
Abstract
Severe pandemic influenza A H1N1 (2009) infection, especially in the lower respiratory tract, is often associated with the virus carrying a D222G substitution in the hemagglutinin (HA) protein of the virus. The mechanism for this association has not been fully explored. In the in vitro binding assay, it was found that clinical isolates carrying D222G substitution exhibit higher binding avidity to 2,3-linked sialic acids than the wild-type virus. The receptor binding pocket of the pandemic influenza (H1N1) HA was found to be smaller than those of other influenza A strains, allowing tighter binding of the virus with the receptor, yet also inducing steric stress for the binding. Our homology modeling and molecular docking calculations implicated that residue 222 may affect the positioning of the conserved Q223 residue, hence modulating flexibility of the binding pocket and steric hindrance during receptor binding. The molecular property of residue 222 can also directly influence the ‘lysine fence’ via the polarity of the amino acid residue where D222G substitution will enhance the electrostatic interactions between the receptor and the protein. The potential importance of residue 222 was illustrated by evolutionary analysis, which showed that this site is under intense selection pressure during adaptation of the virus to human host. Our findings provide a useful reference for follow-up studies in monitoring the ongoing evolution of the pandemic influenza A H1N1 (2009) virus.
Introduction
The influenza A virus has caused seasonal epidemics and some of the biggest pandemics in human history. Given the fact of the devastating impact of the pandemic 1918 ‘Spanish flu’ and the most recent 2009 (H1N1) pandemic, 1,2 preparation for the future influenza pandemic is warranted. Despite the significant effort in worldwide surveillance network for emerging influenza strains, the swine-origin influenza A/H1N1 virus emerged largely undetected until substantial numbers of people were infected and caused the pandemic in 2009. Indeed, transmission of the swine influenza virus strains to humans had already occurred in as early as 1976, and sporadic cases have been detected since 1998. 3–5 Except for the event of 1976, the relatively limited scale of cross-species transmissions and the mild clinical presentation has not drawn much attention from public health authorities and the scientific community. Arguably, the focus on avian influenza viruses, particularly the highly pathogenic A/H5N1 strains, 6 was justified, in view of the significant mortality from reported human cases of influenza A/H5N1 infections. 7,8 The preparedness for a potential pandemic potential of avian influenza viruses had been the major motivation behind international vigilance, which has paid off handsomely in the form of an effective and coordinated global public health response to the novel influenza A/H1N1 (2009) virus.
Earlier studies have identified that the pandemic (H1N1) 2009 virus was a result of multiple re-assortment events with viral genes originating from avian, swine and human influenza strains. 9 Advanced phylogenetic analysis enabled the construction of a temporal profile of viral evolution prior to and around the beginning of the pandemic, and possibly even before it had jumped the species barrier. 10,11 However, these studies could not provide information regarding the particular genetic changes that allow efficient human infection and transmission caused by the pandemic (H1N1) 2009 virus. While there had been three recorded pandemics in the last century, this was the first time we were able to track the pandemic from its early beginning. It is essential to reconcile descriptions of the evolutionary changes with their functional significance for understanding virus adaptation during the early phase of a pandemic. Using influenza hemagglutinin (HA) as an example, functional mutations of this viral protein would be expected to result in variants in antigenicity, receptor binding properties and potentially leading to a change in clinical presentation. We and others found that a variant with D222G (D225G in H3 numbering) in the HA gene of the pandemic (H1N1) 2009 virus is strongly associated with increased disease severity in both humans and mice. 12–15 The D222G variant is more frequently detected from specimens obtained in the lower respiratory tract of severe patients. 12 A receptor binding assay with a mouse-adapted virus containing the D222G substitution in the HA and its parental virus suggested that D222G may lead to an increase in binding to the α2,3-linked sialic acids receptor by the virus. 15 However, the structural basis for an altered receptor binding preference by the D222G substation remains unclear. The D222G mutation in the pandemic influenza (H1N1) virus may be just one of the markers in the adaptation to humans. It is likely that other mutations may exist throughout the influenza genome and act functionally to facilitate the virus' adaptation to the new host environment. Several research groups have described sites in the viral genome under positive selection in an attempt to identify the key functional residues in proteins of the pandemic (H1N1) influenza 2009 virus. 16,17
In the present study, we extended the analysis to delineate the effect of the mutations on viral protein structure and function in sialic acid receptor binding and correlations among functionally important mutations. Our results reveal a potential mechanism for the effect of the widely reported D222G mutation in the HA molecule of the virus on receptor binding, which might help explain its association with clinical severity and tissue tropism.
Materials and methods
Virus-turkey erythrocyte binding assay
Clinical isolates of the pandemic (H1N1) 2009 virus were obtained from the Center for Health Protection of Hong Kong Special Administrative Region and isolates from severe cases were obtained from the Virus Unit of the Queen Mary Hospital, Hong Kong Special Administrative Region. The virus-turkey erythrocyte binding assay was performed as previously described with minor modifications. 18 The optimal condition for neuraminidase treatment of turkey erythrocyte was tested using A/PR/8/34 isolate. Briefly, turkey erythrocytes were treated with different concentrations of Vibrio cholerae neuraminidase (Sigma, St Louis, MO, USA) for 60 min at 37°C to remove the 2,3-linked sialic acid. The erythrocytes were washed twice with phosphate-buffered saline (PBS) and then diluted to 2% (v/v) erythrocytes solutions with PBS. The 2% erythrocyte solution (25 μL) was mixed with eight HA units of influenza viruses (100 μL) and incubated at room temperature for 60 min. Ten isolates with 222D and four isolates with 222G were tested in this study. Hemagglutination was measured and data expressed as the maximal concentration of neuraminidase that allowed for full hemagglutination.
