
Abstract
Hepatic fibrosis, which is characterized by progressive inflammation and deposition of extracellular matrix components, is a common response to chronic liver disease. Hepatic fibrogenesis is a dynamic process that involves several liver cell types including hepatic stellate cells and Kupffer cells. In addition, recent evidence indicates that bile duct epithelial cells (i.e. cholangiocytes) also participate in the progression of biliary fibrosis that is observed during chronic cholestatic liver diseases, such as primary sclerosing cholangitis. To date, there are no effective treatments for hepatic fibrosis. Several recent studies have demonstrated that the renin–angiotensin system (RAS) plays a key role in hepatic fibrosis. Therapies targeting the RAS may represent a promising paradigm for the prevention and treatment of hepatic fibrosis in the setting of chronic liver disease. In this review, we provide a comprehensive update on the role of RAS in the pathogenesis of hepatic fibrosis in both animal models and human studies. We will discuss the profibrotic mechanisms activated by the RAS and the cell types involved. Studies that have utilized angiotensin receptor blockers (ARBs) and angiotensin-converting enzyme (ACE) inhibitors to modulate the RAS in order to ameliorate hepatic fibrosis will also be discussed. Although the cumulative evidence supports the potential for the use of ARBs and ACE inhibitors as treatment for hepatic fibrosis, extensive studies of the effectiveness of RAS therapeutics are necessary in patients with chronic liver disease.
Introduction
Hepatic fibrosis is a dynamic wound healing mechanism that occurs in response to liver injury during the pathogenesis of chronic liver diseases. Hepatic fibrosis is characterized by progressive inflammation, the involvement of several liver cell types and the activation of multiple signaling mechanisms that result in the deposition of extracellular matrix (ECM). 1–3 To date, there is not an effective therapy for hepatic fibrosis despite the fact that chronic liver diseases affect hundreds of millions of people worldwide. 4 In addition, once fibrogenesis has progressed to cirrhosis during chronic liver diseases, there is increased risk of developing hepatocellular carcinoma (HCC). 4–6 Recent evidence in animal models and clinical studies indicate that the renin–angiotensin system (RAS) plays a major role in the progression of liver fibrosis. 7–9 The key components of the RAS are expressed in the liver during chronic injury and activated hepatic stellate cells (HSCs) de novo produce angiotensin II (Ang II), which is the major biologically active peptide of the RAS. 10 In animal studies, lack of angiotensin receptor type 1 (AT1), administration of angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers (ARBs) attenuate the progression of fibrosis during chronic liver injury, highlighting the importance of the RAS in the pathogenesis of hepatic fibrosis. 11–18 In this review, we discuss the current aspects of the RAS and its putative role in the pathogenesis of liver fibrosis. We will also discuss promising avenues for future studies that may lead to the development of therapeutics that modulate the RAS for the prevention and treatment of hepatic fibrosis.
