
Abstract
Alzheimer's disease (AD) is thought to start years or decades prior to clinical diagnosis. Overt pathology such as protein misfolding and plaque formation occur at later stages, and factors other than amyloid misfolding contribute to the initiation of the disease. Vascular and metabolic dysfunctions are excellent candidates, as they are well-known features of AD that precede pathology or clinical dementia. While the general notion that vascular and metabolic dysfunctions contribute to the etiology of AD is becoming accepted, recent research suggests novel mechanisms by which these/such processes could possibly contribute to AD pathogenesis. Vascular dysfunction includes reduced cerebrovascular flow and cerebral amyloid angiopathy. Indeed, there appears to be an interaction between amyloid β (Aβ) and vascular pathology, where Aβ production and vascular pathology both contribute to and are affected by oxidative stress. One major player in the vascular pathology is NAD(P)H oxidase, which generates vasoactive superoxide. Metabolic dysfunction has only recently regained popularity in relation to its potential role in AD. The role of metabolic dysfunction in AD is supported by the increased epidemiological risk of AD associated with several metabolic diseases such as diabetes, dyslipidemia and hypertension, in which there is elevated oxidative damage and insulin resistance. Metabolic dysfunction is further implicated in AD as pharmacological inhibition of metabolism exacerbates pathology, and several metabolic enzymes of the glycolytic, tricarboxylic acid cycle (TCA) and oxidative phosphorylation pathways are damaged in AD. Recent studies have highlighted the role of insulin resistance, in contributing to AD. Thus, vascular and metabolic dysfunctions are key components in the AD pathology throughout the course of disease. The common denominator between vascular and metabolic dysfunction emerging from this review appears to be oxidative stress and Aβ. This review also provides a framework for evaluation of current and future therapeutics for AD.
Introduction
Alzheimer's disease (AD) is thought to begin many years prior to its clinical diagnosis. Recent studies support that metabolic and vascular dysfunctions are involved in pathology and progression of AD. Metabolic and vascular alterations, with impairment of glucose utilization and blood flow changes, occur with and prior to AD diagnosis, 1–15 and may underlie these changes. Furthermore, support for metabolic dysfunction comes from epidemiological studies where diseases characterized by metabolic dysfunction, including diabetes, dyslipidemia and hypertension, are risk factors for AD. 16 Discovery and prevention of metabolic and vascular dysfunction could lead to new strategies to halt or prevent AD therapeutically.
Also closely linked with metabolic and vascular changes in AD is amyloid β (Aβ), one protein involved in disease pathogenesis (Aβ). Tissues that produce the most Aβ are brain and skeletal muscle; both of which possess high metabolisms and well-developed vascular networks. 17 Aβ levels are also reflective of metabolism as Aβ levels increase during synaptic activity. 17,18 Additionally, Aβ may exacerbate initial deficits in glucose utilization and blood flow. For example, exogenous synthetic Aβ reduces glucose utilization and blood flow, 19 which is also observed with endogenous overexpression of Aβ in transgenic mouse models of AD. 20–25 Vascular changes are anticipated as Aβ accumulates in cerebral vessel walls in cerebral amyloid angiopathy (CAA), leading to the death of smooth muscle cells 26 and blockage of the perivascular circulation. 27–29
The general notion that vascular and metabolic dysfunctions contribute to the etiology of AD is becoming accepted. The novelty is that research, both recent and prior, now reveals potential mechanisms by which these processes could possibly contribute to AD pathogenesis. Specifically, oxidative stress is now believed to contribute to vascular dysfunction, and when combined with altered substrate utilization contributes to metabolic dysfunction. This synthesis does not invalidate other pathological mechanisms in AD, but provides a framework to evaluate emerging therapeutic approaches for AD.
Vasculature
It is clear that vascular dysfunction contributes to AD pathology and may represent an early or precedent pathology. 1,13,20,30–34 While under normal conditions vascular flow autoregulates to neuronal activity and nutritional requirements, 20,25 vascular dysfunction compromises the supply of glucose and oxygen to the brain. 35 Vascular dysfunction in AD is characterized by disruption of vascular architecture including lower capillary density, vessel curvature resulting in narrowed blood vessels, reduced blood flow and poor arterial responsiveness. 11,13,25,36,37 Vascular dysfunction precedes cognitive impairment and thus exacerbates underlying AD pathology. 5,8,11,13,20,22,34,38–40 Vascular dysfunction may be related to atherosclerotic lesions and blood flow restriction of arterial vessels leading to the brain. 13,41 The question remains whether this is a cause of pathology or just a vascular response to accumulation of Aβ.
