
Abstract
Hepatitis B x-interacting protein (HBXIP), a co-factor of survivin, was originally identified by its binding with the C-terminus of the hepatitis B virus x protein (HBx). We have recently shown that HBXIP promotes the growth of both normal liver cells and hepatoma cells in vitro, but the molecular mechanisms of this have not been documented. In this study, we investigated the potential effects of HBXIP on the proliferation of HepG2 cells and the intracellular signaling pathway mediating these changes. Over-expression of the HBXIP gene promoted the proliferation of HepG2 cells, as shown by the MTT assay. We also showed that HBXIP induced cellular accumulation in the S phase concomitantly with up-regulation of cyclinD1 and down-regulation of p21 and p53 levels. Moreover, HBXIP over-expression cells showed activation of the PI3K/Akt pathway; this activation was accompanied by an increase in phosphorylation of glycogen synthase kinase 3β. LY294002, a specific inhibitor of PI3K, blocked HBXIP-stimulated Akt phosphorylation and suppressed the cell cycle promotion induced by HBXIP in HepG2 cells. The increase in cyclinD1 protein levels induced by HBXIP was inhibited when cells were incubated with LY294002. In conclusion, our data suggest that the proliferation of HepG2 cells promoted by HBXIP is associated with activation of the PI3K/Akt signaling pathway.
Introduction
Hepatocellular carcinoma (HCC) is one of the most common cancers in the world, and it has been estimated that more than 50% of HCC cases worldwide are associated with hepatitis B virus (HBV). Moreover, research has shown that individuals carrying HBV have a greater than 100-fold increased relative risk of developing HCC. 1 Among the four overlapping genes of HBV, such as S/preS, C/preC, P and X, hepatitis B virus X gene, which encodes a 17-kDa protein known as HBV x protein (HBx), is strongly linked to the development of HCC. 2,3
Hepatitis B x-interacting protein (HBXIP) is a cellular protein which was originally identified by its interaction with the C-terminus of HBx. 4 The sequences of the HBXIP gene, containing a putative leucine zipper motif and two consensus phosphorylation sites for protein kinase C and casein kinase II, are well conserved among mammalian species. HBXIP can reduce the replication of wild-type HBV and suppress the transactivity of activating protein-1 enhanced by HBx expression. HBXIP also forms a complex with survivin, an antiapoptotic protein, and selectively inhibits apoptosis via the mitochondrial/cytochrome c pathway. 5 Recently, another HBXIP interactor named hSuv3p, a human ATP-dependent RNA/DNA helicase, has been identified. The HBXIP-binding domain in the hSuv3p protein is important for its stability and transport into the mitochondria. 6 As a regulator of centrosome duplication, HBXIP is required for bipolar spindle formation in HeLa cells and primary mouse embryonic fibroblast cells. 7 Knockdown of endogenous HBXIP expression arrests the cells in prometaphase with monopolar spindles, and over-expression of HBXIP causes tripolar or multipolar spindles due to excessive centrosome replication. Our previous studies have suggested that HBXIP promotes cellular proliferation in both cancer cells and normal liver cells, 8 but the molecular mechanisms of this are still unclear.
The phosphatidylinositol (PI) 3-kinase (PI3K)/Akt signaling pathway is a central regulator of cell proliferation and cell survival. 9,10 PI3Ks are dual-specificity enzymes that play a pivotal role in the regulation of many cellular processes. 11,12 Activated PI3K phosphorylates PI substrates to produce PI(3)P, PI(3,4)P2 and PI(3,4,5)P3. These molecules act as secondary messengers, recruiting the PI3K-dependent serine/threonine kinases (PDK1) and Akt from the cytoplasm to the plasma membrane. 13 Akt, also known as protein kinase B, is an important target of PI3K. Akt can function to regulate cell proliferation, cell cycle and cell survival by interacting with numerous other regulatory proteins, such as p53, cyclinD1, p21Cip1, glycogen synthase kinase-3 (GSK3) and mammalian target of rapamycin. 14,15 To be fully activated, Akt is phosphorylated on two critical residues: Thr308 (T308-Akt) and Ser473 (S473-Akt), and alterations of the Akt pathway have been detected in a variety of cancerous human malignancies. 16,17
Here, we present evidence that HBXIP promotes hepatoma cell proliferation, partly by activating the PI3K/Akt signaling pathway. Up-regulation of the expression of cyclinD1 by HBXIP over-expression is involved in promoting a G0/G1 to S phase transition in HCC cells. The PI3K inhibitor, LY294002, blocks HBXIP-stimulated Akt phosphorylation and suppresses the cell cycle promotion induced by HBXIP in HepG2 cells.