Sequence and structural data
Complete gene and genome sequence data of the pandemic influenza A/H1N1 (2009) virus were downloaded from the National Center for Biotechnology Information Influenza Virus Resource (
Homology modeling
Homology models of influenza HA protein were constructed using the SWISS-MODEL homology modeling server
20
using established protocols (
Molecular docking
Molecular docking studies were performed on a Dell Dimension 5150 machine with a 3.0 GHz dual core Xenon central processing unit (CPU), 2048 MB RAM and running on Fedora 8 Linux operating system. Autodock Vina (
Sequence analysis
All genes and translated protein sequences were aligned with ClustalW
28
using default parameters. Sequences with slight length variations due to indels were manually edited to maintain consistency of the data-set. Sequences containing ambiguous bases or residues were removed. Identification of sites under positive selection was performed using the following methods as implemented in HyPhy:
29
single likelihood ancestor counting, fixed effects likelihood and directional evolution of protein sequences (DEPS).
30
Phylogenetic reconstruction was performed using the neighbor-joining method within HyPhy. The Consurf webserver (
Co-evolution among residues of the influenza HA protein was examined by mutual information (MI) analysis by using a normalized measure of the MI value to assess the correlation between any two sites. The MI matrix was computed as described in previous studies. 32,33 Hierarchical cluster analysis was then performed with the dChip program to group sites showing highly correlated mutations. Identification of ‘site fixation’ in an attempt to predict future trends in mutation was performed in accordance to methods described in previous studies. 32,34
Statistical analysis
Statistical analysis was performed using R version 2.11.0. All P values reported are for a two-tailed test, and P < 0.05 is considered statistically significant.
Results
The D222G variant in the HA of the pandemic (H1N1) 2009 virus by virus-turkey erythrocyte binding assay
The clinical isolates that contained 222D served as the positive control, which still produced hemagglutination despite treatment of the turkey erythrocyte by 32 μg/mL of neuraminidase, whereas the four clinical isolates with D222G substitution had demonstrated hemagglutination at a neuramindase concentration of not more than 16 μg/mL (Figure 1). Our result suggests that the four clinical isolates carrying D222G substitution were more sensitive to the treatment with neuraminidase in turkey red cells in the hemagglutination assay, suggesting that the D222G variant has a higher binding avidity to 2,3-linked sialic acids than the wild-type virus.
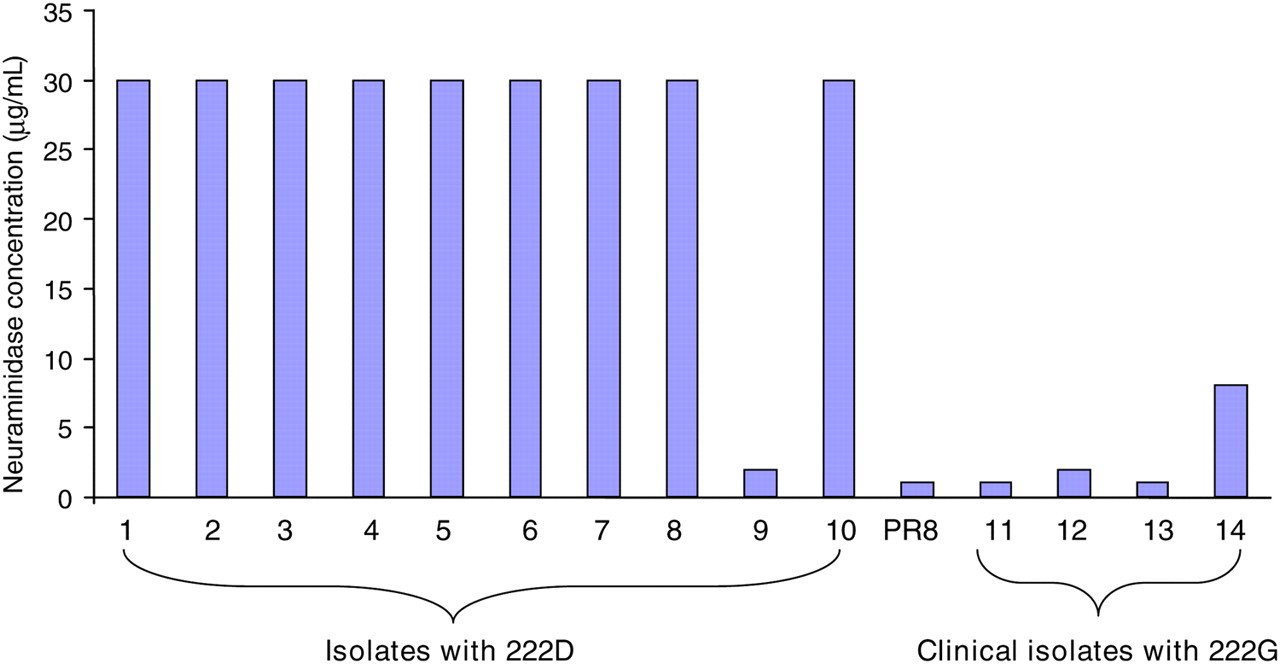
