
Abstract
The present review is focused on the dual role played by platelet-activating factor (PAF) in ischemia and reperfusion (I/R) injury of the heart. Although the involvement of PAF in the pathogenesis of myocardial reperfusion injury is well established, in the last few years it has emerged that very low concentrations of PAF exert cardioprotective effects, comparable to that afforded by ischemic preconditioning (IP). PAF is a potent phosphoglyceride involved in different pathophysiological conditions affecting the cardiovascular system, including the development of myocardial I/R injury. PAF is released from the I/R myocardium in concentrations (1–10 nmol/L) high enough to negatively modulate coronary circulation as well as electrical and contractile activities. PAF may act either directly, via generation of secondary mediators, or through the activation of inflammatory cells like platelets and polymorphonuclear neutrophils, which exacerbate postischemic myocardial injury. The effects of PAF are mediated through specific receptors (PAFRs) that belong to the superfamily of G protein-coupled receptors. Since cardiomyocytes not only produce PAF but also possess PAFRs, it is likely that PAF acts as an autocrine/paracrine mediator. Although the negative effects exerted by high concentrations of PAF are well established, several recent findings from our and other laboratories have demonstrated that very low concentrations (pmol/L) of PAF infused before ischemia induce cardioprotective effects similar to those afforded by IP, and that endogenous PAF production participates in the induction of IP itself. The IP-like action exerted by low concentrations of PAF is due to the activation/phosphorylation of kinases included in the reperfusion injury salvage kinase (RISK) pathway, such as protein kinase C, Akt/PkB and nitric oxide synthase. Together with the activation of mitochondrial KATP channels, these events may allow prevention of mitochondrial permeability transition pores opening at reperfusion. Moreover, the nitric oxide-dependent S-nitrosylation of L-type Ca2+ channels induced by PAF reduces intracellular Ca2+ overload.
Introduction
Platelet-activating factor (1-O-alkyl-2-acetyl-sn-glycero-3-phosphocholine, PAF) is an autacoid with profound effects on many systems, including the muscles, vascular tissue and brain. The term PAF was coined in 1972 by Benveniste et al. 1 to indicate the lipid mediator released from rabbit basophils after IgE stimulation, initially described as ‘a soluble factor’ involved in leukocyte-dependent histamine and serotonin release from platelets (PLT). 2 PAF is a phospholipid with diverse and potent physiological effects that belongs to a family of biologically active, structurally related alkyl phosphoglycerides. 3–7 While PAF is minimally expressed under normal physiological conditions, several cell types (such as neutrophils and monocytes) release significant amounts of PAF in particular conditions, such as oxidative stress or ischemia and reperfusion (I/R). PAF mediates cell-to-cell communication, acting both as an intercellular or an intracellular messenger. Some of its actions are achieved at concentrations as low as 1–10 pmol/L and play a relevant role in the development of several pathological and physiological processes. Numerous cell types, in particular those involved in inflammatory reactions, have been shown to produce PAF upon stimulation. 3–7 In addition, human endothelial cells were found to produce PAF after stimulation by several inflammatory mediators, including leukotrienes C4 and D4, histamine, bradykinin, hydrogen peroxide, interleukin (IL)-8 and IL-1α, or tumor necrosis factor (TNF)-α. 3–7 Cardiomyocytes have also been reported to synthesize PAF under appropriate stimulation. In vitro, PAF promotes the aggregation, chemotaxis, granule secretion and oxygen radical generation from leukocytes and the adherence of leukocytes to the endothelium. PAF increases the permeability of the endothelial cell monolayer, stimulates the contraction of smooth muscle 8 and exerts negative inotropic and arrhythmogenic effects on cardiac muscle. 9–14 PAF decreases the viability of a variety of cell types, including human lymphocytes, corneal epithelial cells, HaCaT cells and human colon carcinoma cells. 3–7
PAF is synthesized by a variety of tissues and cells through two different pathways. The remodeling pathway present in the inflammatory cells includes the activation of phospholipase A2 (PLA2) and acetyl CoA: 1-alkyl-sn-glycero-3-phosphorylcholine 2-O acetyltransferase, and the production of 2-lysophospholipids as metabolic intermediates (1-alkyl-2-lyso-glycero-3-phosphocholine, lyso-PAF). 15–17 The other PAF biosynthetic pathway, which is mainly operative in the kidney and in the central nervous system, has been termed the de novo pathway. 18,19 This pathway involves the synthesis of 1-O-alkyl-2-acetylglycerol, which is then converted to PAF by a specific choline phosphotransferase. A PAF acetylhydrolase (PAF-AH) present in plasma 20 and in various tissues degrades PAF by cleaving the short acyl chain at sn-2 position and originating the biologically inactive lyso-PAF. 21
PAF acts via specific receptors (PAFRs) present on membranes of different cell types, in particular smooth muscle cells, 22 cardiomyocytes 23 and endothelial cells. 24 In these latter cells, PAFRs are expressed not only on the cell surface but also in the large endosomal compartment. 25 It has been suggested that cell surface receptor activation induces immediate effects, whereas long-term responses are mediated by intracellular receptors. In microvascular endothelial cells, intracellular PAFR stimulation modulates several biological functions, such as transcriptional regulation of major genes, namely cyclooxygenase-2 and inducible nitric oxide synthase (NOS). 26 Membrane PAFRs belong to the G protein-coupled receptor (GPCR) superfamily. 27,28 As they are coupled to both Gq and Gi types of G proteins, the stimulation of PAFRs leads to activation of PLC and PLA2. This activation causes the transient production of diacylglycerol, which activates protein kinase C (PKC), and of inositol trisphosphate, which mediates the release of internal calcium stores. Moreover, PAF has been found to stimulate the release of arachidonic acid in various cell types by different mechanisms. 29–32 The activation of PLA2 by PAF occurs through a PKC-dependent mechanism and may be regulated by the intracellular levels of cAMP. 30–32 Arachidonic acid metabolites have been shown to mediate several biological activities of PAF in the cardiovascular system. 29–33 In addition, it has been shown that PAF stimulates tyrosine phosphorylation of several proteins in PLT, 34 neutrophils 35 and macrophages. 36 Because PAFRs contain several tyrosine residues in its intracellular loops and tail, it has been suggested that tyrosine phosphorylation may be involved in receptor downregulation. 36 It has been shown that PAF may activate a mitogen-activated protein kinase (MAPK) 37 and may induce the early tyrosine phosphorylation of focal adhesion kinase in human endothelial cells. 38 Moreover, in human neutrophils, PAF activates MAPK kinase-3, a known activator of p38 MAPK. 39 Further details regarding signal transduction pathways involved in cardiac effects of PAF will be presented in the following sections.
