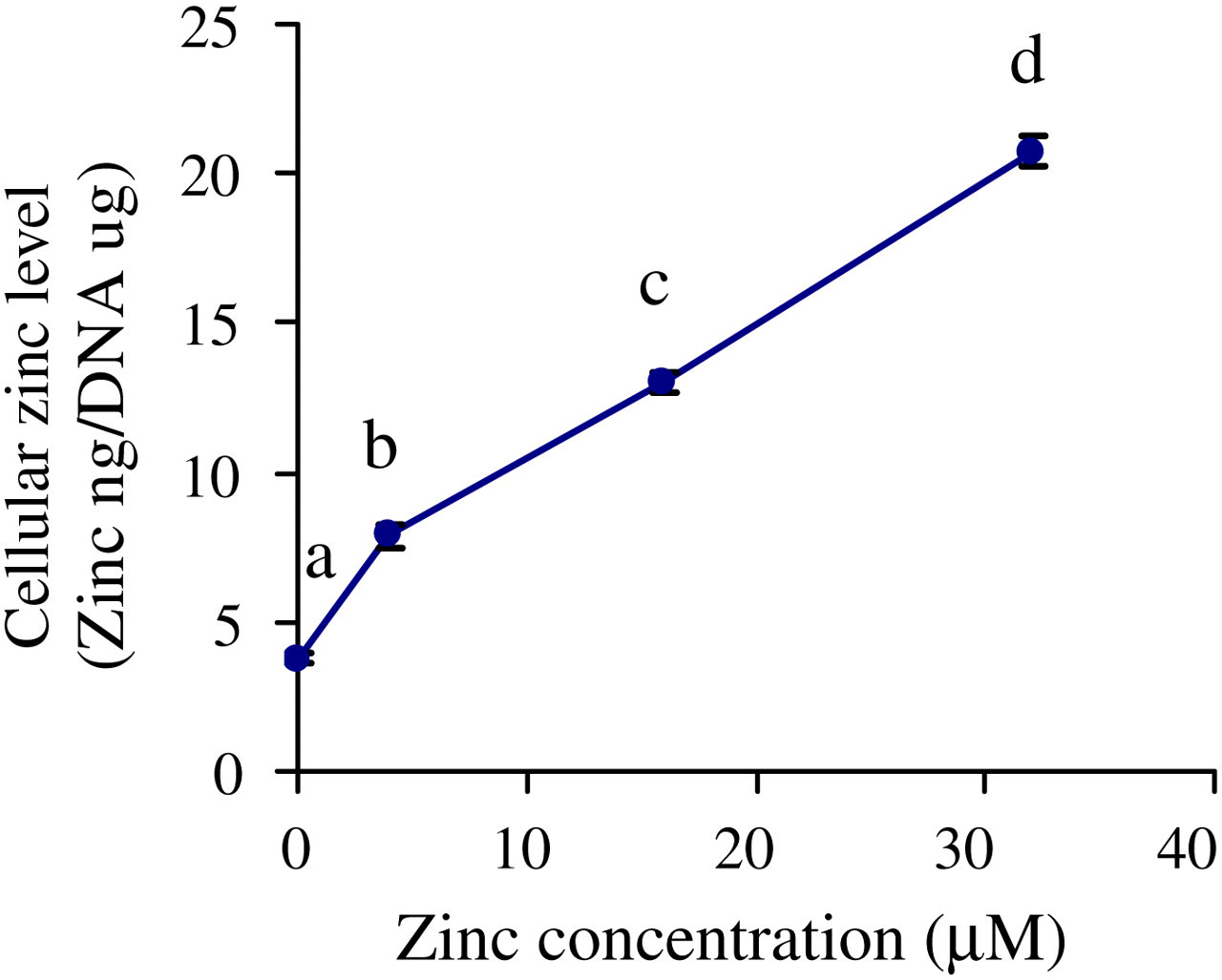
Cellular zinc levels in NHBE cells. Cellular zinc levels in NHBE cells treated with different concentrations of zinc (ZD, ZN, ZA and ZS) for one passage. Cellular zinc was measured by flame atomic absorption spectrophotometry. DNA content per plate in NHBE cells was determined by diphenylamine method. Values are means ± SEM from three experiments. Means with different letters are significantly different (P < 0.05). Similar results were obtained from experiments with cells from another subject. NHBE, normal human bronchial epithelial; ZD, zinc-deficient; ZN, zinc-normal; ZD, zinc-adequate; ZS, zinc-supplemented; SEM, standard error of mean
Zinc supplementation delayed G2/M cell cycle progression in NHBE cells
To uncover the mechanism responsible for the marked cell growth reduction in ZS cells, we next examined the cell cycle progression by flow cytometry. A marked delay in G2/M cell cycle progression was observed in ZS NHBE cells (24.39 ± 0.53%) when compared with ZN cells (13.15 ± 0.12%) (Figure 3). The delay was smaller in magnitude in ZA cells, with 15.59 ± 0.20% of cells in G2/M (Figure 3). In contrast, no change was observed in ZD cells, with 12.98 ± 0.60% cells in G2/M, when compared with ZN cells (Figure 3).

