
Abstract
Numerous infectious agents may trigger autoimmunity or even result in autoimmune diseases. Several mechanisms have been proposed for pathogen-triggered autoimmunity including molecular mimicry, cryptic antigens, epitope spreading, bystander activation and polyclonal activation. In the case of dengue virus infection which causes serious public health problems, the mechanisms regarding the pathogenesis of dengue hemorrhagic syndrome are not fully resolved. Our previous studies suggest a mechanism of molecular mimicry in which antibodies directed against dengue virus non-structural protein 1 (NS1) cross-react with human platelets and endothelial cells and cause their damage and dysfunction, which may be related to the clinical features of dengue disease. Several cell surface proteins recognized by patient serum samples and anti-NS1 antibodies have been identified. Based on proteomic studies and sequence analysis, the C-terminal region of dengue virus NS1 shows sequence homology with target proteins. In addition, different regions of dengue virus proteins including core, prM, E and NS1 proteins show sequence homology with different coagulatory molecules. As an example, the amino acid sequence 101–106 of E protein (WGNGCG) shows sequence homology with factors XI, X, IX, VII, II (thrombin), plasminogen and tissue plasminogen activator. Furthermore, single chain variable region against NS1 can interfere with fibrin formation, which leads to prolonged thrombin time. We hypothesize that molecular mimicry between dengue virus proteins and coagulatory molecules may induce cross-reactive autoantibodies that can interfere with coagulation activation. A molecular mimicry pathogenesis for dengue disease which involves cross-reactivity of dengue virus with human endothelial cells, platelets and coagulatory molecules is proposed.
Infection-induced autoimmunity
Autoimmunity can be triggered by multiple factors, including those of genetic, hormonal and environmental origins. Infections, such as viruses, bacteria, parasites or fungi, are major potential environmental factors of triggering autoimmunity. There are several mechanisms by which pathogens can induce autoimmunity, such as molecular mimicry, cryptic antigens, epitope spreading, bystander activation and polyclonal activation. 1,2
In molecular mimicry, the cross-reactivity between foreign epitopes (including protein, carbohydrate, lipid or DNA) and self-epitopes induces a breakdown of tolerance and leads to autoimmunity. As examples, molecular mimicry plays a role in initiating or promoting the progress of systemic lupus erythematous (SLE) and systemic sclerosis. 3,4
Autoimmunity and molecular mimicry in viral infections
Some examples of virus-associated autoimmune diseases are listed in Table 1. The mechanism of molecular mimicry is described below. Other mechanisms involved in virus-associated autoimmune diseases such as cryptic antigens, polyclonal activation, altered immune responses, bystander activation and epitope spreading are summarized in Table 1.
Examples of viruses that have been implicated in human autoimmune diseases
There are several pieces of evidence for autoimmune mechanisms involved in coxsackievirus (CV)-induced diseases, including molecular mimicry between glutamic acid decarboxylase 65 (GAD65) and p2C protein of CV-B4 in type I diabetes mellitus, 5 and between Ro60 kD autoantigen and CV 2B protein in Sjogren's syndrome. 6 Similarly, T-cells of type I diabetes mellitus patients recognize both rubella virus envelope protein and GAD65 or GAD67 protein determinants. 7
Epstein-Barr virus (EBV) may be a common factor for SLE, rheumatoid arthritis, multiple sclerosis and Sjogren's syndrome. There are both high titers of anti-EBV antibodies and high proportions of EBV-infected cells in patients with these autoimmune diseases. EBV DNA or RNA is found in the lymphocytes and diseased tissues. 4,8 Molecular mimicry has been demonstrated between EBV proteins and self-antigens in these diseases. Patients with multiple sclerosis have predominant EBV nuclear antigen 1 (EBNA1)-specific T-cells which recognize myelin antigens. 9 In addition, antibodies to 60 kDa Ro from SLE patients cross-react with a peptide from EBNA1. 10
An association between human cytomegalovirus (HCMV) and systemic sclerosis derives from the prevalence of anti-HCMV antibodies in patients. 4,11 Autoantibodies against topoisomerase I (Scl-70) from systemic sclerosis patients recognize the UL70 protein of HCMV, 12 although the question as to whether molecular mimicry between viral and Scl-70 antigens accounts for the onset of systemic sclerosis or whether instead there is a causal relationship remains controversial. 11
Herpes stromal keratitis is caused by corneal infection by herpes simplex virus (HSV) and can lead to blindness. Cornea-specific T-cells cross-react with a peptide on HSV-1 protein UL6. A mechanism of molecular mimicry is proposed for the development of this autoimmune disease after viral infection. 13 In addition, parvovirus B19 infection is associated with the production of antibodies against a variety of autoantigens including nuclear antigens, rheumatoid factor, neutrophil cytoplasmic antigens and phospholipids. 14
Autoimmunity in dengue virus infection
