
Abstract
Cell adhesion, mediated by N-cadherin, is critical for embryogenesis since N-cadherin-null embryos die during mid-gestation with multiple developmental defects. To investigate the role of N-cadherin in heart muscle development, N-cadherin was specifically deleted from myocardial cells in mice. The structural integrity of the myocardial cell wall was compromised in the N-cadherin mutant embryos, leading to a malformed heart and a delay in embryonic development. In contrast, cardiac-specific deletion of αE-catenin, found in adherens junctions, or β-catenin, did not cause any morphological defects in the embryonic heart, presumably due to compensation by αT-catenin that is normally found in intercalated disks and γ-catenin (plakoglobin), respectively. Embryos lacking β-catenin in the heart also exhibited a cardiac defect, but only later in development resulting in partial lethality. These genetic studies underscore the importance of the N-cadherin/catenin complex in cardiogenesis.
Introduction
The structural integrity of the heart is necessary for its function and is maintained by the intercalated discs (IDs) that comprise the end-to-end connections between myocytes. IDs consist of three main junctional complexes: adherens junctions (AJs), gap junctions and desmosomes. Each of these junctional complexes is extremely important for maintenance of normal mechanical and electrical coupling between cardiomyocytes, and therefore for normal heart function.
In the heart, the AJs are comprised predominantly of N-cadherin, 1 which is not only highly expressed in the developing and mature myocardium 2 but is also found in extrajunctional sites where it co-localizes with α-actinin in the peripheral Z-disks of the sarcomeres. Classical cadherins are transmembrane proteins that mediate specific cell–cell adhesion. 3 At the cytoplasmic side of the junction, either β-catenin or plakoglobin can interact directly with a core region within the C-terminus of the cadherin cytoplasmic domain. The N-terminal portion of β-catenin or plakoglobin interacts with α-catenin, which links this complex to the cytoskeleton.
Intercellular adhesion is important for the development of any multicellular organism. 4,5 The requirement for E-cadherin and αE-catenin during early embryonic development has previously been reported. 6,7 Both of these proteins are necessary for blastomere adhesion during embryonic compaction, and their absence or compromised function results in embryonic lethality. The complete loss of another cadherin family member, N-cadherin, in the mouse embryo leads to embryonic lethality at mid-gestation. This results from multiple embryonic defects including severe cardiovascular anomalies. 8
As previously described, 4,9,10 the early embryonic phenotypes of E-cadherin-, N-cadherin- and αE-catenin-deficient mutants appear to compromise intercellular adhesion. The phenotypes of β-catenin-null embryos seem to depend on Wnt signaling and may be important not only for embryos and heart development but also for adult cardiomyocyte growth. Homozygous loss of β-catenin leads to embryos with head structure and heart developmental defects. 9–11
Plakoglobin (γ-catenin) is a close relative of β-catenin. 12 In contrast to β-catenin, it can interact with both classical cadherins in AJs and desmosomal cadherins in desmosomes. Thus, plakoglobin loss-of-function experiments have primarily revealed a structural role for this protein. Mouse embryos that lack this protein die in utero, exhibiting defects in cardiac structural integrity and skin blistering. 13,14 Interestingly, plakoglobin is able to maintain the structure of cadherin–catenin complexes after β-catenin loss, and there is some evidence that plakoglobin can participate in Wnt-signaling. 15,16
We have previously demonstrated that ablation of N-cadherin in adult mouse heart leads to lethality within two months after N-cadherin deletion. 17 The loss of N-cadherin resulted in disassembly of the ID structure including AJs and desmosomes. The mutant mice exhibited modest dilated cardiomyopathy and impaired cardiac function. Furthermore, loss of N-cadherin in the adult heart leads to a significant decrease in the gap junction protein connexin 43 (Cx43), which appears to perturb the generation and propagation of electrical impulses. 17 There is a clear relationship between N-cadherin and Cx43 in the heart, which manifests even when N-cadherin is reduced in the heart due to haploinsufficiency, by affecting cardiac gap junctions and susceptibility to arrhythmia. 18
Ablation of β-catenin in the adult heart does not result in cardiomyopathy, presumably due to compensation by plakoglobin. 19 Loss of αE-catenin in adult myocardium does not result in immediate death of the animal immediately after Cre-mediated gene deletion. 20 However, after about 32 weeks following αE-catenin deletion, there are morphological changes in heart tissue, which lead to heart cardiomyopathy. 20 Intercellular connections are clearly important for maintaining normal function of the adult heart. These connections involve critical roles for N-cadherin and its cytoplasmic partners, the catenins, in the formation and functioning of AJs in the adult myocardium.