Overview of hepatic fibrosis
The activated HSC has been identified as the primary cell type responsible for the deposition of excess extracellular matrix in response to chronic liver injury. 2,3,19,20 The current paradigm of the fibrogenic cascade is a multifactoral process that involves: (1) activation of HSC and Kupffer cells (KC); (2) migration and proliferation of HSCs; (3) synthesis and deposition of ECM components; (4) wound contraction; and (5) apoptosis of HSC. 2,3,19 A number of cytokines and growth factors have been shown to play a role in the activation of HSCs, such as platelet-derived growth factor (PDGF), transforming growth factor beta (TGF-β), tumor necrosis factor-α (TNF-α), insulin-like growth factor (IGF-I) and endothelin-1 (ET-1), as well as reactive oxygen species (ROS). 21 However, PDGF and TGF-β are known to be the most potent stimuli for proliferation and fibrogenesis of HSCs, respectively. 3 The key mechanisms regulating HSC activation and the resultant hepatic fibrosis have recently been elegantly reviewed by several authors and will not be covered in this review article. 1–3,19,21
However, accumulating evidence suggests that proliferating cholangiocytes also play a key role in chronic cholestatic liver diseases that are characterized by biliary fibrosis. 22 Recent evidence indicates that proliferating biliary epithelium serves as a neuroendocrine compartment during liver disease pathogenesis, and as such secrete and respond to hormones, neurotransmitters and neuropeptides contributing to the autocrine and paracrine pathways that positively and negatively modulate liver inflammation, fibrosis and cholangiocarcinogenesis. 23 Activation of this neuroendocrine phenotype in cholangiocytes, 24 similar to that observed in HSCs 25 during liver fibrosis, may play a key role in the progression of biliary fibrosis during cholestatic liver diseases (such as, primary biliary cirrhosis [PBC] and primary sclerosing cholangitis [PSC]), which target cholangiocytes as evidenced by increased biliary proliferation and/or apoptosis in these disease states. 26,27 Several studies have demonstrated that proliferating cholangiocytes secrete profibrotic factors, such as, connective tissue growth factor (CTGF), TGF-β2 and hepatic ET-1, which indicates that proliferating cholangiocytes play a key role in the process of biliary fibrosis. 28–31
Recent, compelling evidence suggests that the RAS plays a multifactorial role in fibrogenesis in many organs including the liver. 32–34 HSCs and cholangiocytes both express components of the RAS, implicating the activation of this profibrotic system during the pathogenesis of liver diseases and the resultant liver fibrosis. 7,35 HSCs and cholangiocytes represent a viable target for the inhibition of the progression of liver fibrosis through modulation of the RAS.
Overview of endocrine and local RAS
The endocrine (or classical) RAS is known for modulating physiological functions that play an important role in the regulation of blood pressure, electrolyte balance and fluid homeostasis. 36 Both prorenin and renin (protease) secreted from the kidney cells react with angiotensinogen produced by the liver to release the decapeptide, Ang I, which is further cleaved by ACE that is released from capillaries of the lung to convert Ang I to the octapeptide, Ang II (Figure 1). 36–38 Ang II is considered the major physiologically active component of RAS.

The renin–angiotensin system (RAS). This schematic highlights the important participants both in the classical and alternative RAS. Renin cleaves the decapeptide, Ang I, from angiotensinogen, and Ang I is converted to Ang II by ACE. In the alternative pathway, Ang II can be converted to Ang (1–7) by ACE2. Several additional enzymes that can participate in the conversion of Ang I to Ang II are also shown. ACE, angiotensin-converting enzyme; Ang, angiotensin; AT1, angiotensin receptor type 1; AT2, angiotensin receptor type 2; CAGE, chymostatin-sensitive angiotensin II-generating enzyme; ARB, angiotensin receptor blocker
In recent years, studies have shown that many organs, such as the heart, kidney, liver and pancreas, constitutionally express all the ‘classical’ RAS components required for a functioning local RAS. 39–41 However, not all of the components of the classical RAS are synthesized locally in some tissues. 41 Several alternative enzymatic pathways for the formation of Ang II exist, which include chymostatin-sensitive Ang II-generating enzyme (CAGE), cathepsin G and chymase (Figure 1). 41 One of the functions of the local RAS is to amplify the effects of circulating Ang II especially in tissues that regulated cardiovascular control. 42 The expression of the local RAS may help explain the pleiotropic effects of RAS inhibitors. 42