Aβ and vascular dysfunction
Elevation of Aβ is directly implicated in vascular pathology, and therefore may exacerbate initial deficits in blood flow and affect vascular autoregulation. 20 Furthermore, data suggest a synergistic effect between Aβ deposition and vascular dysfunction on neuronal degeneration. 34 Overexpression of Aβ in mouse models of AD recapitulates vascular dysfunction. 20–25 Vascular dysfunction and reductions in resting cerebral blood flow also occurred with Aβ administered systemically. 34 Additionally, Aβ accumulation on vessels or the cerebral cortex resulted in vascular dysfunction. 11,13,22,34,39,40,42 Conversely, there is evidence that vascular dysfunction and risk factors may promote Aβ deposition. 22,24
The clearance of Aβ occurs via perivascular macrophages, perivasculature 27–29,29,42,43 and by microglia 13 (Figure 1). This clearance is receptor mediated, with the RAGE (receptor for advanced glycation end products) and LPR (lipoprotein receptor-related protein) receptors transporting Aβ into and out of the brain across the blood–brain barrier, respectively. 36,44,45 Modification of such clearance affects pathology. For example, reduced clearance of Aβ across the blood–brain barrier increases CAA and neuritic plaques. 13,27 Also, Aβ antibody therapy clears neuritic plaques with a concomitant increase in the accumulation of Aβ with the vessels (CAA) in AD patients and mouse models of AD. 27,46,47 Finally, a reduction of Aβ clearance and not Aβ production might be the main factor in AD pathology (Holtzman D, unpublished data presented at International Conference on Alzheimer's Disease 2010, Hawaii, 10–15, July, 2010).
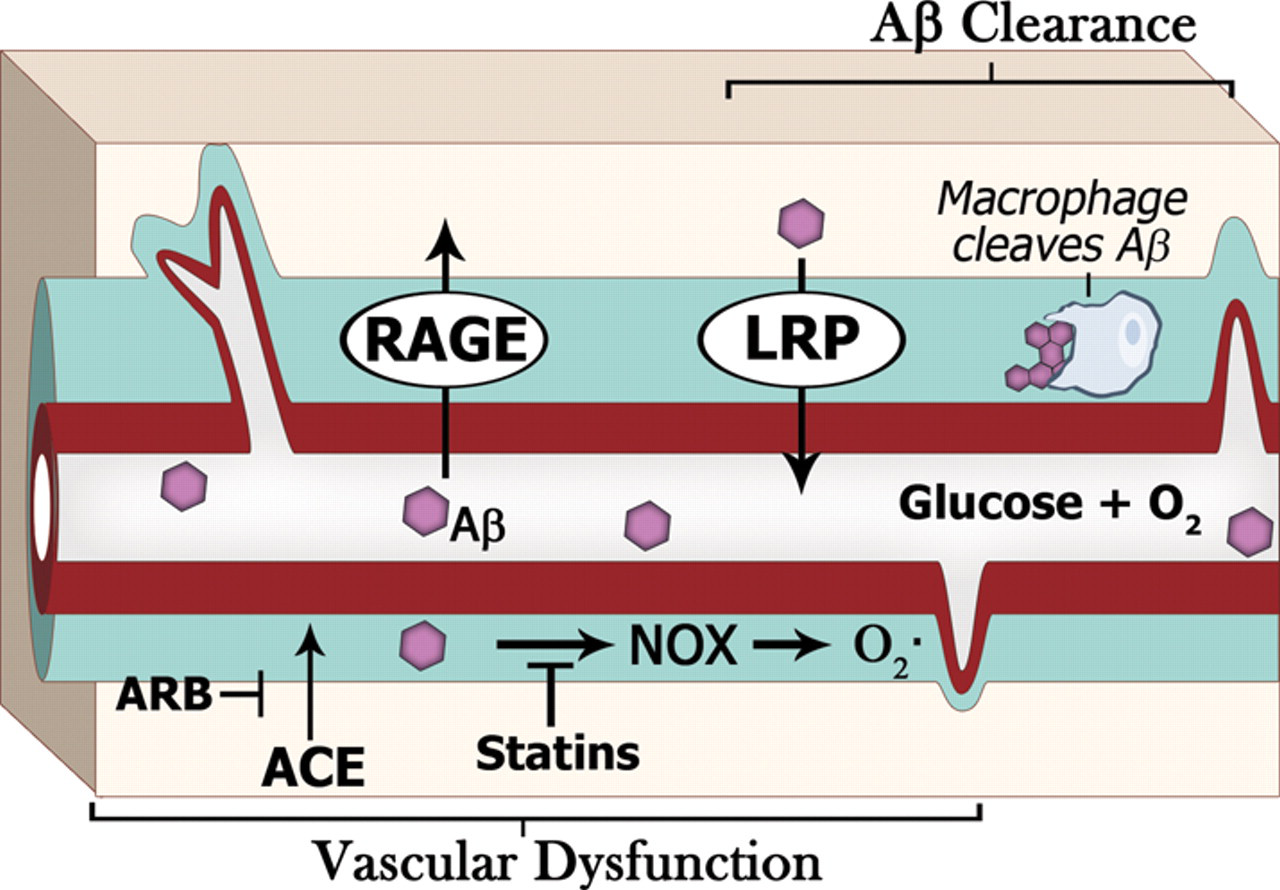
Vascular dysfunction and AD. Vascular dysfunction is an early feature of AD, manifested in part via reduced glucose utilization and blood flow. Aβ is directly implicated in vascular dysfunction, where Aβ increases vasoconstriction, oxidative stress and NOX activity. NOX activity is a key player in vasoconstriction in AD models. The vasculature is not only important for nutrient supply, but also for Aβ clearance. For example, the RAGE (receptor for advanced glycation end products) and LRP (lipoprotein receptor-related protein) are involved in Aβ influx and efflux, while macrophages clear Aβ in the perivascular space. Pharmacological prevention: the angiotensin-converting enzyme (ACE), which is elevated in AD, causes vasoconstriction. Angiotensin receptor blocker (ARB) drugs may be protective in AD as they block ACE activity. Statins alter NOX activity and reduce Aβ production. Aβ, amyloid β; AD, Alzheimer's disease; NOX, NAD(P)H oxidase
In summary, the vascular blood flow brings in nutrients, removes wastes and aids clearance of Aβ. Therefore, impairment of vasodilation and vascular flow may aid in AD pathogenesis by exacerbating aggregation of Aβ plaques.