Role of PAF in I/R injury
Reperfusion injury is defined as the myocardial cellular dysfunction induced by the re-establishment of coronary perfusion, in contrast to myocardial damage brought about during the preceding ischemic episode. Reperfusion injury involves mechanical, extracellular and intracellular processes, which include the following key mechanisms: generation of reactive oxygen species (ROS), reduced availability of NO, Ca2+ overload (Figure 1) and the opening of mitochondrial permeability transition pores (mPTPs; Figure 2). 40–43

Dual role played by platelet-activating factor (PAF) in ischemia and reperfusion (I/R) injury of the heart. Released at high concentrations (≥nmol/L) after prolonged ischemia followed by reperfusion, PAF participates to the pathogenesis of myocardial reperfusion injury. On the other hand, very low concentrations (pmol/L) of PAF may exert a cardioprotective effect, comparable to that afforded by the classical ischemic preconditioning. The negative inotropic and arrhythmogenic effects exerted by PAF are due both to a direct action on cardiomyocytes and to indirect effects consequent to reduced coronary flow and release of other mediators. LVDP, left ventricular developed pressure; LVEDP, left ventricular end-diastolic pressure; mPTP, mitochondrial permeability transition pores; PLT, platelets; PMN, polymorphonuclear neutrophils
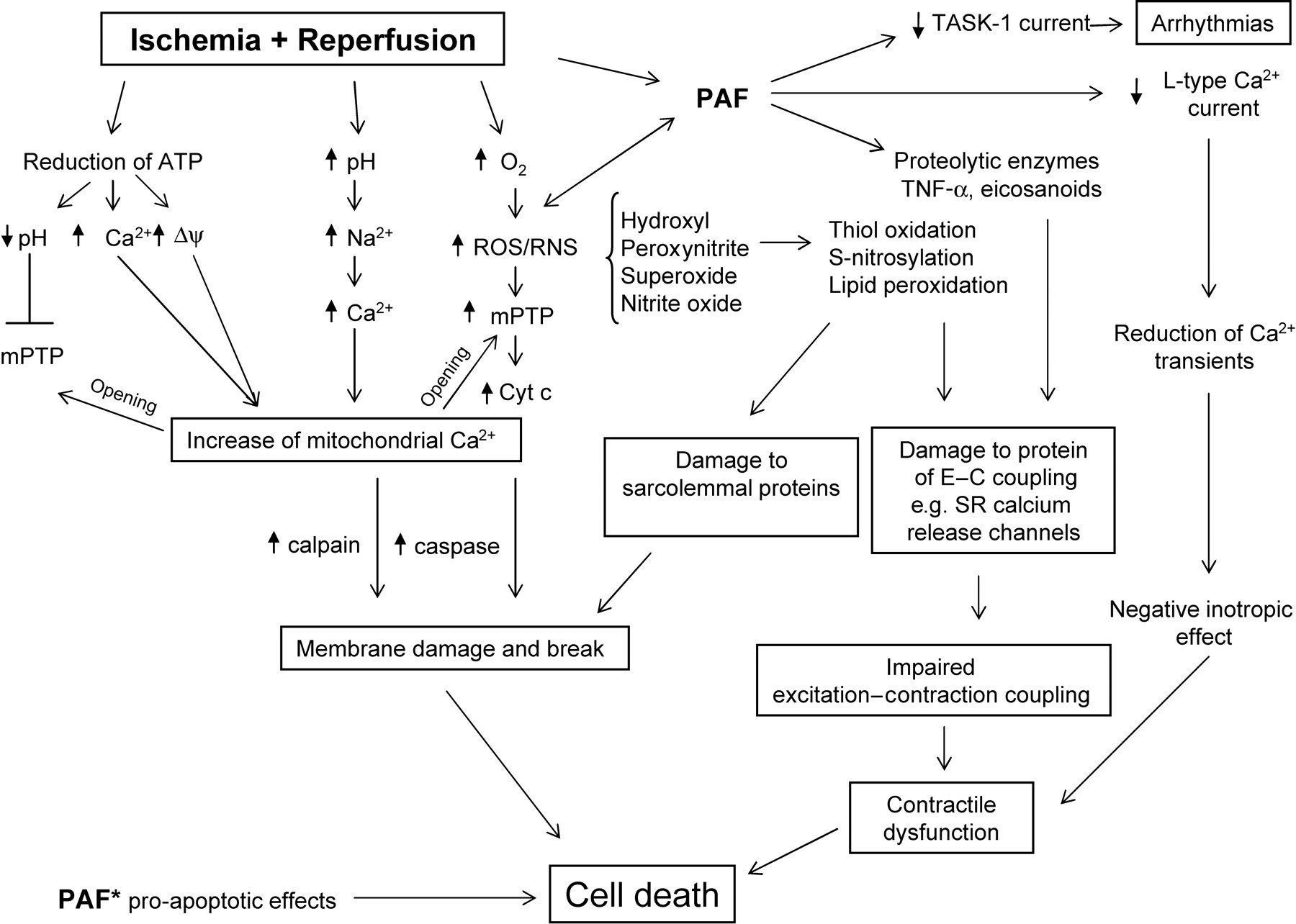
Cross-link between the intracellular pathways responsible for the negative effects of platelet-activating factor (PAF) and those involved in cell damage and contractile dysfunction in ischemia and reperfusion (I/R) heart. PAF* indicates the direct proapoptotic effect exerted by PAF. Cyt c, cytochrome c; Δψ, mitochondrial membrane potential; E–C, excitation–contraction; mPTP, mitochondrial permeability transition pore; RNS, reactive nitrogen species, ROS, reactive oxygen species; SR, sarcoplasmic reticulum; TASK-1, TWIK-related acid-sensitive K+ channels
Release of PAF from I/R heart
Several experiments investigating the production of PAF and the protective effect of PAF receptor antagonists in the I/R heart support the hypothesis that PAF plays a relevant role in these pathophysiological conditions. 4,40 Intravascular release of PAF has been detected in the blood of patients with coronary artery disease undergoing atrial pacing. 43 Increased plasma concentrations of PAF during myocardial infarction have been related in part to increased production of PAF by neutrophils, 44 or to depression of plasma PAF-AH activity, which in turn may allow a prolonged half-life of newly synthesized PAF. In contrast, other studies failed to detect increased PAF concentrations in the peripheral blood after myocardial infarction in humans. 