
Abstract
Chronic low dose of tumor necrosis factor-α (TNF-α) stimulation promotes tumorigenesis by facilitating tumor proliferation and metastasis. The plasma levels of TNF-α are increased in patients with renal cell carcinoma (RCC). Furthermore, high-grade clear cell RCC cell lines secrete more TNF-α than low-grade ones, and allow low-grade cell lines' gain of invasive ability. However, the molecular mechanism of TNF-α in mediating progression of RCC cells remains unclear. In the present study, TNF-α induced epithelial–mesenchymal transition (EMT) of RCC cells by repressing E-cadherin, promoting invasiveness and activating matrix metalloproteinase (MMP) 9 activity. RCC cells underwent promoted growth in vivo following stimulation with TNF-α. In addition, TNF-α induced phosphorylation of extracellular signal-regulated kinase, nuclear factor kappa B (NF-κB) and Akt in a time-dependent manner, and increased nuclear translocation and promoter activity of NF-κB. To investigate the role of NF-κB activation in TNF-α-induced EMT of RCC, we employed chemical inhibitors (NF-κB activation inhibitor and Bay 11-7082) and transfected dominant-negative (pCMV-IκBαM) and overexpressive (pFLAG-p65) vectors of NF-κB. While overexpression of NF-κB p65 alone could induce E-cadherin loss in RCC, EMT phenotypes and MMP9 expressions induced by TNF-α were not reversed by the inhibitors of NF-κB activation. These results suggest that the TNF-α signaling pathway is involved in the tumorigenesis of RCC. However, NF-κB activation is not crucial for invasion and EMT enhanced by TNF-α in RCC cells.
Introduction
In 1863, Rudolf Virchow hypothesized that the lymphoreticular infiltrate in neoplastic tissues reflected the origin of cancer at sites of chronic inflammation. Numerous studies have focused on the inflammatory microenvironment of malignant tissues supporting Virchow's hypothesis. The development of cancer in diverse organs is often associated with chronic inflammation, suggesting a strong relationship between inflammation and tumorigenesis. 1 More recently, research into the impact of inflammation on tumorigenesis has highlighted the importance of studying the complex interactions in the tumor microenvironment. The inflammatory microenvironment of tumors is characterized by the presence of host leucocytes both in the supporting stroma and in tumor areas. 2 Besides inflammatory cells, tumor stroma consists of new blood vessels, connective tissue and a fibrin–gel matrix. In addition, malignant cells themselves secrete proinflammatory cytokines. The tumor cells and/or tumor-associated leucocytes and platelets can produce inflammatory cytokines which may contribute directly to malignant progression. 3 When chronically produced, these cytokines may act as an endogenous tumor promoter, contributing to the tissue remodeling and stromal development necessary for tumor growth and spread. One crucial aspects of the tumor microenvironment is the cytokine-mediated communication between the tumor and stromal cells. 4
One of the key molecules mediating the inflammatory processes in tumor promotion is tumor necrosis factor-α (TNF-α). TNF-α is a key cytokine involved in inflammation, immunity and cellular organization. 5 TNF-α is a proinflammatory cytokine predominantly produced by macrophages. Clinically, elevated serum concentrations and increased expression of TNF-α are present in various preneoplastic and malignant diseases. Although over the last few decades, high-dose administration of TNF-α has been used as a cytotoxic agent, recent preclinical cancer models have provided critical evidence to support the link between chronic, low-level TNF-α exposure and the acquisition of a pro-malignant phenotype (i.e. increased growth, invasion and metastasis). 6 Increasing evidence suggests that TNF-α may regulate many critical processes of tumor promotion and progression. TNF-α may directly contribute to oncogene activation, DNA damage and tumor metastasis. 5,6 TNF-α in serum is significantly higher and correlated with tumor size in renal cell carcinoma (RCC). TNF-α level is also significantly higher as the stage of the RCC increases. In addition, high-grade clear cell RCC cell lines secrete more TNF-α than low-grade ones and allow low-grade cell lines to gain invasive ability. 7 TNF-α may be useful as a marker for the early diagnosis of RCC. 8 However, the molecular mechanism of TNF-α involvement in RCC tumor progression remains unknown.
The epithelial–mesenchymal transition (EMT) is a process in which polarized epithelial cells are converted into motile mesenchymal cells. Alterations in adhesion, morphology, cellular architecture and migration capacity are the major events that occur during this process. 9 Recently, EMT was demonstrated to be a major mechanism responsible for mediating invasiveness and metastasis of cancers. Since nuclear factor kappa B (NF-κB) signaling has a critical role in cancer development and progression, 10,11 we investigated the role of NF-κB in TNF-α-induced EMT of RCC. Our results suggest that overexpression of NF-κB may induce EMT in RCC. However, NF-κB activation is not crucial for invasion and EMT promoted by TNF-α in RCC cells.
Materials and methods
Antibodies and reagents
Anti-E-cadherin mAb was purchased from BD Biosciences (San Jose, CA, USA). Anti-vimentin and anti-β-actin were from Sigma (St Louis, MO, USA). Anti-p-Akt1/2/3 (Ser 473), anti-USF-2, anti-NF-κB p65 and anti-p-extracellular signal-regulated kinase (ERK) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-p-GSK3α/β (Ser21/9), anti-p-NF-κB p65 (Ser536) and anti-p-IKKα(Ser180)/IKKβ(Ser181) were from Cell Signaling (Beverly, MA, USA). The recombinant human TNF-α was obtained from Invitrogen (Camarillo, CA, USA). NF-κB activation inhibitor and Bay 11-7082 were purchased from Calbiochem (San Diego, CA, USA). pcDNA3 was obtained from Invitrogen. pCMV-IκBαM and pNF-κB-luc were from Clontech (Palo Alto, CA, USA). pRL-SV40 was from Promega (Madison, WI, USA). pFLAG-p65 was kindly provided by Dr L P Ting (Department of Microbiology and Immunology, National Yang-Ming University, Taipei, Taiwan). TransIT-LT1 transfection reagent was purchased from Mirus (Madison, WI, USA).