AD, vasculature dysfunction and oxidative stress
Oxidative stress also contributes to vascular dysfunction. Reports show that oxidative damage on the pathogenesis of AD may be directly due to Aβ. 48–51 While neuronal oxidative stress is inversely related to Aβ burden, vascular oxidative damage is positively correlated with oxidative stress. 52 The importance of oxidative stress is made clear with antioxidant therapy and knockout of oxidant generating proteins. A potent source of reactive oxygen species, NAD(P)H oxidase (NOX), is indeed activated by Aβ 25,41,53,54 (Figure 1). Oxidative stress may be involved in Aβ-mediated cell death 55 and NOX generation of superoxide anions contribute to vascular dysfunction. 39 The potential importance of NOX and oxidative stress in AD is illustrated by an increase in NOX levels or activity in the mild cognitively impaired (MCI) and AD brain tissues. 55–59 Furthermore, genetic ablation of NOX prevented the oxidative stress and cerebrovascular dysfunction in transgenic mice. 25,60 Finally, antioxidant therapy abrogated Aβ-mediated cerebrovascular dysfunction in mouse models of AD. 37,39,61 Therefore, a synergistic relationship exists between Aβ and oxidative damage which contributes to vascular dysfunction in AD patients.
AD and stroke
Cerebrovascular disease occurs in AD, 8,62 precedes AD 13 and increases AD risk. Stroke-induced lesions co-exist with markers of dementias, and subcortical infarcts co-incident with AD pathology increase the odds of dementia four-fold. 63 Silent strokes are associated with a doubling of the AD risk. 32 Other cerebrovascular pathologies are also associated with AD. CAA occurs in an overwhelming >80% of AD patients and only 10–40% in controls. 13 Cerebral microbleeds and lobar hemorrhages occur in 30% and 7–18% of AD patients respectively. 13
The high prevalence of CAA in AD, where Aβ deposits in the vessel walls, suggests a pathological role. CAA severely disrupts blood vessels and is strongly associated with cognitive impairment. 13 Mutations in the Aβ sequence (Dutch, Iowa, Flemish, Artic and Italian) lead to increased Aβ in the vessels, and may cause microhemorrhages even in the absence of increased amyloid precursor protein (APP) processing. 13 In addition to weakened blood vessels, CAA can also reduce Aβ clearance 13 and thus increase pathology.
It has been postulated that cerebrovascular diseases lower the threshold for AD pathology. 64,65 Experimentally, focal ischemic lesions in the forebrain increase Aβ levels in a transgenic mouse model 66 and accumulation of intracerebroventricularly infused Aβ. 67 Indeed, ischemic cerebrovascular disease, which may not necessarily cause AD, may underlie the conversion from preclinical AD to AD. 68 However, it is clear that injuries such as stroke or ischemia increase the levels of proteins which elevate Aβ pathology, and thus increase Aβ levels. For example, during ischemia 69,70 proteins which increase Aβ levels such as p25/cdk5 71 , Aβ and BACE are elevated. 72,73 Thus cerebrovascular disease is present in AD, increases AD risk and contributes to the pathology.
Cardiovascular risk factors for stroke are also modified by demographic variables such as advancing age and female sex. Coincidentally, these latter variables also affect the risk for AD. Thus, while the association between vascular injury and AD pathology is under intensive study, critical variables that affect the risk for both AD and stroke such as age and sex are poorly understood. These factors have the potential to affect not only ‘risk’ but also therapeutic outcomes.