Cellular receptor binding avidity assay. Influenza virus cellular receptor binding avidity was estimated with turkey blood cells after treatment with Vibrio cholerae neuraminidase which mainly removes 2,3-linked sialic acids as described in the Materials and methods. The data represent the mean value of two independent experiments. PR8, A/Purto Rico/8/34 is a reference virus used to validate the assay as described in a previous study16
Structural analysis of pandemic influenza A HA
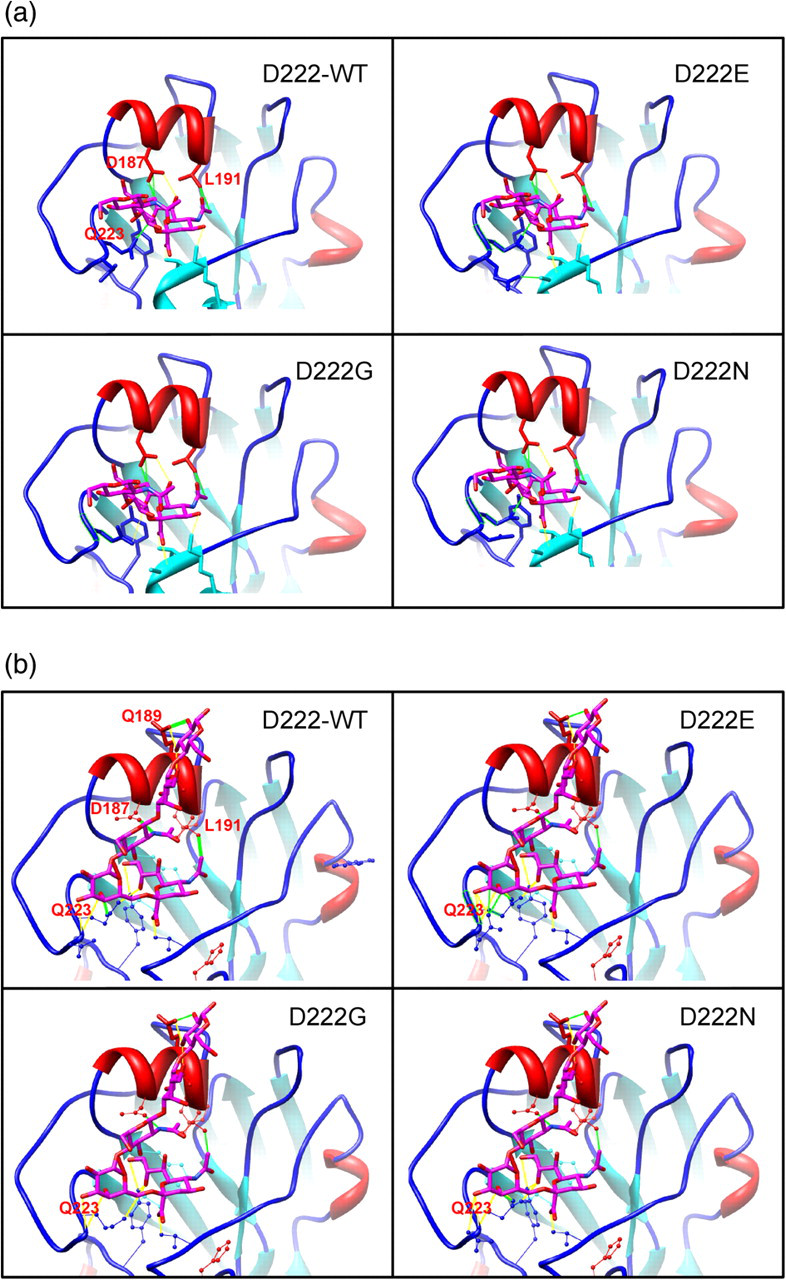
To study the effects of amino acid polymorphism at residue 222 of the pandemic influenza A/H1N1 (2009) HA protein, three homology models were constructed to represent the D222G, D222E and D222N variants using the wild-type HA protein (D222-WT) as the template. As the four proteins are highly similar in sequence (>99% identity) and structure, the structural comparison is mainly focused around the receptor binding pocket which is located in close proximity to residue 222. Among the differences generated by the amino acid substitutions, the change in surface charge has the most apparent effect on the binding of the ligand to the pocket. The ‘lysine fence’ is a long stretch of basic surface charges created by the lysine residues at position 130, 142 and 219 (H1 numbering), which are positioned to anchor the Sia1 and galactose (Gal2) sugars of both α2,3 and α2,6 glycans. 35 This structural feature is considered more significant for the pandemic influenza A/H1N1 (2009) virus, as the lysine residue at position 142 is not found in the other H1N1 strains. Incidentally, the amino acid residue at position 222 is part of the 220-loop which interacts with this lysine fence. A neutral glycine or basic asparagine mutation at this position would extend and enhance the basic charge of the lysine fence, whereas the wild-type aspartic acid residue or the glutamic acid mutation would create a localized patch of acidic charge (Figure 2). The latter effect would result in a diminishing of the attractive effect of the lysine fence towards the sialylated glycans.
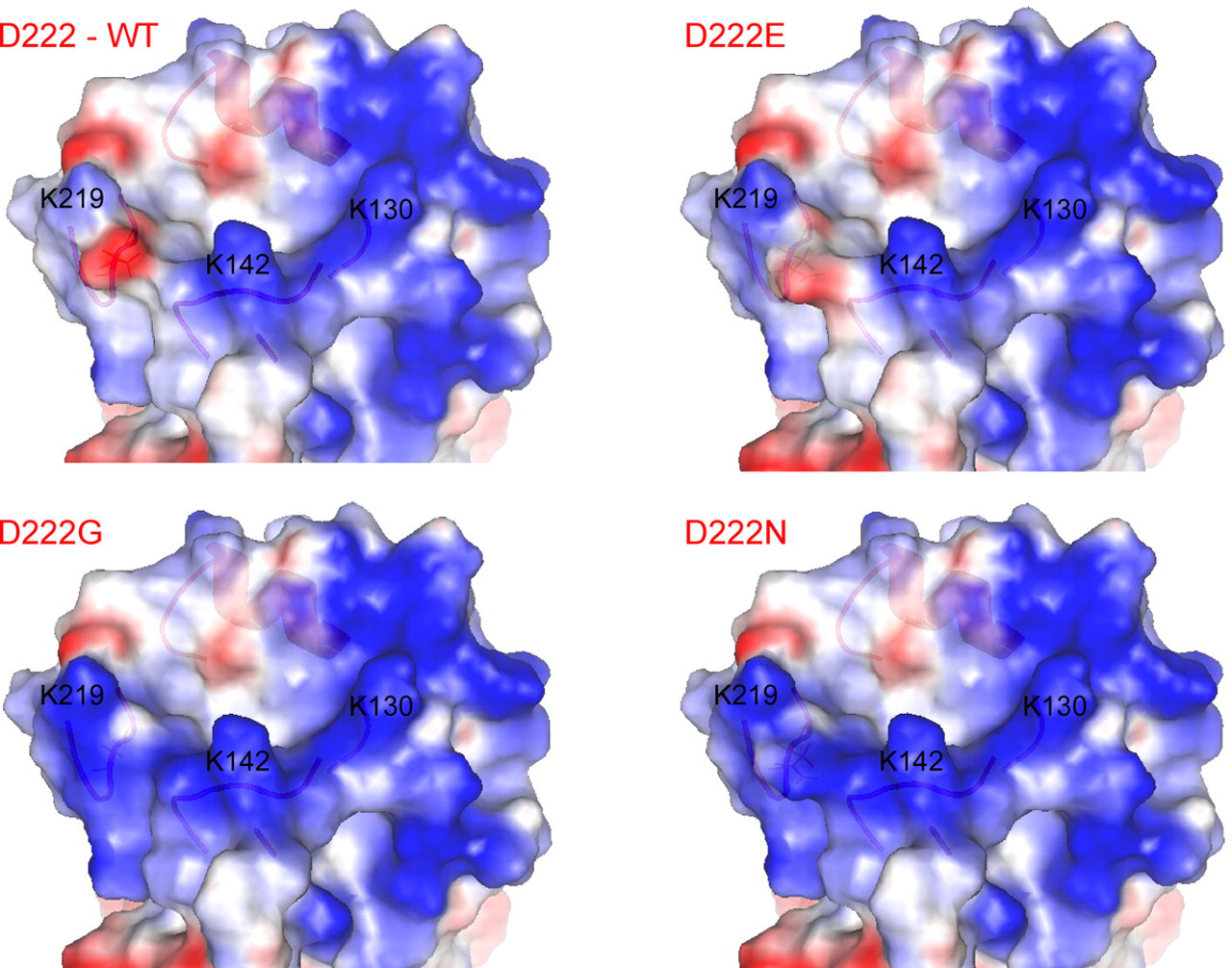
Distribution of surface charges around the sialylated glycan binding site on the pandemic influenza A/H1N1 (2009) hemagglutinin (HA) protein. Blue and red colors represent basic and acidic charges, respectively. Lysine residues at position 130, 142 and 219 (H1 numbering) can be shown to form a stretch of basic charges known as the ‘lysine fence’. D222G and D222N mutations would significantly extend and enhance the basic charge of the lysine fence