Zinc supplementation blocked G2/M progression in NHBE cells. Cell cycle analysis of NHBE cells was assayed by flow cytometry. Cells were cultured in ZD, ZN, ZA and ZS media for one passage. Washed cells were fixed in ethanol and stained with propidium iodide for DNA content. Flow cytometric data files were collected and analyzed using the CELLQuest program. Cell cycle distribution percentages of stained nuclei were calculated using Modfit LT software. The calibration standard LinearFlow green and the DNA QC particle kit were used for verification of instrument performance. Histograms are representative of three independent experiments. The proportions of cells in G0/G1, S phase and the G2/M ratios are indicated for each treatment group. Similar results were obtained from experiments with cells from another subject. NHBE, normal human bronchial epithelial; ZD, zinc-deficient; ZN, zinc-normal; ZD, zinc-adequate; ZS, zinc-supplemented
p53 mRNA abundance was upregulated in ZS NHBE cells
p53 mRNA abundance was upregulated in ZA and ZS NHBE cells to 129 ± 2% and 196 ± 16%, respectively, of ZN control cells (100 ± 5%) (Figure 4). The mRNA abundance in ZD cells was only 17 ± 4% of the ZN cells. Thus, in ZA and ZS cells, the marked elevations in p53 mRNA levels were associated with increases in cellular zinc levels and reductions in cell growth. Also, the p53 mRNA levels in ZD were associated with a decrease in cellular zinc levels and reductions in cell growth.
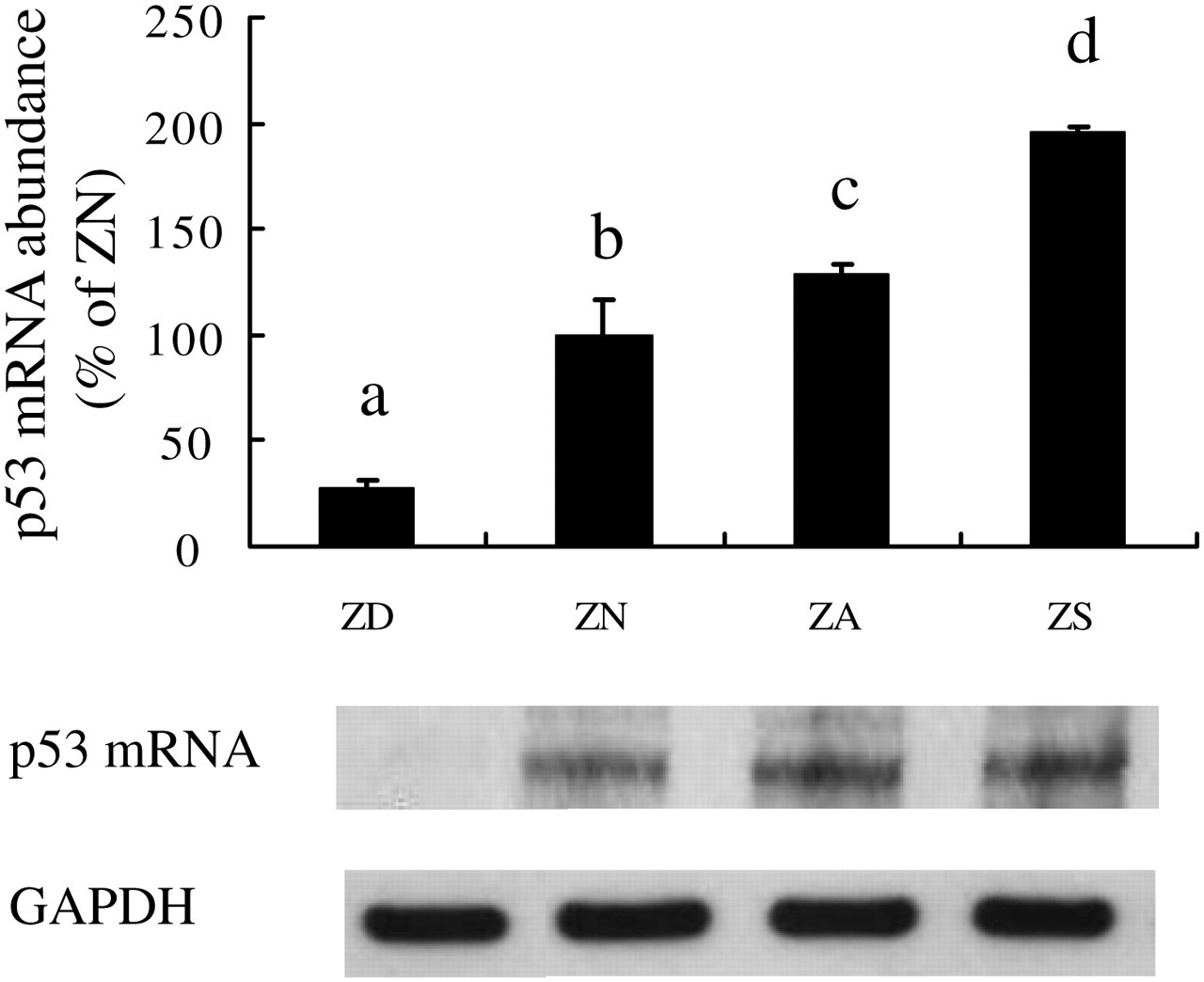