Dengue disease occurs in tropical and subtropical areas of the world. More than 2.5 billion humans are now at risk of dengue virus infection and more than 50 million dengue virus infections occur globally each year. 15,16 Dengue virus infection causes diseases ranging from mild self-limited dengue fever (DF) to severe or even fatal dengue hemorrhagic fever (DHF) and dengue shock syndrome (DSS). The pathogenesis of DHF/DSS is a complex, multifactorial process involving the interplay of viral and host factors. The characteristics of severe dengue disease include thrombocytopenia and plasma leakage. 17,18 A number of mechanisms involved in the disease pathogenesis have been suggested, including virus virulence, antibody-dependent enhancement (ADE) of infection and abnormal host immune responses, such as the production of cytokines and chemokines, the activation of complement and immune cells, and apoptotic cell death. 19–23 A secondary dengue virus infection may result in more severe disease manifestations due to ADE, i.e. non-neutralizing antibodies from the previous infection with different dengue virus serotype enhance subsequent infection. 24–27 Furthermore, there is a link between disease risk and HLA type, race, or receptor and cytokine polymorphisms which has the potential not only to provide important information regarding the pathogenesis of secondary dengue infection but also to identify individuals at increased risk for severe disease. 21 Such host factors may also help explain why some DHF/DSS cases are primary infections in non-endemic areas like Taiwan. 28 It is also not clear why both endothelial cells and platelets are the major targets to be affected in dengue disease.
Substantial evidence supports a role for autoimmunity in dengue disease pathogenesis, particularly involving the generation of autoantibodies against platelets, endothelial cells and coagulatory molecules. 19,22,29 Molecular mimicry between platelets, endothelial cells or coagulatory molecules with dengue virus non-structural protein 1 (NS1), prM and envelope (E) proteins may explain the cross-reactivity of anti-NS1, anti-prM or anti-E antibodies to host proteins and play a role in disease pathogenesis. Cross-reactive antibodies may cause platelet dysfunction, endothelial cell damage, coagulation defects and macrophage activation, which may contribute to some of the clinical features of DHF.
In secondary infection, cross-reactive memory T-cells from previous infection with a different serotype produce cytokines such as interferon gamma (IFN-γ), interleukin 2 (IL-2) and tumor necrosis factor alpha (TNF-α). The complement cascade is also activated by virus–antibody complexes to release C3a and C5a which may cause direct effects on vascular permeability. Synergistic effects of IFN-γ, TNF-α and complement proteins may trigger plasma leakage in secondary dengue virus infection. 20
Important differences are noteworthy between the autoimmune responses induced by dengue virus and the viruses listed in Table 1, including: (1) except for CV, those viruses which are associated with autoimmune diseases cause chronic infections; and (2) for the acute CV infection or chronic infection, the onset of autoimmune disease involves post-infectious sequelae, while dengue virus induces autoimmune-associated pathogenic effects in the acute phase. This suggests that factors in addition to molecular mimicry are necessary to cause the observed clinical symptoms. A recent study showed that two years after infection, an association between persistent symptoms and autoimmune-based disturbance exists. 30 This study showed that having more than one dengue infection could be a trigger for potential autoimmune phenomena. It is hypothesized that after dengue virus infection, cross-reacting antibodies to self-nuclear proteins are generated and a failure to properly clear the immune complexes may further cause an autoimmune post-dengue syndrome. 30
Molecular mimicry between virus and endothelial cells and platelets
Hemorrhagic syndromes of DHF/DSS include thrombocytopenia, coagulopathy and vasculopathy, which are related to dysfunction of endothelial cells and platelets. 20,31 Dengue virus itself does not cause morphological damage to endothelium and vascular leakage is mediated indirectly by host factors induced by virus. 32 ADE-mediated infection of dengue virus in peripheral blood monocytes may modulate endothelial cell function via cytokines such as TNF-α. 33 Previous studies showed that dengue virus can infect endothelial cells, leading to chemokine production, complement activation and apoptosis. 34 In addition to dengue virus effects, it has been shown that mouse anti-dengue virus NS1 antibodies can bind to human endothelial cells. 35 Furthermore, autoantibodies present in dengue patient serum samples can cause endothelial cell damage. The endothelial cell binding activity and apoptosis induced by serum samples from DHF/DSS patients are higher than those induced by serum samples from DF patients. Antibodies against NS1 account, at least in part, for the cross-reactivity and apoptosis induction. 36–39 Furthermore, antibody-binding to liver endothelial cells and a hepatitis-like pathologic effect were also observed in mice after active immunization with NS1 or passive administration with anti-NS1 antibodies. 40
Anti-dengue antibody cross-reactivity to platelets has also been observed. 41,42 Serum samples from DHF/DSS patients show higher binding activity to platelets than do serum samples from DF patients. Furthermore, dengue patient serum samples cause complement-mediated platelet lysis and inhibit ADP-induced platelet aggregation. The findings of transient thrombocytopenia associated with the generation of antiplatelet antibodies in dengue virus-infected mice 43 and the administration of anti-NS1 antibodies to mice 44 further support the hypothesis that cross-reactive antiplatelet antibodies may play a role in dengue pathogenesis.
