
Abstract
This is a Minireview covering landmarks or milestones in the development of erythropoietin (EPO). Thirty-nine landmark advances have been identified, which cover the period 1863–2003. Several reports are included that directly support these original landmark advances. This Minireview also updates some of the advances in EPO research since my last Minireview update on EPO published in this journal in 2003. The areas of EPO research updated are: sites of production; purification, assay and standardization; regulation; action; use in anemias; extraerythropoietic actions; adverse effects; and blood doping. The new reports on the use of EPO in the therapy of myocardial infarction; stroke and other neurological diseases; diabetic retinopathy and other retinal diseases are also covered.
Introduction
The story of the development of erythropoietin (EPO) is one of the most fascinating stories in the history of medicine. A goal of this article is to trace the landmark findings in EPO research, which represent the milestones in an evolving process leading to the development of one of the most important advances in the treatment of anemia and may provide an important therapeutic intervention in stroke, myocardial infarction and perhaps retinopathy. Several reports are also included, which directly support these original landmark findings:
1863 – Jourdanet D 1 – Observed that people living at high altitude in Mexico had more viscous blood than people living at sea level.
1891 – Viault F 2 – Observed an increase in red cell counts in people after traveling from Lima, Peru to Morococha, a mining village at 15,000 feet above sea level.
1906 – Carnot P, DeFlandre Cl 3 – Reported that serum from rabbits after bleeding when injected into normal recipient rabbits stimulated red cell production. They postulated a humoral control of red cell production and named this substance ‘Hemopoietine’.
1932 – Sandor G 4 – Demonstrated a reticulocytosis in recipient rabbits when they were injected with serum from donor rabbits exposed to hypoxia.
1936 – Hjort E 5 – Injected plasma from donor rabbits, following bleeding, into normal recipient rabbits, and produced a reticulocytosis in the recipients.
1943 – Krumdieck N 6 – Demonstrated that serum removed from rabbits after bleeding produced an increase in reticulocytes when injected into normal recipient rabbits.
1948 – Bonsdorff E, Jalavisto E 7 – Gave the humoral factor that stimulates red cell production, previously called ‘Hemopoietine’, the name ‘Erythropoietin’.
1950 – Reissmann K 8 – Reported when one partner in parabiotic rat experiments was exposed to hypoxia, while the other breathed normal air, that an increase in erythroid cells occurred in the bone marrows and an increase in hemoglobin and reticulocytes in peripheral blood of both partners – providing strong support for a humoral factor being produced by the hypoxic partner which passed over through the parabiotic circulation to stimulate erythropoiesis in the partner breathing normal air.
1953 – Erslev A 9 – Reported an increase in hematocrit and reticulocyte counts in normal rabbits following the infusion of large volumes of plasma from donor rabbits, following a bleeding stimulus.
1955 – Plzak L, et al. 10 – Developed the first quantitative and specific assay for EPO when they demonstrated stimulation of radioactive iron incorporation in red cells following injection of plasma from anemic rats into normal rats.
1957 – Jacobson LO, et al. 11 – Found that bilateral nephrectomy in rats prevented the rise in erythropoietin in plasma after a bleeding stimulus.
1959 – Erslev A 12 – Reported an increase in a pool of early erythroid progenitor cells, but not late erythroblasts, in rabbits injected with serum from rabbits made anemic by bleeding followed by retransfusion.
1961 – Fisher JW, Birdwell B 13 – Reported an elevation in erythropoietin levels in perfusates of isolated dog kidneys perfused with blood containing cobalt; Kuratowska Z, et al. 14 – reported an increase in EPO levels in the perfusates of isolated rabbit kidneys perfused with blood at a reduced oxygen tension; providing direct evidence that the kidney produced erythropoietin; and Cotes PM, Bangham DR 15 – developed a quantitative bioassay for EPO in mice made polycythemic by exposure to air at a reduced pressure.
1966 – Cotes PM, Bangham DR 16 – Developed International Reference Standard A Erythropoietin.
1973 – Paulo LG, et al. 17 – Reported stimulation of erythropoietin production by prostaglandins E1.
1974 – Erslev A 18 – Demonstrated that the isolated perfused rabbit kidney produced erythropoietin when perfused with serum-free media and that puromycin blocked EPO production in this system.
1977 – Miyake T, et al. 19 – Reported the purification of human erythropoietin; Zanjani ED et al. 20 – reported the liver as the primary site of EPO production in the fetus.
1979 – Garcia JF, et al. 21 – Developed a radioimmunoassay for erythropoietin using purified EPO.
1985 – Lin FK, et al. 22 – Reported the cloning and expression of the erythropoietin gene; Jacobs K et al. 23 – reported the isolation and characterization of genomic and cDNA clones of human erythropoietin; McGonigle RJS, et al. 24 – reported that anemic uremic end-stage renal disease patients had higher EPO levels than normal yet they remained anemic; whereas a good correlation was seen between hematocrit and serum EPO levels in anemic patients with normal renal function but no correlation was seen in anemic uremic patients.
1986 – Winearls CG, et al. 25 – Found that recombinant human erythropoietin was effective in the treatment of anemic renal disease patients on chronic hemodialysis.
1987 – Eschbach JW, et al. 26 – Reported the correction of the anemia of end-stage renal disease with recombinant human erythropoietin. Results of a combined phase I and II clinical trial.
1988 – Koury ST, et al. 27 – Reported the localization of EPOmRNA in interstitial cells in the mouse kidney; Lacombe C, et al. 28 – found EPOmRNA in peritubular cells in the mouse kidney.
1989 – D'Andrea AD, et al. 29 – Reported the expression cloning of the murine EPO receptor; US FDA 30 – approved recombinant human erythropoietin for use in the treatment of patients with anemia of chronic renal failure.
1990 – D'Andrea AD, Zon D 31 – Reported the elucidation of the structure and activation of the erythropoietin receptor subunit.
1991 – Koury ST, et al. 32 – Reported the presence of EPOmRNA in hepatocytes; Semenza GI, et al. 33 – reported hypoxia-inducible nuclear factors that bind to an enhancer element in the human erythropoietin gene in the regulation of EPO production; Fandrey J, Jelkmann WEB 34 – reported that the cytokines interleukin-1 and tumor necrosis factor-alpha inhibited EPO production.
1995 – Wang GI, Semenza GL 35 – Reported the purification and characterization of hypoxia-inducible factor I; Erslev AJ, Besarab A 36 – reported that despite the same or higher serum EPO levels, the erythrokinetic rates in anemic uremic patients were almost half that of hematologically stable subjects.
1996 – Fisher JW, et al. 37 – Reported EPOmRNA in the interstitial cells of the hypoxic monkey kidney.
1998 – Sakanaka M, et al. 38 – Reported in vivo evidence that erythropoietin protects neurons from ischemic damage.
2003 – Calvillo L, et al. 39 – Reported that recombinant human erythropoietin protects the myocardium from ischemia–perfusion injury.
In addition, an update of my Minireview entitled: ‘Erythropoietin. Physiology and pharmacology update’, published in Experimental Biology and Medicine in 2003 40 is also presented.
