
Abstract
We previously described the death of vascular cells (vascular rhexis) following persistent coronary occlusion. The present study was designed to determine whether non-sustained ischemia can initiate vascular rhexis and if so, whether relatively brief ischemic insults are sufficient. C57BL6 mice were subjected to coronary ligation for 15 min or 3 h followed by reperfusion. Soluble fractions of left ventricular (LV) homogenates were obtained 48 h after the onset of transitory coronary occlusion. They were assayed by Western blotting for quantification of alpha smooth muscle actin (α-SMA) and smooth muscle myosin heavy chain (SM-MHC) that we have shown reflect vascular rhexis delineated immunohistochemically. Non-sustained coronary occlusion for 3 h initiated vascular rhexis evident 45 h after reperfusion, but not earlier, as judged from Western blotting of α-SMA and SM-MHC. The number of small- and medium-sized vessels in the previously ischemic zones was reduced at 45 h after reperfusion as well. Thus, vascular rhexis occurs after ischemia as brief as 3 h but evolves slowly and is not evident for 45 h. The delayed disintegration of the vasculature makes it likely that it can be ameliorated by interventions initiated after non-sustained ischemia, rendering it an attractive target for diminution of phenomena such as late negative LV remodeling, and ‘no reflow.’
Introduction
Sustained myocardial ischemia leads to necrosis of cardiomyocytes and subsequent deposition of fibrous tissue manifested by late negative left ventricular (LV) remodeling. 1–3 Such remodeling after acute myocardial infarction (AMI) is a major contributor to morbidity and mortality. Infarct size 4,5 and persistent occlusion of the infarct-related artery (total ischemic time) 6–8 are among the determinants of such remodeling.
An immediate therapeutic goal for patients with AMI is restoring patency of the occluded epicardial macroscopic infarct-related artery. However, with prolonged myocardial ischemia, even after successful surgical or medical intervention that restores coronary patency, effective microvascular reperfusion may be markedly reduced, a process called the ‘no-reflow’ phenomenon. ‘No reflow’ has been thought to be caused by swelling of capillary endothelial cells, intracardiac edema, aggregation of platelets, deposition of fibrin and platelet leukocyte plugging. 9 We previously reported 10 a loss of functional and structural integrity of coronary arterioles and capillaries in the infarct zones of C57BL6 mice subjected to persistent coronary occlusion, a phenomenon that we termed vascular rhexis. Vascular rhexis was found to be manifested by increases in the concentrations of alpha smooth muscle actin (α-SMA) in soluble fractions of LV homogenates. These changes correlated with loss of intact coronary arterioles quantified immunohistochemically. Vascular rhexis may contribute to both late negative LV remodeling and ‘no reflow’ and its consequences. If vascular rhexis occurs after non-sustained ischemia in patients with AMI who undergo early revascularization procedures and if it evolves more slowly than cardiomyocyte cell death, its attenuation may be possible with interventions initiated at the time of revascularization. Such attenuation appears likely to constitute an attractive therapeutic target. Accordingly, the present study was performed to determine whether vascular rhexis occurs after non-sustained coronary occlusion and to characterize its temporal evolution.
Methods
Experimental animals
C57BL6 mice (The Jackson Laboratory, Bar Harbor, ME, USA) were studied in conformity with a protocol approved by the University of Vermont Institution Animal Care and Use Committee and with adherence to the NIH Principles of Animal Care. The mice were fed normal standard laboratory rodent chow and given water ad libitum. At 10 weeks (wk) of age, they were subjected to either sham operation (control) or non-sustained coronary occlusion for 3 h followed by reperfusion for 21 h (3HIR21H) or 45 h (3HIR45H). Non-sustained ischemia for 15 min was induced in another group of mice followed by reperfusion for 48 h (15MIR48H) to verify that the surgical procedure per se was not inducing the changes we observed in markers of vascular rhexis, following non-sustained ischemia.
Induction of non-sustained ischemia followed by reperfusion
Non-sustained ischemia for selected intervals (15 min or 3 h) followed by reperfusion was induced in mice of 10 wk of age as described previously. 11,12 The mice were anesthetized with 4% isoflurane. The chest cavity was opened at the fourth intercostal space through an 8‐mm skin incision from the left sternal border. The pericardium was removed and the left anterior descending (LAD) coronary artery (technically the middle left in mice) was identified after retraction of the left atrium. With the use of 8-0 sutures on a tapered needle, the artery was ligated 2 mm below the tip of the left atrial appendage after placement of a 0.4‐inch piece of PE10 tubing between the vessel wall and the ligature. 12 The PE10 tubing facilitated cutting of the suture and hence induction of reperfusion. Reperfusion was confirmed visually by the gradual return of color in the previously pale region of myocardium and by prompt resolution of ST segment elevation detected with the use of a base plate (Vevo 770 system, VisualSonics, Toronto, ON, Canada). All mice surviving on the morning after surgery (90% survival) were included in the study. For performance of sham surgery the chest was opened, the pericardium was separated, the LAD was identified and a needle with a suture was passed beneath the LAD. However, the coronary artery was not ligated.