The biological actions of Ang II are mediated by two seven transmembrane G-protein coupled receptors, the AT1 and AT2 (Ang II type 2 receptor). 36,43 Most of the physiological effects of Ang II have been attributed to AT1, which is widely distributed in all organs, including the liver, adrenals, brain, lung, kidney, heart and vasculature. 44 Activation of the AT1 receptor results in increased arterial tone, adrenal aldosterone secretion, renal sodium reabsorption, sympathetic neurotransmission and cellular growth. 13 Several recent studies have also shown that Ang II is involved in the key events of inflammatory processes. 14 AT1 and AT2 have distinct downstream targets that counteract the physiological actions of each other. 45 For example, activation of AT1 stimulates vasoconstriction and cell proliferation whereas AT2 mediates vasodilation, cell growth inhibition and activation of apoptosis (Figure 1). 45
The classic RAS model was challenged in the late 1980s with the discovery of Ang (1–7) and the determination of its diverse biological functions. 46–51 Subsequent studies identified several new components of the RAS, such as ACE2 that catalyzes the generation of Ang (1–7) and the G-protein-coupled Ang (1–7) receptor, Mas (Figure 1). 52–54 These new components, together with Ang (1–7), are now considered as the alternative pathway of the RAS. 55,56 In this alternative pathway, ACE2 converts Ang II to Ang (1–7) directly (Figure 1), and there is also an indirect pathway for the generation of Ang (1–7) from Ang I, which is not discussed here. 57,58 Activation of Mas has been shown to trigger vasodilation and antiproliferative signaling mechanisms in many cell types. 59,60
Expression of the RAS components in the liver
Several groups have examined the expression of classic RAS components in the liver in different animal models of hepatic fibrosis and in human tissue samples. Bataller et al. 61 demonstrated through elegant binding studies that HSCs express the AT1 receptor and that Ang II elicited a marked dose-dependent increase in intracellular calcium levels, cell contraction and proliferation. Paizis et al. 62 demonstrated that gene expression both for ACE and AT1 were upregulated by bile duct ligation (BDL) in total liver and were especially expressed in areas of the liver with active fibrosis. However, AT2 was not detected in normal or diseased liver. 62 In addition to HSC, KCs also expressed AT1 and Ang II stimulated the gene expression of TGF-β1 and fibronectin. 63 In human samples, the expression of AT1 as determined by immunohistochemistry was downregulated in hepatocytes, while expression was increased in HSCs, vascular endothelium and bile duct epithelium. 64 For the alternative ACE2/Ang (1–7)/Mas receptor axis, several groups have demonstrated that there was an increase in ACE2, Ang (1–7) and Mas receptor expression in BDL- and CCl4-induced models of rat liver fibrosis. 65,66 In healthy human livers, ACE2 expression was confined to endothelial cells, occasional bile ducts, and perivenular hepatocytes. 67 In BDL and human cirrhosis samples, there was widespread expression of ACE2 throughout the liver tissue. 67 Others observed a marked increase in the expression of ACE2 in Ang II-treated HSCs. 68 The major source of the Ang II precursor angiotensinogen is the hepatocyte. However, low levels of angiotensinogen have also been detected in KCs and in bile duct epithelium. 69,70 Bataller et al. 10 reported that in vivo activated human HSCs and culture-activated HSCs highly express active renin and angiotensin-converting enzyme and secrete Ang II to the culture media. It is also well known that plasma renin activity and aldosterone levels are elevated in patients with liver cirrhosis, in particular those with hepatorenal syndrome. 71–73 Two recent studies by Vilas-Boas et al. 74,75 have shown that Ang (1–7) is present in the peripheral and splanchnic circulation of patients with advanced liver disease. They reported that plasma renin activity and angiotensins were elevated in advanced liver disease compared with mild-to-moderate liver disease and that Ang (1–7)/Ang II ratios were higher in mild-to-moderate liver disease compared with controls and advanced liver disease. 75 Higher Ang (1–7)/Ang II ratios were observed in the splanchnic compared with the peripheral circulation, which suggest that alterations of Ang (1–7)/Ang II ratio may play a key role in hemodynamic changes of human cirrhosis. 75 Taken together, these findings clearly indicate that the liver possesses an active local RAS in the chronic liver disease setting that may play a key role in the pathogenesis of liver fibrosis. The balance between Ang (1–7) and Ang II may play a critical role during liver diseases pathogenesis. 8,55