Our story begins with a report in 1863 by Denis Jourdanet who found that the blood of people living at high altitude in Mexico was more viscous than that of people living at sea level. 1 This was followed by a finding of Viault 2 in 1890 that people traveling from Lima, Peru to Morococha, a mining village at an elevation of 15,000 feet, had an increase in their red cell count. The first report that postulated a humoral control for red cell production was in 1906 3 when Paul Carnot, Professor of Medicine at The University of Paris (picture shown in Figure 1), working with his colleague Madame Cl DeFlandre, published their work in which they demonstrated that when serum from rabbits following bleeding was injected into normal recipient rabbits, an increase in red cell production occurred in the recipients. They gave the name ‘Hemopoietine’ for this humoral factor. In a paper published in 1918, Paul Carnot made the following comment: ‘The regeneration of new red blood cells after blood letting: We have observed with Mlle DeFlandre that this repair is under the influence of a humoral process and that an animal's blood or serum extracted during a period of active hematic regeneration and injected into a second animal, induces in the latter a hematopoietic flare-up. This is evidenced by increased bone marrow activity and by hematic proliferation. We have given this substance the name “Hemopoietine”’. In 1966, an international conference on erythropoietin in New York was dedicated to Professor Paul Carnot. 41 In a visit to Paris in 1965, and with the assistance of Professor Marcel Bessis, I met Mme Coquin Carnot, daughter of Paul Carnot, at that time a research scientist in The Laboratory of Histology, Faculty of Medicine, University of Paris, who gave me the picture of her father shown in Figure 1.

Paul Carnot. First to propose along with his colleague Mlle DeFlandre that red cell production is regulated by a humoral factor, which they called ‘Hemopoietine’ (Reproduced from an article by Fisher JW.
41
Introduction of an International Conference on Erythropoietin. An N Y Acad Sci 1968;
Carnot and DeFlandre's experiments were difficult to reproduce by several investigators over the years and their work was controversial. An important consideration is that the donor rabbits in Carnot's experiments were not severely anemic after the loss of only 30 mL of blood as Jelkmann 42 has pointed out. The donor rabbits received only 5–9 mL of serum. The donor animals of the investigators who later were successful in confirming Carnot's experiments were more anemic and they also injected larger volumes of plasma or serum into the recipients. In 1932, Sandor 4 reported experiments in which he demonstrated a reticulocyte response in rabbits injected with serum from donor hypoxic rabbits. Experiments by the Norwegian scientist Hjort 5 in 1936, which have been overlooked over the years, demonstrated that when he injected plasma from donor rabbits, following bleeding, into recipient rabbits, a prompt reticulocytosis was observed. In 1997, with the assistance of Sverre Halvorsen, I was fortunate to be able to contact Erling Hjort, who was 98 y old at that time, regarding his experiments. In 1947, Krumdieck 6 also reported an increase in reticulocytes in recipient rabbits injected with serum from donor rabbits following bleeding. Erslev 9 reported in 1953 a series of impressive experiments in which he infused large volumes (50 mL) of plasma from donor rabbits into normal recipient rabbits and observed an increase in reticulocytes and hematocrit in the recipients. The experiments that really re-awakened interest in a humoral control of red cell production were the parabiotic rat experiments reported by Reissmann 8 in 1950. He exposed one partner in the parabiosis to low oxygen tension and the other partner breathed normal air. Both partners were found to have erythroid hyperplasia in their bone marrows and an increase in reticulocytes and hemoglobin in peripheral blood. They concluded that a humoral factor was produced by the hypoxic partner which passed over into the animal breathing normal air to stimulate erythropoiesis. This provided strong support for a humoral factor controlling erythropoiesis. In 1948, this humoral factor was given the name ‘Erythropoietin’ by Bonsdorff and Jalavisto. 7
Sites of production of EPO
Reissmann's 8 classical parabiotic rat experiments in 1950 and Erslev's 9 studies in 1953 confirming Carnot's experiments re-awakened interest in EPO. A search for the site of production of EPO began with the report by Jacobson et al. 11 that bilateral nephrectomy in rats abolished the rise in plasma EPO after bleeding. The removal of the pituitary, adrenal, spleen, gonads and 90% of the liver did not affect the rise in plasma EPO after bleeding. 11 In order to rule out the possibility that the kidney played a ‘permissive’ role in EPO production and to provide more direct proof that the kidney produced EPO, Fisher and Birdwell 13 carried out studies in which they found an elevation in EPO in perfusates from isolated dog kidneys perfused with blood containing cobalt. This was followed by studies reported later that year by Kuratowska et al. 14 in Poland that perfusates from isolated rabbit kidneys perfused with blood at reduced oxygen tension when injected into recipient mice, produced a reticulocytosis. Fisher and Langston 43 in 1967 further confirmed these studies when they reported an increase in EPO levels in the perfusates of isolated dog kidneys perfused with blood containing cobalt or blood at reduced oxygen tension. In 1974, Erslev 18 reported an increase in EPO in perfusates from isolated rabbit kidneys perfused with serum-free medium and that puromycin, an inhibitor of protein synthesis, blocked this increase in EPO in perfusates from the isolated perfused kidneys. This provided further proof that de novo synthesis occurred in the kidney. Mach et al. 44 in 1983 demonstrated mRNA translation from cobalt-treated rat kidneys into biologically active EPO when injected into oocytes from frogs. In 1988, Koury et al. 27 reported EPOmRNA in peritubular interstitial cells of mouse kidneys and LaCombe et al. 28 also found EPOmRNA in the peritubular region of mouse kidneys. Fisher et al. 37 found extremely high levels of EPOmRNA in interstitial cells of the hypoxic monkey kidney using a 645-base pair Kpnl–BglII arrangement of monkey cDNA, which had been subcloned into the plasmid vector PGEM4Z, generating both anti-sense and sense probes, which were labeled with [X-33P] for in situ hybridization studies. Figure 2 shows in situ hybridization studies on hypoxic monkey kidneys 37 and anemic mouse kidneys 27 demonstrating EPOmRNA in peritubular interstitial cells; thus providing proof for EPO production in interstitial cells in both murine and primate kidneys. In 1977, Zanjani et al. 20 demonstrated that the liver was the primary site of EPO production in the fetus and later reported a liver-to-kidney switch in EPO production after birth which was completed by 40 days postpartum. In 1991, Koury et al. 32 reported EPOmRNA in hepatocytes in close proximity to central veins in the livers of anemic transgenic mice.

In situ hybridization studies on hypoxic monkey kidneys (C and D) (reproduced with permission from Wiley-Blackwell Publishing Co. from an article by Fisher JW, et al.