Harvesting of tissue
Mice in each group were killed humanely under 4% isoflurane by harvesting the heart following reperfusion for selected intervals. Blood samples were drawn from the right ventricle (RV) immediately before the hearts were harvested. In some experiments blood samples were collected at selected intervals via tail nicks during the course of reperfusion. After perfusion with phosphate buffered saline (PBS) in situ, the hearts were excised promptly, the LVs were separated from the RVs and atria and weighed.
Immunohistochemistry
Immunohistochemical staining for α-SMA was performed with 10 μm cryostat sections obtained from the LVs after excision and immersion of the tissue into 3% formaldehyde (prepared freshly from paraformaldehyde) in PBS overnight at 4°C. The slides were dried at room temperature for 30 min. After being washed with PBS/1% bovine serum albumin (BSA), they were incubated overnight at 4°C with monoclonal anti-α-SMA Cy3 conjugate (1:300 dilution with PBS/1% BSA; Sigma, St Louis, MO, USA). They were then rinsed with two changes of PBS/1% BSA, five minutes each, in the dark and counterstained and mounted with Vectashield with 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories, Inc, Burlingame, CA, USA) for preservation of fluorescence and visualization of cell nuclei.
For immunohistochemical double staining for CD31, a marker of endothelial cells, and TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling), a marker of cell death slides were rinsed twice with PBS/1% BSA and were blocked with 10% normal goat serum (#191356; MP Biomedicals LLC, Solon, OH, USA) diluted in PBS/1% BSA for 30 min. They were then incubated with purified rat anti-mouse CD31 primary antibody (#550274; BD Bioscience, San Jose, CA, USA) diluted 1:40 in PBS/1% BSA overnight at 4°C. After being washed with PBS/1% BSA, they were treated with goat anti-rat ALEXA-594 (Invitrogen, Carlsbad, CA, USA), diluted 1:600 in PBS/1% BSA, followed by rinsing in two changes of PBS/1% BSA, five minutes each in the dark. Sections were then stained for TUNEL with the use of Apoptag Plus S7111 fluorescence in situ detection kits (Chemicon International, Temecula, CA, USA) to detect necrotic cells. Slides were counterstained and mounted with Vectashield with DAPI.
Photographs were taken with the use of an epifluorescence microscope (Leica DM6000B microscope, Leica CCD camera, and Leica Deblur deconvolution software; Leica Microsystems, Bannockburn, IL, USA). For quantification of α-SMA in the infarct zone, the total number of small- to medium-sized vessels of less than 100 μm in diameter was counted in five separate randomly selected areas with ×10 magnification and averaged. In a corresponding region of equal size, five sections were used for quantification of the number of vessels in normal LVs. The numbers of vessels were counted independently by two different observers masked with respect to each other's results.
Assay of soluble α-SMA and smooth muscle myosin heavy chain released from the infarct zone detected by Western blotting of low salt soluble fractions of LV homogenates
We have shown previously that as vessels undergo degradation, α-SMA is released into soluble fractions of LV homogenates. 10 In the present study we used this marker and vascular smooth muscle myosin heavy chain (SM-MHC) in an analogous fashion to quantify vascular rhesis. α-SMA and SM-MHC in the soluble fraction of LV extracts were detected and quantified by Western blotting and chemiluminescence. Concentrations of protein were determined with the use of the Bradford assay. 13 For blotting, 5 μg of protein were loaded into each well. Coomassie blue staining was used to confirm consistent loading of protein. Samples were incubated with the primary antibodies, monoclonal anti-alpha-SMA clone 1A4 (#A2547; Sigma) and polyclonal anti-rabbit SM-MHC (#250861; Abbiotec, San Diego, CA, USA), diluted in 5% milk overnight at 4°C followed by treatment with the secondary antibody, goat anti-mouse IgG-HRP (sc-2005; Santa Cruz Biotechnology, Santa Cruz, CA, USA) for α-SMA and goat anti-rabbit IgG-HRP (A8275; Sigma) for SM-MHC for 2 h in the dark. Detection of α-SMA and SM-MHC was performed with the use of Pierce ECL Western Blotting Substrate (#32209; Thermo Scientific, Rockford, IL, USA) and quantified by chemiluminescence. Results were expressed as fold increase relative to values seen in LVs from normal mice. In each case normal tissue and tissue from hearts undergoing non-sustained ischemia were compared on the same gel.
