
Abstract
Hepatocytes are one of the important targets in dengue virus (DV) infection. Chemokines produced in DV infection play important immunopathogenic roles. We previously showed that DV infection can directly activate signal transducer and activator of transcription 3 (STAT3) in dendritic cells. In the present study, we examined the possible involvement of the Janus kinase (JAK)/STAT3 pathway in chemokine production from DV-infected hepatocytes. HepG2 cells were infected by DV. The activation of STAT3, nuclear factor-kappaB (NF-κB) and other transcription factors was determined by Western blotting or electrophoretic mobility shift assay. The concentrations of chemokines were measured by enzyme-linked immunosorbent assay. Virus titers were determined by plaque assays. A genetic manipulation with short hairpin RNA (shRNA) was applied to knock-down STAT3. Chemotaxis assays were used to evaluate cell migration. We observed that DV infection induced phosphorylation of STAT3 and its DNA-binding activity and such effects were attenuated by the inhibitor of JAK2 or JAK3. Blocking JAK2 or JAK3 reduced DV-induced cell migration and production of chemokines like interleukin-8 and regulated upon activation, normal T-cell expressed and secreted (RANTES). At high doses, the JAK2 but not JAK3 inhibitor could significantly inhibit DV production. Knocking down STAT3 with shRNA suppressed DV-induced STAT3, NF-κB and AP-1 activation. Furthermore, reduction of STAT3 suppressed DV-induced chemokine production and cell migration but had no effect on virus production. In conclusion, the results show that the JAK/STAT3 pathway is critical in chemokine production from DV-infected hepatocytes. Targeting this pathway may be of benefit in the therapy of DV-induced immunopathologies.
Introduction
Dengue virus (DV) infection affects more than 100 millions people annually, predominantly in tropical and subtropical regions. 1 Patients with DV infection present a wide range of clinical symptoms, from mild dengue fever to life-threatening dengue hemorrhagic fever and dengue shock syndrome (DHF/DSS). 2,3 DV infection causes extensive immunological consequences and a variety of immune effectors and viral factors are responsible for the observed immunopathologies. 4,5 In this regard, several studies have examined the induction of chemokines associated with DV infection and many chemokines such as interleukin-8 (IL-8), monocyte chemoattractant protein-1, macrophage inflammatory protein-1α (MIP-1α), MIP-1β and regulated upon activation, normal T-cell expressed and secreted (RANTES) have been reported to be significantly elevated in DV-infected patients. 6–9 Through recruiting leukocytes, these chemokines may exaggerate the inflammatory responses and lead to the development of severe clinical manifestations.
In addition to dendritic cells and B-cells, hepatocytes have also been shown to be one of the potential targets of DV. 10–13 Clinical observations demonstrate that the elevation of hepatic enzymes is frequently observed in DV-infected patients, especially in those with DHF. 14,15 The liver involvement in DV infection is further supported in liver biopsy specimens that show microvesicular steatosis and hepatocellular necrosis. 14 Targeting of the liver by DV is also demonstrated in mice that show similar histopathological effects indicating virus replication in the hepatic tissue. 16 Clinical observations indicate a bad outcome when liver involvement happens. 17
The well-recognized janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway, including a combination of four JAKs, namely JAK1, JAK2, JAK3 and Tyk2, and seven STAT family members, are signaled through nearly 40 different cytokines and are extensively involved in cell growth, survival, development and differentiation in different tissue cells. 18 STAT family proteins are latent in the cytoplasm and become activated through tyrosine phosphorylation and then move to the nucleus for gene activation. We previously showed that in contrast to other STAT family proteins, STAT3 is quickly activated (within 3 h) after DV infection in an interferon-alpha (IFN-α)-independent manner. 19 Down-regulation of STAT3 provides DV a protection against antiviral effects from IFN-α but not IFN-γ. 19 Given the importance of STAT3 in various cellular responses to virus infection, we wondered whether DV infection in hepatocytes could activate STAT3 and the significance of this event. Using a hepatic cell line HepG2 as the target host for DV, in this report, we demonstrated that DV infection could induce chemokine production through activating the JAK/STAT3 signaling pathway in HepG2 cells.
Materials and methods
Culture medium and reagents
The cell culture medium consisted of RPMI 1640 (Gibco-BRL, Gaithersberg, MD, USA) supplemented with 10% fetal bovine serum, 2 mmol/L glutamine and 1000 U/mL penicillin–streptomycin (Gibco). Antibodies against total STAT3 was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The antityrosine phosphorylated STAT3 (anti-p-STAT3) was purchased from Cell Signaling (Beverly, MA, USA). The JAK2 inhibitor AG490 and JAK3 inhibitor WHI-P131 were purchased from Calbiochem (Darmstadt, Germany). AG490 is a specific JAK2 inhibitor 20 and has been commonly used in different fields of experiments by many researchers. 21,22 WHI-P131, commonly recognized and studied as a specific inhibitor of JAK3, has no effect on JAK1, JAK2, or Zap/Syk or Src tyrosine kinases. 23–26 Unless specified, the rest of the reagents were purchased from Sigma-Aldrich Chemical Company (St Louis, MO, USA).