Cell lines and mice
The human RCC cell lines (A498, 786-O) were purchased from the Bioresource Collection and Research Center (BCRC; Hsinchu, Taiwan) and cultured according to the BCRC instructions. Male NOD/scid (H-2d) mice were purchased from the Animal Center of National Taiwan University (Taipei, Taiwan). The animals were raised under specific pathogen-free conditions in the Animal Center of National Yang-Ming University according to the regulations of the Animal Care Committee of National Yang-Ming University.
Tumor cell invasion assay
The ability of RCC cells to pass through matrigel (BD Biosciences, Bedford, MA, USA)-coated filters with 8-μm pores was performed in 24-well Transwell chambers (Corning Inc, Acton, MA, USA). Briefly, tumor cells were pretreated with or without inhibitors for 18 h and plated in the upper chamber (3 × 104 in 0.5% fetal bovine serum [FBS]-cultured medium) and TNF-α (50 ng/mL) in cultured medium (0.5% FBS) was added in the lower chamber. After incubating at 37°C in a humidified 5% CO2 atmosphere for 24 h, the cells in the upper chamber were fixed with methanol. Then the matrigel was mechanically removed from the filter with a cotton swab. The cells adhering to the underside of the filter were stained with Liu's stain (Muto, Tokyo, Japan). The invading cells were examined, counted and photographed at ×100 magnification under a microscope. Ten fields were counted per filter in each group and each experiment was repeated in triplicate.
Gelatin zymography
The activities of matrix metalloproteinase (MMP) 2 and MMP9 were assayed by gelatin zymography. Briefly, 786-O cells (7 × 105/5 mL) were cultured with or without TNF-α (50 ng/mL) in 0.1% FBS-cultured medium for 24 h. Culture supernatants were collected and concentrated by ultrafiltration (Vivascience Ltd, Stonehouse, UK). Concentrated supernatants (20 μg) were then re-suspended in a sample buffer (2% sodium dodecyl sulfate [SDS], 10% glycerol, 0.00025% bromophenol blue, 0.02 mol/L Tris-HCl, pH 6.8) and loaded without boiling in 10% SDS-polyacrylamide gel containing 1 mg/mL gelatin. After electrophoresis, the gels were soaked twice in 0.25% Triton X-100 for 15 min at room temperature. Then, the gel slab containing gelatin was incubated at 37°C for 20 h in the development buffer containing 0.01 mol/L CaCl2 and 0.05 mol/L Tris-HCl (pH 8.0). The gel was fixed for 15 min in solution containing 50% methanol and 10% acetic acid. The gel was then stained for 2 h in 0.5% (w/v) Coomassie blue R-250, and destained in 10% acetic acid.
RNA isolation and realtime quantitative polymerase chain reaction
786-O cells (2 × 105/mL) were treated with or without TNF-α (50 ng/mL) for the indicated time. Total cellular RNA was extracted using the RNeasy kit (Qiagen, Hilden, Germany). Each extracted RNA sample (5 μg) was reversely transcribed into cDNA. For transcript quantification by realtime polymerase chain reaction (PCR), the SYBR Green Mix containing Thermo-Start DNA Polymerase (Bio-Rad, Hercules, CA, USA) was used according to the manufacturer's instructions in an ABI7700 System (Applied Biosystems, Foster City, CA, USA). All values were normalized against glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA. The forward and reverse primers for MMP9 were 5′ GCAAGCTGGACTCGGTCTTT 3′ and 5′ TGGCGCCCAGAGAAGAAG 3′. The forward and reverse primers for MMP2 were 5′ TTGATGGCATCGCTCAGATC 3′ and 5′ TTGTCACGTGGCGTCACAGT 3′. The forward and reverse primers for Slug were 5′ TGTTGCAGTGAGGGCAAGAA 3′ and 5′ GACCCTGGTTGCTTCAAGGA 3′. The forward and reverse primers for ZEB1 were 5′ GCCAATAAGCAAACGATTCTG 3′ and 5′ TTTGGCTGGATCACTTTCAAG 3′. The forward and reverse primers for GAPDH were 5′ CAACTACATGGTTTACATGTTC 3′ and 5′ GCCAGTGGACTCCACGAC 3′.
In vivo tumor growth
786-O cells (2 × 104/10 mL) were treated with or without TNF-α (50 ng/mL) for six days. Nude/scid mice (n = 3) were subcutaneously injected with TNF-α-treated 786-O cells (1 × 107/100 μL) on day 0. Tumor growth was measured with a caliper and calculated as length × width × height (in mm3) at intervals of three days.
Western blot
786-O cells (7 × 105/5 mL) were cultured in complete medium for 24 h and then pretreated overnight with or without inhibitors. After culturing with or without TNF-α (50 ng/mL) for different times, cells were collected. The cytoplasm and nuclei of cells were separated by using the CNMCS compartmental protein extraction kit (BioChain, Hayward, CA, USA). The protein concentrations of the cell lysates were measured using the Bio-Rad protein assay kit. The proteins (60 μg) of cell lysates were separated by 10% SDS-polyacrylamide gel electrophoresis and Western-blotted with antibodies.
NF-κB reporter assay
786-O cells (1 × 106/5 mL) were transiently co-transfected with pcDNA3 (vector) or pCMV-IκBαM or pFLAG-p65 (2500 ng) and pNF-κB-luc (2500 ng) and pRL-SV40 (250 ng). After 24 h, these cells were treated with TNF-α (50 ng/mL) for 24 h. The firefly and renilla luciferase activities were measured by the dual-luciferase reporter assay system (Promega). The relative fluorescence units (RFU) was calculated as firefly luciferase activity/renilla luciferase activity.
Statistical analysis
Data were expressed as mean ± SD and statistical significance was assessed by the analysis of variance test.