Experimental evidence supporting the role of RAS in animal models of hepatic fibrosis
Numerous studies have clearly defined a role for the involvement of the RAS in the pathogenesis of hepatic fibrosis in animal models (Table 1). Chronic infusion of Ang II by subcutaneously implanted osmotic minipumps to normal rats for four weeks induced HSC activation and a slight increase in collagen deposition. 76 In addition, Ang II infusion induced oxidative stress, increased concentration of proinflammatory cytokines and upregulated the expression of inflammatory proteins, such as inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (Cox2). 76 In a similar fashion, chronic infusion of Ang II to rats with BDL augmented BDL-induced liver injury and increased hepatic levels of proinflammatory proteins, TNF-α and interleukin (IL)-1β. 35 Ang II infusion also increased hepatic TGF-β concentration, collagen deposition, accumulation of smooth muscle alpha-actin (α-SMA) positive cells and lipid peroxidation products. 35 The authors also reported that infusion with Ang II activated c-Jun and extracellular signal-regulated kinase (ERK)-1/2 phosphorylation and enhanced biliary proliferation. 35 In cultured HSCs, Ang II promoted the generation of ROS, cellular proliferation and secretion of proinflammatory cytokines. 35
Studies of the renin–angiotensin system (RAS) in animal models of chronic liver disease
HSC, hepatic stellate cell; BDL, bile duct ligation; Ang II, angiotensin II; TGF-β1, transforming growth factor β-1; CDAA, choline-deficient
Drug therapy started at the initial induction of the model of fibrosis unless otherwise indicated
A number of investigators have utilized both genetic and pharmacological approaches to elucidate the signaling mechanisms that regulate the effects of Ang II-induced fibrosis. Initial studies took the approach of inhibition of ACE and demonstrated that inhibition of Ang II formation could dramatically attenuate the progression of hepatic fibrosis in rats with BDL- and CCl4-induced fibrosis.
11,14
In two different rat models of liver carcinogenesis induced separately by diethylnitrosamine (DEN) and a choline-deficient
There is also considerable evidence supporting an antifibrotic role for Ang (1–7) in animal models of hepatic fibrosis. 32,65,67,89,90 The first evidence of the involvement of the alternative Ang (1–7) RAS pathway in liver fibrosis came from Paizis et al., 67 whom demonstrated that there was an upregulation of ACE2 and its expression was widespread throughout the liver in BDL-induced fibrosis in rats and in human hepatitis C cirrhosis. Herath et al. 65 also reported similar increases in ACE2 and Ang (1–7) expression in rats with chronic BDL. An upregulation of ACE2 expression was also observed in rats with CCl4-induced hepatic fibrosis. 66 In support of the potential protective effects of Ang (1–7), pharmacological inhibition of Mas with A779 accelerated liver fibrosis in the BDL model, which was associated with increased liver collagen content and TGF-β1 expression levels. 32 Most recently, Lubel et al. 90 corroborated the previous studies regarding the protective role of Ang (1–7) against liver fibrosis. In BDL rats, Ang (1–7) not only improved the histological fibrosis stage and reduced hydroxyproline content but also decreased gene expression of collagen 1A1, α-SMA, vascular endothelial growth factor (VEGF), CTGF, ACE and Mas. 90 In addition, cultured hepatic cells expressed both AT1 and Mas, and when treated with Ang (1–7) or the Mas receptor agonist, AVE 0991, produced less α-SMA and hydroxyproline. 90 These effects were reversed by the Mas receptor antagonist, A779. 90 In ACE2 knockout mice, increased liver fibrosis was observed following BDL for 21 d or chronic CCl4 treatment. 91 In human patients with cirrhosis, both plasma Ang (1–7) and Ang II concentrations were markedly elevated. 90 However, non-cirrhotic patients with hepatitis C had elevated Ang (1-7) levels compared with controls, but Ang II concentrations were not increased. 90 Thus, accumulating evidence suggests that the Ang (1–7)/Mas axis represents a counter-regulatory response to RAS-mediated liver injury and may be a fruitful therapeutic target for hepatic fibrosis.
