
Abstract
Angiotensin II (AngII) is important in regulation of vascular resistance and control of blood flow among organs and tissues. The effect of AngII on the cerebral microvasculature may be mediated or altered by endothelial-derived signals. The aim of this study was to test the hypothesis that blood AngII dilates neonatal pial arterioles via an endothelial-dependent mechanism but brain AngII can constrict pial arterioles by activating smooth muscle AT1 receptors. Studies used anesthetized newborn pigs with surgically implanted closed cranial windows. AngII was given either by infusion into the carotid artery ipsilateral to the cranial window or topically. Intracarotid infusion of AngII dilated pial arterioles. The dilation was blocked by systemic administration of the AT1-receptor antagonist, losartan, but unaffected by topical losartan. Topical AngII also caused dilation, but this dilation was converted to constriction by topical losartan. In piglets pretreated with the angiotensin-converting enzyme (ACE) inhibitor, enalapril, topical AngII constricted, rather than dilated, pial arterioles. In enalapril-treated piglets, light/dye endothelial injury blocked dilation to intracarotid AngII but did not affect constriction to topical AngII. Either indomethacin or
Introduction
Angiotensin II (AngII) is important in the regulation of vascular resistance and control of blood flow among organs and tissues. The strong constrictor action of AngII on systemic vascular resistance beds makes manipulation of the renin–angiotensin system a target in the treatment of congenital and acquired heart disease. In patients with biventricular and univentricular heart disease this system is commonly medically antagonized. Inhibition of the vasoconstrictive effects of AngII decreases the amount of work the heart must perform. 1 Conversely, AngII infusion could be employed to increase vascular resistance and mean circulatory filling pressure to elevate both cardiac output and blood pressure, but seldom has been reported. 2 Medical antagonism of the renin–angiotensin system may affect cerebral blood flow differently depending on the extent of blood–brain barrier (BBB) integrity. Cerebral blood flow also factors into treatment of some congenital heart disease. In children born with only a single ventricle, part of the surgical palliation includes routing systemic venous return directly to the lungs, bypassing the heart, 3 to reduce volume loading of the ventricle and increase effective pulmonary blood flow. 4 An intermediate stage in this procedure is the superior cavopulmonary anastomosis in which the superior vena cava is directly anastamosed to the pulmonary arteries providing the only source of pulmonary blood flow. 5 With such circuitry, pulmonary blood flow is largely dependent on cerebral blood flow as the major contribution to superior caval venous return. Thus, understanding actions of the renin–angiotensin system manipulations on cerebrovascular circulation is critical for clinical management of these babies.
Between any circulating agent and the cerebral vascular smooth muscle lie the endothelial cells and internal elastic lamina that comprise the BBB. The effect of any intravascular agent on the cerebral microvasculature may be mediated or altered by endothelial-derived signals. For example, norepinephrine constricts newborn pial arterioles via alpha-adrenergic receptors when applied from the brain side of the BBB, 6 but from the vascular side it dilates cerebral arterioles, increasing cerebral blood flow via beta-adrenergic receptors. 7
There are four identified AngII receptors, although little is known about AT3 and AT4. The AT1 receptor mediates most of the vasoactive effects of AngII. 8 AT2 is involved in organ development and can cause vasodilation. 9 The AT1 receptor antagonist, losartan, causes systemic vasodilation altering vascular resistance and blood pressure. 10 Conversely, intravascular AngII infusion causes vasoconstriction in most systemic vascular beds via direct smooth muscle contraction of resistance arterioles 11 elevating vascular resistance and blood pressure.
As introduced above, there is reason to believe that the endothelium may change the cerebrovascular response to peripherally circulating AngII. AngII has receptors in the brain, 12 is known to release the prostaglandin precursor arachidonic acid from membrane phospholipids and in vivo can result in endothelial-dependent vasodilation of rat cerebral arteries. 13 Nitric oxide (NO) has been reported as a mediator of cerebrovascular dilation to topical AngII via the AT2 receptor in newborn pigs. 14 NO-mediated reduction of systemic vascular resistance is increased with inhibition of angiotensin-converting enzyme (ACE) 15 or AT1 receptor blockade. 16 Vasodilation following blockade of AT1 receptors may be due to the action of unmasked AT2 receptors and/or elevation of bradykinin. 16 In addition, endothelial-derived hyperpolarizing factor(s) (EDHF) has been shown to be involved in AngII dilation of isolated, pressurized, perfused rat cerebral arteries that remain following inhibition of nitric oxide synthase (NOS) and cyclooxgenase (COX). 17
The aim of this study is to test the hypothesis that blood AngII dilates neonatal pial arterioles via an endothelial-dependent mechanism but brain AngII can constrict pial arterioles by activating smooth muscle AT1 receptors. Thus, we anticipated that intravascular AngII would dilate pial arterioles when the endothelial barrier is intact, but cause constriction following endothelial injury. Furthermore, topical AngII was expected to produce constriction via activation of AT1 receptors on vascular smooth muscle, but it could cause dilation via AT2 receptors or endothelial AT1 receptors.
Methods
The University of Tennessee Health Science Center's Animal Care and Use Committee approved all animal procedures.