While the importance of AJs for normal adult heart function is established, the roles of the catenins and cadherins during embryonic development of the heart are less obvious. In this study we have addressed the roles of N-cadherin, αE- and β-catenin function during heart development using Cre-mediated tissue-specific ablation of each of these components in vivo in mice. We performed morphological and immunological analyses of heart tissues at different stages of gestation after deletion of either N-cadherin, αЕ-catenin or β-catenin.
Materials and methods
Generation of mutant embryos
To generate cardiac-specific deletion of N-cadherin N-cadflox/+, α-myosin heavy chain (αMHC)-Cre mice were mated with N-cadflox/flox, Rosa26 reporter R26R/R26R mice and embryos were recovered from timed pregnancies. The embryos harboring a homozygous floxed N-cadherin gene, αMHC-Cre transgene, along with a reporter ROSA gene were designated as mutant and used for the analysis; all other genotypes served as controls. Mice were bred on mixed background. Embryonic heart-specific deletion of β-catenin and αE-catenin was achieved using the same breeding scheme, and appropriate mice carrying floxed β-catenin or αE-catenin genes were selected. αMHC-Cre mice were kindly provided by Dr Michael Schneider, Baylor College of Medicine, Houston, TX, USA; this αMHC-Cre transgene elicited recombination in cardiac muscle, but not other organs. 21 R26R reporter mice, which may be used as a Cre-reporter strain to test the tissue/cellular expression pattern of cre transgenic mice 22 and homozygous conditional αE-cateninflox/flox and β-cateninflox/flox mice, were obtained from Jackson Laboratories (Bar Harbor, ME, USA).
Embryo genotyping
Yolk sac tissues after embryo recovery were used for genotyping. DNA isolation and polymerase chain reaction (PCR) analysis were carried out according to the standard protocols. 23 The following primer sets were used to discriminate floxed, mutant and wild-type alleles: Ncad, 5′ TACAGTTTGGGTGACAAGC 3′ and 5′ CCAAAGCTGAGTGTGACT 3′; αE-catenin, 5′ CATTTCTGTCACCCCCAAAGACAC 3′ and 5′ GCAAAATGATCCAGCGTCCTGGG 3′; and β-catenin, 5′ AGGTAGAGTGATGAAAGTTGTT 3′ and 5′ CACCATGTCCTCTGTCTATTC 3′. The αMHC-Cre transgene was identified with primers 5′ CAGAACCTGAAGATGTTCGC 3 and 5′ TACACCTCGGTGCTAACCAG 3′.
Morphological and molecular analysis of mutant embryos
Whole mount X-gal staining of embryos was performed as previously described. 24 Paraffin embedding and sectioning was carried out according to the standard protocol. Frontal sections through the heart regions of E10.5, E12.5 and E14.5 embryos were analyzed using light microscopy. Indirect immunofluorescence analysis was performed on paraffin sections of embryos as previously described. 25 Sections were incubated with the following primary antibodies in manufacturer-recommended dilutions: N-cadherin, β-catenin and αE-catenin, followed by incubation with Cy3-conjugated goat anti-mouse antibody and visualized using a confocal microscope. All antibodies except αT-catenin antibody were purchased from Zymed Laboratories (South San Francisco, CA, USA). αT-catenin antibody was kindly provided by Dr Frans van Roy from VIB, Ghent, Belgium.