Gadd45 mRNA abundance was upregulated in ZS NHBE cells. Cells were cultured in ZD, ZN, ZA and ZS media. Cells were cultured for one passage in BEGM with zinc added as a supplement to the ZD medium. p53 mRNA level was measured by RPA. RNase protection products were separated on a polyacrylamide gel and quantitated by laser densitometry. L32 was used as an internal reference, and values are expressed as a percentage of ZN controls. Representative samples from each treatment group are shown below the bar graph. Values are means ± SEM from three experiments. Different letters indicate significantly different means, P < 0.05. Treatments with the same letters indicate no significant difference. Similar results were obtained from experiments with cells from another subject. NHBE, normal human bronchial epithelial; BEGM, bronchial epithelial cell growth medium; ZD, zinc-deficient; ZN, zinc-normal; ZD, zinc-adequate; ZS, zinc-supplemented; RPA, RNase Protection Assay; SEM, standard error of mean
p53 protein level was markedly elevated in ZS NHBE cells
In ZS cells, Western blot analysis indicated approximately a four-fold increase in cytoplasmic and a three-fold increase in nuclear p53 protein levels when compared with the ZN cells (Figure 5a and b). The cytoplasmic and nuclear p53 protein levels were increased three- and 2.4-fold, respectively, in ZD cells, as well as 2.9- and 2.8-fold, respectively, in ZA cells, as compared with ZN control cells. These findings indicated that the magnitude of increases in p53 protein among the treatment groups were higher than the increases in mRNA levels, especially for the ZS group. Thus, there may be additional post-transcriptional regulation involved in the enhancement of the protein stability of p53. Because the depression of cell growth induced by an upregulation of p53 protein is well established, the marked elevation in p53 protein in ZS cells may contribute to the observed reduction in cell growth.