Preparation of DV, determination of virus titers and infection of HepG2 cells
The preparation of DV has been described previously. 19 In brief, DV serotype 2 (DV2) New Guinea C strain (American Type Culture Collection [ATCC], Rockville, MD, USA) was propagated in C6/36 mosquito cells in RPMI containing 5% heat-inactivated fetal calf serum (FCS) and maintained at 28°C in a 5% CO2 atmosphere for seven days. The supernatants were collected, virus titers determined and then stored at −70°C until use. To determine virus titers, the culture supernatants were harvested for plaque-forming assays. Various virus dilutions were added to 80% confluent baby hamster kidney (BHK-21) cells and incubated at 37°C for one hour. After adsorption, cells were washed and overlaid with RPMI 1640 containing 1% agarose (SeaPlaque; FMC BioProducts, Philadelphia, PA, USA) and 1% FCS. After incubation for seven days, cells were fixed with 10% formaldehyde and stained with 0.5% crystal violet. The numbers of plaques were counted and the results were shown as plaque-forming unit/mL. HepG2 cells were purchased from ATCC. HepG2 cells, 1 × 106/mL in culture medium, were infected with mock or DV at multiplicity of infections (MOIs) 5 for four hours at 37°C. 19 After viral absorption, cells were then washed, replaced with fresh medium and cultured for further analysis.
Cell toxicity measurement
Evaluation of potential cytotoxic effects from JAK inhibitors was performed by using 3-[4,-dimethylthiazol-2-y]-2, 5-diphenyl-tetrazolium bromide (MTT) colorimetric assays as described. 27 In brief, HepG2 cells at 3 × 104 in 100 μL volume were incubated in the presence or absence of various doses of JAK inhibitors for 24 h. Then, 100 μL of MTT (3 mg/mL in H2O) were added, and cells were incubated at 37°C for six hours followed by the addition of 100 μL of dimethyl sulfoxide. After incubation at 37°C for another 30 min, the content of dissolved reduced MTT crystals was measured with an enzyme-linked immunosorbent assay (ELISA) reader (Dynatech, Chantilly, VA, USA).
Determination of chemokines by ELISA
Standard ELISA methods were used to measure concentrations of chemokines such as RANTES, IL-8, migration inhibitory factor (MIF) and MIP-1α (R&D, Minneapolis, MN, USA or Amersham-Pharmacia, Arlington Heights, IL, USA). All determinants were performed in triplicates and expressed as mean ± SD.
Nuclear extract preparation
Nuclear extracts were prepared according to our published work. 28 Briefly, the treated cells (3–5 × 106 cells in average in each treatment condition) were left at 4°C in 500 μL of buffer A (10 mmol/L 4-(2-hydroxyethyl)-1-piperazinee thanesulfonic acid [HEPES], pH 7.9, 10 mmol/L KCl, 1.5 mmol/L MgCl2, 1 mmol/L dithiothreitol [DTT], 1 mmol/L phenylmethanesulfonyl fluoride [PMSF] and 3.3 μg/mL aprotinin) for 15 min with occasional gentle vortexing. The swollen cells were centrifuged at 15,000 r.p.m. for three minutes. After removal of the supernatants (cytoplasmic extracts), the pelleted nuclei were washed with 50 μL buffer A and subsequently, the cell pellets were re-suspended in 30 μL buffer C (20 mmol/L HEPES, pH 7.9, 420 mmol/L NaCl, 1.5 mmol/L MgCl2, 0.2 mmol/L ethylenediaminetetraacetic acid [EDTA], 25% glycerol, 1 mmol/L DTT, 0.5 mmol/L PMSF and 3.3 μg/mL aprotinin) and incubated at 4°C for 30 min with occasional vigorous vortexing. Then the mixtures were centrifuged at 15,000 r.p.m. for 20 min, and the supernatants were used as nuclear extracts.
Electrophoretic mobility shift assay
The electrophoretic mobility shift assay (EMSA) was performed as detailed in our previous report. 28 The oligonucleotides probe containing the STAT3-binding site were purchased from Santa Cruz. The oligonucleotides probes for NF-κB, AP-1 and Oct-1 were purchased from Promega (Mandison, WI, USA). The DNA probes were radiolabeled with [γ- 32 p] ATP using the T4 polynuclotide kinase (Promega) according to the manufacturer's instructions. For the binding reaction, the radiolabeled STAT3-binding probe was incubated with 5 μg of nuclear extracts. The binding buffer contained 10 mmol/L Tris-HCl (pH 7.5), 50 mmol/L NaCl, 0.5 mmol/L EDTA, 1 mmol/L DTT, 1 mmol/L MgCl2, 4% glycerol and 1 μg poly(dI-dC). The reaction mixture was left at room temperature to proceed with binding reaction for 20 min. Whenever competition assays were performed, 100-fold molar excess of unradiolabeled, competitive oligonucleotides (wild type or mutant probe) were preincubated with nuclear extracts for 30 min before the addition of radiolabeled probes. The sequences of both wild type and mutant probes are given as follows. NF-κB: 5′ AGT TGA GGG GAC TTT CCC AGG C 3′ (wild type) and 5′ AGT TGA GGC GAC TTT CCC AGG C 3′ (mutant); AP-1: 5′ CGC TTG ATG ACT CAG CCG GAA 3′ (wild type) and 5′ CGC TTG ATG ACT TGG CCG GAA 3′ (mutant); Oct-1: 5′ TGT CGA ATG CAA ATC ACT AGA A 3′ (wild type) and 5′ TGT CGA ATG CAA GCC ACT AGA A 3′ (mutant); STAT3: 5′ GAT CCT TCT GGG AAT TCC TAG ATC 3′ (wild type) and 5′ GAT CCT TCT GGG CCG TCC TAG ATC 3′ (mutant).