Results
Cardiac-specific knockout of N-cadherin results in embryonic lethality
To investigate the role of N-cadherin in cardiogenesis, mice harboring a floxed N-cadherin gene 17 were crossed with αMHC-Cre transgenic mice to delete N-cadherin specifically from the myocardial cell lineage. The αMHC promoter is considered to be cardiac specific and known to be expressed in embryonic heart from the early heart tube stage till approximately E16. 26 In addition, the R26R Cre reporter was crossed into the N-cadherin floxed background to confirm the specificity and efficiency of the cardiac-specific deletion. To follow-up N-cadherin function during early cardiogenesis, we utilized embryos at 10.5 d of gestation. Using X-gal staining to detect bacterial β-galactosidase, we have demonstrated that the αMHC promoter drives Cre-dependent deletion in a tissue-restricted manner (Figure 1). The staining associated with deletion of the reporter gene was detected only in embryonic heart and not in other tissues.

α-Myosin heavy chain-Cre transgene activity is restricted to the embryonic myocardium. Cre recombinase activity was monitored using the R26R lacZ reporter allele. Transverse sections of wild-type (a) and N-cadherin CKO (b) X-gal stained E10.5 embryos demonstrated specific lacZ reporter activity in the embryonic heart (arrows) (A color version of this figure is available in the online journal)
Ablation of N-cadherin using αMHC-Cre resulted in embryonic lethality at mid-gestation due to a severe cardiac defect. The morphology of whole mount embryos was studied at embryonic days E10.5. PCR analysis was performed on all embryos to reveal their genotypes. As expected, about 25% of the embryos had an αMHC-Cre+, N-cad Flox/Flox, R26R+ (designated N-cad conditional or CKO) genotype. The N-cad CKO embryos were developmentally delayed