Quantification of LV hemorrhage
To verify the functionality of the biochemical markers of vascular rhexis that we quantified, LV intramural hemorrhage was quantified by assay of LV extracts with enzyme-linked, non-cross-reacting immunosorbent assays (ELISA) for hemoglobin corrected for myoglobin with the use of the QuantiChrom Hemoglobin Assay Kit (DIHB-250; BioAssay Systems, Hayward, CA, USA) and a Myoglobin (Mouse) ELISA kit (Catalog Number 41-MYOMS-E01; ALPCO, Salem, NH, USA).
Assay of angiopoietin 2 in human serum
Unfortunately, we could not detect α-SMA or SM-MHC in the serum spiked with these two candidates for detection of vascular rhexis despite having developed ELISAs that performed well in purified systems. When endothelial cells are activated and in patients with acute coronary syndromes, 14 angiopoietin 2 (ang 2) is released into the blood. We hypothesized that its elaboration could ultimately be used to quantify vascular rhexis in patients by assay of ang 2 in serial serum samples. To assess this possibility we first assayed ang 2 serially in samples obtained from tail nicks in mice subjected to non-sustained ischemia at baseline and 10 min, 4 h and 21 h after the onset of reperfusion and at the time of harvesting of the hearts by Western blotting. Samples were incubated with rabbit anti-mouse ang 2 antibody (#18-732-292088; GenWay, San Diego, CA, USA), diluted 500× in 5% milk followed by goat anti-rabbit IgG, peroxidase conjugate (A8275; Sigma), diluted 2000× in 5% milk. Detection was performed with the use of Pierce ECL Western Blotting Substrate (#32209; Thermo Scientific) and quantified by chemiluminescence. In a separate set of feasibility experiments, ang 2 were assayed in banked samples we had obtained serially from patients with myocardial infarction (MI) with the use of a commercially available Human ang 2 ELISA kit (DANG20; R&D Systems, Minneapolis, MN, USA). These patients had been studied in Dubai in a previous study 15 in accordance with a protocol approved by the Institutional Review Board at Rashid Hospital, Dubai, UAE. All patients had provided informed written consent for analysis of their blood samples for diverse biochemical markers. We had previously measured infarct size based on serial changes in serum creatine kinase activity in the same patients.
Statistical analysis
Results were expressed as means ± SEM. Data were analyzed by Student's t-tests or with Welch's one-way analysis of variance as required, with the use of a SAS program (SAS Institute, Inc, Cary, NC, USA). Significance was assumed to be present when P < 0.05.
Results
Density of vessels in the infarct zones
To detect vascular rhexis, immunohistochemical assays of α-SMA in the infarct zone were performed, and the total numbers of small- and medium-sized vessels of less than 100 μm in size were recorded. Vascular rhexis was evident in mice subjected to 3 h of non-sustained ischemia followed by reperfusion for 45 h as manifested by a significant reduction in the number of vessels in the previously ischemic zones (Figure 1) compared with the number in normal hearts. After 48 h the number of small- and medium-sized vessels was 6.3 ± 0.6 vessels per high powered field, n = 4, compared with 23 ± 4.0, n = 3 in normal hearts. (Figure 1, Table 1). Vascular rhexis was not evident as judged from the immunohistochemical assays after 3 h of ischemia followed by 21 h of reperfusion (20.5 ± 0.75, n = 3).