Results
TNF-α promoted invasive capability and the induction of MMP9
In order to investigate the role of TNF-α in the progression of RCC, two RCC cell lines with von Hippel-Lindau (VHL)-null were used. These were A498 with a high malignancy (grade IV) and 786-O with a low malignancy (grade II). The invasion abilities of 786-O and A498 cells affected by TNF-α were measured by using a matrigel-coated transwell chamber. As shown in Figure 1a, TNF-α strongly promoted the invasive capabilities of RCC. Compared with the untreated cells, the number of RCC cells penetrating to the lower chamber increased almost 200–300-fold after treatment with TNF-α (Figure 1b). To further explore the molecular mechanism of TNF-α in the tumor progression of RCC, low-malignant 786-O cells that expressed higher levels of E-cadherin were used in the following experiments. In order to clarify whether MMPs are involved in the promotion of invasion by TNF-α, the effects of TNF-α on MMP activities and gene expression were examined by gelatin zymography (Figure 1c) and realtime quantitative PCR (Figure 1d). In contrast to MMP2, the expression and activity of MMP9 were time-dependently up-regulated by TNF-α in RCC cells.
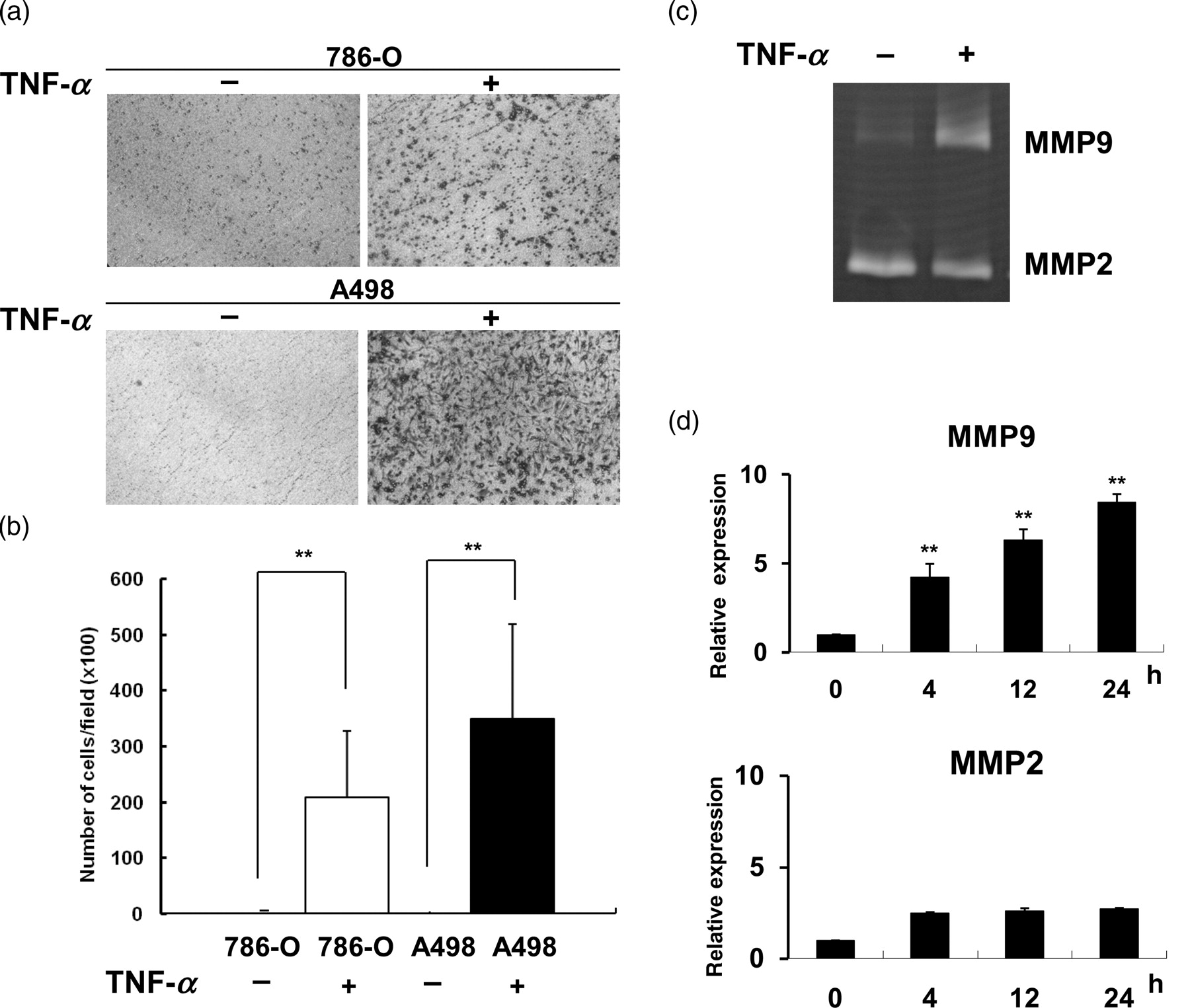
TNF-α promoted invasive capability and the induction of MMP9. (a) Representative images of 786-O and A498 cells that migrated across the transwell membrane to the other side of the filter. (b) Number of 786-O or A498 cells that invaded across the matrigel over a 24-h period in the presence of TNF-α (50 ng/mL) were counted in 10 different microscopic fields (×100 magnification). (c) MMP activity in conditioned media from 786-O cells treated with or without TNF-α (50 ng/mL) for 24 h was analyzed by gelatin zymography. (d) Expression of MMP2 and MMP9 in 786-O cells treated with or without TNF-α were detected by RT-qPCR. All values from RT-qPCR analysis were normalized against the level of GAPDH mRNA. The gene expression as relative expression was plotted relative to the level of TNF-α-treated cells for 0 h. Results are representative of three independent experiments. (**P < 0.01). TNF-α, tumor necrosis factor-α; MMP, matrix metalloproteinase; RT-qPCR, realtime quantitative polymerase chain reaction; GAPDH, glyceraldehyde-3-phosphate dehydrogenase
TNF-α promoted tumor growth in vivo and induced EMT phenotype in vitro
To further prove that TNF-α promoted the progression of RCC, 786-O cells were treated with TNF-α for six days and then injected into mice. As shown in Figure 2a, TNF-α-treated cells grew faster than untreated cells from 18 days to 48 days after injection into mice. In addition, TNF-α induced the conversion of polarized epithelial-like cells into motile mesenchymal-like cells (Figure 2b). The EMT of RCC induced by TNF-α was confirmed by repressing E-cadherin and potentiating vimentin expression (Figure 2c). Also, transcriptional repressors of E-cadherin, Slug and ZEB1, were time-dependently up-regulated (Figure 2d) by TNF-α.