with malformed hearts that were swollen, showing a balloon-like morphology (Figure 2). Cardiac morphogenesis was incomplete and the extraembryonic circulatory system was poorly developed in the N-cadherin CKO embryos. After embryonic stage E10.5, hearts were unrecognizable and the embryos were in the process of being resorbed. Transverse sections through the heart region of mutant embryos and control embryos at E10.5 (Figure 2) confirmed the cardiogenesis after N-cadherin ablation, with thinner myocardial walls than those of controls and compromised intercellular adhesion (Figure 2f).
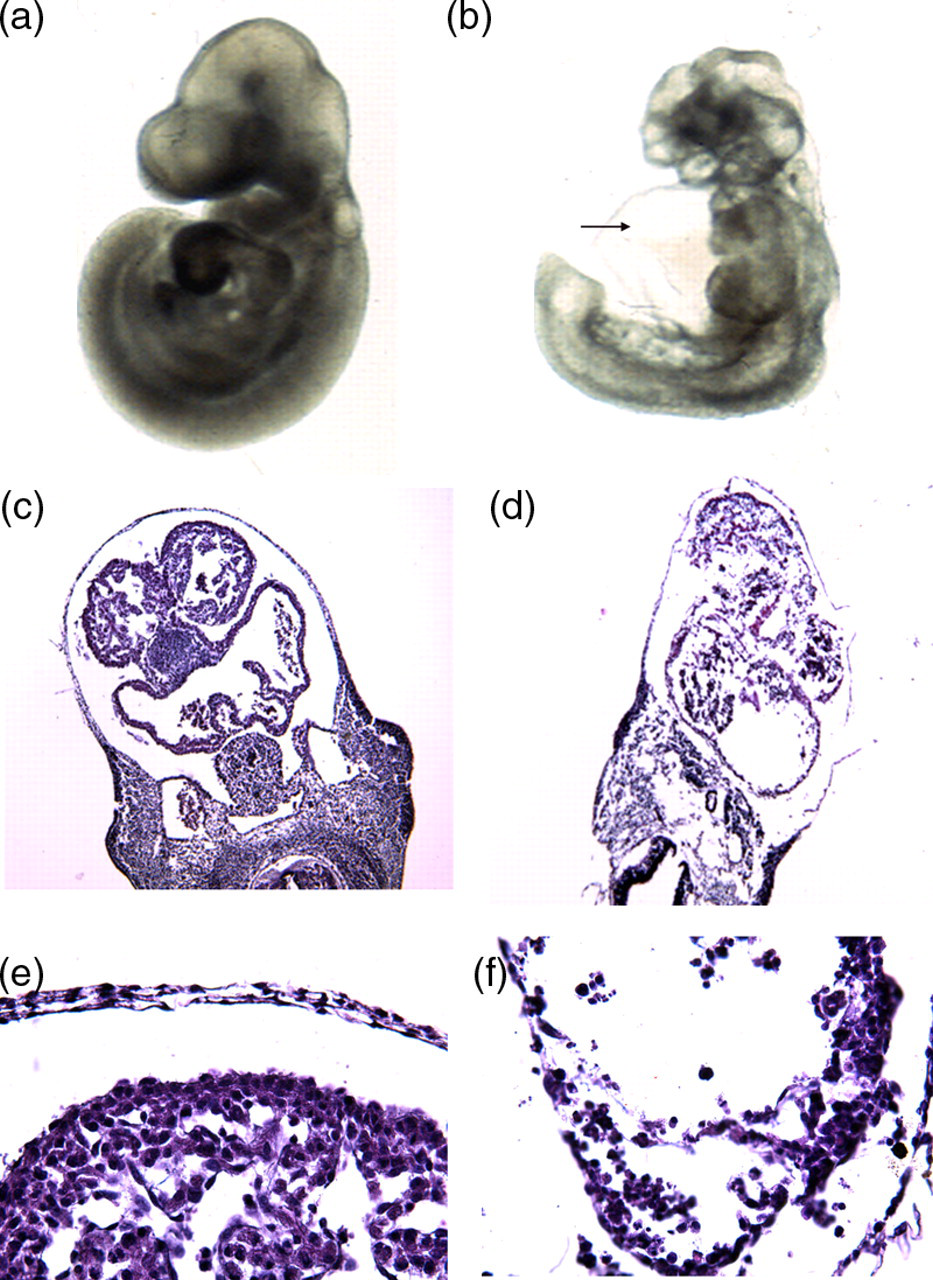
Cardiac-specific deletion of N-cadherin gene leads to cardiomyocyte adhesion defect and embryonic lethality. Whole mount images of wild-type (a) and N-cadherin CKO (b) E10.5 embryos. Note the malformed heart and pericardial edema (arrow) in the mutant embryo. Histological analysis of wild-type (c, e) and N-cadherin CKO (d, f) E10.5 embryos. Note the less compact myocardial cell layer (arrow) in the mutant embryo (f) (A color version of this figure is available in the online journal)
Embryos lacking N-cadherin were immunostained with N-cadherin and β-catenin antibodies at embryonic stage E10.5. As expected, N-cadherin was missing from the heart tissue. Significantly, there was no detectable β-catenin in the N-cadherin-deficient myocardium (Figure 3). These observations support the proposition that N-cadherin plays a critical role in the regulation of β-catenin levels during early cardiogenesis and that N-cadherin may also affect the distribution of β-catenin, especially during the formation of intercalated disks. This analysis confirmed our suggestion that deletion of the N-cadherin gene selectively in heart tissue at early gestational times has a critical role for embryonic development of mice.