Newborn pigs (1–5 days old) (1–3.5 kg) were anesthetized with ketamine hydrochloride (33 mg/kg intramuscular) and acepromazine (3.3 mg/kg intramuscular); sedation was maintained with α-chloralose (50 mg/kg intravenous). The animals were intubated via tracheostomy and ventilated with air. The femoral vein was cannulated for anesthesia, fluid and drug injections. The femoral artery was cannulated for continuous blood pressure monitoring and drawing samples for blood gas and pH analysis. The carotid artery ipsilateral to the cranial window was cannulated for antegrade saline and AngII infusion. Blood gases, pH and body temperature were maintained within reference ranges. Blood gases were obtained prior to initiation of the protocol and following completion of each major segment of the protocol and at protocol completion.
Cranial windows
The scalp was retracted and an opening 2 cm in diameter was created in the skull over the parietal cortex. The dura was cut without touching the brain, and cut edges retracted over the bone so that the periarachnoid space was not exposed to bone or damaged membranes. A stainless steel and glass cranial window was fitted in the hole and cemented in place with dental acrylic. The windows had side needle ports so fluid under the window could be exchanged and test compounds administered topically to the cerebral vessels. The space under the window was filled with artificial cerebrospinal fluid (aCSF) equilibrated with 6% CO2 and 6% O2 producing gases and pH within the reference range for CSF (pH ~7.34, PCO2 and PO2 ~43mmHg). Pial vessels were observed through the window with a dissecting microscope. Arteriole diameters were measured with a video micrometer coupled to a television camera mounted on the microscope and a video monitor. Following cannulation, piglets in most groups were given a single dose of intravenous enalapril (50 μg/kg), an ACE inhibitor, to minimize native AngII and to emulate clinical practice. In another group, responses to topical AngI and AngII were measured in piglets not treated with enalapril. All pharmaceutical agents except losartan (Merck Pharmaceuticals, Whitehouse Station, NJ, USA) were obtained from Sigma Chemicals (St Louis, MO, USA).
Intravascular AngII
AngII was dissolved in normal saline to 50 μg/mL concentration. This solution was infused antegrade into the carotid artery ipsilateral to the cranial window. Initial rate was 5 μg/kg/min and incrementally increased at five-minute intervals to 10, 20 and 50 μg/kg/min. These doses were selected because the 50 μg/kg had been shown to improve cerebral blood flow during cardiopulmonary resuscitation in immature pigs. 11 Blood pressure was continually monitored and kept stable by controlled exsanguination to avoid arteriole diameter changes due to autoregulation. Exsanguinated blood was mixed with heparin and kept warm, and then returned to the animal following completion of AngII infusion. To control for effects of flow and/or pressure due to the infusion, normal saline was infused into the carotid artery prior to administration of AngII. The rate of saline infusion was slightly higher than the maximum rate to be used for infusion of AngII for each animal.
Topical AngII
AngII was dissolved in aCSF to concentrations of 10−8, 10−7 and 10−6 mol/L. Beginning with the 10−8 mol/L, each concentration was sequentially instilled under the cranial window in a volume sufficient to fill the space. Washout with aCSF free of AngII was done after each concentration. Vessel diameter was measured and recorded following each instillation to generate a dose–response curve.
AT1-blockade
To differentiate between effects of AngII receptors, the AT1-specific antagonist, losartan, was administered to two groups of piglets. The first group was administered a single 3 mg/kg dose intravenously; this dose is hemodynamically significant in pigs. 18 Ten minutes were allowed to elapse to stabilize blood pressure and pulse. A separate group had 10−5 mol/L losartan dissolved in aCSF instilled under the cranial window in a volume sufficient to fill the space. During the topical protocol, losartan was re-instilled with the AngII to ensure the presence of both. Intravenous and topical AngII were then administered to both groups as described.
Endothelial injury
To delineate the role of vascular endothelium in effects of AngII in piglets, the endothelium was damaged in vivo. Sodium fluorescein (160 mg/kg in 8 mL/kg volume) was injected intravenously and activated with exposure to appropriately filtered light from a mercury arc lamp producing endothelial injury. 19 Following injection, 30 min elapsed to allow uptake by the endothelial cells and the light was directed through the cranial window for eight minutes. In addition to topical and systemic AngII, piglets in this group were administered topical 10−6 mol/L isoproterenol and 10−6 mol/L bradykinin, before and after light–dye treatment. Washout with aCSF was done prior to each being instilled. Vasodilation due to bradykinin requires a functional endothelium while the effect of isoproterenol is by direct stimulation of smooth muscle beta-receptors, independent of endothelial cells. 20 Loss of dilation to bradykinin showed loss of endothelial function while persistent arteriole reactivity was confirmed with isoproterenol. To ensure fluorescein alone did not affect endothelial function, a group of animals were injected with fluorescein but not exposed to light.
Prostanoids
To block prostanoid synthesis, indomethacin trihydrate was dissolved in normal saline and 10 mg/kg was given intravenously; a dose sufficient to block dilation to topical arachidonic acid 21 and constriction to topical acetylcholine, 22 suggesting sufficient passage across the BBB to be effective. Intravenous and topical AngII were given as above, before and after indomethacin administration. Isoproterenol at 10−6 mol/L was applied as part of the topical protocol to assure generalized alteration of vascular responsiveness had not occurred.
The same protocol was repeated in another group of piglets with indomethacin, at 10−4 mol/L in aCSF, applied topically.
Nitric oxide
Carbon monoxide
Chromium mesoporphyrin (CrMP), dissolved to 2 × 10−5 mol/L in aCSF, was applied topically to inhibit production of CO via heme oxygenase (HO) in another group of piglets. This concentration has been shown to inhibit HO activity in neonatal piglets. 23 This solution was protected from light by wrapping aluminum foil around the syringe during administration to prevent inactivation. The protocol administering AngII and isoproterenol was repeated prior to and following CrMP. CrMP (2 × 10−5 mol/L) inactivated via exposure to light for >24 h was used to act as an additional control.