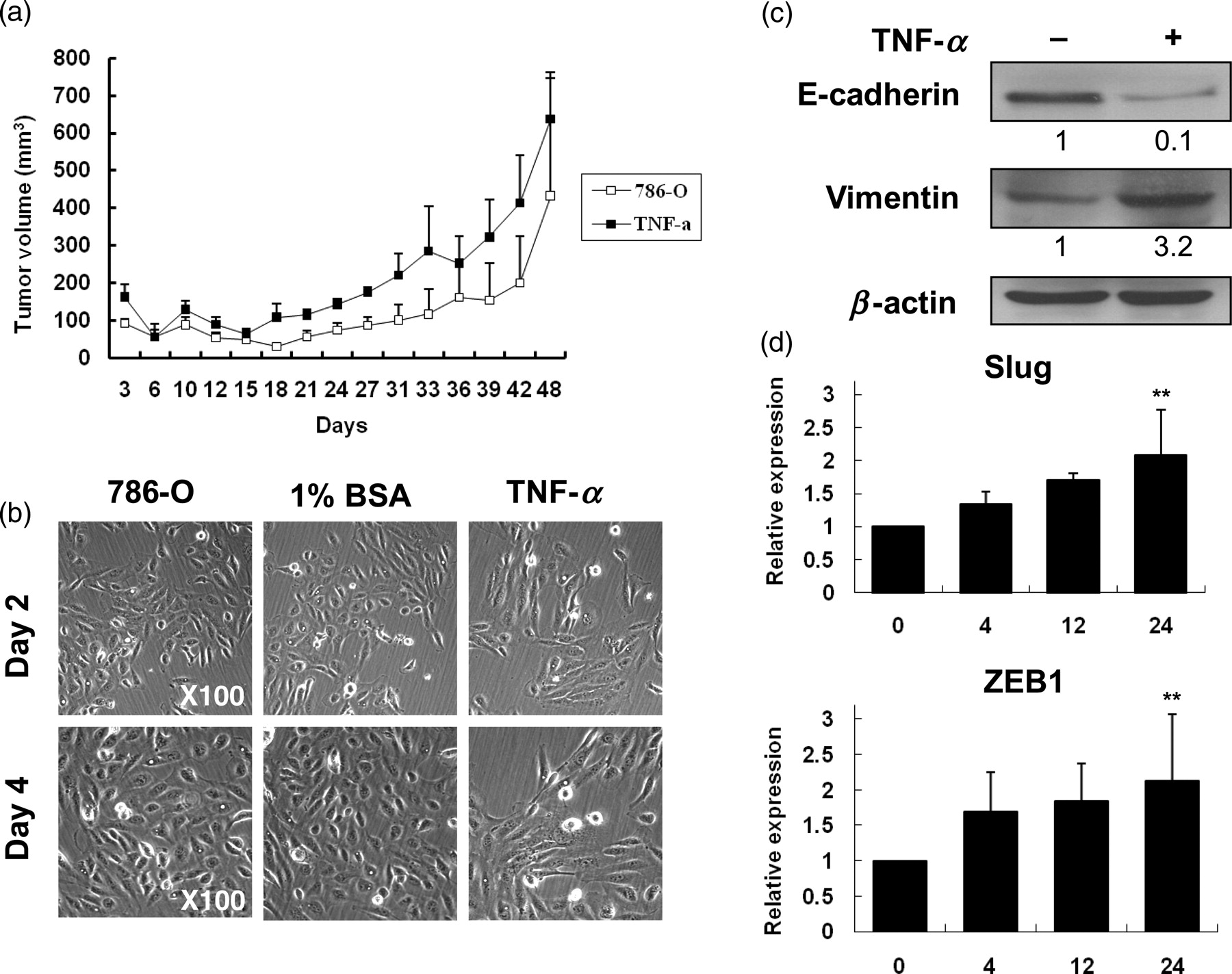
TNF-α promoted tumor growth in vivo and induced EMT phenotype in vitro. (a) 786-O cells were treated with or without TNF-α (50 ng/mL) for six days, then subcutaneously injected into Nude/scid mice (n = 3) and tumor growth was measured. (b) Morphological change in 786-O cells treated with or without TNF-α (50 ng/mL) for two or four days. (c) Western blot analysis of total cell lysates from 786-O cells treated with or without TNF-α (50 ng/mL) for four days. ImageJ 1.44i software (NIH image, Bethesda, MD, USA) was used to quantify the image and the density of each band was normalized to loading control (β-actin). (d) 786-O cells were treated with or without TNF-α (50 ng/mL) for the indicated time and the levels of Slug and ZEB1 mRNA were examined by RT-qPCR. Results are representative of three independent experiments. (**P < 0.01). TNF-α, tumor necrosis factor-α; EMT, epithelial–mesenchymal transition; RT-qPCR, realtime quantitative polymerase chain reaction; BSA, bovine serum albumin
TNF-α activated Akt, NF-κB and ERK signaling pathways
To explore the molecular mechanism of TNF-α-induced EMT of RCC, 786-O cells were treated with TNF-α for different times and the activation of kinases was examined. As shown in Figure 3a, TNF-α time-dependently promoted the phosphorylation and activation of ERK, Akt, IKK and NF-κB p65. Also, activated Akt induced the phosphorylation and inactivation of GSK-3 (panel 3). As we know, the activation of IKK induces phosphorylation and degradation of inhibitor-of-kappaB (IκBα). 11 Then, the deassociation of IκBα and NF-κB leads to the nuclear translocation of NF-κB and transcriptional activation of target genes. Since NF-κB activation has been demonstrated to play a pivotal role in the tumorigenesis of many cancers, 10,11 we investigated the role of NF-κB activated by TNF-α in the EMT of RCC. The activation of NF-κB by TNF-α was further confirmed by time-dependently increasing nuclear translocation of NF-κB (Figure 3b) and dose-dependently potentiating NF-κB promoter activities (Figure 3c). In contrast, control β-actin proteins were not affected by TNF-α in RCC cells (Figure 3b).