Comparison of the crystal structure of the pandemic influenza A/H1N1 (2009) HA protein (3LZG) with available crystal structures of other influenza A HA proteins showed that the conformation of the receptor binding pocket is relatively well conserved. Despite the high overall structural similarity of the HA proteins, we noted a considerable variation in the widths of the opening of their receptor binding pockets. The shortest distance between the 190-helix and the 130-loop across the binding site for the N-acetylneuraminic acid (Sia1) ring is only 9.45 Å in the pandemic influenza A/H1N1 (2009) HA protein, whereas the corresponding distances are 10.54 Å for the avian influenza A/H1N1 HA (pdb 3HTT), 10.58 Å for the avian influenza A/H5N1 HA (pdb 1JSN) and 11.05 Å for the swine influenza A/H9N2 HA (pdb 1JSI) (Figure 3a). Compared with the ligand bound X-ray structures of other HA proteins shown in Figure 3b, in silico molecular docking of an α2,3-linked sialylated oligosaccharide ligand to the conserved binding position onto the pandemic influenza A/H1N1 (2009) HA protein would show at least three regions of steric clashes between the ligand and protein in the absence of conformational flexibility of the binding site. The binding of the α2,6-linked sialylated oligosaccharide ligand to the pandemic influenza A/H1N1 (2009) HA protein would be expected to experience similar steric stress. Therefore, a larger conformational change or flexibility of the receptor binding pocket of the pandemic influenza A/H1N1 (2009) HA protein will be required to allow the effective binding of sialylated oligosaccharide ligands.
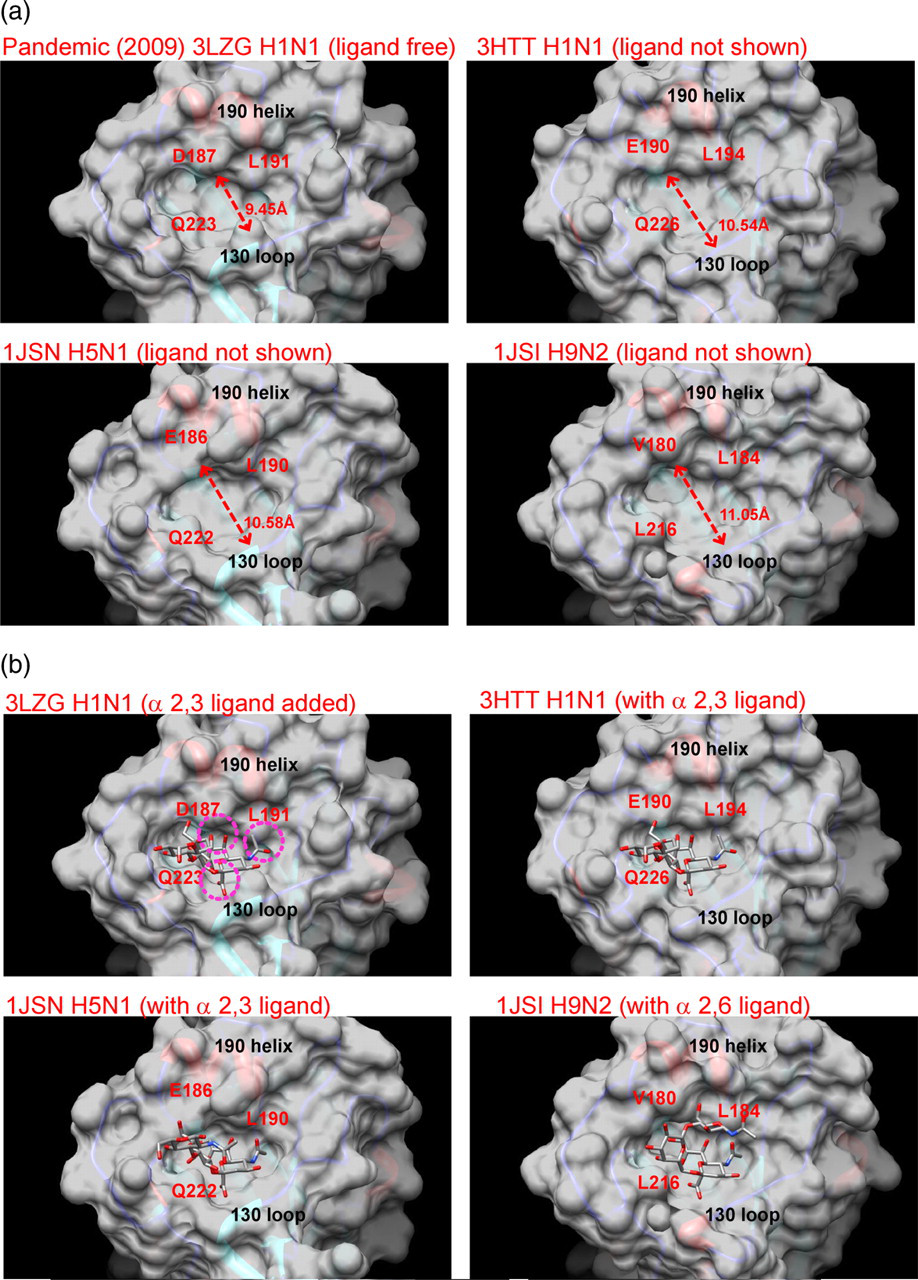
Structural comparison of the receptor binding pocket of the influenza A virus hemagglutinin (HA) protein. (a) The opening of the pocket of the pandemic influenza A/H1N1 (2009) HA protein is appreciable narrower than that of the other influenza viruses shown. Dotted line indicates the shortest distance between the 190-helix and the 130-loop with the distance measurement shown next to the line. (b) Molecular docking of an α2,3-linked sialic acid ligand to the receptor binding pocket of pandemic influenza A/H1N1 (2009) HA protein showed regions of steric clashes between the two molecules. Regions of the steric clashes are highlighted with dotted circles. No such steric clashes are observed with the ligand bound crystal structures of the other influenza HA proteins (3HTT, 1JSN, 1JSI)
The effects of the D222 polymorphisms were further explored by molecular docking of sialylated glycan ligands to the pandemic influenza A/H1N1 (2009) HA variants. Under the condition of no conformational flexibility of the HA molecule, the repertoire of in silico molecular docking results generated by Autodock Vina were mostly suboptimal and did not match the conserved binding position of the Sia1 ring known from crystal structures with bound sialylated glycan ligands (data not shown). Even allowing for flexible side-chains on the residues in the receptor binding pocket, only the α2,3-linked sialic acid was able to produce a docking model with the D222G variant at the conserved Sia1 position, whereas all α2,6-linked sialic acid still failed to dock at the conserved position.