As previously proposed by Rose and Bona, 45 there are three types of evidence for autoimmunity in disease which include direct proof, indirect evidence and circumstantial evidence. The direct transfer of antibody that causes a disease is an example of direct proof. Maternally transferred dengue antibody is a known risk factor for infants who develop DHF on their first infection. This epiphenomenon has been used to justify the theory of ADE, but also may support the observed cross-reactive antibody binding to endothelial cells and platelets 37,41,42 as a cause of DHF. Although IgM in DHF patient serum samples plays a more dominant role than IgG in the cross-reactivity with platelets and endothelial cells, 37,41 antiplatelet IgG is present in DHF patient serum samples (unpublished observations). It will be interesting to determine whether maternally transferred IgG antibody to infants is involved in DHF disease development. Furthermore, the categories of indirect evidence based on reproduction of the autoimmune disease in experimental animals 40,43,44 and circumstantial evidence from clinical clues 30 also support the hypothesis not only of ADE but also of autoimmunity as a contributory factor to DHF.
Several autoantigens recognized by anti-NS1 antibodies have been identified by sequence alignment using NCBI and Swiss-Prot databases, including beta chain of H+-transporter/ATP synthase, protein disulfide isomerase (PDI), vimentin and heat shock protein 60. 46 Of note, anti-prM antibodies cross-reactive with BHK-21 or A549 epithelial cells also recognize heat shock protein 60. 47 Furthermore, anti-NS1-mediated platelet aggregation inhibition is, at least in part, attributable to its binding and inhibitory effect on PDI. 48
The C-terminal amino acid residues 311–352 of dengue virus NS1 shares sequence homology with host cell target proteins. Using a modified NS1 lacking cross-reactive epitopes, the C-terminal region of NS1 may be responsible for cross-reactivity with endothelial cells and platelets. 44,49 In addition, the deletion of the C-terminal region of dengue virus NS1 abolishes anti-NS1-mediated platelet dysfunction and bleeding tendency. 44 Since NS1 is a potential vaccine candidate for dengue, the NS1 epitopes that generate cross-reactive antibodies will likely need to be removed or modified.
Molecular mimicry between virus and coagulatory molecules
Hemostasis is composed of four major events that occur following the loss of vascular integrity: (1) vascular constriction; (2) platelet activation and aggregation; (3) coagulation cascade and clot formation; and (4) clot dissolution. 50 As shown in Figure 1, the coagulation cascade consists of two pathways leading to fibrin formation: the intrinsic and extrinsic pathways. 51 The intrinsic pathway is initiated primarily by exposure of collagen to the blood vessel surface, while the extrinsic pathway is initiated by exposure of tissue factor upon vascular injury. The two pathways converge at the activation of factor X (FX) (generating FXa, ‘a’ signifies active), and FXa forms a complex with factor Va (FVa) to activate prothrombin to thrombin. Thrombin converts fibrinogen to a fibrin network. Fibrinolysis is triggered by activation of plasminogen to plasmin by tissue plasminogen activator (tPA) or urokinase. Both procoagulatory and anticoagulatory molecules are tightly regulated to prevent unwanted thrombosis or bleeding under normal conditions.