Western blotting
Enhanced chemiluminescence Western blotting (Amersham, Arlington Heights, IL, USA) was performed as described. 19 Briefly, after an extensive wash, the cells were pelleted and re-suspended in lysis buffer. After periodic vortexing, the mixture was centrifuged, the supernatant collected and the protein concentration was measured. Equal amounts of whole cellular extracts were analyzed on 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to the nitrocellulose filter. For immunoblotting, the nitrocellulose filter was incubated with Tris-buffered saline with Tween containing 5% non-fat milk (milk buffer) for two hours, and then blotted with antisera against individual proteins overnight at 4°C. After washing twice with milk buffer, the filter was incubated with secondary antibodies conjugated to horseradish peroxidase at a concentration of 1:5000 for 30 min. The filter was then incubated with the substrate and exposed to X-ray film.
Short hairpin RNA knock-down
To reduce STAT3 expression, several STAT3-specific short hairpin RNA (shRNA) constructs were designed, synthesized and obtained from the National RNAi Core Facility in Taiwan, ROC. One of the STAT3-specific shRNA constructs (clone ID: TRCN0000071454 with target sequence CGACTTTGATTTCAACTACAA) or a control green fluorescence protein (GFP) shRNA construct (clone ID:TRCN0000072202 with target sequence GCCACAAC ATCGAGGACGGCA) was co-transfected with the package and envelope plasmids to 293T cells to generate recombinant lentivirus carrying specific shRNA. The virus-containing supernatants were harvested and the relative viral titers were determined by using cell viability assay on 293T cells after puromycin selection according to the protocol from the National RNAi Core Facility. The viruses at MOI 2 were used to infect HepG2 cells. For infection, HepG2 cells were seeded one day before infection and were infected by lentivirus carrying different shRNA constructs in the presence of 8 μg/mL polybrene. After culture for another six days with regular replacement of medium, the cells were used for experiments.
Chemotaxis assay
The chemotaxis assays were performed according to the report. 29 In brief, 600 μL culture supernatants collected from the treated cells as indicated in the figure legends were added to the lower chambers of the transwell cassette (Corning Costar, Lowell, MA, USA). U937 cells, 1 × 105 in 100 μL serum free medium, were loaded in the upper chamber and incubated for two hours at 37°C. Then cells migrating from the upper chamber to the lower chamber were counted by flow cytometry. The acquired events for a fixed time period of 60 s in a FACScan were determined using CellQuest software (BD Biosciences, San Jose, CA, USA).
Statistics
When necessary, the results were expressed as the mean ± SD. The one-way analysis of variance was used to analyze the data; P < 0.05 was considered significant.
Results
DV activated STAT3 through JAK2 and JAK3 in hepatocytes
To determine whether DV infection could activate STAT3 in HepG2 cells like the results observed in dendritic cells, we first investigated the activation status of STAT3 in DV-infected HepG2 cells. Meanwhile, the potential roles of JAK2 and JAK3 in this process were also examined. Both JAK2 inhibitor AG490 and JAK3 inhibitor WHI-P131 were used to test this hypothesis. Hepatocytes infected by DV for various periods of time were collected and the expression of phosphorylated STAT3 was determined in total cell lysates by Western blot. In here, we showed that STAT3 could be activated within six hours after DV infection in hepatocytes (Figure 1a). The kinetics of STAT3 activation in hepatocytes appeared to be similar to those in human dendritic cells, 19 although we could not completely exclude the possibility of secondary effects from the secreted mediators like cytokines. In the presence of JAK2 or JAK3 inhibitor, DV-induced phosphorylation of STAT3 was inhibited (the upper panels of Figures 1b and c). DV infection did not affect total protein levels of STAT3. The EMSA analysis was subsequently performed to examine the DNA-binding activity of STAT3. Consistent with the examination on protein expression, DV infection induced DNA-binding activity of STAT3, which could be suppressed by either JAK2 or JAK3 inhibitor (the lower panels of Figures 1b and c). The binding was considered to be specific because the wild-type STAT3-binding oligonucleotides competed more effectively than the mutant STAT3-binding oligonucleotides on binding to the STAT3-containing protein complex. These results suggest that the activation of JAK2 and JAK3 was in the upstream of DV-induced activation of STAT3 in HepG2 cells.

Activation of STAT3 by DV infection and the effects of blocking JAK2 or JAK3. HepG2 cells at 1 × 106/mL infected by mock or DV at MOI 5 at various time points were collected, lysed and the whole lysates were prepared. Western blotting was performed to determine the levels of phosphorylated STAT3 and β-actin. The representative results from three independent experiments with similar patterns are shown (a). HepG2 cells were pretreated or not with vehicle (V) or different doses of JAK2 inhibitor AG490 (b) or JAK3 inhibitor WHI-P131 (c) as indicated for one hour, then the cells were absorbed with DV for four hours. After adsorption and removal of unbound viruses by gentle washing, the same doses of AG490 or WHI-P131 were freshly added back into the culture medium and incubated for another 20 h. The total cell lysates or nuclear extracts were prepared accordingly. The tyrosine-phosphorylated STAT3, total STAT3 and β-actin were determined by Western blotting. The DNA-binding activity of STAT3 was measured by EMSA. To determine the specificity of STAT3-binding complex, the nuclear extracts from DV-infected cells were preincubated with unradiolabeled wild-type (Wt.) or mutant (Mt.) STAT3-binding oligonucleotides for 30 min before the incubation with radiolabeled oligonucleotides. The representative results out of at least three independent experiments are shown. STAT, signal transducer and activator of transcription; p-STAT3, tyrosine-phosphorylated STAT3; t-STAT3, total STAT3; DV, dengue virus; JAK, Janus kinase; MOI, multiplicity of infection; EMSA, electrophoretic mobility shift assay
Blocking JAK2 or JAK3 inhibited DV-induced production of IL-8 and RANTES
Increased chemokine levels have been observed in patients infected by DV and the possible role of hepatocytes in contributing to this phenomenon was examined. Meanwhile, the effect of inhibition of JAK2 or JAK3 in this event was investigated. The results indicate that DV infection could induce production of IL-8 and RANTES in HepG2 cells and the effect was attenuated by treatment with either JAK2 (Figure 2a) or JAK3 inhibitor (Figure 2b). Regarding the suppression of RANTES secretion in DV-infected cells, JAK2 inhibitor appeared to be more potent than JAK3 inhibitor.