To better access the structural features of receptor-bound HA protein of the pandemic influenza A/H1N1 (2009) in the context of a conformationally widened binding pocket, homology modeling of the wild-type HA protein as well as its D222G, D222E and D222N variants were also carried out using the following glycan bound HA protein crystal structures as templates: 1JSI (swine influenza A/H9N2 [1998] with α2,6-linked sialic acid ligand), 20 1JSN (avian influenza A/H5N1 [1997] with α2,3-linked sialic acid ligand) 22 and 3HTT (avian influenza A/H1N1 [2005] with α2,3-linked sialic acid ligand). 23 The resulting homology models of the pandemic influenza A/H1N1 (2009) HA protein have acquired wider receptor binding pockets resulting at higher successful docking rates for both α2,3- and α2,6-linked sialic acid ligands. In particular, the α2,3-linked sialic acid ligand is able to dock to the conserved position for D222 WT (with 3HTT and 1JSN pockets), D222G (with 3HTT and 1JSN pockets), D222N (with 1JSN pocket) and D222E (with a 1JSN pocket allowing flexible side-chains); whereas the α2,6-linked ligand is able to dock to D222 WT (with 1JSI pocket), D222N (with a 1JSI pocket allowing flexible side-chains) and D222E (with a 1JSI pocket allowing flexible side-chains). These results support that a flexible or wider receptor binding pocket of the pandemic influenza A/H1N1 (2009) viral HA is critical for its binding to sialylated glycans.
Sequence and co-evolutionary analysis of pandemic influenza A HA
A total of 795 non-redundant full-length pandemic influenza HA protein sequences from the period of April 2009 to March 2010 were included for analysis. MI analysis was performed as described above. Using the arbitrary cut-off of 0.5 from the previous study on H3N2 HA protein, 31 45,199 (28.3%) out of 159,895 possible pairs were found to have possible correlations in mutational patterns. This number is reduced to 816 (0.5%) if the cut-off is raised to 2.0 (Figure 4). The amino acid residues found to have correlation with mutations at residue 222 are shown in Table 1. The most significant correlation among these was observed for residue 298 of HA1, although a surprising number of the other residues were located in HA2. Examination of the mutations in residues 222 and 298 identified the D222E and P298S mutations as significantly correlated (φ = 0.62; P < 0.00001 by Fisher's exact test; Table 2). While the two residues are not in close proximity with each other, the location of residue 298 near the C-terminal of HA1 led us to speculate that it may affect the flexibility of the protein. Proline is known to have a rigid conformation and may confer local structural stability and low flexibility. 36,37 Hence, its substitution by serine may permit increased flexibility at the interface between HA1 and HA2, with downstream effects on the receptor binding by the protein.