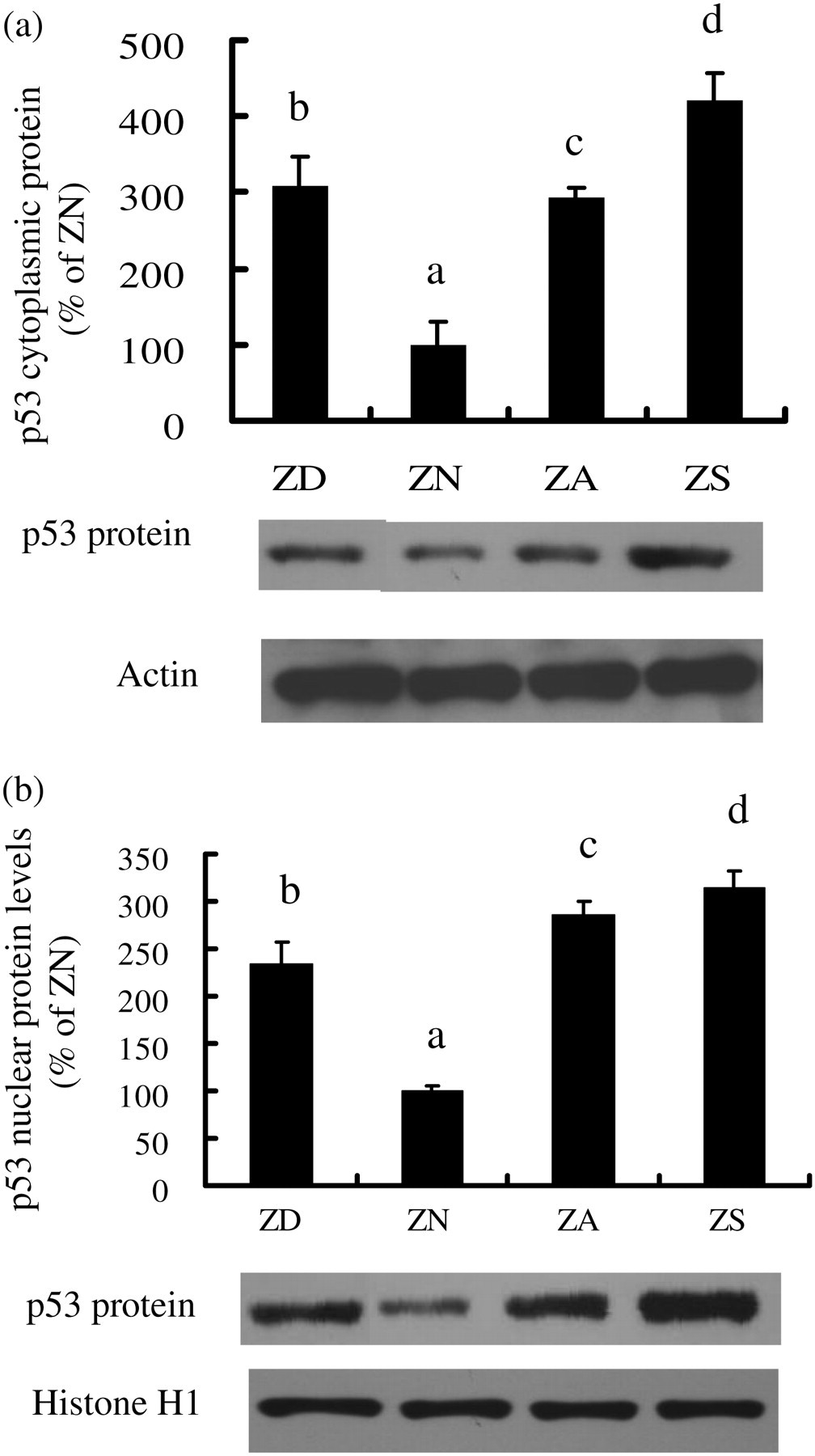
p53 protein level was markedly elevated in ZS NHBE cells. Cells were cultured for one passage in BEGM with zinc added as a supplement to the ZD medium. Nuclear and cytoplasmic protein extracts were separated on 10% polyacrylamide-SDS gels, transferred onto nitrocellulose membranes, and incubated with anti-p53 antibody. Cytoplasmic and nuclear samples were probed with antibodies against Actin and Histone H1, respectively, for normalization. Autoradiography was visualized using enhanced chemiluminescence and quantitated by densitometry. Values are expressed as a percentage of ZN controls. Representative samples, from each treatment group, are shown below the bar graph. Values are means ± SEM from three separated experiments. Different letters indicate significantly different means, P < 0.05. Similar results were obtained from experiments with cells from another subject. NHBE, normal human bronchial epithelial; BEGM, bronchial epithelial cell growth medium; SDS, sodium dodecyl sulfate; ZD, zinc-deficient; ZN, zinc-normal; ZD, zinc-adequate; ZS, zinc-supplemented; SEM, standard error of mean
Usage of p53 transactivation inhibitor PFT alleviated the blockage in G2/M cell cycle progression in ZS NHBE cells
The blockage in G2/M cell cycle progression was alleviated in the ZS-PFT group. Similarly, ZD-PFT and ZN-PFT groups showed the similar amount of cells in G2/M after inhibition of p53 transactivation by PFT (Figure 6). After administration of PFT, the cells in G2/M dropped from 8.91 ± 0.12% to 7.42 ± 0.27% in ZD cells, 10.76 ± 0.98% to 8.82 ± 1.03% in ZN cells and 15.03 ± 0.19% to 8.56 ± 1.04% in ZS cells.