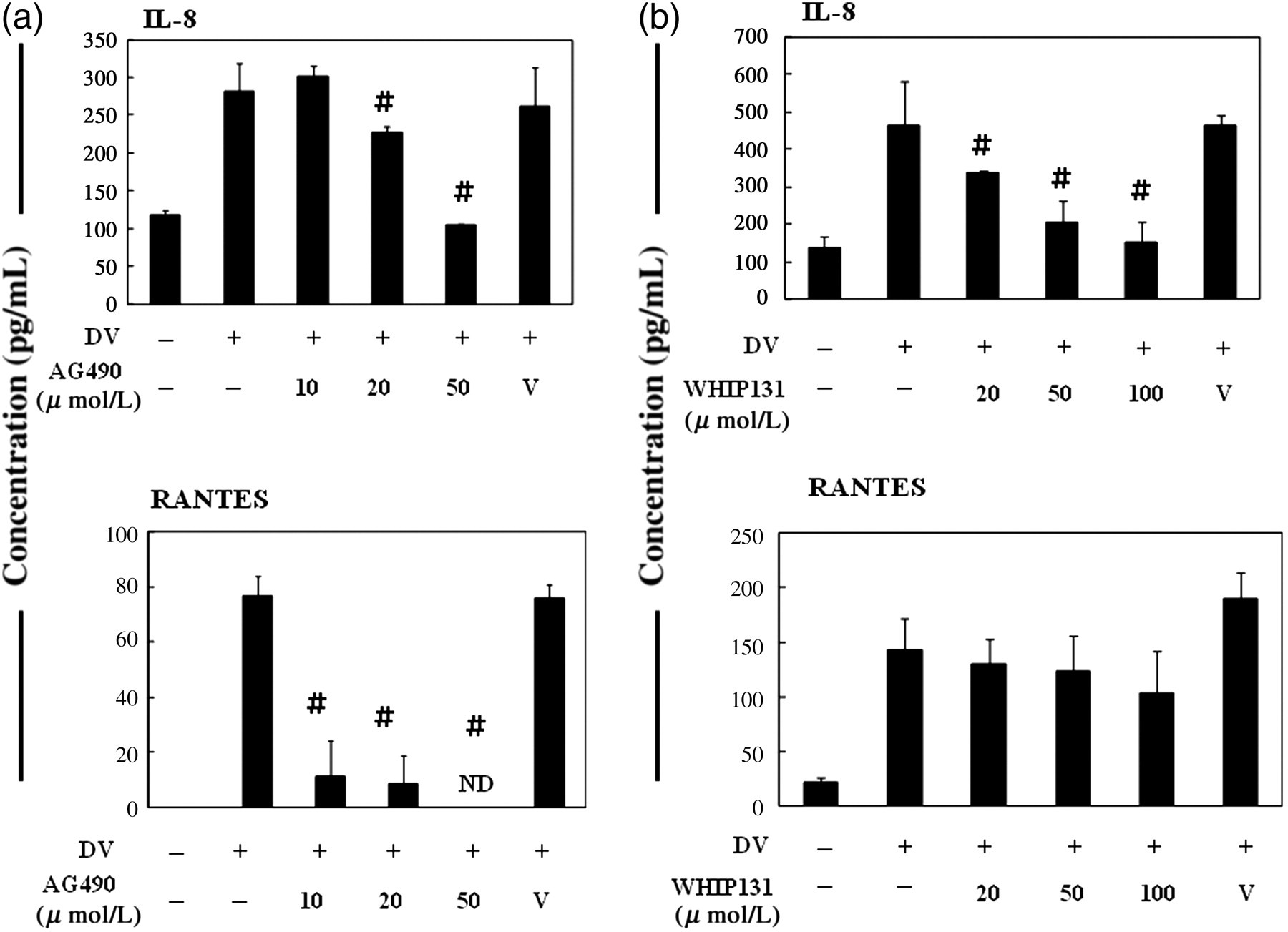
Inhibition of JAK2 (a) or JAK3 (b) suppressed DV infection-induced production of IL-8 and RANTES. HepG2 cells were treated as described in Figure 1 and the supernatants were collected for the measurement of chemokines IL-8 and RANTES. # P < 0.05 compared with the DV-stimulated in the absence of the JAK inhibitor. ND, not detectable; JAK, Janus kinase; DV, dengue virus; IL, interleukin; RANTES, regulated upon activation, normal T-cell expressed and secreted
Inhibition of JAK2 but not JAK3 decreased DV production from HepG2 cells
We next examined whether the suppression of JAK2 or JAK3 might affect virus production in DV-infected hepatocytes. HepG2 cells were pretreated with either JAK2 or JAK3 inhibitor and then infected by DV, and the supernatants were collected for the measurement of virus titers. As shown in Figures 3a and b, treatment with JAK2 but not JAK3 inhibitor could significantly reduce the level of viruses released from DV-infected HepG2 cells and the effect was not due to cytotoxicity of the drug.