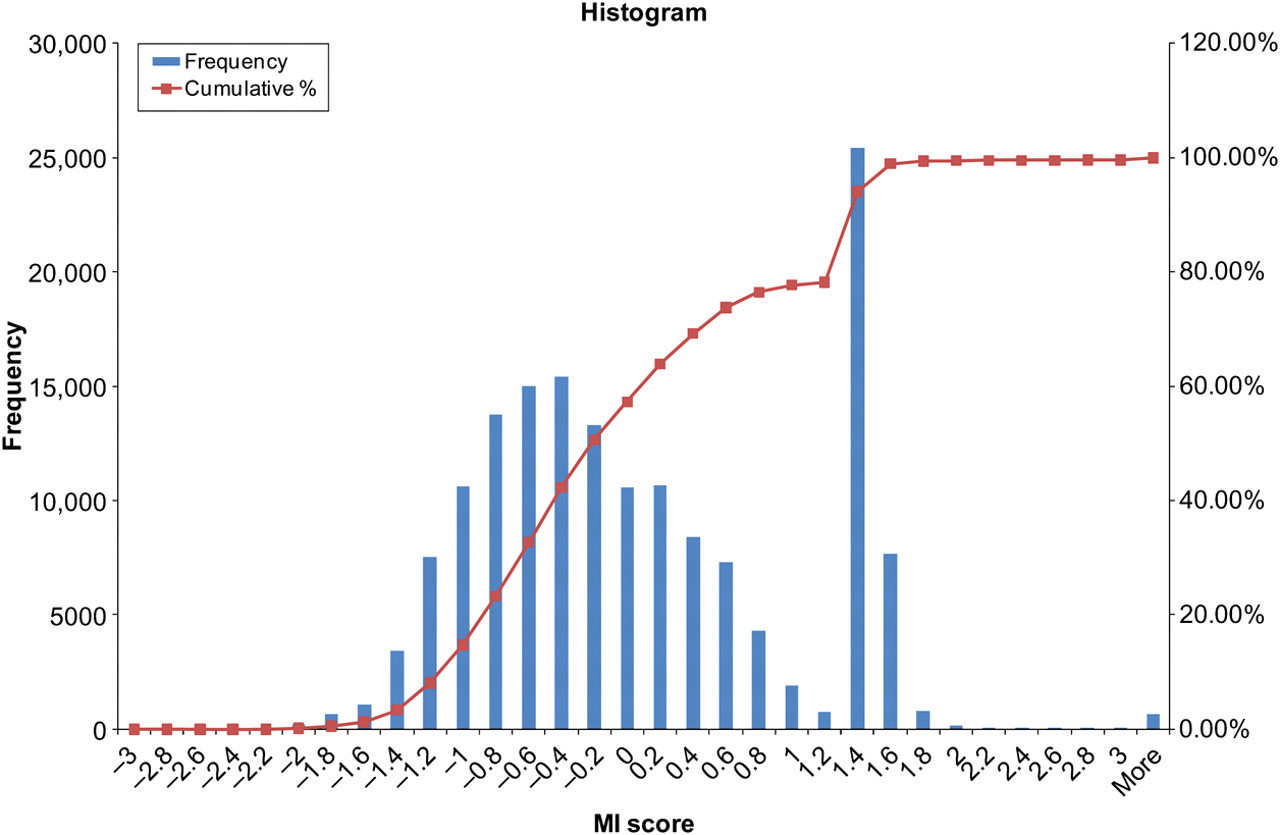
Histogram of the normalized mutual information (MI) scores from MI analysis of the pandemic influenza A/H1N1 (2009) hemagglutinin (HA) protein. It shows a bimodal distribution of MI scores with a broad peak centered around −0.4 and a sharp peak located at around 1.4
Correlation of mutations at amino acid residue 222 (H1 numbering) of the pandemic influenza A/H1N1 (2009) hemagglutinin (HA) protein with other residues of the same protein
Correlation of D222E and P298S mutations in the pandemic influenza A/H1N1 (2009) hemagglutinin (HA) protein
Numbers shown are the sequence counts for the specified polymorphisms
Examination of amino acid residue pairs with the highest MI scores (Table 3) showed that correlations of mutations between residues often occur over relatively large distances. Only four out of the 10 pairs were residues from the same mature peptide fragment. The most intriguing finding is the presence of two amino acid residue pairs with one member located in the signal peptide. Given the temporal and spatial compartmentalization of the peptides, it is implausible for any direct interaction between the signal peptide and the HA1/HA2 molecules. Whether the apparent correlation is an artifact introduced during nucleic acid amplification and sequencing or a result of an uncharacterized mutational bias remains to be seen. Nonetheless, it is a clear reminder that ‘positive’ results from data mining have to be placed in a relevant biological context and be confirmed experimentally.
Top 10 pairs of amino acid residues of pandemic influenza A/H1N1 (2009) hemagglutinin (HA) protein with highest mutual information (MI) scores
Residue positions refer to the full-length consensus HA sequence
Hierarchical cluster analysis of the MI matrix revealed several small clusters of correlated sites which were also closely related in spatial distribution, such as the first eight amino acids of the signal peptide region. The D222 residue did not form a distinct cluster with other residues. A large number of sites were found to be grouped in a significant cluster but all of the sites showed only a weakly positive MI score of less than 2.0. Further examination revealed that most of these sites correspond to the second peak of the histogram shown in Figure 4. The association among these sites could not be confirmed with statistical tests of association such as the Fisher's exact test, and our assessment is that the degree of correlation, if any, does not meet the criteria for statistical significance.