45,46 Besides humans, elevated blood PAF concentrations were also detected in baboons following myocardial infarction 47 and in sheep after coronary occlusion and reperfusion. 48 The presence of increased levels of the PAF metabolite, lyso-PAF, in canine myocardium subjected to permanent ligation of a coronary branch gave indirect evidence of elevated PAF biosynthesis during myocardial ischemia. 49 In vivo findings were supported by in vitro experiments on isolated perfused hearts, demonstrating the release of PAF during reperfusion. In rabbits, PAF was detected in the coronary effluent during the initial reperfusion of ischemic heart. 50,51 Although the precise cellular source of PAF was not identified in this model, likely candidates are endothelial cells and cardiomyocytes. Indeed, it has been reported that cultured endothelial cells, 52 as well as neonatal rat myocytes, synthesize PAF after prolonged hypoxia. 53 In the isolated rabbit heart, in which the effects of PAF are platelet-dependent, the amounts of released PAF (in the nmol/L range) were enough to activate platelets to cause a significant worsening of left ventricular function during reperfusion. 50 A further worsening occurred when both polymorphonuclear neutrophils (PMNs) and PLT were present, as a consequence of PAF-dependent PMN–PLT cooperation. 54 The effects of PAFR antagonists were studied in different in vitro and in vivo experimental models. PAFR antagonists prevented both the PLT-dependent and -independent mechanical and electrical alterations that occurred, respectively, in rabbit, 50,55,56 guinea pig 57 and rat isolated heart 58 after I/R. In the experimental myocardial infarction due to coronary occlusion, PAFR antagonists reduced the hemodynamic alterations as well as the size of the necrotic area and the accumulation of PLT and leukocytes observed in rabbit 59,60 and sheep 61 hearts. In rats, PAFR antagonists reduced infarct size and the occurrence of arrhythmias. 62–64 Although some conflicting results were obtained with different PAFR antagonists, a comparable protective effect was also observed in the dog. 65,66 To date, no clinical trials have investigated the protective effect of PAFR antagonists in cardiac alterations associated with I/R in humans.
In summary, the relevant role of PAF in cardiac dysfunctions caused by I/R is supported by several in vitro and in vivo findings, showing the release of PAF during reperfusion and the protective effect exerted by PAF receptor antagonists. The concentrations of PAF released (1–10 nmol/L) are high enough to activate PLT and PMN, causing a significant worsening of cardiac function during reperfusion.