Materials and methods
Cancer cell culture
The HepG2 cell line was obtained from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China) and cultured in RPMI 1640 medium supplemented with 10% fetal calf serum (Gibco BRL, Gaithersburg, MD, USA), 2 mmol/L glutamine, 100 U/mL penicillin and 100 μg/mL streptomycin at 37°C in humidified 5% CO2.
Stable transfection
HepG2 cells were transfected with pCMV-hbxip 8 or pCMV-tag2B (empty vector) with Lipofectamine 2000 according to the manufacturer's instructions. Twenty-four hours after transfection, the cells were diluted 1:10 and cultured in growth medium containing G418 (700 μg/mL) for three weeks. Stable transfected clones were picked and maintained in a medium containing 350 μg/mL G418 for additional studies.
MTT assay
Cell proliferation was measured using the MTT assay every 24 h. Briefly, 200 μL of cell suspensions (5 × 103 cells/mL) was added to each well of a 96-well plate and incubated at 37°C for six days. A total of 20 μL of MTT (5 mg/mL; Genview, Houston, TX, USA) was added and incubated at 37°C for 4 h. After removing the supernatant, 150 μL of dimethyl sulfoxide was added to dissolve formazan crystals, and the value of the optical density was detected at 570 nm. To explore the role of the PI3K/Akt pathway, HepG2-hbxip cells and control cells were treated with 10 and 40 μmol/L LY294002 (Cell Signaling, Dallas, TX, USA), respectively.
Flow cytometry analysis
Cells (1 × 106) were harvested by trypsinization and washed twice with phosphate-buffered saline (PBS), then fixed in 75% ethanol at 4°C for 18 h. The fixed cells were rinsed twice with PBS and resuspended in propidium iodine (PI) solution containing 50 μg/mL PI and 50 μg/mL RNaseA (Sigma, St Louis, MO, USA) in PBS without calcium and magnesium and incubated at 37°C for 30 min in the dark. Stained cells were passed through a nylon-mesh sieve to remove cell clumps and analyzed by a FACScan flow cytometer and Cell Quest analysis software (Becton Dickinson, San Jose, CA, USA). Flow cytometry analysis was repeated three times.
Western blot analysis
After washing twice with cold PBS, hepatoma cells were lysed with ice-cold lysis buffer (150 mmol/L NaCl, 20 mmol/L Tris-HCl [pH 7.4], 0.1% SDS, 1.0% Nonidet P-40, 0.5% Na-deoxycholate [Na-DOC], 0.2 mmol/L phenylmethylsulfonyl fluoride, protease inhibitor cocktails and phosphatase inhibitors). Lysates were centrifuged at 12,000
Akt kinase assay
Akt kinase activity was measured using the Akt kinase assay kit from Cell Signaling Technology. Briefly, cells transfected with the pCMV-hbxip or pCMV-tag2B plasmid were washed twice with cold PBS and lysed in ice with 600 μL of lysis buffer containing 1% Triton X-100, 1 mmol/L EDTA, 1 mmol/L EGTA, 2.5 mmol/L sodium pyrophosphate, 150 mmol/L NaCl, 20 mmol/L Tris-HCl (pH 7.5), 1 μg/mL aprotinin, 1 μg/mL leupeptin, 1 mmol/L phenylmethylsulfonyl fluoride, 1 mmol/L β-glycerophosphate and 1 mmol/L Na3VO4 according to the protocol. Resuspended, immobilized Akt antibody slurry (20 μL) was added to 200 μL of cell lysate supernatant to selectively immunoprecipitate Akt from the lysate with gentle rocking for 12 h at 4°C. Immunoprecipitations were washed three times with lysis buffer and twice with Akt kinase buffer (25 mmol/L Tris [pH 7.5], 5 mmol/L β-glycerophosphate, 2 mmol/L dithiothreitol, 0.1 mmol/L Na3VO4 and 10 mmol/L MgCl2). The immunoprecipitated pellet was then incubated with kinase buffer containing 200 mmol/L ATP and 1 μg of GSK-3 fusion protein according to the manufacturer's instructions for the non-radioactive Akt kinase assay. Samples were analyzed by Western blot analysis using 15% SDS-PAGE. Akt-induced phosphorylation of GSK-3 was detected by Western blot using the phospho-GSK-3α/β(Ser21/9) antibody. Akt kinase assay was repeated three times.
Statistical analysis
All data are expressed as mean ± SD. Statistical analysis was performed using the Student's t-test and P < 0.05 indicated statistical significance.