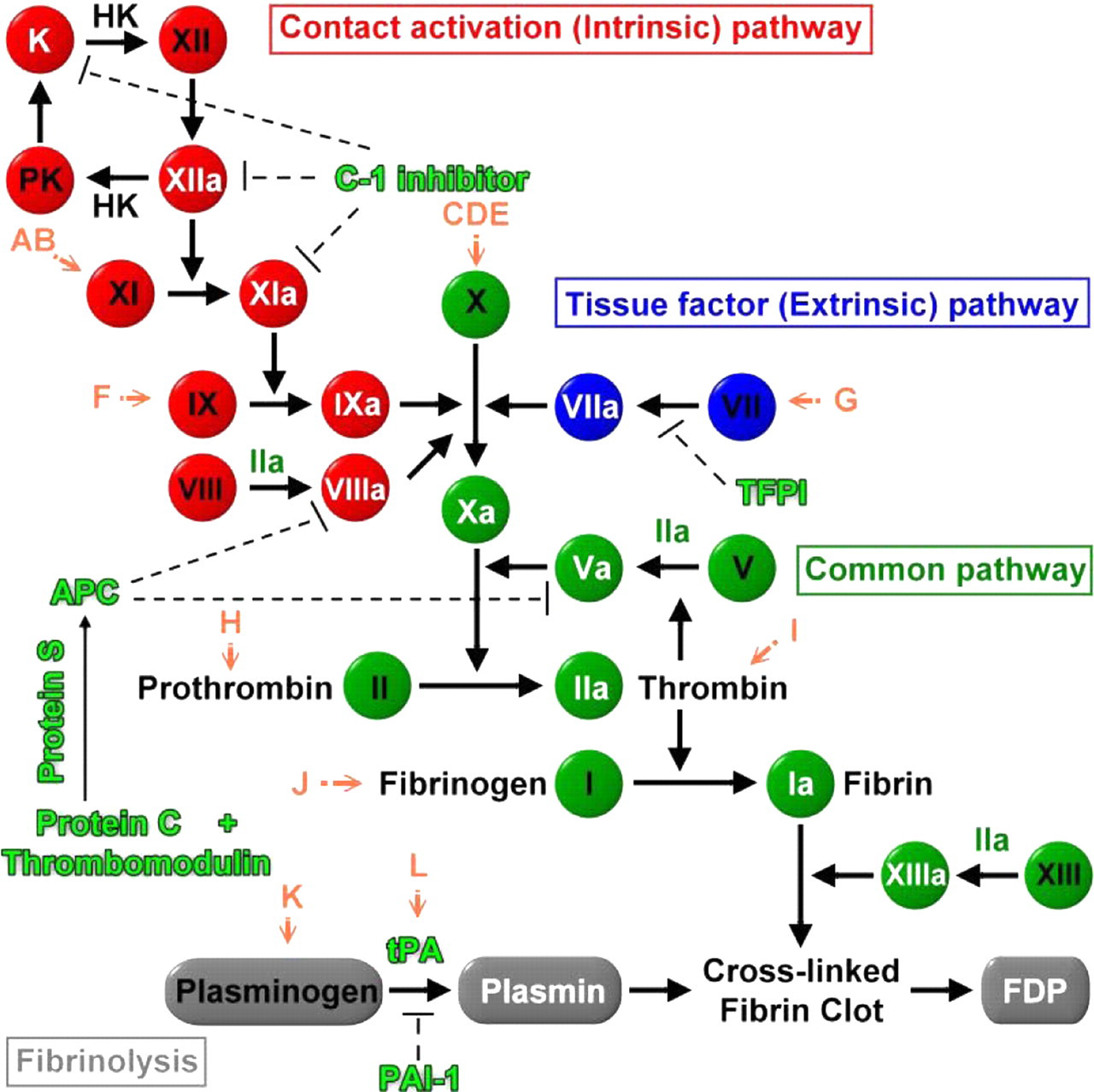
Schematic diagram of the coagulation cascade and possible interference of different autoantibodies against dengue virus in the coagulatory pathway. The letters (A–L) in orange color represent autoantibodies against dengue virus proteins which may cross-react with coagulatory molecules through molecular mimicry as listed in Table 2. K, kallikrein; PK, prekallikrein; HK, high molecular weight kininogen; TFPI, tissue factor pathway inhibitor; APC, activated protein C; PAI-1, plasminogen activator inhibitor 1; FDP, fibrinogen deposition products. Dashed lines indicate inhibition
However, in some pathological conditions, such as antiphospholipid syndrome (APS), this delicate balance is broken by autoantibodies against coagulatory molecules. In APS patients, autoantibodies which cross-react with coagulatory molecules can be found in serum samples which may cause thrombosis and miscarriage. 52–55 It is still unknown by what mechanisms these cross-reactive autoantibodies are induced. However, molecular mimicry through bacterial or viral infection represents a popular and widely accepted hypothesis. 56,57
The presence of pathogen-induced autoantibodies cross-reactive with coagulatory molecules has been confirmed in two ways. One is direct sequence comparison of pathogen proteins and coagulatory molecules to identify a shared linear amino acid sequence. The other is conformational analysis to find a shared motif or domain structure. The homologous sequence or conformation between the host molecules and those of the pathogen must be different enough to be recognized as non-self by host immune cells. In addition, this shared region must be exposed so that antibodies against this region can bind to the target region to cause functional disturbance. 58,59 In the cases of coagulation and fibrinolysis-related molecules, because many of them such as coagulatory factor Xa, IXa, II (thrombin) and plasmin belong to the trypsin-like serine protease superfamily and share similar primary amino acid sequence and catalytic domain, 60 it is not surprising to find antibodies from APS patients that cross-react with many of these coagulatory molecules. 51–55
There are a group of viruses which can cause viral hemorrhage fever (VHF) during infections. 61,62 All these VHF viruses are RNA viruses with different transmission vectors and geographic distribution. However, they all show clinical manifestations of hemorrhage to various degrees. Among VHF, dengue, Marburg and Ebola are the most important ones, 62 and dengue virus is the most prevalent in tropical and subtropical areas inhabited by its mosquito vector. 63 Studies on the pathogenic mechanisms of dengue virus-induced hemorrhage may unveil some clues of the hemorrhagic conditions induced by the other VHF agents.