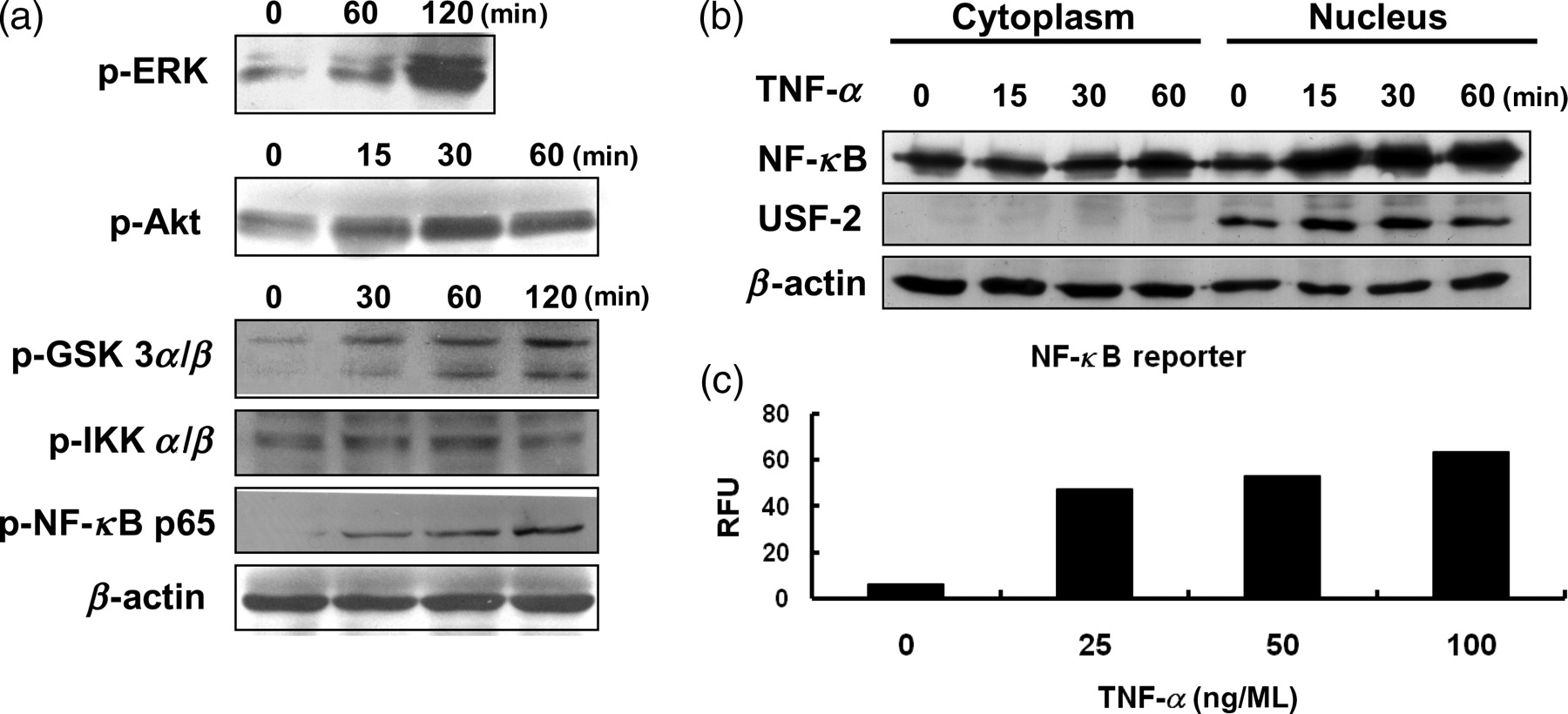
TNF-α activated Akt, NF-κB and ERK signaling pathways. (a) 786-O cells were treated with TNF-α (50 ng/mL) for the indicated period of time. Total cell lysates were extracted and subjected to Western blot analyses. (b) Cytosolic and nuclear fractions were prepared and analyzed by Western blotting. USF-2 was the nuclear marker. (c) 786-O cells were co-transfected with pNF-κB-luc and pRL-SV40. After 24 h, these cells were treated with different concentrations of TNF-α for 24 h. NF-κB reporter activities (RFU) were calculated as firefly luciferase activity/renilla luciferase activity. Results are representative of two independent experiments. TNF-α, tumor necrosis factor-α; NF-κB, nuclear factor kappa B; ERK, extracellular signal-regulated kinase; RFU, relative fluorescence units
EMT induced by TNF-α could not be inhibited by NF-κB activation inhibitor
To further study the role of NF-κB activation in TNF-α-induced EMT of RCC, NF-κB activation inhibitor was used. NF-κB activation inhibitor is a cell-permeable quinazoline compound that acts as a potent inhibitor of NF-κB transcriptional activation. 12 At first, the effect of NF-κB activation inhibitor on NF-κB activated by TNF-α was checked. NF-κB activation inhibitor reduced p-NF-κB activated by TNF-α in a dose-dependent manner (Figure 4a). However, NF-κB activation inhibitor could not inhibit the invasion (Figure 4b) and MMP9 activities (Figure 4c, lane 3) promoted by TNF-α in 786-O RCC cells. Also, E-cadherin loss in TNF-α-treated cells was only slightly reversed by NF-κB activation inhibitor (Figure 4d, lane 3).