Results
Up-regulation of cyclinD1 and down-regulation of p53 and p21 expression are involved in the G0/G1 to S phase transition induced by HBXIP in HepG2 cells
To evaluate the biological function of HBXIP in the proliferation of hepatoma cells, HepG2 cells were engineered to stable express high levels of HBXIP gene and were named HepG2-hbxip. The expression levels of HBXIP in HepG2-hbxip cells were assessed by Western blot assay. Compared with the control (pCMV-tag2B), HepG2-hbxip cells showed high expression of HBXIP (Figure 1a). The results of the MTT assay in Figure 2 indicated that HBXIP can promote HepG2 cells proliferation in vitro. Furthermore, flow cytometry analysis was used to measure the cell cycle distribution in HepG2-hbxip cells. As predicted, HBXIP significantly accelerated the cell cycle progression. The percentage of cells re-entering into the S phase in HepG2-hbxip cells was 39.2 ± 1.32, which was higher than that of pCMV-tag2B cells (32.06 ± 1.46) (Figure 3, P < 0.05), suggesting that HBXIP might promote hepatoma cell growth by accelerating the G0/G1 to S phase transition in the cell cycle.
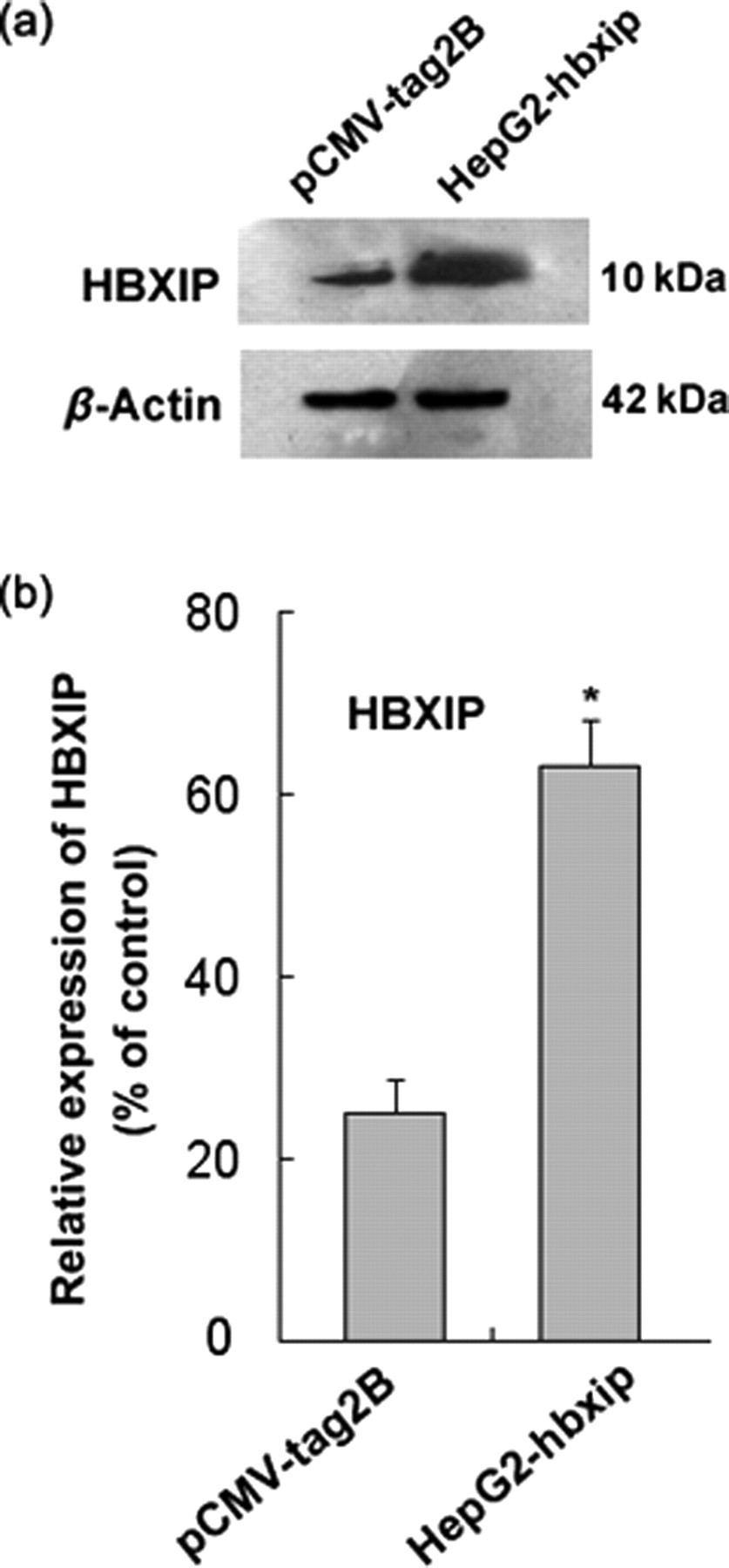
Examination of the levels of HBXIP in HepG2 cells stably transfected with the HBXIP gene or control vector. (a) Western blot analysis of the expression of HBXIP. Detection of β-actin with an anti-β-actin antibody was used as a loading control. (b) Bands were analyzed using Glyco Band-Scan software. Each bar corresponds to the mean ± SD for three independent experiments. *P < 0.05 versus control cells, Student's t-test. HBXIP, hepatitis B x-interacting protein