In DHF/DSS patients, blood platelet numbers drop to less than 105/mm3 (thrombocytopenia) and there are signs of plasma leakage. 17 In addition, DHF patients have abnormal coagulopathy which is also evidenced by prolonged thrombin time (TT) and activated partial thromboplastin time, decreased fibrinogen levels and increased levels of fibrinogen degradation products. 17,64 Hemostatic changes in DHF/DSS involve multifactorial mechanisms which include not only vascular changes and thrombocytopenia but also coagulation disorders. 64–66 Several pieces of evidence suggest that autoantibodies may be involved in causing abnormal hemostasis during dengue virus infection. As mentioned above, antibodies against NS1 from both dengue patients and NS1-immunized mice can bind to endothelial cells and induce activation and apoptosis of endothelial cells. 36–39 In addition, anti-NS1 antibodies can also opsonize normal human platelets and enhance platelet-macrophage engagements in vitro. 67 Murine monoclonal antibodies against NS1 of dengue virus have also been shown to bind to human blood coagulation factors such as fibrinogen. 35 Moreover, like many coagulatory factors, dengue virus can also bind to heparin (although dengue is not unique among viruses in binding to heparin). 68–71 Taken together there is evidence to suggest that molecular mimicry between dengue virus and not only platelets and endothelial cells but also coagulatory molecules may also play important roles in the immunopathogenesis of dengue hemorrhage.
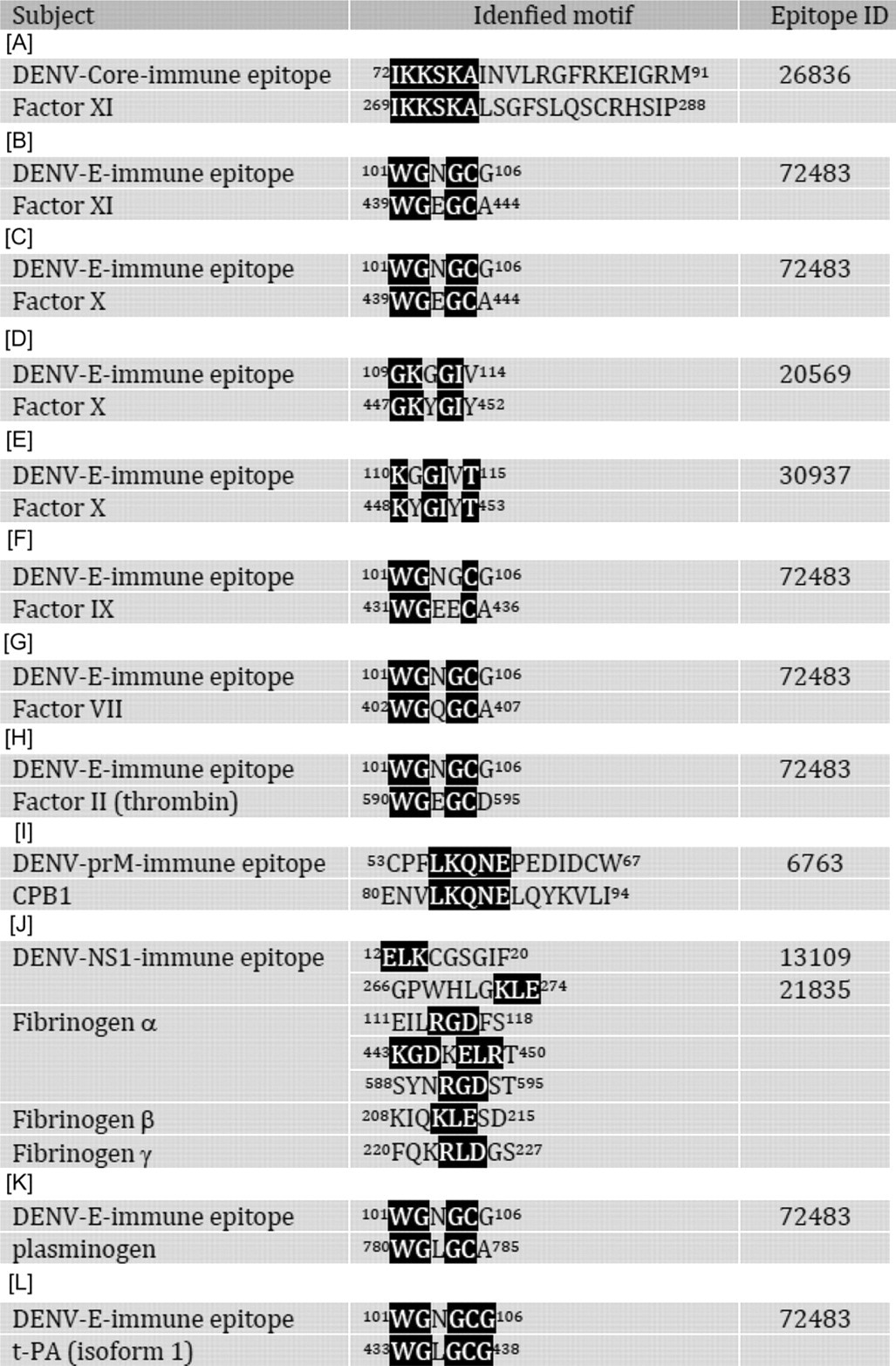
A computer-based sequence comparison between dengue virus proteins and coagulatory molecules shows that at least 12 different regions of dengue virus proteins, including core, prM, E and NS1 proteins, have amino acid sequence similarity with different coagulatory molecules, such as factors