Statistical analysis
Data were analyzed using analysis of variance for repeated measures and Bonferroni post hoc analysis to compare mean arteriolar diameter. Statistical analysis was performed using InStat software (GraphPad Software, Inc, LaJolla, CA, USA).
Results
Control
Infusion of normal saline into the carotid artery, at a rate slightly greater than the maximal infusion rate of AngII, had no effect on the diameter of pial resistance arterioles (Figure 1a ‘Baseline’ versus ‘Control’). Similarly, replacement of aCSF without additional agents under the cranial window did not affect pial arteriolar diameter.
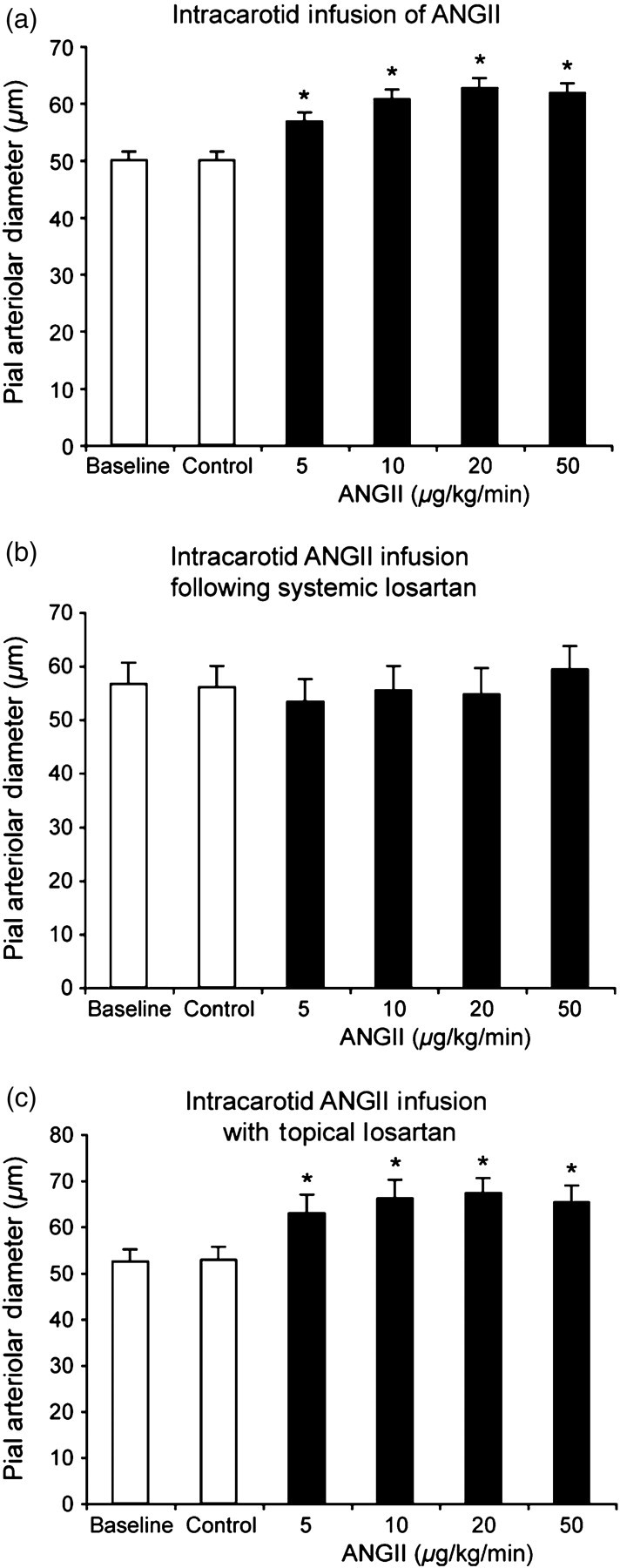
Effect of intracarotid AngII infusion on pial arteriolar diameters of newborn pigs. Baseline is before and Control is during saline vehicle infusion. (a) Without losartan. (b) Following 3 mg/kg losartan, intravenous. (c) During topical losartan (10−5 mol/L). Mean ± SEM. n = 6, 6 and 5 piglets in (a), (b) and (c), respectively. *P < 0.05 from control. Ang, angiotensin
Angiotensin II
Intracarotid infusion of AngII dilated pial arterioles (Figure 1a). The dilation was blocked by systemic administration of the AT1-receptor antagonist, losartan (Figure 1b), but unaffected by topical losartan (Figure 1c).
Conversely, in piglets pretreated with the ACE inhibitor, enalapril, application of AngII directly on surface pial arterioles resulted in vasoconstriction (Figure 2a). In contrast, when piglets were not given enalapril, topical AngII caused vasodilation (Figure 2b). On the other hand, topical losartan not only blocked the constrictor response of pial arterioles to AngII in piglets treated with enalapril but converted the response to concentration-dependent dilation (Figure 2c). All other piglets were studied in the presence of enalapril because we were pursuing mechanisms behind the divergent responses of pial arterioles to intravascular versus topical AngII that were revealed by ACE inhibition.
