
Abstract
DNA sequencing technology has undergone a remarkable and continuous improvement in recent years. The so-called ‘high-throughput DNA sequencers’ can determine hundreds of megabases of DNA sequences per run. We have been applying these new sequencers to the analysis of infectious diseases, especially bacterial infections. We review the efficacy of these sequencers, mainly based on our own experiences. The approach described here can be viewed as a metagenomic analysis of infectious diseases. The approach is in principle a method that does not depend on the type of target pathogens, so that it is possible to analyze various microorganisms, including bacteria, viruses, fungi and parasites, with a single common protocol. Applying this novel approach to cases in which infectious diseases are suspected because of environmental evidence, but the causative agent has not been indentified, may lead to the discovery of unknown, novel pathogens. Also, the approach enables us to conduct an unbiased analysis of dynamics of the pathogen and associated microbiota in human specimens during the course of infectious diseases.
Introduction
DNA sequencing technology has undergone a remarkable and continuous improvement in recent years. So-called ‘high-throughput DNA sequencers’, which have been commercially available since 2005, can determine hundreds of megabases of DNA sequences per run. 1 It is anticipated that such DNA sequencers with a performance that is much superior to that of conventional sequencers can be expected to have a revolutionary impact on the fields of medicine and biology.
We have been applying these new sequencers to the analysis of infectious diseases, especially bacterial infections. In this review, we will describe the efficacy of these sequencers, mainly based on our own experiences.
High-throughput DNA sequencers
In high-throughput DNA sequencing, the principle of sequence determination is fundamentally different from that of conventional DNA sequencers. 1 One major difference is the sequencing chemistry, which is quite different from the Sanger method used until now. Another difference is the use of micro-scale polymerase chain reaction (PCR), including emulsion PCR, which makes it possible to handle massive numbers of DNA templates simultaneously. Three sequencers have achieved widespread market penetration: the Roche 454 platform (454 Life Sciences, Branford, CT, USA), the Solexa/Illunina platform (Illumina, San Diego, CA, USA) and Life Technologies' SOLiD platform (Life Technologies, Carlsbad, CA, USA). These devices yield at least 100 megabases of DNA sequences per run and have therefore been called ‘next-generation sequencers’. However, since DNA sequencers with a newer principle have appeared more recently, 2 the term ‘next-generation sequencers’ is already out of date and should be changed to ‘second-generation DNA sequencers’. The advancement of recent DNA sequencing technology has indeed been remarkable.
Genome analysis of bacterial pathogens, and possibility of rapid characterization of bacteria
Next-generation sequencers have already been used in the genome analysis of many bacterial pathogens. Also, use of next-generation sequencers for large-scale comparative studies of genomic contents using many strains for several pathogenic bacteria have been reported. 3,4 By using the current second-generation DNA sequencers, enough sequence data can be obtained to deduce a draft genome of any kind of bacterium with a single sequencing run. Until recently, identification and characterization at clinical laboratories of bacterial pathogens isolated from patients have required much work. One of the reasons for this is that various pathogens may be encountered in a clinical setting, so one has to examine the specimens with specific methods (culture procedures, genetic detection, etc.) for each of the possible pathogens. However, draft genome analysis using high-throughput DNA sequencers does not depend on the kind of pathogen, and thus constitutes an extremely effective means for species identification and rapid detection of the kinds of genes (especially genes for virulence factors and drug resistance) which the tested bacterial strains possess. Such an approach may well come into wide use as a supplemental method or an alternative method for conventional biochemical and genetic characterization of bacterial pathogens in clinical laboratories in the near future.
Moreover, the application of the high-throughput sequencers to the direct detection of pathogens from human specimens and to the analysis of microbiota surrounding human bodies also deserves special attention (see below).