Vascular diseases and treatment
Hypercholesterolemia
Several studies link hypercholesterolemia to AD 74–76 with elevated cholesterol levels increasing Aβ production. 77–79 Statins inhibit the rate-limiting enzyme in the cholesterol pathway (3-hydroxy-3-methyl-glutaryl-CoA reductase) and are drug therapy for hypercholesterolemia. These drugs are associated with reduced risk of AD. 80 Clinical trials indicate that statins reduce the progression of AD. 80 Modification of other enzymes in the cholesterol pathway also affects Aβ levels. For example, acetyl Co-A acetyl transferase (ACAT – the enzyme which esterifies cholesterol), and ABC transporters (cholesterol efflux) all decrease Aβ levels. 74,75,77,80 Conversely, reduction of LXR (cholesterol sensor) levels resulted in elevated Aβ levels. 77 Cholesterol's effects on Aβ levels is likely due to its effect on β-secretase, one of the enzymes generating Aβ from its precursor protein (APP). 80 The question is how do these drugs reduce AD risk? An additional mechanism by which statins lessen AD progression may be via reduction of NOX and oxidative stress. 81,82 Pharmacological prevention of vascular damage by hypercholesterolemia would allow for optimal brain perfusion, efficient clearance of Aβ via the vasculature and thus reduce pathology. Additionally, the efficacy of statins may also be related to their neuroprotective activity. 80
Hypertension
Hypertension is also a potent risk factor for AD. 41,74,83 It is well known that antihypertensive drugs reduce the risk of AD. One of the main proteins involved in hypertension is angiotensin II (Ang II), which causes vasoconstriction. Ang II is secreted as a proenzyme and undergoes proteolysis to its final active form via angiotensin-converting enzyme (ACE). How is this related to AD? ACE activity and levels are elevated in AD 84 and likely contribute to vascular pathology. ACE inhibitors and angiotensin receptor blockers (ARB), which respectively prevent the production and action of Ang II, also reduce the risk of AD, 33,84 prevent dementia, and reduce cognitive deficits and amyloid plaque load. 33,85,86 The beneficial effect of these drugs may be due to a lowering of hypertension and thus a reduction in vascular pathology.
Antihypertensive drugs have additional effects via the angiotensin pathway. For example, the ACE enzyme both cleaves Aβ and activates peroxisome proliferated-activated receptor γ (PPARγ). The cleavage of Aβ by ACE 84 is important, as such truncations enhance Aβ pathological misfolding. 87 ARB's ability to prevent the above and activate PPARγ 86,88 may further explain the beneficial effects of these drugs on mitochondrial energy production. 89 PPARs are nuclear receptors that induce the proliferation of peroxisomes and are important in metabolism. They are also discussed under insulin resistance in the metabolic diseases and AD section. Finally, Ang II activates NOX, leading to increased reactive oxygen species production. 25,89 Thus, targeting Ang II and NOX signaling elicits beneficial effects with the prevention of deleterious changes, and thus may reduce AD pathogenesis.
Reduced blood flow and vascular dysfunction, in general, are early pathological features of AD. The reviewed literature linked vascular pathology with AD and identified synergism of Aβ and oxidative stress in this vascular pathology. Since vascular diseases are associated with increased risk of AD; hypertension, hypercholesterolemia and their treatment were also evaluated in this review. As such, vascular diseases and their treatments may aid in an understanding of the early stages of AD.
Metabolism
Metabolic dysfunction appears to also be involved in the etiology of AD, and indeed could interact with vascular dysfunction. The decline of glucose utilization is critical in the brain, as glucose is the preferred and primary nutrient source. 4 In fact, the brain utilizes ∼20% of the body's total glucose supply. 4,9 Reduction of glucose utilization (up to 45%) is a well-known pathological feature of AD 2,4,5,10,12,90,91 that is known to occur early in the disease in human patients. 3,7,9,11,14,15,92,93 Impaired glucose uptake is recapitulated in transgenic mouse models of AD. 94,95 During early stages of AD, glucose utilization deficits (50%) are greater than blood flow/oxygen deficits (20%), and only in the later stages of AD do the changes in these factors become similar. 5 This suggests that these initial glucose utilization deficits may be more important in the genesis of pathology than blood flow and oxygen utilization. This decrease in glucose utilization is a feature of cognitive decline, MCI and MCI conversion to AD. 9,10,13,96–98 Thus, glucose utilization and metabolic changes are important factors in AD.
Aβ and metabolism
Aβ may also contribute directly to this metabolic dysfunction in glucose homeostasis. There are several examples of this in the literature. Overexpression of Aβ in transgenic mice results in reduced glucose utilization. 94 Aβ also causes changes characteristic of insulin resistance, such as reduced glucose utilization, 19 decreased insulin receptors 99,100 and promotes insulin resistance 14,92 (Figure 2). Aβ also directly inhibits insulin receptor activity. 99 Conversely, there is significant agreement that insulin stimulates Aβ production. 34 Insulin resistance and poor glucose utilization increase Aβ production and reduces Aβ clearance. Insulin resistance reduces Aβ clearance via a decrease in the expression and activity of an enzyme involved in Aβ clearance, insulin degrading enzyme (IDE) 96,101 (Figure 3). Downregulation of IDE leads to increased Aβ plaque load in AD models. 35,101 Furthermore, the IDE knockout mouse has elevated insulin levels, glucose intolerance and increased Aβ load. 34 Thus, Aβ accumulation may be dependent on insulin resistance and metabolic deficiencies which ultimately feedback to increase Aβ levels.