13
Erythropoietin production by interstitial cells of hypoxic monkey kidneys. Br J Hematol 1996;
Purification, assay and standardization of EPO
The purification of EPO was accomplished for the first time by Miyake et al. 19 in 1977. The big breakthrough apparently occurred following Miyake's arrival in Chicago in 1975 from Kumamoto, Japan with a concentrate of 2550 L of urine from patients with aplastic anemia. These investigators were able to purify human urinary EPO to homogeneity with a specific activity of 70,400 units/mg. Human erythropoietin (huEPO) is a glycoprotein with a molecular mass of 30.4 kDa; half of its molecular weight is sugars. The mature protein in peripheral blood contains 165 amino acids. HuEPO has three N-linked oligosaccharide chains at amino acid positions 24, 38 and 83 and one O-linked oligosaccharide chain at position 126. 19,22,23 The sugar moieties protect EPO from degradation by the liver. Goldwasser and his colleagues at The University of Chicago made this purified EPO available to the National Institutes of Health in Bethesda, MD to be distributed to investigators the world over for their research utilizing the radioimmunoassy for EPO.
The earliest assays for EPO were very crude and involved an increase in peripheral blood reticulocytes. In 1955, Plzak and Fried 10 reported an increase in radioactive iron incorporation in red cells in normal rats injected with plasma from donor rats made anemic by bleeding. This led to the development of the first quantitative assay for EPO which was still rather crude. The sensitivity of the assay was increased several folds after hypophysectomy, starvation, transfusion or hypoxia-induced plethora. A major breakthrough was the work of Cotes and Bangham 15 in 1961 in which they developed a bioassay for EPO using mice made polycythemic by exposure to an atmosphere at a reduced pressure. The posthypoxic or transfusion-induced polycythemic mouse assay became the international reference standard bioassay for EPO. Cotes and Bangham 16 at the National Bureau of Standards, at Mill Hill, London, England, in 1966 developed International Reference Standard (IRP) A Erythropoietin, which was the first IRP for EPO to be used worldwide by investigators to standardize their EPO preparations for their research. IRP A was equivalent to 1.48 mg of freeze-dried crude anemic sheep serum EPO. At the time IRP A was depleted in 1972, the second International Reference Standard B Erythropoietin was developed.
In vivo bioassays are highly variable, lack sensitivity, are difficult to carry out and are expensive. In 1979, with the availability of purified human urinary EPO, Garcia et al. 21 developed a radioimmunoassay (RIA) for EPO. The RIA for EPO is unable to distinguish between sialylated and desialylated EPO. When the National Institute of Health was provided purified human urinary EPO by Eugene Goldwasser's laboratory for distribution, investigators worldwide were able to develop RIAs for EPO in their own laboratories to carry out their research. Using a very sensitive RIA for EPO with EPO purified to homogeneity from an EPO producing baby hamster kidney cell culture, Mason-Garcia et al. 45 reported in 153 hematologically normal human subjects a range of 1–27 mU EPO/mL, which was equivalent to approx. 5 pmol/L. There was no significant difference between male and female subjects. A circadian rhythm was seen with the peak concentration occurring at 16:00 h in the evening and lowest concentration at 08:00 h in the morning.
Regulation of EPO production
One of the most significance advances in the research on EPO over the past 20 y has been the finding that EPO gene expression is a complex process between DNA and nuclear proteins. The finding in 1991 by Semenza et al. 33 and Beck et al. 46 that hypoxia-induced gene expression is due to a 50 bp hypoxia-inducible enhancer that is 120 bp 3′ to the polyadenylation site was very important in elucidating the mechanism of EPO gene expression. Semenza and Wang 47 reported in 1992 that a nuclear factor was induced by hypoxia via de novo protein synthesis that binds to the huEPO gene enhancer at a site required for transcriptional activation. Wang and Semenza 35 reported in 1995 the purification and characterization of hypoxia-inducible factor (HIF)-1, which is involved in the regulation of DNA binding activity induced by hypoxia. This HIF was found to bind to an enhancer element localized 3′ to the EPO gene. 33 When Wang and Semenza 35 purified HIF-1 they found that the DNA complex contained a heterodimeric basic-helix or helix-PAS factors to which they gave the name HIF-1 alpha and HIF-1 beta.
Both HIF-1 and HIF-2 have been postulated to control EPO gene expression. Jiang et al. 48 reported in 1996 the following proof that HIF-1 plays a role in EPO gene transcriptional activation in hypoxic cells: (i) exposure of cells to low O2, cobalt or deferrioxamine induced HIF-1 and EPO in a similar manner; 49–51 (ii) a 3-bp substitution at site 1 eliminated binding and enhancer activity of HIF-1; 33 and (iii) treatment of hypoxic cells with 2-aminopurine, a protein kinase inhibitor, or cycloheximide, a protein synthesis inhibitor, both blocked induction of HIF-1 and EPOmRNA formation. 47,49 On the other hand, as Jelkmann 52 has outlined, HIF-2 (rather than HIF-1) may be the main physiological transcription factor inducing EPO gene expression during hypoxia. First, HIF-2 alpha is found dominantly in EPO-producing peritubular fibroblasts in rat kidney while HIF-1 alpha is mainly present in renal tubular cells and has very different cell protective functions. 53,54 EPOmRNA induction is almost completely abolished by HIF-2 alpha RNA interference (RNA), as shown in studies in human hepatoma cells (Hep3B), Kelly neuroblastoma or mouse astrocytic cell lines. 55,56 HIF-2 alpha also binds preferentially to the hypoxia response elements (HRE) within the native EPO enhancer, whereas HIF-1 alpha may bind to the isolated HREs. 57 Postnatal knockout of HIF-2 alpha, but not HIF-1 alpha, results in anemia in mice. 58 Renal 59 and hepatic 57 EPO production in response to hypoxia is decreased in HIF-2 alpha knockout mice.
Several signal transduction pathways have been postulated to play a role in hypoxia induction of HIF and subsequent EPOmRNA production. The most studied pathways are adenosine and eicosanoids. Paul et al. 60 reported in 1988 that when exogenous adenosine was perfused through an isolated rat kidney in nanamolar concentrations, an increase in EPO levels was seen in the perfusate. An adenosine antagonist and adenosine deaminase diminished EPO levels in the renal perfusates. In the same year, Ueno et al. 61 found that the hemisulfate salt of adenosine and 5′-N-ethylcarboxamideadenosine (NECA), a selective adenosine A2 receptor agonist, both produced a significant increase in radioiron incorporation in red cells of polycythemic mice. Theophylline, a non-selective adenosine antagonist, was found to block the increase in radioiron incorporation in red cells in polycythemic mice induced by adenosine and NECA. Nagashima and Karasawa 62 reported in 1996 that the adenosine agonists NECA, CHA and KW-3902 produced dose-dependent increases in serum levels of EPO in normal rats. KF17837, an adenosine A2a antagonist, inhibited NECA-induced EPO production. Fisher and Brookins 63 in 2001 found that CGS-21580, a selective adenosine A2a agonist, significantly increased levels of EPO in normoxic Hep3B cell cultures and Hep3B cell levels of EPOmRNA. SCH-58261, a selective adenosine A2a antagonist, inhibited the increase in Hep3B cell medium levels of EPO following exposure to hypoxia. Bakris et al. 64 reported in 1990 that theophylline, a non-selective adenosine receptor antagonist, inhibited the elevation in plasma levels of EPO in patients with erythrocytosis following kidney transplantation. Batmunkh et al. 65 reported in 2006 that Il-1, an inhibitor of EPO production, strongly activated NF-kappaB, which is likely a suppressor of the EPO promotor in HepG2 cell cultures. Treating the cells with dibutyryl cyclic adenosine monophosphate (cAMP) inhibited the activation of NF-KB by Il-1. They concluded that NF-KB is probably the primary transcription factor by which cAMP counteracts the inhibition of EPO gene expression by IL-1. On the other hand, Gleiter et al. 66 in 1997 reported that the adenosine A1 receptor blockers, DPCPX and KW-3902, adenosine A1 and A2 agonists CHA and CGS 21680; adenosine reuptake inhibitors, dipyridamole and soluflazine; and the adenosine deaminase inhibitor EHNA, all failed to influence serum levels of EPO in rats following exposure to carbon monoxide. In addition, Grenz et al. 67 in 2007 reported a lack of effect of extracellular adenosine generation and signaling on renal EPO secretion during hypoxia.