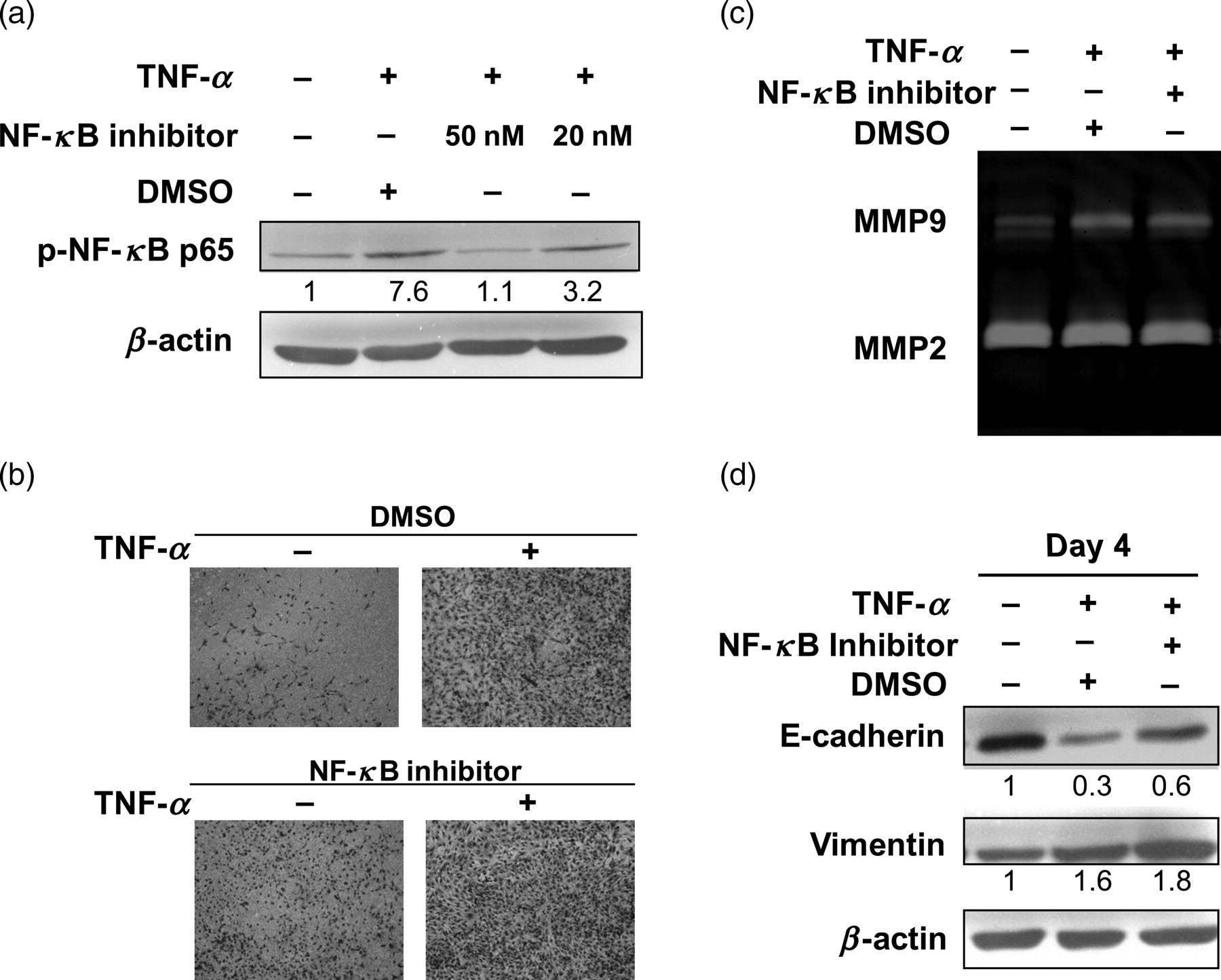
EMT induced by TNF-α could not be inhibited by NF-κB activation inhibitor. (a) 786-O cells were preincubated with or without NF-κB activation inhibitor (20 or 50 nmol/L) for 18 h and then stimulated with TNF-α (50 ng/mL) for two hours. Total cell lysates were harvested and p-NF-κB was determined by Western blot analysis. NF-κB activation inhibitor (50 nmol/L) did not inhibit the invasion activities (b), MMP9 activities (c) and downregulation of E-cadherin (d) induced by TNF-α (50 ng/mL). The density of each band was normalized to loading control (β-actin). Results are representative of three independent experiments. TNF-α, tumor necrosis factor-α; EMT, epithelial–mesenchymal transition; NF-κB, nuclear factor kappa B; MMP, matrix metalloproteinase; DMSO, dimethyl sulfoxide
EMT induced by TNF-α could not be reversed by Bay11-7082
To confirm the results from the NF-κB activation inhibitor study, another chemical inhibitor for NF-κB activation pathway was used. Bay11-7082 selectively and irreversibly inhibits the TNF-α-inducible phosphorylation of IκBα, resulting in a decreased phosphorylation and nuclear translocation of NF-κB. 13,14 The effect of Bay on NF-κB activation was confirmed by the decrease of p-NF-κB (Figure 5a, lane 8 and lane 9) when compared with the control group (lane 4 and lane 5). Then, the effects of Bay on TNF-α-induced EMT were examined. The MMP9 activity (Figure 5b, lane 4) and E-cadherin loss (Figure 5c, lane 3) promoted by TNF-α could not be reversed by Bay 11-7082.