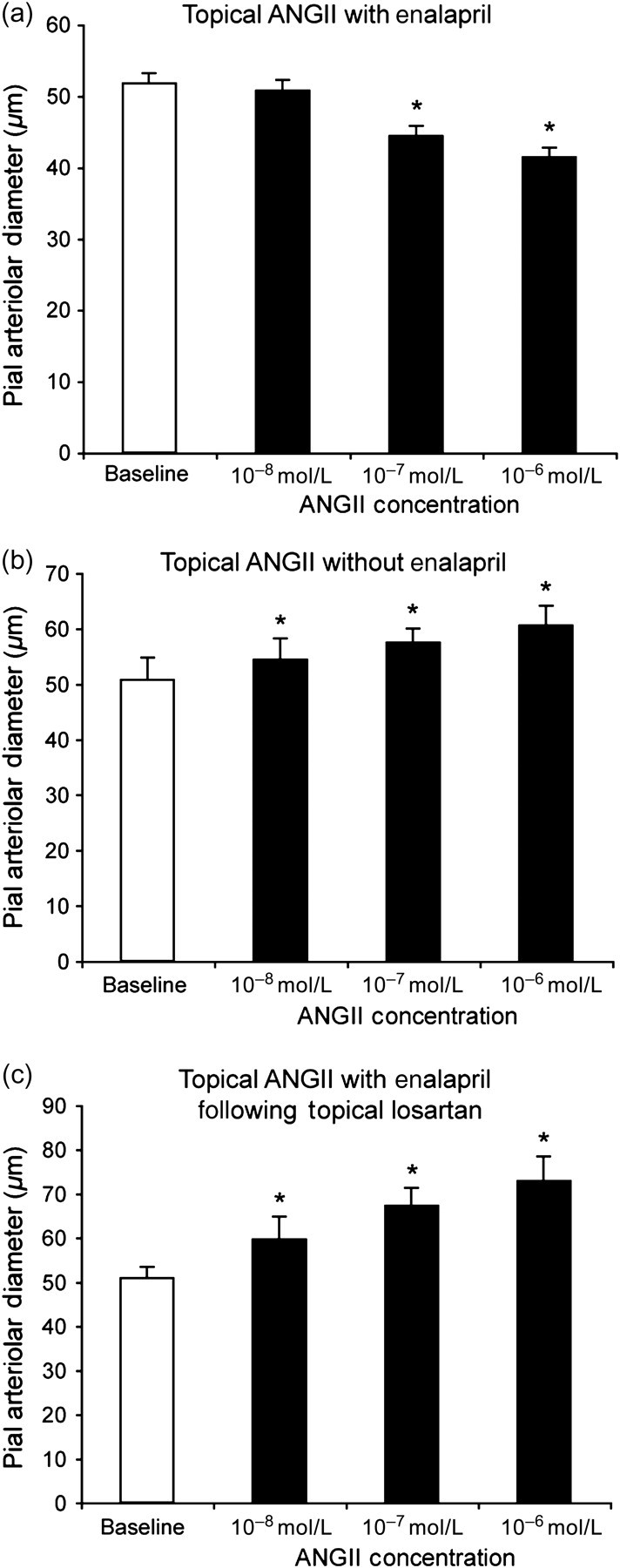
Effect of topical AngII on pial arteriolar diameters of newborn pigs. (a) Piglets pretreated with enalapril (50 μg/kg, intravenous). (b) Untreated piglets. (c) Piglets treated with enalapril (50 μg/kg, intravenous) and topical losartan (10−5 mol/L). Mean ± SEM. n = 6, 4 and 6 piglets in (a), (b) and (c), respectively. *P < 0.05 from baseline. Ang, angiotensin
Topical AngI (10−6 mol/L) had no effect on pial arteriolar diameter even in piglets not treated with enalapril (51 ± 3 and 51 ± 3 μm, before and during AngI, respectively, n = 4 piglets).
Endothelial injury
Both isoproterenol and bradykinin caused pial arteriolar dilation before light–dye treatment (Figure 3). Following light–dye, dilation in response to bradykinin was blocked indicating that the endothelium was non-functional. Dilation to isoproterenol was unchanged indicating the arterioles retained the ability to dilate when given a stimulus that acts directly on the smooth muscle. In the animals given fluorescein dye only (i.e. without light activation), there was no change in the response to either bradykinin or isoproterenol (Figure 3).