Paulo et al. 17 reported in 1973 that prostaglandins E1 increased EPO production. Gross et al. 68 in 1976 found that PGE2, the primary renal eicosanoid, increased radioiron incorporation in red cells in polycythemic mice and increased EPO levels in the perfusates of isolated dog kidneys. Fink and Fisher 69 found that beta-2 adrenergic agonists increased EPO production when injected into polycythemic mice. Jelkmann et al. 70 found in 1979 that indomethacin, an inhibitor of prostaglandin production, blocked the effects of albuterol, a beta-2 adrenergic agonist, on EPO production in the isolated perfused dog kidney. Radtke et al. 71 found that albuterol induced EPO and prostaglandins release in the isolated perfused dog kidney. This provided support for the effects of beta-2 adrenergic activation on EPO production being due to increased prostaglandins production. Nelson et al. 72 in 1983 reported that the eicosanoid PGI-2 and its metabolite 6-ketoPGE-1 produced significant increases in EPO production in polycythemic mice. Fisher and Brookins 63 proposed in 2001 the following model for adenosine and eicosanoids in the regulation of EPO production: first, hypoxia results in the depletion of cellular ATP; this is followed by increased ectonucleotidase activity resulting in increased levels of adenosine in extracellular fluid, which stimulates adenosine A2a and A2b receptors; hypoxia activation of the phospholipase A2 pathway causes increased eicosanoid production; an increase in PKA and PKC; phosphorylaton of HIF-1 and other kinase-C-activated transcriptional factors resulting in increased production of EPOmRNA. Transcription factors ATF-1 and CREB-1 have been reported by Kvietikova et al. 73 to constitutively bind to the HIF-1 DNA recognition site.
Jelkmann et al. 74 reported in 1991 that inhibition of EPO production by phorbol ester is associated with downregulation of protein kinase C isoenzyme in hepatoma cells in culture. Fandrey and Jelkmann 34 also reported in 1991 that interleukin-1 and tumor necrosis factor-alpha inhibit EPO production in vitro in an EPO-producing human hepatoma (HepG2) cell line. Jelkmann et al. 75 postulated in 1994 that inhibition of EPO by cytokines may contribute to the anemia of inflammatory diseases. Frede et al. 76 reported in 1997 that EPO gene expression is suppressed after lipopolysaccharide or interleukin-1 injections in rats. Hellwig-Burgel et al. 77 reported in 1999 that under normoxic conditions interleukin 1-B and tumor necrosis factor-alpha caused a moderate activation of HIF-1 DNA binding. In hypoxia, these two cytokines strongly increase HIF-1 activity compared with hypoxia alone. These investigators concluded that cytokine-induced inhibition of EPO production is not mediated by impairment of HIF-1 function. In contrast to their effects in suppressing EPO production, neither Il-1B nor TNF-alpha decreased vasoactive endothelial cell relaxing factor (VEGF) production. Fandrey and Jelkmann 34 proposed in 1991 that IL-1 and TNF-alpha may be responsible for the defect in EPO production in severe systemic and renal inflammatory diseases. Interleukin-1 and tumor necrosis factor-alpha play a pathophysiological role to suppress EPO production in inflammatory diseases. 78 However, further work is necessary to determine whether these cytokines play a physiological role in a negative feedback system in the day-to-day control of EPO production.
Actions of EPO on erythropoiesis
Jacobson et al. 79 reported in 1957 that mice injected with erythropoietically active serum from anemic donor mice showed the appearance of erythroblasts in the bone marrow in a short period of time. In 1959, Erslev 12 reported that an increase occurred in an early pool of erythroid cells, but not erythroblasts, in rabbits made anemic by bleeding and retransfusion. They observed a wave of erythropoiesis moving through the bone marrow in these rabbits following a bleeding stimulus. After years of research on the mechanism of action of EPO on erythropoiesis, two distinct early erythroid progenitor cells were revealed in the erythroid cell compartment of the bone marrow: the BFU-E (burst forming unit-erythroid), which is not sensitive to EPO, but seems to gain sensitivity to EPO as it matures; 80,81 and the CFU-E (colony forming unit-erythroid), which is a more mature erythroid cell and is the primary target cell for EPO. 82,83 As the CFU-E matures, it gradually loses its sensitivity to EPO. Using purified EPO, investigators found that EPO provides a proliferative signal to the BFU-E 31,84 and a differentiative signal to the CFU-E. 85
Another important advance in the development of EPO was the expression and cloning of the murine EPO receptor by D'Andrea et al. 29 in 1989 and their subsequent elucidation of the EPO receptor sub-unit and activation in 1990. 31 The number of receptors per erythroid cell was estimated to be approx 1000 per cell. 31 Reticulocytes and mature erythroid cells do not contain receptors for EPO. 86 D'Andrea and Zon 31 have reported that the murine EPO receptor has two molecules, one 85 and the other 100 kDa, which are associated with EPO binding. High-affinity (kDa, 1 μmol/L) and low affinity (kDa, 2 μmol/L) binding sites have been reported in the extracellular domain of the murine EPO receptor. 87 When EPO binds to this preformed dimer, a conformational change occurs in this dimer, which causes associated Janus family tyrosine protein kinase 2 (JAK-2) molecules to move in close proximity and stimulates their activation by transphosphorylation. JAK2 molecules then phosphorylate tyrosine residues of the EPO receptor in the cytoplasmic domain. 88,89 These tyrosine residues serve as docking sites for intracellular proteins containing Src homology 2 (SR2). Cellular proliferation after phosphorylation of tyrosines in the cytoplasmic domain and EPO receptor activation involve: the Ras/MAP kinase pathway; 90,91 tyrosine phosphorylation of the adapter protein IRS2; 92 and activation of both STAT5A and STAT 5B. 93 Jones et al. 94 reported that cross-linking EPO to COS cells expressing the human EPO receptor gave apparent molecular weights of 66 and 100 kDa. The overall action of EPO on erythroid cells is to rescue these cells from programmed cell death (antiapoptosis).