Studies utilizing ARBs and ACE inhibitors in patients with liver disease
Inhibitors of the RAS are widely used to treat renal and heart failure and several clinical studies have suggested that the beneficial effects may be at least partially related to the actions of these drugs on cardiovascular and renal fibrosis. 92 There have been several studies of the outcome of treating patients with liver fibrosis with ARBs and ACE inhibitors (Table 2). The majority of these studies have used the ARB, losartan. In one of the earliest studies, Terui et al. 93 evaluated the effects of losartan on liver fibrosis in early stages of chronic hepatitis C and found that losartan decreased both collagen type IV deposition and TGF-β1 expression levels. Improved liver fibrosis was reported in another small pilot study in 14 patients with hepatitis C that were treated with losartan. 94 In a very small pilot study in seven patients with NASH, losartan administration improved aminotransferase levels and decreased TGF-β1 expression levels. 87 Two retrospective studies have also been performed to determine the effects of ACE inhibitors and ARBs on liver fibrosis. 95,96 In one study, 128 patients with graft fibrosis due to hepatitis C recurrence after liver transplantation were evaluated for the beneficial effects of ARB and ACE inhibitors. 95 In this study, 27 patients had received ARBs or ACE inhibitors as antihypertensive treatment and in these patients there was less cirrhosis in the graft and a lower fibrosis progression index compared with the patients that did not receive ARBs or ACE inhibitors. 95 A larger retrospective study of antihypertensive agents in 284 patients with chronic hepatitis C with liver fibrosis has revealed several interesting findings. 96 First, patients with hepatitis C and hypertension have increased fibrosis compared with non-hypertensive patients. 96 Most importantly, hypertensive patients receiving angiotensin-blocking agents had less fibrosis than hypertensive patients who did not receive angiotensin-blocking agents. 96 Another recent study in patients with chronic hepatitis C and liver fibrosis shows that oral losartan therapy for 18 months reduced the expression of profibrogenic and nicotinamide adenine dinucleotide phosphate-oxidase (NADPH oxidase; NOX) genes. 97 Overall, these small studies indicate that utilization of ARBs and ACE inhibitors might be beneficial for patients with hepatic fibrosis. However, a very recent study presents data which indicate that ACE inhibitor/ARB therapy did not retard the progression of hepatic fibrosis. 98 In addition, care should be taken with patients with advanced cirrhosis and ascites. In fact the use of RAS inhibitors, such as captopril to reduce portal pressure have been disappointing and associated with very significant side-effects such as renal impairment and systemic hypotension. 71,89,99–102 Clearly, the jury is out on the effectiveness of ARB and ACE inhibitors on the attenuation of hepatic fibrosis and controlled trials in larger patient populations are warranted.
Studies of angiotensin receptor blockers and angiotensin-converting enzyme inhibitors in humans with chronic liver disease
TGF-β1, transforming growth factor β-1; HSC, hepatic stellate cell; ARB, angiotensin receptor blocker; ACE, angiotensin-converting enzyme; NADPH oxidase, nicotinamide adenine dinucleotide phosphate-oxidase
Several studies have presented data indicating that RAS inhibitors may be a beneficial therapy for HCC. Studies have found that the ACE inhibitor, perindopril, which is a potent inhibitor of experimental HCC growth and angiogenesis, is associated with the suppression of VEGF at a clinically comparable dose in murine hepatocarcinoma cells. 103,104 A small study has shown that the combination treatment of vitamin K and perindopril may suppress the cumulative recurrence of HCC after the curative therapy, which may involve the suppression of the VEGF-mediated neovascularization. 105 These studies indicate that ACE inhibitors might be beneficial for the treatment of HCC due to the suppression of VEGF expression. However, additional clinical studies are necessary in patients with HCC to determine safety and efficacy.