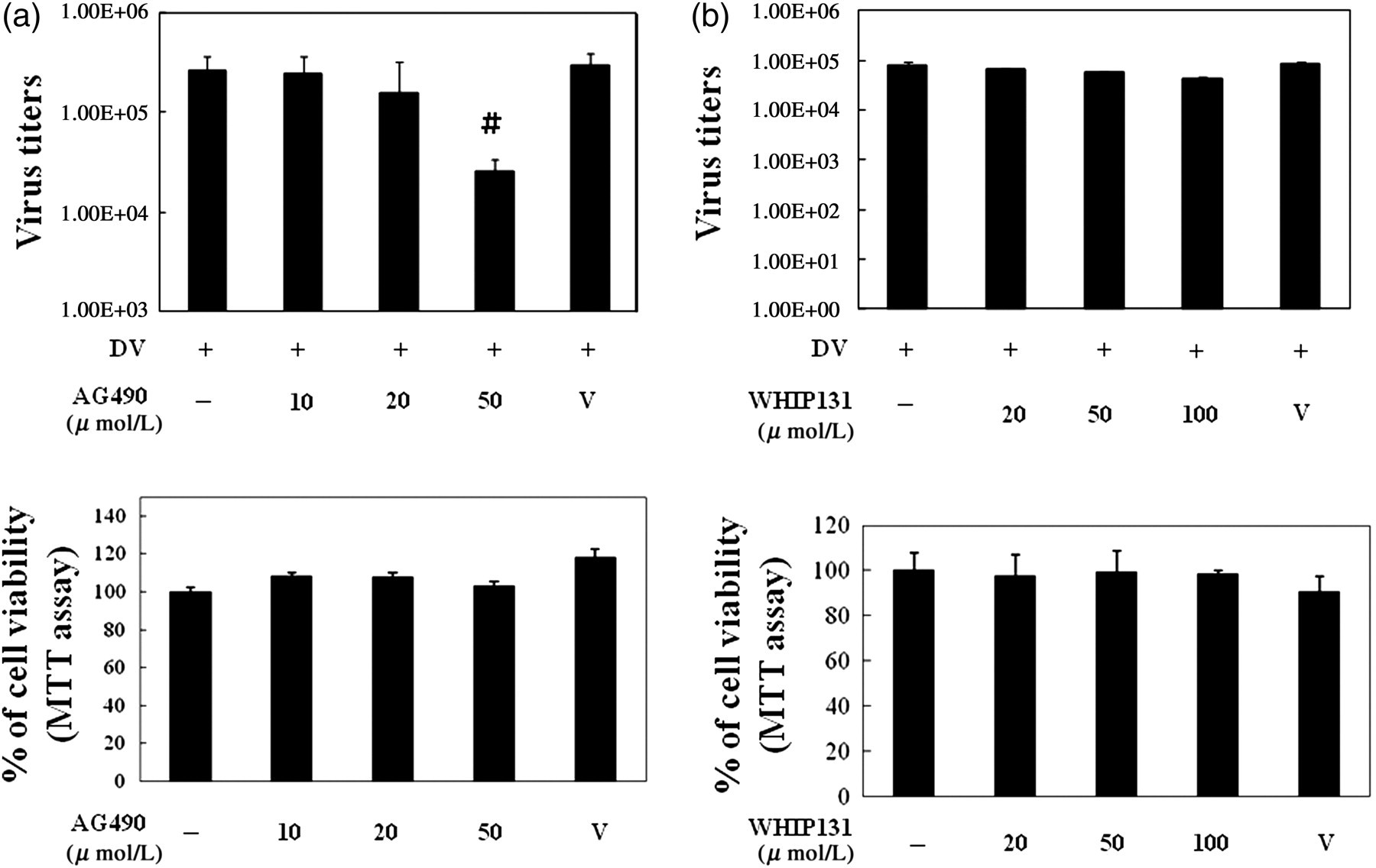
Inhibition of JAK2 (a) but not JAK3 (b) suppressed virus production. HepG2 cells were treated as described in Figure 1 and the supernatants were collected for the measurement of virus titers by plaque assays. To determine potential cytotoxic effects, HepG2 cells at 3 × 104/mL in 100 μL volume were treated with various concentrations of JAK inhibitors for 48 h. The cells and culture supernatants were collected for the evaluation of cell viability with MTT (the lower panels of both Figures 3a and b) as described in Materials and methods. The representative data out of at least three independent experiments are shown. # P < 0.05 compared with the DV-stimulated in the absence of the JAK inhibitor. JAK, Janus kinase; DV, dengue virus; MTT, 3-[4,-dimethylthiazol-2-y]-2,5-diphenyl-tetrazolium bromide
Suppression of JAK2 or JAK3 inhibited DV-mediated cell migration
The production of chemokines is highly related to the process of cell migration that may exaggerate the whole immune responses. We recently observed that dendritic cell infection induces expression of CCR7 on dendritic cells that facilitates their migration toward the chemoattractant MIP-3β. 28 We examined the effects of blocking JAK2 or JAK3 in DV infection-mediated cell migration. By chemotaxis assays, the results demonstrated that the supernatant collected from DV-infected HepG2 cells treated with JAK2 or JAK3 inhibitors attracted less U937 cells migrating from the upper chamber to the lower chamber (Figures 4a and b).