Use of EPO in the anemia of chronic renal failure
The approval of rEPO by the Food and Drug Administration (FDA) in 1989 30 and the introduction of recombinant EPO (rEPO) for the treatment of the anemia of chronic renal failure have been two of the most important therapeutic advances in clinical medicine. The quality of life of the renal-failure patient has been markedly improved. Winearls et al. 25 found in 1986 that recombinant huEPO (rhuEPO) was effective in the treatment of anemic renal disease patients on hemodialysis. Eschbach et al. 26 reported in 1987 the results of a phase I and II clinical trial in which it was found that rhuEPO corrected the anemia of end-stage renal disease (ESRD). There is little doubt that the primary cause of anemia in chronic renal failure is an insufficient amount of EPO to meet the demands for new red cell production created by the decrease in renal function and the uremia. 25,26 In addition, there is some resistance to the patients' own endogenous EPO. 24,36,95,96 As shown in Figure 3, a good correlation has been shown between hematocrit and serum levels of Epo in patients with iron deficiency or sickle cell anemia. 24 However, no correlation was seen between hematocrit and serum levels of EPO in patients with chronic renal failure (Figure 3). 24 Anemic uremic patients with ESRD were found to have higher levels of EPO than hematologically normal human subjects and yet they remained anemic. 24 Several investigators 24,36,95–98 have reported uremic inhibitors of erythropoiesis and increased resistance to the ESRD patient's own endogenous EPO, especially the uremic patient prior to going on dialysis. Radtke et al. 96 have identified spermine as an inhibitor of erythropoiesis in patients with chronic renal failure. In addition, serum from uremic patients that inhibited CFU-E and BFU-E colony growth in normal human bone marrow cultures was fractionated by Sephacryl gel filtration. 99 Fractions in a molecular weight range of 47,000–150,000 Da suppressed CFU-E and BFU-E growth.
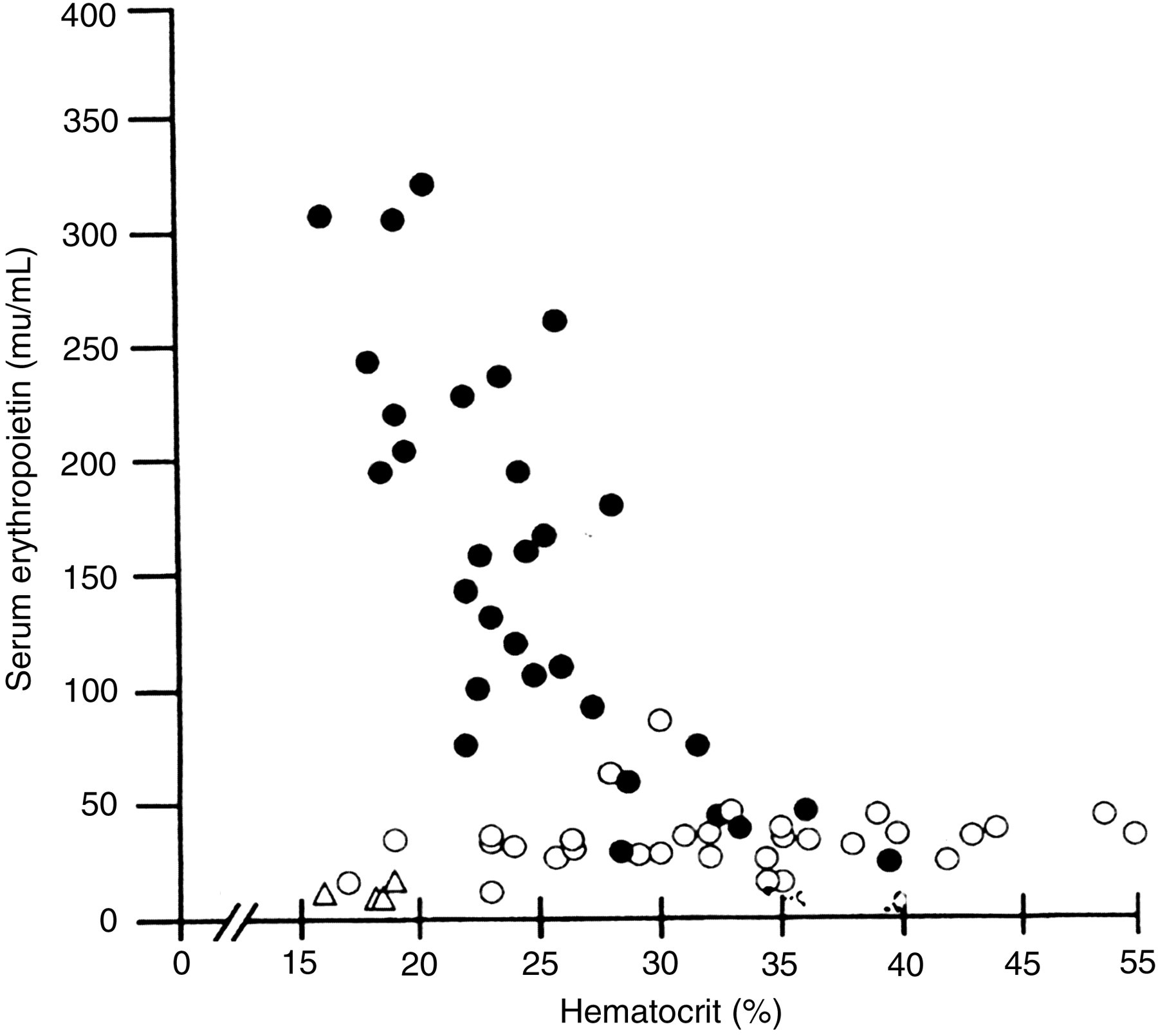
Relation between serum erythropoietin concentration and hematocrit levels in children with normal renal function and varying degrees of anemia due to sickle cell disease or iron deficiency (•), in children with end-stage renal failure (o) and in anephric children (▵). Based on data from McGonigle et al.