X, XI, VII, etc. (Table 2). The possible interference of these antidengue antibodies in the coagulatory activation pathway is shown in Figure 1. Many of these protein motifs are shared by different viruses of the Flaviviridae family which can cause different degrees of hemorrhage, such as Japanese encephalitis virus, Western Nile virus, yellow fever virus, tick-borne encephalitis virus and Omsk hemorrhagic fever virus (Figure 2). Hepatitis C virus which seldom causes hemorrhage, on the other hand, shows no similar motif at this region. Taken together, this region may play important roles in the pathogenesis of hemorrhage induced by these viruses. However, in yellow fever, the hemorrhage occurs during primary infection, while in DHF, most of the cases are secondary infection. Therefore, different mechanisms may be involved in the hemorrhage induced by flaviviruses, particularly with regard to the important role of antibody induced by dengue virus infection in DHF. Whether the protein sequences listed in Table 2 can induce antibodies during dengue virus infection is also largely unknown, although antibodies have been found in dengue patients which identify a sequence homology region between the E protein and plasminogen. 72,73 There is also a correlation between plasminogen cross-reactive antibodies and hemorrhage in dengue patients. 74 In addition to sequence homology with plasminogen, amino acids 101–106 of E protein (WGNGCG) shows sequence homology with factors XI, X, IX, VII, II (thrombin), plasminogen and tPA. In preliminary screening using serum samples from a previous study, 75 our unpublished data show that antibodies against factor Xa are also increased in DHF/DSS patients. Whether these antibodies which cross-react with coagulatory molecules contribute to the hemostatic abnormality and hemorrhage during DHF/DSS needs to be further studied.