EMT induced by TNF-α could not be reversed by Bay 11-7082. (a) 786-O cells pretreated with or without Bay 11-7082 (2.5 μmol/L) for 18 h and then treated with or without TNF-α (50 ng/mL) for the indicated times were subjected to Western blot analyses. Bay 11-7082 could not inhibit the MMP9 activities (b) and E-cadherin loss (c) induced by TNF-α (50 ng/mL). The density of each band was normalized to loading control (β-actin). Results are representative of three independent experiments. TNF-α, tumor necrosis factor-α; EMT, epithelial–mesenchymal transition; MMP, matrix metalloproteinase; DMSO, dimethyl sulfoxide
Dominant-negative mutant (IκBαM) of NF-κB had no effects on TNF-α-induced EMT
To further confirm the results from chemical inhibitor studies, we used a more specific inhibitor, the dominant-negative vector (pCMV-IκBαM) of NF-κB. In addition, an overexpressive vector (pFLAG-p65) was applied to verify the role of NF-κB overexpression in RCC. The IκBαM contains two mutations that prevent the phosphorylation of IκBα, thus leading to persistent association of IκBα with NF-κB and inactivation of NF-κB. 15 At first, the activities of pCMV-IκBαM and pFLAG-p65 on NF-κB promoter activity were examined. As shown in Figure 6a, transfection of NF-κB p65 (p65) tremendously increased the NF-κB promoter activity, and combined TNF-α treatment (p65/T) only slightly promoted the activity. In contrast, transfection of pCMV-IκBαM inhibited the promoter activity induced by TNF-α (IkBaM/T). Then, the effects of transfection of pCMV-IκBαM and pFLAG-p65 on TNF-α-induced EMT were examined. Overexpression of the dominant-negative mutant (IkBaM) could not inhibit the MMP9 activity (Figure 6b, lane 8) and E-cadherin loss promoted by TNF-α (Figure 6c, lane 8) while overexpression of NF-κB p65 alone induced E-cadherin loss (Figure 6c, lane 5).
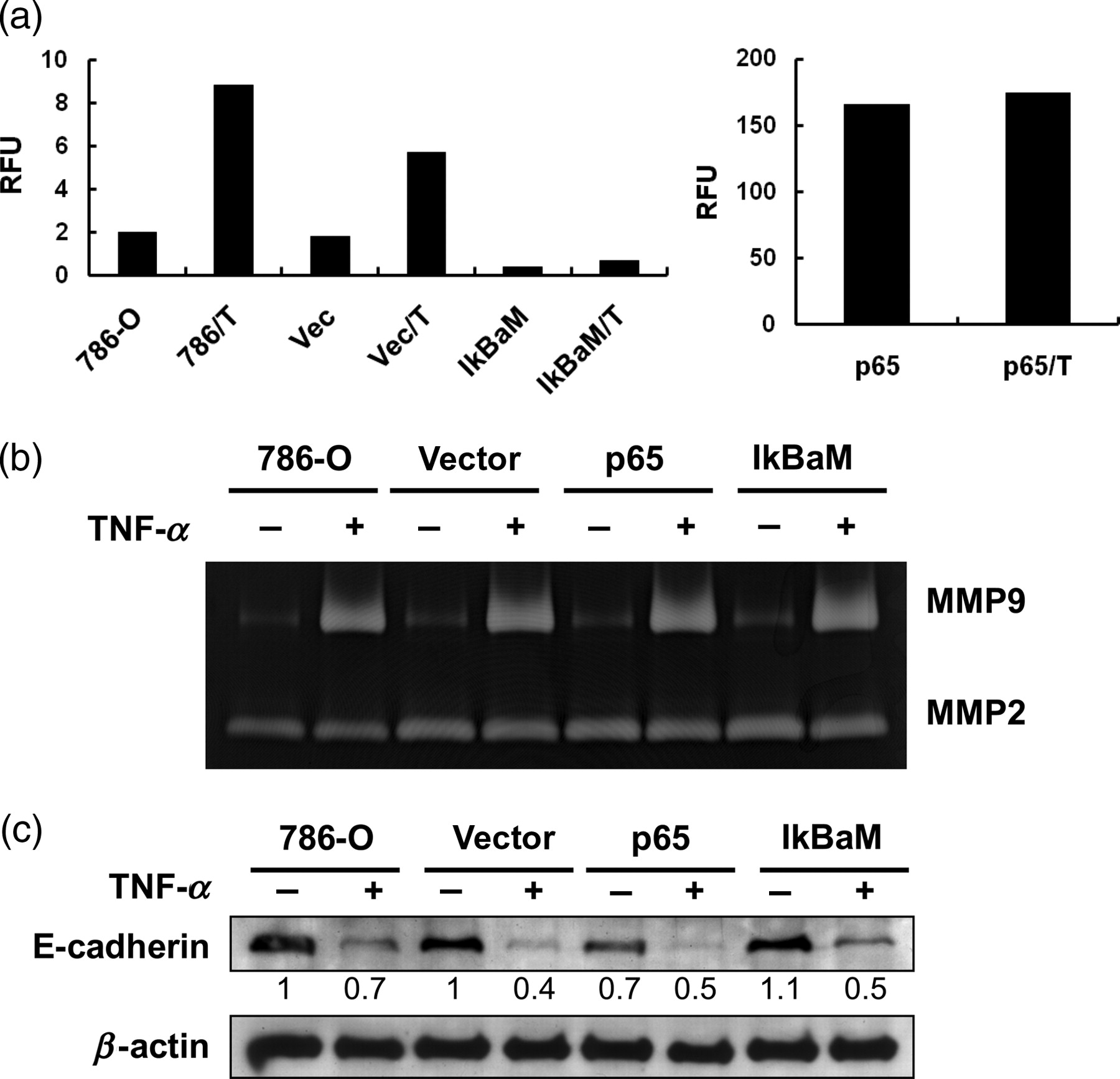
Dominant-negative mutant (IκBαM) of NF-κB had no effects on TNF-α-induced EMT. (a) 786-O cells were co-transfected with control vector (vec) or pCMV-IκBαM (IkBaM) or pFLAG-p65 (p65) and pNF-κB-luc and pRL-SV40. After 24 h, these cells were treated without or with TNF-α (T, 50 ng/mL) for 24 h. NF-κB reporter activities (RFU) were calculated as firefly luciferase activity/renilla luciferase activity. (b) and (c) 786-O cells were transfected with control vector (vec) or pCMV-IκBαM (IkBaM) or pFLAG-p65 (p65). After G418 selection (200 μg/mL), cells were treated with or without TNF-α (50 ng/mL) for two days to evaluate the MMP activities and expression of E-cadherin and vimentin. The density of each band was normalized to loading control (β-actin). Results are representative of two independent experiments. TNF-α, tumor necrosis factor-α; EMT, epithelial–mesenchymal transition; MMP, matrix metalloproteinase
Discussion
The deregulated and sustained production of TNF-α in chronic inflammation could contribute to tumorigenesis. The reduced development of cancers in Tnf −/− or Tnfr1 −/− mice further suggested that TNF-α in the tumor microenvironment enhanced tumor development. 16,17 The tumor suppressor VHL inhibits translation of TNF-α. Cells with mutated VHL in renal cancer produce increased levels of TNF-α. 18 In addition, a chimeric anti-human TNF-α antibody has been demonstrated to be an effective treatment for RCC patients in two phase II studies. 19 However, the pathological mechanism of TNF-α in RCC tumor progression is not known. In the present study, we have demonstrated that TNF-α induced EMT of RCC cells and promoted tumor growth in vivo. TNF-α also activated ERK, NF-κB and Akt. Although overexpression of NF-κB may induce EMT in RCC, NF-κB activation is not crucial for invasion and EMT promoted by TNF-α in RCC cells.