Metabolic dysfunction and AD. Metabolic dysfunction is a key feature in AD, with changes in glycolysis, the tricarboxylic (TCA) cycle and oxidative phosphorylation. In AD, damage to oxidative phosphorylation would result in reduced regeneration of NAD+ used in glycolysis and the TCA cycle. In this situation, alternate processes such as lactate dehydrogenase and NADPH oxidase (NOX) could regenerate NAD+ instead. However, an unfortunate consequence of NOX is increased oxidative damage. Another defect also observed in AD is insulin resistance, where there is reduced glucose uptake, insulin signaling and dysfunction of portions of the TCA cycle. Insulin resistance prompts a switch to ketone body use to provide energy via peroxisomes or the TCA cycle instead of glucose. Pharmacological prevention: several drugs have been used to pharmacologically prevent such pathological processes. For example, drugs such as the angiotensin receptor blocker (ARB, which also activates PPARγ like activity) and PPARy agonists could be of benefit. AD, Alzheimer's disease; PPAR, peroxisome proliferated-activated receptor
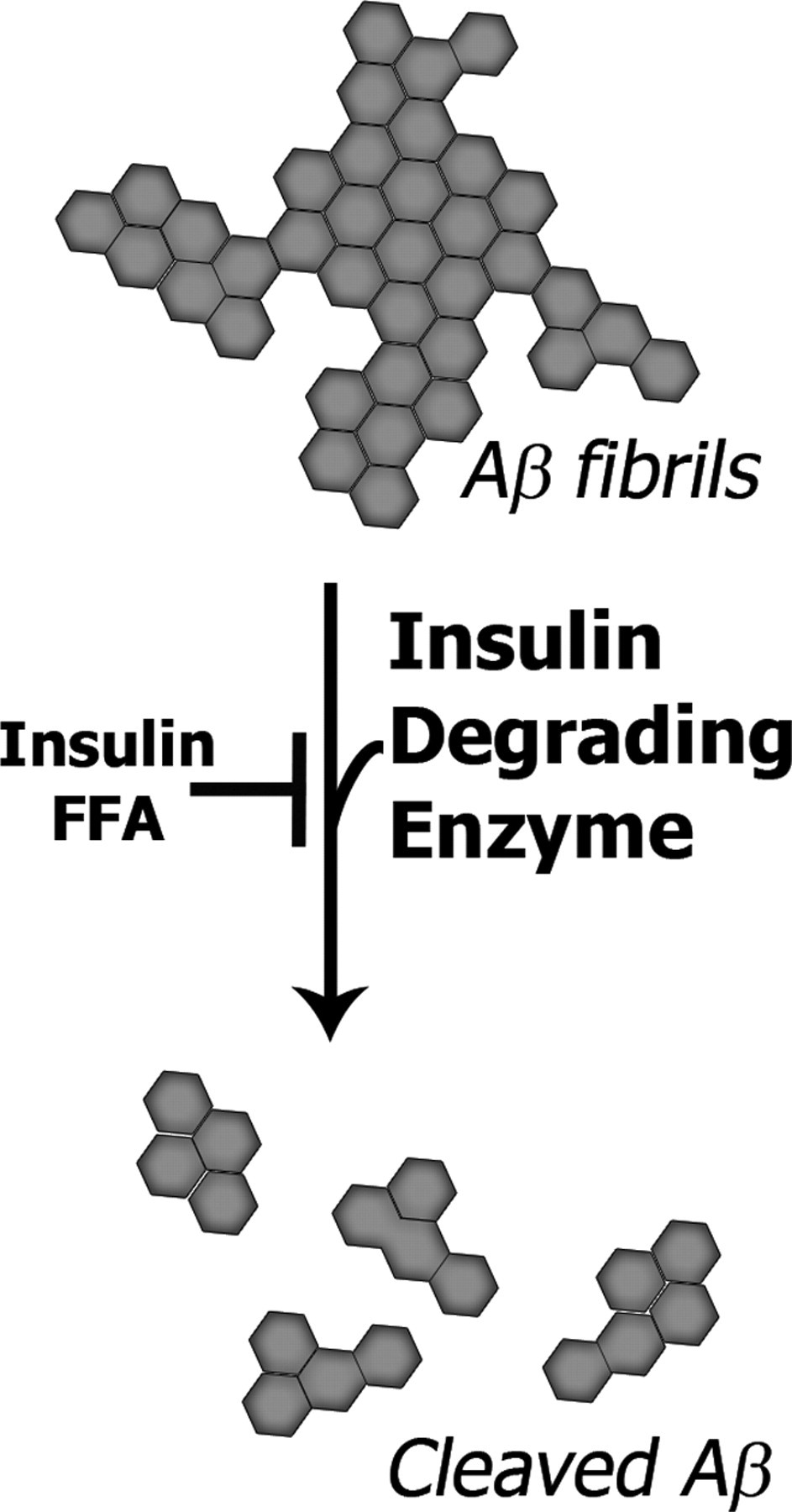
Amyloid plaque clearance by insulin degrading enzyme (IDE). Insulin resistance reduces the IDE cleavage of Aβ via elevated insulin and free fatty acid levels. This is only one of many ways in which insulin resistance contributes to Aβ pathology. Aβ, amyloid β; FFA, free fatty acid
AD, metabolism and insulin resistance (diabetes)
Type II diabetes and AD are linked both epidemiologically and pathologically. 102,103 For example, the epidemiological incidence of both these diseases increases with age, with the incidence of diabetes increased in AD. 102,103 Pathologically, both are associated with altered glucose homeostasis and extracellular accumulation of amyloid proteins, the islet amyloid polypeptide (IAPP) and Aβ protein. 102,103 This suggests that there are common underlying mechanisms such as cross-seeding of amyloid proteins or metabolic dysfunction.