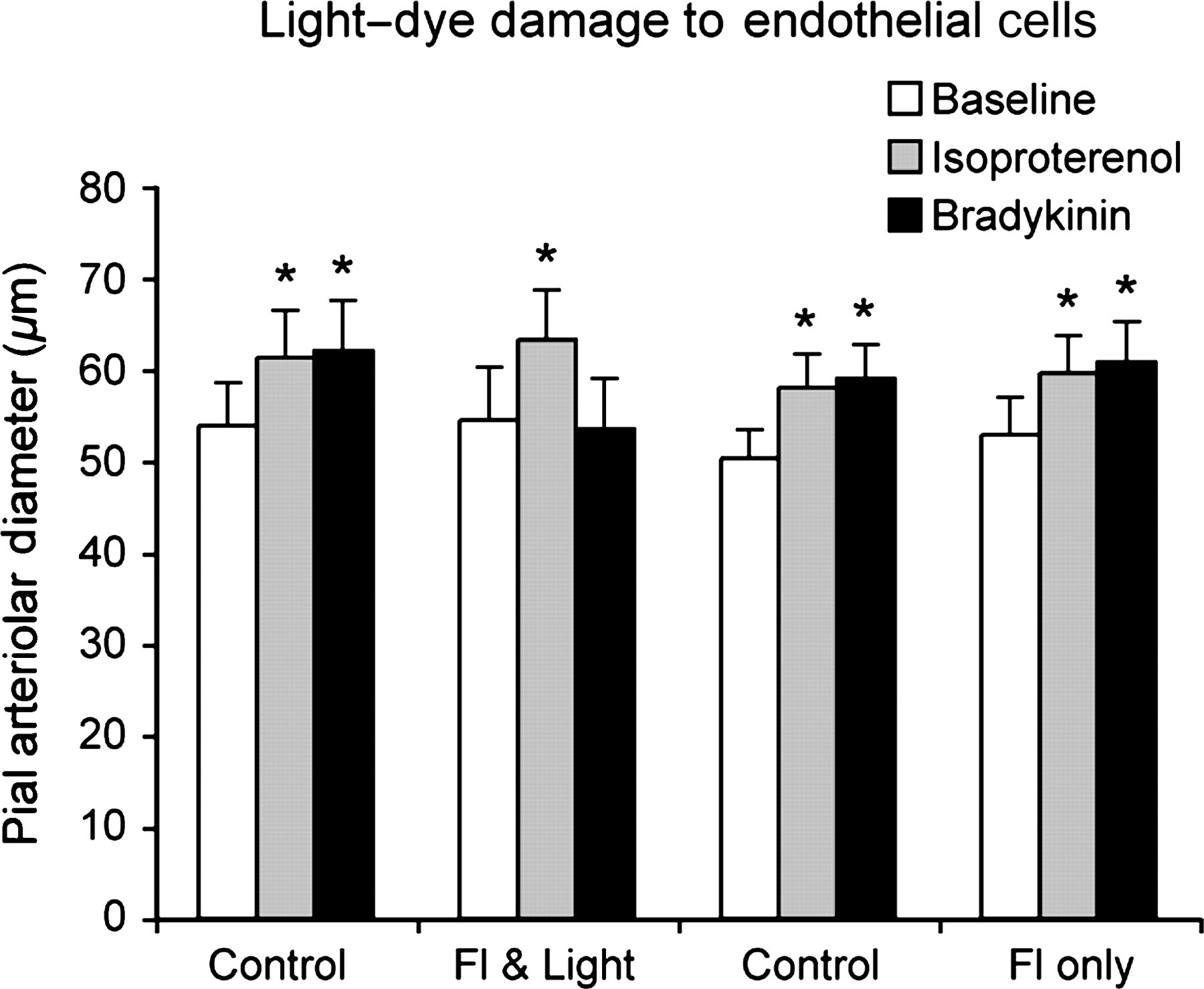
Effect of sodium fluorescein (Fl) (160 mg/kg, intravenous) and light from a 100 W mercury arc lamp filtered for maximal transmission between 420 and 430 nm for eight minutes (light) on pial arteriolar responses to topical isoproterenol (10−6 mol/L) and bradykinin (10−6 mol/L). Controls were before treatment and light/dye (Fl & Light) is following dye and light (n = 3). In separate animals (n = 4), responses were measured before and after fluorescein alone without light activation (Fl only). Mean ± SEM. *P < 0.05 from respective baseline
In piglets with confirmed light–dye injury as in Figure 3, the endothelial injury did not affect the constrictor response to topical AngII (Figure 4a). In contrast, the vasodilatory response to intracarotid infusion of AngII was converted to constriction following light–dye endothelial injury (Figure 4b).

Effect of light–dye endothelial injury on pial arteriolar response to AngII. AngII at increasing concentration was (a) applied topically or (b) infused intracarotidly prior to and following exposure to fluorescein and light. Mean ± SEM. n = 3 piglets. *P < 0.05 from baseline. Ang, angiotensin
To ensure that uptake of fluorescein did not affect the function of the endothelial cells, a separate group of animals was injected with fluorescein intravenously but protected from light to avoid activating the fluorescein (Figure 3). The same period of time was allowed to elapse between injection of flourescein and exposure to AngII as in the animals exposed to light. The response to intracarotid infusion of AngII was no different before and after dye-only treatment (50.8 ± 3.6–61.6 ± 3.5 μm before and 51.4 ± 3.6–62.0 ± 4.1 μm after dye only in response to 20 μg AngII/kg/min infusion and 50.3 ± 3.1–40.6 ± 2.7 μm before and 52.9 ± 4.8–42.5 ± 3.7 μm after dye only in response to topical AngII [10−6 mol/L], respectively).
Characterization of endothelial-derived relaxing factor
Because the data presented above indicate that endothelial-derived relaxing factor(s) (EDRF) cause dilatory responses to AngII in piglet pial arterioles, we investigated potential EDRF(s) involved. Previous data indicate that prostanoids, NO and CO are EDRFs in the newborn cerebral circulation, 14,20,21,23–26 so the COX/prostanoid, NOS/NO and HO/CO systems were addressed.
Prostanoids
Indomethacin, both systemic and topical, blocked the response to intracarotid infusion of AngII (Figures 5a and c). Conversely, the constrictor response to topical AngII was not altered by either systemic or topical indomethacin (Figures 5b and c). There was no change in the vasodilatory response to topical isoproterenol before and after treatment with either systemic or topical indomethacin (data not shown).