Effects of PAF in coronary circulation
PAF infusion into the coronary circulation induced variations in the coronary tone, depending on the doses and the animal species used. Low doses of PAF (0.03–0.3 nmol/L) induced both an increment and decrement of coronary blood flow (CBF) in the pig, in the absence of significant changes in systemic blood pressure. Higher doses (1–10 nmol/L) of PAF caused a reduction in CBF accompanied by a negative inotropic effect and electrocardiogram signs of ischemia (S-T segment depression and arrhythmias). The early increase in CBF is independent from the generation of cyclo- and lipoxygenase-derived metabolites, while the subsequent vasoconstriction is primarily due to the production of thromboxane A2. 67 PAF also induced coronary vasoconstriction and S-T segment depression in rabbits. 68 Conflicting effects were observed in dogs, in which PAF has been reported to reduce CBF and systemic arterial pressure, 69 to produce a PLT-dependent coronary vasodilation 70 or a biphasic vasodilator/vasoconstrictor effect. 71 The effects of PAF strongly depend on the integrity of endothelial cells; indeed, PAF induces a vasodilating effect when the endothelium is intact, whereas vasoconstriction is prominent in the presence of injured endothelium, as it may occur after ischemia. 72
The coronary vasoconstrictor action of high doses of PAF observed in vivo was confirmed by ex vivo experiments performed on isolated perfused hearts. PAF induced a dose-dependent increase in coronary vascular resistances in the isolated guinea pig heart.
9,11,73–75
The vasoconstrictor effect of PAF was completely blocked by PAFR antagonists, thus suggesting the involvement of specific PAF receptors.
75
Comparable coronary vasoconstriction was obtained with high doses of PAF in isolated perfused rat heart.
76–78
However, low doses induced vasodilation alone or vasodilation followed by vasoconstriction, which involves production of both prostaglandins and leukotrienes.
76–78
In the isolated rabbit heart, which is insensitive to this mediator when perfused with physiological saline alone,
50,79
PAF markedly reduced coronary flow when blood cells were added to the perfusion fluid.
79
Thus, the isolated rabbit heart was used as a model to study the in vitro interaction between PLT and PMN in the coronary circulation. In the rabbit heart perfused with PLT alone, the infusion of PAF induced a dose-dependent decrease of coronary flow, while PMN stimulated by PAF had no effect. However, as a result of cooperation of PMN with PLT, a marked reduction of coronary flow occurred when both these cells were stimulated with PAF.
54
A comparable increase in coronary resistances was observed in rabbit hearts perfused with PMN activated by N-formyl-
Taken together, these findings indicate that PAF exerts dose-dependent alterations of coronary tone. At concentrations comparable to those detected in the coronary effluent after I/R (1–10 nmol/L), PAF caused a reduction in coronary flow accompanied by a negative inotropic effect. The vasoconstrictor effect of PAF is due to specific PAFR activation and, depending on the animal species, it may be due to a direct action on smooth muscle cells or, at least in part, to generation of cyclo- and lipoxygenase-derived metabolites, or activation of PLT and PMN.
Electrophysiological and inotropic effects of PAF on cardiac cells
The alterations in cardiac function observed in vivo after infusion of PAF can result either from a direct action on the heart or from indirect effects, such as systemic changes and variations in pre- and afterload pressures. Moreover, alterations in cardiac performance may depend on the effect of PAF on the coronary circulation, on the conduction system and on the contractile properties of myocardium. 84 Experiments performed on the isolated heart perfused at constant flow or isolated atrial and ventricular preparations stimulated at constant frequency indicated that PAF exerts a direct effect on cardiac electrical and contractile activities. The effects of PAF on the isolated heart perfused at constant pressure are quite different, depending on the animal species studied. In guinea pig isolated heart, infusion of PAF induced conduction arrhythmias and reduced both the action potential duration and the force of contraction. 85,86 The negative inotropic effect of PAF was further confirmed by in vitro experiments on isolated guinea pig atrium and papillary muscle, 10,87,88 showing a direct negative inotropic effect of PAF, paralleled by a significant decrease of action potential duration. 10,89 In contrast, the negative effects induced by PAF in the isolated rat heart were mainly dependent on the reduction of coronary flow consequent to production and release of endogenous leukotrienes. 29 PAF induced a biphasic dose-dependent effect on human cardiac muscles, characterized by a transient positive effect followed by a marked prolonged negative inotropic effect and reduction of action potential duration. 12,13 Experiments performed on isolated cardiac cells confirmed the ability of PAF to directly modulate the electrical and contractile activities of cardiac muscle. It has been shown that the decrease in twitch tension induced by PAF in isolated adult cardiomyocytes is accompanied by a reduction of intracellular systolic calcium concentration 90 and of L-type calcium current. 91 On the other hand, the repolarization abnormalities and conduction arrhythmias induced by PAF in cardiac cells may be explained with the observed alterations of inwardly rectifying background potassium channels (IK1) 92 and inhibition of TWIK-related acid-sensitive K+ channels (TASK-1). 93 In addition, PAF stimulates cardiac muscarinic potassium channels, via a PLA2-lipoxygenase dependent mechanism. 32 These results are consistent with the fact that the presence of PAFRs on cardiomyocytes, previously suggested by the protective effect of PAFR antagonists, was directly demonstrated by cloning and characterization of PAF receptor gene in human cardiomyocytes. 23