Metagenomic detection of pathogens
Metagenomics is a method for analyzing the microorganisms and genes present in a certain environment by extracting and sequencing genomic nucleic acids. This is a culture-independent method, and so far has been used mainly in the field of environmental microbiology. Applying high-throughput DNA sequencing to clinical specimens, however, enables the ‘metagenomic diagnosis’ of infectious diseases. 5 Since this is not specific to a limited number of pathogens, but is instead an unbiased detection method, such an approach may make it possible for various pathogens to be detected with a single common protocol. Also, with this method, unforeseen pathogens may be detected, which may lead to the discovery of unknown, novel pathogens.
High-throughput DNA sequencers, especially the 454 platform, are powerful tools for pathogen discovery 6 and have been used to identify new arenaviruses transmitted through solid-organ transplantation, 7 a new hemorrhagic fever in southern Africa, 8 and a new polyomavirus in Merkel cell skin carcinoma samples. 9 The 454 sequencing technique was also used to implicate Israeli acute paralysis virus as a significant marker for colony collapse disorder in honey bees. 10 We reported the feasibility test of metagenomic detection of the influenza virus from nasopharyngeal aspirates of influenza patients and the norovirus from fecal specimens of patients infected with norovirus. 11 In addition, from blood samples, we could detect GB virus C in the undiagnosed cases of disorder of liver function. 12 As mentioned above, the number of reports on viral pathogen detection with high throughput sequencers is rapidly growing.
Diagnosis of bacterial infections
We previously reported the first metagenomic detection of a bacterial pathogen from human specimens using high-throughput DNA sequencing. 5 That was a case of acute diarrhea in which originally the causative agent could not be identified with the conventional microbiological examination. DNA was extracted from fecal specimens from the patient during the diarrhea and convalescence periods, and the DNA samples were sequenced in an unbiased manner with the 454 GS20 sequencer. 13 The DNA sequences obtained were searched with BLAST, the sequences in the database that showed the best matches for the sequences queried were selected and the organisms from which the sequences were derived were investigated.
Of the 96,941 sequences obtained from the fecal specimen during diarrhea, 156 showed best matches to the genome of Campylobacter jejuni, a causative agent of acute gastroenteritis, while no sequences of the convalescence DNA sample (total: 106,327 sequences) showed any such significant matches. These results prompted us to re-examine the fecal sample with more specific and sensitive methods for C. jejuni, including enrichment/selective media and PCR, and finally the case was confirmed to be due to C. jejuni infection. 5
This study demonstrated the possibility that direct unbiased sequencing (or 16S rDNA deep sequencing, see below) of DNA extracted from patient specimens can identify bacterial pathogens. Further studies regarding the experimental conditions, including methods of DNA extraction from specimens and evaluation of the required number of sequence reads, may lead to the establishment of novel diagnostics for bacterial infections.
Dynamics of pathogens and associated microbiota in human specimens during the course of infectious diseases
Another application of the high-throughput sequencing in the area of infectious diseases is its use for the analysis of dynamics of pathogens and associated microbiota in human specimens during the course of infectious diseases. What follows is an example of such use.
A healthy volunteer, who had provided his fecal specimens two times before for our metagenomic study of human gut microbiota, experienced diarrhea after his travel to a developing country (so called traveler's diarrhea). Enterotoxigenic Escherichia coli (ETEC), a bacterial causative agent of acute diarrhea, was isolated by means of conventional microbiological examination. We could obtain fecal samples at several time points (two times before the episode, and at days 4, 5, 8, 13, 18 and 23 after onset of the symptoms) and DNA was extracted from those specimens. This DNA was then subjected 1) to unbiased sequencing, and 2) to PCR targeting the 16S rDNA, after which the PCR amplicons were sequenced with the 454 platform (16S rDNA deep sequencing).