N-cadherin/catenin expression in the embryonic myocardium. Immunofluorescence was performed on wild type (a, c) and N-cadherin CKO (b, d) E10.5 embryos to examine N-cadherin (a, b) and β-catenin (c, d) expression in the embryonic heart. N-cadherin was depleted from the myocardium of the N-cadherin CKO embryo (b), confirming the specificity of the αMHC-Cre transgene. N-cadherin is the only classical cadherin expressed in myocardium; therefore, β-catenin was no longer present at the plasma membrane in the absence of N-cadherin (d) (A color version of this figure is available in the online journal)
Deletion of αE-catenin in the embryonic heart does not affect N-cadherin expression
Conditional deletion of αE-catenin was used to assess its function in the developing heart. Embryos were examined by immunofluorescence at embryonic stages E10.5, E12.5 and E14.5. The mutant embryos did not differ from their normal littermates based upon gross morphology. Immunohistochemical analysis with αE-specific antibody confirmed the absence of αE-catenin in the developing myocardium of mutant mice (Figures 4a and d). Similarly, N-cadherin (Figures 4c and f) was not affected in mutant cardiomyocytes. The ablation of αE-catenin in embryonic heart, however, did affect αT-catenin by elevating its level of expression. Transverse sections through the heart region of mutant embryos also showed no morphological differences compared with control littermates at E14.5 (Figure 5b) or earlier stages of gestation. Our data suggest that αE-catenin in the heart is dispensable, probably due to compensation by αT-catenin for its function in AJ formation in the developing, and possibly in the adult, myocardium.

Loss of αE-catenin does not affect N-cadherin expression in the heart. Immunofluorescence was performed on wild-type (a, b, c) and αE-catenin CKO (d, e, f) E10.5 embryos to examine αE-catenin (a, d), αT-catenin (b, e) and N-cadherin (c, f) expression in the embryonic heart. The loss of αE-catenin is likely compensated by increased αT-catenin at the plasma membrane (e). The expression of N-cadherin did not change in the αE-catenin CKO heart (f), consistent with the lack of a cell adhesion defect (A color version of this figure is available in the online journal)
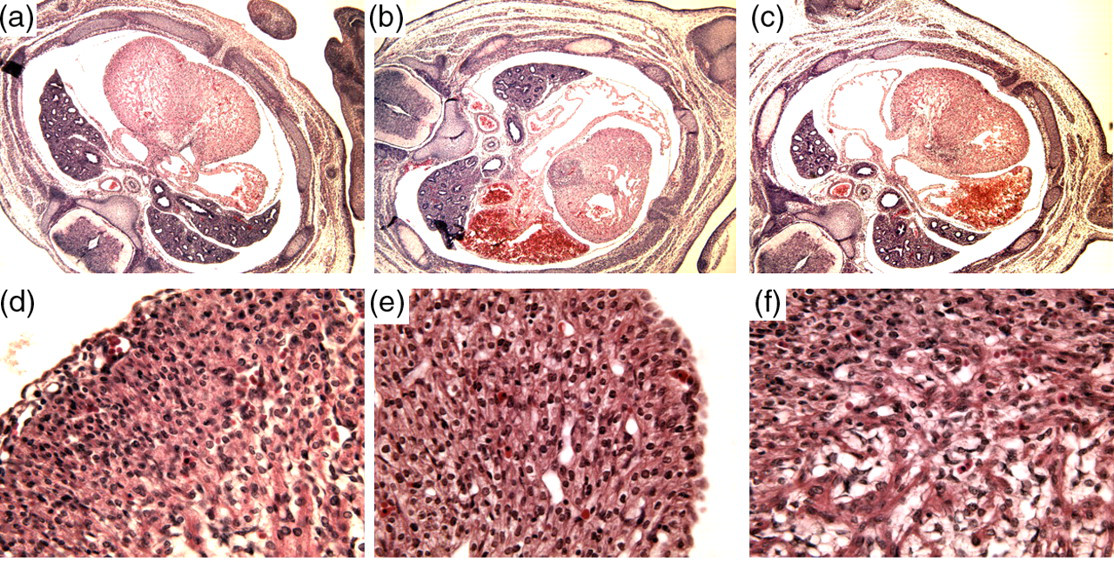
Morphological analysis of transverse heart sections of mutant and control embryos at Е14.5. Hematoxylin and eoxin staining was performed on wild type (a, d), αE-catenin CKO (b, e) and β-catenin CKO (c, f) embryos. Morphological appearance of embryonic myocardium in mutant heart does not differ from control (A color version of this figure is available in the online journal)
β-Catenin is dispensable for development of the embryonic heart
When mice harboring a Cre recombinase transgene under control of an αMHC-Cre promoter were crossed with mice containing a floxed β-catenin allele, the F1 embryos appeared morphologically normal at embryonic stages E10.5 and E14.5 (Figure 5c). To confirm that Cre-mediated deletion of β-catenin gene produces a null phenotype, embryonic heart sections were immunostained with antibodies to β-catenin and N-cadherin. Immunohistochemical analysis confirmed the absence of β-catenin in the developing myocardium of mutant mice (Figures 6a and c) and also demonstrated that N-cadherin (Figures 6b and d) was not affected in β-catenin-deficient cardiomyocytes. Thus, the absence of β-catenin does not perturb N-cadherin expression in the embryonic heart, probably due to functional redundancy between β-catenin and plakoglobin in the formation of AJ formation during early cardiogenesis.