HBXIP induces hepatoma cell proliferation. Cell proliferation was measured using an MTT assay in HepG2-hbxip cells and pCMV-tag2B cells. The y-axis represents the optical density (OD). Mean value and standard deviation are shown (n = 3). *P < 0.05 versus control cells, Student's t-test. HBXIP, hepatitis B x-interacting protein (A color version of this figure is available in the online journal)

Flow cytometry analysis for cell cycle in HepG2 cells stably transfected with HBXIP gene or control vector. Data shown are from representative experiments repeated three times with similar results. HBXIP, hepatitis B x-interacting protein (A color version of this figure is available in the online journal)
To identify molecules responsible for the cell proliferation effect induced by HBXIP, the expression of several activators and inhibitors of the cell cycle was detected by Western blot assay. As shown in Figure 4a, HepG2-hbxip cells contained a greater amount of cyclinD1 and a lower amount of p21 and p53 protein when compared with control cells. The expression level of cyclinB1 was not significantly different between HepG2-hbxip and pCMV-tag2B cells. These data indicated that up-regulation of cyclinD1 and down-regulation of p21 and p53 might contribute to the HBXIP-induced G0/G1 to S phase transition.
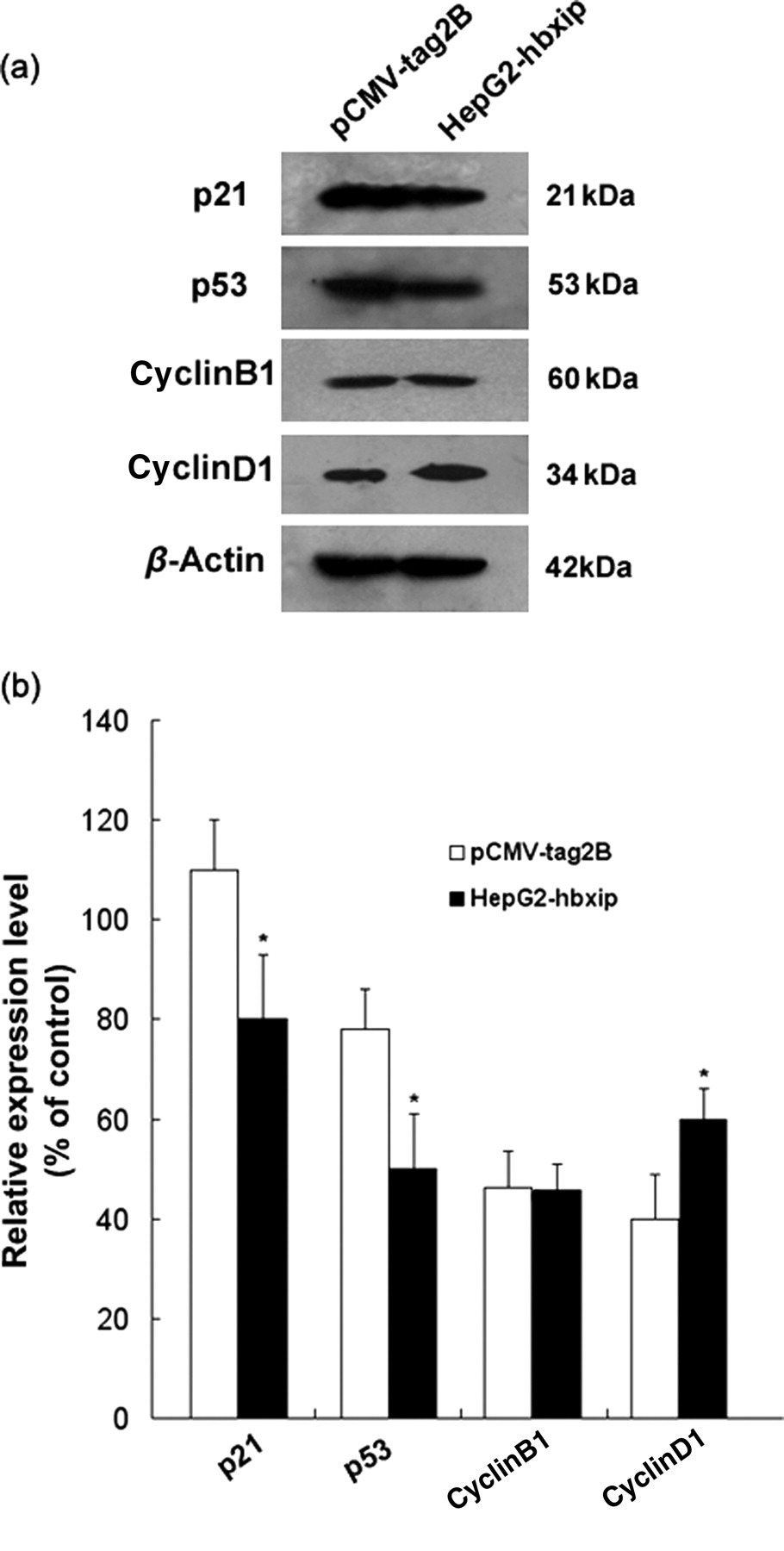
Up-regulation of cyclinD1 and down-regulation of p53 and p21 expression by HBXIP. (a) Examination of expression levels of p21, p53, cyclinB1 and cyclinD1 by Western blot analysis. (b) Bands were analyzed using Glyco Band-Scan software. Each bar corresponds to the mean ± SD for three independent experiments. *Indicates that P < 0.05 versus control using the Student's t-test. HBXIP, hepatitis B x-interacting protein
PI3K/Akt signaling pathway is involved in HBXIP-induced human HCC cell proliferation
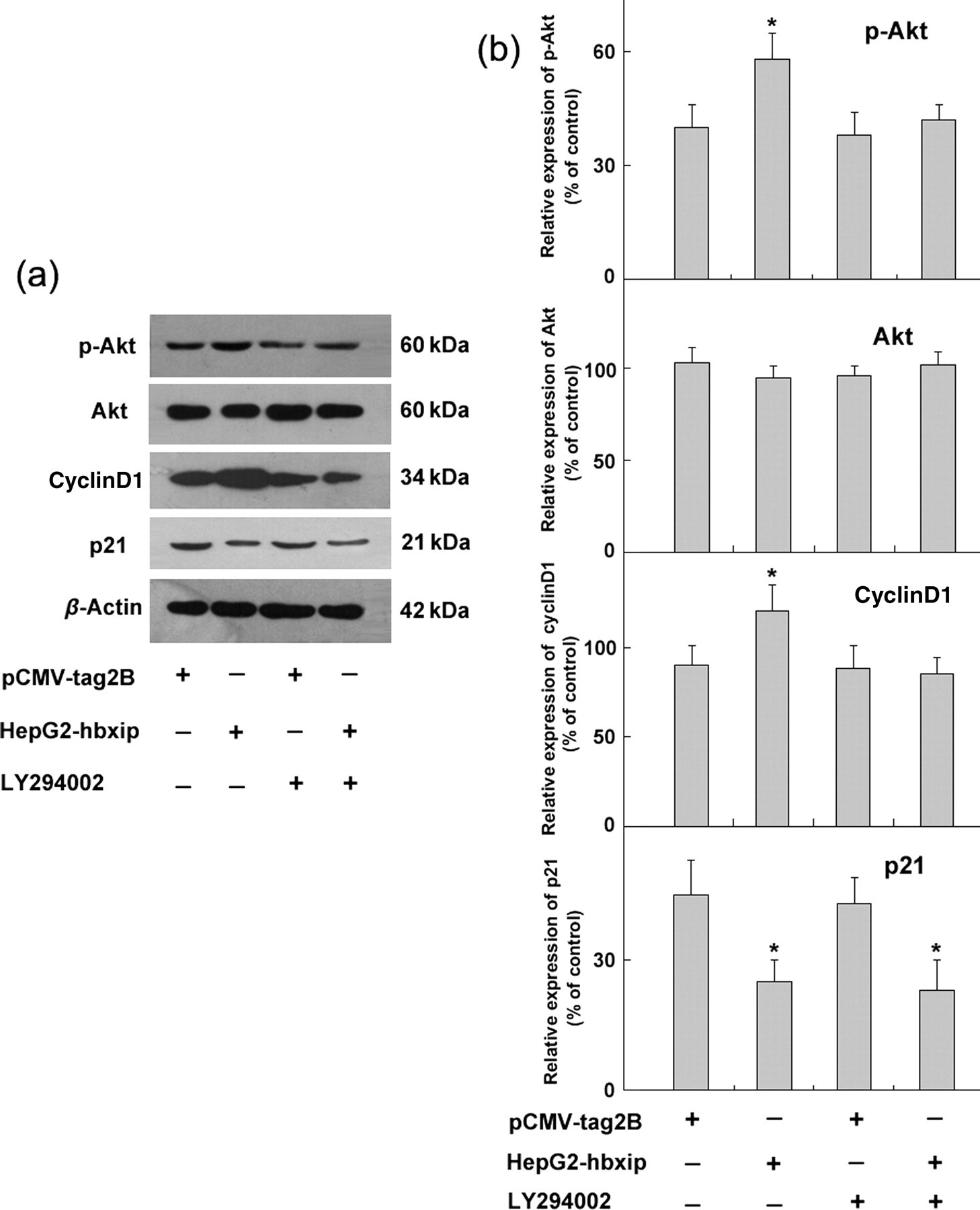
To investigate the possible molecular mechanism of HBXIP-induced cell proliferation, the PI3K/Akt signaling pathway and the MAPK/ERK pathway were examined. Western blot analysis of the phosphorylated Akt (p-Akt) level indirectly showed that Akt activity was increased in HepG2-hbxip cells when compared with the pCMV-tag2B control cells (Figure 5a). No obvious changes for total Akt protein levels were observed, demonstrating that HBXIP has no effect on protein stability. In agreement with the increased phosphorylation of Akt in HepG2-hbxip cells, over-expression of HBXIP in HepG2 cells led to an up-regulation in the phosphorylation of GSK-3β. We also examined the effect of HBXIP on the MAPK/ERK pathway, and no significant changes of phosphorylated ERK were observed in the Western blot assay results.