Indeed, while the amyloid proteins have differing primary sequences, they adopt similar conformations 104 and are recognized by the same conformation specific antibodies. 105,106 As such, one amyloid protein can interact with another to cross-seed misfolding. Such cross-seeding was initially demonstrated in vitro by Lansbury in 1995 and in vivo by several labs. 107–113 However all amyloids may not cross-seed equally. For example, transthyretin fibrils cross-seed IAPP but not vice versa. 114 There is in vitro evidence that IAPP and Aβ interact directly 115,116 and misfolded Aβ is found in the pancreas. 117 It is likely that Aβ and IAPP amyloid proteins can cross-seed each other; however, this has not currently been demonstrated in vivo. However, mitochondrial dysfunction is also a common denominator between several amyloidoses. 118,119
Insulin resistance is a central feature of diabetes and increasing evidence supports that insulin resistance contributes to AD pathology. 92 Insulin resistance in AD has been termed Type 3 diabetes, 56 is characterized by low insulin receptor, brain insulin levels 92,120 and reduced glucose homeostasis. While increased peripheral insulin levels initially result in increased brain insulin, prolonged hyperinsulinemia reduces brain insulin levels via a downregulation of blood–brain barrier insulin receptors and reduced transport of insulin into the brain. 35,121 Given that the brain produces its own insulin, 96 the regulation of cerebral glucose metabolism may be independent of peripheral control. Reduction in insulin and its receptors correlate with the conversion from MCI to AD as well as the severity of AD. 35,77,96,120–122 Insulin resistance also leads to increased oxidative stress, elevated cortisol and mitochondrial dysfunction. 96,98,123
In addition to correlation of insulin factors with AD, insulin resistance also directly affects metabolites. For example, insulin resistance and increased glucocorticoids reduce glucose use 124 and increase dependence on ketone bodies. These non-glucose substrates including ketone bodies and lactic acid are generated for neuronal use by astrocytes. 124 Elevations of free fatty acids (FFAs) are also associated with insulin resistance. These elevated FFAs contribute to insulin resistance, reduce microglia glucose uptake and lactate release for neuronal energy utilization 20,125 and inhibit IDE activity. 98,121 Thus, insulin resistance causes major metabolic changes in utilization of glucose, fatty acids and ketone bodies, which consequently results in altered metabolism and elevated Aβ.
Metabolic pathway dysfunction and AD
From the above, it is well documented that oxidative damage and insulin resistance cause metabolic dysfunction in AD. We now review the known metabolic changes in AD, and we present them in a postulated, temporal three-part sequence with changes in glycolysis, the tricarboxylic acid cycle (TCA) and oxidative phosphorylation pathways as described below.
First, the pentose phosphate pathway (PPP) or hexose monophosphate shunt is activated during oxidative stress and may be the first metabolic response to reduce the oxidative damage in early AD. The PPP is responsible for the production of increased reducing power, which is used for the regeneration of antioxidants such as reduced glutathione (GSH). 126 During oxidative stress the PPP is upregulated, 126,127 and the NADPH generated is used to regenerate antioxidant systems. 9,128,129 The enzyme that controls the entry in this cycle is glucose-6-phosphate dehydrogenase, and its levels are indeed increased in AD. 126–128 Additionally, the PPP and antioxidant defenses are also increased by Aβ. 19 Thus, we have an adaptive, protective cellular response induced by oxidative damage and stimulated by Aβ. This PPP pathway is dependent upon glycolysis and may not be as effective later on in AD when damage occurs to glycolytic enzymes in the PPP pathway such as triosephosphate isomerase, 130–133 glyceraldehyde 3-phosphate dehydrogenase, 19,132,134–137 phosphoglycerate mutase 132,133 and α-enolase 130,132,138–141 . Thus the PPP pathway may initially be upregulated, but then compromised later on in AD.