A total of 1476 non-redundant full-length pandemic influenza HA coding sequences from the period of April 2009 to March 2010 were downloaded for sequence analysis. Positive selection detection was performed using the different algorithms implemented in HyPhy. A total of 12 sites in the pandemic influenza A/H1N1 (2009) HA sequence were identified to be under selection by one or more of the algorithms (Tables 4 and 5). Although directional selection and positive selection act on different levels (amino acids and codons, respectively), a common set of amino acid residues/codon sites was identified to be under both types of selection. The most significant degree of selection was noted for residues 203 and 222 (H1 numbering) of HA1, with estimated dN/dS (ratio of the rate of substitutions at synonymous sites to the rate of substitutions at non-synonymous sites) of greater than 15 and Bayes factors of greater than 10. 16 The dominant mutations at each position by substitution count were T203S and D222G, although the D222E mutation was a close second to D222G for that position. The sites identified generally correspond to regions that are surface accessible or located in close proximity to the receptor binding pocket, and are responsible for an appreciable proportion of variation observed in the HA sequences (Figure 5). In addition, five ‘site fixations’ were identified in HA: V36I, V47A, S220T, E391K and I564K.

Identification of conserved and variable regions on the pandemic influenza A/H1N1 (2009) hemagglutinin (HA) protein. The most conserved regions are colored magneta (HA1)/red (HA2) and the most variable regions are colored cyan (HA1)/blue (HA2). For HA1, the variable regions are distributed throughout the surface of the molecule, while the constant regions can be found as isolated pockets in the internal regions and C-terminal region in contact with HA2. For HA2, regions of high variability can be found to a lesser extent throughout the surface of the molecule
Sites under positive selection as detected using HyPhy
ND, positive selection not detected using this method
Methods used are: single likelihood ancestor counting (SLAC) and two-rate fixed effects likelihood (2FEL). Significance level is set at 0.10 for both methods
Sites under directional selection as detected using the DEPS method as implemented in HyPhy
DEPS, directional evolution of protein sequences; IUPAC, International Union of Pure and Applied Chemistry
A Bayes factor of more than 20 is considered evidence for directional selection. Standard IUPAC abbreviations are used for amino acids
*Directionality of evolution for this site may have been reversed due to an artifact resulting from data sampling. The correct preferred residue should be threonine (T)
Discussion
While the pandemic (H1N1) 2009 virus was found antigenically stable during the first year of circulation in humans, genetic variants were observed. Notably, a variant carrying D222G (H1 numbering) was found mainly in severe cases of patients as reported in several studies from different countries. 12–14 A previous study found D222G substitution associated with change of receptor specificity from Gal 2,6-linked to 2,3-linked sialic acids in the pandemic 1918 virus. 38 We also found a mouse-adapted virus containing a D222G mutation in the HA gene, and which exhibits enhanced virulence in mice. 15 It is suggested that the pandemic (H1N1) 2009 virus with D222G HA may be more likely to infect lower respiratory tract epithelial cells which express predominantly 2,3-linked sialic acids. To further verify if D222G is a phenotypic variant, we examined the receptor binding avidity of wild-type and mutant viruses using turkey blood cells treated with neuraminidase which mainly cleavage 2,3-linked sialic acids. We confirm that D222G variants had a higher preference for binding to 2,3-linked sialic acids, as illustrated by its higher sensitivity to the treatment of neuraminidase, while the wild-type virus, with its preference for binding to 2,6-linked sialic acids, will exhibit resistance to neuraminidase treatment.
The observed narrower receptor binding pocket of the pandemic influenza A/H1N1 (2009) HA protein may have several implications for its function. Firstly, the sialylated glycans binding to the pocket will experience steric hindrance from the neighboring protein residues. This is demonstrated by the molecular docking of an α2,3-linked sialylated oligosaccharide ligand to the pandemic influenza A/H1N1 (2009) HA protein (Figure 3b), which showed at least three regions of steric clashes between the ligand and protein in the absence of conformational flexibility of the binding site. Secondly, the narrower opening of the pocket will likely impact the kinetics of ligand binding in a negative manner, as a larger conformational change of the receptor binding pocket will be required to allow the ligand to adopt the normal bound position and conformation. Thirdly, from a mechanistic point of view, the probability of receptor binding will be decreased but this might be partially offset by a ‘tighter’ grip of the bound sialylated glycan assuming no further conformation changes occur. Therefore our structural analysis of the pandemic influenza A/H1N1 (2009) viral HA suggested that a flexible or wider receptor binding pocket is critical for its binding to sialylated glycans. While it could be difficult to accurately model protein flexibility without performing a computationally expensive molecular dynamics study, it is possible to infer the potential effects of specific amino acid residues on protein–ligand interaction. It has been reported that the HA protein of the pandemic (H1N1) 2009 influenza virus possesses residues that can be positioned to make optimal contacts with both α2,3- and α2,6-linked sialic acid ligands. 35
Visual inspection of the in silico docking results confirmed that the Sia1 ring of the ligand was unable to fit into the narrowed gap between the 190-helix and the 130-loop if a rigid binding pocket was assumed (Figure 6a). The effect was observed to be even greater for α2,6-linked sialic acid because of the additional steric clash between Gal2 of the sialylated oligosaccharide ligand and the 220-loop of HA (Figure 6b), in particular with the Q223 residue, which had been shown to be one of the highly conserved residues involving in anchoring Sia1. 35 From our analysis, the amino acid residue at position 222 of HA1 is postulated to affect the positioning of the Q223 residue and modulate the steric hindrance of sialylated glycan binding. Additionally, the amino acid at position 222 can either reinforce or antagonize the basic charges of ‘lysine fence’ depending on the amino acid substitution, hence providing another means of affecting the receptor binding properties of the pandemic influenza HA protein. It is no surprise that D222G/N mutations were quickly found to be genotypic markers associated with disease severity during the pandemic, and the site was also shown to be under great selection pressure by our sequence analysis. In particular, the D222G mutation in the HA protein of pandemic (H1N1) influenza 2009 virus has reported to link to patients with severe disease. 12 Although it is generally considered that the D222 residue can provide optimal contact of the Gal2 on the α2,6-linked sialic acid ligand, 35 this loss can be compensated with the substitution of a less bulky glycine residue because it can better ease the steric stress on Q223 and also provide more favorable charge attraction towards the sialylated glycan.