The intracellular pathways involved in the myocardial response to PAF were investigated in a series of experiments performed in our laboratory. The role of PI3Kγ-mediated signal transduction in the regulation of myocardial performance in response to PAF was analyzed using p110γ knockout mice. 94 We demonstrated that the negative inotropic effect of PAF in the heart depends on PI3Kγ in response to a Gi-mediated signal transduction pathway. The blocking effect induced by a NOS inhibitor suggests that PKB/Akt-mediated phosphorylation and thus activation of eNOS are the critical events. On the same experimental model, we further studied the relevance of PAF/PI3Kγ signaling for the outcome of I/R in isolated hearts, demonstrating that loss of PI3Kγ resulted in a significantly better postischemic recovery of contractile force during reperfusion than wild-type controls. Postischemic function in wild-type hearts could be improved by both a PAFR antagonist and a NOS inhibitor, indicating the PAF/PI3Kγ/NOS pathway as an important cause of myocardial dysfunction. 94
In addition to exerting direct effects on cardiac cells, it has been shown that PAF stimulates the release of other biologically active mediators such as eicosanoids (leukotrienes and thromboxane), superoxide anions and TNF-α. These mediators can furthermore enhance myocardial dysfunction. 40 In particular, TNF-α, which like PAF acts as cardiodepressant agent released by I/R heart, may cooperate with PAF. Indeed, TNF-α induces release of PAF from several cell types, including cardiac and skeletal muscle cells, and PAF receptor antagonists have been shown to reduce the negative effects of TNF-α. 95,96 Apoptosis has been implicated in myocardial infarction-related cell death. Besides the above-mentioned detrimental actions of PAF, it has been recently shown that high concentrations of PAF (0.2–20 μmol/L) directly induce apoptosis in H9c2 cardiomyocytes via a calcium-dependent p38 MAPK-activated cytochrome c/caspase-3 apoptosis signaling pathway (Figure 2). 97
In summary, it has been shown that, besides altering cardiac performance by reducing CBF, PAF may also exert direct effects on the electrical and contractile activities. The shortening of the action potential duration and the negative inotropic effect induced by PAF have been related to reduction of both L-type calcium current and intracellular systolic calcium concentration. The repolarization abnormalities and conduction arrhythmias induced by PAF may be explained with the observed alterations of IK1 current and inhibition of TASK-1 channels. By using p110γ knockout mice, it has also been shown that the PI3Kγ/NOS pathway plays an important role in myocardial dysfunction induced by PAF.
What causes PAF synthesis and release during I/R?
Selected experiments were performed to investigate the mechanisms responsible for PAF biosynthesis in I/R heart. The occurrence of oxidative stress related to the generation of ROS at the beginning of reperfusion plays an important role in reperfusion-induced cell death. 40,98 Since the generation of oxygen radicals and their release into the coronary effluent occurs in parallel with the release of PAF during the early phases of reperfusion, 50 we hypothesized that oxidative stress may trigger the synthesis of PAF. To test this hypothesis, we treated guinea-pig isolated perfused heart with dihydroxyfumaric acid (DHF), a free radical-generating compound. DHF stimulated PAF production and caused mechanical and electrical alterations that were prevented by superoxide dismutase or by the PAFR antagonist WEB 2170. 99 These results support the hypothesis that the burst of ROS released during early reperfusion may lead to PAF biosynthesis and that, on the other hand, PAF may, at least in part, act as secondary mediator of oxygen radicals in the heart. In line with our observations on multicellular cardiac preparations, it has been already shown that H2O2 stimulates the synthesis of PAF by primary cultures of bovine pulmonary artery and by human umbilical vein endothelial cells (HUVECs). In parallel with PAF synthesis, H2O2 also induced the endothelial-cell-dependent adhesion of neutrophils to HUVEC monolayers. 100 Besides the above-mentioned studies, in which PAF production was induced by acute in vitro treatment with chemicals generating a marked oxidative stress, other observations suggested that the interaction between ROS and PAF may occur also in vivo in certain pathophysiological situations. Inside the cells, the toxic actions of ROS are limited by antioxidant enzyme systems that protect them against peroxidation and control concentrations of intracellular peroxides. One of these, the enzyme phospholipid hydroperoxide glutathione peroxidase, acts as a negative modulator of PAF biosynthesis through inhibition of p38 phosphorylation. Since selenium is required for optimal activity of glutathione peroxidases, it is reasonable that selenium deficiency would result in the accumulation of lipid peroxides and subsequent induction of PAF synthesis. In fact, it has been shown that selenium deficiency causes increased production of PAF in human endothelial cells with enhanced activity of PLA2 and acetyltransferase. 4 Moreover, it has been shown that PAF mediates the adhesion of monocytes to endothelium induced by LDL and oxidized LDL. 101 Cigarette smoking, a factor associated with the pathogenesis of atherosclerosis, causes platelet activation, LDL oxidative changes and increases PAF concentrations. 102 The latter alteration was associated with a compensatory increase of PAF-AH activity. However, in vitro studies demonstrated that cigarette-derived products as well as oxidative changes of LDL, which physiologically carry PAF-AH, inhibit the activity of the enzyme that catabolizes PAF. 103 Furthermore, PAF may also oxidize LDL, via stimulation of human monocytes/macrophages and neutrophils to produce superoxide anions and hydrogen peroxide. 104
In summary, experiments performed to investigate the mechanisms responsible for PAF biosynthesis in I/R heart indicated that the synthesis of PAF is probably triggered by the generation of ROS and the resulting oxidative stress occurring at the beginning of reperfusion.