The second metabolic change in AD is damage to the oxidative phosphorylation pathway. This pathway normally regenerates NAD+ for use in glycolysis and the TCA cycle. Oxidative phosphorylation dysfunction necessitates use of alternate pathways to regenerate this NAD+ (Figure 2). This likely occurs following damage to the oxidative phosphorylation pathway, which normally regenerates NAD+. 142 There is support for damage to the oxidative phosphorylation pathway in AD. For example, oxidative phosphorylation becomes impaired with age in mouse models of AD, 143 with reductions of mitochondrial respiration. 91 Also, enzymes required for oxidative phosphorylation and the TCA cycle are modified in AD: pyruvate dehydrogenase, α-ketoglutarate, ATP synthase, isocitrate dehydrogenase, complex IV and cytochrome c oxidase enzymes. 5,7,14,41,98,134,139,140,144–148 Of note, pyruvate and oxoglutarate dehydrogenases are the rate limiting steps for the TCA cycle. 149 Defective oxidative phosphorylation would thus require the use of alternate pathways, to regenerate NAD+ for these cycles. 142 To compensate for this damage, alternate regeneration of NAD+ can occur via LDH and NOX. 142 LDH can function under anaerobic conditions such as anoxia during stroke and other vascular dysfunctions. However, this use of LDH to generate NAD+ in AD may not predominate as LDH is oxidatively modified in AD. 150 The other compensatory enzyme is NOX, which occurs in almost all tissues and consists of cytosolic and membrane bound subunits. 41,151 The cytosolic subunits translocate to the membrane upon activation, allowing for electron transport, the formation of superoxide and regeneration of NAD(P)+. 41,142,151 Indeed, NOX expression and/or activity is elevated in AD. 41,55,56,58,59,152,153 A significant drawback of the NOX pathway is that it generates significant amounts of reactive oxygen species 25,142,151,153 (Figure 2).
While the previous two processes discussed require glucose, the third utilizes ketone bodies and FFA when glucose levels or utilization are decreased. The reduced brain glucose levels necessitates a bioenergetic switch of nutrient utilization from glucose to FFA or ketone bodies. 95 Thus, we suggest that this part logically occurs last. There is evidence for such changes in vivo. In AD mouse models there is a shift from use of glycolytic energy to use of ketone bodies. 14 Additionally, enzymes involved in ketone body formation (succinate and malate dehydrogenases) are elevated in AD. 98,146 Furthermore, the oversupply of FFA, which occurs during insulin resistance, may overwhelm β-oxidation and the TCA cycle and increase free radical generation from the mitochondria as occurs in muscle tissues. 154 This use of ketone bodies may also provide some amelioration of AD as they can overcome local insulin resistance and inhibition of pyruvate dehydrogenase entry into the TCA cycle. 155 Indeed, such findings led to the evaluation of a ketogenic diet as a therapeutic treatment for AD. 93
Thus, insulin resistance contributes to a switch in metabolites in AD, a change which likely might not satisfy neuronal energy requirements. The hyperdependence on glycolysis and ketone bodies for fuel suggests that mitochondrial dysfunction contributes to insulin resistance, oxidative stress and Aβ accumulation in AD.
Mitochondrial dysfunction and AD
Impairment occurs to all five of the mitochondrial oxidative phosphorylation complexes in the AD brain. 148 Mitochondrial dysfunction, which is associated with metabolic dyshomeostasis and reduced ATP synthesis, occurs early in AD. 44,147,156,157 Additionally, mitochondrial dysfunction is proposed to link amyloid deposition and neuronal and synaptic loss. 156 Thus, the existence of mitochondrial dysfunction is important in AD. Metabolites, oxidative damage and Aβ all affect mitochondria. Oxidative damage and FFA load may increase mitochondrial dysfunction in the brain, as it does in skeletal muscle, 154 resulting in elevated reactive oxygen species production and exacerbation of insulin resistance. 154 Also, Aβ directly interacts with several mitchondrial proteins such as amyloid-binding alchohol dehydrogenase (ABAD) and may inhibit ABAD's detoxification action. 44 ABAD detoxifies aldehydes such as hydroxy-2-nonenal, which are reactive and toxic products of lipid oxidative stress. 44 It is clear that mitochondria, which contain the enzymes for the TCA and oxidative phosphorylation pathways discussed above, directly contribute to metabolic dysfunction in AD (Figure 2).
Thus, we have a scenario of sequential changes in metabolic pathways in AD. There is the initial response to oxidative stress in the PPP. Following damage to oxidative phosphorylation in the mitochondria, alternate pathways (lactate dehydrogenase and NOX) generate NAD+ for glycolysis and the TCA pathway. Lastly, with insulin resistance and an inability to utilize glucose, there is a metabolite switch from glucose to ketone bodies. The resulting changes greatly reduce ATP yield. While numerous publications support each of the proposed steps, further experiments are required to validate the speculated sequence of events.
Test of hypothesis – pharmacological inhibition of metabolism
Does metabolism play a role in AD genesis and progression? One way to answer this question is to directly inhibit metabolism using specific pharmacological agents. Several compounds are available which alter different parts of the various metabolic pathways and affect AD as shown in Table 1.