Suppression of p53 by inhibitor Pifithrin alleviated the blockage in G2/M. Cell cycle analysis after the suppression of p53 by inhibitor Pifithrin. Pifithrin were administrated into the medium in 5 μmol/L for 48 h. Cell cycle analysis of NHBE cells was assayed by flow cytometry as described. Cells were fixed in ethanol and stained with propidium iodide for DNA content. Flow cytometric data files were collected and analyzed using the CELLQuestPro program. Cell cycle distribution percentages of stained nuclei were calculated using Modfit LT software. Histograms are representative of three independent experiments. Similar results were obtained from experiments with cells from another subject. NHBE, normal human bronchial epithelial
Usage of p53 transactivation inhibitor PFT caused knock down of Gadd45 protein expression
The approach of suppressing p53 was used to establish whether the upregulation of Gadd45 was associated with elevated p53. Our data showed that the marked elevation of Gadd45 in ZS cells was normalized after application of PFT. After inhibition of p53 transactivation activity by PFT, the Gadd45 protein levels were knocked down to similar levels in all PFT-treated cells, ZD-PFT (87 ± 8%), ZN-PFT (90 ± 3%) and ZS-PFT (92 ± 2%), as compared with ZN-controls (100 ± 17%) (Figure 7).

Upregulated Gadd45 protein was normalized after suppression of p53 by inhibitor Pifithrin. Pifithrin were administrated into the medium in 2 μmol/L for 48 h. Relative nuclear Gadd45 protein levels in NHBE cells. Nuclear protein extracts were separated on 10% polyacrylamide-SDS gels, transferred onto nitrocellulose membranes and incubated with anti-Gadd45 antibody. Nuclear samples were probed with antibodies against Histone H1 for normalization. Autoradiography was visualized using enhanced chemiluminescence and quantitated by densitometry. Values are expressed as a percentage of ZN controls. Representative samples, from each treatment group, are shown below the bar graph. Values are means ± SEM from three separate experiments. Different letters indicate significantly different means, P < 0.05. NHBE, normal human bronchial epithelial; SDS, sodium dodecyl sulfate; SEM, standard error of mean
Usage of p53 transactivation inhibitor PFT normalized Gadd45 promoter activity