In conclusion, several data comprise evidence for PAF as one of the major GPCR agonists depressing myocardial contractility and thus causing further distress to the ischemic organ in ischemic heart disease. 4,40 Several in vivo and in vitro studies demonstrated that PAF is released from I/R heart, and that activation of PAFRs on smooth muscle cells of coronary vessels or on cardiac cells, results in a relay of signals causing a reduction of coronary flow or direct negative inotropic and arrhythmogenic effects. PAF acts as a potent chemoattractant for PLTs and PMNs and promotes their activation and cooperation, thus exacerbating postischemic myocardial injury by capillary plugging and via damage induced by the formation of ROS or proteolytic enzymes. Furthermore, PAF exerts a direct proapoptotic effect on myocardial cells, and interacts with other inflammatory mediators, like TNF-α and arachidonate derivatives, exerting a detrimental action in the I/R heart (Figure 2).
PAF induces ischemic preconditioning
Ischemic preconditioning
In 1986, Murry et al. 105 introduced the term ischemic preconditioning (IP) to describe the reduction of infarct size obtained in canine hearts after applying four 5-minute alternate episodes of circumflex artery occlusion followed by four days of myocardial reperfusion. The efficacy of IP in limiting the severity of I/R injury was confirmed by repeating the IP protocol in several experimental models. IP not only reduces the extent of the infarct size but also the number and severity of conduction arrhythmias and contractile dysfunction. Three aspects can be distinguished in the process of IP: the initial trigger included in the short periods of I/R activates signaling pathways, which in turn act upon an end-effector, which then induces the delay of lethal ischemic damage during sustained ischemia. 106–108 At first, preconditioning protection lasts a few hours (2–3 h) immediately after the preconditioning procedure. This early classical preconditioning, or first window of protection, is followed by a period without protection lasting about 12–24 h. Then, the protection reappears and lasts 24–72 h in the so-called second window of protection (late preconditioning). The mechanistic components underlying the protection afforded by IP have been conventionally classified as triggers (factors that act before the index ischemic episode and activate downstream signaling mechanisms) and mediators/effectors (factors that act during the index ischemic episode and mediate the protective effect). This separation is not rigid, because certain signaling components have been demonstrated to act both as triggers and as mediators/effectors. The stimulation of cell-surface receptors, specifically GPCR, by endogenous ligands, such as adenosine, opioids and bradykinine, generated by the brief ischemic episodes during the preconditioning phase, provide the initial trigger for the signal transduction pathway of protection. 109 Besides these classical agents, other mediators such as TNF-α, 110 chromogranin A-derived peptides, 111 growth hormone-releasing hormone (GHRH) 112 and GHRH-related peptides 113 have been shown to induce cardioprotection in a dose- and time-dependent manner.
The various surface receptor agonist-mediated signal transduction pathways involved in cell protection include at least the following kinases (singly, or in combination, depending on circumstances): PI3K/Akt, PKCε, PKG, p70S6K, extracellular signal-regulated kinase (ERK) 1/2, MAPK and finally phosphorylation of the residual Ser9, that induces inactivation of the ‘mitochondria-associated’ GSK-3β pool. These kinases, which are activated at the time of myocardial reperfusion, are termed reperfusion injury salvage kinases (RISK). Another important element involved in the IP pathway is mitochondrial ATP-sensitive K+ channels (mitoKATP). Pharmacological activation of mitoKATP was shown to induce a protective state sensitive to inhibition by mitoKATP blockers. In addition, the activation of the RISK pathway inhibits the formation of mPTPs during the reperfusion phase following the infarcting ischemia. In fact, the opening of mPTPs completely disrupts mitochondrial function and invariably leads to cell death by either necrosis or by apoptosis. It is likely that a large number of cells are killed by this mechanism during reperfusion (Figure 3). 109,114