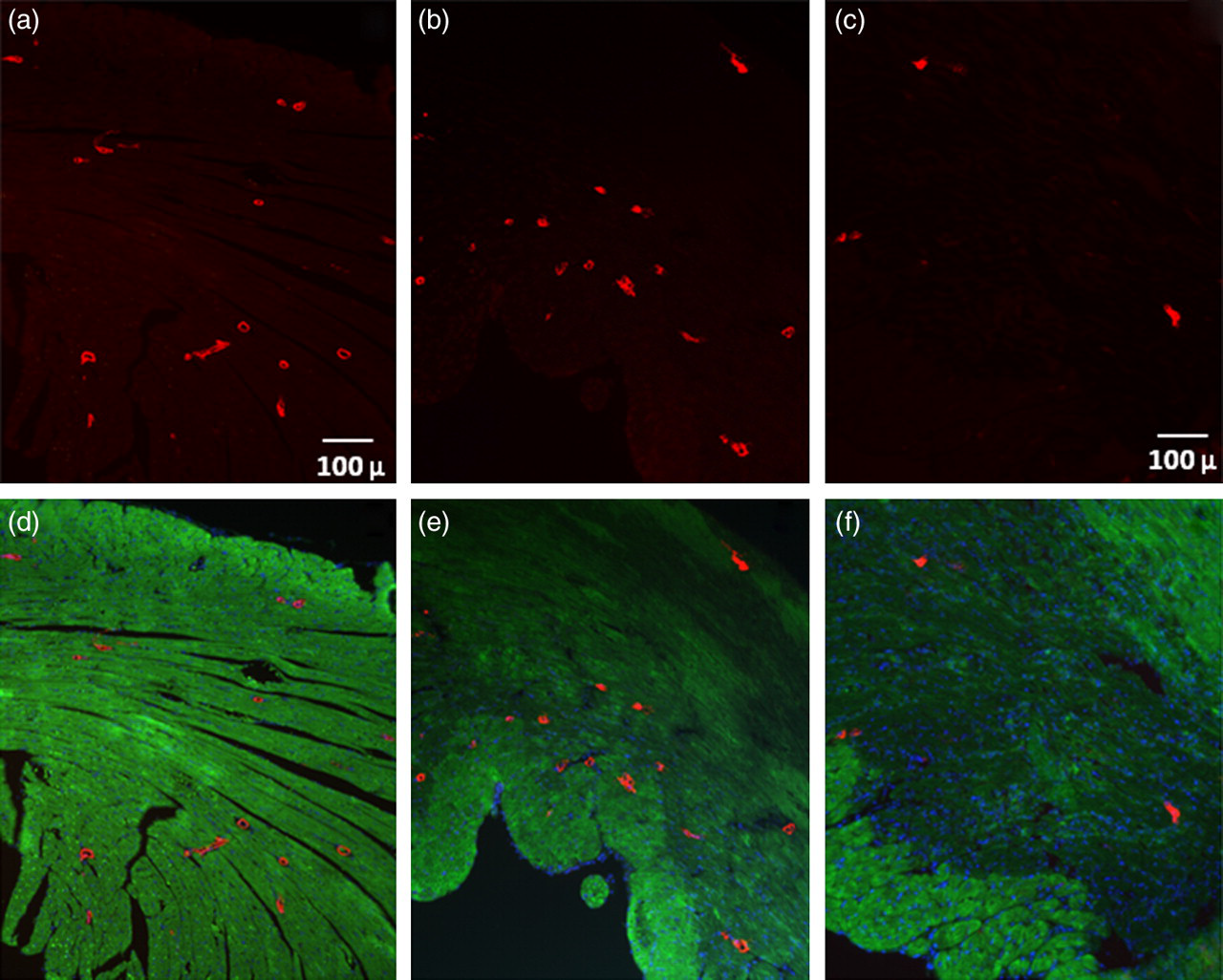
Immunohistochemical staining of alpha smooth muscle actin (α-SMA). Immunohistochemical staining of α-SMA in a normal heart (a,d) and hearts from 3HIR21H (b,e) and 3HIR45H mice (c,f). Staining for α-SMA is shown in (a–c). The merged images are shown in (d–f). The green cast in (d–f) is attributable to autofluorescence
α-SMA in LVs assayed immunohistochemically
HPF, high-power field; LV, left ventricle; α-SMA, alpha smooth muscle actin
The number of small- and medium-sized vessels in the infarct zones in the numbers of mice indicated and in an equivalent region of the LV from a mouse with no myocardial infarction (MI). Values are means ± SEM
*Significantly different from results in hearts with no MI (P < 0.01)
Degradation of vascular smooth muscle cells
To confirm the occurrence of vascular rhexis, α-SMA and SM-MHC in soluble fractions of LV homogenates were assayed by Western blotting. As shown in Figure 2, α-SMA in the soluble fractions of LV extracts was increased by 2.60 ± 0.13-fold in LV harvested from 3HIR45H mice compared with that in normal hearts (1.00 ± 0.07, n = 9, P < 0.05). Soluble SM-MHC liberated from the damaged vascular smooth muscle cells (VSMCs) was increased as well by 1.94 ± 0.15 in 3HIR45H compared with that in normal hearts (1.00 ± 0.07, n = 10, P < 0.05) (Figure 3). Vascular rhexis did not occur as judged from these criteria in mice subjected to very brief cardiac ischemia (15MIR48H mice, α-SMA 0.92 ± 0.16; SM-MHC 1.37 ± 0.23) nor was it evident relatively early after ischemia of 3 h duration (3HIR21H mice, α-SMA 1.44 ± 0.05; SM-MHC 1.24 ± 0.09). Values in both cases were comparable with those in sham‐operated controls (α-SMA 1.29 ± 0.12; SM-MHC 1.35 ± 0.03).