Pharmacological inhibition of metabolic pathways increases Aβ pathology
Aβ, amyloid β; TCA, tricarboxylic acid
Pharmacological inhibition of metabolism does mimic aspects of AD pathology. Several metabolic inhibitors have been used to pharmacologically alter metabolism to study Aβ production or the contribution of metabolic deficiency to pathology in AD mouse models. For example, pharmacological inhibition of metabolism by 2-deoxyglucose, 3-nitroproprionic acid, kainic acid, azide, iodoacetate, rotenone, 3-bromopyruvate, 3-nitropropionic acid, malonate or thiamine deficiency caused increases in β-secretase activity, Aβ accumulation and enhancement of Aβ toxicity. 146,158–163 This occurs possibly as a result of oxidative stress induced increases in both β and γ secretases. 159,164–167 Quinolinic acid, which is elevated in AD and reduces energy metabolism, 168 may also contribute to AD pathology and should also be evaluated in AD models. Thus, metabolic inhibition is amyloidogenic and leads to AD pathology. 161
It can be argued that reduced mitochondrial activity also occurs during caloric restriction, and, unlike the pharmacological treatment above, caloric restriction is not associated with deleterious metabolic function. Numerous adaptive changes occur with caloric restriction; indeed, some of the altered factors are associated with longevity. However, with caloric restriction there is no metabolic or mitochondrial dysfunction 169 as occurs with pharmacological inhibition. This point can be further stressed by a study where results were contrary to the expected; genetic ablation of a mitochondrial enzyme (Cox 10 specific knockout) actually reduced both Aβ plaque load and oxidative stress. 170 These data suggest that mitochondrial COX10 activity does not initiate pathology, and that oxidative stress is important in regulating APP processing and thus Aβ generation. 170 This conclusively demonstrates that mitocohondrial deficits are complex and require more study. In the above transgenic case, it could be that the continual stress occurring since birth resulted in compensatory mechanisms. In other cases, dysfunction resulting in pathological elevation of Aβ occurred following stress (insulin resistance, metabolic changes such as brain insulin resistance) occuring with old age, or sudden pharmacological inhibition of metabolism.
Metabolic diseases and treatments
Metabolic dysfunction
We have reviewed direct tests demonstrating that metabolic changes contribute to AD. Another way of evaluating the contribution of metabolism to AD is to analyze metabolic diseases. Specifically, we review how drugs for metabolic diseases fit into the framework of metabolic dysfunction.
Insulin resistance
The contribution of insulin resistance to metabolic dysfunction and AD has been discussed in detail above. Insulin resistance, along with glucose dyshomeostasis, are features of metabolic diseases such as diabetes and obesity, and are associated with an increased risk of AD. 92,96,123,171–173 Indeed, these metabolic diseases, induced by high fat/sucrose diets or streptozotocin, directly increase the Aβ pathology in AD mouse models. 77,92,95,96,101,174–177 One of the primary anti-diabetic drug classes, PPAR agonists, increase sensitivity to insulin thus enhancing glucose utilization. 61,178 Importantly, PPARγ agonists improve cerebral glucose utilization and blood flow, 61 as well as increase brain mitochondrial metabolic efficiency (Figure 2) and number. 179 Additionally, PPARγ agonists may also enhance IDE and Aβ clearance 180 and thus reduce accumulation of Aβ.
However, PPARγ agonists appear to be more effective in early stages of AD (mild cognitive impairment and early AD) and not in the later stages. 34 This suggests that efficacy occurs within a narrow therapeutic window. Importantly, it indicates that insulin resistance may be involved in the genesis and early progression of AD. For example, PPAR drugs appear ineffective in AD with longstanding insulin resistance, 56,61,181 coinciding with the inception of insulin resistance. 181 This suggests that PPAR agonist restoration of glucose homeostasis is efficacious only within a window of opportunity when glucose is still used as a substrate. With prolonged insulin resistance, later in the disease, glucose homeostasis is likely irrelevant as other substrates such as ketone bodies are used instead of glucose. Thus it is likely that targeting insulin resistance as an early stage intervention will be most effective.
Conclusions
We have outlined how metabolic and vascular dysfunctions contribute to AD in the text and in Figures 1 and 2. This is of importance as undiagnosed AD can occur several years prior to clinical diagnosis. Metabolic and vascular dysfunctions are prominent features of AD, with changes occurring before AD pathology. This is supported by in vivo imaging as well as biochemical and proteomic studies.
This review demonstrates that metabolic and vascular dysfunctions contribute to and exacerbate AD pathology. Vascular dysfunction may represent an early or precedent pathology that, when present, exacerbates AD. Vascular disease is mediated via oxidative damage and Aβ-mediated damage. Metabolic dysfunction is mediated by oxidative stress and insulin resistance, with alterations in the PPP, TCA and oxidative phosphorylation pathways. Since metabolic and vascular dysfunctions are early features of AD pathogenesis, early treatment or therapeutic intervention may have profound effects on the initiation and/or progression of AD.
Footnotes
ACKNOWLEDGEMENTS
We thank Andre Okoreeh and Drs Mendel Rimer, Warren Zimmer and George Perry for critical reading of the manuscript. We also thank Kara Benson for helpful discussions. IVJM is funded by Departmental Startup funds. JML is funded by NIH (AR054084), NSF (055185F) and American Heart Association (0855158F).