Downstream signaling mechanisms triggered by RAS activation in HSCs
The downstream signaling mechanisms activated by RAS components that regulate liver fibrogenesis in all liver cell types have not been explored in fully. However, several studies have shown that Ang II could mediate and exacerbate liver fibrosis through HSC activation and by stimulating TGF-β1 secretion via AT1 receptors. 6,11,20,61,79,106 Most of the signaling mechanisms have been evaluated in the HSCs, and we have summarized the signaling mechanisms that have been elucidated to date in the following text and in Figure 2. Ang II has been shown to induce contraction and proliferation of HSCs and production of TGF-β1. 61 Activated HSCs secrete Ang II, which induces fibrogenic actions through the activation of NADPH oxidase. 107 Bataller et al. 107 demonstrated that Ang II induced phosphorylation of p47phox, a regulatory subunit of NADPH oxidase, and induced ROS formation via NADPH oxidase activity in HSCs. In addition, Ang II phosphorylated AKT and MAPKs and increased AP-1 DNA binding in a redox-sensitive manner. 107 In another study, Ang II also stimulated DNA synthesis, cell migration, procollagen alpha1(I) mRNA expression, and secretion of TGF-β1 and inflammatory cytokines, which were attenuated by N-acetylcysteine and diphenylene iodonium, an NADPH oxidase inhibitor. 107 Other studies have shown that ACE inhibitors and ARBs exert antifibrosis effects through inhibiting NF-κβ activation in liver. Ang II increased NF-κβ activity and NF-κβ target gene TNF-α expression by inhibiting IkBa expression in a redox-sensitive manner in HSCs. 108 In addition, Ang II markedly increased HSC AP-1 activity and AP-1 target gene, a1 (I) procollagen, mRNA expression via ERK-1/2 pathway in a redox-sensitive manner. 108 Finally, Ang II was shown to stimulate the production of monocyte chemotactic protein-1 (MCP-1) by HSCs modulating hepatic inflammation. 109 In contrast to the profibrogenic effects observed for Ang II, evidence suggests that activation of the Ang (1–7)/Mas axis is antifibrogenic. In cultured HSCs, Ang (1–7) has been shown to inhibit Ang II-induced phosphorylation of ERK-1/2. 91
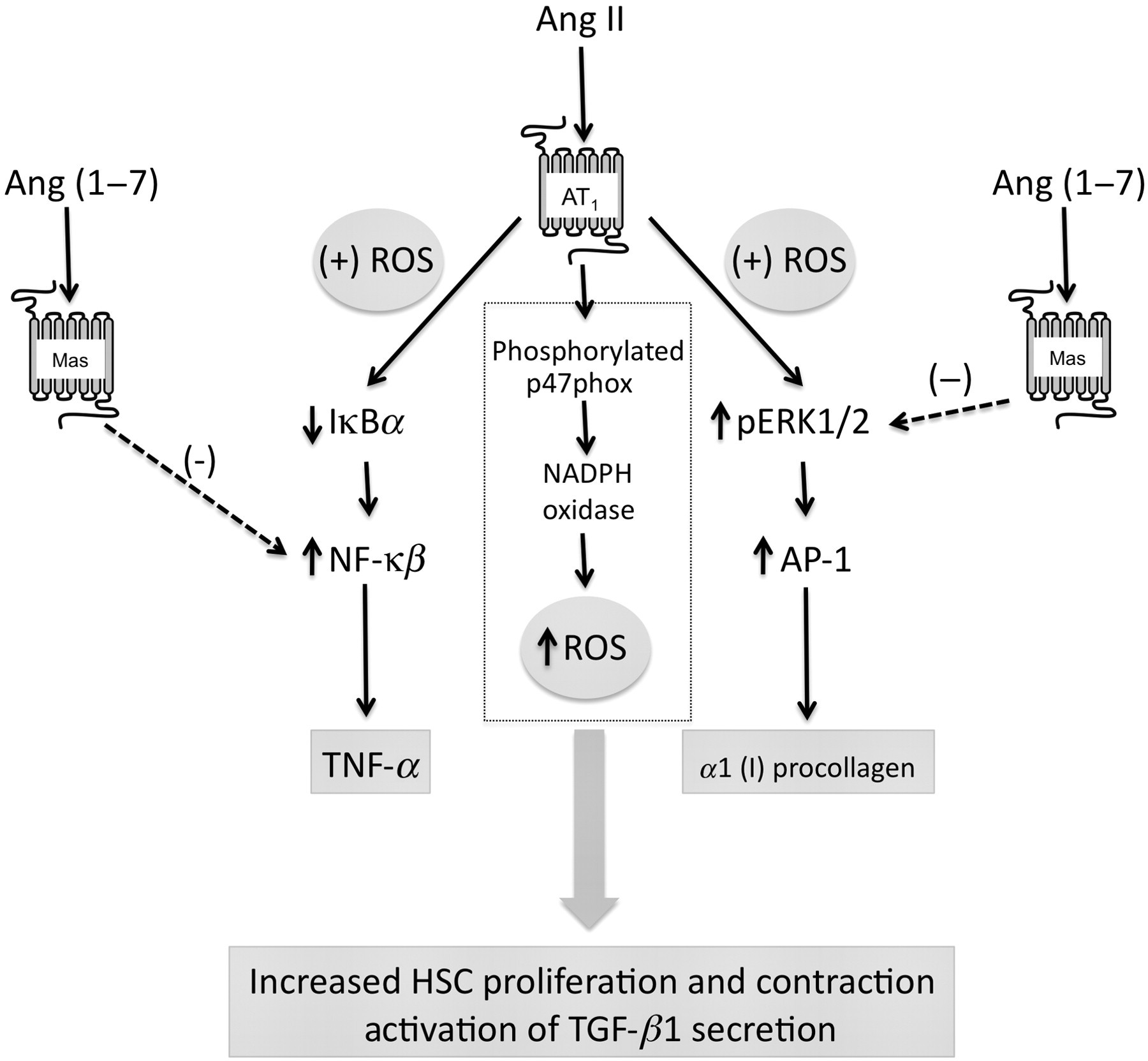