HBXIP activates the PI3K/Akt pathway in HepG2 cells. (a) Western blot analysis of phosphorylation of Akt, GSK3β and ERK. (b) Bands were analyzed using Glyco Band-Scan software. Each bar corresponds to the mean ± SD for three independent experiments. *Indicates that P < 0.05 and **indicates that P < 0.01 versus control cells using the Student's t-test. HBXIP, hepatitis B x-interacting protein; GSK3β, glycogen synthase kinase 3β
Phosphorylation of both Thr308 and Ser473 is required for full activation of Akt, and Akt activity can be measured using an in vitro kinase assay that detects phosphorylation of GSK-3 induced by immunoprecipitated Akt. To evaluate whether Akt enzymatic activity was directly induced by HBXIP over-expression, Akt activity was measured in HepG2 cells after being transiently transfected with either plasmids containing the HBXIP gene or empty plasmid controls. As shown in Figure 6a, HBXIP enhanced Akt enzymatic activity in HepG2 cells.

Kinase reactions and Western blot analysis were performed in anti-Akt immunoprecipitates from the corresponding lysates. (a) HepG2 cells were transiently transfected with either pCMV-hbxip or pCMV-tag2B plasmids. Forty hours after transfection, cells were washed with ice-cold phosphate-buffered saline and ice-cold lysis buffer was added. Akt activity was assessed using GSK-3α/β as a substrate for phosphorylation (p-GSK-3α/β). Phosphorylation of GSK-3 was measured by Western blot using a p-GSK-3α/β (Ser21/9) antibody. (b) Bands were analyzed using Glyco Band-Scan software. Each bar corresponds to the mean ± SD for three independent experiments. **Indicates that P < 0.01 versus control cells using the Student's t-test. HBXIP, hepatitis B x-interacting protein; GSK3β, glycogen synthase kinase 3β
To examine whether the Akt pathway was directly involved in the HBXIP-induced proliferation of HepG2 cancer cells, HepG2-hbxip cells were treated with LY294002, a specific inhibitor of the PI3K/Akt pathway, 18 followed by the MTT assay, cell cycle distribution and Western blot analyses. As expected, MTT results showed that LY294002 treatment reduced HBXIP-enhanced proliferation of HepG2 cells (Figure 7). In comparison with the dimethyl sulfoxide-treated cells, the percentage of HepG2-hbxip cells in the S phase decreased by 10.86 ± 1.42% at 12 h after LY294002 treatment, while that of pCMV-tag2B cells was unaffected (Figure 8). Western blot results showed that the expression of p-Akt in the HepG2-hbxip cells was decreased after treatment of LY294002, and the total levels of Akt were unchanged (Figure 9a). Furthermore, LY294002 potently inhibited the cyclinD1 induction by HBXIP over-expression, but p21 inhibition by HBXIP was unaffected. These data indicated that the PI3K/Akt pathway was involved in the promotion of HBXIP-induced cell proliferation, and that the HBXIP up-regulation of cyclinD1 required PI3K/Akt activation.