NF-κB is a transcriptional factor that provides a mechanistic link between inflammation and cancer. It is a key factor controlling the ability of cancer cells to resist apoptosis-based tumor-surveillance mechanisms. 10 Exposure of cells to cytokines and growth factors results in dissociation of NF-κB from IκBα and subsequent translocation of NF-κB to the nucleus and activation of target genes. NF-κB might regulate genes involved in tumor angiogenesis and invasiveness. 11 Overexpression of NF-κB represses E-cadherin and enhances EMT of mammary epithelial cells. 20 Stabilization of the E-cadherin repressor, Snail, by NF-κB is required for inflammation-mediated EMT and metastastasis of breast cancer. 21 NF-κB activation plays a role in either the initiation or metastastic progression of cancers depending on the cancer cell type and the model system used. For example, NF-κB is required for the progression but not for the initiation of hepatocellar carcinoma (HCC). 22 In breast cancer, NF-κB plays a role in either the initiation or progression of cancer. 20 However, NF-κB may play a negative role in the tumor progression. In the chemically induced HCC model, IKKβ/NF-κB inhibits tumor promotion and progression by preventing oxidative stress-driven STAT3 activation. 23 Furthermore, transforming growth factor-β-induced EMT of pancreatic cancer is controlled by IKKα in a NFκB-independent and SMAD-dependent manner. 24 Therefore, the role of NF-κB in tumorigenesis is complicated, as in some models NF-κB promotes tumorigenesis, whereas in others it blocks tumor development. 25 Functional inactivation of the VHL tumor suppressor protein is responsible for the majority of sporadic RCC. VHL mutation and increased levels of inflammatory cytokines might result in continuous protein kinase C and NF-κB activation in RCC. 8,26 Genetic mutations are diverse among cancers and might account for the complex role of NF-κB in tumorigenesis of different cancers.
Although we also showed that overexpression of NF-κB induces E-cadherin loss in RCC (Figure 6), NF-κB activation was not a major inducer for invasion and EMT promoted by TNF-α in RCC cells. Another signaling pathway may be more important for TNF-α-induced EMT of RCC. In this study, TNF-α also activated Akt. In our other study, the TNF-α-enhanced Akt phosphorylation and EMT of RCC cells was blocked by treatment with PI3K inhibitor (data not shown). The PI3K/Akt pathway is constitutively activated in different types of cancers and plays a critical role in tumor progression. 27 Thus the PI3K/Akt pathway activated by TNF-α may play an important role in TNF-α-induced EMT of RCC. The antithetic effects of Akt isoforms in the tumorigenesis of breast cancer have been demonstrated. 28 Akt1 inhibits EMT whereas Akt2 promotes EMT under conditions of Akt1 downregulation in regulating growth factor-stimulated cells. 29 Akt can directly phosphorylate IKKα to upregulate Snail expression and induce EMT. 30 However, IKKα/NF-κB is not the major signaling pathway for TNF-α-induced EMT of RCC. In an additional pathway, Akt can phosphorylate and inhibit GSK-3β to stabilize nuclear β-catenin, which in turn, can lead to the transactivation of Slug and EMT. 31 The inactivation of GSK-3β can also stimulate transcriptional factor AP-1 and then promote tumorigenesis. 32 The induction of AP-1 responsive genes, including GM-CSF, MMP9 and MMP3, is important for tumor development but these genes are suppressed in Tnf −/− mice. 33 The involvement of Akt isoforms, GSK-3β and AP-1 in the signaling pathway for TNF-α-induced EMT of RCC is now under investigation.
In summary, our data indicate that the TNF-α signaling pathway is involved in the tumorigenesis of RCC. However, NF-κB activation is not crucial for invasion and EMT enhanced by TNF-α in RCC cells. The role of NF-κB in tumorigenesis is complex and dependent on cancer cell type/lineage and model system used. The PI3K/Akt pathway may play a much more important role than NF-κB in TNF-α-induced EMT of RCC. This is the first report that TNF-α-induced renal tumorigenesis is via a NF-κB-independent mechanism that may be different from other cancers.
Footnotes
Acknowledgements
This work was supported by grants from the National Science Council (98-2320-B-010-001-MY3), Cheng-Hsin General Hospital and Taipei City Hospital, ROC.