The approach of suppressing p53 was used to establish whether the upregulation of Gadd45 promoter activity was associated with elevated p53. Our data showed that the marked elevation of Gadd45 promoter activity in ZS cells was normalized after application of PFT. After suppression of p53 transactivation with the PFT inhibitor, the Gadd45 promoter activity was knocked down to similar levels in all PFT-treated cells (Figure 8).
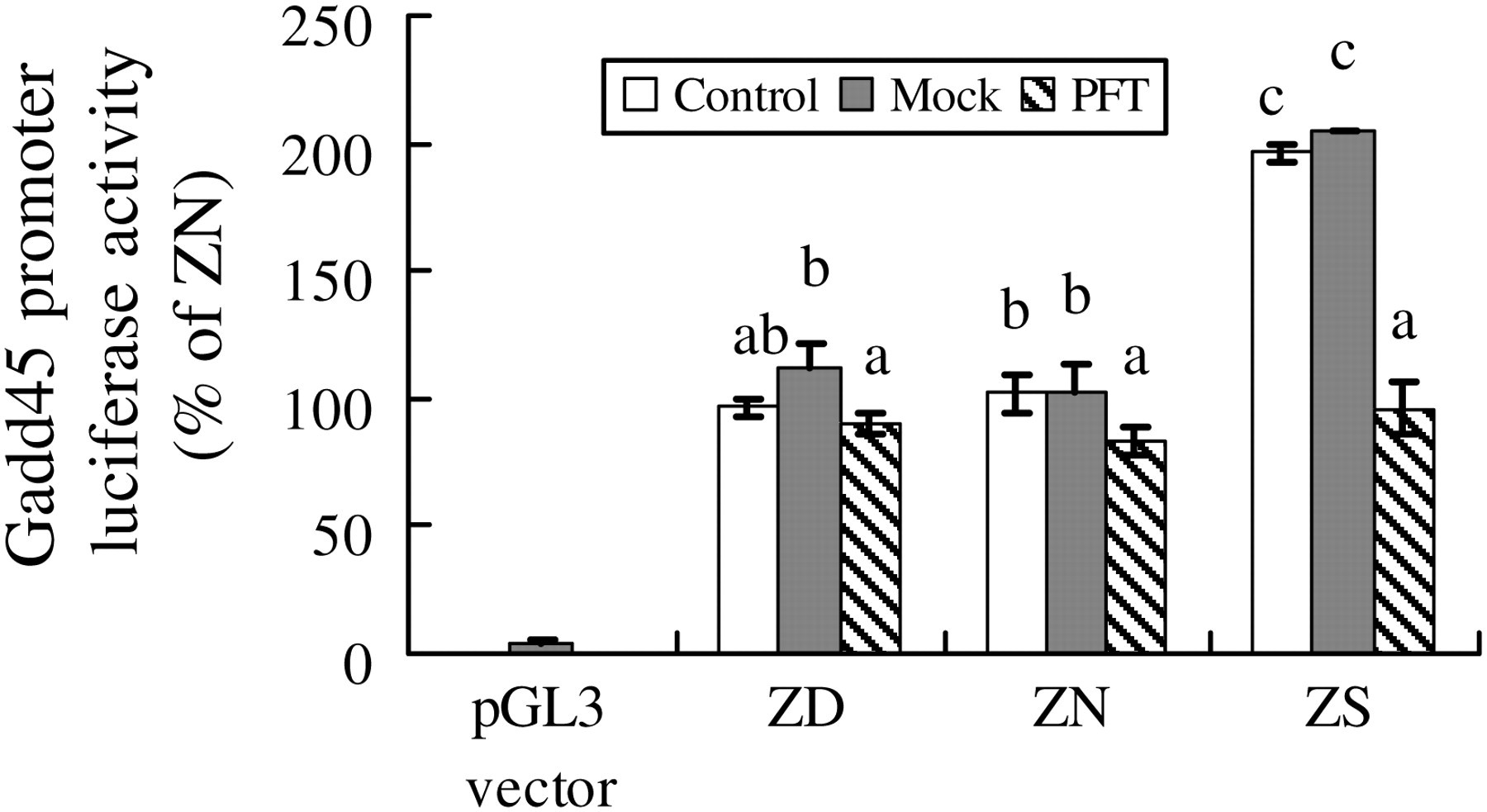
Gadd45 promoter activity after p53 inhibitor Pifithrin. NHBE cells were cultured in media with respective zinc concentration (ZD <0.4 μmol/L; ZN = 4 μmol/L; ZA = 16 μmol/L; and ZS = 32 μmol/L zinc supplemented to the basal medium) for four days. Pifithrin were administrated into the medium in 2 μmol/L for 48 h. Meanwhile, cells were transiently transfected in serum free NHBE media by using Tfx-20 with 500 ng of luciferase reporter construct (pGL3-Gadd45) containing the Gadd45 promoter, together with 10 ng pRL-SV40 as internal control. pGL3 basal vector without carrying any promoter was used as the vector control. After transfection, the cells were cultured in corresponding media for two more days. Cells extracts were then assayed by Dual-Luciferase reporter system and signals were measured by a Luminometer. Values represent means ± SE from three experiments. Data were analyzed by one-way analysis of variance. Different letters indicate significantly different means (P < 0.05); while means with the same letter indicate no significant difference. NHBE, normal human bronchial epithelial; ZD, zinc-deficient; ZN, zinc-normal; ZD, zinc-adequate; ZS, zinc-supplemented; SE, standard error
Dissociation of CDK1/Cyclin B1 in ZS NHBE cells was normalized after administration of p53 transactivation inhibitor PFT
The formation of CDK1/Cyclin B1 complex in ZS NHBE cells was lower compared with ZN control cells (Figure 9): ZS-control (62 ± 4%) and ZS-mock (56 ± 4%), as compared with ZN-control (100 ± 17%) (Figure 9). In ZD cells, the CDK1/Cyclin B1 complex showed no significant changes, ZD-control (110 ± 6%) and ZD-mock (113 ± 7%), as compared with ZN-control (100 ± 6%) (Figure 9). To examine whether this dissociation of the complex was dependent on p53 in ZS NHBE cells, suppression of p53 transactivation by inhibitor PFT was performed. The amount of CDK1/Cyclin B1 complex was quantified by immunoprecipitation in all zinc-treated groups, and the results showed that the depressed CDK1/Cyclin B1 complexes were normalized after suppression of p53 transactivation: ZD-PFT (113 ± 9%), ZN-PFT (129 ± 0%) and ZS-PFT (108 ± 5%), as compared with ZN-control (100 ± 17%).