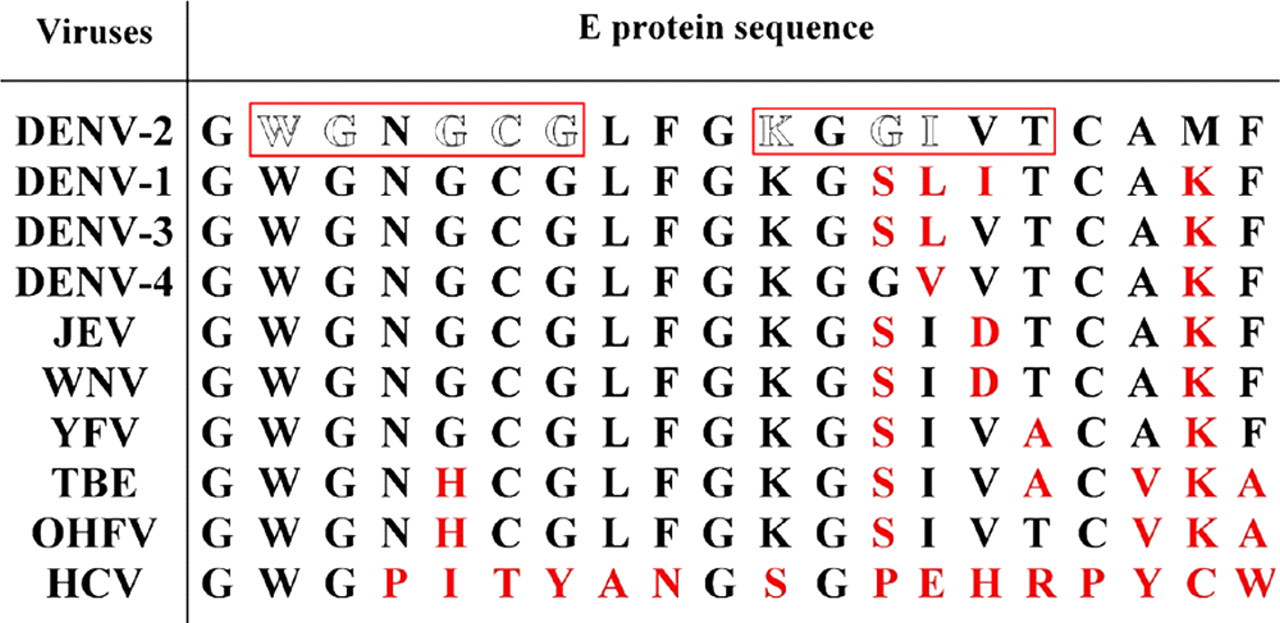
Sequence comparisons between different viruses of the Flaviviridae family. Amino acids in boxes are motifs shared with different coagulatory molecules as shown in Table 2. Amino acids labeled with red color denote non-identical amino acids with dengue virus serotype-2 (DENV-2). JEV, Japanese encephalitis virus; WNV, Western Nile virus; YFV, yellow fever virus; TBE, tick-borne encephalitis virus; OHFV, Omsk hemorrhagic fever virus and HCV, hepatitis C virus
Molecular mimicry between coagulatory factors and dengue virus proteins
In addition to E protein, antibodies against NS1 have also been shown to cross-react with human fibrinogen. 35 However, the influence of these fibrinogen cross-reactive NS1 antibodies on fibrinogen activation remains unknown. Because serum samples generally contain many factors that can interfere with coagulation, it is necessary to separate the contributions of such factors from cross-reactive antibody effects. In this regard, the use of monoclonal antibodies may be of value. Different specificities of monoclonal antibodies may have different effects on coagulation molecules' activation or function. An alternative way to study the influence of antibody on coagulation function is by using recombinant single chain variable region (scFv), which retains the specificity of antibody but can be produced by bacteriophage. We have generated scFv from NS1 immunized mice. Fibrinogen cross-reactive scFv antibodies were selected from the scFv library and tested for their effect on fibrinogen activation by TT. As shown in Figure 3, scFv against NS1 can interfere with fibrin formation which leads to prolonged TT. These data indicate that molecular mimicry between dengue virus and coagulatory molecules may be a result of cross-reactive autoantibodies which can interfere with coagulation activation. However, further studies are required to assess the importance of these autoantibodies in the development of hemorrhage during DHF/DSS.
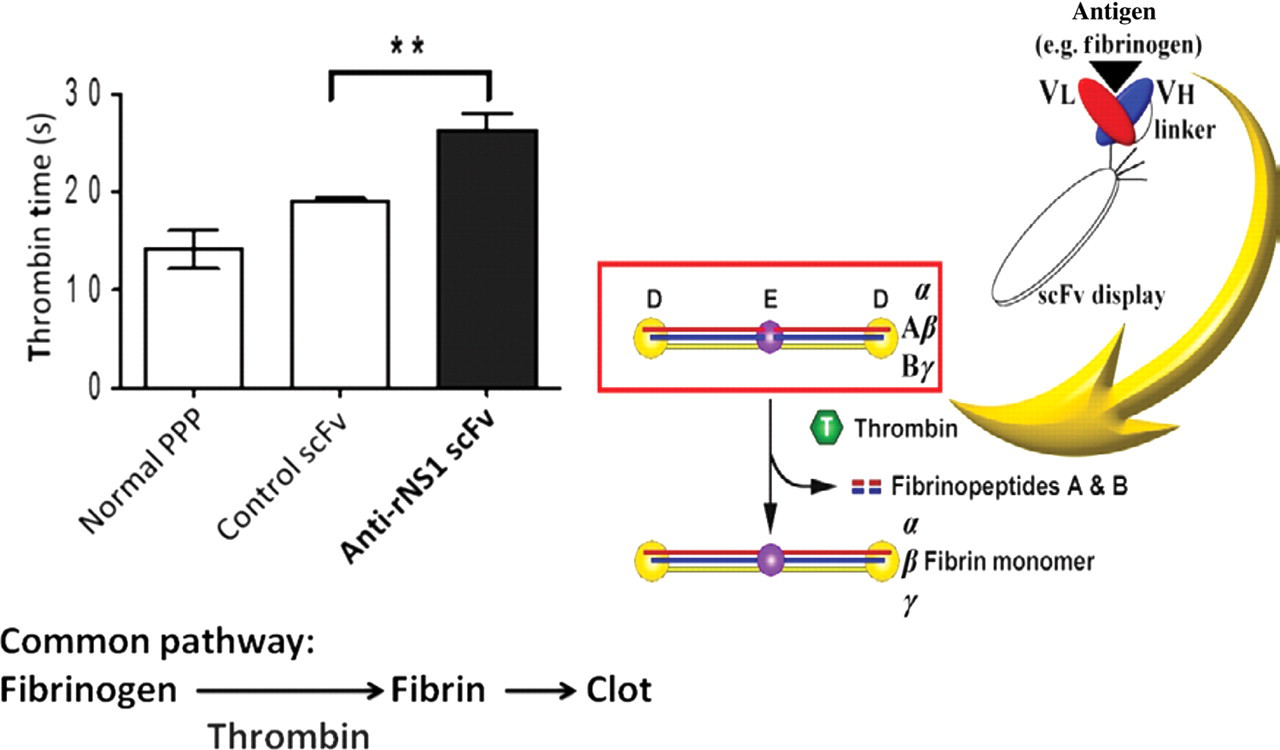
Fibrinogen cross-reactive anti-rNS1 scFv prolong the thrombin time in vitro. NS1, non-structural protein 1; PPP, platelet‐poor plasma; scFv, single chain variable region
In summary, antibodies against dengue virus may interfere with hemostasis due to molecular mimicry between dengue virus proteins and coagulatory molecules. The epitopes recognized by these autoantibodies should be avoided in the design of candidate vaccines against dengue virus infection to avoid unwanted side-effects. Furthermore, the information gathered may provide clues to understand the mechanism of hemorrhage and develop better therapeutic strategies against VHF.
Autoimmunity in dengue disease pathogenesis