Effect of indomethacin on pial arteriolar responses to AngII. Indomethacin was administered intravenously (10 mg/kg) (a and b, n = 6) or topically (10−4 mol/L) (c and d, n = 5). AngII was infused intracarotidly (a and c) or applied topically (b and d). Mean ± SEM. *P < 0.05 from baseline. Ang, angiotensin
Nitric oxide
Similarly to indomethacin, topical
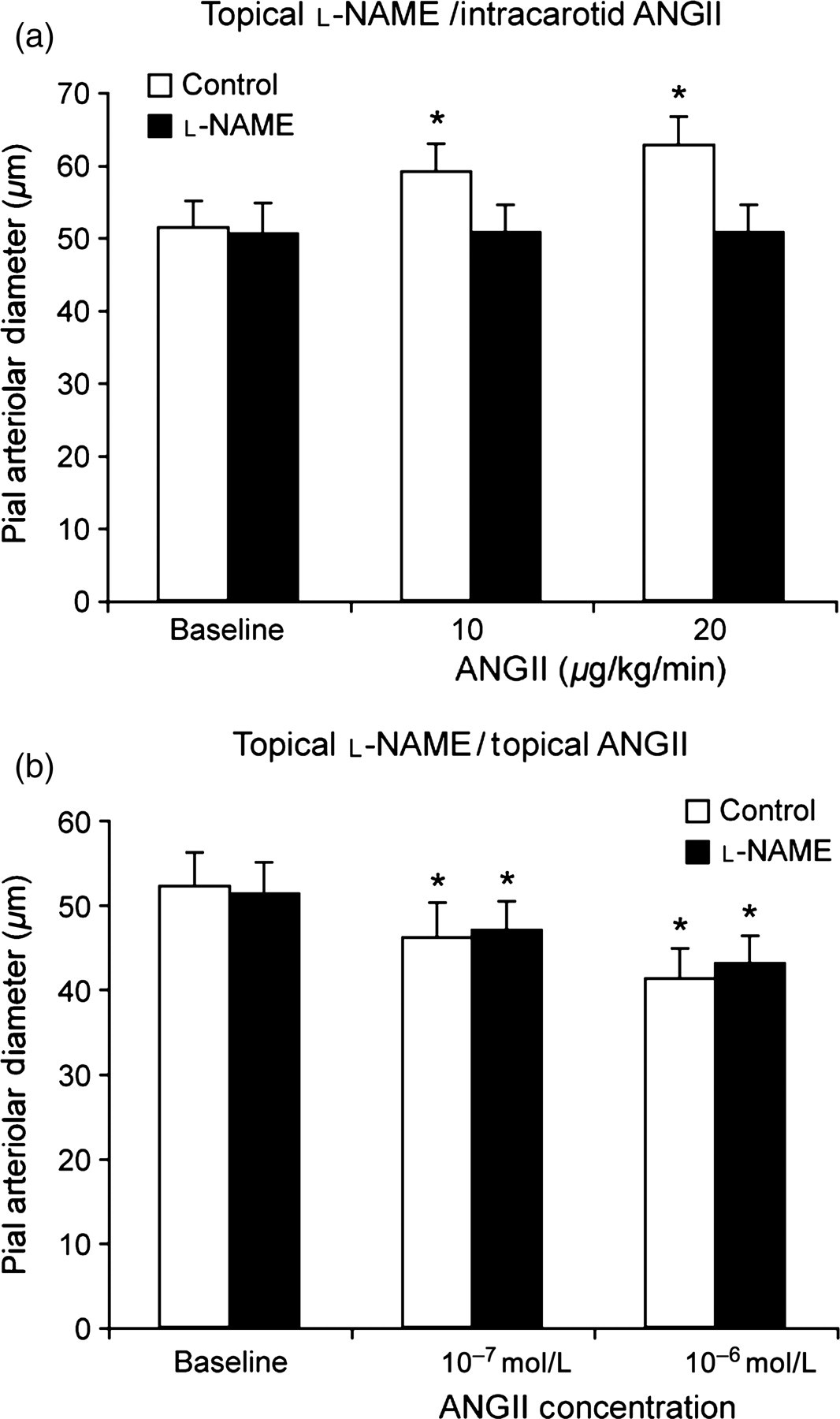
Effect of topical
Pial arteriolar constriction to topical AngII administration was unchanged when NOS was inhibited (Figure 6b).
Carbon monoxide
Inhibition of CO production with CrMP (2 × 10−5 mol/L, topically 23 ) had no effect on pial arteriolar responses to either topical or intracarotid AngII (Figure 7). As a control, CrMP inactivated by light was administered topically. Piglets were administered AngII topically and then systemically, first in the presence of inactivated CrMP, and then following washout with aCSF free of additional substances, with active compound, CrMP.