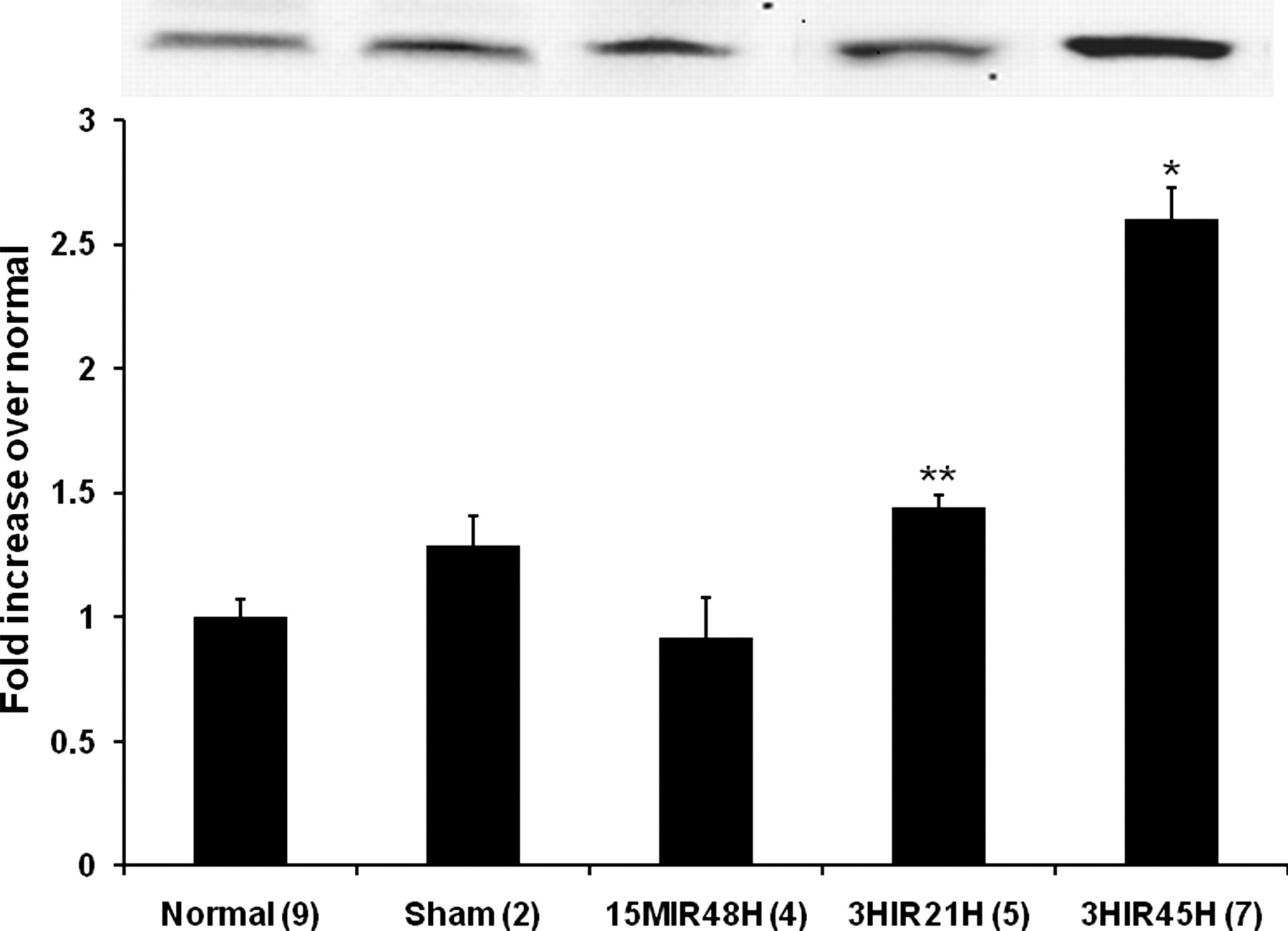
Low salt soluble alpha smooth muscle actin (α-SMA) in homogenates from whole left ventricles. Quantification of α-SMA in normal control and sham‐operated control C57BL6 mice and in mice subjected to various intervals of ischemia followed by reperfusion are shown. Results are means ± SEM. *Significantly different from results in other groups of mice (P < 0.05). **Significantly different from results in 15IR48H mice (P < 0.05). The inset shows the α-SMA band seen on blots
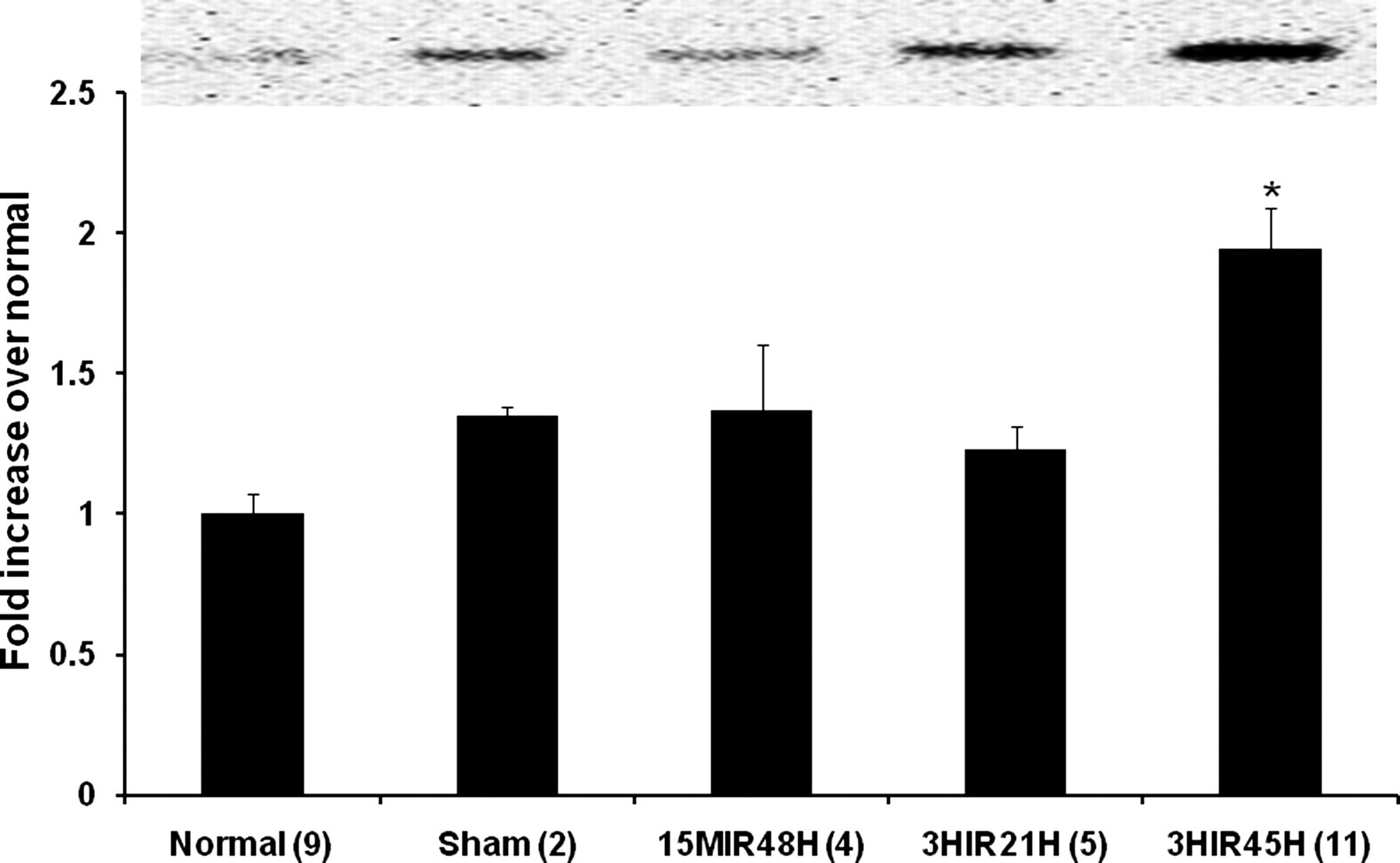
Smooth muscle myosin heavy chain (SM-MHC) in homogenates from whole left ventricles. Quantification of SM-MHC in normal control and sham‐operated control C57BL6 mice and in mice subjected to various intervals of ischemia followed by reperfusion are shown. Results are means ± SEM. *Significantly different from results in other groups of mice (P < 0.05). The inset shows the SM-MHC band seen on blots
Degradation of endothelial cells
In addition to ischemic injury to VSMCs, transitory ischemia initiated degradation of capillary endothelial cells detected by immunohistochemical double staining for CD31, a marker of endothelial cells, and TUNEL, a marker of cell death. As shown in Figure 4, many cells in the infarct zone stained positively for both CD31 and TUNEL 48 h after transitory coronary occlusion in mice in the 3HIR45H group.

Immunostaining for capillary necrosis. Representative double immunostaining is shown for TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) and CD31 in sections of left ventricle from mice in the 3HIR45 group. The endothelial surface marker CD31 positive cells are stained red and necrotic nuclei are stained green (c). Staining for 4′,6-diamidino-2-phenylindole is shown in (b). The merged image is shown in (a). Numerous necrotic endothelial cells are seen in the infarct zone
Myocardial hemorrhage
In the absence of any operative procedures in normal mice or mice subjected to sham surgery, the LV wall contains blood essentially only in the vasculature (approximately 75.1 ± 1.1 μL/g LV). Fifteen minutes of non-sustained ischemia did not induce myocardial hemorrhage (76.7 ± 5.1 μL/g), but 3 h of non-sustained ischemia followed by reperfusion for 45 h led to intramural hemorrhage (83.6 ± 1.9 μL/g, 11.3% increase over control, P < 0.01). This result is consistent with our results obtained by Western blotting of α-SMA and SM-MHC.