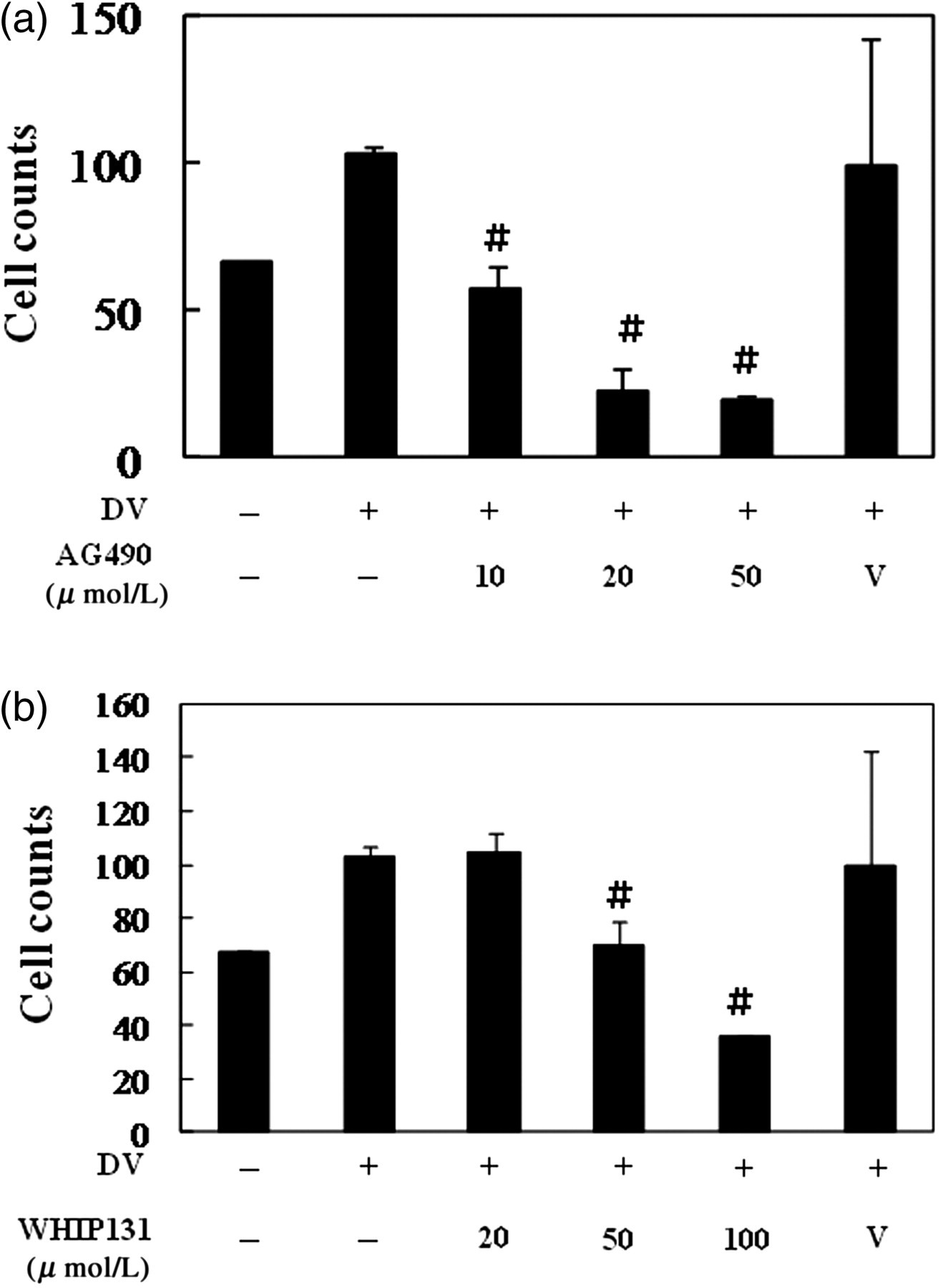
Inhibition of JAK2 (a) or JAK3 (b) suppressed DV infection-mediated cell migration. HepG2 cells were treated as described in Figure 1 and the supernatants were collected. Chemotaxis assays were performed as described in Materials and methods to determine the chemoattractive effects of supernatants collected from different conditions. # P < 0.05 compared with the DV-stimulated in the absence of the JAK inhibitor. JAK, Janus kinase; DV, dengue virus
Suppression of STAT3 inhibited DV-induced NF-κB and AP-1 activation
After confirming the significance of JAK2 and JAK3 in DV-stimulated HepG2 cells, the role of STAT3 was straight-forwardly investigated by knocking down STAT3 expression with shRNA approach. Introduction of shSTAT3 RNA into HepG2 cells successfully reduced the expression of total STAT3 and the tyrosine-phosphorylated STAT3 (Figure 5a, upper panel) and STAT3 DNA-binding activity (Figure 5a, lower panel) induced after DV infection. In contrast, the introduction of GFP shRNA had no effect. Knocking down STAT3 also suppressed DV-induced NF-κB and AP-1 DNA-binding activities but had no effect on Oct-1 DNA-binding activity (Figure 5b). These results further suggest that STAT3 may play upstream in the process of DV-induced activation of NF-κB and AP-1.