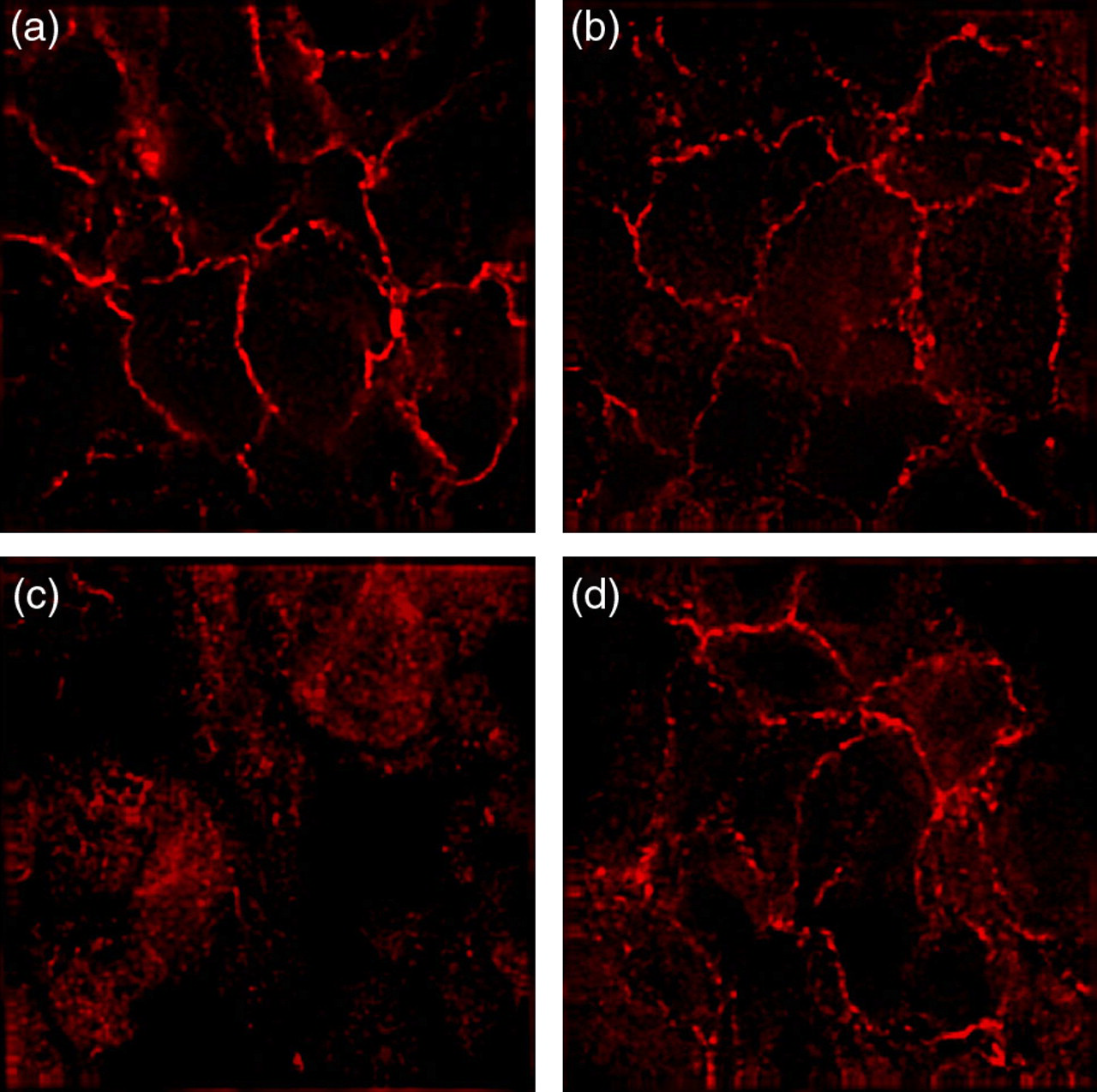
Loss of β-catenin does not affect N-cadherin expression in the heart. Immunofluorescence was performed on wild type (a, b) and β-catenin CKO (c, d) E10.5 embryos to examine β-catenin (a, c) and N-cadherin (b, d) expression in the embryonic heart. The expression of N-cadherin did not change in the β-catenin CKO heart (d), consistent with the lack of a cell adhesion defect (A color version of this figure is available in the online journal)
Analysis of newborn mice
Genotyping of newborns revealed that deletion of the αЕ-catenin gene does not statistically affect the viability of the mice (Table 1). After crossing αЕ-cateninflox/+, αMHC-Cre mice with αЕ-cateninflox/flox mice, we found that 20% of all newborns have an αMHC-Cre+, αE-cateninflox/flox genotype (CKO) which is expected based on Mendelian inheritance.
The affect of cardiac-specific target gene ablation on embryo and newborn survival
Embryos were analyzed at embryonic day 10.5 or 14.5; newborn mice at third–fourth day postnatally. Number in parentheses indicates abnormal/dead animals
*Dead newborn from control group was a β-catenin heterozygote
In contrast, genotyping of β-catenin newborn mice after similar crosses revealed that αMHC-Cre+, β-cateninflox/flox mice were under-represented, suggesting that some mice were dying in utero, probably at late gestation (Table 1). Thus, only 10% of mice born had a genotype of αMHC-Cre+, β-cateninflox/flox, and one of the mutants died shortly after birth (postnatal day 3) (Table 1). These data suggest that β-catenin is disposable during early heart development, probably due to the functional redundancy between β-catenin and plakoglobin. But this functional redundancy is not fully efficient since we observed partial lethality of embryos after β-catenin ablation and low survival of β-catenin CKO newborn mice.