Results of serum ang 2 assays
Ang 2 is selectively expressed by endothelial cells in response to hypoxic tissue injury. 16,17 To assess vascular injury reflected by appearance of a biomarker in serum, ang 2 was measured serially at baseline (before coronary occlusion), and at 10 min, 4 h and 21 h following the onset of reperfusion and in serum samples obtained at the time of harvest of the hearts. In 3HIR45H mice, there was serial increase in serum ang 2 evident at 4 h (129.3 ± 22.9-fold) and 21 h (496.4 ± 27) after the onset of reperfusion compared with that at baseline or 10 min (2.3 ± 8.8) following the onset of reperfusion. Ang 2 peaked 21 h after the onset of reperfusion and then declined, but was still higher than it was at baseline at the time of harvest of the hearts (273.3 ± 31.1). Unfortunately, however, serum ang 2 could not be used as a quantitative marker of vascular rhexis because sham‐operated control mice exhibited considerable increases attributable to the surgical procedures per se. Accordingly, to evaluate potential proof of concept that could serve as a foundation for future studies in patients, we assayed serum ang 2 in serial samples from patients who had sustained MI and undergone prompt coronary thrombolysis but no surgery. Results correlated closely with the infarct size (Figure 5) consistent with the possibility that serial assays of ang 2 in the serum will permit quantification of vascular rhexis and its attenuation in patients treated with putative therapeutic interventions under development.
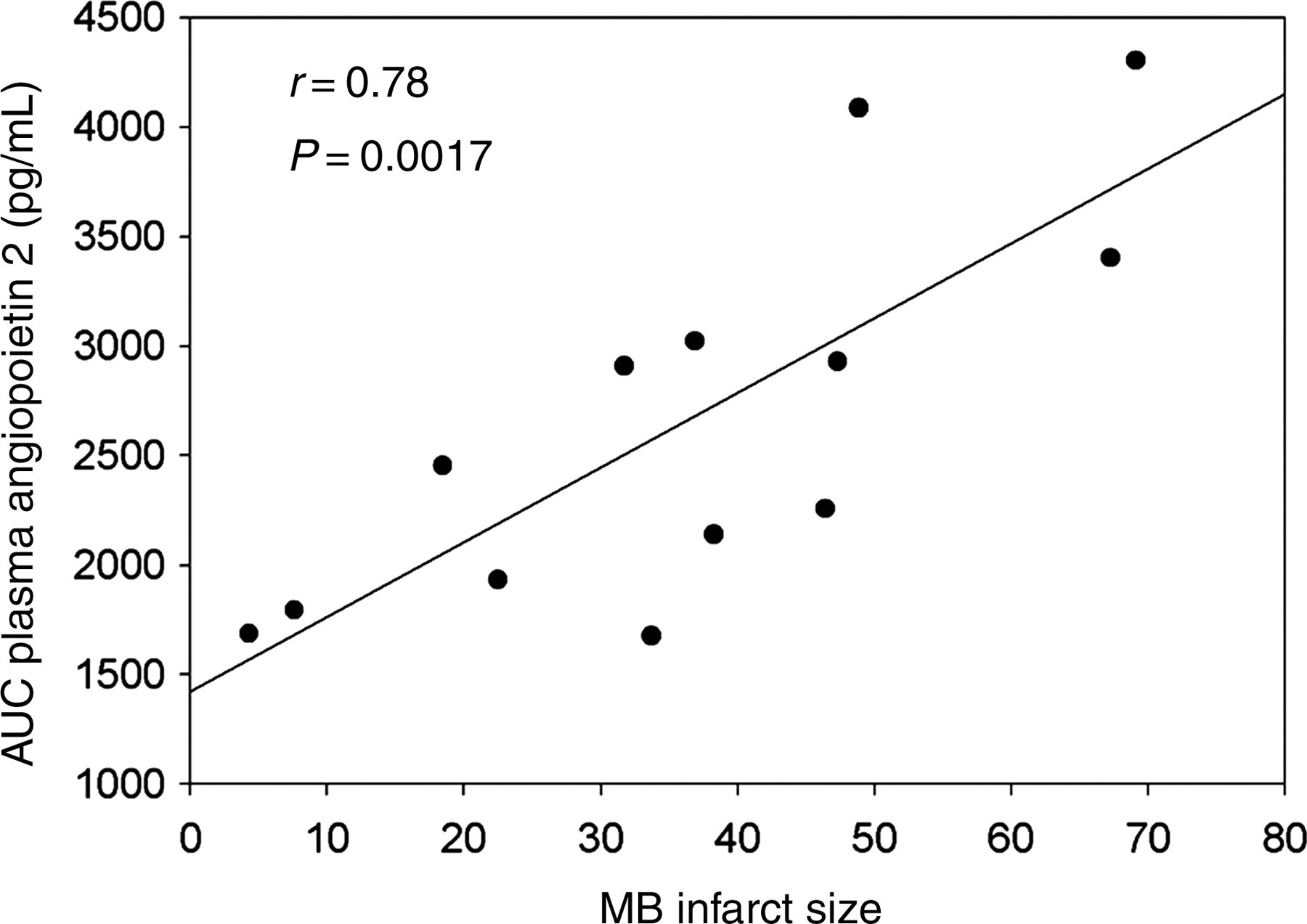
Correlation between infarct size and the area under the 48 h serum ang 2/time curve. Ang 2 in the serum was assayed in samples obtained from patients after myocardial infarction and correlates significantly with the size of infarct measured by MB-creatine kinase analysis. AUC, area under the curve
Discussion
In the present study we demonstrated that vascular rhexis occurs after non-sustained ischemia as brief as 3 h that becomes evident after reperfusion for 45 h. Very brief 15 min of non-sustained ischemia was not sufficient to induce vascular rhexis. Previously, we characterized vascular rhexis in C57BL6 mice in which it became evident 48 h after persistent coronary occlusion. 10 The present results indicate that even brief (3 h) non-sustained ischemia initiates vascular rhexis and that vascular rhexis occurs well after the onset of reperfusion. We and others have previously shown that cardiomyocytes undergo necrosis rapidly. It appears to be virtually complete within as few as 4 h after the onset of ischemia, i.e. well before the onset of vascular rhexis. 10,12 The difference in the temporal evolution of death of cells of the two types and loci probably reflects in part the very high oxygen requirements of cardiomyocytes. Thus, the delay in the onset of vascular rhexis compared with the rapidity of cardiomyocyte death may render vascular rhexis amenable to attenuation with interventions initiated several hours after an ischemic insult.