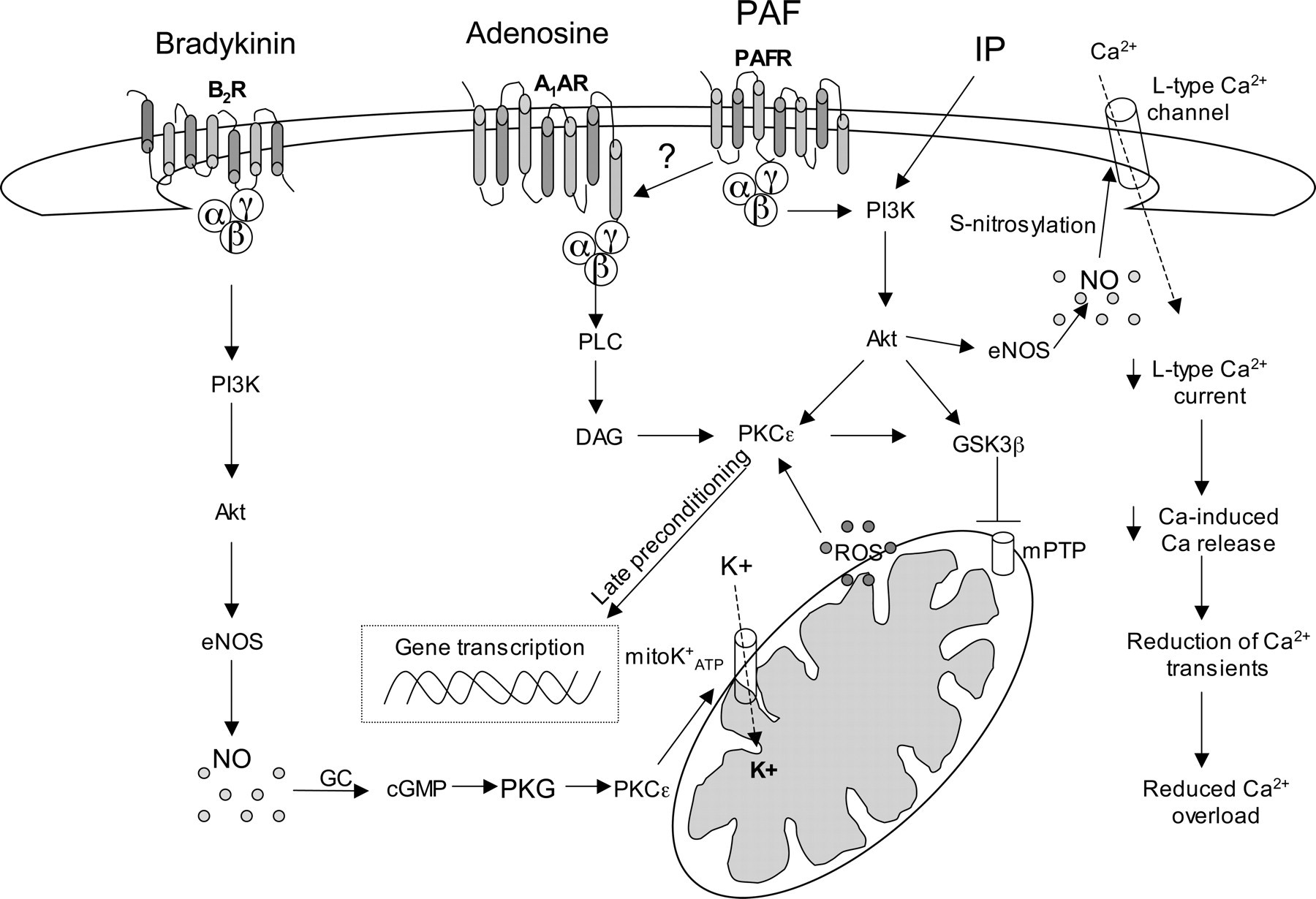
Intracellular signaling pathways involved in the protective effect of classical ischemic preconditioning (IP), bradykinin, adenosine and platelet-activating factor (PAF). PAF-induced protection partially involves adenosine A1 receptors. A1AR, A1 adenosine receptor; Akt, serine/threonine protein kinase; αβγ, G protein subunits; B2R, B2 bradykinine receptor; DAG, diacylglycerol; eNOS, endothelial nitric oxide synthase; GC, guanylyl cyclase; GSK3β, glycogen synthase kinase β; mPTP, mitochondrial permeability transition pores; mitoKATP, mitochondrial ATP-dependent K+channels; NO, nitric oxide; PAFR, PAF receptor; PI3K, phosphatidylinositol 3-kinase; PKC, protein kinase C; PKG, protein kinase G; PLC, phospholipase C; ROS, reactive oxygen species
Low concentrations of PAF induce cardioprotection
Until five years ago, studies performed on the cardiac effects of PAF were mainly devoted to investigating the effects exerted by high concentrations of this mediator, comparable to those released in severe pathophysiological conditions, such as after a long-lasting period of ischemia,
4,40
or acute anaphylactic shock.
11,115
As described in the above section, cardiac tissue undergoing prolonged I/R releases amounts of PAF high enough to activate inflammatory blood cells and to induce severe myocardial dysfunction, including profound electrophysiological alterations and a negative inotropic effect.
4,40
We cannot exclude, however, that brief periods of I/R induce the release of very small quantities of PAF, unable to alter myocardial contractility per se, but enough to activate kinases involved in IP, such as PKC and PI3K. In fact, PAF is thought to be a mediator of cell-to-cell communication, and some of its above-mentioned actions are reported to be achieved at concentrations as low as 1 pmol/L. However, in many species, such a concentration does not appreciably affect heart function.
3–7
We thus hypothesized that the administration of a very low dose of PAF (in the pmol/L range) could be able to induce protective effects akin to IP and that the activation of PAF receptors might play a role in triggering IP. Several findings indeed suggest that, depending on its concentration, PAF may exert biphasic or, in some cases, opposite effects. For instance, while low doses of PAF (pmol/L range) are able to induce both increment and decrement of CBF, it causes a marked vasoconstriction when infused at higher doses (up to 10 nmol/L).
71–79
In the central nervous system, although PAF is required for neuronal survival,
116
it can induce apoptosis when administered at elevated concentrations (μmol/L range).
117
The biphasic or even opposite, both beneficial or detrimental, effects exerted by PAF according to its concentration, somehow resemble the behavior of NO, the main end-effector of PAF action on cardiac muscle.
118
To verify whether PAF is a possible endogenous agent involved in IP, we performed experiments on isolated rat hearts undergoing 30 min of ischemia followed by two hours of reperfusion. A short treatment of the heart before ischemia with a very low concentration of PAF (20 pmol/L), while unable per se to alter cardiac performance, did reduce the extension of infarct size and improved the recovery of left ventricular developed pressure during reperfusion. The cardioprotective effect of PAF was comparable to that observed in hearts in which IP was induced by the classical protocol (brief periods of ischemia separated by reperfusion intervals).