Discussion
Different cadherins are found in the AJs in various cell types, and cardiomyocytes in particular contain only N-cadherin to form myocardium. By using the Cre technique, we found that deletion of N-cadherin in the developing heart leads to embryonic lethality at mid-gestation (around E9.5). As a result, cardiomyocytes dissociate in the primitive heart without the formation of the functional heart tube. This phenotype resembles, in part, heart malformations caused by complete N-cadherin loss, 8 but other systems and organs in the developing embryo remain unaffected in the cardiac-specific N-cadherin knockout. The lethal phenotype resulting from N-cadherin ablation in adult heart has been previously shown. 17 Loss of N-cadherin in the adult heart results in severe perturbations of AJs and disassembly of the ID structure, causing cardiomyopathy and sudden cardiac death.
The delay of embryonic development after loss of N-cadherin, the violation of the cardiovascular system, the disruption of intercellular adhesion and the lethality of mutant embryos confirm the central role of N-cadherin in the development of cardiac pathologies. The data obtained from chimeric embryos derived from N-cadherin-deficient embryonic stem cells support this idea, since N-cadherin-null cardiomyocytes do not participate in the formation of the myocardial wall. 24 Taken together, N-cadherin-mediated adhesion is required for myocardial cell–cell adhesion in the developing heart tube as well as the adult myocardium.