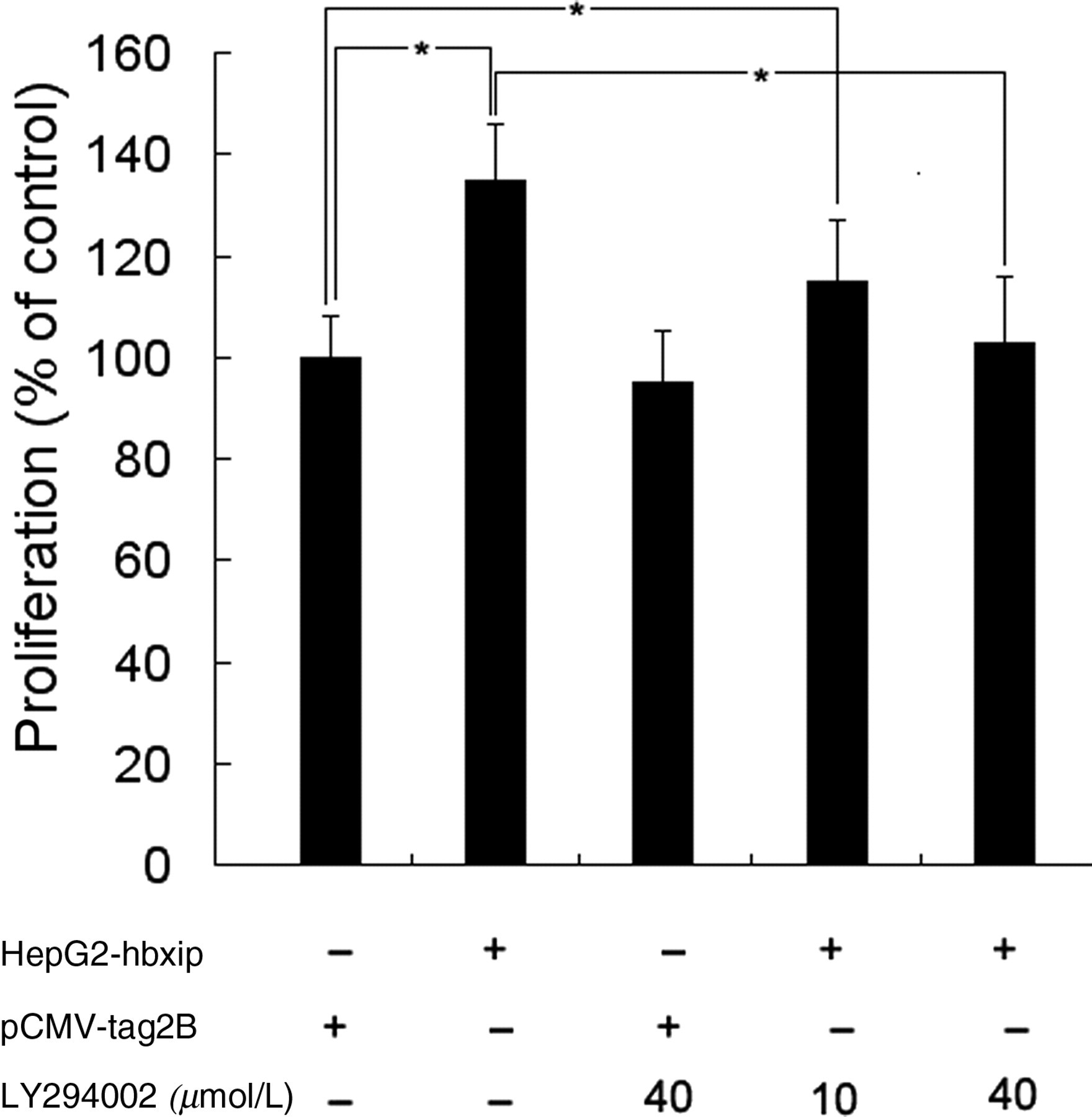
Effects of PI3K inhibitor, LY294002, on HBXIP-induced HepG2 cell proliferation assessed by MTT assay. HepG2 cells stably transfected with HBXIP were treated with LY294002 at 10 and 40 μmol/L and then analyzed by the MTT assay. Values are expressed as percentage of untreated cells, and each bar represents the mean ± SD of triplicate determinations (n = 3). *Indicates that P < 0.05 versus control cells. HBXIP, hepatitis B x-interacting protein

HepG2 cells stably transfected with HBXIP were treated with LY294002 and analyzed by flow cytometry analysis. The data shown are representative of three experiments. HBXIP, hepatitis B x-interacting protein; DMSO, dimethyl sulfoxide (A color version of this figure is available in the online journal)
LY294002 inhibits expression of cyclinD1 induced by HBXIP over-expression in HepG2 cells. (a) HepG2 cells stably transfected with HBXIP were treated with LY294002 (40 μmol/L) and analyzed by Western blot assay. (b) Bands were analyzed using Glyco Band-Scan software. Each bar corresponds to the mean ± SD for three independent experiments. *Indicates that P < 0.05 versus control cells using the Student's t-test. HBXIP, hepatitis B x-interacting protein
Discussion
HBXIP, a co-factor of survivin, was originally identified by its interaction with viral HBx. Although some binding proteins of HBXIP, such as HBx, survivin and hSuv3p, have been identified in the past few years, these interactions are almost irrelevant to cellular proliferation. In hepatoma cells, HBXIP bridges HBx and survivin together to suppress caspase activation, inhibit apoptosis and regulate centrosome dynamics. hSuv3p is another interactor of HBXIP that is mainly involved in the processes of RNA turnover and apoptosis. Previously, we found that HBXIP promoted the growth of both normal liver cells and hepatoma cells in vitro, but the mechanisms were not clearly expatiated; therefore, we speculate that other signaling pathways are involved in HBXIP-regulated cellular proliferation.