24
Erythropoietin and inhibitors of erythropoiesis in the development of anemia in children with renal disease. J Lab Clin Med 1985;
Parathyroid hormone (PTH) in concentrations (7.5–30 U/mL) comparable to those found in the blood of uremic patients produced significant inhibition of BFU-E growth in murine bone marrow cultures. 100 Meytes et al. 100 suggested that PTH excess may be one pathway for the genesis of the anemia of uremia. ESRD patients undergoing ambulatory peritoneal dialysis have also been reported to have a higher hematocrit than hemodialysis patients. 101,102 In 1995, Erslev and Besarab 36 reported that even with the same or higher levels of EPO, the erythrokinetic rates of anemic uremic patients were almost half the rate of hematologically normal human subjects. On the other hand, Eschbach et al. 103 reported that pharmacological doses of EPO improve the anemia in almost all anemic renal dialysis patients. However, most ESRD patients being treated with EPO do not achieve the target level of hemoglobin (120 g/L) recommended by the NKF-DOQI anemia guidelines. 104 Recent guidelines indicate that the hemoglobin level should not exceed 120 g/L. 105 In 1989, the participants in a USA multicenter clinical trial with rEPO suggested the following reasons as causes for the ESRD patient not achieving the target hemoglobin: inflammation, functional iron deficiency, insufficient rEPO dose and co-morbid factors. 106
Clinical uses of EPO in other anemias
In addition to its use in the treatment of the anemia of chronic renal failure, 25,26,102 rhuEPO has been approved to treat HIV patients who have anemia associated with treatment with azidothymidine (AZT); 107 cancer patients who are suffering from anemia associated with treatment with chemotherapy; 108,109 perioperative surgery 110 and autologous blood donation before surgery; 111 clinical trials of rhuEPO have been reported in patients receiving bone marrow transplants; 112 premature infants with anemia; 113 anemia of chronic disease, such as rheumatoid arthritis; 114 the myelodysplastic syndrome 115 ; and the anemia of pregnancy. 116
Extraerythropietic actions of EPO
EPO has been reported to stimulate signaling and mitosis in vitro in cardiomyocytes, 39,117 cardiomyoblasts, 118 astrocytes 119 and endothelial cells. 120–122 rhuEPO, due to its effect in inhibiting programmed cell death (apoptosis), has also been found to prevent ischemia-induced neuronal death. 123–125 In 1998, Sakanaka et al. 38 published the landmark paper reporting in vivo evidence that EPO protects neurons from ischemic damage. These investigators found that infusion of EPO into the lateral ventricles of gerbils prevented ischemia-induced learning disability and rescued hippocampal CA-1 neurons from lethal ischemic damage. This neuroprotective action of EPO was confirmed by counting synapses in the hippocampal CA1 region. These authors 38 postulated that EPO may exert its neuroprotective effect by reducing nitric oxide-induced formation of free radicals or by antagonizing their toxicity. Brines et al. 123 reported in 2000 that rhuEPO crossed the blood–brain barrier to protect against experimental brain injury. These authors 123 found that systemic administration of rhuEPO to mice attenuated injury after blunt trauma (Figure 4). Systemic injections of rhuEPO can protect against traumatic brain injury, 126 acute ischemic injury to the spinal cord 118 or ischemic damage to retinal neurons. 127 In addition, EPO has been reported to produce a protective effect in animal models for multiple sclerosis 128 and status epilepticus. 129 A pilot clinical study was carried out by Ehrenreich et al. 130 in patients with ischemic stroke consisting of a safety part and an efficacy part. In these randomized double-blind studies, 40 patients received either rEPO or saline. Cerebrospinal fluid levels of EPO were 60–100 times higher than that of non-treated patients. These authors 130 concluded that intravenous high doses of rEPO are well tolerated in acute ischemic stroke and are associated with an improvement in clinical outcome at one month. They recommended that a large-scale clinical trial is warranted. Since that time, Ehrenreich et al. 131 have undertaken a larger scale study in 522 patients with acute ischemic stroke and unexpectedly a very high number of patients needed to receive tissue plasminogen activator. These authors concluded that based on analysis of total-intent-to-treat and per-protocol populations, that their study was negative, raised safety concerns and that none of the outcome parameters showed favorable effects of rEPO. Further clinical trials are necessary to determine whether EPO is safe and effective in acute ischemic stroke.
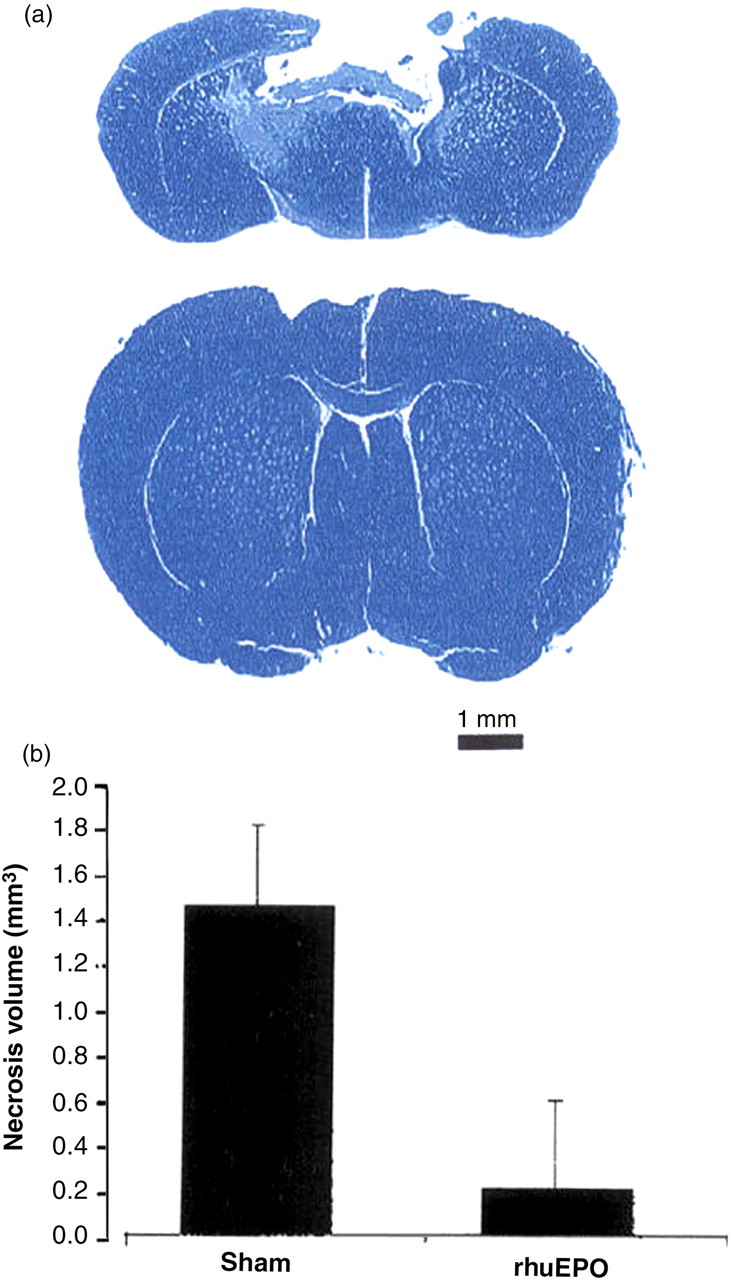
Systemic administration of recombinant human erythropoietin (rhuEPO) attenuates injury after blunt trauma. (a) Mice receiving a non-penetrating blow to the frontal cortex exhibited extensive cavitary necrosis when examined 10 days after injury if treated with saline (upper) in contrast to the minimal injury observed if they had received rhuEPO (lower). Cresyl violet stain of representative brain sections through site of injury. (b) Results of a representative experiment for rhuEPO (5000 units/kg BW) given 24 h before delivery of the impact, n = 6 animals each group; P < 0.05. The experiment was repeated four times with similar results. Reproduced with permission from Proceedings of the National Academy of Sciences, from an article by Brines ML, et al. Erythropoietin crosses the blood-brain barrier to protect against experimental brain injury. 2000;
EPO expression in the retina has been reported and was found to be highly elevated in the vitreous fluid of patients with diabetic macular edema. 132
Grimm et al. 133 in 2002 reported in the adult mouse retina that acute hypoxia dose-dependently stimulated expression of EPO via HIF-1 alpha stabilization. The protective effect of hypoxic preconditioning was mimicked by systemically applied EPO that crosses the blood–retina barrier and prevents apoptosis even when given therapeutically after a light insult. Chung et al. 134 in 2009 reported that the retinal expression of EPO and its receptor (EPOR), as well as Janus kinase 2 phosphorylation, are each tightly linked to a specific time after oxidative stress in anticipation of daily light onset, which is consistent with the physiological protection against daily light-induced, oxidatively mediated retinal apoptosis. On the other hand, because EPO is produced in the eye, increasing EPO levels in the eye could potentially worsen diabetic retinopathy because EPO stimulates angiogenesis. 135 For example, Chen et al. 136 found that inhibiting EPOmRNA expression with siRNA was effective in suppressing retinal neovascularization, suggesting that siRNA is a potententially useful pharmaceutical intervention for treating proliferative retinopathy. Zhang et al. 137 found that intravitreal injection of EPO protects both retinal vascular and neuronal cells in patients with early diabetes. EPO due to its antiapoptotic (inhibition of programmed cell death) effect has been labeled an endogenous retinal survival factor. 138 Further clinical trials are necessary to determine whether intravitreal injections of EPO will prove to be beneficial or will actually worsen diabetic retinopathy as well as age-related macular degeneration and other retinal diseases.