The cell–cell AJ is a plasma membrane structure composed of transmembrane cadherins associated directly with either β-catenin or plakoglobin, which in turn associate with α-catenin. A germline knockout of β-catenin is embryonically lethal, 9–11,27 so the role of β-catenin in cardiomyocyte differentiation and early heart development could only be studied using a conditional knockout approach. To do this, we have deleted β-catenin specifically in the developing heart by crossing loxP-flanked β-catenin mice with transgenic mice expressing Cre protein driven by an αMHC promoter. According to our data, the cardiac-specific loss of β-catenin during embryonic development did not cause morphological malformations of the heart or violation of the cardiovascular system. These data are in complete agreement with earlier findings, 19,28 which demonstrated that deletion of β-catenin in the adult heart results in unchanged intercalated disks with normal distribution of vinculin, N-cadherin, desmoplakin, ZO-1, Cx43, α- and γ-catenins. The expression level of these proteins except plakoglobin is also similar in control and mutant hearts. According to Western blot analysis, the γ-catenin concentration is increased in β-catenin-deficient myocardium and this extra plakoglobin is predominantly associated with IDs, suggesting functional redundancy between β- and γ-catenins. Up-regulation of γ-catenin in β-catenin knockouts may compensate for the absence of β-catenin to maintain normal cardiac structure and function.
β-Catenin and plakoglobin are both members of the Armadillo family of proteins and share about 65% identity. 29 Although these two proteins can directly substitute for each other as structural components of the AJ, they likely play very different roles in cellular signaling pathways. β-Catenin is the major component of the Wnt/Wingless signal transduction pathway, and it is involved in segmental polarity and pattern formation during development. 27,30 Conditional inactivation of β-catenin in endothelial cells prevents endothelial–mesenchymal transformation during cardiac development. 11,27 In our experiments we have not seen any changes in embryonic development after early heart specific deletion of β-catenin, suggesting the functional substitution of plakoglobin for β-catenin function(s). Nevertheless, under-representation of newborns with phenotype Cre + , β-catenin flox/flox means these two proteins are not functionally equal, especially in late embryogenesis. The role of β-catenin in late cardiogenesis remains to be elucidated. But we can speculate that heart-specific ablation of β-catenin negatively influences the survival of newborn mice and can quite probably lead to heart disease development at postnatal period.
α-Catenins also play a key functional role in the cell–cell adhesion complex, mediating the connection of the cadherin–catenin complex to the actin cytoskeleton. Thus, the disruption of this anchoring link should have a devastating effect on intercalated disk formation and function. Nevertheless, in our experiments, the heart-specific deletion of αE-catenin did not appear to affect cardiogenesis and overall embryonic development. This may be another case of functional redundancy among proteins of the adhesion complex, particularly between αE- and αT-catenins, considering that both of them are expressed in cardiomyocytes and are colocalized in IDs. Interestingly, heart-specific ablation of αE-catenin using MLC2v-Cre leads to defects in cardiomyocyte structural integrity, causing cardiomyopathy and heart wall rupture under stress conditions. 20 The difference in the embryonic and adult phenotype suggests the different roles αT-catenin plays in the developing and mature heart. Most likely, in adult myocardium, αT-catenin cannot fully substitute for the loss of αE-catenin as seen in the work of Sheikh et al. 20 Another possibility may be that we are missing some minor developmental anomalies in our analysis that may eventually lead to heart disease development and/or progression.
In summary, we can conclude that perturbation of AJs in the developing heart has a dramatic impact on the proper organization of cardiovascular system. While N-cadherin deletion results in severe cardiac malformations and is embryonically lethal, the loss of cytoplasmic partners of N-cadherin (αЕ-catenin or β-catenin) does not lead to the development of embryonic heart abnormalities at early terms of gestation, presumably due to the functional compensation by other catenin family members. Nevertheless, the life span of mice with developmentally deleted α- or β-catenin genes and their susceptibility to the development of cardiac pathologies is an open question and remains to be elucidated.
Footnotes
ACKNOWLEDGEMENTS
We thank the University of Pennsylvania Biomedical Imaging Core Facility for performing the confocal microscopy analysis. We also thank Dr Michael Schneider from Baylor College of Medicine for providing the αMHC-Cre animals and Dr Frans van Roy from VIB for providing αT-catenin antibody. This study was supported by CRDF (Award number: UK-B2-2577-KV-04).