Signaling mechanisms activated by the renin–angiotensin system in hepatic stellate cells. Summary of signaling mechanisms activated by Ang II and Ang (1–7) in activated hepatic stellate cells. Ang (1–7) induces contraction and proliferation of HSCs and production of TGF-β1 through the activation NADPH oxidase resulting in the production of reactive oxygen species. Ang II stimulates the activation of NF-κβ through the inhibition of IkBa resulting in increased expression of TNF-α in a redox-dependent fashion. Ang II also stimulates the phosphorylation of ERK-1/2 resulting in the activation of AP-1 and AP-1-dependent a1 (I) procollagen gene expression in a redox-dependent mechanisms. Ang (1–7) can inhibit both NF-κβ and ERK-1/2 activation via stimulation of Mas. Ang, angiotensin; HSC, hepatic stellate cell; ROS, reactive oxygen species; NADPH oxidase, nicotinamide adenine dinucleotide phosphate-oxidase
Conclusion and future directions
In conclusion, the evidence summarized in this review clearly indicates that the RAS plays a key role in the modulation of hepatic fibrosis during chronic liver diseases. Evidence in animal models of liver fibrosis has shown that both ARBs and ACE inhibitors are effective at attenuating hepatic fibrosis. Studies in patients with chronic liver disease have also been promising. In addition, several studies have indicated that ACE inhibitors may be a beneficial therapy for HCC. However, large, controlled studies are necessary to determine the effectiveness of ARBs and ACE inhibitors for treating hepatic fibrosis in chronic liver diseases, such as chronic hepatitis C and NASH.
Future studies to evaluate the role of the RAS in other cell types, such as, bile duct epithelial cells (i.e. cholangiocytes) that express RAS components may also reveal novel therapeutic targets for chronic liver diseases that are characterized by biliary fibrosis. Interestingly, a recent study has shown that Ang II stimulates cholangiocarcinoma growth and induces tumor fibrosis through an interaction with HSC in an autocrine and paracrine mechanism. 110 These findings suggest that cholangiocytes may interact with HSC and portal fibroblasts during the pathogenesis of cholestatic liver diseases, such as PSC that is characterized by biliary fibrosis. Future studies are required to evaluate the mechanisms by which the RAS governs hepatic fibrosis during chronic liver diseases, such as PSC.
Footnotes
Acknowledgements
This study was supported by Scott & White Hospital Department of Internal Medicine and a NIH RO1 Grant (DK081442) to SSG.