The unbiased sequencing demonstrated an obvious increase in the number of E. coli in the stool while diarrhea was apparent. In addition, the sequencing of fecal samples obtained during diarrhea detected some virulence factor genes of ETEC including CS6, a colonization factor, thus indicating that the detection of virulence factor genes may also be a potential benefit of this type of sequencing. The 16S rDNA deep sequencing enabled us to trace the dynamics of the bacterial pathogen and associated microbiota over time during the entire diarrheal episode (Figure 1). In this case, bacteria belonging to phylum Fusobacteria showed a similar pattern of dynamics with E. coli, while the Actinobacteria group showed an inverse pattern during the course of infection. Until now, it has not been easy to obtain information regarding the entire intestinal microbiota including difficult-to-culture and unculturable bacteria. The approach presented here should thus constitute a powerful tool for analysis of the dynamics of gut pathogens and microbial flora over time.
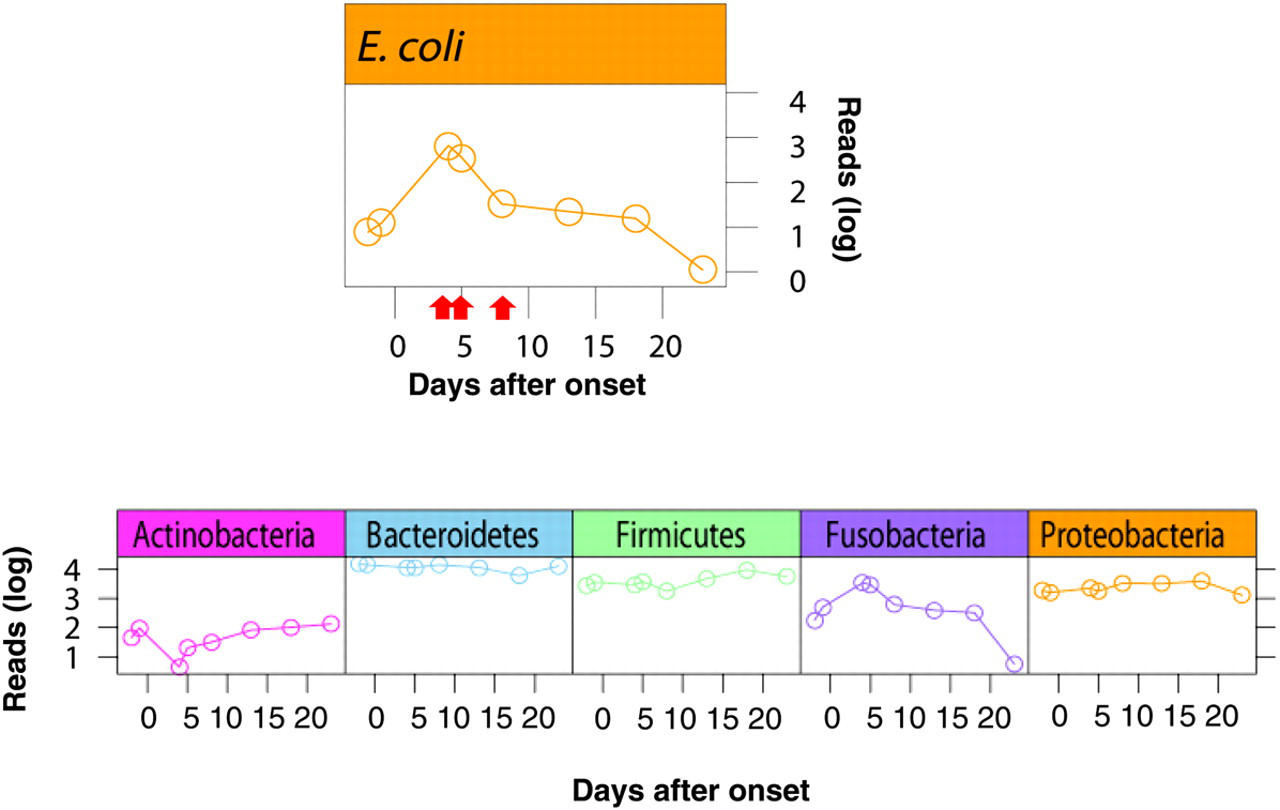
Dynamics of gut Escherichia coli and bacterial microbiota during the course of acute diarrhea due to enterotoxigenic E. coli. Relative abundance of bacterial phyla and E. coli in fecal specimens was estimated by sequencing the polymerase chain reaction (PCR) amplicons targeting 16S rDNA for the DNA samples extracted from each of the fecal specimens. PCR was performed using the primer set (784F: 5′-AGGATTAGATACCCTGGTA-3′ and 1061R: 5′-CRRCACGAGCTGACGAC-3′). The amplified PCR products were then used as a template for pyrosequencing with the GS FLX platform (454 Life Sciences). Sequencing runs yielded an average of 20,066 reads for one sample. The data thus obtained were then subjected to a data analysis pipeline. Data analysis was performed for each read sequence using previously constructed computational tools5 with some modifications. Bacterial rRNA typing was performed by means of BLASTN search against the rRNA database from the All-Species Living Tree Project (LTP). 14 Arrow: enterotoxigenic E. coli was isolated from the fecal sample
It is clear that application of this kind of approach is not limited to the analysis of gut microbiota. The number of studies showing the importance of the various biological aspects of microbiota surrounding human bodies is growing rapidly. The usefulness of this approach for the convenient analysis of microbiota in a variety of settings can thus be expected to increase accordingly.
Conclusions