Reduction of STAT3 by shRNA interference blocked DV-stimulated STAT3, NF-κB and AP-1 activation. HepG2 cells infected by lentivirus carrying STAT3-specific shRNA (sh-STAT3) or GFP shRNA (sh-GFP) were infected by mock or DV and the expression of total STAT3, phosphorylated STAT3 and β-actin was determined by Western blot and the DNA-binding activity of STAT3 was measured by EMSA (a). In (b), similarly, the DNA-binding activities of NF-κB, AP-1 and Oct-1 as in the condition described in (a) were measured by EMSA (b). Wt., wide type; Mt., mutant; STAT, signal transducer and activator of transcription; p-STAT3, tyrosine-phosphorylated STAT3; t-STAT3, total STAT3; shRNA, short hairpin RNA; DV, dengue virus; NF-κB, nuclear factor-kappaB; GFP, green fluorescence protein; EMSA, electrophoretic mobility shift assay
Knocking down STAT3 suppressed DV-induced chemokine production and cell migration
The role of STAT3 in DV-induced chemokine production from HepG2 cells was investigated. The results in Figure 6a showed that decrease of STAT3 expression inhibited DV-induced production of several chemokines, including IL-8, RANTES and MIP-1α. However, decrease of STAT3 did not affect the production of MIF from DV-infected HepG2 cells. These results might indicate certain specificity of STAT3 in regulating the production of chemokines from DV-infected HepG2 cells. By chemotaxis assays, the results demonstrated that the supernatant collected from DV-infected HepG2 cells bearing the reduced level of STAT3 attracted less cells migrating from the upper chamber to the lower chamber (Figure 6b). Interestingly, reducing STAT3 had no effect on the production of viruses from DV-infected HepG2 cells (Figure 6c).
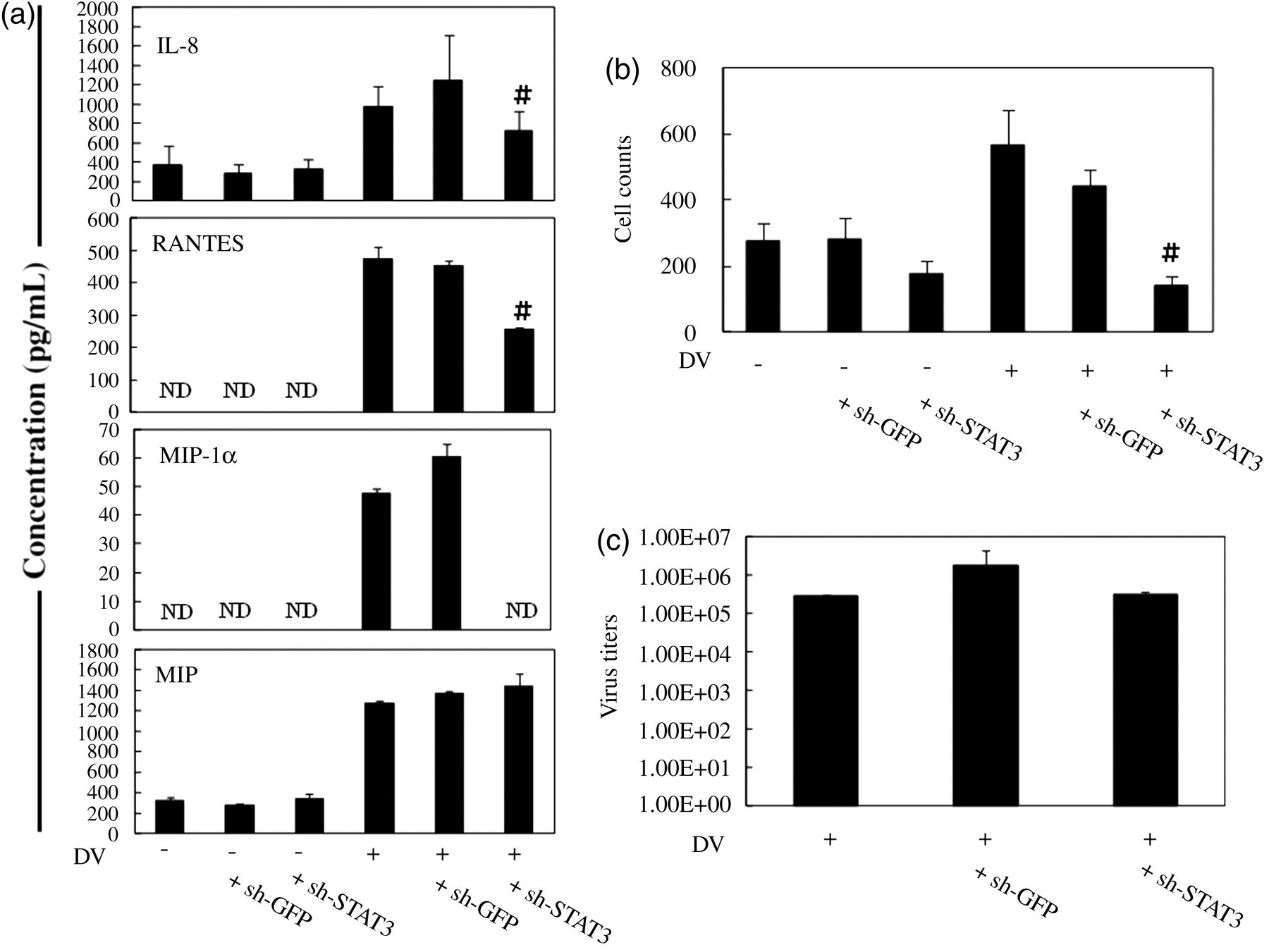
Knock down of STAT3 suppressed DV-induced chemokine production and cell migration but not virus production. HepG2 cells infected by lentivirus carrying STAT3-specific shRNA (sh-STAT3) or GFP shRNA (sh-GFP) were infected by mock or DV and the supernatants were collected for the determination of chemokine concentrations (a). In (b), chemotaxis assays were performed as described in Materials and Methods to determine the chemoattractive effects of supernatants collected from different conditions. In (c), virus production was measured by plaque assays. # P < 0.05 compared with the DV-stimulated. STAT, signal transducer and activator of transcription; DV, dengue virus; shRNA, short hairpin RNA; GFP, green fluorescence protein
Discussion
In facing patients with DV infection, the preservation of hepatic function appears to be critical and has been an important topic for investigation. In this study, we examined the role of STAT3 in chemokine production from DV-infected hepatocytes. We showed that DV infection could activate STAT3 in HepG2 cells and the effect could be decreased by either JAK2 or JAK3 inhibitor. In addition, inhibition of JAK2 or JAK3 suppressed DV-induced IL-8 and RANTES production and cell migration. Importantly, knocking down STAT3 successfully inhibited DV-induced NF-κB and AP-1 DNA-binding activities, two major families of transcription factors that regulate a wide spectrum of immune responses. Decrease of STAT3 effectively reduced chemokine production from DV-infected hepatocytes and inhibited U937 cell migration toward the culture supernatants from DV-infected hepatocytes compared with mock-infected cells.