Results of the molecular docking of (a) α2,3-linked sialic acid ligand and (b) α2,6-linked sialic acid ligand to the receptor binding pocket of the pandemic influenza A/H1N1 (2009) hemagglutinin (HA) protein, showing steric hindrance by the narrowed opening. Solid green and yellow lines represent specific instances of steric clashes and hydrogen bonds, respectively
The findings of the MI analysis are perhaps more surprising with significant correlations found between polymorphisms of distant residues of the HA protein. The number and degree of such correlations are too great to attribute all of them to be experimental or statistical artifacts. Some residue pairs may appear distantly in the same monomer but could be placed more closely in the HA trimer structure. A more rigorous examination of the MI results could be achieved with a hierarchical cluster analysis as correlated residues often form an interaction network, but the results suggest a lack of sufficiently significant clusters to work on. Our interpretation of the situation is that the amount of evolution that had occurred in one year of sampled data was too limited to provide a good description of the interactions within HA. Repeating the analysis with a larger data-set in the future might reveal further details of the nature of the correlations.
We also noted that the direction of selection at residue 203 appears to be inconsistent under different analysis. DEPS suggested that the preferred residue is serine and not threonine while fixation analysis suggested the opposite. The source of this apparent contradiction probably stems from the timing of the occurrence of the selection, as we found that the proportion of sequences with residue S203 was in sharp decline during the first few months of the sampling period. In the absence of a reliable outgroup, this might affect the accuracy of the root placement in the phylogenetic tree supplied for DEPS analysis, which in turn affects the correction determination of directionality. 30 Alternatively, the presence of recombination might confound the analysis although we were unable to detect any recombination signal in the data. We contend that the amino acid selected at residue 203 should be threonine but the DEPS analysis had been skewed by the phylogenetic uncertainty introduced as an artifact of the sampling period cut-off. This illustrates the need for applying multiple independent methods during analysis, examination of the actual data and some form of experimental verification whenever possible. 39
The occurrence of residue 203 in our results is also interesting in another way, as it ranks as the residue under greatest directional and positive selection in our analysis. This residue is located in an accessible area on the surface of the HA1 molecule and could presumably play a role in the antigenic escape from antibody-mediated immunity. 40 The S203T mutation has been reported to be associated with cases of severe and fatal pandemic influenza infections in Greece, 41 although the authors did not clearly establish the proportion of mild cases carrying the same mutation. Most intriguingly, the mutation had been reported to co-occur with two other mutations (V106I and N248D) in the viral neuraminidase. 42 This correlation in polymorphism across different segments of the influenza virus genome deserves to be further investigated, though it is not totally surprising given the extent of long-range correlations within HA shown in the present study.
Our structural analysis is limited by the lack of information on the binding kinetics of the HA protein to the sialylated glycans. Unfortunately, in silico prediction of binding kinetics, especially in cases involving protein flexibility and conformational change, requires computationally expensive molecular dynamics study, much like assessment of protein flexibility itself. On the other hand, experimental determination of binding kinetics requires dedicated equipment to provide quantitative characterization of the interaction, and so far relatively few direct studies had been published. 43 As a narrowed opening of the receptor binding pocket on the HA protein may hinder the approach of the sialylated glycan receptor, we anticipate that the dynamic aspect of the interaction can be an important determining factor in the effective transmission and entry of the pandemic influenza A/H1N1 (2009) virus. Another important structural aspect of the HA protein is its trimer formation, which is essential for the biological function of the triggering of membrane fusion between the virus and endosome to facilitate cell entry. The HA of influenza viruses have evolved extensively to contain antigenically diverse subtypes. For group 2 HAs, which include the H3 subtype but not the H1, H2 and H5 subtypes, a tert-Butylhydroquinone (TBHQ) binding site exists at the interface between HA monomers and the binding of TBHQ at this site can inhibit the conformational change of HA leading to reduced cell entry of the virus. 44,45 The fusion peptide at the N-terminus of HA2 was found to be highly conserved with strong negative selection as expected, but other regions of HA2 contained numerous variable sites under positive selection. It may be possible that polymorphisms at these positions can promote trimer formation, enhance conformational change of the trimer and facilitate membrane fusion. Similar to TBHQ, understanding the functional importance of the other HA2 regions may enable the development of novel antiviral compounds.
In summary, we investigated the flexibility of the receptor binding pocket and the correlation of mutations in influenza HA during viral evolution. Our in silico analysis results are also complemented by the findings of the in vitro assay on cellular receptor binding avidity by HA mutants. Taken together, these results show that a simple mutation at a key site can simultaneously impact different aspects of protein function and evolution, thus potentially contributing to the human adaptation of the pandemic influenza (H1N1) virus. It should be emphasized that our present work does not represent an exhaustive analysis of the functional significance of the D222 residue and associated mutations. Other aspects of host–virus interaction, including adaptive host immunity responses to viral proteins, have not been explored in the present study, and their potential interactions with structural flexibility may yet be discovered. For example, it may be possible for the flexibility of receptor binding pocket in the HA of this virus to facilitate interactions between viral antigen and host immune cells, leading to the rapid establishment of an enhanced immune response. In our opinion, the integration of structural and evolutionary analysis is a powerful and efficient means for the identification of viral determinants important for virulence and host adaptation. Further application of these methods to pandemic influenza and other pathogens can help unravel the mystery behind the appearance of pandemics caused by the novel viruses and hopefully enable us to develop effective preventive strategies.
Footnotes
Acknowledgements
This work is partly supported by the Ted Sun Foundation, Providence Foundation Limited in memory of the late Dr Lui Hac Minh, the Hong Kong Special Administrative Region Research Grant Council, Research Fund for the Control of Infectious Diseases of the Food and Health Bureau of the Hong Kong Special Administrative Region, and The Shaw Foundation. We thank Dr Kelvin To for reading the manuscript.