The approach described here can be viewed as a metagenomic analysis of infectious diseases. The approach is in principle a method that does not depend on the type of target pathogens, so that it is possible to analyze various microorganisms including bacteria, viruses, fungi and parasites with a single common protocol. 5,7,8–12,15 Applying this novel approach to cases in which infectious diseases are suspected because of environmental evidence, but the causative agent has not been identified, may lead to the discovery of unknown, novel pathogens. Also, the approach enables us to conduct an unbiased analysis of dynamics of the pathogen and associated microbiota in human specimens during the course of infectious diseases.
Because the progress of DNA sequencing technology has been rapid, the cost, time, and labor for sequencing have been greatly reduced, and this trend will likely continue in the foreseeable future. 16 There is no doubt that in the near future, new sequencers will become available which greatly exceed the current second-generation DNA sequencers in terms of both performance and cost. 2 Also, more user-friendly DNA sequencers which are suitable for personal use should become available. Such high-throughput DNA sequencing may therefore soon be adopted as the main method for examining microorganisms in major clinical laboratories (Box 1). High-throughput DNA sequencing is a technology that merits close attention for its present and future application in the field of infectious diseases.
Metagenomic analysis of bacterial infections
A variety of microorganisms (including unexpected pathogens) can be detected with a single common protocol.
Taking less time compared to conventional culture-dependent methods.
Possible to detect uncultured or difficult-to-culture organisms.
Relative compositions of microbiota can be estimated.
High costs for sequencers and running.
Improvement for cheaper sequencers and lower-cost running.
Bioinformatics to handle massive amounts of sequence data is needed.
Development of user-friendly pipeline.
Output can differ depending on the extraction method of nucleic acids and PCR primers.
Need to accumulate data for optimization.
The sensitivity of detection is limited by the number of reads.
Improvement of sequencers to read more sequences.
Interpretation on whether detected organisms are the causative agent or not.
Need to accumulate data to establish practical databases.
Footnotes
Acknowledgements
This work was supported by Grants-in-Aid for Scientific Research on Priority Areas and for Scientific Research from the Ministry of Education, Science, Sports Culture and Technology (MEXT), the Program of Founding Research Centers for Emerging and Reemerging Infectious Diseases by the MEXT and the project for the International Research Center for Infectious Diseases, Research Institute for Microbial Diseases, Osaka University from the MEXT. We are grateful to Toshihiro Horii, Teruo Yasunaga, Yoshiyuki Nagai, Yoshihide Hayashizaki, Jun Kawai and Yoshiko Okamoto for valuable discussions, Norihiro Maeda, Michihira Tagami and Hiromi Sano for technical advice, and Kaori Izutsu and Chidoh Kataoka for technical support.