The disintegration of coronary vasculature as a result of non-sustained ischemia for 3 h followed by reperfusion results in the progressive loss of integrity of arterioles and capillaries in the infarct zone. This is associated with LV intramural hemorrhage evident 45 h after the onset of reperfusion. Biochemical manifestations of vascular rhexis also became evident at 45 h after the onset of reperfusion. These changes included increases in LV homogenate soluble α-SMA and SM-MHC and decreases in the numbers of immunohistochemically stained α-SMA positive vessels in the infarct zones. In addition, necrosis of capillaries was demonstrated as judged from colocalization of CD31 and TUNEL staining.
The ‘no-reflow’ phenomenon, manifested by a reduction in myocardial reperfusion distal to a macroscopic coronary obstruction rectified by recanalization affects as many as 42% of patients with acute STEMI (ST elevated MI) treated with percutaneous coronary intervention. 18–21 Mechanisms implicated previously include capillary endothelial cell swelling, intracardiac edema, aggregation of platelets, deposition of fibrin and platelet leukocyte plugging. 9 Our data indicate that disintegration and loss of small arterioles and capillaries following non-sustained ischemia and reperfusion occur slowly. Because vascular rhexis evolves slowly, its attenuation may be possible with interventions implemented after non-sustained myocardial ischemia and offers promise as a therapeutic target. The results we obtained in the pilot experiments with serial ang 2 assays in banked samples from patients with AMI treated with tenectaplase very early after the onset of ischemia 15 correlated with infarct size (Figure 5) and are consistent with the possibility that assay of ang 2 in the serum will permit quantitative estimation of vascular rhexis in patients with and without interventions designed to attenuate it.
Our results show vascular rhexis occurs in mice after non-sustained ischemia as brief as 3 h followed by reperfusion. This phenomenon may contribute to late negative LV remodeling and the ‘no-reflow’ phenomenon seen in patients. Because it evolves slowly, it may be amenable to amelioration relatively soon after an ischemic insult, and therefore its attenuation is an attractive therapeutic target.
Footnotes
Acknowledgements
We appreciate the excellent technical assistance of Patricia Baumann, Keara McElroy-Yaggy and Dagnija Neimane and preparation of the typescript by Lori Dales. CJF was supported in part by a Hemostasis and Thrombosis Training Program, KG Mann T32, HL07594 (CF).