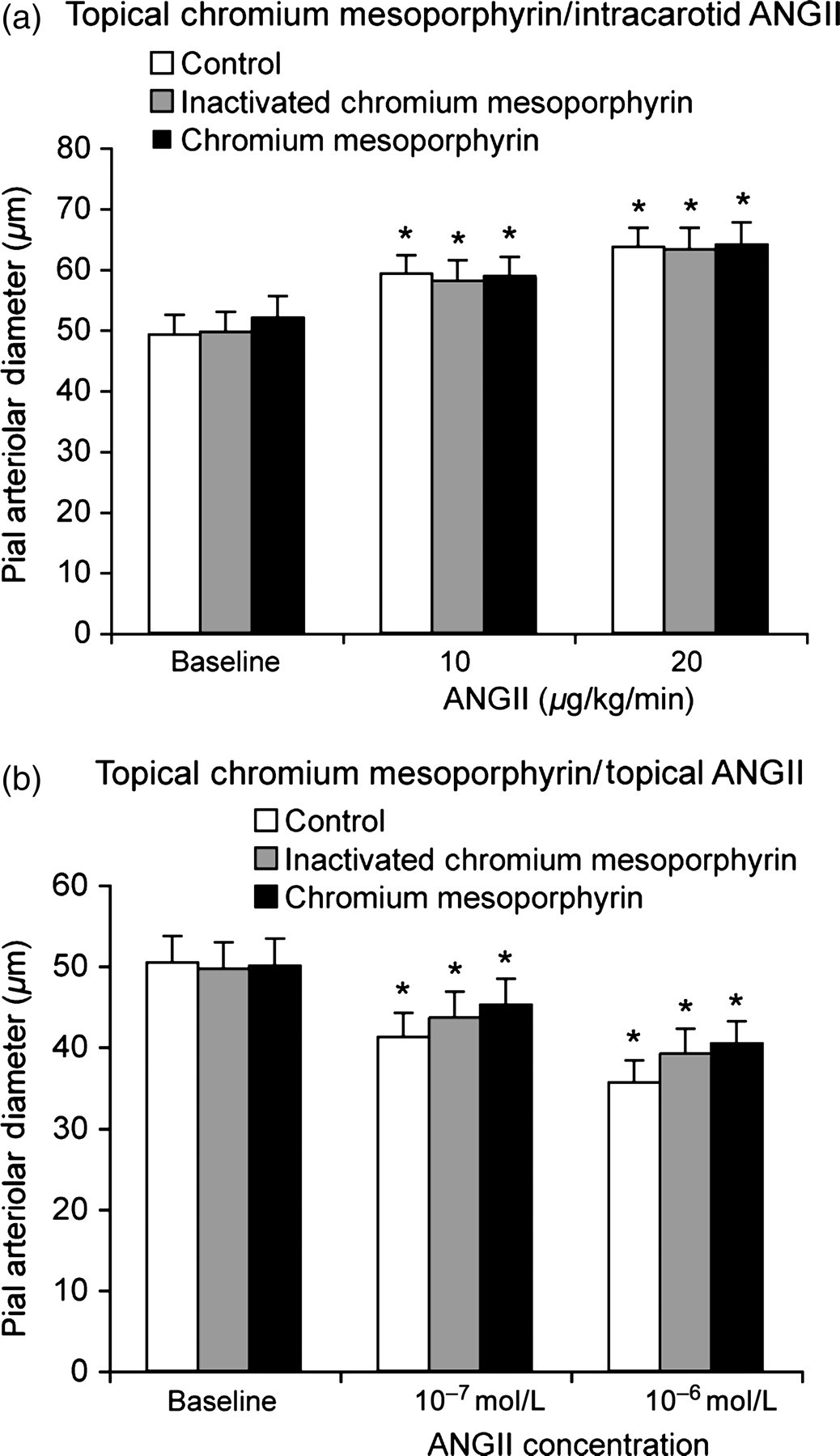
Effect of inhibition of HO with topical chromium mesoporphyrin (2 × 10−5 mol/L, CrMP) on piglet pial arteriolar responses to (a) intracarotid AngII or (b) topical AngII. The effect of inactivated CrMP was determined before active CrMP in the same piglets. Mean ± SEM. n = 5. *P < 0.05 from baseline. Ang, angiotensin; HO, heme oxygenase; CrMP, chromium mesoporphyrin
Discussion
Novel findings of the present investigation in newborn pigs are as follows: (1) intravascular AngII dilates pial arterioles via an endothelial and AT1 receptor-dependent mechanism; (2) topical AngII causes pial arteriole dilation in untreated piglets, but AT1 receptor-dependent constriction following ACE inhibition; (3) either COX or NOS inhibition blocks pial arteriolar dilation to intraluminal AngII, but neither affects vasoconstriction to topical AngII; and (4) heme oxygenase inhibition has no effect on pial arteriolar responses to either intraluminal or topical AngII. These data are consistent with the hypotheses that: (a) circulating AngII dilates pial arterioles via endothelial AT1 receptor-derived relaxing factors, notably prostanoids and NO; and (b) direct AT1 receptor activation on the brain side of the BBB by AngII causes AT1 receptor-mediated constriction that can mask underlying AT1 receptor-independent dilation.
These are the first data regarding the effects of intra-arterial infusion of AngII on cerebrovascular circulation in vivo. Our results suggest that AT1 receptor inhibition to decrease after-load on a failing heart (Introduction) may block AngII-induced cerebral arteriole dilation potentially decreasing cerebral blood flow. This is especially pertinent in children whose pulmonary blood flow is heavily dependent on cerebral venous return. In addition, cerebrovascular endothelial injury, as can be caused by numerous pathological conditions involved in hemodynamic instability in pediatric critical care settings, will likely block the AT1 receptor-induced cerebrovascular dilation, causing AngII-induced constriction and compromising cerebral blood flow.
Previous studies have reported on the effect of topical AngII on pial resistance arterioles. Similarly to the present report in piglets without ACE inhibition, Meng and Busija
27
and Baranov and Armstead
14,24
found that topical AngII caused pial arteriolar dilation. This dilation was blocked by inhibition of AT2 receptors.
14
These studies also reported contributions of prostanoids and NO to AngII-induced pial arteriolar dilation.