Dissociation of CDK1/Cyclin B1 in ZS NHBE cells was normalized after suppression of p53 by inhibitor Pifithrin. NHBE cells were cultured in media with respective zinc concentration (ZD < 0.4 μmol/L; ZN = 4 μmol/L; ZA = 16 μmol/L; and ZS = 32 μmol/L zinc-supplemented to the basal medium) for four days. Pifithrin was administrated into the medium in 2 μmol/L for 48 h. Five hundred proteins from whole cell lysate were added to Protein G. The complex was then centrifuged and 10 μg of CDK1 antibody or rabbit polyclonal antibody were added to the precleared lysate. Protein A/G PLUS agarose was added and centrifuged. The 50 μL of 1× Laemmli sample buffer was added to the immunocomplexes as bead pellet. The immunocomplexes were vortexed, heated and loaded onto the 10% SDS-PAGE gel for immunoblotting analysis using Cyclin B1 antibody. NHBE, normal human bronchial epithelial; ZD, zinc-deficient; ZN, zinc-normal; ZD, zinc-adequate; ZS, zinc-supplemented; SDS, sodium dodecyl sulfate; PAGE, polyacrylamide gel electrophoresis
Discussion
The p53 pathway is composed of a network of genes and their products that are targeted to respond to stress signals that impact upon cellular homeostatic mechanisms that monitor DNA replication, chromosome segregation and cell division. 18 In response to a stress signal, the p53 protein is activated in a specific manner, which leads to either cell cycle arrest or cellular apoptosis. 19 The process of cell division is highly ordered and regulated. Checkpoints exist to delay progression into the next cell cycle phase only when the previous step is fully completed. 20 In addition, checkpoints can become activated due to DNA damage, exogenous stress signals, defects during the replication of DNA or failure of chromosomes to attach to the mitotic spindle. Abrogation of cell cycle checkpoints can result in death for a unicellular organism or uncontrolled proliferation and tumorigenesis in metazoans. 21 Receptor proteins detect DNA damage and initiate signal transducers, mediators and effector proteins that phosphorylate targets, including p53, to produce cell cycle arrest at the G1/S, intra-S or G2/M checkpoint until the lesion undergoes repair.
G2/M progression is triggered by the maturation-promoting factor MPF, which is a complex of CDK1/Cyclin B1. p53 has been shown to regulate G2/M checkpoint by transactivating its target genes, including Gadd45. In addition, Gadd45 has been shown to interact with PCNA. 1,14 Gadd45 also interacts with CDK1 and inhibits its kinase activity by causing dissociation of the Cyclin B1/CDK1 complex. 22 Moreover, induction of Gadd45 resulted in the G2 arrest associated with translocation of cytoplasmic Cyclin B1. 23 Overexpression of Cyclin B1 and Cdc25 have been reported to abrogate the Gadd45-induced G2/M arrest. Furthermore, the induction of G2/M arrest by Gadd45 requires p53 and depends on the type of DNA damage. Human cells with downregulated Gadd45 expression or lymphocytes from Gadd45 knock-out mice failed to arrest at G2/M following UV or radiomimetic MMS. 24
Zinc supplementation inhibits normal human bronchial cell proliferation
Our studies, designed to investigate the influence of zinc depletion and supplementation on the cell signaling and cell cycle regulation in NHBE cells, demonstrated that cell growth was inhibited at optimal zinc concentration, which has been shown to be attainable in the plasma by oral supplementation. In zinc-supplemented NHBE cells, cellular zinc was three-fold higher than that of ZN control cells (Figure 2), and cell growth as measured by cell number was decreased to 60% of ZN cells (Figure 1). Our study is the first to demonstrate that zinc supplementation, within the physiological range, is cytotoxic and reduces the growth of NHBE in primary culture. Thus, the high physiological level of zinc also imposes an adverse effect in normal cells. Controversy towards the efficacy of zinc supplementation in the prevention of cancer growth has been reported. Several studies have indicated that high cellular zinc levels inhibit cancer cell growth. 25–27 Moreover, studies also reported that treatment with high levels of zinc not only inhibited proliferative cancer cells, but also induced G2/M arrest. 28,29 Yet, an epidemiological study has reported an increase in risk for cancer development with usage of high levels of zinc supplement. 30 Even though zinc supplementation can inhibit cancer cell proliferation, the cytotoxic effect of supraphysiological zinc levels (90 and 900 μmol/L) resulting in the induction of DNA fragmentation has been observed in human primary liver cells. 31
G2/M blockage was found in ZS NHBE cells