119
The fact that the PAF receptor antagonist WEB-2170 abrogated the cardioprotective effect induced by both PAF and IP not only suggested that the action of PAF involves a specific receptor-mediated mechanism but also that endogenous PAF is involved in triggering IP. The protective effect of PAF against postischemic injury has been recently confirmed by Leary et al.
120
by studying postischemic functional recovery in isolated hearts from wild-type and PAF receptor-knockout mice. Postischemic performance was reduced in hearts with targeted deletion of the PAF receptor and in wild-type hearts treated with a PAF receptor antagonist. Moreover, perfusion with pmol/L concentrations of PAF improved postischemic function in hearts from wild-type mice. To investigate the pathways involved in the preconditioning effect of PAF, the role of several kinases was studied by using pharmacological agents and by Western blot analysis in the isolated rat heart.
119,121
In a first series of experiments, we observed that the cardioprotective effect of PAF was reduced by both PKC and PI3K inhibitors. In agreement with these findings, Western blot analysis revealed that PAF infusion enhanced the phosphorylation of PKCε and Akt (the downstream target of PI3K) to levels higher than those measured in control hearts, and comparable to those observed after IP treatment.
119
However, since the actual protection of IP occurs in the reperfusion phase, an additional series of experiments was performed to study whether the activation of kinases involved in PAF-induced preconditioning persisted also in this latter phase.
121
To test this hypothesis, we studied whether PKC and PI3K inhibitors are able to block the protective effect of PAF, even when the treatment occurs during the initial reperfusion, as well as phosphorylation of PKCε, PKB/Akt, GSK-3β and ERK1/2 at the beginning of reperfusion. The reduction of infarct size and contractile dysfunctions induced by PAF treatment was abolished by postischemic infusion of chelerythrine or LY294002. A comparable effect was observed in the heart pretreated with PAF and infused with the mPTP opener atractyloside.
122
During reperfusion, phosphorylation/activation of PKCε, PKB/Akt and the phosphorylation/inactivation of GSK-3β were enhanced in PAF-treated hearts. We thus concluded that the cardioprotective effect exerted by PAF pretreatment involves activation of PKC and PI3K also in postischemic phases and might be mediated by the prevention of mPTP opening in reperfusion via GSK-3β inactivation. The role of mitoKATP channels and ROS in PAF-induced cardioprotection was studied by using N-acetyl-
In summary, recent findings demonstrated that brief periods of I/R induce the release of very small quantities (pmol/L range) of PAF, which are unable to alter myocardial contractility per se, but enough to activate kinases included in the RISK pathway, such as PKCε, PKB/Akt, GSK-3β and ERK1/2. Together with the activation of mitochondrial KATP channels, these events may allow prevention of mPTPs opening at reperfusion, leading to a reduction of infarct size and postischemic contractile dysfunctions, comparable to that afforded by the IP. In parallel to activation of RISK pathways, PAF-dependent NO production and reduction of intracellular Ca2+ overload due to NO-dependent S-nitrosylation of L-type Ca2+ channels may participate towards the protective effect of PAF.
Conclusions: physiological significance of PAF in cardioprotection
In conclusion, like NO and ROS, PAF may too act as a ‘double edged sword’ agent on the I/R heart, being able to exert a cardioprotective activity when released at very low concentrations, as occurs after brief ischemic periods. Indeed, low concentrations of exogenous PAF are able to trigger preconditioning-like effects without evident alteration of cardiac function. The signaling events downstream of PAFR stimulation involve the activation of several kinases included in the RISK pathway, thus preventing mPTP opening at reperfusion. Moreover, endogenous synthesis and release of low quantities of PAF, induced by brief I/R periods, might play a crucial role in the triggering of IP. Evidence for preconditioning in humans derives from in vitro studies 125 and observations made during coronary angioplasty and cardiac surgery. 126–128 It is known that exercise can mimic the protective effect of IP. 129,130 Indeed, it has been recently demonstrated that exercise delays the appearance of ST segment depression during a subsequent effort in the early and late periods of protection after exercise-induced ischemia in patients with stable angina. 131 Since a very low increase in PAF concentrations occurs during exercise in normoalbuminuric diabetic patients, 132 it may be argued that low levels of PAF participate in exercise-induced cardioprotection. The fact that low levels of PAF may be released in certain conditions, such as in non-infarcting ischemia 119 and atrial pacing, 43 or during exercise, 132 and the fact that these low levels of PAF participate in IP, may be taken into account when strategies of myocardial protection are considered. In particular, these data emphasize the potential importance of a moderate release of PAF, such as that occurring during exercise, as an attempt by the cardiovascular system to protect itself against I/R damages. On the other hand, since inhibition of the PAF protective pathway reduces myocardial postischemic function, the fact that clinical therapies for inflammatory diseases that lead to complete blockade of PAFR may eliminate a significant, endogenous cardioprotective pathway must be taken into account.
Footnotes
Acknowledgements
The authors were supported by funding from Regione Piemonte (CP and GA), the National Institute for Cardiovascular Research (INRC; CP and GA), and from the Italian Ministry of the University and Research (MIUR) and Progetti di Rilevanza Nazionale (PRIN, 2008) (CP).