14,24,27
We found dilation mediated by relaxing factors also, but vasoconstriction in response to topical AngII application following ACE inhibition with enalapril. Present data indicate that this constriction is not mediated by the endothelium. Neither indomethacin nor
The appropriate dose of
The responses of pial arterioles to agonists, inhibitors and receptor blockers are dependent on the side of the BBB to which they are applied and the integrity of the barrier. Thus, intravascular AngII dilated pial arterioles and this dilation could be blocked by systemic losartan but not by topical losartan, suggesting the AT1 receptors providing the dilatory signal may be on the luminal endothelial face. While losartan can cross the BBB with chronic administration, it crosses the BBB more slowly than other more lipophilic AT1 receptor blockers, requiring hours not minutes for efficacy. 32–34 Losartan has no partial agonist effect and approximately 1000-fold greater affinity for AT1 than AT2. 35 The constriction to topical AngII seen in enalapril-pretreated piglets was reversed to dilation with topical losartan, a response identical to that seen in piglets not pretreated with the ACE inhibitor. The action of enalapril to convert constriction to dilation must be on the endothelium because enalapril does not cross the BBB. 36,37 That the endothelium generates the dilator signals was confirmed by converting the vasodilator response to intravascular Ang II to constriction by selective endothelial injury. Efficient endothelial injury in the pial arterioles under the cranial window was confirmed by total loss of dilation to the endothelial-dependent dilator, bradykinin, 20 but no effect on the dilation to endothelial-independent dilator, isoproterenol, 19 that dilates via activating of vascular smooth muscle β-adrenergic receptors. Consistent with the present functional studies, Wackenfors et al. 17 detected AT1 and AT2 receptors on the endothelium. AngII receptors appear to be on the vascular smooth muscle also because AngII causes constriction following endothelial injury (Figure 4).
The mechanism(s) by which enalapril alters the pial arteriolar responses to AngII on the brain side of the BBB are not known. ACE inhibitors are known to effect cerebral autoregulation in the short term, decreasing the lower limit and decreasing or leaving unchanged the upper limit. 38 Enalapril increases cerebral blood flow in adult patients in heart failure without an increase in cardiac output. 39 While we controlled for the effect of autoregulation by keeping systemic mean arterial pressure constant, enalapril may effect local renin–angiotensin action that is somehow involved in the dilation. However, as the effect of ACE would be in the amount of AngII available to receptors, it seems unlikely that enzyme inhibition would change the response of a receptor when stimulated. The activation of AT2 receptors can cause vasodilation independent of AT1 and prostanoids in piglets. 14 Therefore, stimulation of the AT2 receptor may cause vasodilation in contrast to AT1-mediated constriction, as found in the present study with topical application of AngII in lostartan-treated piglets. Similarly, in adult mice, topical AngII caused AT1 receptor-mediated pial arterial constriction, but vasodilation could be evoked by AT2 receptor stimulation. 40 AngII can attenuate endothelial-derived vasodilator responses, 40 but the effect of the endothelium on action of AngII had not been previously clarified. ACE inhibition has been shown to amplify the endothelial-derived dilator responses in hypertensive rats reducing reactive oxygen species 15 and could also involve elevations of bradykinin 16 that dilate piglet pial arterioles via endothelial-produced prostacyclin that increases cAMP. 20
In the search for EDRFs involved in the dilatory action of intraluminal AngII, we found pathways either of whose inhibition totally blocked the pial arteriolar dilation to AngII, COX/prostanoid (presumably prostacyclin) and NOS/NO. The role of NO in dilation of piglet pial arterioles to AngII has been described before, and that was via AT2 receptors.
14
There are several reasonable possibilities for either COX or NOS inhibition alone to completely block dilation to AngII. COX products could stimulate NOS to elevate NO that produces dilation or conversely NO may stimulate COX. While possible, we have no evidence for this hypothesis. Also, the newborn cerebral circulation is very much prostanoid dominant with NO becoming of more significance with age.
20
It is conceivable that the effect of removal of one pathway is masked by increased activity of the other. We do have evidence that removal of COX elevates the role of NO in the newborn cerebral circulation, but on a timescale of days not minutes.
41
A third possibility has more supporting evidence. In the newborn pig cerebrovascular circulation CO is a functionally highly significant dilator.
26
However, either indomethacin or
In summary, the endothelium alters the response of pial arterioles to AngII in neonatal piglets. Topical AngII can cause either constriction or dilation depending on whether ACE is blocked or not, respectively. Intraluminal AngII produces AT1 receptor-mediated, endothelium-dependent dilation that is totally blocked by either COX or NOS, but not HO, inhibition. Clinical manipulation of the renin–angiotensin system will have disparate actions on cerebral circulation depending on the integrity of the BBB and ACE that requires further attention.
Footnotes
Acknowledgements
The authors thank Alex Fedinec, Dan Wilkinson and Christine Petrin for technical assistance, Greg Short for the graphics and Courtnie Holliday for clerical support. The current address of Kenneth Knecht is Division of Pediatric Cardiology, Department of Pediatrics, University of Arkansas for the Medical Sciences, 1 Children's Way, Slot 512-3, Little Rock, AR, 72201. These studies were supported by Award Numbers HL042851 and HL34059 from the National Heart, Lung, and Blood Institute and a grant from LeBonheur Children's Medical Center, Memphis. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health, LeBonheur Children's Medical Center, or the University of Tennessee.