There are several examples suggesting a connection between STAT3 activation and chemokine production. For example, in arterial injury, the release of tumor necrosis factor-alpha (TNF-α) can activate the transcription and production of RANTES in a STAT3-dependent manner in vascular smooth muscle cells. 30 Applying a novel STAT3 blockade agent, WP1066, demonstrates that this treatment reduces the production of RANTES from melanoma cells. 31 In the example of IL-8, the results indicate that STAT3 activation can up-regulate IL-8 production through transcriptional regulation in melanoma cells. 32 In addition, the activation of STAT3 with oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine can regulate transcription of IL-8 in human endothelial cells. 33 Using genetic manipulation, expressing a dominant-negative isoform of STAT3 reduces Src-mediated expression of IL-8 in pancreatic adenocarcinoma cells. 34 In contrast, the increase of IL-8 is detected in cells expressing the activated isoform of STAT3. Consistent with these reports, by interfering STAT3 expression, the DV-induced production of several chemokines, including RANTES and IL-8, was reduced.
Activation of NF-κB/AP-1 is a hallmark of many viral infections. 35,36 Cytokines stimulated by NF-κB/AP-1, such as TNF-α and IL-1β, are potent NF-κB inducers, hence establishing a positive auto-regulatory loop that can amplify the inflammatory response and lead to chronic infection. It is clear that viruses can directly activate NF-κB/AP-1 and utilize it in different ways for their own advantages. 35,37 Both NF-κB and AP-1 have also been shown to be critical in chemokine production triggered by a variety of stimuli. 38 Furthermore, interactions between STAT3 and NF-κB in regulating chemokine production have been observed previously by different groups. Unphosphorylated STAT3 can compete more effectively with IκB for unphosphorylated NF-κB and together form a new transcription factor complex that through binding to the κB element of the RANTES promoter induces RANTES expression. 39 An additive effect by both STAT3 and p65 in regulating RANTES expression is also demonstrated. 39 Evidence also suggests that IL-1 stimulation results in a complex formation comprising of STAT3 and p65 on a κB element of the human IL-8 promoter. 40 The synergistic cooperation among STAT3, NF-κB and C/EBPβ effectively enhances transcription of the C-reactive protein gene. 41 In this regard, that knocking down STAT3 could suppress DV-induced NF-κB and AP-1 DNA-binding activities, suggests wider application of blocking STAT3 in controlling the overwhelming inflammatory responses. In addition, many observed effects from inhibiting STAT3 may also be counted by the inhibition of both NF-κB and AP-1 signaling events. Although the activation of both STAT3 and NF-κB by certain virus infections has been observed, how the inter-regulation of these transcription factors affects viral replication is currently not clear. 42,43
Efforts aiming to develop a vaccine against DV infection have not yet been successful. 44 Part of the reason is the complicated set of mechanisms in the immunopathogenesis of DV infection. 45,46 Furthermore, the presence of four DV serotypes has complicated vaccine design because incomplete protection against one serotype may influence the disease outcome once infection is established by a distinct serotype. 47 In addition to vaccine design efforts, there has been a growing interest in discovering drugs against DV infection. JAK/STAT activation is an active cellular process, and it is possible that some viruses like DV may derive certain replication advantages by activating JAK/STAT-dependent genes in infected cells. Given that JAK2 inhibition could reduce virus production, it might suggest that DV required the JAK2 pathway for replication in infected hepatocytes. However, there remain concerns because the suppressive effects on viral production were only observed when a high dosage (50 μmol/L) of JAK2 inhibitor was used. It is currently not clear why the inhibition of virus production could not be seen in suppression of JAK3 or knock down of STAT3. There is indeed evidence suggesting that inhibition of JAK2 activity through pharmacological inhibitor AG490 can suppress parvovirus B19 replication in human erythroid progenitor cells. 21 Because in addition to STAT3, JAK2 can activate several other STAT molecules and STAT3-independent downstream signaling molecules, any effects observed in blocking JAK2 may not necessarily be reflected in inhibition of STAT3. A similar explanation goes to the different results reported in suppressing either JAK2 or JAK3. Even more complicated is the presence of both canonical and non-canonical JAK–STAT signaling pathways and effects as reviewed recently. 48 Altogether, this study suggests that targeting the JAK2/3–STAT3 pathway may be one of the potential approaches in pharmacological attenuation of the inflammatory responses in DV infection.
Footnotes
ACKNOWLEDGEMENTS
This paper was supported by grants from the Institutional Collaboration Projects raised by both Chi Mei Medical Center and National Defense Medical Center (CMNDMC99; CMNDMC10008) as well as from the Institute of Preventive Medicine, National Defense Medical Center.