In this study, we first provide evidence that HBXIP promotes HepG2 cell proliferation by facilitating the G1 to S phase transition (Figure 3) that is commonly seen in cancer cells. Many kinases and kinase inhibitors in the cell cycle are involved in mediating the G1/S transition. 19,20 p21 is central to cell cycle arrest, and down-regulation or loss of p21 expression has been found in a variety of human cancers. 21,22 In HepG2 cells, high expression of HBXIP decreased the levels of p21 (Figure 4a). Because p21 protein levels are directly stimulated by the p53 protein, 23 we then examined the effect of HBXIP on p53 expression. Western blot results indicated that HBXIP down-regulated the levels of p53. A recent study showed that HBXIP was a substrate of ATM involved in the cellular response to DNA damage, 24 and that activation of p53 was mainly dependent on signaling by ATM kinase when DNA damage occurred; thus, regulation of p53 expression by HBXIP may be involved in the cellular response to DNA damage. We also determined the expression levels of cyclinB1 and cyclinD1 by Western blot. As shown in Figure 4a, HBXIP induced the expression of cyclinD1 in HepG2 cells, and the cyclinB1 level remained unaffected.
The PI3K/Akt signaling pathway is a key regulator of numerous physiological cellular processes, including proliferation, apoptosis and metabolism. Recent studies indicated that PI3K activation was required for the progression of mitosis, promoting the entry of quiescent cells into the S phase. 25 In tumor cells, the regulation of Akt is complex, and phosphorylation of Akt at Ser473 is still an unclear mechanism. Here, we have shown that PI3K/Akt is induced by HBXIP over-expression. GSK-3β, a downstream target of PI3K/Akt, has been implicated in diverse biological processes including cell proliferation and cell survival signaling in various cell types. Therefore, we also investigated whether HBXIP enhanced the phosphorylation of GSK-3β in HepG2 cells. As shown in Figure 5a, HBXIP over-expression stimulated a significant increase in the phosphorylation of GSK-3β. It has been previously reported that activation of MAPK/ERK occurs in human HCC, 26 so we also examined the effect of HBXIP on the activation of ERK. Western blot results showed that HBXIP did not have a significant impact on the phosphorylation of ERK (Figure 5a).
This study further investigated whether the activation of the PI3K/Akt pathway induced by HBXIP was specific. HepG2-hbxip cells and control cells were treated with LY294002, a widely used PI3K inhibitor. The MTT assay showed that pretreatment with LY294002 blocked the cell proliferation promoted by HBXIP (Figure 7). Inhibition of the PI3K/Akt signaling pathway partially reversed HBXIP-induced cell cycle progression (Figure 8). Both p21 and cyclinD1 are downstream factors of the PI3K/Akt signaling pathway, and are involved in the regulation of cell cycle progression via the Akt pathway. 27,28 Moreover, we observed that HBXIP inhibited the level of p21 and induced the expression of cyclinD1, so Western blot assay was performed to detect whether or not p21 and cyclinD1 were involved in Akt activation. As shown in Figure 9a, the PI3K chemical inhibitor LY294002 suppressed the induction of cyclinD1 but not p21 in HepG2 cells, suggesting that the PI3K/Akt pathway is involved in HBXIP-induced cell proliferation, possibly through the induction of cyclinD1.
In summary, our findings suggest that one of the functions of HBXIP is associated with cell proliferation regulation. Over-expression of HBXIP targets cyclinD1, promoting HCC cell proliferation through activation of the PI3K/Akt pathway. Blocking the PI3K/Akt pathway using LY294002 decreases the p-Akt protein level as well as the fraction of cells in the S phase. Up-regulating the level of cyclinD1 is involved in the Akt activation, and the down-regulation of p21 and p53 by HBXIP may be relevant to other signaling pathways in HepG2 cells.
Footnotes
ACKNOWLEDGEMENTS
This work was supported by grants from the National Natural Science Foundation of China (No. 30800422).