Calvillo et al. 39 reported in 2003 that EPO inhibits apoptosis in adult rat cardiomyocytes exposed to 28 h of hypoxia (3% O2). In addition, employing a rat model for coronary–ischemia reperfusion, it was demonstrated that the administration of rhuEPO (5000 units/kg, intraperitoneaaly daily for 7 days) reduces cardiomyocyte loss by approx. 50%, which was sufficient to normalize hemodynamic function within one week. 39 These authors 39 found that animals administered vehicle alone exhibited an increase in left ventricular end diastolic pressure (LVEDP) as a function of infarct size (Figure 5). In contrast, LVEDP of animals receiving rhuEPO was unrelated to infarct size (Figure 5). EPO has also been reported to reduce myocardial infarction and left ventricular function decline after coronary ligation in rats. 139 These studies suggested a potential therapeutic role for rhuEPO in the treatment of myocardial ischemia and infarction by preventing apoptosis and postinfarct deterioration in hemodynamics. Namiuchi et al. 140 reported that high serum levels of EPO are associated with smaller infarct size in patients with acute myocardial infarction who undergo successful primary percutaneous coronary intervention. Klopsch et al. 141 found that intracardiac injection of EPO induces stem cell recruitment and improves cardiac functions in a rat myocardial infarction model. Pittenger and Martin 142 suggest that human bone marrow stem cells (hMSCs) isolated from adult human bone marrow provide an excellent model for development of stem cell therapeutics. Phase I and II studies of EPO were carried out by Andreotti et al. 143,144 in patients with acute myocardial infarction with a primary endpoint of infarct size, quantified by CK-MB time–concentration curve, left ventricular wall median score index and pattern of contrast-enhanced magnetic resonance imaging. We will need to await the results of these studies before a recommendation can be made to administer EPO to all patients following an acute myocardial infarction.
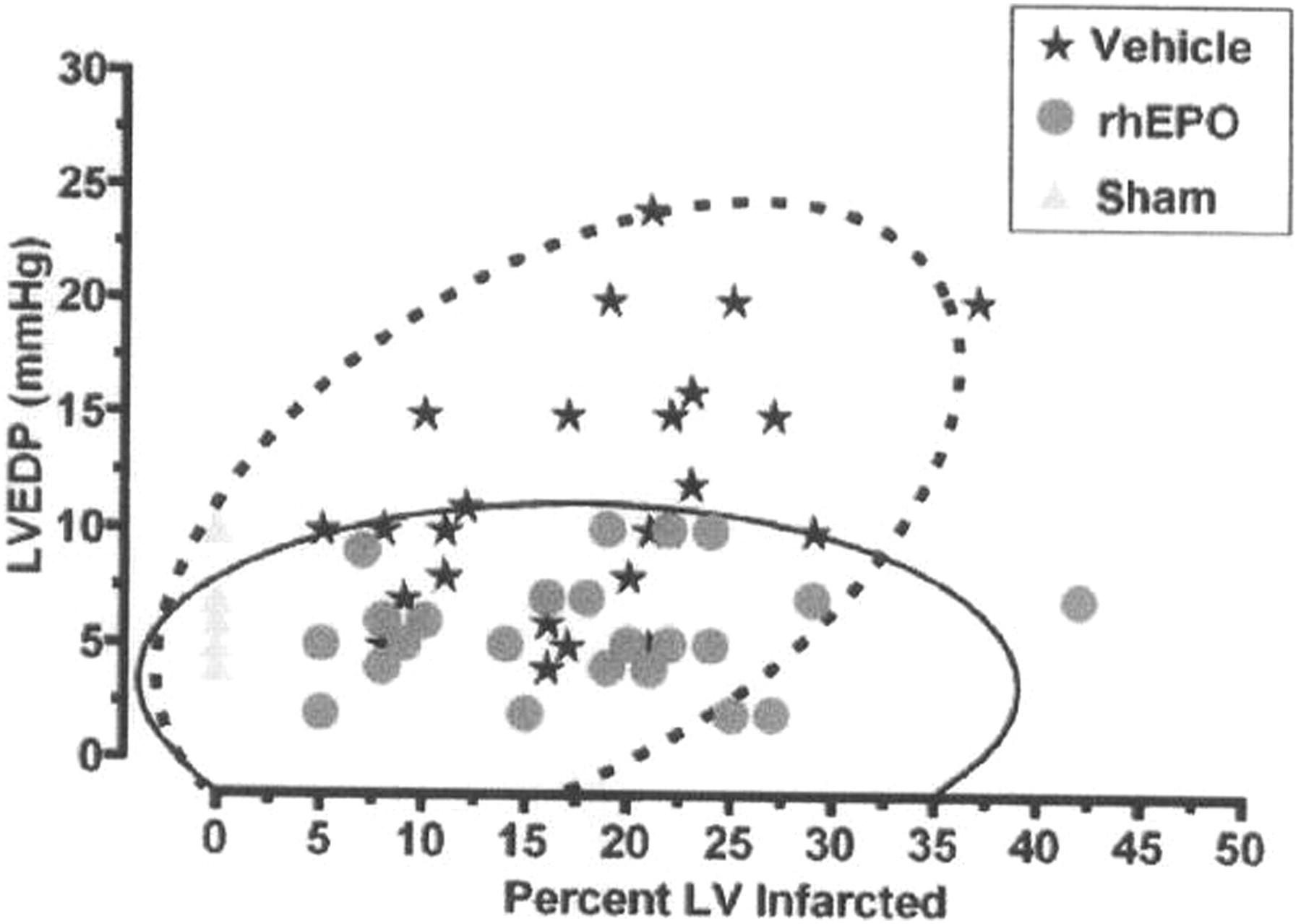
Correlation between left ventricular end diastolic pressure (LVEDP) and infarct size after seven days of reperfusion. Animals administered vehicle alone exhibited an increase in LVEDP as a function of increasing infarct size. In contrast, LVEDP of animals receiving recombinant human erythropoietin (rhuEPO) was unrelated to infarct size. Bivariate normal distribution of LVEDP versus infarct size at the 95% level is indicated by the ellipses, saline treatment by the dashed lines, and rhEPO by the solid lines, respectively. Reproduced with permission from Proceedings of the National Academy of Sciences, from an article by Calvillo L, et al. Recombinant human erythropoietin protects the myocardium from ischemia–reperfusion injury and promotes beneficial remodeling. 2003;
Adverse effects of erythropoiesis-stimulating agents
The adverse effects of erythropoiesis-stimulating agents (ESAs) that have been reported are: dose-dependent increases in blood pressure or worsening of an existing hypertension (seen in 30–35% of patients receiving ESAs); usually seen during the first three months of treatment. It is recommended that the physician give attention to fluid status and use of antihypertensive medication in patients experiencing an elevation in blood pressure during therapy with ESAs. A moderate dose-dependent increase in platelet counts has also been reported. 145,146 Thromboses may occur in the shunts of dialysis patients during ESA therapy. The revision of the shunts early and prophylaxis against thromboses is recommended. 147–149 Patients with pre-existing vascular diseases on dialysis should be monitored carefully since they may be more prone to develop thromboembolic events. 148,150
Side-effects that have only occasionally been seen are: seizures, hypertensive crisis, transient increases in serum potassium and phosphate levels, skin reactions and palpebral edema. Rare cases of anaphylactoid reactions have been observed. About 5% of patients receiving ESAs have been shown to have flu-like symptoms but these usually resolve with continued treatment. 149,151
Henke et al. 152 in 2003 carried out randomized, double-blind, placebo-controlled trials to treat the anemia in patients with head and neck cancer and concluded that EPO corrects the anemia but does not improve cancer control or survival. Disease control might even be impaired. Pirker et al. 153 carried out studies with Darbepoetin alfa in 600 previously untreated extensive-stage small-cell lung cancer treated with platinum plus etoposide. Treatment with darbepoetin alfa maintained hemoglobin levels significantly higher than placebo controls. However, there was no statistically significant difference between overall survival between the treatment groups. These authors concluded that their studies reinforce the benefit of ESAs in reducing transfusions and their neutral impact on survival in patients with chemotherapy-induced anemia. LaMontagne et al. 154 reported that recombinant epoetins do not evoke a physiological response on epoetin positive receptor (EPOR)-bearing tumor cells in two preclinical models for breast cancer (MDA-MB-231 and MDF-7 cell lines) assessed by