As mentioned above, antibodies against dengue virus proteins such as NS1, prM or E can cross-react with platelets, endothelial cells and coagulatory molecules. 19,22,28 Molecular mimicry between dengue virus antigens and self-antigens may break self-tolerance. Plasma leakage in dengue patients is likely due to increased vascular permeability mediated by endothelial cells. Endothelial cell perturbation occurs by a variety of mechanisms, such as cytokine-mediated effects, direct virus cytopathy and antiendothelial cell antibody-mediated pathology. In addition, platelets are destroyed or functionally impaired by cross-reactive antibodies.
Macrophages are activated by dengue virus or cytokines such as IFN-γ during the acute phase of infection. Activated macrophages may then phagocytose autoantibody-opsonized platelets and endothelial cells, and thus contribute to the development of thrombocytopenia and plasma leakage in DHF/DSS. 19,28,76 Antidengue cross-reactive antibodies to platelets and endothelial cells provide an explanation for the target specificity and unique feature of thrombocytopenia and plasma leakage during the development of DHF/DSS. In addition, antibodies against dengue virus may interfere with hemostasis due to the molecular mimicry between dengue virus proteins and coagulatory molecules, which lead to interference with coagulation activation. A schematic model of molecular mimicry-based autoimmunity in dengue disease is illustrated in Figure 4.

Schematic model of autoantibody-mediated immunopathogenesis in dengue virus infection. DV, dengue virus; NSI, non‐structural protein 1; DHF, dengue hemorrhagic fever
Conclusion and perspectives
Dengue disease has increased globally and is now reported in more than 100 countries. The prevention and management of dengue disease is crucial. Nevertheless, the onset of vascular leakage and hemorrhage is one of the life-threatening complications that occur in severe dengue patients. Its pathogenic mechanisms are not yet fully resolved. The concept of autoantibody-associated immunopathogenesis in DHF raises concerns for vaccine development, since autoimmune memory may confer susceptibility to more severe disease in a subsequent dengue infection. 77 Several candidate vaccines are currently being evaluated, 78,79 most of which use live attenuated or chimeric attenuated virus. Autoimmune complications may need to be considered for future development of a safe and effective dengue vaccine.
Footnotes
Acknowledgements
The research was supported by grants NSC99-2321-B006-008, NSC98-3112-B006-003 and NSC-96-2628-B-006-006-MY3 from the National Science Council, Taiwan, and NHRI-98A1-PDCO-0209115 from the National Health Research Institutes, Taiwan.