In our studies, cell cycle analysis by flow cytometry indicated a G2/M blockage in ZS cells (Figure 3). In ZS cells, G2/M blockage was accompanied by three- to four-fold induction of nuclear and cytoplasmic p53 proteins levels, respectively (Figure 5a and b), and by a two-fold increase in mRNA expression (Figure 4) as compared with ZN control cells. These results suggest that zinc supplementation may cause DNA fragmentation which induces p53 expression. With the increase in p53 expression in ZS cells, G2/M blockage occurs to delay the transition and hinder cell growth. The underlying mechanism by which p53 blocks the G2/M cell cycle progression needs to be further investigated. This action is probably caused by the downstream p53 regulator, Gadd45, binding to CDK1, which decreases CDK1 kinase activity, and hinders the cell progression from G2 to M. 22 High amounts of zinc in supraphysiological levels have been reported to induce DNA fragmentation in human cells, implying that DNA damage may occur with high zinc supplementation. Gadd45 induces G2/M arrest through p53-dependent and -independent pathways, decreases CDK1 kinase activity and changes subcellular Cyclin B1 levels. These are all events that have been observed in many cell types exposed to stresses. 24,32–35 The effect of Gadd45 on the G2/M transition may be due to its ability to dissociate complexes of Cyclin B1 and CDK1 as well as inhibit the activity of CDK1/Cyclin B1 in vitro. 22,36 In the present study, no G1 arrest was observed. This is consistent with data from another study which showed that microinjected Gadd45 did not efficiently inhibit CDKk2/Cyclin E or cause G1 arrest. 22
Upregulation of p53 is associated with cell growth suppression and G2/M block in ZS NHBE cells
Our results demonstrate that cell growth suppression was accompanied by marked induction of p53 expression, suggesting that p53 may be the main regulator in cell growth inhibition in zinc-supplemented NHBE cells. To determine whether p53 is the single regulator of p53-dependent pathway in response to cellular stress, we administrated p53 inhibitor PFT to suppress p53 transactivation activity. Our cell cycle analysis findings demonstrated that the G2/M blockage was alleviated by PFT and normalized to similar levels as in all zinc-treated cells (ZD-PFT, ZN-PFT and ZS-PFT in Figure 6), which suggested G2/M blockage is dependent on enhanced p53 expression in ZS NHBE cells.
Upregulation of Gadd45 is p53-dependent in ZS NHBE cells
To test whether Gadd45 expression was dependent on p53 pathway, we quantified Gadd45 protein amount and assayed the promoter activity after the p53 inhibitor PFT treatment. Results from these treatments showed that the Gadd45 expression and promoter activity were dependent on the p53 pathway (Figures 7 and 8). This finding is consistent with results obtained with p53 null cells, which failed to arrest at G2/M. Our finding suggested that the upregulation of Gadd45 is dependent on p53 at both transcriptional and translational levels.
p53 dissociated CDK1/Cyclin B1 complex in ZS NHBE cells
Cell division relies on the expression of cyclins that bind and activate cyclin-dependent kinases to promote cell cycle progression to initiate mitosis. Association of Cyclin B with the Cyclin-dependent kinase CDK1 induces CDK1 kinase activity to promote progression through mitosis. 37 However, during a G2/M cell cycle checkpoint, Cyclin B dissociates from CDK1, producing a decrease in the complex and delaying the progression through mitosis. 38 To test whether p53 dissociates CDK1/Cyclin B1 complex and leads to G2/M delay in ZS NHBE cells, we examined the complex formation of the CDK1 and Cyclin B1 by immunoprecipitation. The amount of CDK1 and Cyclin B1 complex was decreased in ZS NHBE cells with the treatment of p53 inhibitor PFT (Figure 9). Thus, our results suggest that the upregulation of p53 induced by zinc supplementation dissociates the formation of the Cdk1 and Cyclin B1 complex and delays the G2/M transition.
In summary, the present novel findings indicate that zinc supplementation, in a physiologically relevant system, exerted an adverse effect in normal cells, markedly elevated Gadd45 and p53 expressions, depressed CDK1/Cyclin B1 complex formation and inhibited G2/M progression in NHBE cells. A better understanding of the mechanisms involved in the protection of normal cells from zinc cytotoxicity is essential for the identification of potential targets, and may contribute to the development of future therapeutic approaches for the treatment of human lung diseases associated with zinc toxicity.
Footnotes
ACKNOWLEDGEMENTS
The assistance provided by Ms Noella A Bryden in the analyses of trace minerals in Dr Richard A Anderson's Lab at the Diet, Genomics and Immunology Laboratory, Beltsville Human Nutrition Research Center, Agricultural Research Service, United States Department of Agriculture is gratefully acknowledged. This work was supported by US Department of Agriculture National Research Initiative Grant 2002-35200-12241 (KYL) and USDA ARS CRIS Project 1235-51530-052-00D (NWS).