growth, migration, and cytotoxic challenge in these preclinical in vivo models. Sinclair et al. 155 reported the lack of functional EPO receptors (EpoR) on non-hematopoietic cells. These authors 155 reported that functional EPO receptor is undetectable in endothelial, cardiac, neuronal and renal cells. Swift et al. 156 reported that EpoRmRNA levels in 209 human cells lines representing 16 tumor types were low compared with ESA-positive controls. EpoR protein production was evaluated in a subset of 66 cells lines using a novel anti-EpoR antibody. EpoR positive control cells had an estimated 10,000–100,000 dimers/cell. In contrast, 54 of 61 lines had EpoR protein levels below 100 dimers/cell. Cell lines with the highest EpoR protein levels (400–3200 dimers/cell) were studied further and though one cell line bound detectable levels of [I-125]-rHuEpo, none showed evidence of ESA-induced EpoR activation. In a recent update on ESAs and clinical trials in oncology, Aapro and Spivak 157 stated that: ‘ESAs decrease RBC transfusion needs and sustain targeted hemoglobin levels, and this ESA response does not significantly impact overall survival or mortality when ESAs are used within guidelines and labeling. However, based on the currently available data and meta-analysis, the use of ESAs has to be carefully balanced against any possible risk for higher mortality.’
The US Food and Drug Administration issued a drug safety communication on February 20, 2010 ‘requiring all drugs called erythropoiesis stimulating agents (ESAs), listing Procrit, Epogen and Aranesp, to be prescribed and used under a risk management program known as a risk-evaluation and mitigation strategy (REMS). 158 FDA required Amgen, the manufacturer of these products, to develop a risk management program because studies show that ESAs can increase the risk of tumor growth and shorten survival in patients with cancer who use these products. Studies also show that ESAs can increase the risk of heart attack, heart failure, stroke or blood clots in patients who use these drugs for other conditions’.
Blood doping and ESAs
Blood doping was introduced in the 1970s and is defined as the use of blood transfusions to increase the total circulating red cell volume artificially for the purpose of enhancing both maximal oxygen uptake and performance in endurance sports. Cazzola 159 has proposed a global strategy for prevention and detection of blood doping with EPO and related drugs to provide a test to differentiate between endogenous and exogenous EPO. As reported by CON (Conitato Olimpico Nazionale Italiano) (Italian National Olympic Committee), blood doping with rhuEPO is very common in professional cross-country skiing and cycling. 160 Hematocrit values above 60% have been reported in cyclists in the Netherlands in the 1990s with the use of rhuEPO. 159
Some authors 161 have speculated that rhuEPO could have been involved in the death of some of these cyclists in the Netherlands early in the 1990s. Erythrocytosis at this level compounded by dehydration during exercise predisposes athletes to thromboembolic disorders and has caused deaths in some athletes. Thus, the medical risks of blood doping with rhuEPO to increase red cell mass in order to enhance athletic performance are very great and their use for this purpose is to be condemned.
The World Anti-Doping Agency has established Athlete Biological Passport Operating Guidelines, 162 which they first proposed in 2002. This doping control approach is based on the detection of markers of a substance or its metabolites. It does have limitations when an athlete is using substances on an intermittent and low-dose basis which may go undetected. Monitoring and identifying possible doping is the objective of the Athlete Biological Passport. The requirements for the hematological module, such as enhancement of oxygen transport, including rEPO abuse and any form of blood transfusion, should collect markers of erythropoiesis. The following markers were recommended by the World Anti-Doping Agency as part of the hemogram if blood doping is suspected: hematocrit, hemoglobin, red cell count, reticulocytes, mean corpuscular volume and mean corpuscular hemoglobin concentration. Gore et al. 163 have recommended an index of stimulation with a blood profile score if EPO abuse is suspected.
Summary and conclusions
Thirty-nine landmark discoveries in the development of EPO have been identified during the period of 1863–2003. These advances represent milestones in the history of EPO. Each advance opened new horizons in the treatment of erythropoietic, neurological, cardiovascular and retinal diseases. In addition to the anemia of chronic renal failure and the anemia associated with chemotherapy of malignant diseases or HIV infection, several other anemias have been reported to respond to EPO. Clinical trials currently underway should clarify whether EPO is effective in the treatment of stroke or myocardial infarction. New findings on the production of EPO in the retina and EPO receptors on neurons in the retina could lead to new treatments for retinopathy. The adverse effects of EPO on some biological systems need to be closely monitored, especially in patients with cancer and cardiovascular disease.